

SUMMARY
Diverse biological systems utilize fluctuations (‘noise’) in gene-expression to drive lineage-commitment decisions. However, once a commitment is made, noise becomes detrimental to reliable function, and the mechanisms enabling post-commitment noise suppression are unclear. Here, we find that architectural constraints on noise suppression are overcome to stabilize fate commitment. Using single-molecule and time-lapse imaging, we find that—after a noise-driven event—human immunodeficiency virus (HIV) strongly attenuates expression noise through a non-transcriptional negative-feedback circuit. Feedback is established through a serial cascade of post-transcriptional splicing, whereby proteins generated from spliced mRNAs auto-deplete their own precursor unspliced mRNAs. Strikingly, this auto-depletion circuitry minimizes noise to stabilize HIV’s commitment decision, and a noise-suppression molecule promotes stabilization. This feedback mechanism for noise suppression suggests a functional role for delayed splicing in other systems and may represent a generalizable architecture of diverse homeostatic signaling circuits.
Keywords: stochastic noise, pulse chase, fate selection, negative feedback, post-transcriptional splicing
In Brief
Noise helps drive fate decisions, and a mechanism rooted in alternative splicing allows cells to stop dithering and commit
INTRODUCTION
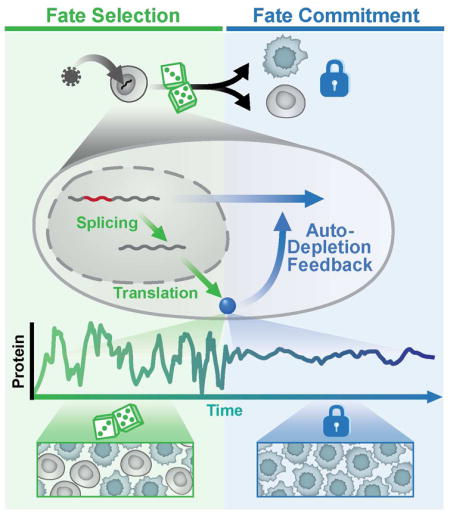
Stochastic fluctuations (noise) in gene expression—inescapable at the single-cell level—can be beneficial in certain contexts (Balazsi et al., 2011; Symmons and Raj, 2016) by enhancing signaling and fitness in variable environments (Acar et al., 2008; Kellogg and Tay, 2015), but are also detrimental to reliable cellular function, consistent with the observation that noise is minimized by evolutionary selection (Metzger et al., 2015). This tradeoff manifests during lineage-commitment decisions where transcriptional noise promotes selection between alternate cell fates (Chang et al., 2008; Weinberger et al., 2005), but, must subsequently be attenuated (Schmiedel et al., 2015) to enable reliable cell function and stability (Figure 1A). The mechanisms enabling mammalian cells to transition from noise-enhancing to noise-suppressing states remain unclear.
Figure 1. The Selection-Stability Tradeoff in Noise-Driven Fate Regulation and the HIV-Specific Case.

(A) Regulatory circuits that amplify fluctuations in gene-expression (noise; e.g., as measured by σ2/μ2) enable probabilistic fate selection but at the cost of reduced stability of the selected fate, whereas low-noise circuits are stable but have limited capacity for probabilistic fate selection (upper panel). The ideal scheme for enabling both probabilistic fate selection and stability (i.e., commitment) to the selected state is for a fate circuit to exhibit high noise during early times but subsequently attenuate noise once a fate has been selected (lower panel).
(B) HIV’s fate decision in CD4+ T cells: early after infection, infected cells commit to either latent or active infection with actively infected cells producing virus after a delay of ~1 day and then dying after ~2 days. Inset: the HIV Tat positive-feedback circuit regulates the active and latent states with Tat transactivation of the LTR promoter (HIV’s only promoter) being obligate for active replication and low Tat levels enabling viral latency. As long as positive feedback persists, it generates large fluctuations in gene expression, enabling entry to latency (green) but potentially destabilizing the active state (purple).
(C) Two models of HIV gene regulation during active replication. Left: The established model of co-transcriptional RNA alternative splicing; US, SS, and MS RNAs are independently generated and thus Rev-mediated export of US and SS does not deplete MS RNA. Right: The precursor auto-depletion model relies on a serial cascade of post-transcriptional splicing where US and SS RNAs are precursors for MS RNA and Rev-mediated export of US and SS precursors depletes MS RNA establishing negative feedback. Lower panels indicate noise and stability dynamics for each model.
An archetypal example of this tradeoff during a fate-commitment decision is found in the human immunodeficiency virus type 1 (HIV): the virus undergoes a noise-driven binary fate decision (Weinberger, 2015) (Figure 1B), leading to either an active replication program—producing viral progeny and cell death—or a long-lived quiescent state called proviral latency (Pai and Weinberger, 2017; Siliciano and Greene, 2011). While T-cell activation strongly biases latency establishment and reversal, the phenotype is inherently probabilistic, and cellular activation is insufficient to deterministically regulate latency (Chavez et al., 2015; Ho et al., 2013). This probabilistic nature of HIV latency appears largely driven by stochastic ‘bursts’ in the activity of HIV’s long terminal repeat (LTR) promoter (Singh et al., 2010) that are amplified by positive feedback from HIV’s Tat protein and sufficient to shift cells between active replication and latency (Razooky et al., 2015). If transcriptional fluctuations are subsequently attenuated after fate commitment is not known.
The virus may encode components of a putative negative-feedback loop (Felber et al., 1990), but models of co-transcriptional alternative splicing in HIV (Fong and Zhou, 2001; Taube and Peterlin, 2013) appear inconsistent with a proposed negative-feedback mechanism, which requires a precursor-product relationship and post-transcriptional splicing (Figure 1C). Specifically, HIV pre-mRNA is spliced into three classes of transcripts: a 9-kb unspliced (US) transcript, a 4-kb singly spliced (SS) class, and a 2-kb multiply spliced (MS) class. US and SS transcripts (encoding capsid, envelope, and genomic RNA) are initially retained in the nucleus of infected cells, but the MS transcripts, which encode the regulatory proteins Tat, Rev, and Nef, are efficiently exported. Nuclear export of the US and SS transcripts is facilitated by binding of Rev proteins to the Rev-Responsive Element (RRE) within the US and SS transcripts, which enables export through cellular chromosome region maintenance 1 (CRM1) (Malim et al., 1988; Ossareh-Nazari et al., 1997). The essential consequence of the co-transcriptional model is that a transcript’s splicing fate is sealed during transcriptional elongation, such that differentially spliced transcripts are uncoupled from each other and share no precursor-product relationship (Figure 1C, left). Thus, upon Rev-mediated export of US RNA, the co-transcriptional splicing model precludes any effect on MS RNA (i.e., no negative feedback). Conversely, if HIV splicing occurs post-transcriptionally (Figure 1C, right), it would establish a precursor-product relationship and enable negative feedback upon Rev-mediated export of US RNA (i.e., precursor auto-depletion). Such feedback could attenuate transcriptional noise and stabilize commitment to the active-replication state. Alternatively, direct Rev inhibition of splicing could generate negative feedback, but this does not appear to occur (Malim et al., 1989b).
Here, we probe what mechanisms may attenuate noise after HIV fate-selection in order to stabilize viral fate. We theoretically analyze noise properties of the proposed post-transcriptional precursor depletion feedback versus the common transcriptional auto-repression mechanism. Surprisingly, we find that feedback via precursor auto-depletion can surpass limits on noise suppression that constrain transcriptional auto-repression. Using single-molecule mRNA imaging and time-lapse imaging, we find that HIV encodes an RNA precursor-depletion circuit that is mediated by post-transcriptional splicing. This auto-depletion feedback circuit substantially attenuates gene-expression noise to stabilize viral fate commitment. We propose that the noise-suppression properties of precursor auto-depletion represent a functional role for the widespread occurrence of post-transcriptionally spliced introns (Boutz et al., 2015; Mauger et al., 2016; Ninomiya et al., 2011) and for the feedback motif’s presence in diverse alternative splicing pathways, such as nonsense-mediated decay (Lareau et al., 2007).
RESULTS
Architectural Constraints on Noise Suppression and the Associated Tradeoffs Can Be Overcome by RNA Precursor-Depletion Feedback
To understand the constraints on transitioning from a noise-enhancing to a noise-suppressing regime, we theoretically analyzed the common negative-feedback noise-attenuation mechanism, (Black, 1934). The effects of feedback on gene-expression noise are typically analyzed by comparing the standard two-state ‘random-telegraph’ model with and without feedback (Alon, 2007). Using this approach, we compared the two-state model to models with generic transcriptional auto-repression feedback or RNA precursor auto-depletion feedback (Figure 2A). In these models, ON and OFF represent the active and repressed promoter states, respectively, R represents mRNA, P represents protein, and all parameters are ‘lumped’ rate parameters representing promoter toggling between ON and OFF states (kon and koff), transcription (α), translation (β), mRNA degradation (δ), protein degradation (γ), and feedback strength (ε). Feedback by transcriptional auto-repression is modeled by the protein interacting with the promoter to reduce transitioning from OFF to ON (Alon, 2007), whereas feedback by precursor-depletion is modeled by introducing a pre state variable (representing a pre-mRNA that is spliced into R at rate θ) that can be removed upon a 2nd-order interaction with its protein product P.
Figure 2. RNA Precursor-Depletion Feedback Could Overcome Architectural Constraints on Noise Suppression, Mitigating the Selection-Stability Tradeoff. See also Figure S1 and Supplementary Methods S1.

(A) Schematics of simplified gene-circuit models used for comparing effects of negative feedback on noise suppression.
(B) Outputs of Gillespie simulations for each model. Mean ± one standard deviation (shaded) shown for each model. Transcriptional auto-repression and precursor auto-depletion are matched in feedback strength (i.e., loop transmission) such that both generate an ~2-fold reduction in mean expression compared to the two-state model and such that response-time (i.e., rise time) is approximately the same for both. Precursor-depletion feedback exhibits significantly less noise than transcriptional auto-repression for this parameter set. Inset: mean protein levels (μ) and noise magnitude (σ2/μ2) for each model.
(C) Analytical calculations of noise (Eqs. [1–2]) showing how precursor depletion (blue) surpasses the noise-suppression capabilities of transcriptional auto-repression (red). T is the same for both transcriptional auto-repression and precursor auto-depletion and T=0 for the two-state model with no feedback (black). Solid lines were calculated from Eqs. [1–2] in the form . The numerical simulation results from panel B are superimposed on the analytical results as colored circles and can be explained using Eq. [1–2] (for detailed explanation see Supplementary Methods S1).
Numerical solutions (Gillespie, 1977) of these models show that, as expected, transcriptional auto-repression reduces mean protein levels (μ) to half while avoiding the increases in noise (σ2/μ2) that Poisson scaling necessarily generates (Figure 2B and S1A). However, for the same parameters and feedback strength, precursor auto-depletion reduces noise substantially more for the same reduction in mean (σ2/μ2 = 0.06 for precursor auto-depletion vs. σ2/μ2 = 0.12 for transcriptional auto-repression). The improved noise attenuation achieved by precursor auto-depletion is particularly interesting, given the added noise source introduced by adding the extra pre-mRNA pre species. Analytical results below explain this result.
Moreover, in parameter regimes where auto-repression has the perverse effect of amplifying noise (Austin et al., 2006; Hornung and Barkai, 2008), precursor auto-depletion feedback avoids this noise amplification issue (Figure S1B). Importantly, the observed noise-suppression does not depend upon the specific molecular details of the model and the pre-mRNA species (pre) is dispensable—models that omit pre show somewhat improved noise attenuation (Figure S1C), consistent with this intermediate state acting as an additional noise source.
We next examined generality using an analytical approach. Previous analyses examining the idealized case where noise is dominated by mRNA birth-death events (i.e., the minimal-noise Poissonian expression limit (σ2 = μ)) had predicted that reducing the relative proteolysis rate by post-transcriptional feedback could efficiently control noise (Swain, 2004). However, such Poisson-regime analyses often break down when transcriptional bursting (i.e., promoter ON-OFF toggling) is the dominant noise source (Austin et al., 2006; Hornung and Barkai, 2008), and it was unclear if post-transcriptional feedback attenuates noise in the substantially noisier transcriptional-bursting regime. To analyze the transcriptional-bursting case (koff ≫ kon), which generates super-Poissonian noise, we built off analyses of transcriptional bursting (Dar et al., 2015; Simpson et al., 2003) that show that intrinsic σ2 = bRbPμ (where bR is the transcriptional burst size [α/koff] and bP is the translational efficiency [β/δ]). Using this relation, the mean and intrinsic noise can be calculated as:
| (Equation 1) |
| (Equation 2) |
where f is the transcriptional burst frequency (1/kon +1/koff)−1, γ is the protein degradation rate, and T is the modification due to net-feedback strength, which is the combination of positive and negative feedback (Simpson et al., 2003).
As detailed in Supplementary Methods S1, Eqs. 1–2 are derived assuming exponential protein decay, and Eq. 2 strictly holds only for small deviations from steady state when koff≫kon (outside of this regime Eq. 2 is corrected by the multiplier bpeff breff/brbp). Nevertheless, Eqs. 1–2 identify general architectural constraints on noise suppression for the two-state, auto-repression, and auto-depletion models. For example, one constraint exposed by Eqs. 1–2 is the perverse effect on noise by auto-repression: when transcriptional auto-repression reduces μ by decreasing f (the most common version of auto-repression), noise increases if T is not sufficiently strong. Moreover, in the linear feedback approximation (Simpson et al., 2003) when transcriptional auto-repression decreases f to reduce the mean, T becomes larger and causes noise to remain constant (Figure 2C, red arrow “i” and Figure S1A). However, if reducing the mean causes feedback to saturate (T → 0), noise increases (Figure 2C, red arrow “ii”, and Figure S1A). This stasis and amplification of noise upon induction of transcriptional auto-repression have been experimentally noted in both prokaryotic (Dublanche et al., 2006) and eukaryotic circuits (Nevozhay et al., 2009). In contrast, precursor auto-depletion acts on bP (and possibly bR) without perturbing f, and is thus predicted to break the inverse relation, attenuate noise substantially better for equivalent reductions in mean protein level, and avoid the perverse noise-amplification effect (Figure 2C, blue arrow “ii” and Figure S1A). Notably, auto-repression facilitated through modulating α can also change bR and achieve noise reductions approaching auto-depletion but only when koff ≫kon whereas auto-depletion reduces noise irrespective of koff and kon values (Figure S1D and Supplementary Methods S1). Importantly, the analysis also explains how precursor auto-depletion surpasses these constraints on noise attenuation despite the added noise source from the pre species: the added noise from pre is high frequency and effectively filtered out at the protein level (Supplementary Methods S1). Thus, the precursor auto-depletion motif effectively attenuates noise even in the presence of intermediate species that typically act as additional noise sources.
Intuitively, the enhanced noise minimization results because precursor auto-depletion acts on RNA, which can have a large number of molecules that can be reduced in an analog-like fashion (from x molecules to x – n molecules). In contrast, transcriptional auto-repression acts on a single-molecule, DNA, which can only toggle between fully active (ON) and repressed (OFF). Thus, transcriptional auto-repression inevitably affects promoter toggling (the major driver of noise), but precursor auto-depletion does not. This analysis is general and holds beyond two-state models (Supplementary Methods S1).
Post-Transcriptional Splicing Enables RNA Precursor-Depletion Feedback in HIV
To test if HIV exploits these noise-attenuation properties, we used single-molecule mRNA florescence in situ hybridization (smFISH) (Figure 3A and Table S1). First, to determine if splicing occurs post-transcriptionally, we performed a transcriptional pulse-chase experiment (Noble and Guthrie, 1996) at single-molecule resolution. HIV transcription was activated by tumor necrosis factor alpha (TNF) for 14 minutes, after which the promoter was shut off using the transcriptional elongation inhibitor actinomycin D (ActD) in the chase phase (Figure 3B). smFISH quantification of mRNAs per cell (2000 cells total, ~100 cells per time point) shows that during the 14-minute TNF pulse, US RNA substantially increases (whereas neither SS nor MS RNA increases); however, during the ActD chase, US RNA is depleted, whereas SS RNA peaks (increases then decreases), and MS RNA continually increases (Figure 3C). Since mRNA detection was limited to the nucleus and all mRNAs are detected via a probe at their 3′ end (~10 % of detected mRNAs lack the 3′ probe), neither XRN1-mediated decay (cytoplasmic 5′ to 3′ degradation) nor exosome-mediated mRNA decay (nuclear 3′ to 5′ degradation) readily account for these results. Instead, the results are most consistent with SS and MS RNA being spliced post-transcriptionally from an US RNA precursor, since both species increase after transcriptional elongation is halted.
Figure 3. Post-Transcriptional Splicing in HIV Enables RNA Precursor-Depletion Feedback. See also Figure S2 and Table S1.

(A) Schematic of smFISH probes used to quantify US, SS and MS mRNAs shown relative to the HIV genome with representative splice donor (SD) and splice acceptor (SA) sites and representative US, SS, and MS HIV genes.
(B) Representative smFISH images of transcriptional pulse-chase in HIV-infected Jurkat cells (cells are polyclonal for HIV-integration sites). TNF activation of the HIV promoter was chased 14 minutes later with the transcriptional elongation inhibitor Actinomycin D (ActD).
(C) Quantification of mRNA molecules during the TNF pulse (blue) and the ActD chase (red).
(D) smFISH spatial distributions of HIV mRNAs within the nucleus. Radial-distance distributions for each RNA species from the TC in that cell shows that US RNA is maximal at TC and decays with distance from TC while MS and SS RNAs are absent at TC and peak 1.5–3 μm from TC, respectively, corresponding to post-transcriptional splicing.
(E) Nuclear RNA kinetics after TNF activation as measured by smFISH. MS RNA exhibits a negative-feedback overshoot while US RNA continually increases. (C–E) Each data point represents the mean mRNA count for ~100 cells; error bars represent SEM.
We also used a spatial version of smFISH (Waks et al., 2011) that quantifies the radial distance of individual mRNA molecules from the transcriptional center. If splicing were co-transcriptional and the results from the transcriptional pulse-chase assay were due to nuclear 5′ to 3′ mRNA degradation (e.g., by XRN2), SS and MS mRNAs would be expected to be maximal at the transcriptional center (TC). However, the data are inconsistent with these hypotheses and show that SS and MS mRNAs are maximal ~1.5 and 3 μm from the TC, respectively, and only US mRNAs are observed at the TC (Figure 3D and S2A–B).
To further test whether transcription is dispensable for splicing, we also developed an assay termed Splicing After Transfection of Unspliced pre-mRNA into the Nucleus (SATURN) in which synthetic, in vitro transcribed full-length HIV mRNA was transfected into cellular nuclei. This assay relies upon a reporter expressed only from a multiply-spliced transcript and tests whether it is possible for splicing to occur without in situ transcription within cells. Over half of transfected T cells exhibited MS reporter expression 4 hours after transfection of full-length unspliced HIV mRNA (Figure S2D–F), demonstrating that HIV mRNA can be spliced post-transcriptionally within cells.
Finally, as would be predicted for negative feedback (Figure S3A), the temporal kinetics of cytoplasmic MS RNA exhibit a stereotypical ‘overshoot’ signature: when US RNA begins to increase substantially in the cytoplasm at ~90 minutes after TNF activation, MS RNA begins to reciprocally decrease (Figure 3E and S2C), consistent with Rev-mediated export of an US RNA precursor of MS RNA.
RNA Precursor-Depletion Feedback Occurs in Individual Cells and Manifests in Protein Dynamics
To verify that the observed smFISH overshoot was not a population-level phenomenon brought on by differential responses in different cellular subpopulations (i.e., to verify that individual cells exhibit negative-feedback dynamics), we used live-cell time-lapse imaging. This approach had the added benefit of allowing quantification of protein expression kinetics, which drive HIV’s fate decision.
We stably transduced cells with inducible HIV-reporter constructs for MS and US gene expression (Figure 4A). The MS reporter is based on a single-round (Δenv) HIV (Jordan et al., 2003) modified to encode a short-lived green fluorescent protein with a 2-hour half-life (d2GFP) within the HIV nef reading frame. Each transduced cell contains a single latent integration of HIV d2GFP that is inducible by TNF, and the Δenv mutation ensures no subsequent transmission of the virus between cells after treatment with TNF. Longitudinal single-cell imaging over 25 hours after induction shows that virtually all cells (~86%) show the characteristic ‘overshoot’ signature of negative feedback: GFP expression increases, reaching a maximum at 5 to 8 hours, followed by a decay in GFP intensity (Figure 4B, S3B and Movie S1). This maximum in GFP expression at 5–8 hours is predicted by the mRNA maximum at ~60 minutes, given the rates of GFP proteolysis and fluorophore maturation (Figure S3A). To directly quantify feedback strength, we performed temporal auto-correlation function (ACF) analysis of the single-cell trajectories—a sensitive reporter of feedback strength (Austin et al., 2006; Weinberger et al., 2008)—that confirmed negative feedback during the overshoot (Figure S3C) and showed that inhibiting HIV Rev-mediated nuclear export, via the CRM1 inhibitor Leptomycin B (Fukuda et al., 1997) or a Rev mutant that acts as a competitive inhibitor of nuclear export (Malim et al., 1989a), diminished the negative-feedback (Figure S3C). Similar overshoot trajectories were observed after direct infection of human donor-derived primary CD4+ T cells (Figure S3D) and human donor-derived primary monocyte-derived macrophage cells (Figure S3E). Thus, precursor-depletion feedback appears to operate both during infection of primary cells and during latent reactivation.
Figure 4. Time-Lapse Imaging Verifies that Precursor-Depletion Feedback Occurs in Individual Cells and Transmits to Protein Expression Kinetics. See also Figure S3 and S4, and Movie S1.

(A) Schematics of reporter constructs. The MS reporter encodes a destabilized GFP (d2GFP) in nef reading frame and the US reporter encodes mCherry whose expression is dependent on Rev-mediated nuclear export.
(B) Time-lapse imaging of Jurkat cells stably transduced with the MS reporter (cells are monoclonal for the HIV-integration site) after transcriptional activation with TNF. Mean and standard error for ~200 cells shown.
(C) Time-lapse imaging data of Jurkat cells stably transduced with both the MS and US reporters shows direct coupling of negative feedback to Rev-mediated export of unspliced RNA export. GFP and mCherry mean trajectories from 100 representative cells.
(D) Quantification of slopes of GFP and mCherry trajectories: (i) during the early phase after TNF stimulation (0–4 h), GFP production rate is positive and increasing while mCherry production is near zero, consistent with Rev needing to accumulate and multimerize for its function (Malim and Cullen, 1991); (ii) as mCherry production increases (4–12 h), GFP production begins to taper off, consistent with depletion of MS RNA; (iii) in the late phase (after 12 h), the GFP production rate is negative only when the mCherry production rate is high.
To verify that the overshoot dynamics were not due to transcriptional silencing or nuclear factor kappa B (NF-κB)-induced oscillatory dynamics (Hoffmann et al., 2002), we performed three tests. First, we tested a pair of HIV-reporter constructs lacking Rev; both constructs generate monotonic increases in GFP, rather than overshoots, in cells activated by the NF-κB pathway (Figure S4A–C). Second, we tested whether re-stimulation of cells with TNF, during the overshoot, or with the histone deacetylase inhibitor trichostatin A, during the overshoot, would re-stimulate the HIV LTR and abrogate the overshoot, as transcriptional silencing predicts (Pearson et al., 2008). No abrogation of the overshoot occurred upon re-stimulation (Figure S4D). Third, to confirm that the precursor-product relationship is responsible for the overshoot, we tested whether strengthening Rev export would enhance auto-depletion. Simulations predicted that increasing the nuclear-export rate of US RNA would enhance negative feedback, accelerating the response time and intensifying the overshoot (Figure S4E), and based on previous results that additional nuclear-export elements can enhance HIV-1 export (Wodrich et al., 2000), we encoded a second distal RRE in gag and found that this construct exhibited faster 50% response time during the overshoot decay (Figure S4F).
To determine if the overshoot was directly coupled to export of unspliced RNA precursors, we generated a two-color reporter system (Figure 4A) requiring Rev nuclear export (Wu et al., 2007a). Analysis of individual cells (Figure 4C) shows that the GFP overshoot occurs concurrently with increasing expression from the US mCherry reporter (the reduced GFP overshoot relative to the one-color system is consistent with Rev having to export an extra species of US mCherry RNA). Plotting the relative slopes (i.e., net-expression rates) verifies that expression from US transcripts precisely coincides with the decay in expression from MS transcripts (Figure 4D).
Modeling Predicts that HIV Precursor-Depletion Feedback Requires Inefficient Post-Transcriptional Splicing
Next we developed and tested a series of ordinary differential equation (ODE) models of HIV gene-regulation (Figure 5A), screened out models that failed to fit the time-lapse imaging data, and performed parameter sensitivity analysis. As expected from the smFISH analysis, nonlinear least squares fitting showed that post-transcriptional splicing is sufficient to fit the single-cell time-lapse data (Figure 5B, S5A and Table S2). Parameter sensitivity analysis indicated that Hill coefficient (h) of Rev/RRE-dependent RNA nuclear export (i.e., self-cooperativity of Rev) was a critical parameter, with h ≥ 3 required (Figure S5B), in agreement with biochemical studies (Cook et al., 1991; Daly et al., 1993; Daugherty et al., 2010; Mann et al., 1994; Pond et al., 2009). The model was further verified by experimentally testing a counter-intuitive model prediction that over-expression of Rev (an obligate nuclear-export factor) would reduce the level of late transcripts and their gene products (Figure S5C–D). This prediction arises because increased Rev exports late gene products too quickly, precluding sufficient accumulation of Tat for positive feedback and accordingly leads to decreased levels of both MS and US gene products (e.g., p24 and Nef). As the model predicted, Rev over-expression—from an expression plasmid (Malim et al., 1988) in cells stably expressing the HIV provirus (Jordan et al., 2003)—led to decreased levels of both GFP and intracellular p24 at 24 hours, as measured in individual cells by flow cytometry (Figure S5E–F).
Figure 5. Kinetic Modeling Predicts that Precursor-Depletion Feedback Requires Inefficient HIV Splicing. See also Figure S5, Table S2 and Supplementary Methods S1.

(A) ODE models of HIV co-transcriptional and post-transcriptional splicing.
(B) Nonlinear least squares fitting of ODE models to single-cell trajectory data (Supplementary Methods1 and Table S2). The post-transcriptional splicing model can recapitulate the experimental data whereas the co-transcriptional splicing model cannot.
(C) ODE model predictions for enhancement of HIV splicing. As the splicing rate increases, the overshoot from precursor auto-depletion feedback diminishes.
The most physiologically relevant insight from the model was that negative–feedback strength depended on the relative rates of protein-mediated depletion and splicing. This insight can be intuited from the minimal precursor auto-depletion model (Figure 2A) by noticing that negative feedback is only active when the depletion rate is substantially greater than the splicing rate. Numerical simulations predicted that enhancing the efficiency of HIV splicing would diminish precursor auto-depletion, which could be observed by loss of the negative-feedback overshoot signature (Figure 5C).
Enhancing Splicing Abrogates Precursor-Depletion Feedback and Dramatically Amplifies HIV-Expression Noise Despite Increasing the Mean
To test the prediction that enhancing the efficiency of HIV splicing would diminish precursor auto-depletion, we focused on the HIV splice acceptor SA7, which is used by all three MS transcripts (Tat, Rev, and Nef) but lacks the canonical sequence elements required for efficient splicing (i.e., SA7 exhibits neither a consensus branch-point A nor a poly-pyrimidine tract) (Dyhr-Mikkelsen and Kjems, 1995). A series of SA7 HIV mutants was generated to enhance the poly-pyrimidine tract and create a consensus branch-point A sequence (Figure 6A and S6A).
Figure 6. Inefficient Splicing Enables HIV’s Precursor-Depletion Feedback Circuit to Surpass Apparent Noise-Suppression Constraints. See also Figure S6 and Table S3.

(A) Schematic of HIV A7 splice-acceptor site sequence together with sequence mutations cloned to correct A7 to a more consensus splice acceptor and enhance splicing.
(B) Time-lapse microscopy of wild-type HIV d2G (n=592) and three SA7 mutants (A7–4 n=665, A7–5 n=774, A7–7 n=788) with enhanced splicing (populations are polyclonal for viral integration sites). Insets: mean trajectories normalized to max (to examine overshoot).
(C) Decoupling between MS and US expression in the SA7 mutants. The two-color reporter assay shows that, in the SA7 mutants, US expression rate (i.e., mCherry slope) increases without the wild-type-like decrease in MS expression rate (i.e., d2GFP slope) and MS expression rate does not enter the negative (i.e., negative feedback) regime. Slopes are normalized to max for comparison purposes. Insets: mean intensity trajectories for GFP and mCherry.
(D) Dynamic CV2 versus mean for data in panel B (0–12 h) showing that wild-type HIV (left y axis) decreases in mean and CV2 coincident with negative feedback being initiated whereas mutants (right y axis) exhibit monotonic increase in CV2 and mean.
(E) Steady-state CV2 versus mean for data in panel B (12–18 h) plus two additional repeats (Errors bars represent standard deviation). Mutants exhibit a large increase in CV2 compared to wild-type HIV despite increased mean expression. The dotted line is the Poisson-model prediction.
These optimized SA7-mutant viruses exhibit enhanced RNA splicing by qRT-PCR and flow cytometry (Figure S6B and Table S3) and increased GFP expression, but lack the overshoot kinetics characteristic of negative feedback (Figure 6B). The optimized SA7 mutants exhibit substantial decoupling of MS and US expression kinetics by two-color analysis, consistent with negative feedback being diminished in the mutants (Figure 6C and Figure S6C).
Strikingly, the precursor auto-depletion circuit in the wild-type virus reduces mean expression and noise, whereas the optimized SA7 mutants exhibit a monotonic increase in noise (Figure 6D), and despite final expression levels being increased for GFP, the optimized SA7 mutants show substantially amplified noise (Figure 6E). This noise increase, although contrary to simple Poissonian models—in which noise decreases as the mean-expression level increases—is consistent with diminution of negative feedback, which suppresses both noise and mean-expression level. The data indicate that inefficient splicing is an important molecular determinant of precursor auto-depletion and its resultant noise minimization.
Noise Suppression by Precursor-Depletion Feedback Stabilizes HIV’s Active State
To test how noise attenuation from auto-depletion feedback might affect fate commitment, we computationally analyzed the rate of transitioning between alternate states by developing a stochastic version of the HIV auto-depletion model (Supplementary Methods S1). Monte-Carlo simulations showed that noise suppression by auto-depletion circuitry stabilizes the active transcriptional state, whereas removing auto-depletion feedback increases noise levels, resulting in increased transitioning from the active to a transcriptionally silent state (Figure 7A) and accumulation of trajectories in the silent/off state (Figure 7B). Notably, when auto-depletion feedback is removed, decay into the off state is accelerated despite a 3-fold increase in expression level (including expression of the Tat transactivator from multiply spliced transcripts). This result is the opposite of the deterministic case (Figure S7A–B) where increased expression levels would buffer against and decelerate decay into a silenced/off state. Thus, the model predicts that fluctuations, rather than mean levels, are the dominant factor driving decay to the silenced state.
Figure 7. Noise Suppression by Precursor-Depletion Feedback Stabilizes the HIV Active State. See also Figure S7 and Supplementary Methods S1.

(A) Representative Gillespie simulations of the HIV precursor auto-depletion model (blue trajectories) showing that noise in Tat expression is attenuated and stochastic ON–OFF switching is minimized. Abrogating precursor auto-depletion (red trajectories) results in increased Tat expression noise and stochastic ON–OFF switching despite an ~3-fold increase in mean-expression level of the ON state.
(B) Histograms of 1000 simulations of the HIV precursor auto-depletion model (blue) and abrogated precursor auto-depletion mutant (red) at end of simulation run (i.e., t = 300 h). In agreement with the flow cytometry data, simulations of the mutant exhibit substantially more trajectories in the GFP OFF state compared to wild type.
(C) Flow cytometry analysis of active-state stability following a pulse of TNF reactivation for wild-type HIV d2G and SA7 mutants (cells are polyclonal for the viral integration sites); cells were removed from TNF induction at time 0 (n>15000 per sample).
(D) Flow cytometry analysis of active-state stability following a pulse of TNF reactivation (TNF removed at time 0) in the presence (black) and absence (red) of an LTR transcriptional noise-suppressor molecule (Dar et al., 2014) (n>15000 per sample). As predicted, due to the intact negative feedback in the wild type, the noise suppressor minimally affected stability of wild-type HIV d2G (Figure S7C). Insets: Representative flow cytometry histograms (48 h time point).
We next compared stability of the active-replication state in wild-type HIV and the SA7 mutants by flow cytometry (Figure 7C). The SA7-enhanced splicing mutants exhibit dramatically reduced stability of the active-replication state showing accelerated decay to a transcriptionally latent state. While the distribution of A7-7 appears slightly more similar to wild type and exhibits a third high-expression peak, this mutant’s decay into the off/latent state remains faster than A7-4 and A7-5 at 9 hours and faster than wild type at 20 and 58 hours. Experiments below (Figure 7D, insets) show this more clearly. As mentioned above, destabilization of the active state cannot be explained by the deterministic model (Figure S7A–B) since the deterministic model predicts that SA7 mutants, which have increased Tat (Figure S6B), require more time to turn off Tat and establish a latent state. Moreover, establishment of latency appears to occur as an abrupt ‘exchange of mass’ process between bimodal peaks—a hallmark of stochastic transitioning (Weinberger et al., 2005)—as opposed to gradual shifting of a single unimodal peak.
Based on these results, we hypothesized that attenuating transcriptional noise, through orthogonal means, might reduce decay into the transcriptionally latent state in the mutants. To test this, we used a published small-molecule transcriptional noise-suppressor of the HIV LTR, manidipine (Dar et al., 2014); this molecule attenuates transcriptional fluctuations without significantly altering mean-expression levels of the HIV LTR. Flow cytometry analysis of active-state stability after a pulse of TNF reactivation shows that, as predicted, chemical noise suppression slows the dynamics of transitioning to the transcriptionally latent state in the mutants, and partially stabilizes the active transcriptional state (Figure 7D). As predicted, due to the intact negative feedback in the wild type, the noise suppressor molecule has minimal effects on stability of wild-type HIV (Figure S7C)—intact auto-depletion feedback dominates to suppress noise in the wild type and supersedes the effect from the noise-suppressor molecule.
DISCUSSION
HIV’s precursor auto-depletion circuit appears to surpass the noise attenuation properties of transcriptional auto-repression circuits in herpesviruses (Teng et al., 2012) or synthetic transcriptional auto-repression circuits (Dublanche et al., 2006). In HIV, precursor auto-depletion circuitry requires post-transcriptional splicing; co-transcriptional splicing could not generate Rev-mediated negative feedback as it precludes a precursor-product relationship. smFISH and time-lapse imaging data support post-transcriptional splicing and negative feedback, respectively. Notably, exotic mRNA-degradation mechanisms would be needed to explain the smFISH data in Figure 2, particularly given the polyclonal nature of the HIV-1 integrations used for smFISH (each cell carries a single integration of HIV-1 at a unique pseudo-random position in the human genome such that each TC is at a different 3D position in the nucleus of each cell).
Interestingly, Eqs. 1–2 show that auto-repression through α (transcriptional elongation), can, in principle, approach auto-depletion’s noise attenuation, though only in the transcriptional bursting parameter regime (koff ≫ kon). One speculative mechanism to achieve such α feedback regulation could be through modulation of RNAPII ‘backtracking’ (Churchman and Weissman, 2011) but such feedback regulation has not yet been reported. Further, elongation and splicing rates are linked for many genes such that artificially changing the elongation rate alters alternative splicing patterns (Kornblihtt, 2005), so modulating α could alter splicing rates to either enforce or cancel auto-depletion.
Implications for Evolutionary Selection
The coupling of noise-amplifying (Tat positive-feedback) circuitry, followed downstream by strongly noise-suppressing (Rev auto-depletion) circuitry, appears to be expensive regulatory architecture but has the benefit of optimizing fitness for probabilistic bet-hedging strategies (Rouzine et al., 2015). Interestingly, the auto-depletion motif appears to be the equivalent of “parametric control” in electrical signal processing—in which additional parameters are embedded in a circuit to provide more layers of regulation—a concept also exploited by kinetic proofreading in biochemical networks (Hopfield, 1974).
For HIV, it remains unclear whether this circuitry evolved specifically as an adaptation to suppress noise or is tied to the virus’s need to retain full-length unspliced genomic RNA for packaging. However, inefficient splicing at SA7 would not be sufficient to retain full-length genomic RNA—all HIV splice sites would need to be less efficient than export to satisfy the feedback criterion. Either way, the elements of this circuitry appear conserved (Figure S7D–E) in lentiviruses and its noise buffering effects appear required to stabilize active replication. Interestingly, enhanced splicing in the A7-4 mutant did not substantially reduce the levels of intracellular p24 capsid protein (Figure S6B), or extracellular genomic RNA packaged in virions (Figure S7F)—whereas it abrogated precursor auto-depletion and destabilized HIV’s active state—arguing that genomic RNA packaging and auto-depletion feedback can be uncoupled.
How Widespread Might Auto-Depletion Feedback Be?
The noise-buffering effects of auto-depletion circuitry may offer a new regulatory scheme adding to comprehensive analysis of dynamic RNA-regulatory strategies (Rabani et al., 2014). While auto-depletion feedback requires that RNA splicing be post-transcriptional and relatively inefficient relative to RNA synthesis, HIV’s post-transcriptional splicing rates—15–30 minutes after transcriptional activation (Figure S2)—appear to fall within reported regimes. Specifically, while slightly slower than some genes, whose splicing occurs within ~8 minutes of 3′ exon synthesis (Singh and Padgett, 2009), HIV-1 is not dissimilar to splicing rates at other NFkB-induced genes (Pandya-Jones et al., 2013), and close to the genome-wide median of ~14 minutes. Previous comparisons of transcriptional elongation rates and splicing times (Coulon et al., 2014) report 1.35–2.75 kb/min and 4–14.5 minutes, respectively, so HIV’s transcriptional elongation rate of ~1.8 kb/min (Boireau et al., 2007) and splicing time of ~15 minutes (Figure 2C and S2B) fall within this range.
The noise-buffering effects of auto-depletion circuitry also suggest a functional basis for similar regulatory motifs in other systems that ‘license’ splicing of precursors (Li et al., 2016). For example, a central mechanism for the activation of alternative splicing is via serine- and arginine-rich (SR) proteins that regulate their homeostasis by splicing their pre-mRNAs to variants that are shunted to the non-sense mediated decay (NMD) pathway (Lareau et al., 2007; Ninomiya et al., 2011). Thus, generalized alternative splicing appears to be regulated by auto-depletion of SR RNAs and the auto-depletion motif’s noise-buffering properties suggest a functional reason. Moreover, the ability of neurons to post-transcriptionally regulate splicing and nuclear export (Mauger et al., 2016), as well as the widespread occurrence of post-transcriptionally spliced introns (Boutz et al., 2015), and the delayed splicing of introns in multiple systems (Hao and Baltimore, 2013; Pandya-Jones et al., 2013; Rabani et al., 2014) indicates that the criteria for auto-depletion circuitry may exist in diverse cellular subsystems.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Leor S. Weinberger (leor.weinberger@gladstone.ucsf.edu). The dHIV-d2G construct (Figure S4F) is shared under an MTA, to be discussed in good faith with the recipient.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cloning
Lentiviral vectors HIV-1 Δenv-d2GFP A7-4, A7-5 and A7-7 were created by amplifying the backbone of WT HIV-1 Δenv-d2GFP (Jordan et al., 2003) using primers and standard PCR techniques. G-blocks containing splice acceptor mutations A7-4, A7-5, and A7-7 were inserted into the backbone using the Gibson Annealing kit (New England Biolabs.) All vectors were verified by sequencing. G-Block sequences and primers used to amplify the viral vector backbone used for Gibson cloning of the mutants are listed in Table S3.
Lentiviral vector dHIV-BlaM was created by inserting the PCR amplified MluI/XhoI fragment of BlaM (Cavrois et al., 2002; Landowski et al., 2014; Zlokarnik et al., 1998) into the MluI/XhoI restriction–digested backbone of dHIV-d2G. The dHIV-BlaM MSD mutant (dHIV-dMSD-BlaM) was created using PCR site-directed mutagenesis. To obtain the DNA templates for in vitro transcription, XmaI site and T7 promoter sequence were inserted into the upstream of TSS in the 5′ LTR, and XmaI site was inserted into the downstream of PolyA signal in the 3′ LTR of dHIV-BlaM and dHIV-dMSD-BlaM to create the XmaI-dHIV-BlaM and XmaI-dHIV-dMSDBlaM plasmids. For insertion of XmaI in the 5′ LTR, the AgeI/SalI-XmaI-T7 fusion PCR fragment was amplified using primer sets of dHIV-AgeI F, dHIV-XmaIT7TSS R, dHIV-XmalIT7TSS F, and dHIV-SalI R, and then inserted into the AgeI/SalI restriction digested backbone of dHIV-BlaM or dHIV-dMSD-BlaM. To insert the XmaI restriction site into the 3′ LTR, the XhoI/PacI-PolyAXmaI fragment was first amplified by fusion PCR with primer sets of dHIV-XhoI F, dHIV-PolyAXmaI R, dHIV-PolyAXmaI F, and dHIV-PacI R. The XhoI/PacI-PolyAXmaI fusion PCR fragment was later inserted into the XhoI/PacI restriction digested dHIV-BlaM or dHIV-dMSD-BlaM (already XmaI/T7 modified). Primers used for cloning of dHIV-BlaM are listed in Table S3.
Lentiviral vectors Ld2G, Ld2GT, dHIV-d2G and HIV-d2G were as described (Razooky et al., 2012). To create the mCherry-RRE lentiviral vector, BamHI-IRES2 fragment was PCR amplified and fused to mCherry (Clonetech)-XbaI PCR fragment. The BamHI/XbaI IRES2-mCherry PCR fusion fragment was inserted into the BamHI/XbaI restriction digested backbone of pNL-GFP-RRE (Wu et al., 2007b). Primers used for cloning of pNL-mCherry-RRE are listed in Table S3.
Cell Culture and Cell Lines
VSV-G pseudo-typed mCherry-RRE, Ld2G, Ld2GIT, HIVΔ-d2G, HIV-1 env-d2G (WT and A7 mutants) and HIV-d2G lentiviruses were made and packaged in 293T cells (ATCC CRL-3216: human, female, embryonic kidney, epithelial, adherent, fetus, not authenticated) by transient transfection with pCMVΔR8.91; after 24 hours, medium was replaced, and an additional 24 hours later, supernatants containing viral particles were harvested (Jordan et al., 2003; Weinberger et al., 2005). The harvested supernatant containing the lentiviral vectors was cleared by low-speed centrifugation and filtered through 0.22-μm-pore-size cellulose acetate filters, yield was determined by flow cytometry analysis of infected Jurkat (ATCC TIB-152: human, male peripheral blood, T lymphocytes, lymphoblast, suspension, not authenticated) cells after TNFα incubation (Jordan et al., 2003; Weinberger et al., 2005). Isoclonal and polyclonal Jurkat populations were developed by infecting cells with mCherry-RRE, Ld2G, Ld2GIT, dHIV-d2G, HIV-1Δenv-d2G (WT and A7 mutants) or HIV-d2G lentivirus at a low MOI and performing single-cell and bulk sorting on a FACS Aria for a range of different GFP fluorescence regions (Jordan et al., 2003; Weinberger et al., 2005). Jurkat cells were cultured in RPMI-1640 medium + L-glutamine, 10% FBS, and 1% PenStrep at 37°C, 5% CO2, in humidified conditions at between 2 × 105 to 2 × 106 cells/mL. To activate HIV-1 transduced Jurkat cells, 10 ng/mL TNF (Sigma, catalog # T0157) or 400 nM TSA (Sigma, catalog # T8552) was added into the culture. To inhibit the CRM1 nuclear export, 0.3 ng/mL Leptomycin B (Sigma, catalog # L2913) was added into TNF treated culture. Primary CD4+ T lymphocytes were isolated from peripheral blood by negative selection method. Cells were then activated from the resting state for 48 h with Dynabeads Human T-Activator conjugated with anti-CD3/CD28 antibodies from Invitrogen and infected with the constructs described above (Razooky et al., 2012). For pharmacological noise-suppression experiments, the transcriptional noise-suppressor molecule, Manidipine (Microsource Discovery Systems, Inc., catalog # 01502388), which attenuates noise without altering the mean LTR expression level (Dar et al., 2014), was used. Cells were incubated at a final concentration of 10 μM Manidipine dissolved in culture media after TNF-α stimulation was halted by washing (the 10 μM final concentration of Manidipine was replaced daily by media exchange). Primary CD4+ T lymphocytes were cultured in RPMI 1640 + L-glutamine, 10% FBS, and 1% PenStrep at 37°C, 5% CO2, in humidified conditions at between 1×106 to 5×106 cells/mL. Positively selected CD14+CD4+ primary monocytes were isolated from buffy coats of healthy donors (Roan et al., 2009), which were generously provided by Dr. Warner Greene. Primary monocytes were cultured at 5×105 cells/mL in DC medium (RPMI-1640 medium with 10% FBS, 1% PenStrep, 2 mM L-glutamine, and 1 mM sodium pyruvate) (Ramos et al., 2011). To induce the differentiation, 1000 U/mL human GM-CSF (Peprotech) was added to CD14+CD4+ primary monocytes culture every 2 or 3 days after isolation (Ramos et al., 2011). Fully differentiated primary monocyte-derived macrophages (primary MDMs) were used for imaging purpose at day 10 after isolation.
METHOD DETAILS
Infection and imaging of cells for protein expression time-courses
Live-cell imaging of Jurkat cells was performed in humidified conditions at 37°C and 5% CO2 for 12–24 h with a 40X (1.3 N.A.) oil-immersion objective on an Zeiss Observer Z1 microscope equipped with 488nm laser as described (Dar et al., 2012). After isolation from peripheral blood, activation and infection (see above) primary CD4+ T lymphocytes were loaded into a microwell device (Razooky et al., 2012) and imaged. Infection and imaging of primary MDMs was performed by differentiating ~200,000 primary monocytes (see above) in 8-well chambered cover-glass dish (Nunc Lab-Tek #155411). On day 10 post isolation, GM-CSF was washed off with cold DC medium. Cells were pre-chilled to 4°C, and a high titer (MOI > 10) of HIV-d2G lentivirus was added on ice for 30 minutes to synchronize infection. Infected primary MDMs were imaged in humidified conditions at 37°C and 5% CO2 for 72~100 h with a 20X/0.8 APO air objective on a Zeiss Observer Z1 microscope. Jurkat and primary CD4+ T lymphocyte images were segmented as described using a custom MATLAB code (Dar et al., 2012; Razooky et al., 2012). Single cell trajectories of infected primary MDMs were manually tracked and calculated using in-house MATLAB code (available upon request). The general trend lines were obtained by synchronizing the initial GFP rise of each single-cell trajectory in silico.
In vitro transcription for SATURN assay
The DNA template for in vitro transcription was obtained by gel purification of XmaI restriction digested XmaI-dHIV-BlaM and XmaI-dHIV-dMSD-BlaM. mMESSAGE mMACHINE® T7 ULTRA Kit (Life Technologies) was used to in vitro transcribe and poly-A tail the RNA. Approximate 0.3 μg of DNA template was used in each reaction, and cleaned up by turbo DNase, following the manufacturer’s instructions. The RNA product was further purified with RNeasy Mini Kit (QIAGEN).
Transfection of RNA into the nucleus for SATURN assay
Cell Line Nucleofector® Kit R (Lonza) was used to transfect in vitro transcribed mRNA into the nucleus of naive Jurkat cells. For each nucleofection, 2 μg of each RNA or equivalent molar amounts, and 1.6 ×106 Jurkat cells were used. For the electroporation step in the nucleofection, pre-set program O-028 from the Nucleofector device was used. Control transfection efficiency was estimated by using 2 μg of pmaxGFP (Lonza) plasmid per nucleofection.
BlaM assay and flow cytometry analysis for SATURN assay
LiveBLAzer™ FRET-B/G Loading Kit with CCF2-AM (#K1032, Life technologies) was used to detect β-lactamase positive (BlaM+) cells. Jurkat cells were harvested 2 hours after RNA nucleofection, by centrifuging at 800xg for 5 minutes, and washing once with CO2-independent media (#18045-088, Life Technologies). Because the fluorescent substrate CCF2-AM is light sensitive, all the following procedures were performed with limited exposure to light. The pellet was resuspended in CCF2-AM loading medium (1 μM CCF2-AM, 1 mg/mL pluornic-F127, 0.001% acetic acid in CO2-independent medium), followed by a 1-hour incubation on the bench top. Cells were spun down and the pellet was resuspended in the development medium (2.5 mM probenecid, 10% FBS in CO2-independent medium). The β-lactamase enzymatic reaction was performed by incubating the cells for 16 hours at room temperature. Cells were later washed once with PBS, and resuspended in fixation buffer (2% paraformaldehyde in PBS) at 4°C overnight. An LSRII cytometer (BD Biosciences) with 404-nm laser was used to detect the green-to-blue shift in emission fluorescence. The cytometry data were analyzed using FlowJo (http://www.flowjo.com/).
qPCR analysis to determine cellular RNA levels of wt HIV versus A7 mutants
Abundances of different RNA species in the HIV-1Δenv-d2GFP A7 mutants were quantified and compared to RNA levels found in WT HIV-1Δenv-d2GFP. Stable cell lines WT HIV-1Δenv-d2GFP, HIV-1Δenv-d2GFP A7-4, HIV-1Δenv-d2GFP A7-5, HIV-1Δenv-d2GFP A7-7 were activated with TNFα. After 4 hours, cells were collected and washed. Cellular RNA was extracted using the RNaesy kit (Qiagen) and quantified using a Nanodrop (ThermoFisher). 1 μg of RNA from each cell line was reverse transcribed into cDNA, using the Quantitect Reverse Transcription kit (Qiagen). cDNA content was analyzed using the Fast SYBR Green Master Mix (Applied Biosystems) in 384-well plate format in a 7900 HT Fast Real-Time PCR System (Applied Biosystems) using a standard protocol. Primers used for qPCR are listed in Table S3.
Immunofluorescence analysis to determine protein levels of WT HIV versus A7 mutants
To quantify HIV-1 protein expression levels for different RNA species in the HIV-1Δenv-d2GFP A7 mutants as compared to RNA levels found in WT HIV-1Δenv-d2GFP, direct immunofluorescence by flow cytometry was used. At 20 hours after TNF-alpha induction, cells were fixed with a final of 2% formaldehyde (Tousimis. cat#1008A) for 15 min at room temperature. Cells were then centrifuged and supernatant aspirated. Cells were permeabilized with 1:1 methanol:acetone and incubated on ice for 10 min. Cells were then washed with Stain Buffer (DPBS without calcium and magnesium, 2% FBS, 2 mM EDTA, 0.1% IGEPAL CA-630) with centrifugation and supernatant aspiration between washes. Cells were centrifuged and supernatant aspirated and resuspended in 50 μL of Stain Buffer and 1 μL of KC57-RD1 (Beckman Coulter. cat#6604667) and incubated at room temperature in the dark for 15 minutes. Cells were then washed with 1 mL of Flow Buffer (DPBS without calcium and magnesium, 2% FBS, 2 mM EDTA) and centrifuged and supernatant aspirated. Cells were then resuspended in 200 μL of Flow Buffer. Fluorescence was detected using a LRSII flow cytometer (BD Biosciences) excitation at 531 nm, filtered at 585/42 nm, and recorded at 568–590 nm.
smFISH
Probe design
Probes were developed using the designer tool from http://www.singlemoleculefish.com/. Three sets of probes were designed to detect regions the Pol/Vif splice junction (4548–5414 bp of pNL4-3), Env (7251–8251 bp of pNL4-3), and GFP (corresponding to Nef, 8799–8887 bp of pNL4-3) in HIV-d2G. Each set contained 27–30 probes, each probe was 18 nt long, using a masking level of 3–5, and at least 2 bp spacing between single probes (see probe sequence in Table S1). Probes were conjugated with Quasar 670 or TAMRA.
Drug treatment and smFISH
Jurkat cells stably transduced with a full-length HIV reporter provirus (Δenv with d2GFP in nef reading frame) were washed with 10 mL of PBS solution and then immobilized on Cell-Tak coated eight-well chambered imaging dishes (Weinberger et al., 2008). Cells were treated with 10 ng/ml TNF (Sigma, catalog # T0157) for time periods ranging for 7–140 minutes. For the transcriptional pulse-chase cells were subsequently treated with 6 μg/ml of actinomycin D (Sigma, catalog # A9415). Cells were then fixed with PBS in 3.4% paraformaldehyde for 10 minutes. Fixed cells were stored in 70% EtOH at 4 °C for a minimum of 1 hour to permeabilize the cell membranes. Probes were diluted to a final concentration of 50 nM, in 1g/mL dextran sulfate (Sigma, catalog # 42867), 2xSSC and 10% formamide (ThermoFisher, catalog # AM9342) and allowed to hybridize at 37°C overnight. Wash steps and DAPI staining were performed as described (https://www.biosearchtech.com/support/applications/stellaris-rna-fish).
Imaging of smFISH
Cells were imaged in a previously described buffer to minimize photo-bleaching (50% glycerol, 75 μg/mL glucose oxidase, 520 μg/mL catalase, and 0.5 mg/mL Trolox) (Waks et al., 2011). Images were taken on a Nikon Ti-E microscope equipped with a W1 Spinning Disk unit, an Andor iXon Ultra DU888 1k x 1k EMCCD camera and a Plan Apo VC 100x/1.4 oil objective in the UCSF Nikon Imaging Center. Approximately 10 xy locations were randomly selected for each condition. For each xy location, Nyquist sampling was performed by taking ~30, 0.4 μm steps along the z-plane. The exposure times for Quasar 670 (100% laser power), TAMRA (50% laser power), and DAPI (50% laser power) channels were 500 ms, 500 ms, and 50 ms for single mRNA analysis and 50 ms, 50 ms, and 50 ms for transcriptional center (TC) analysis. For each z-plane in a 3-D stack images for both single mRNA analysis and TC analysis were taken. Cells were segmented manually. DAPI image stacks were used for nuclear segmentation, spot/TC identification and counting was performed using in-house MATLAB programs (available upon request).
Probes for smFISH
US probe sets: Detecting Pol-Vif region
Unspliced target sequence: agatggccagtaaaaacagtacatacagacaatggcagcaatttcaccagtactacagttaaggccgcctgttggtgggcggggatcaagcaggaatttggcattccctacaatccccaaagtcaaggagtaatagaatctatgaataaagaattaaagaaaattataggacaggtaagagatcaggctgaacatcttaagacagcagtacaaatggcagtattcatccacaattttaaaagaaaaggggggattggggggtacagtgcaggggaaagaatagtagacataatagcaacagacatacaaactaaagaattacaaaaacaaattacaaaaattcaaaattttcgggtttattacagggacagcagagatccagtttggaaaggaccagcaaagctcctctggaaaggtgaaggggcagtagtaatacaagataatagtgacataaaagtagtgccaagaagaaaagcaaagatcatcagggattatggaaaacagatggcaggtgatgattgtgtggcaagtagacaggatgaggattaacacatggaaaagattagtaaaacaccatatgtatatttcaaggaaagctaaggactggttttatagacatcactatgaaagtactaatccaaaaataagttcagaagtacacatcccactaggggatgctaaattagtaataacaacatattggggtctgcatacaggagaaagagactggcatttgggtcagggagtctccatagaatggaggaaaaagagatatctgctataagaaataccatat
Probe sequence: One version of this US probe set is conjugated to TAMRA, and the other is conjugated to Quasar 670. The US-TAMRA probe set is used with SS-Quasar 670, and the US-Quasar 670 probe set is used with MS-TAMRA. For sequences, see Table S1.
SS probe set: Detecting Env region
Singly-spliced target sequence: ctagcaaattaagagaacaatttggaaataataaaacaataatctttaagcaatcctcaggaggggacccagaaattgtaacgcacagttttaattgtggaggggaatttttctactgtaattcaacacaactgtttaatagtacttggtttaatagtacttggagtactgaagggtcaaataacactgaaggaagtgacacaatcacactcccatgcagaataaaacaatttataaacatgtggcaggaagtaggaaaagcaatgtatgcccctcccatcagtggacaaattagatgttcatcaaatattactgggctgctattaacaagagatggtggtaataacaacaatgggtccgagatcttcagacctggaggaggcgatatgagggacaattggagaagtgaattatataaatataaagtagtaaaaattgaaccattaggagtagcacccaccaaggcaaagagaagagtggtgcagagagaaaaaagagcagtgggaataggagctttgttccttgggttcttgggagcagcaggaagcactatgggcgcacggtcaatgacgctgacggtacaggccagacaattattgtctgatatagtgcagcagcagaacaatttgctgagggctattgaggcgcaacagcatctgttgcaactcacagtctggggcatcaaacagctccaggcaagaatcctggctgtggaaagatacctaaaggatcaacagctcctggggatttggggttgctctggaaaactcatttgcaccactgctgtgccttggaatgctagttggagtaataaatctctggaacagatttggaataacatgacctggatggagtgggacagagaaattaacaattacacaagcttaatacactccttaattgaagaatcgcaaaaccagcaagaaaagaatgaacaagaattattggaattagataaatgggcaagtttgtggaattggtttaacataacaaattggct
Probe sequences: Probes are all conjugated to Quasar 670. For sequences, see Table S1
MS probe set: Detecting d2GFP (replacing Nef reading frame)
Multiply-spliced target sequence: atggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagaagcttagccatggcttcccgccggaggtggaggagcaggatgatggcacgctgcccatgtcttgtgcccaggagagcgggatggaccgtcaccctgcagcctgtgcttctgctaggatcaatgtgtag Probe sequences: Probes are all conjugated to TAMRA. For sequences, see Table S1.
Image Processing and Data Analysis of smFISH images
Cellular and nuclear segmentation
The cellular masks for each xy location were manually created in ImageJ™ using a 2D projection of the image stacks from the TAMRA channel, only cells with diameters between approximately 9 and 15 μm were analyzed (area ≈ 4000–11000 pixels). Individual cell boundaries, the numbers of spots and transcription centers (TCs) for each single cell, and the x-y-z location of spots and TCs were manually selected and segmented using in-house MATLAB programs (available upon request). The center position of TCs were determined at the pixel position where the intensity of the US probe was greatest. In-house MATLAB programs (available upon request) were used to determine the boundary between the nucleus and cytoplasm of individual cells per z slice, using the DAPI images.
Spot classification and quality controls
Using naive cells, a threshold was determined above which pixels were considered as possible spots. A possible spot which fell within the same xy position in consecutive z slices was counted a single possible spot. A Gaussian filter was used to reduce the amount of local maxima caused by pixel to pixel noise. Then for each z slice, local maxima which were brighter than the threshold were determined. Each local maximum was considered as one mRNA molecule. If the local maximum from one spot fell within the area of a spot from a different channel, they would be considered co-localized. By comparing the number of co-localized spots with the number of spots from the US channel only, the number of false positives was gauged to be less than 10%. The distance between each spot and TC was described by the 3D Euclidian distance from the respective local maxima.
Transcriptional Center Analysis
Unspliced and spliced (either SS or MS) mRNA at the TC was determined using the US and MS probe sets. The unspliced mRNA was quantified by dividing the individual TC intensity by the median spot intensity and multiplying the outcome by the respective TC’s size divided by the median spot size.
Though the full-length US transcript is ~9 kb long, the US probes start binding approximately 4 kb into the transcript. Thus, it takes roughly 2 minutes for the remaining (5 kb) transcript to be transcribed (assuming a transcription rate of 2.6 kb/min) (Coulon et al., 2014). Fully transcribed transcripts will reside at the TC for approximately 116 seconds (Coulon et al., 2014). Therefore, half of the mRNA at the TC would be tethered to the DNA, and the other half of the mRNA would be residing. The MS signal is expected to be equal to the US signal for the mRNA that is residing at the TC. The MS probe set is towards the end of the gene, so for every US mRNA tethered to the DNA 0.08 MS signals are expected (Bahar Halpern and Itzkovitz, 2016). This is calculated from the ratio of ½ MS target sequence to the full length US transcript (422.5/5103). The expected MS signal can therefore be calculated as follows:
and, the Spliced mRNA at each TC is:
where MS signal is the raw mRNA count, calculated by dividing the individual TC intensity from the MS channel, by the median spot intensity and multiplying the outcome by the respective TC’s size divided by the median spot size.
MATHEMATICAL MODELING METHODS (see also Supplementary Methods S1)
We developed ordinary differential equation (ODE) models and simulated them using Berkeley Madonna™ and MATLAB™. General trend lines obtained from microscopic experiments were fit in Berkeley Madonna. Sensitivity analysis and parameter plots for the ODE model were performed in MATLAB or Berkeley Madonna. For stochastic simulations, chemical reaction schemes were coded in programming language C using the Gillespie algorithm (Gillespie, 2007; Gillespie et al., 2013). The simulation results were analyzed using MATLAB or Mathematica™.
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of cells analyzed for each experiment (n), definition of center, and dispersion and precision measures can be found in the respective figure legends.
Supplementary Material
Detailed method for construction of co-transcriptional, post-transcriptional and “mixed” mathematical models followed by parameter sensitivity analysis and experimental model validation. Stochastic simulations as well as supporting analytical arguments are also included.
Model parameters used, related to Figure 5
Table of primers and G-block Sequences, related to Figure 6 and STAR methods
(A) Analytical predictions shown together with statistics of Gillespie numerical simulations for the two-state, auto-repression, and auto-depletion models (red and blue dashed arrows “i” and “ii” correspond to dashed arrows in Figure 2C).
(B) Outputs of Gillespie simulations for each model in parameter regimes where auto-repression has the perverse effect of amplifying noise. 500 simulation runs were performed (in Mathematica™ using the xSSAlite™) for each model to calculate the mean and CV2. P indicates simulated protein levels. The two-state model is shown in grey, transcriptional auto-repression is shown in red and precursor auto-depletion in blue (mean ± standard deviation).
(C) Corresponding analytical predictions and statistics of Gillespie simulations for panel B.
(D) Fano factor of Gillespie simulations comparing the two-state, auto-repression, α auto-repression and auto-depletion feedback models. As predicted (see Supplementary Methods S1, page 17) the simulations show that in the koff ≫ kon regime, α auto-repression nearly matches the noise suppression capability of auto-depletion (Fano ~2000→680 vs. 612), however in the kon > koff regime, α auto-repression only gets one halfway to the noise suppression capability of auto-depletion (Fano 61→49 for auto-repression vs. 61→41 for auto-depletion).
(A) Representative smFISH images for HIV-infected Jurkat cells. Three sets of probes, US, SS, and MS probes, were designed to bind to the Pol/Vif, Env, and d2GFP reading frames on HIV-d2G, respectively. Each probe set contains at least 27 single probes that are conjugated with fluorophores. Due to the inherent limitations of the technique and available fluorophore probes, only any two of the three RNA species could be imaged at once. Using the US together with the SS probe set and the SS together with the MS probe set, we detected all three alternative splicing variants.
(B) TC analysis showing US mRNA versus spliced mRNA (either SS or MS) at each TC. Each dot represents one TC. The full lines represent the expected spliced versus US behavior for 80% 50% and 0% spliced transcripts at the TC. Most transcriptional centers fall along the line representing 0% spliced transcripts at the TC. The shaded area shows the expected variability for 0% spliced transcripts at the TC. Spliced RNAs are quantified by subtraction of the intron signal from the exon signal (Bahar Halpern and Itzkovitz, 2016). Since the intron is at the 5′ end of the transcript and the exon is at the 3′ end of the transcript (see left inset), during transcriptional elongation a subset of transcripts will exhibit only the intron signal (as the exon has not yet been transcribed). Thus, upon subtraction (exon – intron), the numbers of RNAs will appear negative for the subset of elongating transcripts that have partially completed transcription and only contain the 5′ intron region (where the US probes bind) but haven’t yet transcribed the 3′ exon region (where the MS probes bind). Right inset: Mean increase of US, SS and MS transcripts in the nucleus after transcriptional activation with TNF. Error bars represent SEM.
(C) Mean increase of unspliced (US) and spliced (MS + SS) transcripts in the nucleus (top) and cytoplasm (bottom) after transcriptional activation with TNF. Error bars represent SEM.
(D) 5′-Capped, and 3′-polyadenylated dHIV-BlaM (β-lactamase) pre-mRNA was in vitro transcribed, purified, and nucleofected into naïve Jurkat cells. After a 2-hour recovery post-nucleofection, CCF2-AM, the fluorescent substrate of β-lactamase, was loaded into cells. Cells that successfully took up unspliced pre-mRNA into their nuclei and spliced the pre-mRNA post-transcriptionally, express BlaM and convert the green fluorescent substrate into the blue fluorescent product. Otherwise, the cells remain green due to uncleaved CCF2-AM substrate which cannot be cleaved due to lack of BlaM expression. Flow cytometry was later utilized to determine the percentage of cells capable of post-transcriptional splicing.
(E–F) Cells nucleofected with different RNA constructs show that unspliced HIV-1 pre-mRNA, as well as β-globin, a gene known to be co-transcriptionally spliced, recruit spliceosome machinery post-transcriptionally in vivo. The positive control consisted of an RNA segment containing the full β-lactamase gene, which did not require splicing to express BlaM. Similar results were obtained upon nucleofection of an equivalent mass of RNA (2 μg) or equivalent molar amounts of RNA (1.11 pmoles) into cells.
(A) Numerical simulations of post-transcriptional splicing ODE model showing cytoplasmic MS and US mRNA (green and orange line respectively), and GFP (black line). See Supplementary Methods S1 and Table S2 for equations and parameters.
(B) Statistics of trajectories for the spliced reporter HIV-d2G. More than 80% of cells exhibit an overshoot (i.e., overshoot or oscillating) trajectory during an 18 hour imaging experiment while more than 40% of cells display oscillating features in the later hours, after the overshoot peak.
(C) Direct measurement of negative-feedback regulation by temporal autocorrelation function (ACF) analysis of single-cell trajectories, as described in (Weinberger et al., 2008), after transcriptional activation with TNF. Feedback strength is calculated from the ACF half-correlation time (CT50) for a given 5-hour window normalized to the initial 5-hour time window (i.e., [CT50window/CT500–5hrs − 1] such that negative values correspond to negative feedback). Each ACF is calculated from at least 100 single-cell trajectories. The HIV MS reporter (green) exhibits significant negative feedback beginning at ~5 hours. Overexpression of the Rev-M10 mutant in the same cells (red) or Leptomycin B treatment (blue) abrogate negative feedback.
(D–E) Infection and time-lapse single-cell imaging of human donor-derived primary CD4+ T cells and primary monocyte-derived macrophages (primary MDMs). Primary CD4+ T cell imaging was conducted in micro-well chambers, as described in (Razooky et al., 2012). The general trend line shows an overshoot similar that seen in Jurkat cells. Primary MDMs were obtained by incubating primary CD17+/CD4+ monocytes with GM-CSF for 10 days. Differentiated primary MDMs were infected with VSV-G pseudo-typed HIV-1-d2G and imaged at 10-minute intervals for 40 hours. Averaged trend line (n = 489 cells) exhibits the negative-feedback overshoot. (Error bars represent the standard error.)
(A) Single-cell time-lapse imaging of Jurkat cells transduced with an HIV-1 LTR-d2GFP (Ld2G) lentiviral vector after induction with TNF. General trend line (n=255) does not exhibit the overshoot in GFP at 5–10 hours, as compared to full-length HIV. (Error bar represents the standard error.)
(B) Single-cell time-lapse imaging of cells transduced with an HIV-1 LTR-d2GFP-IRES-Tat (Ld2GT) lentiviral vector after TNF induction. Ld2GT represents a minimal Tat positive-feedback circuit (i.e., lacking Rev). The general trend line (n=135) does not exhibit the characteristic negative-feedback overshoot. (Error bar represents the standard error.)
(C) Single-cell time-lapse imaging of primary CD4+ T cells transduced with either Ld2G (black) or Ld2GT (grey) lentiviral vectors and transcriptionally activated by TNF induction.
(D) Flow cytomtery analysis of cells stably transduced with an HIV spliced (MS) reporter (dHIV-d2G) after serial induction with TNF. 12 hours after first induction (i.e., after GFP expression has peaked), a second TNF (or TSA) stimulation was added to the culture, and the GFP expression was quantified by flow cytometry. The results show that the second reactivation with TNF or TSA (black or grey bars, respectively) does not rescue the decaying GFP level (compare to white bars), indicating that the turnover of GFP is not due to the silencing of LTR promoter.
(E) ODE model predictions for enhancement of Rev-dependent export (kr). As the export rate increases, the overshoot from precursor auto-depletion feedback increases as exhibited by the accelerated response time (a measure of feedback strength) during the post-peak decay phase.
(F) Single-cell time-lapse imaging of GFP expression in Jurkat cells stably transduced with the HIV spliced (MS) reporter dHIV-d2G (n=97), which encodes the wild-type RRE within env as well as a second distal RRE within the Δgag/RRE. The overshoot is enhanced and the post-peak response time is accelerated (50% response time of decay is accelerated by ~5 hours, compared to Figure 4B). This construct indicates that a Tat/Rev/Env cassette is sufficient to generate the same overshoot dynamics as the full-length construct, compared to the Ld2G and Ld2GT constructs. (Error bars represent ± standard error.)
(A) Nonlinear least–squares fitting of ODE models to single-cell imaging data. Only the post-transcriptional splicing/Rev negative feedback model provides a good fit. Although the “mixed” model does not fit the data as well as the post-transcriptional model, it is still capable of generating an overshoot trajectory, indicating that post-transcriptional splicing is essential to generate negative feedback and the feedback is robust to potential RNA “leakage” into the system from co-transcriptional splicing. Inset: nonlinear least-squares fitting of GFP signal from single-cell time-lapse imaging data to the ODE model for post-transcriptional splicing when active Tat positive feedback is included (i.e., Tat feedback does not “saturate”). (B) Hill coefficient (h) of Rev/RRE nuclear export should be larger than 3 to fit HIV single-cell time-lapse imaging data. When the h is lower than 3, the R-squared value of post-transcriptional model fitting is less than 0.9. When the h is large (e.g., 12), other parameters can be made to fit the reference/physiologically reasonable range.
(C–D) The post-transcriptional (Rev negative feedback) model predicts how the dynamics of HIV gene expression respond to exogenous Rev over expression. Surprisingly, expression of US (C) and MS (D) gene products decrease upon Rev over expression. This prediction contradicts the common conclusion that Rev upregulates the expression of US RNA.
(E–F) Overexpression of Rev decreases the level of p24 expression in full-length HIV-1 (E) Jurkat cells latently infected with HIV-d2G were transfected with pcRev plasmid DNA. At 24 hours after transfection, TNF-α was added to activate viral replication. GFP and p24 levels were measured 24 hours after TNF-α treatment, using immunostaining and flow cytometry. The levels were normalized to the background fluorescence of naïve Jurkat cells. As Rev overexpression was increased, GFP levels decreased. (F) The intracellular p24 level also decreased as Rev levels were increased.
(A) Diagram showing where the respective RT-PCR primer sets bind.
(B) Right panel: RT-PCR data from HIV and mutant transduced cells after TNF induction. Total cellular RNA was collected 4 hours after TNF activation. D1 to A3, D1 to A4c and D1 to A5 are the first splicing events used for Tat, Rev and Nef, respectively. D4 to A7 is a splicing event shared by Tat, Rev and Nef. D4 to A7 splicing is enhanced by the mutations shown in Figure 6A. Data represent the mean and SEM of two biological replicas. Left panel: Quantification of intracellular protein expression for proteins translated from spliced and unspliced HIV-1 transcripts in the HIV-1 A7 mutants relative to the A7 wild-type HIV-1 by direct immunofluorescence (for p24) and GFP fluorescence. These data were quantified by flow cytometery.
(C) Raw slope data corresponding to Figure 6C showing the GFP and mCherry slopes for A7 mutants and wild-type HIV (WT). The increased mCherry slope in the mutants is consistent with reduced HIV US RNA that acts as RRE-export competitor for the mCherry-RRE in the mutants (i.e., less US HIV RNA in the mutants).
Cellular GFP expression, related to Figure 4
(A–B) The ODE model used is described in detail in Supplementary Methods S1, specifically see section entitled: “Modeling circuit-relaxation dynamics after a TNF pulse-chase experiment (Figure 7, main text)”. Briefly, gray trajectory represents wild-type HIV (sp = 1.5) and red trajectory represents A7 mutants (sp = 15); blue trajectory (sp = 0.15) represents a hypothetical slower-splicing virus shown to illustrate the trend of the dynamics. At time = 48 h, basal LTR expression is instantaneously reduced by threefold to simulate TNF removal. In contrast to the data in Figure 7A, more efficient splicing results in slower turning off (i.e., crossing of GFP = 0.005 occurs at a later time the more efficient the splicing). (B) An expansion of panel A (expansion of x axis from time = 48 to 96 h). “simulated GFP” represents GFP.
(C) Active-state stability by flow cytometry for wild-type HIV d2G after a pulse of TNF reactivation (TNF removed at time 0) in the presence (black) and absence (blue) of the LTR transcriptional noise-suppressor molecule (Dar et al., 2014), corresponding to Figure 7D. As predicted, due to the intact negative feedback in the wild type, the noise suppressor has minimal effect on the stability of wild-type HIV d2G. Histogram corresponds to the 48-hour time point.
(D) Summary analysis of the number of mutations in the 41-nucleotide sequence region surrounding the A7 splice-acceptor across HIV and SIV sequences representing global diversity in the Los Alamos HIV database compendium (alignment is of nucleotides 8337–8378 in the HIV-1 HXB2 reference genome, which is identical to the ‘wild-type’ NL4-3 molecular clone used in this study). The analysis summarizes sequence alignments obtained using the AnalyzeAlign tool (http://www.hiv.lanl.gov/) of HIV-compendium sequences (200 HIV-1 sequences and 100 HIV-2/SIV sequences representing global diversity) and indicates a relatively low mutation rate, with most sequences having two to four differences from consensus.
(E) Top: Sequence logo from alignments of the HIV compendium sequences, using the Los Alamos AnalyzeAlign tool, for the A7 splice-acceptor region (nucleotides 8337-8378 in the HXB2 HIV-1 reference genome. For reference, the consensus sequence for ‘wild-type’ HIV-1 NL4-3 (8337-8378) is: GTGAATAGAGTTAGGCAGGGATATTCACCATTATCGTTTCAG. Bottom: Corresponding sequence logo for mutations from consensus.
(F) Ratio of viral genomic RNA (gRNA), collected from supernatants of A7-4 and wild-type infected cells after 48 hours. (qPCR was used to quantify packaged US RNA in the supernatant of A7-4 infected cells versus wild-type cells at 48 hours post infection). Data represent the mean and SEM of two biological replicas.
HIGHLIGHTS.
Post-transcriptional splicing enables feedback via auto-depletion of precursor RNA
RNA auto-depletion attenuates noise better than transcriptional auto-repression
Auto-depletion counterbalances noisy fate-selection circuitry stabilizing HIV fate
Disrupting RNA auto-depletion amplifies transcriptional noise promoting HIV latency
Acknowledgments
We are grateful to H. Madhani, P. Walter, P. Sharp, C. Guthrie, A. Frankel, W. Greene, J. Karn, R. Tsien, A. Hoffmann, J. Guatelli, L. Tsimring, M. Cavrois, T. Notton, J. Young, and S. Chunda for providing reagents, equipment, and helpful discussions; K. Thorn and D. Larsen (Nikon Imaging Center, UCSF, funded through S10 1S10OD017993-01A1) and the UCSF-Gladstone Center for AIDS Research flow core, funded through (P30 AI027763), (S10 RR028962-01) and the James B. Pendleton Charitable Trust for invaluable technical expertise. M.M.K.H is supported by the Netherlands Organization of Scientific Research (NWO) through a Rubicon fellowship (No. 019.153LW.028). M.L.S acknowledges support from the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. L.S.W. acknowledges support from the Alfred P. Sloan Research Fellowship, NIH awards R01AI109593, P01AI090935, and the NIH Director’s New Innovator Award (OD006677) and Pioneer Award (OD17181) programs.
Abbreviations
- CV
coefficient of variation
- TNF
Tumor Necrosis Factor alpha
- NFκB
Nuclear Factor kappa B
- SATURN
Splicing After Transfection of Unspliced RNA into Nucleus
Footnotes
AUTHOR CONTRIBUTIONS
L.S.W., W.Y.W., E.I., M.M.K.H., M.L.S., and B.R. conceived and designed the study. W.Y.W., E.I., B.R., M.M.K.H., C.E.T and L.S.W. designed and performed the experiments. W.Y.W., C.C., M.L.S., E.I., B.R., R.D.D., M.M.K.H. and L.S.W. analyzed the data and models. M.M.K.H., W.Y.W., E.I., and L.S.W. wrote the paper.
DECLARATION OF INTERESTS
LSW is a cofounder of Autonomous Therapeutics, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acar M, Mettetal JT, van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet. 2008;40:471–475. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- Alon U. An introduction to systems biology: design principles of biological circuits. Boca Raton, FL: Chapman & Hall/CRC; 2007. [Google Scholar]
- Austin DW, Allen MS, McCollum JM, Dar RD, Wilgus JR, Sayler GS, Samatova NF, Cox CD, Simpson ML. Gene network shaping of inherent noise spectra. Nature. 2006;439:608–611. doi: 10.1038/nature04194. [DOI] [PubMed] [Google Scholar]
- Bahar Halpern K, Itzkovitz S. Single molecule approaches for quantifying transcription and degradation rates in intact mammalian tissues. Methods. 2016;98:134–142. doi: 10.1016/j.ymeth.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Balazsi G, van Oudenaarden A, Collins JJ. Cellular decision making and biological noise: from microbes to mammals. Cell. 2011;144:910–925. doi: 10.1016/j.cell.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black HS. Stabilized Feedback Amplifiers*. Bell System Technical Journal. 1934;13:1–18. [Google Scholar]
- Bohan CA, Kashanchi F, Ensoli B, Buonaguro L, Boris-Lawrie KA, Brady JN. Analysis of Tat transactivation of human immunodeficiency virus transcription in vitro. Gene expression. 1992;2:391–407. [PMC free article] [PubMed] [Google Scholar]
- Boireau S, Maiuri P, Basyuk E, de la Mata M, Knezevich A, Pradet-Balade B, Backer V, Kornblihtt A, Marcello A, Bertrand E. The transcriptional cycle of HIV-1 in real-time and live cells. J Cell Biol. 2007;179:291–304. doi: 10.1083/jcb.200706018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Bhutkar A, Sharp PA. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015;29:63–80. doi: 10.1101/gad.247361.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nature biotechnology. 2002;20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez L, Calvanese V, Verdin E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS Pathog. 2015;11:e1004955. doi: 10.1371/journal.ppat.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KS, Fisk GJ, Hauber J, Usman N, Daly TJ, Rusche JR. Characterization of HIV-1 REV protein: binding stoichiometry and minimal RNA substrate. Nucleic Acids Res. 1991;19:1577–1583. doi: 10.1093/nar/19.7.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan AM, Tunnacliffe E, Cannon D, Chubb JR. A continuum model of transcriptional bursting. Elife. 2016:5. doi: 10.7554/eLife.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon A, Ferguson ML, de Turris V, Palangat M, Chow CC, Larson DR. Kinetic competition during the transcription cycle results in stochastic RNA processing. eLife. 2014;3:e03939. doi: 10.7554/eLife.03939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CD, McCollum JM, Austin DW, Allen MS, Dar RD, Simpson ML. Frequency domain analysis of noise in simple gene circuits. Chaos. 2006;16:026102. doi: 10.1063/1.2204354. [DOI] [PubMed] [Google Scholar]
- Daly TJ, Doten RC, Rennert P, Auer M, Jaksche H, Donner A, Fisk G, Rusche JR. Biochemical characterization of binding of multiple HIV-1 Rev monomeric proteins to the Rev responsive element. Biochemistry. 1993;32:10497–10505. doi: 10.1021/bi00090a028. [DOI] [PubMed] [Google Scholar]
- Dar RD, Hosmane NN, Arkin MR, Siliciano RF, Weinberger LS. Screening for noise in gene expression identifies drug synergies. Science. 2014;344:1392–1396. doi: 10.1126/science.1250220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar RD, Razooky BS, Singh A, Trimeloni TV, McCollum JM, Cox CD, Simpson ML, Weinberger LS. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc Natl Acad Sci U S A. 2012;109:17454–17459. doi: 10.1073/pnas.1213530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar RD, Razooky BS, Weinberger LS, Cox CD, Simpson ML. The Low Noise Limit in Gene Expression. PLoS One. 2015;10:e0140969. doi: 10.1371/journal.pone.0140969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Natl Acad Sci U S A. 2010;107:12481–12486. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dublanche Y, Michalodimitrakis K, Kummerer N, Foglierini M, Serrano L. Noise in transcription negative feedback loops: simulation and experimental analysis. Mol Syst Biol. 2006;2:41. doi: 10.1038/msb4100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyhr-Mikkelsen H, Kjems J. Inefficient spliceosome assembly and abnormal branch site selection in splicing of an HIV-1 transcript in vitro. J Biol Chem. 1995;270:24060–24066. doi: 10.1074/jbc.270.41.24060. [DOI] [PubMed] [Google Scholar]
- Edelstein-Keshet L. Mathematical models in biology. 1. New York: Random House; 1988. [Google Scholar]
- Felber BK, Drysdale CM, Pavlakis GN. Feedback regulation of human immunodeficiency virus type 1 expression by the Rev protein. Journal of virology. 1990;64:3734–3741. doi: 10.1128/jvi.64.8.3734-3741.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong YW, Zhou Q. Stimulatory effect of splicing factors on transcriptional elongation. Nature. 2001;414:929–933. doi: 10.1038/414929a. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Gillespie DT. Exact stochastic simulation of coupled chemical reactions. The Journal of Physical Chemistry. 1977;81:2340–2361. [Google Scholar]
- Gillespie DT. Stochastic simulation of chemical kinetics. Annual review of physical chemistry. 2007;58:35–55. doi: 10.1146/annurev.physchem.58.032806.104637. [DOI] [PubMed] [Google Scholar]
- Gillespie DT, Hellander A, Petzold LR. Perspective: Stochastic algorithms for chemical kinetics. The Journal of chemical physics. 2013;138:170901. doi: 10.1063/1.4801941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Baltimore D. RNA splicing regulates the temporal order of TNF-induced gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11934–11939. doi: 10.1073/pnas.1309990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proceedings of the National Academy of Sciences of the United States of America. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung G, Barkai N. Noise propagation and signaling sensitivity in biological networks: a role for positive feedback. PLoS Comput Biol. 2008;4:e8. doi: 10.1371/journal.pcbi.0040008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg RA, Tay S. Noise facilitates transcriptional control under dynamic inputs. Cell. 2015;160:381–392. doi: 10.1016/j.cell.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Kornblihtt AR. Promoter usage and alternative splicing. Curr Opin Cell Biol. 2005;17:262–268. doi: 10.1016/j.ceb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Landowski M, Dabundo J, Liu Q, Nicola AV, Aguilar HC. Nipah virion entry kinetics, composition, and conformational changes determined by enzymatic virus-like particles and new flow virometry tools. Journal of virology. 2014;88:14197–14206. doi: 10.1128/JVI.01632-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446:926–929. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- Lestas I, Vinnicombe G, Paulsson J. Fundamental limits on the suppression of molecular fluctuations. Nature. 2010;467:174–178. doi: 10.1038/nature09333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK. RNA splicing is a primary link between genetic variation and disease. Science. 2016;352:600–604. doi: 10.1126/science.aad9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Bohnlein S, Hauber J, Cullen BR. Functional dissection of the HIV-1 Rev trans-activator--derivation of a trans-dominant repressor of Rev function. Cell. 1989a;58:205–214. doi: 10.1016/0092-8674(89)90416-9. [DOI] [PubMed] [Google Scholar]
- Malim MH, Cullen BR. HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell. 1991;65:241–248. doi: 10.1016/0092-8674(91)90158-u. [DOI] [PubMed] [Google Scholar]
- Malim MH, Hauber J, Fenrick R, Cullen BR. Immunodeficiency virus rev trans-activator modulates the expression of the viral regulatory genes. Nature. 1988;335:181–183. doi: 10.1038/335181a0. [DOI] [PubMed] [Google Scholar]
- Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989b;338:254–257. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- Mann DA, Mikaelian I, Zemmel RW, Green SM, Lowe AD, Kimura T, Singh M, Butler PJ, Gait MJ, Karn J. A molecular rheostat. Co-operative rev binding to stem I of the rev-response element modulates human immunodeficiency virus type-1 late gene expression. J Mol Biol. 1994;241:193–207. doi: 10.1006/jmbi.1994.1488. [DOI] [PubMed] [Google Scholar]
- Mauger O, Lemoine F, Scheiffele P. Targeted Intron Retention and Excision for Rapid Gene Regulation in Response to Neuronal Activity. Neuron. 2016;92:1266–1278. doi: 10.1016/j.neuron.2016.11.032. [DOI] [PubMed] [Google Scholar]
- Metzger BP, Yuan DC, Gruber JD, Duveau F, Wittkopp PJ. Selection on noise constrains variation in a eukaryotic promoter. Nature. 2015;521:344–347. doi: 10.1038/nature14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevozhay D, Adams RM, Murphy KF, Josic K, Balazsi G. Negative autoregulation linearizes the dose-response and suppresses the heterogeneity of gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5123–5128. doi: 10.1073/pnas.0809901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya K, Kataoka N, Hagiwara M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol. 2011;195:27–40. doi: 10.1083/jcb.201107093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, Guthrie C. Transcriptional pulse-chase analysis reveals a role for a novel snRNP-associated protein in the manufacture of spliceosomal snRNPs. EMBO J. 1996;15:4368–4379. [PMC free article] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Pai A, Weinberger LS. Fate-Regulating Circuits in Viruses: From Discovery to New Therapy Targets. Annu Rev Virol. 2017;4:469–490. doi: 10.1146/annurev-virology-110615-035606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, Bhatt DM, Lin CH, Tong AJ, Smale ST, Black DL. Splicing kinetics and transcript release from the chromatin compartment limit the rate of Lipid A-induced gene expression. RNA. 2013;19:811–827. doi: 10.1261/rna.039081.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. Journal of virology. 2008;82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SJ, Ridgeway WK, Robertson R, Wang J, Millar DP. HIV-1 Rev protein assembles on viral RNA one molecule at a time. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:1404–1408. doi: 10.1073/pnas.0807388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo Deborah J, Pauli A, Hacohen N, Schier Alexander F, Blackshear Perry J, Friedman N, et al. High-Resolution Sequencing and Modeling Identifies Distinct Dynamic RNA Regulatory Strategies. Cell. 2014;159:1698–1710. doi: 10.1016/j.cell.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos I, Bernal-Rubio D, Durham N, Belicha-Villanueva A, Lowen AC, Steel J, Fernandez-Sesma A. Effects of receptor binding specificity of avian influenza virus on the human innate immune response. Journal of virology. 2011;85:4421–4431. doi: 10.1128/JVI.02356-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razooky BS, Cao Y, Hansen MMK, Perelson AS, Simpson ML, Weinberger LS. Nonlatching positive feedback enables robust bimodality by decoupling expression noise from the mean. PLOS Biology. 2017;15:e2000841. doi: 10.1371/journal.pbio.2000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razooky BS, Gutierrez E, Terry VH, Spina CA, Groisman A, Weinberger LS. Microwell Devices with Finger-like Channels for Long-Term Imaging of HIV-1 Expression Kinetics in Primary Human Lymphocytes. 2012 doi: 10.1039/c2lc40170c. Under Revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razooky BS, Pai A, Aull K, Rouzine IM, Weinberger LS. A hardwired HIV latency program. Cell. 2015;160:990–1001. doi: 10.1016/j.cell.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roan NR, Munch J, Arhel N, Mothes W, Neidleman J, Kobayashi A, Smith-McCune K, Kirchhoff F, Greene WC. The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. Journal of virology. 2009;83:73–80. doi: 10.1128/JVI.01366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine IM, Weinberger AD, Weinberger LS. An evolutionary role for HIV latency in enhancing viral transmission. Cell. 2015;160:1002–1012. doi: 10.1016/j.cell.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9:676. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Bluthgen N, Marks DS, van Oudenaarden A. Gene expression. MicroRNA control of protein expression noise. Science. 2015;348:128–132. doi: 10.1126/science.aaa1738. [DOI] [PubMed] [Google Scholar]
- Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med. 2011;1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson ML, Cox CD, Sayler GS. Frequency domain analysis of noise in autoregulated gene circuits. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4551–4556. doi: 10.1073/pnas.0736140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson ML, Cox CD, Sayler GS. Frequency domain chemical Langevin analysis of stochasticity in gene transcriptional regulation. J Theor Biol. 2004;229:383–394. doi: 10.1016/j.jtbi.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Singh A, Razooky B, Cox CD, Simpson ML, Weinberger LS. Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression. Biophys J. 2010;98:L32–34. doi: 10.1016/j.bpj.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain PS. Efficient attenuation of stochasticity in gene expression through post-transcriptional control. J Mol Biol. 2004;344:965–976. doi: 10.1016/j.jmb.2004.09.073. [DOI] [PubMed] [Google Scholar]
- Symmons O, Raj A. What’s Luck Got to Do with It: Single Cells, Multiple Fates, and Biological Nondeterminism. Mol Cell. 2016;62:788–802. doi: 10.1016/j.molcel.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube R, Peterlin M. Lost in transcription: molecular mechanisms that control HIV latency. Viruses. 2013;5:902–927. doi: 10.3390/v5030902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MW, Bolovan-Fritts C, Dar RD, Womack A, Simpson ML, Shenk T, Weinberger LS. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell. 2012;151:1569–1580. doi: 10.1016/j.cell.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waks Z, Klein AM, Silver PA. Cell-to-cell variability of alternative RNA splicing. Mol Syst Biol. 2011;7:506. doi: 10.1038/msb.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger LS. A minimal fate-selection switch. Curr Opin Cell Biol. 2015;37:111–118. doi: 10.1016/j.ceb.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Weinberger LS, Dar RD, Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat Genet. 2008;40:466–470. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- Wodrich H, Schambach A, Krausslich HG. Multiple copies of the Mason-Pfizer monkey virus constitutive RNA transport element lead to enhanced HIV-1 Gag expression in a context-dependent manner. Nucleic acids research. 2000;28:901–910. doi: 10.1093/nar/28.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Beddall MH, Marsh JW. Rev-dependent indicator T cell line. Current HIV research. 2007a;5:394–402. doi: 10.2174/157016207781024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Beddall MH, Marsh JW. Rev-dependent lentiviral expression vector. Retrovirology. 2007b;4:12. doi: 10.1186/1742-4690-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science. 1998;279:84–88. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed method for construction of co-transcriptional, post-transcriptional and “mixed” mathematical models followed by parameter sensitivity analysis and experimental model validation. Stochastic simulations as well as supporting analytical arguments are also included.
Model parameters used, related to Figure 5
Table of primers and G-block Sequences, related to Figure 6 and STAR methods
(A) Analytical predictions shown together with statistics of Gillespie numerical simulations for the two-state, auto-repression, and auto-depletion models (red and blue dashed arrows “i” and “ii” correspond to dashed arrows in Figure 2C).
(B) Outputs of Gillespie simulations for each model in parameter regimes where auto-repression has the perverse effect of amplifying noise. 500 simulation runs were performed (in Mathematica™ using the xSSAlite™) for each model to calculate the mean and CV2. P indicates simulated protein levels. The two-state model is shown in grey, transcriptional auto-repression is shown in red and precursor auto-depletion in blue (mean ± standard deviation).
(C) Corresponding analytical predictions and statistics of Gillespie simulations for panel B.
(D) Fano factor of Gillespie simulations comparing the two-state, auto-repression, α auto-repression and auto-depletion feedback models. As predicted (see Supplementary Methods S1, page 17) the simulations show that in the koff ≫ kon regime, α auto-repression nearly matches the noise suppression capability of auto-depletion (Fano ~2000→680 vs. 612), however in the kon > koff regime, α auto-repression only gets one halfway to the noise suppression capability of auto-depletion (Fano 61→49 for auto-repression vs. 61→41 for auto-depletion).
(A) Representative smFISH images for HIV-infected Jurkat cells. Three sets of probes, US, SS, and MS probes, were designed to bind to the Pol/Vif, Env, and d2GFP reading frames on HIV-d2G, respectively. Each probe set contains at least 27 single probes that are conjugated with fluorophores. Due to the inherent limitations of the technique and available fluorophore probes, only any two of the three RNA species could be imaged at once. Using the US together with the SS probe set and the SS together with the MS probe set, we detected all three alternative splicing variants.
(B) TC analysis showing US mRNA versus spliced mRNA (either SS or MS) at each TC. Each dot represents one TC. The full lines represent the expected spliced versus US behavior for 80% 50% and 0% spliced transcripts at the TC. Most transcriptional centers fall along the line representing 0% spliced transcripts at the TC. The shaded area shows the expected variability for 0% spliced transcripts at the TC. Spliced RNAs are quantified by subtraction of the intron signal from the exon signal (Bahar Halpern and Itzkovitz, 2016). Since the intron is at the 5′ end of the transcript and the exon is at the 3′ end of the transcript (see left inset), during transcriptional elongation a subset of transcripts will exhibit only the intron signal (as the exon has not yet been transcribed). Thus, upon subtraction (exon – intron), the numbers of RNAs will appear negative for the subset of elongating transcripts that have partially completed transcription and only contain the 5′ intron region (where the US probes bind) but haven’t yet transcribed the 3′ exon region (where the MS probes bind). Right inset: Mean increase of US, SS and MS transcripts in the nucleus after transcriptional activation with TNF. Error bars represent SEM.
(C) Mean increase of unspliced (US) and spliced (MS + SS) transcripts in the nucleus (top) and cytoplasm (bottom) after transcriptional activation with TNF. Error bars represent SEM.
(D) 5′-Capped, and 3′-polyadenylated dHIV-BlaM (β-lactamase) pre-mRNA was in vitro transcribed, purified, and nucleofected into naïve Jurkat cells. After a 2-hour recovery post-nucleofection, CCF2-AM, the fluorescent substrate of β-lactamase, was loaded into cells. Cells that successfully took up unspliced pre-mRNA into their nuclei and spliced the pre-mRNA post-transcriptionally, express BlaM and convert the green fluorescent substrate into the blue fluorescent product. Otherwise, the cells remain green due to uncleaved CCF2-AM substrate which cannot be cleaved due to lack of BlaM expression. Flow cytometry was later utilized to determine the percentage of cells capable of post-transcriptional splicing.
(E–F) Cells nucleofected with different RNA constructs show that unspliced HIV-1 pre-mRNA, as well as β-globin, a gene known to be co-transcriptionally spliced, recruit spliceosome machinery post-transcriptionally in vivo. The positive control consisted of an RNA segment containing the full β-lactamase gene, which did not require splicing to express BlaM. Similar results were obtained upon nucleofection of an equivalent mass of RNA (2 μg) or equivalent molar amounts of RNA (1.11 pmoles) into cells.
(A) Numerical simulations of post-transcriptional splicing ODE model showing cytoplasmic MS and US mRNA (green and orange line respectively), and GFP (black line). See Supplementary Methods S1 and Table S2 for equations and parameters.
(B) Statistics of trajectories for the spliced reporter HIV-d2G. More than 80% of cells exhibit an overshoot (i.e., overshoot or oscillating) trajectory during an 18 hour imaging experiment while more than 40% of cells display oscillating features in the later hours, after the overshoot peak.
(C) Direct measurement of negative-feedback regulation by temporal autocorrelation function (ACF) analysis of single-cell trajectories, as described in (Weinberger et al., 2008), after transcriptional activation with TNF. Feedback strength is calculated from the ACF half-correlation time (CT50) for a given 5-hour window normalized to the initial 5-hour time window (i.e., [CT50window/CT500–5hrs − 1] such that negative values correspond to negative feedback). Each ACF is calculated from at least 100 single-cell trajectories. The HIV MS reporter (green) exhibits significant negative feedback beginning at ~5 hours. Overexpression of the Rev-M10 mutant in the same cells (red) or Leptomycin B treatment (blue) abrogate negative feedback.
(D–E) Infection and time-lapse single-cell imaging of human donor-derived primary CD4+ T cells and primary monocyte-derived macrophages (primary MDMs). Primary CD4+ T cell imaging was conducted in micro-well chambers, as described in (Razooky et al., 2012). The general trend line shows an overshoot similar that seen in Jurkat cells. Primary MDMs were obtained by incubating primary CD17+/CD4+ monocytes with GM-CSF for 10 days. Differentiated primary MDMs were infected with VSV-G pseudo-typed HIV-1-d2G and imaged at 10-minute intervals for 40 hours. Averaged trend line (n = 489 cells) exhibits the negative-feedback overshoot. (Error bars represent the standard error.)
(A) Single-cell time-lapse imaging of Jurkat cells transduced with an HIV-1 LTR-d2GFP (Ld2G) lentiviral vector after induction with TNF. General trend line (n=255) does not exhibit the overshoot in GFP at 5–10 hours, as compared to full-length HIV. (Error bar represents the standard error.)
(B) Single-cell time-lapse imaging of cells transduced with an HIV-1 LTR-d2GFP-IRES-Tat (Ld2GT) lentiviral vector after TNF induction. Ld2GT represents a minimal Tat positive-feedback circuit (i.e., lacking Rev). The general trend line (n=135) does not exhibit the characteristic negative-feedback overshoot. (Error bar represents the standard error.)
(C) Single-cell time-lapse imaging of primary CD4+ T cells transduced with either Ld2G (black) or Ld2GT (grey) lentiviral vectors and transcriptionally activated by TNF induction.
(D) Flow cytomtery analysis of cells stably transduced with an HIV spliced (MS) reporter (dHIV-d2G) after serial induction with TNF. 12 hours after first induction (i.e., after GFP expression has peaked), a second TNF (or TSA) stimulation was added to the culture, and the GFP expression was quantified by flow cytometry. The results show that the second reactivation with TNF or TSA (black or grey bars, respectively) does not rescue the decaying GFP level (compare to white bars), indicating that the turnover of GFP is not due to the silencing of LTR promoter.
(E) ODE model predictions for enhancement of Rev-dependent export (kr). As the export rate increases, the overshoot from precursor auto-depletion feedback increases as exhibited by the accelerated response time (a measure of feedback strength) during the post-peak decay phase.
(F) Single-cell time-lapse imaging of GFP expression in Jurkat cells stably transduced with the HIV spliced (MS) reporter dHIV-d2G (n=97), which encodes the wild-type RRE within env as well as a second distal RRE within the Δgag/RRE. The overshoot is enhanced and the post-peak response time is accelerated (50% response time of decay is accelerated by ~5 hours, compared to Figure 4B). This construct indicates that a Tat/Rev/Env cassette is sufficient to generate the same overshoot dynamics as the full-length construct, compared to the Ld2G and Ld2GT constructs. (Error bars represent ± standard error.)
(A) Nonlinear least–squares fitting of ODE models to single-cell imaging data. Only the post-transcriptional splicing/Rev negative feedback model provides a good fit. Although the “mixed” model does not fit the data as well as the post-transcriptional model, it is still capable of generating an overshoot trajectory, indicating that post-transcriptional splicing is essential to generate negative feedback and the feedback is robust to potential RNA “leakage” into the system from co-transcriptional splicing. Inset: nonlinear least-squares fitting of GFP signal from single-cell time-lapse imaging data to the ODE model for post-transcriptional splicing when active Tat positive feedback is included (i.e., Tat feedback does not “saturate”). (B) Hill coefficient (h) of Rev/RRE nuclear export should be larger than 3 to fit HIV single-cell time-lapse imaging data. When the h is lower than 3, the R-squared value of post-transcriptional model fitting is less than 0.9. When the h is large (e.g., 12), other parameters can be made to fit the reference/physiologically reasonable range.
(C–D) The post-transcriptional (Rev negative feedback) model predicts how the dynamics of HIV gene expression respond to exogenous Rev over expression. Surprisingly, expression of US (C) and MS (D) gene products decrease upon Rev over expression. This prediction contradicts the common conclusion that Rev upregulates the expression of US RNA.
(E–F) Overexpression of Rev decreases the level of p24 expression in full-length HIV-1 (E) Jurkat cells latently infected with HIV-d2G were transfected with pcRev plasmid DNA. At 24 hours after transfection, TNF-α was added to activate viral replication. GFP and p24 levels were measured 24 hours after TNF-α treatment, using immunostaining and flow cytometry. The levels were normalized to the background fluorescence of naïve Jurkat cells. As Rev overexpression was increased, GFP levels decreased. (F) The intracellular p24 level also decreased as Rev levels were increased.
(A) Diagram showing where the respective RT-PCR primer sets bind.
(B) Right panel: RT-PCR data from HIV and mutant transduced cells after TNF induction. Total cellular RNA was collected 4 hours after TNF activation. D1 to A3, D1 to A4c and D1 to A5 are the first splicing events used for Tat, Rev and Nef, respectively. D4 to A7 is a splicing event shared by Tat, Rev and Nef. D4 to A7 splicing is enhanced by the mutations shown in Figure 6A. Data represent the mean and SEM of two biological replicas. Left panel: Quantification of intracellular protein expression for proteins translated from spliced and unspliced HIV-1 transcripts in the HIV-1 A7 mutants relative to the A7 wild-type HIV-1 by direct immunofluorescence (for p24) and GFP fluorescence. These data were quantified by flow cytometery.
(C) Raw slope data corresponding to Figure 6C showing the GFP and mCherry slopes for A7 mutants and wild-type HIV (WT). The increased mCherry slope in the mutants is consistent with reduced HIV US RNA that acts as RRE-export competitor for the mCherry-RRE in the mutants (i.e., less US HIV RNA in the mutants).
Cellular GFP expression, related to Figure 4
(A–B) The ODE model used is described in detail in Supplementary Methods S1, specifically see section entitled: “Modeling circuit-relaxation dynamics after a TNF pulse-chase experiment (Figure 7, main text)”. Briefly, gray trajectory represents wild-type HIV (sp = 1.5) and red trajectory represents A7 mutants (sp = 15); blue trajectory (sp = 0.15) represents a hypothetical slower-splicing virus shown to illustrate the trend of the dynamics. At time = 48 h, basal LTR expression is instantaneously reduced by threefold to simulate TNF removal. In contrast to the data in Figure 7A, more efficient splicing results in slower turning off (i.e., crossing of GFP = 0.005 occurs at a later time the more efficient the splicing). (B) An expansion of panel A (expansion of x axis from time = 48 to 96 h). “simulated GFP” represents GFP.
(C) Active-state stability by flow cytometry for wild-type HIV d2G after a pulse of TNF reactivation (TNF removed at time 0) in the presence (black) and absence (blue) of the LTR transcriptional noise-suppressor molecule (Dar et al., 2014), corresponding to Figure 7D. As predicted, due to the intact negative feedback in the wild type, the noise suppressor has minimal effect on the stability of wild-type HIV d2G. Histogram corresponds to the 48-hour time point.
(D) Summary analysis of the number of mutations in the 41-nucleotide sequence region surrounding the A7 splice-acceptor across HIV and SIV sequences representing global diversity in the Los Alamos HIV database compendium (alignment is of nucleotides 8337–8378 in the HIV-1 HXB2 reference genome, which is identical to the ‘wild-type’ NL4-3 molecular clone used in this study). The analysis summarizes sequence alignments obtained using the AnalyzeAlign tool (http://www.hiv.lanl.gov/) of HIV-compendium sequences (200 HIV-1 sequences and 100 HIV-2/SIV sequences representing global diversity) and indicates a relatively low mutation rate, with most sequences having two to four differences from consensus.
(E) Top: Sequence logo from alignments of the HIV compendium sequences, using the Los Alamos AnalyzeAlign tool, for the A7 splice-acceptor region (nucleotides 8337-8378 in the HXB2 HIV-1 reference genome. For reference, the consensus sequence for ‘wild-type’ HIV-1 NL4-3 (8337-8378) is: GTGAATAGAGTTAGGCAGGGATATTCACCATTATCGTTTCAG. Bottom: Corresponding sequence logo for mutations from consensus.
(F) Ratio of viral genomic RNA (gRNA), collected from supernatants of A7-4 and wild-type infected cells after 48 hours. (qPCR was used to quantify packaged US RNA in the supernatant of A7-4 infected cells versus wild-type cells at 48 hours post infection). Data represent the mean and SEM of two biological replicas.