

Rotavirus replicates exclusively in the cytoplasm. Knowledge on the relocalization of nuclear proteins to the cytoplasm or the role(s) of host proteins in rotavirus infection is very limited. In this study, it is demonstrated that rotavirus infection induces the cytoplasmic relocalization of a large number of nuclear RNA-binding proteins (hnRNPs and AU-rich element-binding proteins). Except for a few, most nuclear hnRNPs and ARE-BPs, nuclear transport proteins, and some cytoplasmic proteins directly interact with the viroplasmic proteins NSP2 and NSP5 in an RNA-independent manner and become sequestered in the viroplasms of infected cells. The host proteins differentially affected viral gene expression and virus growth. This study demonstrates that rotavirus induces the relocalization and sequestration of a large number of host proteins in viroplasms, affecting host processes in both compartments and generating conditions conducive for virus growth in the cytoplasm of infected cells.
KEYWORDS: rotavirus, viroplasm, nuclear transport, nuclear-cytoplasmic relocalization, colocalization, hnRNPs, AU-rich element-binding proteins (ARE-BPs), pulldown assay, immunofluorescence confocal microscopy, sequestration, immune confocal microscopy
ABSTRACT
Rotavirus replicates in the cytoplasm of infected cells in unique virus-induced cytoplasmic inclusion bodies called viroplasms (VMs), which are nucleated by two essential viral nonstructural proteins, NSP2 and NSP5. However, the precise composition of the VM, the intracellular localization of host proteins during virus infection, and their association with VMs or role in rotavirus growth remained largely unexplored. Mass spectrometry analyses revealed the presence of several host heterogeneous nuclear ribonucleoproteins (hnRNPs), AU-rich element-binding proteins (ARE-BPs), and cytoplasmic proteins from uninfected MA104 cell extracts in the pulldown (PD) complexes of the purified viroplasmic proteins NSP2 and NSP5. Immunoblot analyses of PD complexes from RNase-treated and untreated cell extracts, analyses of coimmunoprecipitation complexes using RNase-treated infected cell lysates, and direct binding assays using purified recombinant proteins further demonstrated that the interactions of the majority of the hnRNPs and ARE-BPs with viroplasmic proteins are RNA independent. Time course immunoblot analysis of the nuclear and cytoplasmic fractions from rotavirus-infected and mock-infected cells and immunofluorescence confocal microscopy analyses of virus-infected cells revealed a surprising sequestration of the majority of the relocalized host proteins in viroplasms. Analyses of ectopic overexpression and small interfering RNA (siRNA)-mediated downregulation of expression revealed that host proteins either promote or inhibit viral protein expression and progeny virus production in virus-infected cells. This study demonstrates that rotavirus induces the cytoplasmic relocalization and sequestration of a large number of nuclear and cytoplasmic proteins in viroplasms, subverting essential cellular processes in both compartments to promote rapid virus growth, and reveals that the composition of rotavirus viroplasms is much more complex than is currently understood.
IMPORTANCE Rotavirus replicates exclusively in the cytoplasm. Knowledge on the relocalization of nuclear proteins to the cytoplasm or the role(s) of host proteins in rotavirus infection is very limited. In this study, it is demonstrated that rotavirus infection induces the cytoplasmic relocalization of a large number of nuclear RNA-binding proteins (hnRNPs and AU-rich element-binding proteins). Except for a few, most nuclear hnRNPs and ARE-BPs, nuclear transport proteins, and some cytoplasmic proteins directly interact with the viroplasmic proteins NSP2 and NSP5 in an RNA-independent manner and become sequestered in the viroplasms of infected cells. The host proteins differentially affected viral gene expression and virus growth. This study demonstrates that rotavirus induces the relocalization and sequestration of a large number of host proteins in viroplasms, affecting host processes in both compartments and generating conditions conducive for virus growth in the cytoplasm of infected cells.
INTRODUCTION
Rotaviruses, a major cause of acute gastroenteritis in infants and young children (1), have a genome composed of 11 segments of double-stranded RNA (dsRNA) enclosed in an icosahedral triple-layered protein capsid. The viral genome encodes six structural viral proteins (VP1 to VP4, VP6, and VP7) and six nonstructural proteins (NSP1 to NSP6) (2, 3). During virus entry into the cell, removal of the outer layer from the triple-layered particle (TLP) activates the synthesis and extrusion of the viral positive-strand single-stranded RNAs (ssRNAs) through dedicated class I channels of the double-layered particles (DLPs) into the cytoplasm of the infected cell (4–7). The positive-strand RNAs function as mRNAs for viral protein synthesis and as pregenomic templates for negative-strand synthesis.
Key steps in the virus replication cycle, such as viral genome replication and the assembly of immature DLPs, take place exclusively in the cytoplasm of infected cells in specialized non-membrane-bound electron-dense cytoplasmic inclusion bodies called viroplasms (VMs). These bodies are nucleated by two essential nonstructural proteins, NSP2 and NSP5, and the inner virion capsid protein VP2, with NSP5 being crucial for both the recruitment of viroplasmic proteins and the architectural assembly of VMs (8–14). VP2 appears to be associated with the formation of replication complexes consisting of a positive-strand ssRNA segment, VP1, VP3, and a pentamer of VP2 rather than with VM formation (7, 15, 16). Viroplasm-like structures (VLSs) are formed in transfected cells coexpressing NSP5 with either NSP2 or VP2 (9, 10, 12, 14). At early times (3 to 5 h) postinfection, the number of small VMs increases, but by 8 h postinfection (hpi), several small VMs join to form large VMs, resulting in a decrease in the overall number of VMs (17, 18). The temporal transition of small VMs into large VMs and their concomitant movement to the perinuclear region depend on NSP2 and VP2 and appear to be stabilized by the acetylated microtubular network (19, 20). The formation of VMs seems to be initiated by a specific interaction of the cytoplasmic disperse form of NSP2 (dNSP2) with acetylated tubulin and hypophosphorylated NSP5. The maturation of these nascent VMs is associated with the phosphorylation of NSP5 and the viroplasmic form of NSP2 (vNSP2) (21).
As obligate intracellular parasites with small genomes, many RNA viruses are highly dependent on cellular factors, particularly the heterogeneous nuclear ribonucleoproteins (hnRNPs), for the completion of productive replication, coopting them into new roles, modulating their functions through the induction of nuclear-cytoplasmic relocalizations and/or posttranslational modification and cleavage (22–24). Cellular hnRNPs (25–30) and AU-rich element-binding proteins (ARE-BPs) (31–33) are predominantly nuclear, but many, with the exception of hnRNP C1/2 and hnRNP U (25–27), shuttle between the nucleus and the cytoplasm and often exhibit stress-induced or stimulus-dependent cytoplasmic relocalization. hnRNPs and ARE-BPs function in both cellular compartments at multiple levels of mRNA metabolism, encompassing transcription through translation and stability.
The cytoplasmic relocalization of the nuclear protein RoXaN and the nuclear accumulation of the cytoplasmic poly(A)-binding protein PABPC1 in rotavirus-infected cells (34, 35) are the only examples reported to date of cellular factors being affected by rotavirus infection. However, the significance of these changes for virus infection is as yet not clearly understood (36–38). To date, there have been no studies on the cytoplasmic relocalization of hnRNPs, ARE-BPs, and other host proteins; their association with VMs; or their possible role(s) in virus replication in infected cells. To investigate the possible relocalization of nuclear proteins to the cytoplasm during rotavirus infection and the association of host factors with viroplasmic complexes, the interactions of cellular proteins with the viral VM proteins NSP2 and NSP5 were examined by using mass spectrometry, pulldown (PD) assays using control cell extracts, coimmunoprecipitation (co-IP) analyses using virus-infected cell extracts, and immunofluorescence confocal microscopy (ICM) analyses. The influence of the relocalized host proteins on virus infection was examined by their ectopic overexpression and small interfering RNA (siRNA)-mediated downregulation of expression.
RESULTS
Identification of host cell proteins in PD complexes with NSP2 and NSP5 by mass spectrometry analyses.
Mass spectrometry analyses of PD complexes of N-terminal hexahistidine (NH)-tagged NSP2 and NSP5 from human rotavirus IS2 (39) with extracts from uninfected rotavirus-permissive MA104 cells revealed the presence of several nuclear hnRNPs (see Tables S1 and S2 in the supplemental material). PD complexes with both NSP2 and NSP5 contained hnRNPs D, I (polypyrimidine tract-binding protein [PTBP]), K/J, F/H, L, and U. The coat protein complex II (COPII) secretory vesicle component Sec31A was also detected in both complexes, whereas hnRNPs A1/3, E1/2 [poly(rC)-binding proteins 1 and 2 (PCBP1/2)], and M; the cytoplasmic vacuolar protein sorting-associated protein VPS35; and the small-subunit ribosomal protein RPS8 were detected only in NSP5 complexes, and β-tubulin, which has been reported to interact with NSP2 (19–21), was detected only in the NSP2 complexes (Tables S1 and S2).
In addition, pre-mRNA cleavage polyadenylation specificity factor 6 (CPSF6), HSP90, Hsc70, HSPA1A (Hsp70), and HSPA5 (GRP78/BiP) in NSP2 and NSP5 complexes and kinesin heavy chain 1 and eIF4A in NSP5 complexes were observed (Tables S1 and S2) but were not further investigated in this study.
It is important to note that control uninfected cell extracts were deliberately used in the PD assays, as NSP2 and NSP5 are known to interact with each other as well as with other viral proteins (9, 10, 12, 14, 40, 41). Consequently, had extracts been made from virus-infected cells, it would not have been possible to establish whether cellular proteins captured in the PD complexes were directly interacting with bead-bound NSP2 or NSP5 or being recruited to the PD complex through their interaction with another viral protein(s) and viroplasm assemblies present in virus-infected cells.
The presence of some cellular proteins in the PD complexes could not be unequivocally established using mass spectrometry. Consequently, where the necessary immunological reagents were available, the possible binding of hnRNPs and other well-studied ARE-BPs (31–33), besides hnRNP D, was examined by immunoblotting of the PD complexes. This revealed that hnRNPs A1, C1/2, and M, which were not detected by mass spectrometry in the NSP2 PD complexes, and the ARE-BPs human antigen R (HuR), butyrate response factor 1 (BRF1), T cell-restricted intracellular antigen 1 (TIA1), TIA1-like 1 (TIAL1), KH-type splicing regulatory protein (KSRP), and Staufen1, which were not detectable in the PD complexes of both NSP2 and NSP5, were detectable by immunoblotting (Fig. 1a).
FIG 1.

Interaction of cellular hnRNPs and ARE-BPs in RNase-treated and untreated cell extracts with purified recombinant viral nonstructural proteins NSP2 and NSP5 demonstrated by a pulldown assay. The recombinant NH-tagged viral proteins NSP5 and NSP2 were purified from E. coli by affinity chromatography using Ni2+-NTA-agarose beads. Control Ni2+-NTA-agarose beads, which were prepared by passing the lysate from E. coli harboring the pET22-NH vector lacking the viral gene, were used for mock binding. Both the experimental and control beads were further incubated in binding buffer containing 0.5% BSA to minimize the nonspecific binding of cellular proteins. (a and b) The RNase-treated purified recombinant NSP2 and NSP5 proteins bound to Ni2+-NTA-agarose beads, and the control beads (mock binding) were incubated with equal amounts (500 μg) of control MA104 cell extracts that were either not treated with RNase (a), similar to what was done for mass spectrometry, or treated with RNase (b). The cellular proteins bound to the beads were resolved by SDS-PAGE, and the interacting cellular proteins were detected by immunoblotting. In the lane representing 10% input, 50 μg of the RNase-treated or untreated cell extracts was loaded. The same blot was used to detect two or three host proteins by sequential deprobing and reprobing depending on clear differences in the molecular weights of the proteins. Each PD assay was repeated at least 3 to 4 times to confirm reproducibility. (c) The cell extracts (1 mg/ml) were incubated with 100 μg of RNase A for 45 min at room temperature, and 100 μg of the RNase-treated and untreated cell extracts was resolved by agarose gel electrophoresis and visualized by ethidium bromide staining. Note the complete digestion of cellular RNA in the RNase-treated extract. M, molecular marker. (d) Expression and purification of GST-tagged recombinant host proteins. The bacterial cell extracts were incubated with RNase A (100 mg/ml) prior to purification. (e) Demonstration of direct interactions of purified NH-NSP2 and NH-NSP5 with glutathione bead-bound GST-tagged nuclear proteins. Ten micrograms of purified NH-NSP2 or NH-NSP5 was incubated with approximately 5 μg of the bead-bound recombinant GST-tagged hnRNPDp40 isoform and hnRNP K (top) and hnRNP F and RPS8 (bottom) treated further with RNase A (10 mg/ml), and the bound viral protein was detected by Western blotting (WB).
To both complement and extend the mass spectrometry data, immunoblot assays were carried out. The MA104 cell extracts used for the mass spectrometry-based analyses were not treated with RNase in order to not lose the possible RNA-mediated interactions. Therefore, in the immunoblot-based assays, comparative analyses using both RNase-treated and untreated cell extracts were performed to distinguish the possible RNA-mediated interactions from RNA-independent interactions (Fig. 1a and b). The results shown in Fig. 1a and b independently confirmed the interacting cellular proteins identified by using mass spectrometry of PD complexes. In addition, while all the ARE-BPs, including hnRNP D; HuR; the TIS11 family proteins BRF1, TIA1, and TIAL-1; KSRP; and Staufen1, from RNase-untreated extracts bound to NSP5 and NSP2, the interactions of BRF1 and TIAL-1 with NSP2 were abolished upon RNase treatment of the extracts (Fig. 1b and c). Of note, RNase treatment of the extracts resulted in enhanced binding of HuR to NSP5 but reduced binding to NSP2 (Fig. 1b). These results suggest that while the interactions of the majority of the host proteins with both NSP5 and NSP2 are RNA independent, those of BRF1 and TIAL-1 with NSP2 are RNA dependent. Finally, the results in Fig. 1b show that RNase treatment of cell extracts resulted in a significant reduction in the binding of hnRNP A1 to NSP2. The results on RNA-independent and -dependent interactions are summarized in Table 1.
TABLE 1.
Summary of interactions between the viroplasmic proteins NSP2 and NSP5 and host proteinsa
| Host protein | Interaction by mass spectrometry |
Interaction by PD-Western blotting |
Colocalization with VM | Total protein level (8 hpi) | ||
|---|---|---|---|---|---|---|
| NSP5 | NSP2 | NSP5 | NSP2 | |||
| hnRNPs | ||||||
| hnRNP A1 | + | − | + | +c | − | ↑ |
| hnRNP C1/2 | + | − | + | + | + | ↑ |
| hnRNP D | + | + | + | + | + | ↓ |
| hnRNP E | + | + | + | + | + | ↑ |
| hnRNP F | + | + | + | + | + | ↑ |
| hnRNP H | + | + | + | + | + | ↑ |
| hnRNP I | + | + | + | + | + | ↑ |
| hnRNP K | + | + | + | + | + | ↑ |
| hnRNP L | + | + | + | + | + | ↑ |
| hnRNP M | + | − | + | + | + | ± |
| hnRNP Q | − | − | − | − | − | ↑ |
| hnRNP U | + | + | + | + | + | ↑ |
| ARE-BPs | ||||||
| BRF1 | − | − | + | +b | + | ↓ |
| HuR | − | − | − | +c | + | ↑ |
| hnRNP D | + | + | + | + | + | ↓ |
| KSRP | − | − | NT | NT | + | ↑ |
| Staufen | − | − | + | + | + | ± |
| TIA1 | − | − | + | + | + | ↑ |
| TIAL-1 | − | − | − | +b | + | ↓ |
| TTP | − | − | NT | NT | + | ± |
| Other nuclear/cytoplasmic proteins | ||||||
| RPS8 | + | − | + | − | + | ↑ |
| VPS35 | + | + | + | − | + | ↑ |
| Sec31A | + | + | + | + | + | ↑ |
| α/β-Tubulin | − | + | NT | NT | NT | ± |
| G3BP1 | − | − | − | − | − | ↓ |
| Transport proteins | ||||||
| Transportin1 | − | − | − | − | + | ↓ |
| Exportin1 | − | − | + | − | + | ↑ |
| Importin-β | − | − | − | + | + | ↑ |
| Ran | − | − | − | + | + | NT |
The interactions identified between the cellular proteins and viral proteins NSP2 and NSP5 by a PD assay followed by immunoblotting and colocalization by ICM (Fig. 1a and b and 4) are summarized. +, positive interaction/colocalization; −, no interaction/colocalization. ↑ and ↓ indicate increased and decreased host protein levels in virus-infected cells in comparison to those in uninfected cells, respectively; ± indicates that there was no significant change in the protein levels between virus-infected and uninfected cells. NT, not tested.
Loss of binding of NSP2 with BRF1 and TIAL-1 in RNase-treated cell extracts.
Reduced binding of NSP2 with hnRNP A1 and HuR in RNase-treated cell extracts.
That the host proteins interact directly with NSP2 and NSP5 was further demonstrated by a direct binding PD assay using a few purified recombinant host proteins and the two viral viroplasmic proteins (Fig. 1d and e).
The proteins listed in Tables S1 and S2 in the supplemental material are only those that were exclusively detected in the NSP5- or NSP2-bound PD complexes, and those that were also found in PD complexes generated using control Ni-nitrilotriacetic acid (NTA)-agarose beads were excluded. However, it is important to note that neither mass spectrometry nor immunoblot analyses allowed any assessment to be made of the relative affinities with which cellular proteins bound to control and viral protein-bound beads; consequently, the proteins listed in Tables S1 and S2 in the supplemental material are likely to represent only a partial cataloguing of the complete interactome.
Cell compartment relocalization of cellular proteins detected in PD complexes during rotavirus infection.
Rotavirus replication is confined to the cytoplasm of the virus-infected cell, and neither NSP2 nor NSP5 is known to localize to the nucleus. Consequently, the detection of a large number of host cell nuclear proteins in the PD complexes of NSP2 and NSP5 (Fig. 1a and b) suggested the possibility of their cytoplasmic relocalization following rotavirus infection. As nothing is known about the localization of hnRNPs in rotavirus-infected cells, the intracellular distribution of NSP5- and NSP2-interacting hnRNPs and ARE-BPs during the course of rotavirus infection was investigated by immunoblotting.
The analysis of the nuclear and cytoplasmic fractions at different time points after infection of rhesus rotavirus (RRV)-infected MA104 cells (Fig. 2a, right) and serum-starved control mock-infected cells (Fig. 2a, left) revealed that a number of the hnRNPs analyzed were specifically enriched in the cytoplasmic fraction of virus-infected cells during the course of infection. For clarity, the proteins in Fig. 2 are discussed according to their relative pattern of intracellular localization during infection. The cytoplasmic relocalizations of hnRNPs A1, K/J, and U (Fig. 2a) and of HuR, hnRNP D, and BRF1 (Fig. 2b) were specifically induced by virus infection and not by simple serum starvation-induced stress in mock-infected MA104 cells. In contrast, hnRNPs C1/2, F/H, L, and M (Fig. 2a) and the ARE-BPs KSRP and TIA1 (Fig. 2b) showed a partial relocalization to the cytoplasm in mock-infected cells following serum starvation-induced stress, and virus infection caused further increases in their cytoplasmic relocalization. To date, both hnRNPs C1/C2 and U have been considered to be nonshuttling proteins (25–27), but rotavirus infection clearly induces their cytoplasmic relocalization. hnRNP E levels increased in both compartments in both mock-infected and virus-infected cells (approximately 2.5-fold at 8 hpi), which appeared to be primarily due to serum starvation but not rotavirus infection. Of note, both isoforms of hnRNP E were observed in the cytoplasm, but only one was observed in the nucleus.
FIG 2.

Analysis of nuclear and cytoplasmic distributions of hnRNPs, ARE-BPs, and other proteins in mock-infected and rotavirus RRV-infected MA104 cells during the course of infection. (a) Time course immunoblot analysis of the levels of hnRNPs in the nuclear and cytoplasmic fractions. The samples in the lane from 0 hpi represent nuclear and cytoplasmic fractions from serum-grown control cells. Lanes from 2 to 10 hpi on the left represent mock-infected cells incubated for the indicated time periods in medium lacking serum, similar to the conditions under which virus infections were performed, and those on the right represents lysates prepared from RRV-infected cells (2 to 10 hpi). NSP5 was detected using protein A-agarose affinity-purified rabbit PAb generated against purified recombinant NSP5, and VP6 was detected using subgroup I MAb 631/9, which are very specific to the viral proteins, with no cross-reactivity to host proteins. β-Actin and PCNA were used to determine the purity of the cytoplasmic and nuclear fractions, respectively, and as internal loading controls. At each time point, 50 μg of the protein was analyzed. C.E., cytoplasmic extract; N.E., nuclear extract. (b) Analysis of the altered nuclear and cytoplasmic distributions of ARE-BPs during rotavirus infection. NSP2 and VP6 were detected using affinity-purified rabbit PAbs generated against purified NSP2 and RRV DLPs, respectively. For other details, see the legend to panel a. (c) Analysis of intracellular levels of host cytoplasmic proteins that interact with NSP5 and/or NSP2. See the legends to panels a and b for details. (d) Nuclear-cytoplasmic levels of nuclear transport proteins during rotavirus infection. Note the enhanced cytoplasmic retention of nuclear transport proteins during rotavirus infection in MA104 cells. ns, not significant. Other details are described in the legend to panel a. See Table S3 in the supplemental material for quantification of the changes in the protein levels in the nucleus and cytoplasm during the course of infection, and see Fig. S1 for changes in total protein levels.
The increased cytoplasmic accumulation of hnRNPs A1, K/J, L, I, and M and the ARE-BPs HuR, hnRNP D, and BRF1 (Fig. 2a and b) during virus infection correlated with concomitant decreases in the nucleus. Note that for hnRNPs A, D, F/H, L, K, Q, and U; HuR; and BRF1, no signal could be detected in the cytoplasm of serum-grown control and/or serum-starved cells during the course of the experiment using 50 μg of protein in Western blot analyses (Fig. 2a and b; see also Table S3 in the supplemental material). The ARE-BPs FXR1, TIAL-1, tristetraprolin (TTP), and Staufen1 were abundant and stable in the cytoplasm. While there was no change in the levels of TTP and Staufen1 in the nucleus, those of FXR1 and TIAL-1 decreased during the course of infection (Fig. 2b and Table S3). Staufen1 showed cleavage during the late period of infection. The relative increases or decreases in the levels of hnRNPs and ARE-BPs in the nuclear and cytoplasmic fractions in infected cells (Table 1) correlated with the total protein levels between control and infected cells (Fig. S1a and S1b).
Immunofluorescence confocal microscopy (ICM) analysis reveals the predominant nuclear localization of the majority of the hnRNPs (Fig. S2a) and ARE-BPs (Fig. S2b) in serum-grown control MA104 cells. TTP and TIAL-1 showed significant presences in both compartments, correlating with the results of the analysis of the nuclear and cytoplasmic fractions (Fig. 2b and Fig. S2b).
hnRNP Q showed no change in its localization and was detected only in the nucleus under all conditions. No change in either the localization or abundance of PCNA and β-actin, used as nuclear and cytoplasmic control proteins, respectively, was observed under any condition (Fig. 2a and b and Table S3), suggesting that the cytoplasmic relocalization of nuclear proteins is selective but not global.
The cytoplasmic proteins RPS8 and VPS35, which were detected in the PD complexes with NSP5, and Sec31A, which was detected in complexes with NSP2 and NSP5, showed significant increases (4- to 10-fold) in their levels in the cytoplasm of virus-infected MA104 cells (Fig. 2c). Of note, while RPS8 showed a significant increase in its level in the nucleus in both mock- and virus-infected cells, a small proportion of Sec31A was detectable in the nucleus during late stages of infection. In contrast, VPS35 was detected only in the cytoplasm. The total protein levels of RPS8, Sec31A, and VPS35 showed approximately 13-, 1.4-, and 3.9-fold increases, respectively, correlating with the changes observed in their cytoplasmic and/or nuclear levels (Fig. 2c and Fig. S1c). PABPC1, which has been previously shown to relocalize to the nucleus during rotavirus infection, showed 30% and 90% reductions in the cytoplasm of mock-infected and virus-infected cells but exhibited increases in the nucleus of approximately 5.5- and 9.5-fold, respectively, at 8 hpi (Fig. 2c and Table S3), suggesting that its relocalization to the nucleus is partly due to serum starvation during virus infection. The total protein levels of both PABPC1 and G3BP1 were marginally reduced in infected cells (Fig. S1c).
Intracellular distribution of nuclear transport proteins in rotavirus-infected cells.
The relocalization of some host cell proteins from the nucleus to the cytoplasm observed following rotavirus infection raised the possibility that the nuclear-cytoplasmic transport processes may suffer dysregulation following virus infection. To explore this possibility, the nuclear-cytoplasmic abundance of a few known nuclear transport factors (42, 43) was examined by immunoblotting. This revealed 2- to 3-fold increases in the levels of Exportin1, importin-β, and Ran in the cytoplasm of rotavirus-infected cells compared to those seen in either control or serum-starved mock-infected MA104 cells, but there was no significant change in the cytoplasmic levels of Transportin1 (Fig. 2d and Table S3). In contrast, in the nucleus, the level of Transportin1 decreased by approximately 3-fold following virus infection, while there was no significant change in the level of either Exportin1 or importin-β. The steady-state level of the Ran transporter increased to the same extent in both cellular compartments following virus infection. Comparison of the total protein levels in control and infected cells revealed a good correlation with the changes observed in the nuclear and cytoplasmic fractions (Fig. S1d).
Relocalized host proteins are associated with the viroplasmic complexes in rotavirus-infected cells.
Coimmunoprecipitation (co-IP) studies on RNase-treated virus-infected cell lysates were undertaken to investigate the possible physical association of cytoplasmically relocalized host proteins with the key viral constituents (NSP2, NSP5, and VP6) of viroplasmic complexes. Figures 3a and b reveal that NSP2, NSP5, and VP6 could be detected in the co-IP complexes with the majority of the relocalized host proteins examined.
FIG 3.

Co-IP analyses using virus-infected cell extracts demonstrate the association of host proteins with viroplasmic complexes/proteins. RNase-treated lysates from mock-infected and RRV-infected (MOI of 10) MA104 cells were prepared at 8 hpi. A total of 500 μg of the lysate was used to immunoprecipitate the cellular proteins using specific MAbs/PAbs. The immune complexes from mock-infected (C) and virus-infected (Inf) cell lysates were captured using BSA-saturated protein A-Sepharose beads and were resolved by SDS-PAGE. The lanes under 10% input (C and Inf) contain 50 μg of the lysates from control and infected cells, respectively. The viral proteins were coimmunoprecipitated using antibodies against the interacting host protein, and the viral proteins were detected by Western blotting using PAbs against NSP2, NSP5, and VP6 or MAb against VP6.
Immunofluorescence confocal microscopy analyses reveal selective sequestration of relocalized host proteins to VMs in rotavirus-infected cells.
In all the ICM analyses, the cells were infected at a multiplicity of infection (MOI) of approximately 0.5 so that the localization statuses of the host proteins in both infected and uninfected cells could be compared in the same field under identical experimental conditions. ICM analyses demonstrated the cytoplasmic relocalization of the majority of the hnRNPs and ARE-BPs, confirming the results of immunoblot analyses of the nuclear and cytoplasmic fractions of virus-infected cells (Fig. 2a and b). Surprisingly, the majority of the relocalized hnRNPs (hnRNPs C1/C2, E, F/H, I, K/J, L, and U) and ARE-BPs (hnRNP D, BRF1, HuR, TIA1, TIAL-1, TTP, Staufen1, and KSRP) showed colocalization with the viroplasmic proteins in VMs in virus-infected cells (Fig. 4a and b).
FIG 4.




Immunofluorescence confocal microscopy analyses of relocalization and colocalization of host proteins with VS in rotavirus RRV-infected MA104 cells. (a) ICM analyses of the nuclear-cytoplasmic distribution and colocalization with VMs of endogenous hnRNPs. MAbs against different hnRNPs (see Table S4 in the supplemental material), affinity-purified PAbs against NSP5 and NSP2, anti-RRV DLP PAbs against VP6, and Cy3-tagged anti-mouse (for cellular proteins) and Cy5-tagged anti-rabbit secondary antibodies (for viral proteins) were used. After mounting in DAPI, the proteins were visualized by fluorescence under a LSM Zeiss 710 immunofluorescence confocal microscope (63× oil immersion). Infections of cells grown on coverslips for confocal imaging were done at an MOI of 0.5 for 8 h. In ICM analyses, fields having relatively fewer infected cells were selected to clearly distinguish the localization status of host proteins in uninfected cells. Note the colocalization of hnRNPs with the VM. In all the confocal images, uninfected cells, infected cells, and the plot profile path on the VM are indicated by white and red arrowheads and a white broken line, respectively. The plot profiles and quantification of the cytoplasmic/nuclear (C/N) ratios of the proteins from 50 infected and uninfected cells were done using Image J software. The fluorescence quantification data represent the average C/N ratios for 50 infected cells normalized to the average C/N ratio for 50 control cells from one of three independent experiments and are expressed as averages ± standard deviations. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, nonsignificant (as determined by a t test). (b) Cytoplasmic relocalization and colocalization with VMs of ARE-BPs in RRV-infected cells. MAbs against different ARE-BPs (Table S4) were used. See the legend to panel a for more details. See Fig. S2 in the supplemental material for the predominant nuclear localization of hnRNPs and ARE-BPs in the nuclei of control cells. (c) Colocalization of nuclear transport proteins and cytoplasmic proteins in the VMs. MAbs against Exportin1, importin-β, and Transportin1 and rabbit PAb against Ran were used. See the colocalization of the transport proteins with viroplasmic NSP5 and VP6. The plots of cytoplasmic/nuclear ratios of the transport proteins are shown. See the legend to panel a for details. (d) Nuclear and cytoplasmic proteins that do not interact or colocalize with viroplasmic proteins. The virus-encoded NSP2, NSP5, and VP6 proteins and the ectopically expressed NH-tagged NSP5 protein colocalize in the VM. The panel showing NH-NSP5 represents HEK293T cells transfected with pc-NH-NSP5 expressing NH-tagged IS2-NSP5. Other panels represent MA104 cells. See Fig. S3 in the supplemental material for time course ICM analysis of the nuclear (TIA1) and cytoplasmic (RPS8) proteins, showing their colocalization with VM starting from 4 hpi, and see Fig. S4 for Z-stack analysis of the colocalization of hnRNP D with NSP2 and of hnRNP L with NSP5.
These analyses further confirmed the enhanced levels of the nuclear transport proteins in the cytoplasm of virus-infected cells in comparison to those in uninfected cells, as demonstrated by immunoblotting of the nuclear and cytoplasmic fractions (Fig. 2d and 4c). Moreover, all four transport proteins exhibited enrichment/colocalization with the VMs (Fig. 4c).
The cytoplasmic proteins VPS35, Sec31A, and RPS8 also showed colocalization with VMs (Fig. 4c). In contrast, no detectable colocalization of hnRNP A1 and hnRNP M with the VM was observed, and the splicing factor Sc35 and hnRNP Q remained solely in the nucleus (Fig. 4a and d). In addition, cytoplasmic PABPC1 relocalized to the nucleus in infected cells, as previously reported (34, 35, 38), and there was consequently no significant colocalization with VMs (Fig. 4d). The abundant cytoplasmic protein G3BP1, the best-characterized stress granule (SG)-specific marker, neither colocalized with the VM nor formed G3BP-specific SGs in infected cells (Fig. 4d), confirming a previous report that SGs are not formed in rotavirus-infected cells (35).
It is important to note that the colocalization of hnRNPs and ARE-BPs with the VM is not due to their release into the cytoplasm as a result of nuclear lysis caused by an ultimately cytocidal viral infection, as 4′,6-diamidino-2′-phenylindole dihydrochloride (DAPI)-stained virus-infected cells clearly showed that nuclei remained intact at 8 hpi (Fig. 4a to d). The cytoplasmic relocalization of host cell hnRNPs and ARE-BPs is a gradual process that commences at between 2 and 4 hpi, and significant cytoplasmic relocalization and colocalization of the nuclear proteins with VM could be detected at approximately 4 hpi, with their enrichment in the VM increasing with time, as shown by time course confocal image analysis of nuclear TIA1 and cytoplasmic RPS8 (see Fig. S3 in the supplemental material). In addition, β-actin and VPS35 were detected only in the cytoplasm, and PCNA localized to the nucleus (Fig. 2 and 4). As expected, the viral proteins NSP2, NSP5, and VP6 and ectopically expressed NH-NSP5 were observed only in the cytoplasm and colocalized with each other (Fig. 2 and 4).
It may be argued that colocalization with the VM of some of cytoplasmic proteins such as RPS8, Sec31A, and VPS35 and ARE-BPs such as TIAL-1 and TTP could be nonspecific due to their high abundance in the cytoplasm. However, the distinct punctate structures observed at identical positions for both the viral and host proteins in infected cells suggest that their colocalization is specific (Fig. 4a to d), which was also verified by Z-stack analysis, shown, for example, for two of the host proteins, hnRNP D and hnRNP L, in Fig. S4 in the supplemental material.
Ectopically expressed nuclear and cytoplasmic proteins colocalize with rotavirus VMs.
To further confirm the cytoplasmic relocalization and colocalization of host cell proteins with the VM, viral and selected host cell proteins tagged with a small amino-terminal NH or Myc tag or a larger fluorescent protein (FP) tag, green FP (GFP), enhanced cyan FP (ECFP), or mCherry (mCH), were ectopically expressed by transfection of cDNA constructs into HEK293T cells, followed by virus infection. Figure 5a shows the colocalization of a number of Myc-tagged hnRNPs and ARE-BPs (hnRNPs D, I, K, and L; BRF1; and TIA-1), NH-HuR, and Myc-RPS8 with virus-expressed NSP5 and NSP2 in the VM. Conversely, NH-tagged NSP5 also colocalized with the endogenous host proteins in the VM (Fig. 5b). As expected, ectopically expressed ECFP-NSP5 also colocalized with virus-expressed NSP2 and VP6 in the VM (Fig. S5). Ectopically expressed host nuclear proteins carrying larger fluorescent tags (ECFP-hnRNP D, ECFP-hnRNP C1, and ECFP-hnRNP K) also showed colocalization with viral proteins in the VMs (Fig. S5). However, unlike the ectopically expressed host proteins carrying the smaller Myc tags, a significant proportion of the ECFP-tagged nuclear proteins was retained in the nucleus (Fig. S5). In contrast, the mCH-tagged cytoplasmic protein VPS35 (mCH-VPS35) was present only in the cytoplasm and colocalized with the VM (Fig. S5).
FIG 5.


Colocalization of tagged ectopically expressed hnRNPs and ARE-BPs with viral proteins in the VM. (a) Ectopically expressed Myc- and NH-tagged hnRNPs and ARE-BPs colocalize with the VM. HEK293T cells on coverslips were transfected with plasmid DNA constructs expressing Myc- or NH-tagged host proteins for 36 h, followed by RRV infection (MOI of 0.5 for 8 h). Proteins were visualized by using anti-Myc or anti-His tag antibodies (mouse), PAb against NSP5 (rabbit), and the corresponding Cy3-tagged anti-mouse and Cy5-tagged anti-rabbit secondary antibodies. The transfected-infected cells and uninfected cells are indicated with orange and white arrowheads, respectively. The plot profile path on the VM is indicated by a white dashed line. See the legend to Fig. 4a for more details. (b) Ectopically expressed NH-NSP5 colocalizes with endogenous hnRNPs and ARE-BPs in the VM. Ectopically expressed NH-NSP5 was detected using anti-His tag antibody (mouse), and the endogenous cellular proteins were detected using rabbit PAbs, along with the corresponding fluorescent dye-tagged secondary antibodies. Transfected-infected HEK293T cells are shown by orange arrowheads. For other details, see the legend to Fig. 4a. See Fig. S5 in the supplemental material for colocalization of the ectopically expressed fluorescent protein-tagged host proteins and NSP5 in the VM, and see Fig. S6 for a demonstration that ectopic expression of the viroplasmic proteins does not induce the relocalization of host proteins, as observed in infected cells.
To investigate if the simple ectopic expression of the key VM proteins (NSP2, NSP5, or VP6) alone is able to induce the relocalization of host proteins seen in virus-infected cells, the subcellular localization of some hnRNPs and ARE-BPs was examined in HEK293T cells transiently expressing the VM viral proteins. BRF1 was present at detectable levels in the cytoplasm of control HEK293T cells, and the ectopic expression of the viral proteins did not further induce its cytoplasmic localization (Fig. S6a to S6c). However, the ectopic expression of NSP2 and VP6 induced the partial cytoplasmic relocalization of hnRNP D, HuR, and hnRNP K but not of the other host cell proteins examined (Fig. S6a to S6c). These results suggest that the expression of just the key viral VM proteins (NSP2, NSP5, or VP6) alone does not induce the drastic changes in the nuclear-cytoplasmic localization of hnRNPs and ARE-BPs seen following rotavirus infection.
Influence of relocalized host cell proteins on viral gene expression and infectious virus yield.
To assess the influence of host cell proteins relocalized following rotavirus infection on viral gene expression and progeny virus production, the effect of the ectopic overexpression and/or siRNA-mediated knockdown of selected host cell proteins was examined in HEK293T cells. The positive or negative effect of relocalized host proteins on viral gene expression was assessed by looking at the expression of the viroplasmic proteins NSP2, NSP5, and VP6. The ectopic expression of BRF1 and TIA1 had a negative effect on the levels of NSP5 expression but did not affect either NSP2 or VP6 expression levels (Fig. 6a). hnRNP C1, G3BP1, and VPS35 affected the expression of all three viral proteins. While hnRNP C1 exhibited a significant negative effect on the expression of NSP2, hnRNP E1 affected those of both NSP2 and VP6. The p40 and p42 isoforms of hnRNP D did not support the expression of NSP2, but the p37 and p45 isoforms enhanced the expression of the three viral proteins (Fig. 6a), and NSP5 expression was highly enhanced by hnRNP L (Fig. 6a). The siRNA-mediated downregulation of hnRNP F significantly affected the expression of NSP5 (Fig. 6b). While the knockdown of hnRNP C1 resulted in a significant increase in the NSP2 level, that of hnRNP E1 enhanced the expression of both NSP2 and NSP5 (Fig. 6c). The downregulation of G3BP1 markedly increased the levels of not only NSP5 but also NSP2 and VP6 in virus-infected cells (Fig. 6c).
FIG 6.

Effect of ectopic expression and siRNA-mediated knockdown of cellular proteins on viral protein expression. (a) Differential influence of ectopic expression of hnRNPs, ARE-BPs, and other host proteins on viral protein expression. HEK293T cells, transfected for 48 h with the plasmid DNA constructs expressing the cellular proteins, were infected with rotavirus strain RRV (MOI of 5.0), and lysates were prepared at 8 hpi from virus-infected and mock-infected cells. Fifty micrograms of total protein was used for the detection of NSP2 and NSP5 using purified PAbs and for the detection of VP6 using a MAb by Western blotting. Since the tagged ectopically expressed host proteins are detected using antibodies specific to the tag (Myc, GFP, mCH, or His tag), the endogenous protein is not detectable. C refers to serum-grown control cells. The levels of the endogenous host proteins can be seen in lane C for control cells that were treated with Accell nontarget siRNAs in panels b and c. The numbers at the bottom of each blot refer to the fold changes in the protein levels in transfected-infected cells in comparison to the levels in serum-grown control cells. (b and c) Differential effect of siRNA-mediated downregulation of expression of host proteins on viral protein expression. HEK293T cells were transfected with 30 pmol of siRNA against the host protein for 48 h and infected with the RRV strain of rotavirus, and at 8 hpi, 50 μg of total protein was analyzed by Western blotting. Note that the hnRNP D siRNA targets all four isoforms. Fold changes in the protein levels in siRNA-transfected–virus-infected cells in comparison to the levels in serum-grown control cells are indicated at the bottom of each blot.
Turning next to the effects of regulating the synthesis of relocalized host cell proteins on infectious virus yields, Fig. 7 shows that while the siRNA-mediated downregulation of HuR, hnRNP D45, hnRNP I, and hnRNP K decreased progeny virus titers by almost 2 orders of magnitude, their overexpression resulted in only a 6- to 8-fold increase in the progeny virus titers, as determined in MA104 cells by an enzyme-linked immunoperoxidase focus assay (ELIFA) (44–46). In contrast, the downregulation of G3BP1, TIA1, and hnRNP C1 increased the progeny virus yields by 3, 2.4, and 2.5 orders of magnitude, respectively, while ectopic overexpression resulted in reductions of virus yields by 3.2, 1.6, and 2.0 orders of magnitude, respectively (Fig. 7). Of note, the ectopic expression and siRNA-mediated knockdown of host proteins in HEK293T cells resulted in only a 1.2- to 2-fold increase or decrease in the number of infected cells, as examined by a direct ELIFA on transfected cells, but differences in progeny virus titers of 1.4 to 2.0 orders of magnitude, as assessed in MA104 cells. This suggests that host proteins could influence either the number of mature viroplasms, the size of the viroplasms, or both, impacting the infectious progeny virus yield, which was not examined in this study.
FIG 7.
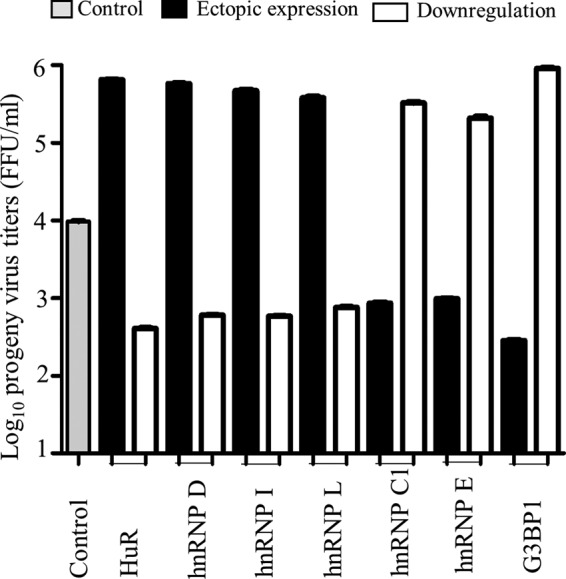
Effect of ectopic expression and downregulation of expression of selected host proteins on infectious virus yield. HEK293T cells in duplicate wells in a 24-well plate were transfected with the Myc-tagged hnRNP D45 isoform, hnRNP C1, hnRNP E1, hnRNP I, hnRNP L, NH-HuR, and ECFP-G3BP1 plasmid DNA constructs (1 μg/well) or the corresponding siRNAs (25 pmol/well) for 48 h, followed by infection with 400 FFU of the RRV strain of rotavirus. Nontarget fluorescent Accell siRNA was used as a control. At 8 hpi, cell lysates were prepared in DMEM by the freeze-thaw method. MA104 cells were infected with serially diluted lysates from the transfected-infected HEK293T cells, and the progeny virus titers in two wells each from two independent experiments were determined by an ELIFA as described in Materials and Methods. MAb 631/9 against VP6 was used for detection, and deep-brown foci were counted under a microscope.
DISCUSSION
Since virtually all viruses are totally dependent on host translational machinery for their protein synthesis, they employ a variety of mechanisms to subvert and hijack the system to promote the rapid and selective translation of viral mRNAs by manipulating one or more processes associated with host mRNA metabolism and/or translation during their short life cycle (22–24, 47, 48). An increasing number of studies in recent years reported the cytoplasmic relocalization of either one or a few hnRNPs and/or ARE-BPs during infection by different viruses and their influence on viral gene expression and/or replication (22–24, 49–55). However, very little is known about the intracellular localization of hnRNPs, ARE-BPs, and other host proteins during rotavirus infection. The results presented here demonstrate that rotavirus infection induces the relocalization of a large number of nuclear proteins, the majority of which associate with the viroplasmic proteins NSP2 and NSP5 in the VM.
The most unexpected finding was the direct interaction of large numbers of hnRNPs, ARE-BPs, and nuclear transport proteins and some cytoplasmic proteins with the viroplasmic proteins NSP2 and NSP5 (Table 1). It could be argued that the interaction of several host proteins with both NSP2 and NSP5 could be because of some kind of nonspecific binding due to an improper folding of the recombinant viral proteins. It is also possible that most of the host proteins could be recruited through one or two common proteins that interact with NSP2 and NSP5 or a common motif in the viral proteins, which requires further investigation by deletion mapping analyses. In this context, it may be noted that some hnRNPs are known to interact with each other (25, 56, 57). The VM contains a large number of NSP2 and NSP5 molecules depending on the state of maturation. It is unlikely that a single NSP2 or NSP5 molecule interacts with many host proteins simultaneously. Different NSP2 and NSP5 molecules could be interacting with one or two host proteins. The lack of one or two viral proteins in the co-IP complexes (Fig. 6) with a few host proteins could be due to the binding of the host protein to subviroplasmic complexes or a soluble form of the viral proteins. Furthermore, NSP2 and NSP5 are known to undergo phosphorylation, glycosylation, methylation, and/or SUMOylation in virus-infected cells (10, 12, 14, 58–62). Since proteins expressed in Escherichia coli lack these modifications, the interactome of the native viral proteins could differ from that of the recombinant proteins. It may be argued that some of the interactions observed in the in vitro pulldown analyses could still be RNA mediated in spite of the use of RNase-treated viral proteins and cell extracts due to incomplete RNA digestion. Note that hnRNPs and ARE-BPs as well as NSP2 and NSP5 are RNA-binding proteins, and the majority of the host proteins interacted with the viral proteins in an RNA-independent manner. However, in spite of the observed apparent RNA-independent interactions in vitro, the association of host proteins with the VM inside the cell, as revealed by ICM analyses, in reality could be facilitated by RNA by virtue of their inherent RNA-binding properties, rendering the argument that the interactions between viral and cellular proteins in the PD and co-IP complexes in vitro are nonspecific to be irrelevant and untenable.
Rotaviral mRNAs, in general, are 57 to 68% rich in A+U content (63), with UU, UA, and AU sequences being uniformly distributed along the length, and could be subject to AU-rich sequence-mediated translational repression and/or destabilization mediated by the interacting ARE-BPs (31–33, 64, 65). In contrast, cellular mRNAs generally contain AREs in the 3′ untranslated regions but not in the protein-coding regions (31, 66, 67). It is intriguing to understand why a cytoplasmic virus with an AU-rich genome imports ARE-BPs on a large scale into the cytoplasm, the site of its replication and protein synthesis (Fig. 2b and 4b), and how the virus circumvents the ARE-mediated translational repression and degradation of the AU-rich viral mRNAs to promote its protein synthesis, genome replication, and productive infection. This aspect needs further investigation to be better understood.
For productive rotavirus infection, the sustained selective nuclear import and accumulation of specific cellular proteins, such as PABPC1 (34–36, 38), appear to be important. PABPC1, a nuclear-cytoplasmic shuttling protein (68), is predominantly cytoplasmic at steady state but accumulates in the nucleus in response to several types of cellular stress (69–71), the depletion of paxillin (72) and cytoplasmic poly(A) RNA in the cytoplasm (73), and infection by many viruses, including rotavirus (34, 38, 74–79). The cytoplasmic depletion and nuclear accumulation of PABPC1 by rotavirus (Fig. 2c and 4d) and other viruses result in hyperadenylation and nuclear retention of host mRNAs and their global degradation and/or inhibition of host protein synthesis in the cytoplasm (38, 73, 75, 78, 79).
Furthermore, the significant increase in hnRNP C1/2, hnRNP U, hnRNP F/H, KSRP, and exportin1 levels in both cell compartments (Fig. 2a, b, and d and 4a to c) suggests selective modulation of the expression of these genes at the transcriptional, posttranscriptional, translational, and/or protein stabilization level in rotavirus-infected cells. The molecular basis for the selectively increased levels of these host proteins during rotavirus infection remains to be addressed. In this context, rotavirus was reported to antagonize the cellular antiviral response by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-κB by a mechanism after STAT1 binding to importin-α (80, 81). All these findings suggest that the impairment of nuclear transport processes in rotavirus infection is selective, but not global, and the import pathways involving different members of the importin families or other karyopherins affected by rotavirus need to be understood.
RNA-binding proteins undergo posttranslational modifications, such as phosphorylation, methylation, acetylation, and SUMOylation, in response to environmental stimuli, stress, and virus infection, which influence their functions associated with nuclear-cytoplasmic localization, RNA binding, protein-protein interactions, and RNA metabolism (82–87). In this context, hnRNP A1, which showed interactions with both NSP2 and NSP5 in the PD assay, showed a diffuse distribution in the cytoplasm and did not show significant colocalization with VMs, unlike other hnRNPs. Whether posttranslational modifications affect protein-protein interactions between some host proteins and viral proteins requires further investigations. Since several signaling pathways have been reported to be activated in rotavirus-infected cells (88–91), it will be of interest to understand how rotavirus, which is not known to encode a protease, kinase, or phosphatase, affects the selective subversion of nuclear transport processes.
Recently, VMs were shown to be associated with lipids and components of lipid droplets (92, 93), and VMs assemble in association with membrane components. Interruption of lipid/lipid droplet homeostasis was shown to decrease the formation, number, and size of VMs and the infectivity of rotavirus yields (94). Furthermore, due to the accumulation of the viral viroporin NSP4 in the endoplasmic reticulum (ER) and other membrane structures and the maturation of the virus in the ER lumen, the membrane structures are highly compromised in infected cells (95–97). The present finding that VPS35 and Sec31A, associated with vesicular traffic, interact with the cytoplasmic viroplasmic proteins suggests a disruption of cytoplasmic transport pathways in rotavirus infection (98).
The dramatic modulation of the intracellular environment in both the nuclear and cytoplasmic compartments by the relocalization of a large number of host proteins appears to be a potent viral strategy to subvert nuclear processes, including the splicing, processing, polyadenylation, and export of host mRNAs to the cytoplasm, leading to the cytoplasmic depletion of host mRNAs and generating a conducive environment in the cytoplasm for hijacking the host translational machinery for the selective translation of the nonpolyadenylated viral mRNAs. Since each of the nuclear proteins might influence the expression of individual viral proteins differentially, it will be of interest to investigate the role of each host protein in the expression of different viral genes in future studies. In conclusion, the present study lays the foundation for future detailed explorations of the influence, at different levels, of the large number of nuclear and cytoplasmic proteins on the expression of individual viral proteins, viral genome replication, the role of posttranslational modifications in intracellular localization, functions and association with VMs of host proteins in virus-infected cells, virus morphogenesis, infectious virus yield, and the architectural organization of viral and host proteins in viroplasms.
MATERIALS AND METHODS
Cells, viruses, and infection of cells.
The African green monkey kidney cell line MA104 (provided by Harry B. Greenberg, Stanford University, USA) and the human embryonic kidney cell line HEK293T (from Stuart A. Aaronson, Mount Sinai School of Medicine, New York, NY, USA) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Life Technologies–Gibco-BRL). The virus strains used were rhesus monkey rotavirus RRV (G3[P3]; subgroup I [SGI]), simian rotavirus SA11 (G3[P2]; SGI), and human rotaviruses KU (G1[P8]; SGII) (obtained from Harry B. Greenberg) and IS2 (G2[P4]; SGI), an Indian isolate from a diarrheic child (39). The viruses were grown in MA104 cells in the presence of 0.5 μg/ml trypsin and purified by isopycnic centrifugation in cesium chloride gradients (99).
Enzymes and reagents.
Restriction endonucleases, nucleotides, DNA purification kits, T4 DNA ligase, and 2× PCR reagent mix were obtained from Fermentas and Roche Applied Science. Laboratory reagents were purchased from Sigma-Aldrich and Promega. Oligonucleotides were obtained from Eurofins and Sigma-Aldrich, India. O'gene ruler DNA ladder mix (catalogue no. SM1173) and protein molecular weight markers (catalogue no. SM1811) were obtained from Fermentas, Lithuania, or from Genetix, India (catalogue no. PG-PMT2922 or PG-PMT2962).
Plasmid vectors.
The protein-coding regions (open reading frames [ORFs]) of the genes encoding NSP2, NSP5, and VP6 from the IS2 strain of rotavirus and that for NSP2 from the SA11 strain were all amplified by one-step reverse transcription-PCR (RT-PCR) (Invitrogen), and the BamHI- and XhoI-digested cDNAs were cloned into the pBS-bluescript KS(+) (pBS) vector. The SA11-NSP2 and IS2-NSP5 cDNAs, excised from the recombinants, were cloned into a modified bacterial expression vector, pET22-NH (99), between the NdeI and XhoI sites to generate fusion constructs (pET-NH-NSP5 and pET-NH-NSP2) with a His tag at the N terminus. The IS2-NSP5, IS2-VP6, and SA11-NSP2 ORFs were also cloned into the pcDNA3 mammalian expression vector between the BamHI and XhoI sites, generating pc-NSP5, pc-VP6, and pc-NSP2. ORFs from a number of cellular genes (hnRNP Dp40, hnRNP K, hnRNP F, and RPS8) were cloned into the pGEX-5X-2 vector (GE Healthcare), and the expressed glutathione S-transferase (GST) fusion proteins were purified by using glutathione-Sepharose beads and elution with reduced glutathione. Plasmid DNAs containing ORFs of the four isoforms of cellular hnRNP D with a Myc tag fused to the N terminus (100) were kindly provided by Ann-Bin Shyu, University of Texas Health Medical School, Houston, TX, USA. The sequences of all the constructs were verified by nucleotide sequencing at Eurofins or Medauxin, Bangalore, India.
The ECFP and mCherry ORFs (Clontech) lacking the translational termination codon were amplified by PCR and cloned into pcDNA3 between the HindIII and BamHI sites, generating pc-ECFP (pc-EC) and pc-mCH, respectively, or into pc-NSP5 and pc-NSP2 upstream of the rotaviral gene ORF between the HindIII and BamHI sites in translational fusion with the downstream IS2-NSP5 or SA11-NSP2 ORF for the expression of the rotaviral protein with a fluorescent tag, generating pc-EC-NSP5 and pc-EC-NSP2, respectively. The sequence-verified clones containing the genes for hnRNP C1, hnRNP K, hnRNP L, hnRNP U, hnRNP E1, hnRNP I, TIA1, G3BP1, VPS35, and RPS8 were obtained from Thermofisher (Dharmacon). The HuR ORF was amplified by RT-PCR using poly(A) mRNA from HEK293T cells. The ORFs were amplified by PCR and cloned in translational fusions downstream of the fluorescent reporter ORF into pc-EC or pc-mCH or in fusions with the N-terminal Myc or His (NH) tag into the pCMV-Myc and pCMV-His vectors (Clontech or Sigma-Aldrich) between the BamHI and XhoI sites, generating pc-EC-HuR, pc-EC-p40hnRNPD, pc-EC-hnRNP K, pc-EC-hnRNP L, pc-EC-hnRNP I, pc-EC-hnRNP C1, pc-EC-G3BP1, pc-mCherry-VPS35, pMyc-hnRNP C1, pMyc-hnRNP K, pMyc-HuR, and pNH-HuR, etc. hnRNP E1 was cloned between the EcoRV and XhoI sites. HuR was also cloned into the pET-NH vector (pET-NH-HuR) and purified as an NH-tagged protein. The plasmid clone for BRF1 was kindly provided by C. Moroni, Institute for Medical Microbiology, Basel, Switzerland. pMyc-BRF1 was generated by cloning the PCR fragment into the pCMV-Myc vector between the BamHI and XhoI sites. The pCMV6-myc-TIA-1 (pMyc-TIA1) clone was kindly provided by S. P. Somasekharan, BC Cancer Centre, Vancouver, Canada. The Sec31A expression clone pQCXIH-Sec31 expressing GFP-Sec31 was a kind gift from Ayano Satoh, Okayama University, Okayama, Japan. Plasmid DNAs purified using a Qiagen kit were used for transfection. All the constructs were verified by nucleotide sequencing at Eurofins or Medauxin, Bangalore, India.
Antibodies.
Horseradish peroxidase (HRP)-conjugated rabbit and mouse secondary antibodies were obtained from GE Healthcare. Antibodies against purified recombinant NSP2 and NSP5 from the IS2 strain were generated in rabbits and affinity purified using viral proteins immobilized either on an agarose matrix or by using protein A-agarose. SGI- and SGII-specific monoclonal antibodies (MAbs) were kindly provided by Harry B. Greenberg, Stanford University, and anti-NSP5 and -NSP2 antibodies raised in guinea pigs were generously provided by O. Burrone, ICGEB, Trieste, Italy. Information on the large number of antibodies to host cell proteins used in this study is given in Table S4 in the supplemental material. Mouse monoclonal antibodies against the majority of the host proteins were used in this study. When rabbit polyclonal antibodies (PAbs) to cellular proteins had to be used, they were screened for their cross-reactivity with rotaviral proteins by Western blotting and an enzyme-linked immunosorbent assay (ELISA) using recombinant VP6 or infected cell lysates prior to their use. Any PAbs that showed cross-reactivity with viral proteins were not used in this study. The PAbs generated in rabbits against NSP5, NSP2, and RRV DLPs are very specific to viral proteins and did not show detectable cross-reactivity with cellular proteins. These PAbs can detect NSP2 and NSP5 from a large number of human and animal group A rotaviruses, similar to those generated against SA11 and RRV proteins and used extensively by others, as they are essential proteins and their structure and function are conserved among group A rotaviruses.
Oligonucleotide primers.
The NSP2 and NSP5 gene-specific oligonucleotide primers used corresponded to the 5′ and 3′ ends of the viral gene ORFs; the corresponding GenBank accession numbers for the two genes from the IS2 strain are EF185863 and X94562, and that for SA11 NSP2 is NC_011502. ECFP- and mCherry-specific forward and reverse primers corresponded to the 5′ and 3′ ends of the ORFs (Clontech), except that the nuclear localization signal sequence at the 3′ end of ECFP was not included. The primers for the 5′ and 3′ ends of the cellular gene ORFs were synthesized based on their GenBank accession numbers, which are U38175 for HuR, NM_002138 for hnRNP D, NM_004926 for BRF1, NM_022037 for TIA1, NM_006196 for PCBP1, NM_005016 for PCBP2, NM_002140 for hnRNP K, NM_0031844 for hnRNP U, NM_017452 for Staufen, NM_031314 for hnRNP C1, BC013694 for PTBP1, BC108278 for G3BP1, BC070875 for RPS8, and BC093036 for VPS35. For sequencing, EC3′Forward, mCherry 3′Forward, SP6, and M13 forward and reverse primers were used.
Preparation of nuclear, cytoplasmic, and whole-cell extracts; SDS-PAGE; and Western blotting.
Nuclear and cytoplasmic extracts of control, serum-starved mock-infected, and rotavirus-infected (MOI of 10) MA104 and HEK293T cells were prepared at the desired time points postinfection using NE-PER nuclear and cytoplasmic extraction reagents from Thermoscientific, as described by the supplier. Whole-cell extracts were prepared by lysing the cells in ice-cold 1× radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris-HCl (pH 7.4), 1% NP-40 or Triton X-100, 0.25% sodium deoxycholate, 150 mM NaCl, and a protease inhibitor cocktail (Thermoscientific). After vortexing and centrifugation at 4°C, the protein concentration was estimated by using the Bradford protein assay reagent (Bio-Rad), and the cell extracts at 1 mg/ml were treated with 100 μg/ml of RNase (GE Healthcare) for 45 min at room temperature. RNA digestion was assessed by analysis of the RNase-treated and untreated cell extracts (100 μg) by agarose gel electrophoresis followed by ethidium bromide staining.
Enzyme-linked immunoperoxidase focus assay.
Virus titers in MA104 and HEK293T cells and the effects of the siRNA-mediated knockdown and overexpression of the cellular proteins on rotavirus replication in HEK293T cells were determined by using an immunoperoxidase-based focus assay or ELIFA (44–46). The virus titers determined by a PFU assay and by an ELIFA (focus-forming units [FFU]) were comparable, with the ELIFA method showing about 2% more foci than the PFU method. The focus assay has been extensively used in the laboratory of C. F. Arias and S. Lopez (44, 45, 101). Recently, this method was successfully adapted for enterovirus detection and enumeration (46). Because of the ease and rapidity of the ELIFA over the plaque assay, the former method has been extensively used during the Indian rotavirus vaccine trials during the last 20 years by us (102). Primary antibodies generated against the purified RRV DLP in rabbits or a MAb against the RRV DLP and secondary anti-rabbit or anti-mouse HRP-conjugated IgG (GE Healthcare) were used in the assay. The cells in each well were infected with 400 FFU (determined in MA104 cells, which corresponds to approximately 200 FFU in HEK293T cells). At 8 hpi, the cells were washed with phosphate-buffered saline (PBS) twice, fixed in 3.7% formaldehyde in a buffer containing 10 mM Tris-HCl (pH 7.4) and 100 mM NaCl for 30 min, permeabilized with 1% Triton X-100 in the same buffer for 3 min, washed, and incubated with a 1:2,000 dilution of the primary antibody solution. After 1 h of incubation at 37°C, the antibody was removed, and cells were washed and incubated for 1 h with HRP-conjugated secondary antibody. Cells were washed and stained for 15 to 30 min, until dark brown foci of infected cells were visible, using an AEC (3-amino-9-ethylcarbazole)-staining kit (Sigma-Aldrich). The AEC reagent was discarded, and the cells were left in PBS containing 0.01% sodium azide. The foci were counted under a microscope.
Coimmunoprecipitation.
Co-IP experiments were performed using 0.5 mg per co-IP assay of the RNase-treated total cell extract prepared from RRV-infected cells or control uninfected MA104 cells in a buffer containing 150 mM NaCl, 10 mM Tris-Cl (pH 7.5), and 0.05% NP-40 in a reaction volume of 0.7 to 1.0 ml. The appropriate antibody (2 to 8 μl per IP reaction depending on the antibody concentration) was incubated with the precleared lysate (cell extract incubated with protein A/G-Sepharose beads, which were presaturated with bovine serum albumin [BSA] [0.5 mg/ml] for 1 h at 4°C) for 3 h at 4°C with continuous mixing. Following incubation, 100 μl of a washed BSA-saturated protein A/G-Sepharose (3 mg) slurry was added to each IP reaction mixture and incubated for 4 h under the same conditions. The protein-bound beads were extensively washed five times in wash buffer, with 5 min of mixing in between. The bead-bound complexes were resolved by SDS-PAGE. In the control reaction, the uninfected cell lysate was incubated with the antibody.
Immunofluorescence confocal microscopy.
Confluent MA104 cells grown on coverslips were infected with the rhesus monkey RRV strain at an MOI of approximately 0.5. At 4 to 8 hpi or at the desired time points, cells were fixed in 3.7% formaldehyde in PBS for 10 min at room temperature, washed twice with PBS–0.1 M glycine buffer (pH 7.3) for 5 min each, permeabilized with a PBS–0.5% Triton X-100 solution, and blocked in a 5% BSA solution for 30 min, followed by incubation with the primary antibody overnight in a cold room. In the majority of experiments, an appropriate combination of MAbs against cellular proteins, rabbit or guinea pig PAbs against NSP2 and NSP5, and rabbit PAb or MAb against RRV DLPs was used to visualize the proteins in the same cell. After washing, cells were incubated with the appropriate fluorophore-tagged secondary antibodies for 2 h at room temperature. Anti-rabbit secondary antibodies tagged with Alexa Fluor 488 or Cy5 (emission, 670 nm), anti-mouse secondary antibody conjugated to Cy3 (emission, 570 nm), and anti-guinea pig secondary antibody conjugated to Alexa Fluor 633 were used in this study, at a dilution of 1:200. After washing, the cells were mounted in ProLong Gold antifade mountant with DAPI (Invitrogen). DAPI emission was monitored at 461 nm. Images were taken at a ×63 magnification using an LSM Zeiss 710 microscope. Images were processed with Adobe Photoshop (Adobe Systems, Inc., San Jose, CA) and ImageJ freeware (http://rsb.info.nih.gov/ij/index.html). Plotting of the plot profile curves and quantification of band intensities in Western blots were also done using Image J software. Quantification of the ratio of cytoplasmic to nuclear abundance of proteins in the cells was done using Image J software. The corrected total cell fluorescence (CTCF) was calculated using the formula CTCF = integrated density − (area of selected cell × mean fluorescence background reading). The ratio of cytoplasmic to nuclear fluorescence was quantified using Image J software for 50 virus-infected and 50 uninfected cells in each of three independent experiments. The normalized ratios from one experiment were plotted in the graph next to the confocal images to show the relative fold changes. Statistical analyses were performed using Student t test.
Expression and affinity purification of recombinant proteins.
E. coli BL21(DE3) was transformed with pET-NH-NSP5 and pET-NH-NSP2 plasmid DNAs. The cells were grown at 37°C to an optical density at 600 nm (OD600) of 0.8, protein expression was induced with 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h, and proteins were purified by Ni2+-nitrilotriacetic acid (NTA)-agarose affinity chromatography as described previously (99). Since the proteins were found mostly in the insoluble fraction, they were purified after solubilization in 8 M urea. The cells were lysed by sonication in urea buffer (20 mM Tris-Cl [pH 7.4], 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 8 M urea) and incubated at room temperature for 30 min, followed by centrifugation at 17,000 rpm (35,000 × g) for 45 min to remove cellular debris. The supernatant was passed through Ni2+-NTA-agarose resin (Qiagen). The protein-bound beads were washed with 10 column volumes (approximately 120 ml) of wash buffer containing 20 mM Tris-Cl (pH 7.4), 100 mM NaCl, 40 mM imidazole, and 2 M urea, followed by washing with wash buffer lacking urea. Although the proteins are likely to be free of RNA when solubilized in urea, the matrix-bound recombinant proteins were treated with RNase A for 30 min (10 μg/ml) before the extensive final wash. The proteins, eluted in 500 μM imidazole, were renatured by step dialysis, gradually reducing the concentration of urea in the buffer, and final dialysis in the absence of urea. The GST-tagged host proteins were expressed in a soluble form in E. coli and purified using glutathione-Sepharose beads and elution with reduced glutathione, according to the supplier's protocol (GE Healthcare). The homogeneity of the protein was monitored by SDS-PAGE.
Pulldown of NSP2- and NSP5-interacting cellular proteins for mass spectrometry and for Western blotting.
The Ni2+-NTA-agarose bead-bound viral proteins were used to pull down the interacting cellular proteins from control MA104 cell extracts. The control Ni2+-NTA-agarose beads were also saturated with extracts from E. coli transformed with the empty pET-NH vector and finally stored in 0.5% BSA for use in mock pulldowns. For mass spectrometry, the host cell extract was not treated with RNase. The cell extract (2 to 3 mg) at 0.5 to 1.0 mg/ml in lysis buffer was precleared by incubation with control Ni-NTA-agarose beads (prepared as described above) for 2 h at 4°C in the presence of a protease inhibitor cocktail prior to passing it 2 to 3 times through the column containing the NSP2- or NSP5-bound beads at room temperature. Prior to passing the cell extract, the bead-bound viral proteins were allowed to renature in PBS overnight. For pulldown assays, 10 μl of control Ni2+-NTA beads and NH-NSP5- or NH-NSP2-bound beads (containing 10 μg of soluble recombinant protein) was incubated in 0.5 ml of buffer A (150 mM NaCl, 10 mM Tris-Cl [pH 7.5], 0.05% NP-40, and 0.5% BSA) for 2 h at 4°C and washed in buffer B (150 mM NaCl, 10 mM Tris-Cl [pH 7.5], and 0.05% NP-40), and the Ni2+-NTA-agarose control beads (mock) and protein-bound beads were incubated with 0.5 mg of the precleared cell extract for 4 h at 4°C with mixing, followed by extensive washing of the beads five times with RIPA buffer containing 0.25% Triton X-100 and 300 mM NaCl. PD experiments were also performed using control cell extracts that were not treated with RNase. The proteins bound to the beads were directly released into SDS sample buffer by heating, resolved by SDS-PAGE on a 12% gel, and detected by Western blotting using antibodies against cellular proteins. Proteins bound to control beads were also run in a separate lane.
Western blot analysis.
Proteins separated by SDS-PAGE were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, USA) by a wet-transfer method. The membranes were blocked with a 5% skimmed milk solution in TBST (20 mM Tris-HCl [pH 7.4], 137 mM NaCl, and 0.1 to 0.2% Tween 20) for 2 to 3 h to reduce the nonspecific binding of antibodies. The blots were incubated with primary antibody at the appropriate dilution given in Table S4 in the supplemental material. After incubation with primary antibody, the membrane was washed three times for 15 min each in 15 ml of TBST and incubated with anti-rabbit or anti-mouse HRP-conjugated secondary antibody (1:5,000 dilutions) in 10 ml of blocking buffer, with gentle agitation for 1 to 2 h at room temperature. The blots were washed three times for 15 min each in 15 ml of TBST and developed by a chemiluminescence method using the Luminata Forte Western HRP substrate (Millipore). For reprobing of the blots, the primary and secondary antibodies were removed by incubating the membrane in stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl [pH 6.8]), and the membrane was incubated at 60°C for 15 min with moderate agitation, followed by extensive washing with TBST buffer for 30 to 40 min and incubation in blocking buffer (5% skimmed milk) for 1 h. The membrane was then used for the detection of another protein. The blots were reused two to three times for the detection of two or three proteins depending on significant differences in their molecular weights without a significant loss of the signal to confirm the presence of other proteins in the complex.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of cellular proteins interacting with NSP2 and NSP5.
The polyacrylamide gel stained with colloidal Coomassie blue was rinsed a few times with distilled water, and the lanes were cut into 3 pieces. The three regions were further cut into pieces in a microcentrifuge tube and destained by incubation in 100 mM ammonium bicarbonate-acetonitrile (1:1, vol/vol) buffer (pH 8.4) for 30 min. The buffer was removed after centrifugation, and the gel pieces were incubated with neat acetonitrile until they became white and shrunk. The gel pieces were dried in a vacuum centrifuge, followed by incubation with trypsin in buffer containing ammonium bicarbonate at 37°C overnight (103). After centrifugation at 10,000 rpm (9,400 × g), the supernatant was transferred to a new tube, and mass spectrometry analysis of the peptides was carried out at the C-CAMP mass spectrometry facility, National Centre for Biological Sciences, Bangalore, India, using high-performance liquid chromatography (1D nano-LC) and an LTQ-Orbitrap Discovery system (Thermoscientific). Results were analyzed with MASCOT using proteome discoverer 1.3. Functional annotation of the cellular proteins interacting with the viral proteins was done using the Web-based tools DAVID (104) and Gene SeT Analysis Toolkit (105).
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by grants from the SERB, Department of Science and Technology (grant no. SR/SO/BB-30/2010), and the Department of Biotechnology (grant no. BT/PR11549/MED/29/865/2014), Government of India, and by the DBT-IISc Partnership Programme during 2011 to 2016. We acknowledge financial support from the Indian National Science Academy for an INSA senior scientist fellowship to C.D.R.
We acknowledge the services of the departmental and institutional confocal imaging facilities. We are grateful to M. A. McCrae, University of Warwick, United Kingdom, for his critical comments on the PhD thesis of P.D. and suggestions on the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00612-18.
REFERENCES
- 1.Dennehy PD. 2012. Rotavirus infection: an update on management and prevention. Adv Pediatr 59:47–74. doi: 10.1016/j.yapd.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Estes MK, Greenberg HB. 2013. Rotaviruses, p 1347–1401. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Smith ML, Lazdins I, Holmes IH. 1980. Coding assignments of double-stranded RNA segments of SA11 rotavirus established by in vitro translation. J Virol 33:976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawton JA, Estes MK, Prasad BV. 1997. Three-dimensional visualization of mRNA release from actively transcribing rotavirus particles. Nat Struct Biol 4:118–121. doi: 10.1038/nsb0297-118. [DOI] [PubMed] [Google Scholar]
- 5.Prasad BV, Wang GJ, Clerx JP, Chiu W. 1988. Three-dimensional structure of rotavirus. J Mol Biol 199:269–275. doi: 10.1016/0022-2836(88)90313-0. [DOI] [PubMed] [Google Scholar]
- 6.Periz J, Celma C, Jing B, Pinkney JN, Roy P, Kapanidis AN. 2013. Rotavirus mRNAs are released by transcript-specific channels in the double-layered viral capsid. Proc Natl Acad Sci U S A 110:12042–12047. doi: 10.1073/pnas.1220345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. 2011. Atomic model of an infectious rotavirus particle. EMBO J 30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campagna M, Eichwald C, Vascotto F, Burrone OR. 2005. RNA interference of rotavirus segment 11 mRNA reveals the essential role of NSP5 in the virus replicative cycle. J Gen Virol 86:1481–1487. doi: 10.1099/vir.0.80598-0. [DOI] [PubMed] [Google Scholar]
- 9.Contin R, Arnoldi F, Campagna M, Burrone OR. 2010. Rotavirus NSP5 orchestrates recruitment of viroplasmic proteins. J Gen Virol 91:1782–1793. doi: 10.1099/vir.0.019133-0. [DOI] [PubMed] [Google Scholar]
- 10.Fabbretti E, Afrikanova I, Vascotto F, Burrone OR. 1999. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J Gen Virol 80:333–339. doi: 10.1099/0022-1317-80-2-333. [DOI] [PubMed] [Google Scholar]
- 11.López T, Rojas M, Ayala-Bretón C, López S, Arias CF. 2005. Reduced expression of the rotavirus NSP5 gene has a pleiotropic effect on virus replication. J Gen Virol 86:1609–1617. doi: 10.1099/vir.0.80827-0. [DOI] [PubMed] [Google Scholar]
- 12.Mohan KV, Muller J, Som I, Atreya CD. 2003. The N- and C-terminal regions of rotavirus NSP5 are the critical determinants for the formation of viroplasm-like structures independent of NSP2. J Virol 77:12184–12192. doi: 10.1128/JVI.77.22.12184-12192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patton JT, Silvestri LS, Tortorici MA, Vasquez-Del Carpio R, Taraporewala ZF. 2006. Rotavirus genome replication and morphogenesis: role of the viroplasm. Curr Top Microbiol Immunol 309:169–187. [DOI] [PubMed] [Google Scholar]
- 14.Eichwald C, Rodriguez JF, Burrone OR. 2004. Characterization of rotavirus NSP2/NSP5 interactions and the dynamics of viroplasm formation. J Gen Virol 85:625–634. doi: 10.1099/vir.0.19611-0. [DOI] [PubMed] [Google Scholar]
- 15.Estrozi LF, Settembre EC, Goret G, McClain B, Zhang X, Chen JZ, Grigorieff N, Harrison SC. 2013. Location of the dsRNA-dependent polymerase, VP1, in rotavirus particles. J Mol Biol 425:124–132. doi: 10.1016/j.jmb.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClain B, Settembre E, Temple BR, Bellamy AR, Harrison SC. 2010. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 Å resolution. J Mol Biol 397:587–599. doi: 10.1016/j.jmb.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carreno-Torres JJ, Gutierrez M, Arias CF, Lopez S, Isa P. 2010. Characterization of viroplasm formation during the early stages of rotavirus infection. Virol J 7:350. doi: 10.1186/1743-422X-7-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eichwald C, Arnoldi F, Laimbacher AS, Schraner EM, Fraet C, Burrone OR, Ackerman M. 2012. Rotavirus viroplasm fusion and perinuclear localization are dynamic processes requiring stabilized microtubules. PLoS One 7:e47947. doi: 10.1371/journal.pone.0047947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cabral-Romero C, Padilla-Noriega L. 2006. Association of rotavirus viroplasms with microtubules through NSP2 and NSP5. Mem Inst Oswaldo Cruz 101:603–611. doi: 10.1590/S0074-02762006000600006. [DOI] [PubMed] [Google Scholar]
- 20.Martin D, Duarte M, Lepault J, Poncet D. 2010. Sequestration of free tubulin molecules by the viral protein NSP2 induces microtubule depolymerization during rotavirus infection. J Virol 84:2522–2532. doi: 10.1128/JVI.01883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Criglar JM, Hu L, Crawford SE, Hyser JM, Broughman JR, Prasad BVV, Estes MK. 2014. A novel form of rotavirus NSP2 and phosphorylation-dependent NSP2-NSP5 interactions are associated with viroplasm assembly. J Virol 88:786–798. doi: 10.1128/JVI.03022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lloyd RE. 2015. Nuclear proteins hijacked by mammalian cytoplasmic plus strand RNA viruses. Virology 479–480:457–474. doi: 10.1016/j.virol.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol 9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yarbrough ML, Mata MA, Sakthivel R, Fontoura BMA. 2013. Viral subversion of nucleocytoplasmic trafficking. Traffic 15:127–140. doi: 10.1111/tra.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han SP, Tang YH, Smith R. 2010. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J 430:379–392. doi: 10.1042/BJ20100396. [DOI] [PubMed] [Google Scholar]
- 26.Piñol-Roma S, Dreyfuss G. 1993. hnRNP proteins: localization and transport between the nucleus and the cytoplasm. Trends Cell Biol 3:151–155. doi: 10.1016/0962-8924(93)90135-N. [DOI] [PubMed] [Google Scholar]
- 27.Nakielny S, Dreyfus G. 1996. The hnRNP C proteins contain a nuclear retention sequence that can override nuclear export signals. J Cell Biol 134:1365–1373. doi: 10.1083/jcb.134.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geuens T, Bouhy D, Timmerman V. 2016. The hnRNP family: insights into their role in health and disease. Hum Genet 135:851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krecic AM, Swanson MS. 1999. hnRNP complexes: composition, structure, and function. Curr Opin Cell Biol 11:363–371. doi: 10.1016/S0955-0674(99)80051-9. [DOI] [PubMed] [Google Scholar]
- 30.Pichon X, Wilson LA, Stoneley M, Bastide A, King HA, Somers J, Willis AE. 2012. RNA binding protein/RNA element interactions and the control of translation. Curr Protein Pept Sci 13:294–304. doi: 10.2174/138920312801619475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C-YA, Shyu A-B. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20:465–470. doi: 10.1016/S0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 32.Baou M, Jewell A, Murphy JJ. 2009. TIS11 family proteins and their roles in posttranscriptional gene regulation. J Biomed Biotechnol 2009:634520. doi: 10.1155/2009/634520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barreau C, Paillard L, Osborne HB. 2005. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res 33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harb M, Becker MM, Vitour D, Baron CH, Vende P, Brown SC, Bolte S, Arnold ST, Poncet D. 2008. Nuclear localization of cytoplasmic poly(A)-binding protein upon rotavirus infection involves the interaction of NSP3 with eIF4G and RoXaN. J Virol 82:11283–11293. doi: 10.1128/JVI.00872-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montero H, Rojas M, Arias CF, Lopez S. 2008. Rotavirus infection induces the phosphorylation of eIF2α but prevents the formation of stress granules. J Virol 82:1496–1504. doi: 10.1128/JVI.01779-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold MM, Brownback CS, Taraporewala ZF, Patton JT. 2012. Rotavirus variant replicates efficiently although encoding an aberrant NSP3 that fails to induce nuclear localization of poly(A)-binding protein. J Gen Virol 93:1483–1494. doi: 10.1099/vir.0.041830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keryer-Bibens C, Legagneux V, Namanda-Vanderbeken A, Cosson B, Paillard L, Poncet D, Osborne HB. 2009. The rotaviral NSP3 protein stimulates translation of polyadenylated target mRNAs independently of its RNA-binding domain. Biochem Biophys Res Commun 390:302–306. doi: 10.1016/j.bbrc.2009.09.115. [DOI] [PubMed] [Google Scholar]
- 38.Rubio RM, Mora SI, Romero P, Arias CF, Lopez S. 2013. Rotavirus prevents the expression of host responses by blocking the nucleocytoplasmic transport of polyadenylated mRNAs. J Virol 87:6336–6345. doi: 10.1128/JVI.00361-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aijaz S, Gowda K, Jagannath MR, Reddy RR, Maiya PP, Ward RL, Greenberg HB, Raju M, Anandababu M, Rao CD. 1996. Epidemiology of symptomatic human rotaviruses in Bangalore and Mysore, India, from 1988 to 1994 as determined by electropherotype, subgroup and serotype analysis. Arch Virol 141:715–726. doi: 10.1007/BF01718329. [DOI] [PubMed] [Google Scholar]
- 40.Arnoldi F, Campagna M, Eichwald C, Desselberger U, Burrone OR. 2007. Interaction of rotavirus polymerase VP1 with nonstructural protein NSP5 is stronger than that with NSP2. J Virol 81:2128–2137. doi: 10.1128/JVI.01494-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torres-Vega MA, González RA, Duarte M, Poncet D, López S, Arias CF. 2000. The C-terminal domain of rotavirus NSP5 is essential for its multimerization, hyperphosphorylation and interaction with NSP6. J Gen Virol 81:821–830. doi: 10.1099/0022-1317-81-3-821. [DOI] [PubMed] [Google Scholar]
- 42.Cautain B, Hill R, de Pedro N, Link W. 2014. Components and regulation of nuclear transport processes. FEBS J 282:445–462. doi: 10.1111/febs.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soniat M, Chook YM. 2015. Nuclear localization signals for four karyopherin-β nuclear import systems. Biochem J 468:353–362. doi: 10.1042/BJ20150368. [DOI] [PubMed] [Google Scholar]
- 44.Arias CF, Lizano M, López S. 1987. Synthesis in Escherichia coli and immunological characterization of a polypeptide containing the cleavage sites associated with trypsin enhancement of rotavirus SA11 infectivity. J Gen Virol 68:633–642. doi: 10.1099/0022-1317-68-3-633. [DOI] [PubMed] [Google Scholar]
- 45.Pando V, Isa P, Arias CF, López S. 2002. Influence of calcium on the early steps of rotavirus infection. Virology 295:190–200. doi: 10.1006/viro.2001.1337. [DOI] [PubMed] [Google Scholar]
- 46.Rao CD, Reddy H, Naidu JR, Raghavendra A, Radhika NS, Karande A. 2015. An enzyme-linked immune focus assay for rapid detection and enumeration, and a newborn mouse model for human non-polio enteroviruses associated with acute diarrhea. J Virol Methods 224:47–52. doi: 10.1016/j.jviromet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Gustin KE. 2003. Inhibition of nucleocytoplasmic trafficking by RNA viruses: targeting the nuclear pore complex. Virus Res 95:35–44. doi: 10.1016/S0168-1702(03)00165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Narayanan K, Makino S. 2013. Interplay between viruses and host mRNA degradation. Biochim Biophys Acta 1829:732–741. doi: 10.1016/j.bbagrm.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barnhart MD, Moon SL, Emch AW, Wilusz CJ, Wilusz J. 2013. Changes in cellular mRNA stability, splicing, and polyadenylation through HuR protein sequestration by a cytoplasmic RNA virus. Cell Rep 5:909–917. doi: 10.1016/j.celrep.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brunetti JE, Scolaro LA, Castilla V. 2015. The heterogeneous nuclear ribonucleoprotein K (hnRNP K) is a host factor required for dengue virus and Junín virus multiplication. Virus Res 203:84–91. doi: 10.1016/j.virusres.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Dechtawewat T, Songprakhon P, Limjindaporn T, Puttikhunt C, Kasinrerk W, Saitornuang S, Yenchitsomanus PT, Noisakran S. 2015. Role of human heterogeneous nuclear ribonucleoprotein C1/C2 in dengue virus replication. Virol J 12:14. doi: 10.1186/s12985-014-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Friedrich S, Schmidt T, Geissler R, Lilie H, Chabierski S, Ulbert S, Liebert UG, Golbik RP, Behrens SE. 2014. AUF1p45 promotes West Nile virus replication by an RNA chaperone activity that supports cyclization of the viral genome. J Virol 88:11586–11599. doi: 10.1128/JVI.01283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jagdeo JM, Dufour A, Fung G, Luo H, Kleifeldm O, Overall CM, Jan E. 2015. Heterogeneous nuclear ribonucleoprotein M facilitates enterovirus infection. J Virol 89:7064–7078. doi: 10.1128/JVI.02977-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monette A, Ajamian L, López-Lastra M, Mouland AJ. 2009. Human immunodeficiency virus type 1 (HIV-1) induces the cytoplasmic retention of heterogeneous nuclear ribonucleoprotein A1 by disrupting nuclear import: implications for HIV-1 gene expression. J Biol Chem 284:31350–31362. doi: 10.1074/jbc.M109.048736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pettit Kneller EL, Connor JH, Lyles DS. 2009. hnRNPs relocalize to the cytoplasm following infection with vesicular stomatitis virus. J Virol 83:770–780. doi: 10.1128/JVI.01279-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim JH, Hahm B, Kim YK, Choi M, Jang SK. 2000. Protein-protein interaction among hnRNPs shuttling between nucleus and cytoplasm. J Mol Biol 298:395–405. doi: 10.1006/jmbi.2000.3687. [DOI] [PubMed] [Google Scholar]
- 57.Park HG, Yoon JY, Choi M. 2007. Heterogeneous nuclear ribonucleoprotein D/AUF1 interacts with heterogeneous nuclear ribonucleoprotein L. J Biosci 32:1263–1272. doi: 10.1007/s12038-007-0135-8. [DOI] [PubMed] [Google Scholar]
- 58.Gonzalez SA, Burrone OR. 1991. Rotavirus NS26 is modified by addition of single O-linked residues of N-acetylglucosamine. Virology 182:8–16. doi: 10.1016/0042-6822(91)90642-O. [DOI] [PubMed] [Google Scholar]
- 59.Poncet D, Lindenbaum P, L'Haridon R, Cohen J. 1997. In vivo and in vitro phosphorylation of rotavirus NSP5 correlates with its localization in viroplasms. J Virol 71:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Afrikanova I, Fabretti E, Miozzo MC, Burrone OR. 1998. Rotavirus NSP5 phosphorylation is up-regulated by interaction with NSP2. J Gen Virol 79:2679–2686. doi: 10.1099/0022-1317-79-11-2679. [DOI] [PubMed] [Google Scholar]
- 61.Campagna M, Marcos-Villar L, Arnoldi F, de la Cruz-Herrera CF, Gallego P, Gonzalez-Santamaria J, Gonzalez D, Lopitz-Otsoa F, Rodriguez MS, Burrone OR, Rivas C. 2013. Rotavirus viroplasm proteins interact with the cellular SUMOylation system: implications for viroplasm-like structure formation. J Virol 87:807–817. doi: 10.1128/JVI.01578-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sotelo PH, Schumann M, Krause E, Chnaiderman J. 2010. Analysis of rotavirus non-structural protein NSP5 by mass spectrometry reveals a complex phosphorylation pattern. Virus Res 149:104–108. doi: 10.1016/j.virusres.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 63.Estes MK, Cohen J. 1989. Rotavirus gene structure and function. Microbiol Rev 53:410–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kruys V, Marinx O, Shaw G, Deschamps J, Hues G. 1989. Translational blockade imposed by cytokine-derived UA-rich sequences. Science 245:852–855. doi: 10.1126/science.2672333. [DOI] [PubMed] [Google Scholar]
- 65.Zhang T, Kruys V, Huez G, Gueydan C. 2002. AU-rich element-mediated translational control: complexity and multiple activities of transactivating factors. Biochem Soc Trans 30:952–958. doi: 10.1042/bst0300952. [DOI] [PubMed] [Google Scholar]
- 66.Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. 1986. Identification of a common nucleotide sequence in the 3′ untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci U S A 83:1670–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shaw G, Kamen R. 1986. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- 68.Eliseeva IA, Lyabin DN, Ovchinnikov LP. 2013. Poly(A)-binding proteins: structure, domain organization, and activity regulation. Biochemistry (Mosc) 78:1377–1391. doi: 10.1134/S0006297913130014. [DOI] [PubMed] [Google Scholar]
- 69.Afonina E, Stauber R, Pavlakis GN. 1998. The human poly(A)-binding protein 1 shuttles between the nucleus and the cytoplasm. J Biol Chem 273:13015–13021. doi: 10.1074/jbc.273.21.13015. [DOI] [PubMed] [Google Scholar]
- 70.Burgess HM, Gray NK. 2012. An integrated model for the nucleo-cytoplasmic transport of cytoplasmic poly(A)-binding proteins. Commun Integr Biol 5:243–247. doi: 10.4161/cib.19347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma S, Bhattacharjee RB, Bag J. 2009. Expression of poly(A)-binding protein is upregulated during recovery from heat shock in HeLa cells. FEBS Lett 276:552–570. doi: 10.1111/j.1742-4658.2008.06803.x. [DOI] [PubMed] [Google Scholar]
- 72.Woods AJ, Kantidakis T, Sabe H, Critchley DR, Norman JC. 2005. Interaction of paxillin with poly(A)-binding protein 1 and its role in focal adhesion turnover and cell migration. Mol Cell Biol 25:3763–3773. doi: 10.1128/MCB.25.9.3763-3773.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar GK, Shum L, Glaunsinger BA. 2011. Importin α-mediated nuclear import of cytoplasmic poly(A)-binding protein occurs as a direct consequence of cytoplasmic mRNA depletion. Mol Cell Biol 31:3113–3125. doi: 10.1128/MCB.05402-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blakqori G, van Knippenberg I, Elliot RM. 2009. Bunyamwera orthobunyavirus S-segment untranslated regions mediate poly(A) tail-independent translation. J Virol 83:3637–3646. doi: 10.1128/JVI.02201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee YJ, Glaunsinger BA. 2009. Aberrant herpesvirus-induced polyadenylation correlates with cellular mRNA destruction. PLoS Biol 7:e1000107. doi: 10.1371/journal.pbio.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salaun C, MacDonald AI, Larralde O, Howard L, Lochtie K, Burgess HM, Brook M, Malik P, Gray NK, Graham SV. 2010. Poly(A)-binding protein 1 partially relocalizes to the nucleus during herpes simplex virus type 1 infection in an ICP27-independent manner and does not inhibit virus replication. J Virol 84:8539–8548. doi: 10.1128/JVI.00668-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Copeland AM, Altamura LA, Van Deusen NM, Schmaljohn CS. 2013. Nuclear relocalization of polyadenylate binding protein during Rift Valley fever virus infection involves expression of the NSs gene. J Virol 87:11659–11669. doi: 10.1128/JVI.01434-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kumar GK, Glaunsinger BA. 2010. Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol Cell Biol 30:4996–5008. doi: 10.1128/MCB.00600-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Richner JM, Clyde K, Pezda AC, Cheng BY, Wang T, Kumar GR, Covarrubias S, Coscov L, Glaunsinger B. 2011. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog 7:e1002150. doi: 10.1371/journal.ppat.1002150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holloway G, Truong TT, Coulson BS. 2009. Rotavirus antagonizes cellular responses by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-κB. J Virol 83:4942–4951. doi: 10.1128/JVI.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holloway G, Dang VT, Jans DA, Coulson BS. 2014. Rotavirus inhibits interferon-induced STAT nuclear translocation by a mechanism that acts after STAT binding to importin-α. J Gen Virol 95:1723–1733. doi: 10.1099/vir.0.064063-0. [DOI] [PubMed] [Google Scholar]
- 82.Blee TK, Gray NK, Brook M. 2015. Modulation of the cytoplasmic functions of mammalian post-transcriptional regulatory proteins by methylation and acetylation: a key layer of regulation waiting to be uncovered? Biochem Soc Trans 43:1285–1295. doi: 10.1042/BST20150172. [DOI] [PubMed] [Google Scholar]
- 83.Dickson AM, Anderson JR, Barnhart MD, Sokoloski KJ, Oko L, Opyrchal M, Galanis E, Wilusz CJ, Morrison TE, Wilusz J. 2012. Dephosphorylation of HuR protein during alphavirus infection is associated with HuR relocalization to the cytoplasm. J Biol Chem 287:36229–36238. doi: 10.1074/jbc.M112.371203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Habelhah H, Shah K, Huang L, Ostareck-Lederer A, Burlingame AL, Shokat KM, Hentze MW, Ronai Z. 2001. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat Cell Biol 3:325–330. doi: 10.1038/35060131. [DOI] [PubMed] [Google Scholar]
- 85.Vassileva MT, Matunis MJ. 2004. SUMO modification of heterogeneous nuclear ribonucleoproteins. Mol Cell Biol 24:3623–3632. doi: 10.1128/MCB.24.9.3623-3632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu Q, Dreyfuss G. 1995. In vivo and in vitro arginine methylation of RNA-binding proteins. Mol Cell Biol 15:2800–2808. doi: 10.1128/MCB.15.5.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.García-Mauriño SM, Rivero-Rodríguez F, Velázquez-Cruz A, Hernández-Vellisca M, Díaz-Quintana A, De la Rosa MA, Díaz-Moreno I. 2017. RNA binding protein regulation and cross-talk in the control of AU-rich mRNA fate. Front Mol Biosci 4:71. doi: 10.3389/fmolb.2017.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Holloway G, Coulson BS. 2006. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J Virol 80:10624–10633. doi: 10.1128/JVI.00390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martin-Latil S, Cotte-Laffitte J, Beau I, Quéro AM, Géniteau-Legendre M, Servin AL. 2004. A cyclic AMP protein kinase A-dependent mechanism by which rotavirus impairs the expression and enzyme activity of brush border-associated sucrase-isomaltase in differentiated intestinal Caco-2 cells. Cell Microbiol 6:719–731. doi: 10.1111/j.1462-5822.2004.00396.x. [DOI] [PubMed] [Google Scholar]
- 90.Rossen JW, Bouma J, Raatgeep RH, Büller HA, Einerhand AW. 2004. Inhibition of cyclooxygenase activity reduces rotavirus infection at a postbinding step. J Virol 78:9721–9730. doi: 10.1128/JVI.78.18.9721-9730.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sen A, Rott L, Phan N, Mukherjee G, Greenberg HB. 2014. Rotavirus NSP1 protein inhibits interferon-mediated STAT1 activation. J Virol 88:41–53. doi: 10.1128/JVI.01501-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheung W, Gill M, Sposito A, Kaminski CF, Courousse N, Chwetzoff S, Trugnan G, Keshavan N, Lever A, Desselberger U. 2010. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. J Virol 84:6782–6798. doi: 10.1128/JVI.01757-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gaunt ER, Zhang Q, Cheung W, Wakelam MJ, Lever AM, Desselberger U. 2013. Lipidome analysis of rotavirus-infected cells confirms the close interaction of lipid droplets with viroplasms. J Gen Virol 94:1576–1586. doi: 10.1099/vir.0.049635-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crawford SE, Desselberger U. 2016. Lipid droplets form complexes with viroplasms and are crucial for rotavirus replication. Curr Opin Virol 19:11–15. doi: 10.1016/j.coviro.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Altenburg BC, Graham DY, Estes MK. 1980. Ultrastructural study of rotavirus replication in cultured cells. J Gen Virol 46:75–85. doi: 10.1099/0022-1317-46-1-75. [DOI] [PubMed] [Google Scholar]
- 96.Tinari A, Ammendolia MG, Superti F, Donelli G. 1996. Tubuloreticular structures induced by rotavirus infection in HT-29 cells. Ultrastruct Pathol 20:571–576. doi: 10.3109/01913129609016361. [DOI] [PubMed] [Google Scholar]
- 97.Bhowmick R, Halder UC, Chattopadhyay S, Chanda S, Nandi S, Bagchi P, Nayak MK, Chakrabarti O, Kobayashi N, Chawla-Sarkar M. 2012. Rotaviral enterotoxin non-structural protein 4 targets mitochondria for activation of apoptosis during infection. J Biol Chem 287:35004–35020. doi: 10.1074/jbc.M112.369595. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Xu A, Bellamy AR, Taylor JA. 2000. Immobilization of the early secretory pathway by a virus glycoprotein that binds to microtubules. EMBO J 19:6465–6475. doi: 10.1093/emboj/19.23.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jagannath MR, Kesavulu MM, Deepa R, Sastri PN, Kumar SS, Suguna K, Rao CD. 2006. N- and C-terminal cooperation in rotavirus enterotoxin: novel mechanism of modulation of the properties of a multifunctional protein by a structurally and functionally overlapping conformational domain. J Virol 80:412–425. doi: 10.1128/JVI.80.1.412-425.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu N, Chen CY, Shyu AB. 2001. Versatile role for hnRNP D isoforms in the differential regulation of cytoplasmic mRNA turnover. Mol Cell Biol 21:6960–6971. doi: 10.1128/MCB.21.20.6960-6971.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Díaz-Salinas MA, Silva-Ayala D, López S, Arias CF. 2014. Rotaviruses reach late endosomes and require the cation-dependent mannose-6-phosphate receptor and the activity of cathepsin proteases to enter the cell. J Virol 88:4389–4402. doi: 10.1128/JVI.03457-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bhandari N, Sharma P, Glass RI, Ray P, Greenberg H, Taneja S, Saksena M, Rao CD, Gentsch JR, Parashar U, Maldonado Y, Ward RL, Bhan MK. 2006. Safety and immunogenicity of two live attenuated human rotavirus vaccine candidates, 116E and I321, in infants: results of a randomised controlled trial. Vaccine 24:5817–5823. doi: 10.1016/j.vaccine.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 103.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 104.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 105.Zhang B, Kirov S, Snoddy J. 2005. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.