

Abstract
Human papillomavirus type 16 (HPV16) is the etiologic factor for cervical cancer and a subset of oropharyngeal cancers. Although several prophylactic HPV vaccines are available, no effective therapeutic strategies to control active HPV diseases exist. Tumor implantation models are traditionally used to study HPV-associated buccal tumors. However, they fail to address precancerous phases of disease progression and display tumor microenvironments distinct from those observed in patients. Previously, K14-E6/E7 transgenic mouse models have been used to generate spontaneous tumors. However, the rate of tumor formation is inconsistent, and the host often develops immune tolerance to the viral oncoproteins. We developed a preclinical, spontaneous, HPV16+ buccal tumor model using submucosal injection of oncogenic plasmids expressing HPV16-E6/E7, NRasG12V, luciferase, and sleeping beauty (SB) transposase, followed by electroporation in the buccal mucosa. We evaluated responses to immunization with a pNGVL4a-CRT/E7(detox) therapeutic HPV DNA vaccine and tumor cell migration to distant locations. Mice transfected with plasmids encoding HPV16-E6/E7, NRasG12V, luciferase, and SB transposase developed tumors within three weeks. We also found transient anti-CD3 administration is required to generate tumors in immunocompetent mice. Bioluminescence signals from luciferase correlated strongly with tumor growth, and tumors expressed HPV16-associated markers. We showed that pNGVL4a-CRT/E7(detox) administration resulted in antitumor immunity in tumor-bearing mice. Lastly, we demonstrated that the generated tumor could migrate to tumor draining lymph nodes. Our model provides an efficient method to induce spontaneous HPV+ tumor formation, which can be used to identify effective therapeutic interventions, analyze tumor migration, and conduct tumor biology research.
Keywords: Human Papillomavirus, preclinical model, spontaneous tumor, buccal tumor, immune therapy
Introduction
Human papillomaviruses (HPVs) are small DNA viruses responsible for a multitude of diseases ranging from benign warts to anogenital and oropharyngeal malignancies (1–3). In most cases, HPV infections spontaneously regress or remain subclinical. However, persistent infections with high-risk HPV types (i.e. HPV 16 and 18) may progress into precancerous lesions, and eventually cancer (4,5). In the case of HPV+ head and neck squamous cell carcinoma (HNSCC), which is mainly driven by HPV16, patients do not usually display symptoms until an advanced stage, during which tumor metastasis, particularly to tumor draining lymph nodes, is common (3,6). Typically, surgery, radiation therapy, and chemotherapy are used for the treatment of HNSCC, but these therapeutic methods often result in irreversible damage to anatomical structures and reduced quality of life (7–9). It has been suggested that antitumor immunity and immune microenvironments are important for tumor prognosis. As such, many attempts have been made to generate HPV tumor models useful for the evaluation of candidate immunotherapies against HPV-associated HNSCC that may be better tolerated and more effective than conventional therapies (10).
Traditionally, preclinical HPV+ oral tumor models are generated by implanting tumor cell lines that express the HPV oncoproteins into the oral cavity of mice (11–13). One major drawback of these tumor challenge models is their inability to capture major aspects of tumorigenesis caused by HPV infection, including the processes of cellular transformation after viral entry and the disease progression from precancerous lesion to invasive cancer (14). An alternative method to create a preclinical HPV tumor model involves the generation of immunocompetent transgenic mice that expresses the HPV oncogenes E6 and E7 (15–20). Nonetheless, due to continuous HPV16 E6 and E7 expression, the transgenic mice often become immunotolerant to these oncogenes, rendering the model less effective at evaluating immunotherapy (10). Zhong et al. has published an improved inducible transgenic HPV+ oral tumor model that may be able to circumvent this issue of immune tolerance (21). However, because HPV16-E6/E7 becomes universally expressed in the epithelium after induction, the antitumor immune response is expected to be different from the clinically localized tumor. Therefore, more research is required to improve preclinical HPV oral tumor models. Ideally, a preclinical HPV oral tumor model should possess the following characteristics: (i) forms spontaneous, localized, HPV+ oral carcinomas; (ii) displays an immune microenvironment that closely resembles those of HPV+ oral tumors in patients; (iii) the tumor-bearing mice should respond appropriately to immune therapies and generate antitumor immunity; and (iv) tumor formation should follow clinical progression starting from precancerous to invasive and metastatic states.
HPV infects basal epithelial cells through wounding. After entering the host cell and uncoating its capsid, viral DNA is transferred into the cell nucleus and maintained in low copy numbers, ranging from 50 to 100 copies per basal cell (22). Under normal circumstances, these basal cells divide, migrate off the basement membrane, change shape, and terminally differentiate into cornified cells at the epithelial surface. Once the basal cells detach from the basement membrane, they silence the cell cycle and stop dividing. Occasionally, HPV DNA integrates into the genomic DNA of the infected cell, during which, several HPV DNA encoded genes, including L1, L2, E1, E2, E4, and E5, may be lost. Importantly, E2 proteins serve as inhibitors for E6 and E7 oncogenes. Oncoproteins E6 and E7 can degrade p53, disrupt the interaction between retinoblastoma protein (pRb) and E2 transcription factors (E2Fs), and reinitiate the cell cycle, resulting in rapid cell proliferation (23,24). Thus, integration of the HPV viral genome into the host genome and the continual expression of E6/E7 are important for HPV oncogenesis.
In order to simulate HPV oncogenesis, we hypothesized that local introduction of high-risk HPV16-E6/E7 DNA can mimic viral DNA entering the cells after HPV infection. The introduction of plasmids encoding HPV16 oncogenes into epithelial cells can be achieved with the use of electroporation, a physical transfection method that uses an electrical pulse to create temporary pores in cell membranes through which substances like nucleic acids can pass into cells. It has been shown that electroporation is effective in introducing foreign nucleic acids into many cell types in mice (25). However, although electroporation achieves very stable transfections, the gene expression is typically transient in mice. Therefore, an additional method is required to reproduce the persistent expression of E6/E7 oncogenes resulting from HPV DNA integration.
Sleeping beauty (SB) is a synthetic transposable element composed of a transposon DNA substrate and a transposase enzyme. SB transposase regulates excision and insertion of transposon DNA into a TA-dinucleotide of the host genomic DNA in a cut-and-paste manner (26,27). The SB transposable element has been previously used to introduce anti-angiogenic genes for long-term therapeutic effects and to generate spontaneous tumor models with persistent oncogene expression (28–30). We hypothesized that the utilization of the SB transposase system could reproduce the essentially random integration process of HPV oncogenes into the host genome and result in continual expression of E6/E7 in the transfected cells.
Previously, it has been demonstrated that HPV16-E6/E7 and mutant Ras oncogenes cooperate to form tumors in transgenic mice (31). This synergistic, carcinogenic effect is restricted to anatomic sites with epithelial transition zones classically affected by HPV16-E6/E7 oncogene expression, which, in the buccal area, is located between the outer skin of the lip and the inner buccal mucosa. Therefore, introduction of viral oncogenes HPV16-E6/E7 with mutant Ras oncogene locally in the buccal area may also result in efficient tumor formation in mice.
In this study, we developed a spontaneous, HPV+ buccal tumor model by injecting mice with plasmids encoding oncogenes HPV16-E6/E7, mutant Ras, Luciferase (as a reporter gene), and SB transposase into the buccal area followed by electroporation. Using a candidate therapeutic HPV DNA vaccine, pNGVL4a-CRT/E7(detox), we evaluated responses to immunotherapy and examined the ability of the transformed tumor cells to migrate to distant locations.
Materials & Methods
Mice and Animal Care
Six- to eight-week old female C57BL/6NCr (strain #556), FVB/NCr (strain #559), BALB/cAnNCr (strain #555) and athymic nude (Athymic NCr-nu/nu, strain #553) mice were purchased from Charles Rivers Laboratories (Frederick, MD, USA). All mice were maintained at the Johns Hopkins University School of Medicine Animal Facility (Baltimore, MD) under specific pathogen-free conditions. All procedures were performed according to protocols approved by the Johns Hopkins Institutional Animal Care and Use Committee and in accordance with recommendations for the proper use and care of laboratory animals.
Plasmid Vectors
To generate pcDNA3-Luc, luciferase was cloned into the XbaI/XhoI sites of pcDNA3 by using the following primers: 5′-aaatctagaatggaagacgccaaaaacat-3′ (forward) and 5′-aaactcgagcacggcgatctttccgccct-3′ (reverse).
To generate pcDNA3-Luc-T2a-E7, T2a-E7 was cloned into the Xho I/EcoRI sites of pcDNA3-Luc by using the following primers: 5′-aaactcgaggagggcagaggaagtcttctaacatgcggtgacgtggaggagaatcccggccctatgcatggagatacacct-3′ (forward) and 5′-tttgaattctggtttctgagaacagatgg-3′ (reverse).
To generate the luciferase E7 and E6 oncogene-fusion construct, pcDNA3-Luc-T2a-E7-T2a-E6), T2a-E6 was cloned into the EcoRI/BamHI sites of pcDNA3-Luc-T2a-E7 by using the following primers: 5′-aaagaattcgagggcagaggaagtcttctaacatgcggtgacgtggaggagaatcccggccctatgcaccaaaagagaact-3′ (forward) and 5′-tttggatcccagctgggtttctctacgtg-3′ (reverse).
To generate Pkt2-Luc-T2a-E7-T2a-E6, Luc-T2a-E7-T2a-E6 was removed from pcDNA3-Luc-T2a-E7-T2a-E6 using XbaI/BamHI sites, and the sequence was cloned into the XbaI/Bgl II sites of the Pkt2/clp-akt vector, as previously performed (30).
To generate Pkt2-Luc-T2a-E7, T2a-E7 was cloned into the XhoI and BstXI sites of Pkt2-Luc-T2a-E7-T2a-E6 by using the following primers: 5′-aaactcgaggagggcagaggaagtcttct-3′ (forward) and 5′-tttccagctagctggttatggtttctgagaacaga-3′ (reverse).
To generate Pkt2-Luc-T2a-E6, T2a-E6 was cloned into the XhoI and BstXI sites of Pkt2-Luc-T2a-E7-T2a-E6 using the primers 5′-aaactcgaggagggcagaggaagtcttct-3′ (forward) and 5′-tttccagctagctggttacagctgggtttctctacg-3′ (reverse).
In the DNA constructs, T2a (EGRGSLLTCGDVEENPGP), a self-cleavage peptide from the T. asigna virus, was inserted between each gene, allowing for the production of each protein individually from a single fusion protein (32). Cleavage of the T2a sequence occurs due to a ribosomal skip mechanism at the point between the final glycine and proline residues (32).
The construction of the pT/Caggs-NRasV12 plasmid, pT2/C-Luc//PGK-SB13 plasmid, and the pKT2/CLP-AKT plasmid has been described previously (30). The plasmids were purchased from Addgene (plasmid #20205, plasmid #20207, and plasmid #20281, respectively). The construction of the pCMV(CAT)T7-SB100 plasmid has also been described previously (33) and was purchased from Addgene (plasmid #34879).
DNA Vaccine
The DNA vaccine, pNGVL4a-CRT/E7(detox), has been previously described (34) and expresses a fusion of human calreticulin (CRT) and detoxified HPV16-E7 antigen. CRT is a 46-kDa protein located in the lumen of the cell’s endoplasmic reticulum and has been shown to strengthen MHC class I presentation for the activation of CD8+ T cells. E7(detox) represents a HPV16-E7 gene with mutations at positions 24 and 26, which disrupt the Rb-binding site of E7 and, thus, abolishes the capacity of E7 to transform cells (35). The plasmid backbone, pNGVL4a, was obtained from the NIH National Gene Vector Laboratory and has been used in several clinical studies (36).
In Vivo Tumor Formation
For the generation of tumors in immunodeficient mice, athymic nude mice (5 mice/group) were submucosally injected with (i) plasmids encoding luciferase and HPV16-E6/E7, mutant Ras, and SB100; (ii) plasmids encoding luciferase and HPV16-E6, mutant Ras, and SB100; (iii) plasmids encoding luciferase and HPV16-E7, mutant Ras, and SB100; (iv) plasmids encoding luciferase, HPV16-E6/E7, and SB100; or (v) plasmids encoding mutant Ras, SB13, and luciferase into the buccal area (10 μg/plasmid, 30 μL/injection), followed by electroporation (8 pulses, 72v for 20ms / pulse, 20 ms intervals between each pulse).
For the generation of tumors in immunodeficient mice with Akt expression, athymic nude mice (5 mice/group) received submucosal injection of DNA plasmids encoding luciferase, HPV16-E6/E7, constitutively active AKT, and SB100 into the buccal area, followed by electroporation as described (10 μg/plasmid, 30 μL/injection).
For the generation of tumors in immunocompetent mice, C57BL/6NCr, Balb/c, or FVB/NCr mice (5 mice/group) were submucosally injected with plasmids encoding luciferase and HPV16-E6/E7, mutant Ras, and SB100 into the buccal area, followed by electroporation (10 μg/plasmid, 30 μL/injection).
For the generation of tumors in immune-suppressed mice, C57BL/6NCr mice (5 mice/group) received one of the following treatments:
(i) intraperitoneal (IP) injection of 100 μg monoclonal anti-CD3 (Clone: 17A2; Catalog #BE0002, Bio X Cell) per day for three consecutive days, and on the day after the final anti-CD3 injection, submucosally injected with plasmids encoding luciferase and HPV16-E6/E7, mutant Ras, and SB100 into the buccal area, followed by electroporation; (ii) IP injection of 100 μg monoclonal anti-CD4 (Clone: GK1.5; Catalog #BE0003-1, Bio X Cell) per day for three consecutive days, and on the day after the third anti-CD4 injection, submucosally injected with plasmids encoding luciferase and HPV16-E6/E7, mutant Ras, and SB100 into the buccal area, followed by electroporation. Anti-CD4 injections were continued at weekly intervals; (iii) IP injection of 200 μg monoclonal anti-CD8 (Clone: 2.43; Catalog #BP0061, Bio X Cell) per day for three consecutive days, and on the day after the third anti-CD8 injection, submucosally injected with plasmids encoding luciferase and HPV16-E6/E7, mutant Ras, and SB100 into the buccal area, followed by electroporation. Anti-CD8 injections were continued at four-day intervals; (iv) submucosal injection of plasmids encoding luciferase and HPV16-E6/E7, mutant Ras, and SB100 into the buccal area, followed by electroporation with no antibody treatment; or (v) IP injection of both 100 μg monoclonal anti-CD4 and 200 μg monoclonal anti-CD8 per day for three consecutive days, and on the day after the final anti-CD4 and anti-CD8 injection, submucosally injected with plasmids encoding luciferase and HPV16-E6/E7, mutant Ras, and SB100 into the buccal area, followed by electroporation. 10μg/plasmid and 30 μL/injection was used for all treatment arms.
Following oncogene plasmid transfection, buccal tumor growth was monitored 1–2 times per week via luminescence imaging (37). In addition, tumor size was measured with digital calipers. To reduce mouse suffering due to the negative impacts of the presence of buccal tumors, mouse death due to tumor burden was defined as: (i) display of a bioluminescence signal of 109 p/sec/cm2/sr; (ii) a tumor volume of 150 mm3; or (iii) a 10% reduction of weight compare to the average weight of healthy mice of the same age. Mice were subsequently euthanized via CO2 asphyxiation when any of these three characteristics were observed.
In Vivo Bioluminescence Image
In vivo bioluminescence imaging was performed using the IVIS Spectrum in vivo imaging system series 2000 (PerkinElmer). In summary, mice were anesthetized by intramuscular (IM) injection of a ketamine/xylazine solution (5:1 ratio). After IP injection of substrate D-luciferin (GoldBio), bioluminescence imaging for luciferase expression was conducted on a cryogenically cooled IVIS system using Living Image acquisition and analysis software (Xenogen). Images were acquired 10 minutes after D-luciferin administration. Bioluminescence was detected by the IVIS imager, integrated, and digitized. The region of interest from displayed images was quantified as total photon counts using Living Image 2.50 software (Xenogen).
Tumor Treatment Experiments
C57BL/6NCr mice (5 mice/group) received IP injections of 100 μg monoclonal anti-CD3 per day for three consecutive days, and on the day after the last anti-CD3 injection, submucosally injected with plasmids encoding luciferase and HPV16-E6/E7, mutant NRas, and SB100 into the buccal area, followed by electroporation as described. Ten days after plasmid injection, mice were vaccinated with pNGVL4a-CRT/E7(detox) or empty pNGVL4a vector only (20 μg/dose/mouse) via IM injection in the hind leg, followed by electroporation. The mice continued to receive the same vaccination at four-day intervals three additional times. Following vaccination, tumor growth was monitored twice a week by in vivo bioluminescence imaging, and the survival and weight of mice were monitored for up to 50 days after oncogenic plasmids transfection. Mice were euthanized via CO2 asphyxiation on day 50 or when any of the following conditions were observed: (i) display of a bioluminescence signal of 109 p/sec/cm2/sr; (ii) a tumor volume of 150 mm3; or (iii) a 10% reduction of weight compare to the average weight of healthy mice of the same age.
Histology and Immunohistochemistry Staining
After euthanizing the tumor-bearing mice, buccal tumors were surgically isolated and placed into 10% neutral buffered formalin solution for adequate fixation with a minimum of 48 hours at room temperature. The tumor samples were then formalin fixed and paraffin embedded. Hematoxylin and eosin (H&E) staining of tissue sections was performed by the Johns Hopkins University Oncology Tissue Services. Immunohistochemical (IHC) staining was performed for the following markers: proliferation marker Ki-67 (Clone: D3B5; Cell Signaling, Catalog #12202), oncoprotein NRas (Santa Cruz Biotechnology, Catalog #sc-519), and HPV marker p16 (Abcam, Catalog #ab51243), secondary antibody (poly-HRP anti-rabbit IgG; Leica, Catalog #PV6119) was used for all IHC staining. The histology slides were reviewed by a board certified gynecologic pathologist (Dr. T.-C. Wu) of the Pathology Department in the Johns Hopkins University School of Medicine.
Preparation of Tumor-Infiltrating Lymphocytes
Seven days after the last vaccination, mice were euthanized, and the tumor tissues were surgically removed, minced into 1- to 2mm pieces, digested with serum-free RPMI-1640 medium containing collagenase I (0.05 mg/mL; Sigma-Aldrich, Catalog #C9891), collagenase IV (0.05 mg/mL; Sigma-Aldrich, Catalog #C5138), hyaluronidase IV (0.025 mg/mL; Sigma-Aldrich, Catalog #H3506), DNase I (0.25 mg/mL; Sigma-Aldrich, Catalog #DN25-g), and penicillin (100 U/mL) and streptomycin (100 μg/mL) (Thermo Fisher, Catalog #15140122), and incubated at 37°C with periodic agitation. The tumor digest was then filtered through a 70 μm nylon filter mesh to remove undigested tissue fragments. The resultant single tumor cell suspensions and tumor-infiltrating lymphocytes were washed with PBS containing 0.5% BSA, and viable cells were determined using trypan blue dye exclusion.
In Situ hybridization to Detect HPV16 Oncogenes
In situ hybridization was performed using the RNAscope® 2.0 HD Brown Chromogenic Reagent Kit (Advanced Cell Diagnostics, Hayward, CA) using the supplied protocol and a target probe against HPV16-E6/E7 (Advanced Cell Diagnostics #311521). Briefly, fresh-cut, formalin-fixed, paraffin-embedded slides were baked at 60°C overnight. After deparaffinization, slides were air-dried, circled with a hydrophobic barrier pen (Vector labs, ImmEdge pen Catalog # H-4000), and then exposed to pretreatment solutions 1, 2, and 3. Target probes were hybridized for 2 hours at 40°C in hybridization chambers, followed by a series of signal amplification and washing steps. The signals were detected by chromogenic reactions using DAB chromogen followed by undiluted Gill’s hematoxylin (Sigma-Aldrich, St. Louis, MO) counterstaining.
Tetramer Staining and Flow Cytometry
To evaluate the systemic immune response, peripheral blood was collected into 1.5 mL EDTA-coated Eppendorf tubes from the ventral arteries of mice tails. After red blood cell lysis with ACK lysing buffer (Quality Biological, Gaithersburg, MD, USA), peripheral blood mononuclear cells (PBMCs) were then stained with fluorescein isothiocyanate (FITC)-conjugated monoclonal rat anti-mouse CD3 (BD Pharmingen, Catalog # 555274), allophycocyanin (APC)-conjugated monoclonal rat anti-mouse CD8a (Biolegend, Catalog # 100712), and phycoerythrin (PE)-conjugated H-2Db tetramer loaded with HPV16-E7aa49–57 peptide (provided by NIH Tetramer Core Facility) at 4°C for at least 30 minutes. After washing with FACS washing buffer (0.5% BSA in PBS), the cells were stained with 7-AAD before flow cytometry analysis to exclude dead cells.
To evaluate the presence of E7-specific CD8+ T cells in the tumor microenvironment (TME) following vaccination, tumor-infiltrating lymphocytes (TILs) obtained from the tumor tissue of mice were stained with FITC-conjugated anti-mouse CD8a (BD Pharmingen, Catalog # 553031) and PE-conjugated HPV16-E7aa49-57 peptide-loaded H-2Db tetramer. To evaluate the presence of regulatory T cells in the TME, TILs were stained with FITC-conjugated rat anti-mouse CD4 (eBioscience, clone RM4-5) and APC-conjugated anti-mouse CD25 (eBioscience, Catalog # 17-0251-82), permeabilized and fixed (Fixation/Permeablization Solution Kit, BD Biosciences, Catalog #555028), and stained with PE-conjugated anti-mouse Foxp3 (eBioscience, clone FJK-16s). To evaluate the presence of myeloid derived suppressor cells in TME, TILs were stained with FITC-conjugated anti-mouse Gr-1 (eBioscience, Catalog #11-5931-82) and PE-conjugated anti-mouse CD11b (eBioscience, Catalog #12-0112-82). After staining and washing, the cells were acquired with a FACS Calibur flow cytometer (BD Biosciences) and analyzed with FlowJo® V.10 software.
Evaluation of Metastasis Capability
When the tumor-bearing C57BL/6 mice experienced one of the following conditions: (i) display of a bioluminescence signal of 109 p/sec/cm2/sr; (ii) a tumor volume of 150 mm3; or (iii) a 10% reduction of weight compare to the average weight of healthy mice of the same age, mice were statistically considered to have died from tumor burden, and euthanized according to the protocol. Enlarged tumor-draining lymph nodes (TDLNs) and nontumor-draining lymph nodes (NTDLNs) were surgically remove for ex vivo bioluminescence assays. The TDLNs were minced, treated with tissue digestion buffer (collagenase [0.05 mg/mL], collagenase IV [0.05 mg/mL], hyaluronidase IV [0.025 mg/mL], and DNase I [0.25 mg/mL] in deionized water), filtered, and then cultured in 6-well plates with RPMI 1640 medium (Thermo Fisher, Catalog #11875093) supplemented with penincillin (100 U/mL) and streptomycin (100 μg/mL), 1X MEM non-essential amino acids (Thermo Fisher, Catalog #11140050), 1 mM sodium pyruvate (Thermo Fisher, Catalog #11360070), 2 mM L-glutamine (Thermo Fisher, Catalog #25030081), 55 nM 2-mercaptoethanol (Thermo Fisher, Catalog #21985023), and 10% fetal bovine serum (Thermo Fisher, Catalog #16000036). After one week of culturing, whole cell mixtures were transferred to T75 tissue culture flasks. Once the cells expanded to cover more than 70% of the flask surface, the cells were trypsinized, PBS-washed, and 5x105 cells were resuspended in 40 μL PBS solution, followed by submucosal buccal injection into immunodeficient athymic mice. After injection, the presence and growth of tumor cells in mice were monitored via in vivo bioluminescence imaging.
Ex Vivo Luciferase Bioluminescence Assay
Athymic nude mice received submucosal injection of plasmid DNA in the buccal area, followed by electroporation. The plasmids included luciferase, E6/E7, NRasG12V, and SB100. When the tumor size exceeded 7 mm in diameter, mice were considered to have died from tumor burden, and euthanized according to protocol. Tumor tissue, TDLNs, and spleens were harvested for luciferase assays. Tissues from naïve mice were used as negative controls. The tissue was homogenized by a Mini-Beadbeater-1 with 1 mm and 0.1 mm glass beads (Bio Spec Products Inc.) in 50000. Bioluminescence assays of the tissue samples were conducted using the superficGloMax®-Multi Detection System, according to the manufacturer’s instructions (Promega Corporation). Relative light units (RLU) were adjusted to each gram of tissue.
Statistical Analysis
The statistical analyses were performed with GraphPad Prism V.6 software (La Jolla, CA, USA). Data were expressed as means ± standard deviation (SD). Kaplan-Meier survival plots were constructed to estimate either tumor-free rate or survival percentage. The log-rank test was used to compare survival times between treatment groups. Comparisons between individual data points were analyzed by two-tailed Student’s t tests. A p-value < 0.05 was considered statistically significant.
Results
Optimal plasmid combination to generate HPV16 spontaneous buccal tumors
To generate spontaneous oral tumors that continuously express HPV16-E6 and E7, different combinations of plasmids (Fig. 1A) were submucosally injected into the buccal area of athymic nude mice, followed by electroporation. The luciferase enzymes encoded in the plasmids resulted in the generation of bioluminescence from viable, transfected cells that could be measured quantitatively using an IVIS imager. After plasmid injection, tumor growth was monitored through in vivo bioluminescence imaging. We observed that a combination of plasmids encoding sleeping beauty transposase (SB100), luciferase, HPV16-E6/E7, and NRasG12V oncogenes resulted in the fastest tumor formation and growth rate as measured by the luminescence intensity (Fig. 1B–C). Based on our results, the combination of E6, E7, and mutant NRas oncogenes is the most efficient at causing oral tumor formation in mice.
Figure 1. Generation of spontaneous HPV buccal tumors in immunocompromised mice.

(A) Schematic of plasmids used to induce oral tumors by oncogene transfection. (B) Immunocompromised, athymic nude mice (NCr strain, n=5/group) received plasmids once through submucosal injection followed by electroporation in the buccal area. Different combinations of plasmids are indicated in the legend. The bioluminescence image was recorded by IVIS Spectrum after IP injection of luciferin solution. (C) Percent tumor-free survival. Three independent experiments were performed. P values were calculated by log-rank test (C). P values < 0.05 were considered significant. Data are presented as mean ± SD.
Initial CD3 depletion is necessary for the formation of spontaneous buccal tumors
After demonstrating the ability to generate spontaneous oral tumors in immunodeficient mice by oncogene-encoded plasmid transfection with SB transposase, we sought to apply the same strategy for the generation of spontaneous oral HPV tumors in immunocompetent mice. Plasmids encoding luciferase, HPV16-E6/E7, NRasG12V, and SB100 were submucosally injected into the buccal area of C57BL/6NCr, Balb/c, and FVB/NCr mice. However, in contrast to our observation in immunodeficient mice, the luciferase gene expression decreased steadily after transfection, as indicated by bioluminescence intensity, indicating the clearance of transfected cells by immunocompetent mice, regardless of genetic strain (Fig. 2A). Thus, we hypothesized that an immunosuppressed environment is necessary for the spontaneous formation of tumors following plasmid transfection. To test this hypothesis, we treated C57BL/6NCr mice with or without anti-CD3, anti-CD4, or anti-CD8 antibodies to deplete various T-cell populations prior to plasmid transfections (illustrated in Fig. 2B). Mice that received anti-CD3 (total T-cell depletion) experienced a rapid increase in luminescence signal after plasmid transfection, suggesting the expansion of plasmid-transfected cells. In contrast, mice that received anti-CD4 or anti-CD8 antibodies (i.e. CD4+ or CD8+ T-cell depletion, respectively) generated similar luminescence signals as the control group (no antibody depletion) (Fig. 2C). When co-treated with anti-CD4 and anti-CD8, plasmid-transfected mice showed an initial increase in luminescence signal, which decreased back to baseline by day 28 (Supplementary Fig. S1).
Figure 2. Generation of spontaneous HPV buccal tumors in immunocompetent mice.

(A) C57BL/6NCr, Balb/c, or FVB/NCr mice (n=5/group) received submucosal plasmid injections, followed by electroporation. The plasmids contained HPV16-E6/E7, luciferase, NRasG12V, and SB100 transposase. Bioluminescence intensity was recorded by an IVIS Spectrum. (B) Schematic diagram of treatment protocol. Control C57BL/6NCr mice (n=5): plasmid transfection only; CD3-depleted C57BL/6NCr mice (n=5): 100 μg anti-CD3 IP for a total of three doses at one-day intervals before plasmid transfection; CD4-depleted C57BL/6NCr mice (n=5): began with 100 μg anti-CD4 IP for a total of three doses at one-day intervals before receiving one dose weekly after plasmid transfection; CD8-depleted C57BL/6NCr mice (n=5): began with 200 μg anti-CD8 IP for a total of three doses at one-day intervals before receiving two doses weekly after plasmid transfection. (C) Bioluminescence kinetics of buccal tumors in different groups. Three independent experiments were performed. Data are presented as mean ± SD.
In addition to the observed increase in luminescence intensity, visible tumors were observed in CD3-depleted mice around 17 days after oncogene transfection. To examine the relationship between T-cell depletion and the formation of tumors, total T-cell populations in CD3-depleted, plasmid-transfected mice was measured. As shown in Fig. 3A, the amount of systemic CD3+ T cells in mice declined sharply following anti-CD3 injection. On day 9, the luminescence intensity in mice began to increase and continued to increase even as the CD3+ T-cell population began to recover. The volume of the spontaneous tumors formed after plasmid transfection was strongly correlated with luminescence intensity (Fig. 3B). The presence of growing tumor in plasmid-transfected mice did not influence the rate of T-cell recovery when compared to that of CD3-depleted mice without oncogenic plasmid transfection (Supplementary Fig. S2).
Figure 3. Bioluminesence signal as index for spontaneous HPV buccal tumor growth.

C57BL/6NCr mice (n=5) were CD3-depleted using 100 μg anti-CD3IP daily for three consecutive days, followed by submucosal plasmid injection and electroporation in the buccal area one day after the final antibody injection. Plasmids contained HPV16-E6/E7, luciferase, NRasG12V, and SB100 (10 μg/plasmid). (A) Relationship between bioluminescence kinetics of buccal tumor and percentage of CD3+ T cells over time. Black star: visualized overt tumor. Peripheral blood from tail arteries was taken for flow cytometry to check CD3+ T cells percentage over time. (B) The bioluminescence intensity plotted against tumor volume. Three independent experiments were performed. Data are presented as mean ± SD.
To assess the influence of growing oral tumors on the health status of mice, the formation and growth of tumors, as well as the weight of oncogenic plasmids-transfected mice with or without CD3 depletion, were monitored and evaluated. Similar to those observed in Fig. 2C, the luminescence signal in control mice (without CD3 depletion) decreased rapidly following oncogenic plasmid transfection, resulting in no tumor formation, as well as a steady weight gain (Supplementary Fig. S3A). In comparison, a steady increase in luminescence signal was observed with oncogenic plasmid-transfected, CD3-depleted mice, which corresponded to formation of tumors and reduction in mouse weight (Supplementary Fig S3B). Four weeks after oncogenic plasmid transfection, significant differences in mouse weight (~10%) were observed between oncogenic plasmid-transfected mice with or without initial CD3 depletion (Supplementary Fig. S3C).
Together, these results suggest that initial total T-cell depletion, as compared to partial depletion, is required for the formation of spontaneous oral HPV tumors in immunocompetent mice and that luminescence intensity is an effective and reliable measurement for monitoring tumor growth. These data demonstrate that the size of spontaneous HPV buccal tumors negatively impacts the health status of mice. Thus, to minimize mouse suffering due to the presence of growing buccal tumor, we subsequently defined mouse death due to tumor burden as discussed in the methods section, and mice were euthanized when any of the listed characteristics were observed.
Characteristics of the HPV16 spontaneous oral tumor model
To characterize the spontaneous buccal tumors formed after the transfection of oncogene-encoding plasmids, we treated C57BL/6NCr mice with anti-CD3 daily for three days, followed by transfection of plasmids encoding E6/E7, NRasG12V, SB100, and luciferase at the buccal area. When the resultant spontaneous tumors reached 7 mm in diameter, the mice were sacrificed, and the buccal tumors were excised, fixed with formalin, and embedded in paraffin. The tissue sections of the tumors were prepared and stained for various tumor markers. The spontaneous tumors originated from the buccal mucosa area appeared as solid papillary masses with bleeding ulcers (Fig. 4A). H&E staining showed that the tumors were composed of spindle-shaped tumor cells with high mitotic activity (Fig. 4B). IHC staining revealed high expressions of Ki-67, a cell proliferation biomarker, as well as positive NRas and HPV biomarker p16 expression (Fig. 4B–C). RNA in situ hybridization with HPV16-E6/E7 probes also showed strong staining in the tumor (Fig. 4C).
Figure 4. Characteristics of spontaneous HPV buccal tumors.

(A) Representative pictures of gross buccal tumors arising from oncogene cotransfection in C57BL/6NCr mice. Plasmids contained HPV16-E6/E7, luciferase, NRasG12V, and SB100. (B) Representative histology sections with H&E stain and special IHC staining with Ki-67 and NRas. (C) Representative images for RNAscope targeting HPV16 RNA and IHC staining with HPV-associated biomarker p16. Three independent experiments were performed.
HPV DNA vaccination inhibited the growth of spontaneous buccal tumors
After we successfully demonstrated the ability to repeatedly generate the HPV16+ spontaneous buccal tumors in C57BL/6NCr mice, we sought to verify whether this model could be used to assess the efficacy of immune therapy. Initial CD3-depleted, oncogene plasmid-transfected C57BL/6NCr mice were vaccinated with either a therapeutic HPV DNA vaccine, pNGVL4a-CRT/E7(detox), or empty pNGVL4a vector only 10 days after the plasmid transfection. The mice received the same vaccination a total of four times at four-day intervals (Fig. 5A). As shown in Fig. 5B, mice vaccinated with pNGVL4a-CRT/E7(detox) had significantly better survival percentages compared to mice vaccinated with empty pNGVL4a control. 80% of the mice that received pNGVL4a-CRT/E7(detox) DNA vaccination survived more than eight weeks after plasmid transfection, whereas all mice that received empty pNGVL4a vector vaccination died in less than four weeks after plasmid transfection. When the experiment was repeated over an extended time frame, we observed that all tumor-bearing mice treated with pNGVL4a-CRT/E7(detox) survived more than twenty weeks after oncogenic plasmid transfection, whereas all mice treated with empty pNGVL4a vector died in less than six weeks (Supplementary Fig. S4). The control of spontaneous HPV buccal tumors in vaccinated mice corresponded with the generation of a potent systemic E7-specific CD8+ T-cell response (Fig. 5C–D). Mice vaccinated with pNGVL4a-CRT/E7(detox) showed lower luminescence signals compared to the mice treated with empty pNGVL4a vector, demonstrating the clearance of plasmid-transformed tumor cells (Fig. 5E–F).
Figure 5. DNA vaccination generated E7-specific CD8+ T-cell responses against spontaneous HPV oral tumors.

(A) Schematic diagram of treatment regimens, and the overall time course. C57BL/6NCr mice (n=5) were CD3-depleted, as described, for three consecutive days, followed by submucosal injection and electroporation in the buccal area with plasmids containing HPV16-E6/E7, luciferase, NRasG12V, and SB100. Ten days after plasmid injection, mice were vaccinated with pNGVL4a-CRT/E7 DNA vaccine or empty pNGVL4a plasmid vector control for a total of four times at four-day intervals. (B) Kaplan-Meier survival analysis of mice. (C) Peripheral blood from tail arteries were examined by flow cytometry for E7-specific CD8+ T cells. Percentages were checked over time from two weeks after plasmid injection at one-week intervals. (D) Representative flow cytometry images of the percentage of E7-specific CD8+ T cells in PBMCs of tumor-bearing mice treated with pNGVL4a control (top) and pNGVL4a-CRT/E7(detox) (bottom) on day 21 after plasmid injection. A total number of 120,000 cells were acquired. (E) Real-time bioluminescence image of tumor-bearing mice. (F) Line graph depicting the change in tumor volume of tumor-bearing mice after plasmid transfection. Three independent experiments were performed. P values were calculated by log-rank test (B) or two-tailed Student t-test (C). P values < 0.05 were considered significant. Data are presented as mean ± SD.
In addition to evaluating the systemic E7-specific CD8+ T-cell response elicited in tumor-bearing mice following vaccination, the influence of vaccination on the immune cell populations in the TME was assessed using the tumor tissues harvested from mice one week after the last vaccination. In comparison to tumor-bearing mice treated with empty pNGVL4a plasmid vector, a significantly higher population of E7-specific CD8+ T cells was observed in the tumors of mice treated with pNGVL4a-CRT/E7(detox) (Fig. 6A). In contrast, no differences in the abundance of regulatory T cells or myeloid-derived suppressor cells in the tumor were observed among mice treated with various regimens (Fig. 6B–C). These results indicate that therapeutic vaccination can lead to the generation of a potent antigen-specific immune response against the spontaneous HPV buccal tumors, resulting in effective tumor control and prolonged the survival of tumor-bearing mice.
Figure 6. Analysis of the TME.
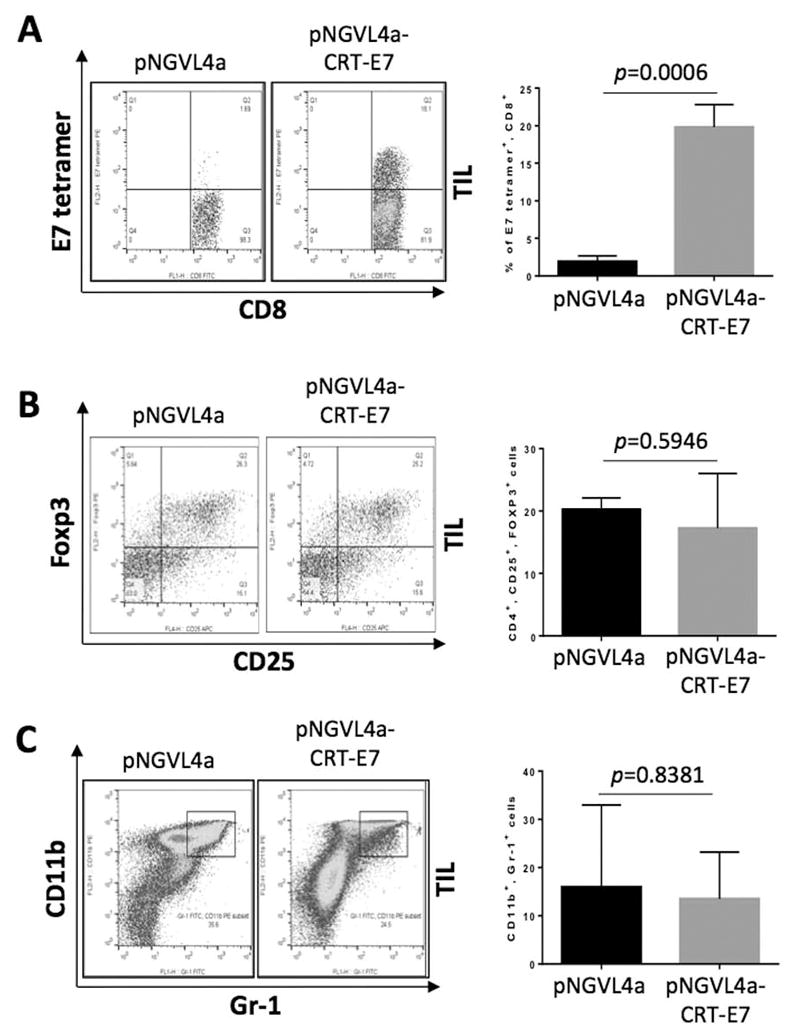
C57BL/6NCr mice (n=3) were CD3-depleted using 100 μg anti-CD3 IP injected daily for three consecutive days, followed by submucosal plasmid injection and electroporation in the buccal area one day after the final antibody injection. The plasmids contained HPV16-E6/E7, luciferase, NRasG12V, and SB100 (10 μg/plasmid). Ten days after plasmid transfection, mice were treated with pNGVL4a-CRT/E7(detox) or empty pNGVL4a vector only (20 μg/dose/mouse) via IM injection, followed by electroporation for a total of four times at four-day intervals. Seven days after last vaccination, the mice were euthanized, and the tumor tissues were harvested and analyzed for the presence of various immune cell populations. (A) Representative flow cytometry images (left) and bar graph summary (right) of E7-specific CD8+ T cells in the TME. (B) Representative flow cytometry images (left) and bar graph summary (right) of regulatory T cells in TME. (C) Representative flow cytometry images (left) and bar graph summary (right) of myeloid-derived suppressor cells in TME. Three independent experiments were performed. P values were calculated by two-tailed Student t-test. P values < 0.05 were considered significant. Data are presented as mean ± SD.
Continued growth of spontaneous HPV16 oral tumors results in metastasis to LNs
In our initial experiments involving the use of immunodeficient mice, a substantial amount of luciferase activity was observed in the tumor, tumor-draining lymph nodes (TDLNs), and spleens of the plasmid-transfected, athymic nude mice as compared with naïve mice (Fig. 7A). Likewise, in C57BL/6NCR mice bearing spontaneous HPV buccal tumors generated from plasmid transfection, enlarged and superficial TDLNs were observed with detectable luciferase activity as compared to non- tumor-draining lymph nodes (NTDLNs) (Fig. 7B–C). We hypothesized that these TDLNs contained transformed tumor cells that metastasized from the buccal area. To test our hypothesis, when the mice were euthanized due to tumor burden, luciferase-positive TDLNs were surgically removed and subsequently minced, digested, and cultured. After cell proliferation was observed, the cell mixture was collected and implanted into the buccal area of immunodeficient nude mice. The re-implanted LN cells showed positive luciferase activity, and the formation of buccal tumors in implanted nude mice was observed (Fig. 7D). IHC staining revealed that the tumors generated by LN cell implantation were positive for HPV marker p16, as well as NRas (Fig. 7E). These histological analyses of the tumors formed from the LN cell implantation revealed characteristics similar to that of HPV16 spontaneous buccal tumors originating from plasmid transfection (Fig. 4), suggesting that the HPV16 tumor cells can migrate to different locations inside tumor-bearing mice, replicating the metastatic process of late-stage HPV-associated cancer.
Figure 7. Metastatic capability in the spontaneous tumor model.

Representative mice were presented. (A) Ex vivo bioluminescence assay for the tissues of tumor (left), tumor draining lymph node (TDLN; middle), and spleen (right) of athymic nude mice receiving buccal plasmid injections containing HPV16-E6/E7, luciferase, NRasG12V, and SB100 followed by electroporation. (B-C) C57BL/6NCr mice received anti-CD3 to deplete T cells for three consecutive days, followed by buccal plasmid injection with HPV16-E6/E7, NRasG12V, and SB100. (B) Representative image showing the TDLNs and non-TDLNs (NTDLN) of plasmid-transfected mice at the time of euthanization (C) Representative image depicting the luciferase activity of TDLNs and NTDLNs. (D) Luciferase activity after re-implantation of 5 × 105 harvested, proliferating TDLN cells, as well as tumor formation. (E) Representative histology of a tumor formed by re-implantation of TDLN cells. Three independent experiments were performed. P values were calculated by two-tailed Student t-test. P values < 0.05 were considered significant. Data are presented as mean ± SD.
Generation of a spontaneous HPV16+ oral tumor model with AKT oncogenes
Although we demonstrated that transfection of plasmids encoding HPV16-E6/E7, NRasG12V, luciferase, and SB100 lead to the spontaneous formation of tumors in mice, Ras mutation is not typically observed in the HPV+ oral cancers presented by patients in the clinic (38). In comparison, many clinical HPV+ oral tumors are associated with mutations in the PI3K signaling pathway (38). Thus, we explored the ability to generate spontaneous HPV+ buccal tumors in immunocompromised mice by changing the plasmid encoding NRasG12V to plasmid encoding constitutively active AKT, a downstream molecule of the PI3K pathway. We observed that the introduction of AKT with HPV16 E6 and E7 oncogenes could also lead to the spontaneous formation of oral tumors in athymic nude mice (Supplementary Fig. S5).
Discussion
In this study, we developed a preclinical, spontaneous HPV buccal tumor model through transfection of plasmids encoding oncogenes HPV16-E6/E7, NRasG12V, as well as SB transposase and luciferase reporter genes. Plasmid transfection resulted in the formation of spontaneous tumors in immunodeficient, athymic nude mice and in immunocompetent C57BL/6NCr mice subjected to early immune suppression. We demonstrated that these spontaneous tumors expressed relevant HPV16 biomarker p16 and detectable HPV16 RNA, could metastasize and migrate to distant locations, such as TDLNs, and could be controlled through immunotherapeutic treatments.
The spontaneous HPV buccal tumor model developed in the current study complements existing models. As mentioned in the introduction, although prior HPV+ HNSCC tumor models can result in consistent tumor formation and have the ability to utilize human-derived HPV+ tumor cells, they fail to replicate several crucial characteristics of HPV cancers (11–13). Likewise, conventional HPV transgenic mice require multiple rounds of selection to ensure successful genetic modification, which can take months to a year or more for tumors to form and often result in immune tolerance (10,15–19,39–41). The improved, Cre-LoxP–inducible, transgenic HPV oral model addressed some issues of earlier models, making it more clinically relevant compared to conventional transgenic models (21). However, the modified transgenic model requires the integration of three transgenes, which further complicates the selection process. Mammalian genomes have been shown to contain many ‘pseudo’ LoxP sites that can potentially be recombined by Cre-recombinase with unknown effects (42). The spontaneous HPV tumor model developed in the current study does not require xenograft implantations of pre-existing tumor cells but instead utilizes the SB transposon system to induce oncogene integration and long-term expression (26,28–30). The method used in our current study resulted in the formation of spontaneous tumors in both athymic nude mice and wildtype mice within a few weeks following plasmid transfection, with a tumor formation rate of greater than 80%, allowing for time-efficient and time-flexible experimental designs compared to the transgenic models mentioned previously. The high tumor formation rate upon plasmid transfection can potentially reduce the mouse turnover while ensuring the generation of a sufficient amount of tumor-bearing mice to be used for subsequent evaluations. Therefore, our model provides a cost-, time-, and labor-efficient method to generate spontaneous HPV oral tumors that mimic localized viral gene integrations and cellular transformations that occur during natural HPV disease progressions.
In our spontaneous tumor model, we demonstrated that initial CD3 T-cell depletion in immunocompetent C57BL/6NCr mice is necessary for tumor formation to occur. This likely reflects the natural immunogenicity of HPV oncoproteins E6 and E7, as the generation of antigen-specific immune responses following exposure to HPV antigens, either through infection or through introduction of DNA encoding the HPV antigenic proteins, has been well documented in both human and murine settings (43–48). It is likely that the initial anti-CD3 injection depleted the CD3+ T cells in mice at the time of plasmid transfection and prevented the generation of rapid antitumor immune responses, including the priming of antigen-specific T cells. As a result, no generation of systemic or local E7-specific CD8+ T-cell responses were observed in initial CD3-depleted, plasmid-transfected control mice, which did not receive subsequent antigen-specific immunization. In clinical settings, it is also observed that patients experiencing immune suppression, such as those infected with human immunodeficiency virus (HIV), have elevated risks for persistent HPV infections and cervical cancers, further emphasizing the importance of the immune system in mediating HPV-associated disease progression (49–55). Nonetheless, we demonstrated that despite the decrease in total T-cell population caused by early CD3 depletion, vaccination with a therapeutic HPV DNA vaccine, pNGVL4a-CRT/E7(detox), still effectively enhanced HPV16/E7-specific CD8+ T-cell responses in tumor-bearing mice, leading to potent tumor control and prolonged mouse survival. These findings suggest that a potent therapeutic HPV vaccine can potentially trigger effective HPV-specific immune responses, even in immune-suppressed patients. This is in-line with our previous preclinical study demonstrating that HPV DNA vaccination is capable of controlling HPV-associated tumors in the absence of CD4+ T cells (56), and results in the clearance of HPV infection. Although the current study utilized immune therapy as the modeled therapeutic method, the spontaneous HPV+ buccal tumor model may also be used to evaluate other forms of treatment, including chemotherapy and radiotherapy.
In addition to CD4, CD8, or CD3 depletion, we also tested the co-administration of anti-CD4 and anti-CD8 for the induction of tumor formation. However, although the CD4/CD8-depleted mice experienced an initial increase of luciferase signal, the signal eventually decreased to baseline levels, indicating clearance of transfected cells in CD4/CD8 depleted mice as compared to that in CD3-depleted mice. The discrepancy between the experimental outcomes in oncogenic plasmid-transfected mice treated with anti-CD3 versus anti-CD4 anti-CD8 suggests the potential involvement of CD3+ CD4− CD8− immune cells, such as natural killer T (NKT) cells, γδ T cells, and double-negative αβ T (DNT) cells, in preventing the formation of tumors following oncogenic plasmid transfection, in addition to the antigen-specific CD4+ and CD8+ T cells. NKT cells have been reported to recognize α-galactosylceramide in conjunction with the MHC class I-like CD1d molecule and produce a large amount of IFNγ to activate NK cells and CD8+ T cells to eliminate MHC-negative and MHC-positive tumors, respectively (57). NKT cells have been suggested to contribute to the immune surveillance of chronic lymphocytic leukemia (58), prevent metastasis of preclinical breast cancer model (59), and it has been demonstrated to be well-tolerated and potentially efficacious as an antitumor therapy in patients with non-small cell lung cancers, head and neck tumors, and advanced melanoma (for review see (60,61)). γδ T cells have been suggested as an early source of IFNγ in tumor immune surveillance, and they regulate the function of tumor-specific CD8+ T cells (62), as well as to exert direct cytotoxicity against various tumor cells (for review see (63,64)). γδ T cell-based immunotherapy has been suggested to have better efficacy than current second-line therapies for a few types of cancers (for review see (65)). Lastly, DNT cells has been shown to have the capacity to restrict the growth of a lymphoma tumor cell line in mice (66), to have potent cytotoxic effects against allogeneic and autologous primary leukemic blasts isolated from AML patients (67), and to inhibit the growth of pancreatic carcinoma in vivo via MHC class 1 chain-related molecules A and natural killer group 2 member D (NKG2D) mediated manner (68). It would be of particular interest to examine the potential roles of these immune cell populations in regulating the formation and growth of spontaneous HPV+ oral tumors following oncogenic plasmid transfection in future studies. Further studies are also warranted to explore additional methods to induce an immunosuppressive environment in mice for the spontaneous formation of tumors.
At around four weeks after oncogene injection, when the mice were sacrificed, enlarged TDLNs were observed. These LN cells had positive luciferase activity and, when implanted into the buccal area of immunodeficient, athymic nude mice, resulted in the formation of gross tumors. Based on the difference in the luminescence intensity observed in the buccal tumor tissue and TDLNs, our data suggests that the transformed tumor cells detected in the LNs originated in the buccal region and subsequently metastasized to the TDLNs. Previous studies also support the notion that buccal tumor formation precedes metastasis in the oral TDLNs and, thereby, resulting in tumor-positive TDLNs (69–72). We hope to potentially utilize our model in the future to study and analyze in vivo tumor migration and metastasis, and future studies are warranted to evaluate how the metastasis of these tumor cells influences the immune microenvironment of TDLNs and to correlate the results with those in clinical settings.
Despite all of the potential benefits that our preclinical tumor model may introduce, we recognize that our model still has room for further improvements. In particular, while our spontaneous tumor model expresses several relevant markers that are present in clinical HPV oral tumors, the morphology of the tumors derived through our approach is closer to that of a sarcoma rather than carcinoma. This may be due to the fact that submucosal injection delivers the plasmid below the epithelial layer, causing transfection and transformation of non-epithelial cells. This issue could potentially be addressed through the use of the K14 promoter. Although the conventional promoters currently used allow for more efficient expression of the oncogenes, K14 promoters can restrict the oncogene expression within transfected epithelial cells, increasing the likelihood for the generation of spontaneous HPV16+ buccal carcinomas. Our current spontaneous HPV tumor model involved the transfection and integration of HPV16-E6/E7 and NRasG12V oncogenes into the mice. Although it has been well-documented that this combination is effective in triggering carcinogenesis in vivo (31), clinically-reported HPV+ oral cancers are not typically associated with Ras mutation (38). In comparison, mutations in PI3K-AKT signaling pathway is observed in over 50% of HPV+ head and neck squamous cell carcinomas (38). We have demonstrated that transfection of HPV16-E6/E7, AKT, luciferase, and SB100 could also lead to the spontaneous formation of oral tumors in athymic nude mice, although the tumor growth rate of AKT tumors was slower than that formed from transfection of plasmid encoding NRasG12V. Further investigation is warranted for the generation and optimization of the HPV AKT tumor model, which may further enhance the clinical relevance of the spontaneous HPV+ oral tumor model.
Although the incidence of HPV-associated oropharyngeal cancer has been on the rise in developed countries (73), a significant portion of oropharyngeal cancer cases worldwide are still attributed to causes independent of HPV infections such as tobacco smoking (74). Unlike HPV+ oral cancers that are associated with HPV-E6/E7 and mutations in the PI3K signaling pathway, HPV− oral cancers are mainly characterized by mutations in TP53 (38). Previous studies utilized the administration of carcinogen 4-nitroquinoline 1-oxide (4-NQO) in mice to generate HPV− oral cancer (75–78). However, the induction of carcinogenesis by administration of 4-NQO alone is very slow, requiring up to 36 to 40 weeks before the tumor becomes visible. We believe our method to generate spontaneous HPV+ oral tumors may also be applied for the generation of spontaneous HPV− oral tumors. By integrating mutant TP53 genes into cells located at the buccal area of mice via the SB transposase system and exposing the mice to carcinogen 4-NQO, we believe we can generate clinically relevant spontaneous HPV− tumors at a much faster rate, allowing for rapid evaluation of therapeutic options against HPV− oral tumors.
In conclusion, our model provides a fast, efficient, and simple option to induce spontaneous formation of HPV+ tumors, which can be used to assess therapeutic strategies. This model can potentially be used to identify effective therapeutic intervention, analyze tumor migration, and conduct tumor biology and TME research.
Supplementary Material
Acknowledgments
We thank Dr. Richard B.S. Roden for helpful discussions and critical review of this manuscript.
Funding: This work was supported by the United States National Institutes of Health (NIH) Cervical Cancer Specialized Program of Research Excellence (SPORE) (P50CA098252), R21 grant (R21AI109259), R21 grant (R21CA194896), and R01 (R01CA114425).
Footnotes
Disclosure of Potential Conflicts of Interest
T.-C. Wu is a co-founder of and has an equity ownership interest in Papivax LLC. He also owns Papivax Biotech Inc. stock options and is a member of Papivax Biotech Inc.’s Scientific Advisory Board. Additionally, under a licensing agreement between Papivax Biotech Inc. and the Johns Hopkins University, Dr. Wu and Dr. Hung are entitled to royalties on an invention described in this article. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. Yung-Nien Chang is the Chief Scientific Officer of Papivax Biotech Inc. and owns stock options in Papivax Biotech Inc. Other co-authors have declared that no conflict of interest exists.
Authors’ contributions
Conception and design: Y.H. Lin, C.F. Hung, T.C. Wu
Development of methodology: Y.H. Lin, M.C. Yang, S. Peng, C.F. Hung, T.C. Wu
Acquisition of data: Y.H. Lin, M.C. Yang, S.H. Tseng, R. Jiang, S. Peng, T. Henkle, C.F. Hung
Analysis and interpretation of data: Y.H. Lin, M.C. Yang, S.H. Tseng, R. Jiang, T. Henkle
Write, review, and/or revision of the manuscript: Y.H. Lin, A. Yang, E. Farmer, S. Peng, Y.N. Chang, C.F. Hung, T.C. Wu
Administrative, technical, or material support: Y.H. Lin, A. Yang, E. Farmer, S. Peng, C.F. Hung, T.C. Wu
Study supervision: C.F. Hung, T.C. Wu
References
- 1.Ljubojevic S, Skerlev M. HPV-associated diseases. Clinics in dermatology. 2014;32(2):227–34. doi: 10.1016/j.clindermatol.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV. The causal relation between human papillomavirus and cervical cancer. Journal of clinical pathology. 2002;55(4):244–65. doi: 10.1136/jcp.55.4.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehanna H, Beech T, Nicholson T, El-Hariry I, McConkey C, Paleri V, et al. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer--systematic review and meta-analysis of trends by time and region. Head & neck. 2013;35(5):747–55. doi: 10.1002/hed.22015. [DOI] [PubMed] [Google Scholar]
- 4.Ostor AG. Natural history of cervical intraepithelial neoplasia: a critical review. International journal of gynecological pathology : official journal of the International Society of Gynecological Pathologists. 1993;12(2):186–92. [PubMed] [Google Scholar]
- 5.Ghittoni R, Accardi R, Chiocca S, Tommasino M. Role of human papillomaviruses in carcinogenesis. Ecancermedicalscience. 2015;9:526. doi: 10.3332/ecancer.2015.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garbuglia AR. Human papillomavirus in head and neck cancer. Cancers. 2014;6(3):1705–26. doi: 10.3390/cancers6031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monk BJ, Sill MW, McMeekin DS, Cohn DE, Ramondetta LM, Boardman CH, et al. Phase III trial of four cisplatin-containing doublet combinations in stage IVB, recurrent, or persistent cervical carcinoma: a Gynecologic Oncology Group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(28):4649–55. doi: 10.1200/JCO.2009.21.8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lutz ST, Chow EL, Hartsell WF, Konski AA. A review of hypofractionated palliative radiotherapy. Cancer. 2007;109(8):1462–70. doi: 10.1002/cncr.22555. [DOI] [PubMed] [Google Scholar]
- 9.Tewari KS, Sill MW, Long HJ, 3rd, Penson RT, Huang H, Ramondetta LM, et al. Improved survival with bevacizumab in advanced cervical cancer. The New England journal of medicine. 2014;370(8):734–43. doi: 10.1056/NEJMoa1309748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doorbar J. Model systems of human papillomavirus-associated disease. The Journal of pathology. 2016;238(2):166–79. doi: 10.1002/path.4656. [DOI] [PubMed] [Google Scholar]
- 11.Williams R, Lee DW, Elzey BD, Anderson ME, Hostager BS, Lee JH. Preclinical models of HPV+ and HPV− HNSCC in mice: an immune clearance of HPV+ HNSCC. Head & neck. 2009;31(7):911–8. doi: 10.1002/hed.21040. [DOI] [PubMed] [Google Scholar]
- 12.Kimple RJ, Harari PM, Torres AD, Yang RZ, Soriano BJ, Yu M, et al. Development and characterization of HPV-positive and HPV-negative head and neck squamous cell carcinoma tumorgrafts. Clin Cancer Res. 2013;19(4):855–64. doi: 10.1158/1078-0432.CCR-12-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paolini F, Massa S, Manni I, Franconi R, Venuti A. Immunotherapy in new pre-clinical models of HPV-associated oral cancers. Hum Vaccin Immunother. 2013;9(3):534–43. doi: 10.4161/hv.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strati K, Pitot HC, Lambert PF. Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(38):14152–7. doi: 10.1073/pnas.0606698103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. Journal of virology. 1996;70(3):1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. Journal of virology. 1999;73(7):5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer research. 2003;63(16):4862–71. [PubMed] [Google Scholar]
- 18.Schaeffer AJ, Nguyen M, Liem A, Lee D, Montagna C, Lambert PF, et al. E6 and E7 oncoproteins induce distinct patterns of chromosomal aneuploidy in skin tumors from transgenic mice. Cancer research. 2004;64(2):538–46. doi: 10.1158/0008-5472.can-03-0124. [DOI] [PubMed] [Google Scholar]
- 19.Brake T, Lambert PF. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2490–5. doi: 10.1073/pnas.0409883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jabbar SF, Abrams L, Glick A, Lambert PF. Persistence of high-grade cervical dysplasia and cervical cancer requires the continuous expression of the human papillomavirus type 16 E7 oncogene. Cancer research. 2009;69(10):4407–14. doi: 10.1158/0008-5472.CAN-09-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong R, Pytynia M, Pelizzari C, Spiotto M. Bioluminescent imaging of HPV-positive oral tumor growth and its response to image-guided radiotherapy. Cancer research. 2014;74(7):2073–81. doi: 10.1158/0008-5472.CAN-13-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS pathogens. 2009;5(10):e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chellappan S, Kraus VB, Kroger B, Munger K, Howley PM, Phelps WC, et al. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10):4549–53. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yim EK, Park JS. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer research and treatment : official journal of Korean Cancer Association. 2005;37(6):319–24. doi: 10.4143/crt.2005.37.6.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potter H. Transfection by electroporation. Current protocols in molecular biology / edited by Frederick M Ausubel [et al] 2003;Chapter 9(Unit 9):3. doi: 10.1002/0471142727.mb0903s62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91(4):501–10. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 27.Ivics Z, Izsvak Z. Transposons for gene therapy! Current gene therapy. 2006;6(5):593–607. doi: 10.2174/156652306778520647. [DOI] [PubMed] [Google Scholar]
- 28.Ohlfest JR, Demorest ZL, Motooka Y, Vengco I, Oh S, Chen E, et al. Combinatorial antiangiogenic gene therapy by nonviral gene transfer using the sleeping beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Molecular therapy : the journal of the American Society of Gene Therapy. 2005;12(5):778–88. doi: 10.1016/j.ymthe.2005.07.689. [DOI] [PubMed] [Google Scholar]
- 29.Carlson CM, Frandsen JL, Kirchhof N, McIvor RS, Largaespada DA. Somatic integration of an oncogene-harboring Sleeping Beauty transposon models liver tumor development in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(47):17059–64. doi: 10.1073/pnas.0502974102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer research. 2009;69(2):431–9. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schreiber K, Cannon RE, Karrison T, Beck-Engeser G, Huo D, Tennant RW, et al. Strong synergy between mutant ras and HPV16 E6/E7 in the development of primary tumors. Oncogene. 2004;23(22):3972–9. doi: 10.1038/sj.onc.1207507. [DOI] [PubMed] [Google Scholar]
- 32.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nature biotechnology. 2004;22(5):589–94. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 33.Mates L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nature genetics. 2009;41(6):753–61. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- 34.Kim JW, Hung CF, Juang J, He L, Kim TW, Armstrong DK, et al. Comparison of HPV DNA vaccines employing intracellular targeting strategies. Gene therapy. 2004;11(12):1011–8. doi: 10.1038/sj.gt.3302252. [DOI] [PubMed] [Google Scholar]
- 35.Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. The EMBO journal. 1989;8(13):4099–105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jurk M, Vollmer J. Therapeutic applications of synthetic CpG oligodeoxynucleotides as TLR9 agonists for immune modulation. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2007;21(6):387–401. doi: 10.2165/00063030-200721060-00006. [DOI] [PubMed] [Google Scholar]
- 37.Kim D, Hung CF, Wu TC. Monitoring the trafficking of adoptively transferred antigen- specific CD8-positive T cells in vivo, using noninvasive luminescence imaging. Hum Gene Ther. 2007;18(7):575–88. doi: 10.1089/hum.2007.038. [DOI] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunn LA, Evander M, Tindle RW, Bulloch AL, de Kluyver RL, Fernando GJ, et al. Presentation of the HPV16E7 protein by skin grafts is insufficient to allow graft rejection in an E7-primed animal. Virology. 1997;235(1):94–103. doi: 10.1006/viro.1997.8650. [DOI] [PubMed] [Google Scholar]
- 40.Azoury-Ziadeh R, Herd K, Fernando GJ, Lambert P, Frazer IH, Tindle RW. Low level expression of human papillomavirus type 16 (HPV16) E6 in squamous epithelium does not elicit E6 specific B- or T-helper immunological responses, or influence the outcome of immunisation with E6 protein. Virus Res. 2001;73(2):189–99. doi: 10.1016/s0168-1702(00)00241-0. [DOI] [PubMed] [Google Scholar]
- 41.Frazer IH, De Kluyver R, Leggatt GR, Guo HY, Dunn L, White O, et al. Tolerance or immunity to a tumor antigen expressed in somatic cells can be determined by systemic proinflammatory signals at the time of first antigen exposure. J Immunol. 2001;167(11):6180–7. doi: 10.4049/jimmunol.167.11.6180. [DOI] [PubMed] [Google Scholar]
- 42.Thyagarajan B, Guimaraes MJ, Groth AC, Calos MP. Mammalian genomes contain active recombinase recognition sites. Gene. 2000;244(1–2):47–54. doi: 10.1016/s0378-1119(00)00008-1. [DOI] [PubMed] [Google Scholar]
- 43.Chen CH, Ji H, Suh KW, Choti MA, Pardoll DM, Wu TC. Gene gun-mediated DNA vaccination induces antitumor immunity against human papillomavirus type 16 E7-expressing murine tumor metastases in the liver and lungs. Gene therapy. 1999;6(12):1972–81. doi: 10.1038/sj.gt.3301067. [DOI] [PubMed] [Google Scholar]
- 44.Shi W, Bu P, Liu J, Polack A, Fisher S, Qiao L. Human papillomavirus type 16 E7 DNA vaccine: mutation in the open reading frame of E7 enhances specific cytotoxic T-lymphocyte induction and antitumor activity. Journal of virology. 1999;73(9):7877–81. doi: 10.1128/jvi.73.9.7877-7881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masterson L, Lechner M, Loewenbein S, Mohammed H, Davies-Husband C, Fenton T, et al. CD8+ T cell response to human papillomavirus 16 E7 is able to predict survival outcome in oropharyngeal cancer. Eur J Cancer. 2016;67:141–51. doi: 10.1016/j.ejca.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 46.Allen CT, Judd NP, Bui JD, Uppaluri R. The clinical implications of antitumor immunity in head and neck cancer. Laryngoscope. 2012;122(1):144–57. doi: 10.1002/lary.21913. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa M, Gupta SK, Coleman HN, Sellers MA, Banken JA, Greenfield WW. A favorable clinical trend is associated with CD8 T-cell immune responses to the human papillomavirus type 16 e6 antigens in women being studied for abnormal pap smear results. J Low Genit Tract Dis. 2010;14(2):124–9. doi: 10.1097/LGT.0b013e3181c6f01e. [DOI] [PubMed] [Google Scholar]
- 48.Stanley M. Immune responses to human papillomavirus. Vaccine. 2006;24(Suppl 1):S16–22. doi: 10.1016/j.vaccine.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Strickler HD, Burk RD, Fazzari M, Anastos K, Minkoff H, Massad LS, et al. Natural history and possible reactivation of human papillomavirus in human immunodeficiency virus-positive women. J Natl Cancer Inst. 2005;97(8):577–86. doi: 10.1093/jnci/dji073. [DOI] [PubMed] [Google Scholar]
- 50.Engels EA, Pfeiffer RM, Goedert JJ, Virgo P, McNeel TS, Scoppa SM, et al. Trends in cancer risk among people with AIDS in the United States 1980–2002. AIDS. 2006;20(12):1645–54. doi: 10.1097/01.aids.0000238411.75324.59. [DOI] [PubMed] [Google Scholar]
- 51.Angeletti PC, Zhang L, Wood C. The viral etiology of AIDS-associated malignancies. Adv Pharmacol. 2008;56:509–57. doi: 10.1016/S1054-3589(07)56016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187–94. doi: 10.1002/ijc.23487. [DOI] [PubMed] [Google Scholar]
- 53.Dempsey AF. Human papillomavirus: the usefulness of risk factors in determining who should get vaccinated. Rev Obstet Gynecol. 2008;1(3):122–8. [PMC free article] [PubMed] [Google Scholar]
- 54.Chaturvedi AK, Madeleine MM, Biggar RJ, Engels EA. Risk of human papillomavirus-associated cancers among persons with AIDS. J Natl Cancer Inst. 2009;101(16):1120–30. doi: 10.1093/jnci/djp205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gross ND, Hanna EY. The Role of Surgery in the Management of Recurrent Oropharyngeal Cancer. Recent Results Cancer Res. 2017;206:197–205. doi: 10.1007/978-3-319-43580-0_15. [DOI] [PubMed] [Google Scholar]
- 56.Peng S, Song L, Knoff J, Wang JW, Chang YN, Hannaman D, et al. Control of HPV-associated tumors by innovative therapeutic HPV DNA vaccine in the absence of CD4+ T cells. Cell Biosci. 2014;4(1):11. doi: 10.1186/2045-3701-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McEwen-Smith RM, Salio M, Cerundolo V. The regulatory role of invariant NKT cells in tumor immunity. Cancer Immunol Res. 2015;3(5):425–35. doi: 10.1158/2326-6066.CIR-15-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gorini F, Azzimonti L, Delfanti G, Scarfo L, Scielzo C, Bertilaccio MT, et al. Invariant NKT cells contribute to chronic lymphocytic leukemia surveillance and prognosis. Blood. 2017;129(26):3440–51. doi: 10.1182/blood-2016-11-751065. [DOI] [PubMed] [Google Scholar]
- 59.Gebremeskel S, Clattenburg DR, Slauenwhite D, Lobert L, Johnston B. Natural killer T cell activation overcomes immunosuppression to enhance clearance of postsurgical breast cancer metastasis in mice. Oncoimmunology. 2015;4(3):e995562. doi: 10.1080/2162402X.2014.995562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin Cancer Res. 2017;23(14):3510–9. doi: 10.1158/1078-0432.CCR-16-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujii S, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, et al. NKT cells as an ideal anti-tumor immunotherapeutic. Front Immunol. 2013;4:409. doi: 10.3389/fimmu.2013.00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH, et al. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J Exp Med. 2003;198(3):433–42. doi: 10.1084/jem.20030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chitadze G, Oberg HH, Wesch D, Kabelitz D. The Ambiguous Role of gammadelta T Lymphocytes in Antitumor Immunity. Trends Immunol. 2017;38(9):668–78. doi: 10.1016/j.it.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 64.Silva-Santos B, Serre K, Norell H. gammadelta T cells in cancer. Nat Rev Immunol. 2015;15(11):683–91. doi: 10.1038/nri3904. [DOI] [PubMed] [Google Scholar]
- 65.Fisher JP, Heuijerjans J, Yan M, Gustafsson K, Anderson J. gammadelta T cells for cancer immunotherapy: A systematic review of clinical trials. Oncoimmunology. 2014;3(1):e27572. doi: 10.4161/onci.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Young KJ, Kay LS, Phillips MJ, Zhang L. Antitumor activity mediated by double-negative T cells. Cancer research. 2003;63(22):8014–21. [PubMed] [Google Scholar]
- 67.Merims S, Li X, Joe B, Dokouhaki P, Han M, Childs RW, et al. Anti-leukemia effect of ex vivo expanded DNT cells from AML patients: a potential novel autologous T-cell adoptive immunotherapy. Leukemia. 2011;25(9):1415–22. doi: 10.1038/leu.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu H, Zhu XX, Chen J. DNT cell inhibits the growth of pancreatic carcinoma via abnormal expressions of NKG2D and MICA in vivo. Biochem Biophys Res Commun. 2016;469(2):145–50. doi: 10.1016/j.bbrc.2015.11.085. [DOI] [PubMed] [Google Scholar]
- 69.Shah JP. Patterns of cervical lymph node metastasis from squamous carcinomas of the upper aerodigestive tract. Am J Surg. 1990;160(4):405–9. doi: 10.1016/s0002-9610(05)80554-9. [DOI] [PubMed] [Google Scholar]
- 70.Li XM, Wei WI, Guo XF, Yuen PW, Lam LK. Cervical lymph node metastatic patterns of squamous carcinomas in the upper aerodigestive tract. J Laryngol Otol. 1996;110(10):937–41. doi: 10.1017/s0022215100135406. [DOI] [PubMed] [Google Scholar]
- 71.Sanderson RJ, Ironside JA. Squamous cell carcinomas of the head and neck. BMJ. 2002;325(7368):822–7. doi: 10.1136/bmj.325.7368.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Civantos FJ, Zitsch RP, Schuller DE, Agrawal A, Smith RB, Nason R, et al. Sentinel lymph node biopsy accurately stages the regional lymph nodes for T1–T2 oral squamous cell carcinomas: results of a prospective multi-institutional trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(8):1395–400. doi: 10.1200/JCO.2008.20.8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gillison ML, D’Souza G, Westra W, Sugar E, Xiao W, Begum S, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–20. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 75.Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin Cancer Res. 2004;10(1 Pt 1):301–13. doi: 10.1158/1078-0432.ccr-0999-3. [DOI] [PubMed] [Google Scholar]
- 76.Kanojia D, Vaidya MM. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol. 2006;42(7):655–67. doi: 10.1016/j.oraloncology.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 77.Schoop RA, Noteborn MH, Baatenburg de Jong RJ. A mouse model for oral squamous cell carcinoma. J Mol Histol. 2009;40(3):177–81. doi: 10.1007/s10735-009-9228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Visscher SA, Witjes MJ, van der Vegt B, de Bruijn HS, van der Ploeg-van den Heuvel A, Amelink A, et al. Localization of liposomal mTHPC formulations within normal epithelium, dysplastic tissue, and carcinoma of oral epithelium in the 4NQO-carcinogenesis rat model. Lasers Surg Med. 2013;45(10):668–78. doi: 10.1002/lsm.22197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.