

Abstract
Purpose
Mercaptopurine (MP) is one of the main chemotherapeutics for acute lymphoblastic leukemia (ALL). To improve treatment outcomes, constant MP dose titration is essential to maintain steady drug exposure, while minimizing myelosuppression. We performed two-stage analyses to identify genetic determinants of MP-related neutropenia in Korean pediatric ALL patients.
Materials and Methods
Targeted sequencing of 40 patients who exhibited definite MP intolerance was conducted using a novel panel of 211 pharmacogenetic-related genes, and subsequent analysis was performed with 185 patients.
Results
Using bioinformatics tools and genetic data, four functionally interesting variants were selected (ABCC4, APEX1, CYP1A1, and CYP4F2). Including four variants, 23 variants in 12 genes potentially linked to MP adverse reactions were selected as final candidates for subsequent analysis in 185 patients. Ultimately, a variant allele in APEX1 rs2307486was found to be strongly associated with MP-induced neutropenia that occurred within 28 days of initiating MP (odds ratio, 3.44; p=0.02). Moreover, the cumulative incidence of MP-related neutropenia was significantly higher in patients with APEX1 rs2307486 variants, as GG genotypes were associated with the highest cumulative incidence (p < 0.01). NUDT15 rs116855232 variants were strongly associated with a higher cumulative incidence of neutropenia (p < 0.01), and a lower median dose of tolerated MP throughout maintenance treatment (p < 0.01).
Conclusion
We have identified that APEX1 rs2307486 variants conferred an increased risk of MP-related early onset neutropenia. APEX1 and NUDT15 both contribute to cell protection from DNA damage or misincorporation, so alleles that impair the function of either gene may affect MP sensitivities, thereby inducing MP-related neutropenia.
Keywords: NUDT15, APEX1, 6-Mercaptopurine, Neutropenia, Acute lymphoblastic leukemia, Pediatrics
Introduction
Acute lymphoblastic leukemia (ALL) is the most common hematologic malignancy in children [1]. Prolongation of therapy by incorporating a maintenance phase, containing mercaptopurine (MP) has improved outcomes in pediatric ALL [1]. However, MP has a narrow therapeutic index with common dose-limiting toxicities in hematopoietic tissues [2]. During the maintenance phase that continues for 1 to 2 years, patients with ALL must take MP daily. MP-related toxicities are most likely to occur during the maintenance phase if a patient has a susceptible genotypic trait for low metabolism, so treatment interruption can be frequent. Because high-intra individual variability in responses to thiopurine exposure can negatively affect ALL treatment outcomes [3], constant MP dose titration to maintain steady thiopurine exposure while minimizing toxicity is important.
In previous s2018-02-09tudies, thiopurine methyltransferase (TPMT), which is involved in methylating MP metabolites, is known to be one of the most critical genes in the pharmacogenetics of MP [4]. Patients with a nonfunctional variant allele of TPMT have lower TPMT enzyme activity, and consequently 6-thioguanine nucleotide (6-TGN) excessively accumulates in hematopoietic tissues and frequently causes hematopoietic toxicity. These findings lead to the concept of individualized MP dosing according to TPMT genotype; however, the frequency of the variant allele of TPMT is lower in Asian than Caucasian patients [5]. Therefore, determination of TPMT polymorphisms has limited clinical benefit for children with ALL in most Asian countries [6]. In practice, most of East Asian patients still show sensitivity to MP after adjusting for TPMT variants, suggesting the existence of other yet unknown factors that are related to MP sensitivity.
Recently, a single nucleotide polymorphism (SNP) of nucleoside diphosphate-linked moiety X-type motif 15 (NUDT15) has been associated with thiopurine-induced myelosuppression in pediatric patients with ALL and inflammatory bowel disease [2,7-9]. Although recent studies have shown a strong correlation between the NUDT15 rs1168-55232 SNP and major MP toxicity, careful evaluations of potential additive effects with other candidate genetic loci are still needed.
In our present study, we used targeted sequencing with bioinformatics analysis to identify genetic determinants of MP-related adverse effects in Korean pediatric patients with ALL.
Materials and Methods
1. Patients and treatment
Among patients diagnosed with ALL at Seoul National University Children’s Hospital, 185 patients for whom samples and informed consent was available were included. Bone marrow aspirates or peripheral blood at complete remission were obtained for sequencing. Patients were assigned to the standard-risk group if they were 1 to 9 years of age at the time of diagnosis with a white blood cell (WBC) count of less than 50×109/L; all other patients were assigned to the high-risk group. Patients underwent hematopoietic stem cell transplantation (HSCT) if they had one or more of the following criteria: age younger than 1 year, hypodiploidy, the presence of t(9;22), a WBC count equal to or greater than 200×109/L, or the 11q23 rearrangement.
At our institution, the planned dose of MP was modified from 75 to 50 mg/m2 based on our institutional experiences, as many patients who had been given the same dose under the original CCG protocol exhibited moderate to severe toxicities during MP administration [10]. Doses of MP during maintenance were adjusted to maintain a WBC count of 2.0-3.5×109/L with an absolute neutrophil count (ANC) over 500/μL, and hepatotoxicity related dose modifications were performed at the discretion of the treating physician. All doses of drugs were not prospectively adjusted based on patient genotypes.
Medical records from all patients were retrospectively reviewed, including complete blood cell counts, total bilirubin, serum liver enzyme levels, and the frequency and first date of neutropenia noted during maintenance. Records of MP and methotrexate (MTX) doses were collected on a cyclical basis (12 weeks) and the dose in the final maintenance cycle was considered to be the maximum tolerated dose for each patient.
2. Primary targeted sequencing and analysis
Subjects were selected for targeted sequencing among those patients who could not tolerate a full dose MP, as their final dose at the last maintenance cycle was less than 25% of the initial planned dose at the maintenance phase (50 mg/m2/day). Finally, a total of 40 patients were selected as a representative cohort for prominent MP intolerability. Targeted sequencing was conducted using a novel pharm-gene panel to screen for suspected genetic determinants of MP-related toxicities. This panel included the exomes of 147 pharmacogenetic-related genes and 84 SNPs that were known to be involved in the metabolism of various drugs; the size of the total targeted region was 464,798 base pairs (S1 and S2 Tables). The range of amplicons amplified by these oligo primer pairs ranged from 125 to 275 bp, and the rate of “on target” coverage for this panel was 96.74%.
Polymerase chain reaction (PCR) assays were performed directly to amplify 10 ng of genomic DNA samples to collect the target regions using the oligo primer pairs of the panel. After amplification, Library construction was performed by using the Ion Ampliseq library kit 2.0 as described in the manufacturer's instructions (Life Technologies, Carlsbad, CA). Libraries were diluted to ~10 pM. Subsequently, 2.27 μL of the barcoded libraries were combined in sets of 40 barcodes.
The combined libraries were sequenced using the Ion Proton platform, following the manufacturer's instructions (Life Technologies). A mean depth of sequencing that ranged from 450× to 470× was obtained, which was a sufficient depth to interrogate the target regions for mutations. Raw reads were initially mapped to the human reference genome build (GRCh37) using the Torrent Mapping Alignment Program (TMAP). Genome Analysis Toolkit 2.8-1, HaplotypeCaller, was used to call single nucleotide variants and short insertions/deletions [11].
After sequencing, variants that fulfilled the following criteria were selected (Fig. 1). First, variants predicted to have functional alterations on gene products (i.e., nonsense, missense, frameshift, or splice site) were selected. Second, variants predicted to be deleterious by at least one of three bioinformatics tools, which are scale-invariant feature transform (SIFT; http://sift.jcvi.org/) [12], PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) [13], and combined annotation dependent depletion (CADD; http://caxdd.gs.washington.edu/) [14], were selected. Next, for all variants identified, the odds ratio (OR) of those variants for cases compared with controls were calculated considering the effect of the NUDT15 rs116855232 variant. Those individuals who did not carry the rs116855232 variant were subgrouped, and variants present at a higher frequency (OR > 1) were obtained among these cases. Finally, the relative risk of those variants for cases compared with an East Asian population (1000 Genomes project, Phase 3, http://www.1000genomes.org/) [15] was calculated in the same way. Only those positions that were significantly associated (p < 0.05) with higher occurrence (OR > 1) among all cases were selected. We reviewed read-level alignment status of Binary Alignment Map files to identify false-positive calls which can be introduced by technical and/or algorithmic issues like misalignment and true-negative calls due to off-target regions.
Fig. 1.
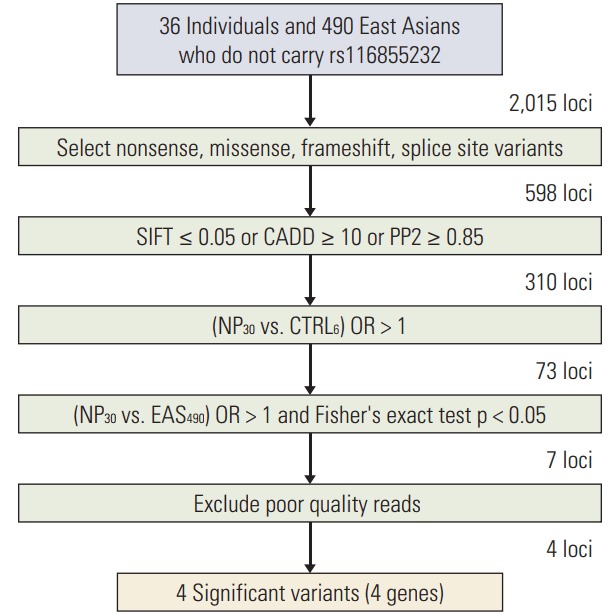
Primary targeted sequencing data analysis steps. SIFT, scale-invariant feature transform; CADD, combined annotation dependent depletion; PP2, PolyPhen2; NP30, 30 patients with neutropenia; CTRL6, 6 patients without neutropenia; EAS490, 490 East Asian data.
3. Subsequent candidate gene analysis
Subsequent analysis of suspected determinant genes based on the primary sequencing results was conducted using a candidate gene approach. Candidate genes were also selected by considering previous clinical studies in which genes exhibited polymorphisms that were reported to be associated with the pharmacokinetics and pharmacodynamics of MP [4]. We analyzed 185 samples by an array-based high throughput method to simultaneously detect multiple positions using SNPtype and SNaPshot assays.
In the SNPtype assay, 40 ng of genomic DNA flanking the interested SNP was amplified by PCR with a STA primer set. PCR was performed as described in the manufacturer's instructions (Fluidigm, San Francisco, CA). After amplification, the SNPtype assay reaction was carried out as described in the manufacturer's instructions. SNP analysis was performed using Fluidigm SNP Genotyping Analysis software ver. 4.0.1 (S3 Table).
The SNaPshot assay was performed according to the manufacturer’s instructions (ABI PRISM SNaPShot Multiplex kit, Foster City, CA). Analyses were carried out using Genemapper software ver. 4.0 (S4 Table).
We analyzed 185 samples using a SNPtype assay for 21 positions in 12 genes and a SNaPshot assay for two positions in two genes.
4. Statistical analysis
Significant associations between categorical variables, such as the frequency of neutropenia and SNPs, were identified using chi-square or Fisher exact tests. Odds ratios and 95% confidence intervals (CI) associated with each SNP were determined using logistic regression assuming log-additive and dominant genetic models of inheritance. Comparisons of the administered dose percentage according to each SNP group were analyzed using the T-test or ANOVA for parametric data or the Mann-Whitney U or Kruskal-Wallis test for nonparametric data. Cumulative incidence curves were made using the method of Fine and Gray [16], comparisons were performed by Gray’s test [17]. Cox-regression test was conducted for the multivariate analysis. Sequence data analysis, competing risk estimations, Cochran-Armitage trend test, and Cox-regression analysis were conducted using R (3.2.2, http://www.r-project.org/), and other analyses was carried out with SPSS ver. 20.0 (IBM Corp., Armonk, NY) with a threshold of statistical significance of p < 0.05.
5. Ethical statement
This study was approved by the Institutional Review Board of Seoul National University Hospital (H-0611-021-189). Informed written consents for blood sampling and analyses were obtained from all participants.
Results
1. Patients and treatments
A total of 185 patients were analyzed (Table 1), including 110 males and 75 females. The median follow-up duration was 74.2 months (26.6-235.7 months). The median age of diagnosis was 4.9 years (1.1-17.3 years), and 45 patients were greater than 10 years old at the time of diagnosis. There were 57 patients who were allocated to the standard-risk group, and one and 56 patients were treated following the modified CCG-1891 [18] and CCG-1952 [19] protocols. A total of 128 patients were allocated to the high-risk group, and 2, 59, 20, and 47 patients were treated with the modified CCG-1882A, CCG-1882B, CCG-1882C [20], and 0601 protocols for Korean multicenter studies, respectively.
Table 1.
Characteristics of the study patients
| Characteristic | No. (n=185) |
|---|---|
| Age at diagnosis, median (range, yr) | 4.9 (1.1-17.3) |
| 1 year to less than 10 years | 140 |
| At least 10 years | 45 |
| Sex | |
| Male | 110 |
| Female | 75 |
| Risk group | |
| Standard-risk patients | 57 |
| Modified CCG 1891 | 1 |
| Modified CCG 1952 | 56 |
| High-risk patients | 128 |
| Modified CCG 1882A | 2 |
| Modified CCG 1882B | 59 |
| Modified CCG 1882C | 20 |
| 0601 protocol Korea | 47 |
CCG, Children’s Cancer Group.
2. Primary targeted sequencing findings
Targeted sequencing was performed on a total of 40 individuals and those individuals with neutropenia (ANC < 500/μL) were defined as cases for with neutropenia (34 of 40, 85%) (Fig. 1). The median dose percentage of last cycle MP in these patients was 17.9%. After sequencing, a total of 2,015 variants were identified in 36 individuals who do not carry rs116855232, which is strongly associated with drug toxicity. First, we selected 598 variants that potentially had strong effects on gene function, including frameshift, nonsense, missense, and splice site variants. Next, three different bioinformatics tools were applied and 310 positions were selected that were predicted to be deleterious by at least one of the three prediction tools (SIFT ≤ 0.05, PolyPhen2 ≥ 0.85, or CADD ≥ 10). To determine whether nonrandom associations existed between cases and controls, allele/genotype-based association tests were carried out for those positions that were identified more than once in each case group. For 310 positions, the OR between the neutropenia cases (NP) and the matched controls was calculated using Fisher exact test. A total of 73 loci showed higher associations in NP cases than in controls, although no significant difference was detected, possibly because of limited statistical power. To increase the sample size, we estimated the relative risk for those variants as compared with 490 East Asians (1000 Genomes Project, Phase 3) who did not carry the rs116855232 variant. As a result, seven significantly associated (p < 0.05) potential risk variants were identified in NP. We manually reviewed Binary Alignment Map files in order to identify false-positives and excluded three variants which are called due to misalignment. From these studies, four significant variants in ABCC4, apurinic/apyrimidinic endonuclease 1 (APEX1), CYP1A1, and CYP4F2 were selected. Ultimately, 23 variants in 12 genes plausibly linked to or previously implicated in MP adverse reactions, including four selected variants, were determined as final candidates (S5 Table).
3. Candidate gene analysis results
The results of candidate gene analysis are summarized in Table 2. The distribution of patients with variant alleles were as follows: NUDT15 rs116855232 (22.4%), APEX1 rs2307486 (14.8%), ABCC4 rs2274407 (37.3%), ABCC4 rs3765534 (14.7%), ABCC4 rs11568658 (25.0%), CYP4F2 rs2108622 (55.9%), CYP1A1 rs4646422 (31.3%), SLCO1B1 rs11045879 (59.4%), SLCOB1 rs4149056 (29.5%), ITPA rs1127354 (28.4%), ITPA rs7270101 (0%), MTHFR rs1801131 (30.8%), MTHFR rs180-1133 (66.4%), MTHFR rs1901133 (54.1%), GRIA1 rs4958351 (3.8%), MOCOS rs594445 (50.3%), and PACSIN2 rs2413739 (14.8%). The TPMT allele type was *1/*1 in most patients (96.2%), while the variant allele was only detected in seven patients (*1/*3C, 3.8%).
Table 2.
Frequencies of candidate gene loci subjected to subsequent validation
| Gene | rsID | Genotype | No. of patients (%) (n=185) |
|---|---|---|---|
| NUDT15 | rs116855232 | CC | 142 (77.6) |
| CT | 37 (20.2) | ||
| TT | 4 (2.2) | ||
| APEX1 | rs2307486 | AA | 155 (85.2) |
| AG | 26 (14.3) | ||
| GG | 1 (0.5) | ||
| ABCC4 | rs2274407 | GG | 116 (62.7) |
| GT | 62 (33.5) | ||
| TT | 7 (3.8) | ||
| rs3765534 | CC | 156 (85.2) | |
| CT | 24 (13.1) | ||
| TT | 3 (1.6) | ||
| rs11568658 | CC | 138 (75.0) | |
| CA | 41 (22.3) | ||
| AA | 5 (2.7) | ||
| CYP4F2 | rs2108622 | CC | 81 (44.0) |
| CT | 77 (41.8) | ||
| TT | 26 (14.1) | ||
| CYP1A1 | rs4646422 | CC | 125 (68.7) |
| CT | 51 (28.0) | ||
| TT | 6 (3.3) | ||
| SLCO1B1 | rs11045879 | TT | 74 (40.7) |
| TC | 74 (40.7) | ||
| CC | 34 (18.7) | ||
| rs4149056 | TT | 129 (70.5) | |
| TC | 51 (27.9) | ||
| CC | 3 (1.6) | ||
| ITPA | rs1127354 | CC | 131 (71.6) |
| CA | 47 (25.7) | ||
| AA | 5 (2.7) | ||
| rs7270101 | AA | 185 (100) | |
| MTHFR | rs1801131 | TT | 128 (69.2) |
| TG | 54 (29.2) | ||
| GG | 3 (1.6) | ||
| rs1801133 | GG | 61 (33.5) | |
| GA | 92 (50.5) | ||
| AA | 29 (15.9) | ||
| rs1901133 | GG | 84 (45.9) | |
| GA | 87 (47.5) | ||
| AA | 12 (6.6) | ||
| GRIA1 | rs4958351 | GG | 178 (96.2) |
| AG | 7 (3.8) | ||
| MOCOS | rs594445 | CC | 92 (49.7) |
| AC | 79 (42.7) | ||
| AA | 14 (7.6) | ||
| PACSIN2 | rs2413739 | CC | 155 (85.2) |
| CT | 27 (14.8) | ||
| TPMT | *1/*1 | 178 (96.2) | |
| *1/*3C | 7 (3.8) |
4. MP-related neutropenia during maintenance and genotype associations
Absolute neutrophil counts below 500/μL developed in 121 patients after MP administration during the maintenance phase (65.4%). When the frequency of MP-related neutropenia was compared with the genetic variant frequencies, neutropenia was significantly more frequent in carriers of a variant T allele in ABCC4 rs3765534, and was associated with a 4.64-fold increased risk of prominent neutropenia compared with homozygous wild-type patients (OR, 4.64; 95% CI, 1.32 to 25.12, p < 0.01) (Table 3). This significant association was also present within patients without NUDT15 rs1168552532 variant alleles (n=140), while the odds ratio was even greater (OR, 6.36; 95% CI, 1.43 to 58.73; p=0.02) in those patients without the NUDT15 variant.
Table 3.
Genotypes associated with mercaptopurine-related neutropenia (ANC < 500/μL, n=179)
| Neutropenia | Allelic frequency |
Genotype (Dominant model) |
CATT p-value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | T | χ2 p-value | Fisher p-value | OR (95% CI) | CC | CT | TT | χ2 p-value | Fisher p-value | OR (95% CI) | ||||
| ABCC4 rs3765534 | Total (n=179) | Yes | 213 | 27 | 0.01 | 0.004 | 4.84 (1.44-25.48) | 96 | 21 | 3 | 0.016 | 0.008 | 4.64 (1.32-25.12) | 0.02 |
| No | 115 | 3 | 56 | 3 | 0 | |||||||||
| No NUDT15 rs116855232 (n=140) | Yes | 160 | 20 | 0.01 | 0.005 | 6.1 (1.43-54.9) | 71 | 18 | 1 | 0.014 | 0.006 | 6.36 (1.43-58.73) | 0.02 | |
| No | 98 | 2 | 48 | 2 | 0 | |||||||||
| Early-onset Neutropenia | A | G | χ2 p-value | Fisher p-value | OR (95% CI) | AA | AG | GG | χ2 p-value | Fisher p-value | OR (95% CI) | CATT p-value | ||
| APEX1 rs2307486 | Total (n=179) | Yes | 24 | 6 | 0.03 | 0.02 | 3.44 (1.04-9.89) | 10 | 4 | 1 | 0.09 | 0.06 | 3.18 (0.78-11.43) | 0.003 |
| No | 304 | 22 | 141 | 22 | 0 | |||||||||
ANC, absolute neutrophil count; OR, odds ratio; CI, confidence interval; CATT, Cochran-Armitage trend test.
Considering the first date of neutropenia after MP intake, early onset neutropenia (within 28 days) was detected in 15 of 121 patients who exhibited neutropenia during maintenance (12.4%), while neutropenia was detected in 106 patients later than 28 days after beginning maintenance (87.6%). In cases of APEX1 rs2307486 derived from our primary targeted sequencing, variants in APEX1 rs2307486 showed no significant correlations with the total frequencies of MP-related neutropenia. However, considering the onset of neutropenia, the G allele in APEX1 rs2307486 was strongly related to early onset MP-related neutropenia (OR, 3.44; 95% CI, 1.04 to 9.89; p=0.02) (Table 3).
The cumulative incidence of MP-related neutropenia was significantly higher in patients with variants of ABCC4 rs3765534 (p=0.01) (Fig. 2A) and APEX1 rs2307486 (p < 0.01) (Fig. 2B). Three patients with a homozygous variant (TT) of ABCC4 developed neutropenia < 500/μL, yielding a cumulative incidence of 100%. There was a correlation with the CT genotype of ABCC4 rs3765534 and increased cumulative incidence of neutropenia as 91.7%, and patients with CC genotype (wild type) showed cumulative incidence of 60.8%.
Fig. 2.

Estimated cumulative incidence of mercaptopurine-related neutropenia according to the ABCC4 rs3765534 (A), APEX1 rs2307486 (B), and NUDT15 rs116855232 (C) genotypes. ANC, absolute neutrophil count.
Regarding APEX1 rs2307486, there was one individual with a homozygous variant (GG) of APEX1 who developed neutropenia < 500/μL at the 21st day of maintenance. The estimated cumulative incidence of neutropenia at 2 years of maintenance phase treatment was 73% (95% CI, 50.5 to 86.6) in those patients with an AG genotype versus 66.3% (95% CI, 57.9 to 73.5) in patients with a wild-type.
NUDT15 rs116855232 variants were also strongly associated with a greater cumulative incidence of neutropenia, as it was 100%, 72.2% (95% CI, 53.9 to 84.3), and 65.0% (95% CI, 56.2 to 72.4) in patients with a TT, CT, or CC genotype, respectively (p < 0.01) (Fig. 2C). All three patients with the homozygous variant genotype (TT) developed neutropenia at 19, 35, or 42 days in the first cycle of maintenance.
According to the multivariate analysis using Cox-regression, GG genotype in APEX1 rs2307486 and TT genotype in NUDT15 rs116855232 showed statistically significant hazard ratios so that those variants conferred a remarkable 12.03, and 11.42-fold increase in neutropenia risk (Table 4).
Table 4.
Genotypes affecting mercaptopurine-related neutropenia according to multivariate analysis
| Variable | HR | 95% CI | p-value |
|---|---|---|---|
| APEX1 rs2307486 | |||
| AA genotypes (wild type) | 1.00 | - | - |
| AG genotypes | 1.49 | 0.87-2.53 | 0.139 |
| GG genotypes | 12.03 | 1.43-101.20 | 0.022 |
| NUDT15 rs116855232 | |||
| CC genotypes (wild type) | 1.00 | - | - |
| CT genotypes | 1.25 | 0.78-2.01 | 0.358 |
| TT genotypes | 11.42 | 3.35-38.97 | < 0.001 |
| ABCC4 rs3765534 | |||
| CC genotypes (wild type) | 1.00 | - | - |
| CT genotypes | 1.72 | 1.05-2.80 | 0.031 |
| TT genotypes | 2.11 | 0.62-7.20 | 0.232 |
HR, hazard ratio; CI, confidence interval.
5. MP and MTX doses and genotype associations
Because the doses of MP and MTX were reduced in response to toxicity that occurred during maintenance, doses were gradually adjusted during the first cycle of maintenance in most patients. The majority of patients generally reach their tolerable doses during the second cycle, and the doses of MP and MTX during the last maintenance cycle are considered as the maximum tolerated doses for each patient to maintain a WBC count without apparent toxicity.
In our present study, protocol-based doses were 50 mg/m2/day for MP, and 20 mg/m2/wk for MTX. The median doses of MP at the second and last cycles in our study cohort were 22.8 and 23.5 mg/m2/day, respectively, while those of MTX were 10 and 13 mg/m2/wk (Table 5). We observed that non-synonymous rs116855232 variants in NUDT15 predisposed patients to significant MP dose reduction (Fig. 3A), likely because of increased hematological toxicity. Four patients with variant homozygous (TT) for NUDT15 were highly sensitive to MP with a tolerated dose at the second cycle of only 8.9 mg/m2/day, compared with patients with a CT genotype (n=36) or wild-type (n=131) who tolerated an average dose of 15.6 and 27.4 mg/m2/day, respectively (p < 0.01). This effect persisted throughout the entire maintenance phase, so the average dose during the final cycle was 5, 17.6, and 26.9 mg/m2/day for patients with TT, CT, and CC genotypes, respectively (p < 0.01).
Table 5.
Doses of mercaptopurine and methotrexate administered at the second and last maintenance cycles (n=185)
| Second cycle |
Last cycle |
|||
|---|---|---|---|---|
| No. of patients (%) | Median dose (mg/m2/day) | No. of patients (%) | Median dose (mg/m2/day) | |
| MP dose (mg/m2/day) | ||||
| MP < 12.5 | 38 (20.5) | 7.8 | 51 (27.6) | 7 |
| 12.5 ≤ MP < 25 | 58 (31.4) | 17.9 | 41 (22.2) | 17.4 |
| 25 ≤ MP < 37.5 | 37 (20) | 27.8 | 48 (25.9) | 31.3 |
| 37.5 ≤ MP < 50 | 41 (22.2) | 41.7 | 24 (13) | 42.7 |
| MP ≥ 50 | 5 (2.7) | 52.1 | 14 (7.6) | 55.7 |
| Total patients | 179a) | 22.8 | 178b) | 23.5 |
| Methotrexate dose (mg/m2/wk) | ||||
| MTX < 5 | 38 (20.5) | 3.3 | 29 (15.7) | 7 |
| 5 ≤ MTX < 10 | 49 (26.5) | 7.9 | 32 (17.3) | 17.4 |
| 10 ≤ MTX < 15 | 42 (22.7) | 11.8 | 51 (27.6) | 31.3 |
| 15 ≤ MTX < 20 | 46 (24.9) | 17.7 | 39 (21.1) | 42.7 |
| MTX ≥ 20 | 4 (2.2) | 21.3 | 25 (13.5) | 55.7 |
| Total patients | 179a) | 10 | 176c) | 13 |
MP, mercaptopurine; MTX, methotrexate.
Data was not available in six patients,
Data was not available in seven patients,
Data was not available in nine patients.
Fig. 3.

Average doses of mercaptopurine (MP) (A) and methotrexate (MTX) (B) administered during the second and last maintenance cycles according to the NUDT15 rs116855232 genotype.
The risk allele of rs116855232 was also highly associated with a lower dose of MTX (Fig. 3B). The average MTX dose during the second maintenance therapy for patients with TT, CT, and CC genotypes was 5.4, 7.8, and 11.7 mg/m2/wk, respectively (p < 0.01). Additionally, during the final cycle, tolerance for MTX in patients with the T allele was significantly lower than in patients with the C allele (TT:CT:CC=1.7:10.6:13.1 mg/m2/wk, respectively; p < 0.01).
Discussion
Maintenance therapy in ALL treatment is needed to prevent relapse. Generally, all patients with ALL who are not candidates for HSCT receive maintenance chemotherapy for 2-2.5 years [1]. Daily MP and weekly MTX with periodic vincristine, prednisone, and intrathecal therapy represent the backbone of maintenance regimens. The importance of compliance with MP and constant dose titration has been illustrated in a previous cohort study that revealed an association between reduced adherence rates and an increased risk of relapse [21].
Therefore, for appropriate dose optimization of MP, many studies have been conducted on the various genetic factors associated with MP-related toxicities [4]. Among these factors, TPMT has been considered to be a major locus in determining susceptibility to toxicity [1].
Recently, a non-synonymous mutation in NUDT15 was identified as a strong risk factor for myelotoxicity of MP during maintenance therapy for ALL [2,8]. However, most of the patients who could not tolerate full dose MP did not have a known genetic factors.
To determine the genetic factors associated with MP-related neutropenia, it is necessary to determine whether neutropenia is associated with the drug. In the later phase of maintenance therapy, the tolerable dose is continuously administered for a while. Therefore, it is difficult to confirm the association of MP with sudden neutropenia. However, early onset neutropenia immediately after the start of MP administration is likely to have been caused by the new drug, and thus is of greater value as a phenotype associated with toxicity.
In this present study, we developed a novel pharm-gene panel and conducted targeted sequencing of patients who exhibited MP-related severe toxicity. We carried out candidate gene validation to identify genetic determinants of MP-related adverse effects in Korean pediatric patients with ALL. Our analyses revealed that APEX1 SNP rs2307486 conferred an increased risk of MP-related early onset neutropenia. Additionally, the minor allele of APEX1 rs2307486 was linked to a significantly higher cumulative incidence of MP-related neutropenia.
Human APEX1 is the major enzyme in the DNA base excision repair pathway [22] where its main role is to create a nick in the phosphodiester backbone of the apurinic/apyrimidinic site that is created when DNA glycosylase removes a damaged base [23]. Therefore, it is not surprising that APEX1 has been implicated in sensitivities or resistances to a wide range of chemotherapeutic agents. Indeed, previous studies that used small molecule inhibitors established that APEX1-deficient cells exhibited hypersensitivity to various DNA-damaging agents, including alkylators and antimetabolites. In the previous study of McNeill et al. [22], APEX1-deficient cells were shown to exhibit the greatest sensitivity to antimetabolites among various anticancer drugs in an in vivo setting, and consequently apoptotic cell death was profoundly increased, supporting a role for mutant APEX1 in thiopurine-induced neutropenia.
The rs2307486 variant in APEX1 is a missense mutation, leading to an A to G change that results in an amino acid change from isoleucine [Ile] to valine [Val]. The allele frequencies of the variant alleles in rs2307486 were near 0% in Europeans, 0.08% in Africans, and 1.73% in Americans, whereas the G allele frequency in East Asian individuals was 4.66% according to the 1000 Genomes database. Thus, we considered that those East Asians who have a higher frequency of APEX1 variant alleles might be more susceptible to the variant related effects, whereas Caucasian or European populations might not be as affected by these variant-associated toxicities because of minor allele frequency differences between ethnic groups. Although we could not identify a correlation between APEX1 SNP rs2307486 and the total frequency of neutropenia in our patient cohort, the correlation was significantly strong within the larger East Asian population according to the 1000 Genomes database (S6 Table). This effect of APEX1 variants on MP-related neutropenia should be replicated in a larger patient cohort in the future.
NUDT15 encodes a nucleoside diphosphatase, which is involved in degrading oxidized purine nucleoside triphosphates (e.g., 8-oxo-dGTP) via dephosphorylation to prevent DNA incorporation and to protect cells from damage and apoptosis [24]. The rs116855232 variant in NUDT15 results in a missense mutation, and a C to T change in rs116855232 results in an amino acid change from arginine [Arg] to cysteine [Cys], which is thought to be as a loss-of-function mutation. A recent genome-wide association study in Korean patients with inflammatory bowel disease reported that the same NUDT15 variant was associated with thiopurinerelated leucopenia, with each copy of the T allele conferring a remarkable 35.6-fold increase in neutropenia risk [7]. In our present study, we established that NUDT15 variants were significantly associated with MP-induced early neutropenia.
Another interesting finding of our present study was the correlation between NUDT15 rs116855232 variants and the average dose of MTX. Chiengthong et al. [9] and Yang et al. [2] reported previously that NUDT15 rs116855232 variants were associated with a lower median cumulative MP dose, which accords with our present data. In ALL maintenance, MP and MTX doses are usually alternatively reduced in response to observed drug toxicity. Therefore, those patients who exhibit frequent myelosuppression should receive lower doses of both drugs, and the correlation between the dose (percent) of MP and MTX in each patient should be linear [10]. In this regard, patients who have NUDT15 variants show early and frequent myelotoxicity, so that they had concurrently reduced doses of both MP and MTX.
The genetic variant in TPMT was not a significant factor for MP-related toxicities in our present study. Previous studies confirmed that variant allele frequencies of TPMT in Asian populations are lower than those in other ethnic groups. In Caucasians, the most common variant allele is *3A with an allele frequency of 3.54%, followed by *3C with a frequency of 0.42% [25]. However, in Asian populations, the *3C allele has only been reported at a rate of 0.9%-1.8%, while other variant alleles, such as *2, *3A, or *3B, are rarely observed [26-28]. Additionally, the homozygous variant is observed in ~0.33% of Caucasians, but the homozygous variant is rarely reported in Asian individuals. Therefore, determining TPMT polymorphisms has only minor clinical implications in children with ALL in most Asian countries.
In our present study cohort, only five patients were heterozygous for the TPMT variant, and the average MP doses of patients with TPMT *1/*1 compared with *1/*3C were not significantly different (S7 Fig.). The *1/*3C diplotype is known to have intermediate TPMT activity, so the recommended starting dose for MP is a 30%-70% reduced dose compared with the full dose [25]. This explains our finding that there was no significant association with TPMT genotypes because a starting dose of 50 mg/m2/day of MP was already identical to the reduced dose level recommend by the Clinical Pharmacogenetics Implementation Consortium guidelines [25].
In our present study, the ABCC4 rs3765534 variant was found to be associated with MP-induced neutropenia during maintenance. ATP-binding cassette sub-family C member 4 (ABCC4), also known as multidrug resistance protein 4 (MRP4), protects cells against thiopurine-induced hematotoxicity by actively exporting thiopurine nucleotides [29]. Reduced expression of MRP4 would be predicted to interrupt efflux of active metabolites of MP, so patients with ABCC4 mutations would show increased sensitivity for MP.
According to Krishnamurthy et al. [29], a mutation in ABCC4 rs3765534 dramatically reduces MRP4 function by impairing its localization in the cell membrane. Ban et al. [30] also reported clinical data for 279 Japanese inflammatory bowel disease patients, and showed that the WBC counts of patients with the rs3765534 variant were significantly lower than in patients with the wild-type allele. They also showed that 6-TGN levels were significantly higher in patients with the rs3765534 variant, suggesting that these patients had reduced ABCC4 function and increased sensitivity to MP.
Our present study had several limitations of note, including the limited number of validated loci, a modest number of enrolled patients, and the absence of measurements of serum levels of drugs or metabolites. Moreover, 8.1% of the patients in our present cohort who could tolerate less than 25% of the planned dose of MP had wild-type alleles for the TPMT, APEX1, NUDT15, and ABCC4 variants. Therefore, other genetic and non-genetic factors remain to be discovered to improve the patient-specific tailoring of MP dosing in children with ALL.
In conclusion, we have identified a novel genetic risk factor for MP-induced early onset neutropenia using an extensive pharm-gene panel for sequencing. APEX1 and NUDT15 both contribute to protecting cells from DNA damage or misincorporation, so defective functions in either gene may affect cellular drug sensitivities and toxicities. Further validation studies will be needed to confirm the frequency and pharmacogenetic consequences of APEX1 variants in other ethnic groups, including additional Asian populations.
Acknowledgments
This study was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health, & Welfare, Republic of Korea Government (1420250), by a grant of the Korean Health Technology R&D Project, Ministry of Health and Welfare (HI13C2164), by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2012-0000994), by ChungAng University Research Grants in 2015 and by a grant from Ministry of Food and Drug Safety in 2016 (16183MFDS541).
Footnotes
Conflict of interest relevant to this article was not reported.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (http://www.e-crt.org).
References
- 1.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–43. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 2.Yang JJ, Landier W, Yang W, Liu C, Hageman L, Cheng C, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33:1235–42. doi: 10.1200/JCO.2014.59.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhatia S, Landier W, Hageman L, Chen Y, Kim H, Sun CL, et al. Systemic exposure to thiopurines and risk of relapse in children with acute lymphoblastic leukemia: a children's oncology group study. JAMA Oncol. 2015;1:287–95. doi: 10.1001/jamaoncol.2015.0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheok MH, Evans WE. Acute lymphoblastic leukaemia: a model for the pharmacogenomics of cancer therapy. Nat Rev Cancer. 2006;6:117–29. doi: 10.1038/nrc1800. [DOI] [PubMed] [Google Scholar]
- 5.Chang JG, Lee LS, Chen CM, Shih MC, Wu MC, Tsai FJ, et al. Molecular analysis of thiopurine S-methyltransferase alleles in South-east Asian populations. Pharmacogenetics. 2002;12:191–5. doi: 10.1097/00008571-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Cho HG, Baek HJ, Han DK, Bae SY, Kook H, Hwang TJ. Genetic polymorphism of thiopurine methyltransferase in children with acute lymphoblastic leukemia. Clin Pediatr Hematol Oncol. 2008;15:1–9. [Google Scholar]
- 7.Yang SK, Hong M, Baek J, Choi H, Zhao W, Jung Y, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet. 2014;46:1017–20. doi: 10.1038/ng.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka Y, Kato M, Hasegawa D, Urayama KY, Nakadate H, Kondoh K, et al. Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol. 2015;171:109–15. doi: 10.1111/bjh.13518. [DOI] [PubMed] [Google Scholar]
- 9.Chiengthong K, Ittiwut C, Muensri S, Sophonphan J, Sosothikul D, Seksan P, et al. NUDT15 c.415C>T increases risk of 6-mercaptopurine induced myelosuppression during maintenance therapy in children with acute lymphoblastic leukemia. Haematologica. 2016;101:e24–6. doi: 10.3324/haematol.2015.134775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim H, Kang HJ, Kim HJ, Jang MK, Kim NH, Oh Y, et al. Pharmacogenetic analysis of pediatric patients with acute lymphoblastic leukemia: a possible association between survival rate and ITPA polymorphism. PLoS One. 2012;7:e45558. doi: 10.1371/journal.pone.0045558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 17.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–54. [Google Scholar]
- 18.Lange BJ, Bostrom BC, Cherlow JM, Sensel MG, La MK, Rackoff W, et al. Double-delayed intensification improves event-free survival for children with intermediate-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood. 2002;99:825–33. doi: 10.1182/blood.v99.3.825. [DOI] [PubMed] [Google Scholar]
- 19.Stork LC, Matloub Y, Broxson E, La M, Yanofsky R, Sather H, et al. Oral 6-mercaptopurine versus oral 6-thioguanine and veno-occlusive disease in children with standard-risk acute lymphoblastic leukemia: report of the Children's Oncology Group CCG-1952 clinical trial. Blood. 2010;115:2740–8. doi: 10.1182/blood-2009-07-230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nachman JB, Sather HN, Sensel MG, Trigg ME, Cherlow JM, Lukens JN, et al. Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med. 1998;338:1663–71. doi: 10.1056/NEJM199806043382304. [DOI] [PubMed] [Google Scholar]
- 21.Bhatia S, Landier W, Hageman L, Kim H, Chen Y, Crews KR, et al. 6MP adherence in a multiracial cohort of children with acute lymphoblastic leukemia: a Children's Oncology Group study. Blood. 2014;124:2345–53. doi: 10.1182/blood-2014-01-552166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNeill DR, Lam W, DeWeese TL, Cheng YC, Wilson DM., 3rd Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol Cancer Res. 2009;7:897–906. doi: 10.1158/1541-7786.MCR-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Stubbe J. Bleomycins: towards better therapeutics. Nat Rev Cancer. 2005;5:102–12. doi: 10.1038/nrc1547. [DOI] [PubMed] [Google Scholar]
- 24.Yoshimura D, Sakumi K, Ohno M, Sakai Y, Furuichi M, Iwai S, et al. An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J Biol Chem. 2003;278:37965–73. doi: 10.1074/jbc.M306201200. [DOI] [PubMed] [Google Scholar]
- 25.Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89:387–91. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheon JH, Kim JH, Kim BY, Kim SW, Hong SY, Eun CS, et al. Allele frequency of thiopurine methyltransferase and inosine triphosphate pyrophosphatase gene polymorphisms in Korean patients with inflammatory bowel diseases. Hepatogastroenterology. 2009;56:421–3. [PubMed] [Google Scholar]
- 27.Ban H, Andoh A, Tanaka A, Tsujikawa T, Sasaki M, Saito Y, et al. Analysis of thiopurine S-methyltransferase genotypes in Japanese patients with inflammatory bowel disease. Intern Med. 2008;47:1645–8. doi: 10.2169/internalmedicine.47.1268. [DOI] [PubMed] [Google Scholar]
- 28.Zhang LR, Song DK, Zhang W, Zhao J, Jia LJ, Xing DL. Efficient screening method of the thiopurine methyltransferase polymorphisms for patients considering taking thiopurine drugs in a Chinese Han population in Henan Province (central China) Clin Chim Acta. 2007;376:45–51. doi: 10.1016/j.cca.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Krishnamurthy P, Schwab M, Takenaka K, Nachagari D, Morgan J, Leslie M, et al. Transporter-mediated protection against thiopurine-induced hematopoietic toxicity. Cancer Res. 2008;68:4983–9. doi: 10.1158/0008-5472.CAN-07-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ban H, Andoh A, Imaeda H, Kobori A, Bamba S, Tsujikawa T, et al. The multidrug-resistance protein 4 polymorphism is a new factor accounting for thiopurine sensitivity in Japanese patients with inflammatory bowel disease. J Gastroenterol. 2010;45:1014–21. doi: 10.1007/s00535-010-0248-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.