

Abstract
Alu elements, the short interspersed element numbering more than 1 million copies per human genome, can mediate the formation of copy number variants (CNVs) between substrate pairs. These Alu/Alu-mediated rearrangements (AAMRs) can result in pathogenic variants that cause diseases. To investigate the impact of AAMR on gene variation and human health, we first characterized Alus that are involved in mediating CNVs (CNV-Alus) and observed that these Alus tend to be evolutionarily younger. We then computationally generated, with the assistance of a supercomputer, a test data set consisting of 78 million Alu pairs and predicted ∼18% of them are potentially susceptible to AAMR. We further determined the relative risk of AAMR in 12,074 OMIM genes using the count of predicted CNV-Alu pairs and experimentally validated the predictions with 89 samples selected by correlating predicted hotspots with a database of CNVs identified by clinical chromosomal microarrays (CMAs) on the genomes of approximately 54,000 subjects. We fine-mapped 47 duplications, 40 deletions, and two complex rearrangements and examined a total of 52 breakpoint junctions of simple CNVs. Overall, 94% of the candidate breakpoints were at least partially Alu mediated. We successfully predicted all (100%) of Alu pairs that mediated deletions (n = 21) and achieved an 87% positive predictive value overall when including AAMR-generated deletions and duplications. We provided a tool, AluAluCNVpredictor, for assessing AAMR hotspots and their role in human disease. These results demonstrate the utility of our predictive model and provide insights into the genomic features and molecular mechanisms underlying AAMR.
Alu elements are repetitive sequences originally described by reassociation kinetics (Schmid and Jelinek 1982). The term “Alu” derives from these sequences sharing a cut site for the restriction endonuclease AluI (Houck et al. 1979). Alu repetitive sequences comprise ∼11% of the human genome and number more than 1 million copies per haploid genome (Lander et al. 2001). They belong to the primate-specific short interspersed element (SINE) family of mobile DNA. Alu elements can be grouped into distinct subfamilies based on sequence divergence. AluJs are the oldest Alu dimeric subfamily; AluSs are the most numerous, and are younger than AluJ, while AluYs are the youngest of this family of repetitive elements (Shen et al. 1991; Batzer and Deininger 2002). Monomeric Alus also exist in the human genome, such as FRAM and FLAM elements (Quentin 1992). Full-length Alu elements are ∼300 bp in size and consist of two monomeric repeats derived from 7SL RNA, an adenosine-rich connector, and a poly(A) tail. The left monomer contains an internal RNA polymerase III promoter, A Box, and B Box; the right monomer has an A′ box (Fig. 1A; Deininger et al. 2003; Beck et al. 2011). Alu sequences are often found at the endpoints of segmental duplications (SDs) and the breakpoints of genomic rearrangements and are associated with genome instability (Bailey et al. 2003; Shaw and Lupski 2005; Vissers et al. 2009). Copy number variants (CNVs) differ from a normal diploid state by deletion or amplification of genomic segments. When a pair of Alus mediate a genomic rearrangement (i.e., Alu/Alu-mediated rearrangement [AAMR]), a chimeric Alu hybrid will form at the junction (Fig. 1B). Microhomologies are the sequences surrounding the breakpoint junctions that are identical between the CNV-Alu elements within a pair. The first observed AAMR event was described 30 years ago in a patient with hypercholesterolemia and a 7.8-kb deletion of LDLR (Lehrman et al. 1987); similar AAMR-mediated exonic events have been elucidated during the decades that followed in association with different diseases, including spastic paraplegia 4 (MIM 182601) (Boone et al. 2011, 2014), Fanconi anemia (MIM 227650) (Flynn et al. 2014), and von Hippel–Lindau syndrome (MIM 193300) (Franke et al. 2009). Alu-associated CNVs have been estimated to cause ∼0.3% of human genetic diseases (Deininger and Batzer 1999). In spite of this fairly large potential impact of AAMR events on gene variation and human health, to date fewer than 300 independent events have been experimentally characterized at nucleotide-level resolution (Supplemental Table S1). Array comparative genomic hybridization (aCGH) is a robust experimental procedure for CNV detection, including CNVs at the exonic level; however, array techniques have non-nucleotide-level breakpoint junction resolving capability. Although the combination of aCGH and PCR can achieve breakpoint junction sequence resolution, such an approach is not currently scalable. Thus, it is impractical and costly to map the breakpoints for all detected disease-associated rare CNVs using this approach. Moreover, many studies utilizing genome-wide variant assays, including whole-genome sequencing (WGS) and whole-exome sequencing (WES), are limited by sequence coverage and alignment difficulties inherent to the relatively short length of sequencing reads and high degree of Alu sequence identity (Treangen and Salzberg 2011).
Figure 1.
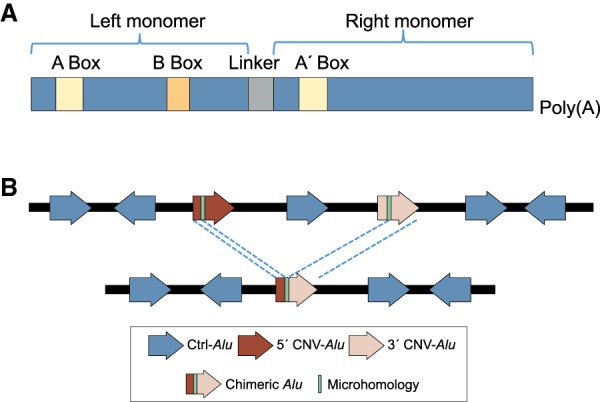
Alu structure and Alu/Alu-mediated rearrangement (AAMR) event formation. (A) A consensus Alu element is depicted, with both left and right 7SL monomers indicated. A Box, B Box, and A′ Box are internal Pol III promoter elements; the linker is an A-rich sequence; and the element ends in a poly(A) tail. (B) A diagram of an AAMR event is shown: A genomic rearrangement is mediated by a substrate pair of Alu elements followed by the formation of a relatively complete chimeric Alu. Block arrows represent Alu elements on the + (forward arrow) and − (reverse arrow) strand. The 5′ CNV-Alu is colored maroon, and the 3′ CNV-Alu is pink. Ctrl-Alu elements not involved in AAMR are in blue. The microhomology generated at the breakpoint junction after the AAMR event is shown in green.
CNVs and other structural variants (SVs) can result from distinct molecular mechanisms, including DNA recombination–associated processes, DNA repair–associated processes, and DNA replication–associated processes (Carvalho and Lupski 2016), and lead to human diseases often termed genomic disorders (Lupski 1998). Previously, repeated sequences (e.g., paralogous genes/pseudogenes, low-copy repeats [LCRs], etc.) and repetitive elements (e.g., SINEs, long interspersed nuclear elements [LINEs]) that are involved in the formation of genomic rearrangements have been posited to undergo nonallelic homologous recombination (NAHR). For example, duplications and deletions of the same genomic segment can be flanked by similar human endogenous retroviral sequences (HERVs) (Sun et al. 2000; Campbell et al. 2014), LINEs (Higashimoto et al. 2013; Startek et al. 2015), or LCRs (Lupski 1998; Sharp et al. 2005). The PRDM9 binding motif is a cis-acting sequence motif associated with allelic homologous recombination (AHR) and NAHR hotspots (Lupski 2004; Lindsay et al. 2006; Myers and McCarroll 2006; Berg et al. 2010; Dittwald et al. 2013). PRDM9 targeting sites are associated with ∼40% of recombination hotspots ascertained through studies of historical recombinants (Myers et al. 2008; Webb et al. 2008). Deletions mediated by LCRs and HERVs in human genomes are enriched for PRDM9 binding motifs proximal to the junctions, further implicating NAHR as the mechanism for their formation (Repping et al. 2002; Campbell et al. 2014). Classically, NAHR has been proposed as the mechanism underlying AAMR events (Cordaux and Batzer 2009); however, the minimal processing segment required for NAHR is generally thought to be longer than an individual Alu sequence (Reiter et al. 1998). It has recently been proposed that Alu repetitive elements may participate in aberrant rearrangement of the genome by mediating template switching (TS) during replication-based repair mechanisms (Boone et al. 2011, 2014; Gu et al. 2015) such as MMBIR (Hastings et al. 2009) or by undergoing SSA (single-strand annealing) or MMEJ (microhomology-mediated end joining) mechanisms given that they provide multiple regions of microhomology (Elliott et al. 2005; Morales et al. 2015).
Results
To better understand the mechanism(s) of AAMR and potentially identify human genes that are prone to instability due to these events, we conducted a machine learning–based analysis of Alu and the human genome reference; the steps of which are described below and summarized in Figure 2.
Figure 2.

Diagram of the workflow used for predicting CNV-Alu pairs and AAMR hotspot genes in this study. Approximately 1.2 million Alus are documented in the “Repeating Elements by RepeatMasker” track at the UCSC Genome Browser. CNV-Alus are those with experimental evidence supporting their role in AAMR (Supplemental Table S1), and all the others are Ctrl-Alus. We selected Alu pairs that are in the same orientation, span at least one exon, and are located <250 kb from each other. Both the individual Alu sequence features and genomic architectural features were characterized, and a subset of features were utilized in model training. The QDA (quadratic discriminant analysis) model achieved the highest sensitivity and was applied for predicting CNV-Alu pairs. The amount of predicted CNV-Alu pairs is significantly correlated with the number of observed AAMR events for known hotspot genes. Therefore, we further determined the relative risk of AAMR in 12,074 human genes that have a MIM entry using the count of predicted CNV-Alu pairs. Finally, we experimentally validated this prediction with 89 samples selected by correlating predicted hotspot genes with a database of approximately 54,000 chromosomal microarrays (CMAs) by performing aCGH and mapping the breakpoint junctions of detected CNVs. We achieved an 87% positive predictive value overall.
Collection and characteristics of CNV-Alu pairs
We define the Alu pairs involved in AAMR events as CNV-Alus and all the other non CNV-Alus as Ctrl-Alus (Fig. 1B). To build a classifier for predicting Alu pairs that may be more likely to mediate genomic rearrangements, we utilized a positive training data set composed of 219 CNV-Alu pairs, 218 of which were collected from deletions published in 58 articles and one that is currently unpublished (Supplemental Table S1). Each breakpoint of the 219 deletions has been mapped at nucleotide-level resolution within the resultant chimeric Alu in the original studies, enabling determination of microhomology at the breakpoint junction of AAMR. The deletions vary in size from ∼800 bp to ∼4 Mb, and 75% of the deletions are <57 kb (Supplemental Fig. S1A). We have determined each of the Alu elements involved and their genomic coordinates, orientation, and the information of subfamily using the RepeatMasker track of the UCSC Genome Browser (Supplemental Table S1; Kent et al. 2002).
To determine whether CNV-Alu pairs are enriched for a specific subfamily, we first calculated the relative frequency of each subfamily composition in CNV-Alu pairs. Considering the different frequency of Alu subfamilies, we further calculated the expectation of the relative frequency of each composition in Ctrl-Alu pairs that were documented in the RepeatMasker database (Smit et al. 2013–2015). We found that AAMR events are more likely to be mediated by younger Alu elements, such as AluS-AluY and AluY-AluY (P < 0.001, one-tailed binominal test) (Fig. 3A). This could be potentially explained by a higher possibility of aligning better with each other due to a less divergent sequence in the younger families (Batzer and Deininger 2002). Of note, younger subfamilies are more active in retrotransposition assays (Bennett et al. 2008; Konkel et al. 2015). Thus, one potential explanation for younger Alus being involved in AAMR is that open chromatin exists over active Alus during transcription, which could allow them to function as better substrates for DNA repair.
Figure 3.

Features of CNV-Alu pairs and microhomology preferences. (A) The relative frequency of Alu subfamilies is shown. For example, the AluS-AluY indicates CNVs mediated by Alus from family AluS and AluY respectively, and “Other” indicates monomeric Alus such as FRAMs. We compared the relative frequency of a given subfamily composition of CNV-Alu pairs (in maroon) with that of the expected relative frequency of observing a given subfamily pair (in blue) using the one-tailed binomial test. (**) P ≤ 0.01; (***) P ≤ 0.001. (B) The histogram describes the distribution of microhomology length at breakpoint junctions. (C) The histogram indicates the %GC content within the stretch of microhomology. (D) The figure depicts the collected 219 microhomologies from disease-related studies in human with respect to their relative position on an Alu consensus sequence (lower panel). The peak in the histogram indicates an enrichment of breakpoint junctions on the specific locus. The light blue shading shows a 26-bp core sequence detected by a previous compilation study of Alu-involved gene rearrangements (Rudiger et al. 1995). (E) Adapted from a comparative genomic study on chimpanzee and human reference genome (Han et al. 2007). The blue line describes 492 human-specific breakpoint junctions of Alu/Alu-mediated deletions, and the red line depicts 663 chimpanzee-specific events. The dashed horizontal line indicates the average percentage of breakpoints across the entire Alu element. (F) The schematic shows the construct utilized to detect template switches in yeast. Two human Alu pairs were inserted into Chr II separately with the same distal AluSx element. URA3 and TRP1 are the markers for selecting colonies with successful transformation. We induced a single-strand DNA break at the FRT site using a mutation of FLP recombinase. (G,H) The relative positions of microhomologies generated by mapping junctions from the yeast assay are depicted in relation to an Alu consensus sequence. (G) Data from 503 AAMR events observed in the first AluSx-AluSp strain. (H) Distribution of 114 events from the AluSx-AluY construct.
We next analyzed the properties and characteristics of AAMR breakpoint junctions. The majority of AAMR microhomologies are <25 bp (Fig. 3B), and have a higher GC content than that of whole–CNV-Alu element sequences (P < 0.0001, one-tailed t-test) (Fig. 3C). The higher GC percentage might facilitate strand annealing by a stronger hydrogen bonding than A-T. We mapped the sequence of each breakpoint microhomology generated during AAMR formation to an Alu consensus sequence (Fig. 3D), and noted enrichment downstream from RNA pol III promoters (A Box, B Box, and A′ Box). The location of breakpoint junctions in 18 rearrangements with at least one Alu element involved was previously described by Rudiger et al. (1995); a 26-bp core sequence was at or close to the breakpoint junctions (Fig. 3D–H, shaded light blue). With the 219 AAMR events, we further showed that more breakpoint junctions (n = 62) are located at or near the first 26-bp core sequence. There is no significant enrichment of a specific subfamily in these 62 events compared with all of the 219 junctions (P > 0.05, one-tailed binomial test) (Supplemental Fig. S2).
Of note, the microhomology distribution we observed is consistent with Alu/Alu-mediated evolutionary deletions previously identified by comparing human and chimpanzee genomes (Fig. 3E; Sen et al. 2006; Han et al. 2007). To infer the underlying mechanism for AAMR, we adapted a yeast TS assay to examine human Alu pairs that occur more often in AAMR events, where AluS-AluS and AluS-AluY are most numerous; the elements were chosen from experimentally determined events at the SPAST locus (Boone et al. 2014). We induced a nick downstream from a replication origin to generate a single-ended, double-strand DNA break (seDSB), inducing a substrate that is repaired via TS during break-induced replication (BIR). The previously published 74 events displayed an enrichment pattern of breakpoint junctions (Supplemental Fig. S3A,C; Mayle et al. 2015); this pattern was robust and was supported by an additional 429 events (Supplemental Fig. S3C–E). To test if this pattern is peculiar to the construct used, we made a strain with the same distal Alu and a different proximal Alu element (AluSx-AluY, 88.4% similar) (Fig. 3F). Despite the distinctive pattern of available microhomologies (Supplemental Fig. S3A,B), the experimentally detected junctions from the two yeast assays differ from the observed pattern in human events and display preferences for similar regions (Fig. 3G,H). The yeast junctions favor blocks of nearly identical sequence that are minimally interrupted by single-base-pair mismatches, therefore representing the largest sequence homeology (highly similar but not identical sequences) between each pair. These data support the contention that AAMR can be mediated by TS and potentially occurs by MMBIR (Hastings et al. 2009). The divergent frequency distribution patterns observed between human and yeast experimental data might be due to the distribution of homeology between the two Alu pairs in the TS assay and species differences.
Predicting CNV-Alu pairs
The skewed distribution of the properties of AAMR breakpoint junctions discussed above indicates that AAMR events could be generated by Alu pairs enriched for particular features. This finding motivated characterization of a group of factors that can potentially distinguish CNV-Alu pairs from their genomic milieu. We investigated whether features of an individual Alu and its surrounding genomic region may potentially influence genomic instability and the choice of which elements serve as templates for repair. There are more than 1.1 million Alu elements in the human genome. If one also considers interchromosomal recombination events, more than 6 × 1011 Ctrl-Alu pairs exist in the human genome. As the number of CNV-Alu pairs is grossly unequal to that of Ctrl-Alu pairs, factors were selected before utilization in our machine learning model to abrogate concerns of overfitting the model.
The distribution across the genome of the 219 events in our positive training set was biased because of the limited number of studies and the focus on several disease genes (Supplemental Fig. S1B,C). To maximize the knowledge that one could potentially gain from these events, we treated each deletion as an individual locus and have characterized each locus independently.
Generating a control, i.e., a negative training data set, is challenging because the absence of evidence for an Alu to be involved in CNV formation does not mean that the element cannot be used in a heretofore-uncharacterized event. To overcome this issue, we constructed distinct control data sets for analyzing individual features and genomic features (see Methods; Supplemental Fig. S4). To decrease the possibility of choosing false-negative CNV-Alu pairs, 1000 Ctrl-Alu pairs with the same orientation as each other (either plus or minus) in each region were randomly collected as a negative control data set. Since the local environmental genomic features can be variable simply because of the relative position within the gene, the negative control data sets for these features were generated analogously to the collected CNV-Alu pairs, which are intragenic exonic CNVs in/near disease-associated genes. Of note, the CNV sizes for these experimentally determined AAMR events tend to be <250 kb (Supplemental Fig. S1A). There are only 27 deletions spanning >250 kb, and none were between 250 and 500 kb in size. We chose “<250 kb” as a cutoff to include the majority of the known events and generate a comparable test data set to the training data. For each of the 192 CNV-Alu pairs that are <250 kb apart, we selected 1000 directly oriented Ctrl-Alu pairs that could delete at least one exon and that have the same distance between them as the CNV-Alu pair.
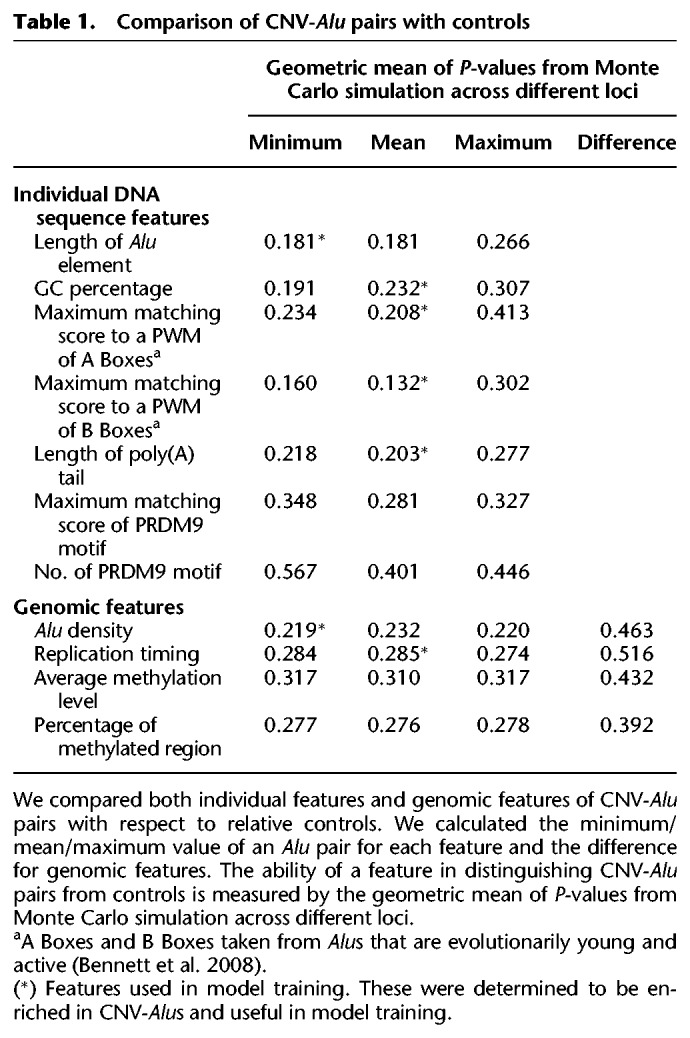
As it was not clear whether each Alu element within a pair would contribute equally to AAMR events, we calculated the pairwise value for each feature by computing the minimum, mean, and maximum values for each pair; and we also considered the difference between the two elements for genomic features. We then characterized each pairwise feature for Alu pairs in the training data set. At each locus, we plotted the Ctrl-Alu pair distribution as a boxplot and marked the CNV-Alu pair as a red dot. We also calculated a P-value using a Monte Carlo approach (see Methods). For example, at most loci, the red dots showing the value of pairwise alignment of CNV-Alu pairs are located above the medians of Ctrl-Alu pairs (Fig. 4A). The distribution of P-values across different loci is plotted in Figure 4B. The geometric mean of P-values for the feature of pairwise alignment is 0.102. Therefore, CNV-Alu pairs in the majority of loci have a higher sequence similarity with each other than the control pairs. We next examined the region of 500 bp upstream of and downstream from each Alu element to identify putative PRDM9 binding motifs that match >85% to a position weight matrix (PWM) (Campbell et al. 2014). There is no significant difference between CNV-Alu pairs and their locus-specific controls with respect to the number of surrounding PRDM9 binding motifs (Fig. 4C,D; Table 1). We summarized the Monte Carlo–based P-values for this and other tested parameters, including both individual Alu sequence features and genomic features surrounding CNV-Alu pairs, in Table 1 and Supplemental Figure S5. After conducting these analyses and combining results from the plots and P-values, we noted that not all the features distinguish CNV-Alu pairs from control elements and that distinct pairwise values also perform differently. Therefore, we utilized the following parameters to train our model for prediction: minimum Alu element length and Alu density, mean GC percentage, sequence similarity of A Boxes/B Boxes, poly(A) tail length, replication timing, and the pairwise alignment score (marked with asterisks in Table 1).
Figure 4.

Determining feature enrichment for CNV-Alu pairs with respect to Ctrl-Alu pairs. (A) The comparison of pairwise alignments between CNV-Alus (n = 219) and the corresponding Ctrl-Alu pairs (n = 1000 per CNV-Alu) is shown. The y-axis is a score showing the alignment performance, a higher value of which indicates a better alignment between two sequences. As shown in the key, at each locus, we displayed the alignment score of CNV-Alu with a red dot and showed the distribution of the Ctrl-Alus with a boxplot. The information of all the 219 events is summarized in an increasing order of the median value of the Ctrl-Alus. (B) The distribution of P-values calculated using Monte Carlo simulation for pairwise alignment is shown. (C,D) The same strategy was adopted for analyzing the mean value of the maximum matching score of the PRDM9 targeting motif within an Alu pair.
Table 1.
Comparison of CNV-Alu pairs with controls
We applied these features to train a model using quadratic discriminant analysis (QDA) with the CNV-Alu pairs and 10,000 control pairs. The prior probability was set to 0.3, which provided the highest sensitivity in testing the known 192 CNV-Alu pairs (sensitivity = 0.83, 10-fold cross validation) (Supplemental Fig. S6). Our test data set, consisting of approximately 78 million Ctrl-Alu pairs, was built to enrich potential intragenic exonic events but efficiently decrease the computational burden by choosing Alu pairs that have the same orientation, span at least one exon, and are located within 250 kb of each other. We utilized the BlueGene supercomputer at Rice University to calculate the selected features for each pair, as calculating pairwise alignment scores of the 78 million pairs was computationally intensive. These analyses predicted 17.6% of the Ctrl-Alu pairs to be more likely to mediate CNVs.
Of note, the features utilized in the QDA model training share some degree of codependence. For example, evolutionarily young Alus tend to have a higher sequence similarity and be more active in retrotransposition (Bennett et al. 2008). The QDA model explicitly accounts for codependence (covariance) between variables in making its predictions; in this way, the influence of correlation is statistically controlled and accounted for in the model. Any unwanted or erroneous impact of covariance on prediction should be minor. In addition, to better understand the level of feature codependence, we measured the impact of feature codependence by training a series of models with all selected features and removing one feature at a time and evaluating the performance of each model. The model that was trained with all selected features performed best in terms of having the lowest error rate as shown in Figure 5A; however, the feature of Alu length seems to be redundant based on the same error rate as the overall model. The feature of Alu density contributed tremendously to the prediction as the error rate increased dramatically when this feature was removed; removing the other features could increase the error rate to some degree as well.
Figure 5.

Comparing and selecting machine learning models and the result of a gene-level prediction. (A) The measurement of feature codependency in model training. We tested the error rate for models trained with all selected features (Table 1) as well as by removing one feature at a time (see Methods). (B) The frequency distribution of the gene-level AAMR risk scores for 12,074 OMIM genes. (C) The frequency distribution of the gene-level AAMR risk scores for 133 genes that have been involved in AAMR more than once.
Predicting hotspot genes for AAMR events
We developed a risk score to estimate the relative effect of AAMR events at a gene level. We first tested the correlation between the count of predicted CNV-Alu pairs from our QDA analysis and the count of reported CNV-Alu pairs in known AAMR hotspot genes (Supplemental Table S1): They are significantly correlated (linear regression, P < 0.001). We then utilized this model to predict the risk score for 12,074 human genes that are collected in the OMIM database (https://www.omim.org/) with a gene MIM number and have complete information for each involved Alu element (Supplemental Table S2). We focused on a disease-related gene set because we only chose the potentially pathogenic Alu pairs in the test data set. In addition, we could implement validation experiments using a clinical microarray database, which mainly interrogates disease-related genes. Although the prediction might not be as accurate as the OMIM genes, we also performed the same gene-level prediction on a total of 23,637 RefSeq genes with available tested Alu pairs (Supplemental Table S2).
An AAMR risk score of one indicates a prediction of 10 (101) CNVs intersecting this gene were expected. Using a score greater than one as a cutoff determined a subset of 329/12,074 OMIM genes, or ∼3% of the total. Given the left skewed distribution of AAMR scores (Fig. 5B), we regarded a AAMR risk score greater than one as an extremely strict cutoff. In addition, we showed the distribution of the genes that have been affected by AAMR more than once (n = 133) (Fig. 5C; Supplemental Table S1). These genes have a median score of 0.601, and the scores enrich around 0.6; we suggest a score of 0.6 as a loose “cutoff” for at-risk genes. Genes with a score of less than 0.6 might be more susceptible for AAMR.
Furthermore, Alu density alone, a parameter previously suggested to influence AAMR-mediated genomic instability (Boone et al. 2014), did not achieve the same performance in prediction compared with the model trained with all the selected features (P < 0.001, χ2 test). To clarify the potential effect of gene size, we performed two analyses: First, we fit a model treating count divided by gene size as a predictor and compared this model to our reported model; a goodness of fit test shows that these two models are not significantly different (χ2 P = 0.331). This result suggests that accounting for gene size does not alter the predictive power of the model. As a second analysis, we created a multiple regression model that examined predicted count and gene size as independent contributors to the model (Supplemental Table S3). The results analyzing this model show that although count is a significant variable, there is no residual variation significantly associated with gene size. Taken together, we conclude that the gene size does not significantly contribute to our results. Therefore, we trained this gene-level prediction only using the count of predicted CNV-Alu pairs.
Validating the computational prediction
We tested our pairwise prediction results against a list of 663 human Alu pairs that recombined during evolution as the corresponding positions in the chimpanzee genome only contain one chimeric Alu element at the breakpoint junction (Han et al. 2007). However, only one Alu pair was collected in our test data set and was not predicted as a likely CNV-Alu pair. The narrow overlap of the two data sets might be due to the pathogenicity and constraint on AAMR events between predicted pairs in humans that could delete at least one exon.
We then experimentally tested our prediction by cross-referencing the list of 329 predicted hotspot genes (risk score > 1) (Supplemental Table S2) with an anonymized database of exon-targeted clinically applied CMAs performed in approximately 54,000 individuals. We selected subject samples that have one or more CNVs that intersect with genes in this hotspot gene list. To investigate any enrichment of AAMR versus variants mediated by other repeats/repetitive elements, we chose genes with three or more samples available. We developed a high-density aCGH platform targeted to 15 regions (covering 18 predicted hotspot genes), and performed custom-designed high-density array CGH against gender-matched controls on the obtained 89 DNA samples from the CMA laboratory to both validate the original clinical array findings and facilitate our breakpoint junction analyses (Supplemental Table S4). We found that two of the duplications were complex genomic rearrangements (CGRs). About 95% (83/87) of the samples contain simple deletions or duplications that have at least an Alu located within the uncertain region defined by oligonucleotide probes at one or both ends. Furthermore, 81.0% (51/63) of the samples that potentially intersect with Alu pairs at both ends likely terminate within predicted CNV-Alu pairs.
After determining breakpoints at a higher resolution, we next mapped the nucleotide-level breakpoint junctions for 52 out of the 87 simple CNVs with long-range PCR and Sanger sequencing (Supplemental Fig. S7). The reasons for an inability to experimentally map the remaining breakpoints include a low quality of genomic DNA, complex genome structure (e.g., tandem Alus), and a low resolution of uncertain breakpoint intervals in some regions. For each of those resolved CNVs, we have tabulated the resultant sequence and the exons affected by the SV; an example detailing a duplication intersecting CLIP1 mediated by an Alu pair is shown as Figure 6, A through C. Seventy-three percent (38/52) of the experimentally solved breakpoint junctions were meditated by Alu pairs (including 19 unique events) (Fig. 6D). As shown in Supplemental Figure S8A, 14 unique events were detected, i.e., found only once in the validation data set. Two junctions were observed four times each (eight events in total), and another three Alu pairs were used twice, five times, and nine times separately (16 events in total). To answer whether the frequently observed events are truly independent or due to a founder effect, we compared five of these events with SVs in the DGV database (Database of Genomic Variants) (MacDonald et al. 2014). As the resolution of DGV variants varies from each other due to the different detection methods/assays utilized, we could only roughly indicate whether an apparent same variant was also observed in the DGV. As shown in Supplemental Figure S8B,C, two recurrent variants were recaptured in the DGV. We hypothesized these two variants are polymorphisms in the human genome. For the remaining three, we were unable to obtain family information, and therefore, they could come from a common founder or could be independent but contribute to the same clinical phenotypes.
Figure 6.

Experimental and computational validation of AAMR hotspot prediction. (A) High-density aCGH results from one individual selected from the CMA database shows a duplication of the two terminal exons of CLIP1, a predicted AAMR hotspot gene. Red dots signify probes that indicate relative copy number gain (the region indicated contains a duplication); black dots, a region unaffected by CNV; and green dots, deletion. (B) The UCSC Genome Browser image depicts RefSeq genes and RepeatMasker annotations within the same genomic interval as shown in the aCGH result. The red block represents the duplicated region. The two SINE elements, AluSc8 and AluSx, in which the breakpoints of this CNV are located are marked with red arrows. (C) The first line of sequence shows the reference sequence of the AluSx; the middle line, the sample sequence; and the bottom line, the sequence of the AluSc8. The sequences are on the plus strand, and both Alus are in the plus orientation. The sequence of microhomology at the breakpoint junction is highlighted in red. The gray sequence starts from the first mismatching base. The genomic coordinates of the microhomologies are annotated in the hg19 assembly. (D) A chart summarizing 52 breakpoint junctions mapped at nucleotide level is depicted. The CNVs are grouped into three types: Alu-Alu, CNVs mediated by an Alu pair; Alu-Other, Alu pairing with a non-Alu sequence, including LINE, LCR, and nonrepeat/repetitive sequence, mediates the CNV formation; and Other, no Alu elements were involved. For those mediated by an Alu pair, the QDA prediction result is shown to the right. True prediction indicates these Alu pairs were predicted as high risk for AAMR. (E) A box plot showing the enrichment of genes within different risk score tertiles among three classes of the count of susceptible AAMR CNVs in the CMA database.
Alu elements participated in another 21% (11/52) of junctions with a LINE element (four of 52, two unique events) or nonrepetitive sequences (seven of 52, four unique events) at the other end. One unique Alu-LINE mediated event and two unique Alu-nonrepetitive sequence-mediated duplications have microhomology detected at the breakpoint junction. Only 6% (3/52) of these events were mediated by non-Alu elements, including one LINE-LCR and two LCR-LCR events. We also resolved the two breakpoint junctions in one complex rearrangement event (Sample88, Supplemental Fig. S7), both of which contain one Alu element, indicating an involvement of Alus in potential TS events.
Validation of the performance of our score for predicted low-risk genes using wet-bench experimental studies is challenging, because these studies would be directed toward the absence of observations. Therefore, we analyzed the association between lower-scoring genes and their relative risk for potential absence of AAMR-mediated genomic instability. First, we examined the CMA database and collected the susceptible AAMR CNVs, which have at least a pair of predicted Alus located within the uncertain region. Second, we then counted the overlapping susceptible CNVs for each gene and assigned zero for the well-targeted genes but with no potential AAMR CNV detected. We established another category for those genes with exactly one, and finally a third category for genes with more than one likely AAMR CNV. There are 2433 well-targeted genes with no potential CNV, 1260 genes with one, and 824 genes with more than one. Third, to examine the association of the observed CMA data with the scores, we independently divided the genes (N = 4517) into three categories using the tertiles of the predictive risk score. Fourth, we tested the enrichment of genes with variable risk score in each subset based on real data from CMA.
As shown in Figure 6E, in the group of genes with zero AAMR CNV, we observed a significant overrepresentation of low-scoring genes and a depletion in genes with a high score (binomial test, P < 1 × 10−16), consistent with a lower score being predictive of lower rates of AAMR CNVs. Correspondingly, the genes with at least one observed susceptible AAMR CNV are highly enriched among the high-scoring group (binomial test, P < 1 × 10−16). Overall, a test of independence between the risk score tertiles and the potentially AAMR CNV count classes shows a highly significant association (P < 1 × 10−16) for both Fisher's exact test and a χ2 test.
Assessing the role of AAMR in causing genome instability and human disease
We further developed a tool, AluAluCNVpredictor, to help query the data. With this tool, investigators can query a gene and receive output for the gene rank in the total 12,074 OMIM genes or 23,637 RefSeq genes, the relative gene-level risk score, the count of predicted CNV-Alu pairs, and a plot showing the AAMR risk score of this gene on the total score distribution. AluAluCNVpredictor may also assist a query for any predicted CNV-Alu pairs intersecting a genomic interval pair of interest, e.g., a pair of uncertain regions from aCGH data. This tool is publicly available (see Supplemental File S1; http://alualucnvpredictor.research.bcm.edu:3838).
We annotated the hotspot genes for human disease phenotypes using OMIM. The known disease relevant genes (77 out of 329) are listed in Table 2 (in alphabetical order). Of the 77 OMIM entries, 33 are associated with recessive disease traits, in which a CNV-Alu mediated exonic deletion may contribute to a carrier state (Boone et al. 2013; Harel et al. 2016). Twenty-five of the OMIM entries represent dominant disease traits, and 19 are either lacking documentation or have cases in which both inheritance patterns were observed. Although not all of these 77 genes have been associated with Alu/Alu CNVs, we emphasize a potentially underappreciated role of Alu in causing variants in these genes as AAMR events are easily missed by routine short-read genomic sequencing techniques.
Table 2.
Human disease-associated genes in predicted AAMR hotspots

Finally, we tested the correlation between the AAMR risk score and the number of events of CNVs (<250 kb) within each hotspot gene in the CMA database by Poisson regression. At the relatively low resolution of the CMA CNVs, we could not determine directly whether they are mediated by Alu pairs. The two scores are positively correlated with each other (P < 1 × 10−16), indicating that the Alu pairs around these genes make the region genomically unstable, supporting the contention that these genes may be more susceptible to exonic CNVs.
Discussion
We identified characteristics of CNV-Alu pairs, predicted Alu pairs that are more likely to mediate genomic rearrangements, and determined the relative risk of AAMR for 12,074 OMIM human genes with machine learning methods. We further evaluated the validity of our score for predicted high-risk genes by performing molecular biology experiments combining custom-designed high-resolution aCGH, breakpoint junction PCR and Sanger sequencing and performed an association analysis to explore more carefully the observed abundance of potential AAMR events for genes with a lower score. We provided a tool, AluAluCNVpredictor, for assessing the predicted potential susceptibility of a gene or genomic interval to AAMR events.
The potential biological and clinical utility for delineating genomic instability due to AAMR hotspots is indicated from studies focusing on disease loci. For example, 45.5% (20/43) of the CNVs described in 17p13.3 were mediated by Alu pairs (Gu et al. 2015), 56% (9/16) in the FOXF1 locus (Szafranski et al. 2016), 68% (39/57) in the SPAST locus (Boone et al. 2014), 88% (29/33) in the VHL locus (Franke et al. 2009), and 100% (45/45) in EPCAM (Kuiper et al. 2011). Previously, the prevalence of AAMR in disease was estimated based on observed events (Deininger and Batzer 1999). However, this earlier approximation of ∼0.3% is likely an underestimate due to the inherent challenges in the detection of repetitive element–mediated events genome-wide. Batzer and colleagues (Sen et al. 2006; Han et al. 2007) analyzed human-specific Alu/Alu-mediated deletions by comparing the haploid genomic reference sequences of chimpanzee and human. Alus have also been shown to be enriched at or near the junctions of SDs/LCRs, a correlation consistent with a potential role in genomic instability (Bailey et al. 2003). These studies indicate a potentially important role for AAMR events during evolution. However, the genes/genomic regions potentially susceptible to a pathogenic mutagenic effect due to AAMR have not been elucidated. An alternative in silico approach to estimate the distribution of putative AAMR events could be analyzing next-generation sequencing data (NGS) from large cohorts with a multitude of disease phenotypes, such as samples recruited to the Centers for Mendelian Genomics (CMG) (Chong et al. 2015). However, the range of read length in widely used NGS platforms is 50–150 bp (Goodwin et al. 2016), shorter than the length of an Alu sequence (∼300 bp). The highly repetitive and interspersed nature of Alus further challenges read alignment and local assembly (Treangen and Salzberg 2011). Although longer read sequencing methods such as 454 sequencing and more extended long-read sequencing technologies potentially overcome this limitation, they are more expensive and can be error-prone (Wheeler et al. 2008; Goodwin et al. 2016). Accumulating genome-wide long-read sequencing data from large disease cohorts is currently cost prohibitive. Moreover, WES does not resolve AAMR events due to most of Alus mapping in intergenic regions and introns.
Our machine learning method for discerning gene/variant genomic instability susceptibility due to AAMR-mediated intragenic exonic CNV may find utility in genome-wide studies: (1) The machine learning model directly adopts the current knowledge of AAMR events in the human genome, and (2) this method examined all possible disease-associated genes instead of focusing on a specific locus. We validated the reliability of the risk score in predicting hotspots by performing “wet-bench” experiments on variants in 18 out of 329 genes that have an AAMR risk score greater than one. We examined a total of 52 breakpoint junctions, 73% of which were mediated by Alu pairs and another 21% of which were partially Alu mediated. Overall, we achieved an 87% positive rate in predicting at risk Alu pairs. We suggest a cutoff of 0.6 based on the risk score frequency distribution of genes involved in AAMR more than once (Fig. 5C). NXN is a potential hotspot gene given a risk score of 0.87 and being ranked among the top 6% of OMIM genes (663/12,074) (Supplemental Table S2). An Alu/Alu-mediated exonic deletion interrupting NXN together with a variant on the other allele was recently shown to cause recessive Robinow syndrome (MIM 268310) (White et al. 2018). In contrast, CFTR has a risk score of 0.43 and ranks at 10,285 out of the 12,074 genes, suggesting a “nonhotspot gene” for AAMR; none of the 18 successfully mapped unique breakpoint junctions were mediated by Alu pairs (Quemener et al. 2010). These experimental results demonstrate the utility and performance of our predictive model.
The limitations of our study include the small training data set and the fact that most CNVs are pathogenic; however, these are the only published, experimentally determined AAMR events available at the time of initiation of this study. Alus close to genes might be conserved in particular features, especially genomic environmental features, during evolution. Therefore, we only chose Alu pairs into our test data set that share similar characteristics with the 219 CNV-Alu pairs, including internal size, pair orientation, and covering at least one exon. We have obtained evidence to support the contention that the 219 pairs can capture as least some of the features enriched in AAMR events given the similar microhomology enrichment pattern with both a smaller compilation study and a human–chimpanzee comparative study; as shown in Figure 3D,E. This approach was not a whole-genome level analysis and did not cover intergenic or intronic only events, which could cause diseases by affecting transcription modifiers, and ignores AAMRs that are larger in size (>250 kb).
Of note, when we collected the CNV-Alu pairs from the literature, we retrieved 219 Del-Alu pairs but only nine Dup-Alu pairs. To minimize the potential impact of a mixture of deletion and duplication in feature preference analyses, we recruited only the deletions in the training data set but tested both in the experimental validation step. As a result, we successfully mapped the breakpoint junctions of six Alu/Alu-mediated duplications and made a correct prediction for four events (Supplemental Table S4). Although this performance (66.7% accuracy, four of six) is not comparable to the 100% accuracy in predicting Del-Alu pairs, the prediction could still be helpful considering only ∼17.6% of Alu pairs were predicted as potential CNV-Alu pairs.
We lack a true-negative data set and have a small training data set from the literature and therefore could not precisely test the overall performance of our score, especially for interpreting the genes with a lower score and whether that implied genomic stability, or absence of instability by AAMR. Instead, we performed an indirect analysis to provide a brief thread using aCGH data, which are of low resolution compared with NGS data. The association analysis between the risk scores and susceptible AAMR CNVs in the CMA database suggests a likely good performance of the risk score in predicting both potential hotspot and nonhotspot genes. Nevertheless, the pattern is not perfect, suggesting that some high-scoring genes have zero susceptible AAMR CNVs, and some low-scoring genes have counts greater than zero.
Our results could also improve the understanding of AAMR mechanisms. AAMR events result in the formation of a chimeric Alu element at the breakpoint junction; therefore, imprecise repair mechanisms, e.g., nonhomologous end joining (NHEJ), are less likely (Inoue et al. 2002; Lieber et al. 2003). The minimum requirement for efficient homologous recombination is thought to be ∼300–500 bp (Waldman and Liskay 1988; Metzenberg et al. 1991; Reiter et al. 1998; Gu et al. 2008), which is longer than the length of an Alu element. PRDM9 targeting sites are associated with ∼40% of recombination hotspots (Myers et al. 2008; Webb et al. 2008). We did not observe an enrichment of these sites surrounding CNV-Alus. Eighteen out of 219 AAMR loci have no PRDM9 binding motif present surrounding either Alu involved in the rearrangement (Supplemental Fig. S5). SSA was also implicated as a potential mechanism underling AAMR events (Morales et al. 2015), in which double-strand breaks (DSBs) could be repaired by end resection and RAD52-mediated strand annealing (Bhargava et al. 2016). Efficient SSA requires at least 15-bp-long microhomology and is inhibited by sequence divergence (Villarreal et al. 2012). However, microhomologies that are <15 bp were found in 82 out of the 219 AAMRs. Therefore, neither NAHR nor SSA can explain all of the reported AAMR events alone.
Replication-based mechanisms can utilize iterative TSs between Alu elements to generate CGRs (Gu et al. 2015; Liu et al. 2017). The microhomologies present at the breakpoint junctions of our AAMR events are <50 bp. Replication-based mechanisms were preferred when the homologies at junctions are <150 bp in yeast (Mehta et al. 2017). Furthermore, in a yeast model of seDSB repair via TS during BIR, Alu elements involved in CNV at the human SPAST locus were effective at mediating TSs (Mayle et al. 2015); these results were extended in this work (Fig. 3F,G). After introducing a DSB in mammalian cell lines, MMEJ was the predominant pathway to mediate the repair between two heterologous Alu elements (Elliott et al. 2005; Morales et al. 2015); this also results in an intact and full-length chimeric Alu. We hypothesize that both MMBIR and MMEJ could contribute to AAMR events, but they might be utilized to repair different DNA DSBs. It appears that seDSBs, which can result from a collapsed fork generated during replication through a nick, may be much more common than double-ended DSBs, indicating a potentially more universal use of MMBIR. In our study, we cannot completely exclude the possibility of a cooperative model with multiple mechanisms involved in AAMR, which could be potentially dependent on the type of damage (e.g., seDSB, DSB, or single-strand break), cell fate, and sequence divergence. Future studies could experimentally attempt to elucidate which DNA repair pathways are responsible for generating the AAMR events observed in human and which are predominant.
We show the power of our model to predict genes susceptible to AAMR-associated genomic instability by correlating our risk score with the CNV frequency in a database of clinical array results. These data further underscore the importance of Alu elements in gene and genome evolution and in mediating human disease and point to a potentially underappreciated source of CNVs, particularly those resulting in exonic deletions. Such information may help elucidate novel disease–gene associations, assist molecular diagnosis, and reveal further insights into genomic instability and human gene and genome evolution.
Methods
Building a positive training data set with CNV-Alu pairs
We collected 219 CNV-Alu pairs (218 from the literature and one currently unpublished) (Supplemental Table S1). Inclusion criteria included that all breakpoint loci were determined at nucleotide-level resolution in the original studies; microhomologies were observed at the breakpoints, and resultant Alus are intact. We collated the coordinates of each Alu pair and each microhomology in the GRCh19/hg19 assembly. We downloaded the “Repeating Elements by RepeatMasker” track at the UCSC Genome Browser, which was last updated in April 2009 and built in GRCh37/hg19, with which the information of subfamily and orientation for each Alu element was annotated.
A TS assay performed in S. cerevisiae
We previously generated a derivative of the yeast W303 strain with two Alu elements from the SPAST locus, AluSx1 (Chr 2: 32378388–32378684, GRCh37/hg19) and AluSp (Chr 2: 32381110–32381405, GRCh37/hg19), that are known to mediate an AAMR event (Boone et al. 2014; Mayle et al. 2015). Colonies were screened on 5-FOA plates, and 74 Alu-mediated deletion events were determined by PCR and sequencing. Details of the assay can be found in work by Mayle et al. (Nielsen et al. 2009; Mayle et al. 2015). Here, we further mapped 429 breakpoint junctions from colonies with the same construct to determine the minimum number of events required to reveal a consistent and persistent frequency distribution pattern of the breakpoint junctions. A second strain was constructed with the same AluSx1 paired with an AluY (Chr 2: 32403014–32403315, GRCh37/hg19). We mapped 114 Alu-mediated deletions in this construct. We aligned each CNV-Alu sequence with a consensus Alu utilizing EMBOSS Water, an online tool based on the Smith–Waterman algorithm (McWilliam et al. 2013). The position of each microhomology on the consensus sequence was manually curated. The frequency that each nucleotide is involved in the microhomology was calculated, and the distribution was plotted as histogram using an R script (R Core Team 2016).
Analyzing features of Alu elements
The element length and GC percentage were analyzed using Biostrings package in R (http://bioconductor.org/packages/Biostrings/). We calculated the poly(A) tail length from the 3′ end to the 5′ end of each Alu in + orientation (from 5′ end to 3′ end for Alu in −) and stopped counting when two continuous nonadenines were read that are not followed by at least five continuous adenines. The PWM for the A Box and B Box were developed from active Alu element sequences (Bennett et al. 2008). Each Alu sequence was searched for the PWM pattern using the matchPWM function of the Biostrings package, and the maximum PWM matching score was returned. The PRDM9 binding motif was queried in 500 bp upstream of and downstream from each Alu by matching a previously developed PWM (Campbell et al. 2014). The maximum matching score (>0.4) and the count of matches >0.85 were both determined. We tested the alignment performance using global alignment with default penalties of Biostrings package. We characterized the Alu density by calculating the percentage of Alu sequences in the ±60 kb surrounding each Alu element. The replication timing data were generated by the McCarroll group (Koren et al. 2012) and converted to hg19 coordinates using liftOver (Hinrichs et al. 2006). We assigned the replication timing value for each Alu element by choosing the closest peak within ±2 kb. The methylation level was estimated by both the greatest signal value and the count of methylated bases as determined in the DNA methylation track from ENCODE in H1 hESC cell line (Meissner et al. 2008).
Feature selection
The Monte Carlo simulation method was utilized to generate control data sets for analyzing both individual and genomic features. As shown in Supplemental Figure S4A, 1000 Ctrl-Alu pairs in the same orientation (either plus or minus) were randomly selected within ±57 kb of each CNV-Alu pair as one local control set. We generated 219 local control sets for analyzing individual features. Genomic control sets were determined by first selecting the Alu pairs across the whole human genome satisfying three criteria as shown in Supplemental Figure S4B: (1) same orientation, (2) at least one exon would be deleted, and (3) the Alu elements are <250 kb apart. In total, 78,291,946 Alu pairs were selected. Next, for each CNV-Alu pair, 1000 Alu pairs with the same distance size (difference <1 kb) between them were randomly chosen from the approximately 78 million pairs as one genomic control set. By using this approach, we developed 192 genomic control sets; 27 of the CNV-Alu pairs were not utilized for determining genomic features, as they were >250 kb apart.
We calculated scores for each CNV-Alu element and control elements and then determined for each pair of Alu sequences the minimum, mean, and maximum value of each feature. We also calculated the absolute difference for genomic features. For every pairwise feature, we plotted the control sets in boxplots in an order of increasing median values and labeled the values of CNV-Alu pairs as red dots. The P-value for each locus was defined as
where f was a pairwise value of one feature. We integrated the P-values of the same pairwise feature from different loci by calculating the geometric mean of P-values across different loci. We selected the pairwise features that could distinguish CNV-Alu pairs and the corresponding control pairs by P-value. Calculating pairwise alignment of the 78 million pairs is computationally intensive; therefore, we used the BlueGene supercomputer at Rice University.
Predicting hotspot genes for AAMR
We predicted CNV-Alu pairs by performing QDA using the MASS package in R (Venables and Ripley 2002). We used a prior probability of 30% of all Alu pairs being capable of mediating CNVs, as the 0.3 prior settings gave the highest prediction sensitivity as shown in Supplemental Figure S6. We further evaluated the potential impact of feature codependency by first choosing Alu pairs that are most likely to be non CNV-Alu pairs given the lowest posterior value. The data set consists of 192 CNV-Alu pairs and 448 controls that fit the 0.3 prior setting in the QDA model. We then trained a series of models with all selected features as well as removing one feature at a time. We evaluated the performance of each model using the error rate from the 10-fold cross validation.
To predict loci-level AAMR hotspots, we built a linear regression model by utilizing the lm function in the R stats package (R Core Team 2016) as follows:
where y is the number of CNV-Alu pairs with experimental evidence for each gene, and n is the number of predicted CNV-Alu pairs that overlap with the same gene. We next applied this model to assign a risk score of AAMR events for 12,074 genes, as well as 23,637 RefSeq genes with available tested Alu pairs. To answer whether gene size has an impact on the gene-level prediction, we build two additional models as follows:
where m is gene size. We compared the fit of model2 to model1 with a χ2 test and showed the results of model3 in Supplemental Table S3.
We developed the AluAluCNVpredictor using R package Shiny (http://CRAN.R-project.org/package=shiny) for querying the prediction results in both hg19 and hg38. We remapped the coordinates of predicted CNV-Alu pairs to GRCh38/hg38 using R package rtracklayer (Lawrence et al. 2009). We cross-referenced the coordinates after liftOver with the hg38 version of RepeatMasker track at the UCSC Genome Browser, which was updated in January 2014, and only used the overlapping Alu pairs (99.6%) that remained.
Verifying hotspot genes by correlating with a CMA database
The CMA database at Baylor Genetics includes the genome-wide custom-designed oligonucleotide arrays from approximately 54,000 individuals. We cross-referenced the gene list of 329 AAMR hotspot OMIM genes with the CMA database. We selected samples that have one or more CNV intersecting hotspot genes and chose genes with three or more samples available. We acquired 89 samples from Baylor Genetics. This study was approved by the institutional review board for human subject research at BCM (IRB No. H-37586). The DNA samples were de-identified in our analyses. We verified the CNVs with a custom-designed 8 × 60K aCGH chip with ∼90 bp per probe coverage and mapped the breakpoint to nucleotide level by long-range PCR (see Supplemental Table S4) and Sanger sequencing.
We cross-referenced each CMA CNV with predicted AAMR Alu pairs using BEDTools (Quinlan and Hall 2010) to ascertain a data set of potential AAMR events. We calculated an odds ratio to estimate the enrichment of genes with variable risk score in each subgroup based on the CMA data as follows:
where i is determined by the tertiles of the predictive risk score—genes with a score ≤0.485, genes with a score >0.485 but ≤0.643, and genes with a score >0.643; j is the count of genes that have potential AAMR CNVs of zero, one, or more than one; and N is the total number of genes (4517); this represents genes that are well targeted but lack a putative AAMR CNV or have at least one potential CNV.
Statistics
The statistical analysis was performed with R version 3.2.5. We calculated the frequency expectation of every Alu family composition by using the combination value of Alus from the specific family composition as recorded in RepeatMasker. The difference in this relative frequency of CNV-Alus and the expectation was tested using a one-tailed binominal test. The one-tailed t-test was applied to compare the GC percentage of microhomology (n = 219) and the CNV-Alu element (n = 438). We selected the features for training the Alu pair prediction model using a Monte Carlo approach (see details in Methods described above). We tested the linear association between the number of experimentally verified CNV-Alu pairs per gene and different parameters (e.g., Alu density versus all the chosen features) by linear regression using lm function in R. To compare the performance of these models, we performed a χ2 test. The enrichment of low-scoring genes was compared with genes with a higher score in each subset based on the CMA data by binomial test. We applied both Fisher's exact test and a χ2 test to test of independence between the risk score tertiles and the potentially AAMR CNV count classes.
Data access
The microarray data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE100590. The Sanger traces from this study have been submitted to the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP130889. The source script of AluAluCNVpredictor is available as Supplemental File S1 and can also be accessed at https://github.com/BCM-Lupskilab/AluAluCNVpredictor.
Competing interest statement
J.R.L. has stock ownership in 23andMe and Lasergen, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis and clinical exome sequencing offered in the Baylor Genetics Laboratory (http://bmgl.com/).
Supplementary Material
Acknowledgments
We thank Claudia M.B. Carvalho and Jennifer E. Posey for critical review of the manuscript. This work was funded in part by the US National Human Genome Research Institute (NHGRI)/National Heart Lung and Blood Institute (NHLBI) grant UM1HG006542 to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG), National Institute of Neurological Disorders and Stroke (NINDS) grants R01 NS058529 and R35 NS105078, and National Institute of General Medical Sciences (NIGMS) grant GM106373 to J.R.L. and GM080600 to G.I. The work was further supported by NIGMS grant K99GM120453 and an HHMI Damon Runyon Cancer Foundation fellowship DRG-2155 to C.R.B. and by NINDS grant F31 NS083159 to I.M.C.
Author contributions: X.S., C.R.B., G.I., C.A.S., and J.R.L. designed the study; C.A.S. and J.R.L. supervised all aspects of the study; and X.S., C.R.B., and J.R.L. wrote the manuscript. X.S., I.M.C., and C.A.S. performed the bioinformatics analyses. X.S., R.D., and C.R.B. conducted the wet-bench experiments. S.G., A.M.B., and P.S. contributed to CMA data generation and DNA sample collection. C.A.S. and Z.C.A. supervised the statistical and computational analyses. All contributing coauthors have read, edited, and agreed to the contents of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.229401.117.
References
- Bailey JA, Liu G, Eichler EE. 2003. An Alu transposition model for the origin and expansion of human segmental duplications. Am J Hum Genet 73: 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379. [DOI] [PubMed] [Google Scholar]
- Beck CR, Garcia-Perez JL, Badge RM, Moran JV. 2011. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet 12: 187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EA, Keller H, Mills RE, Schmidt S, Moran JV, Weichenrieder O, Devine SE. 2008. Active Alu retrotransposons in the human genome. Genome Res 18: 1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Lam KW, Sarbajna S, Odenthal-Hesse L, May CA, Jeffreys AJ. 2010. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet 42: 859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava R, Onyango DO, Stark JM. 2016. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 32: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Liu P, Zhang F, Carvalho CM, Towne CF, Batish SD, Lupski JR. 2011. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet Med 13: 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Campbell IM, Baggett BC, Soens ZT, Rao MM, Hixson PM, Patel A, Bi W, Cheung SW, Lalani SR, et al. 2013. Deletions of recessive disease genes: CNV contribution to carrier states and disease-causing alleles. Genome Res 23: 1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Yuan B, Campbell IM, Scull JC, Withers MA, Baggett BC, Beck CR, Shaw CJ, Stankiewicz P, Moretti P, et al. 2014. The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. Am J Hum Genet 95: 143–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Gambin T, Dittwald P, Beck CR, Shuvarikov A, Hixson P, Patel A, Gambin A, Shaw CA, Rosenfeld JA, et al. 2014. Human endogenous retroviral elements promote genome instability via non-allelic homologous recombination. BMC Biol 12: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Harrell TM, McMillin MJ, Wiszniewski W, Gambin T, et al. 2015. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet 97: 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. 1999. Alu repeats and human disease. Mol Genet Metab 67: 183–193. [DOI] [PubMed] [Google Scholar]
- Deininger PL, Moran JV, Batzer MA, Kazazian HH Jr. 2003. Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 13: 651–658. [DOI] [PubMed] [Google Scholar]
- Dittwald P, Gambin T, Szafranski P, Li J, Amato S, Divon MY, Rodriguez Rojas LX, Elton LE, Scott DA, Schaaf CP, et al. 2013. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res 23: 1395–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Jasin M. 2005. Chromosomal translocation mechanisms at intronic Alu elements in mammalian cells. Mol Cell 17: 885–894. [DOI] [PubMed] [Google Scholar]
- Flynn EK, Kamat A, Lach FP, Donovan FX, Kimble DC, Narisu N, Sanborn E, Boulad F, Davies SM, Gillio AP III, et al. 2014. Comprehensive analysis of pathogenic deletion variants in Fanconi anemia genes. Hum Mutat 35: 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke G, Bausch B, Hoffmann MM, Cybulla M, Wilhelm C, Kohlhase J, Scherer G, Neumann HP. 2009. Alu-Alu recombination underlies the vast majority of large VHL germline deletions: molecular characterization and genotype-phenotype correlations in VHL patients. Hum Mutat 30: 776–786. [DOI] [PubMed] [Google Scholar]
- Goodwin S, McPherson JD, McCombie WR. 2016. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17: 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. 2008. Mechanisms for human genomic rearrangements. Pathogenetics 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Yuan B, Campbell IM, Beck CR, Carvalho CM, Nagamani SC, Erez A, Patel A, Bacino CA, Shaw CA, et al. 2015. Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum Mol Genet 24: 4061–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Lee J, Meyer TJ, Wang J, Sen SK, Srikanta D, Liang P, Batzer MA. 2007. Alu recombination-mediated structural deletions in the chimpanzee genome. PLoS Genet 3: 1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel T, Yoon WH, Garone C, Gu S, Coban-Akdemir Z, Eldomery MK, Posey JE, Jhangiani SN, Rosenfeld JA, Cho MT, et al. 2016. Recurrent de novo and biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am J Hum Genet 99: 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimoto K, Maeda T, Okada J, Ohtsuka Y, Sasaki K, Hirose A, Nomiyama M, Takayanagi T, Fukuzawa R, Yatsuki H, et al. 2013. Homozygous deletion of DIS3L2 exon 9 due to non-allelic homologous recombination between LINE-1s in a Japanese patient with Perlman syndrome. Eur J Hum Genet 21: 1316–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. 2006. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res 34: D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck CM, Rinehart FP, Schmid CW. 1979. A ubiquitous family of repeated DNA sequences in the human genome. J Mol Biol 132: 289–306. [DOI] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Thurston VC, Clarke JT, Yoneyama A, Rosenbarker L, Bird TD, Hodes ME, Shaffer LG, Lupski JR. 2002. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet 71: 838–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel MK, Walker JA, Hotard AB, Ranck MC, Fontenot CC, Storer J, Stewart C, Marth GT, Genomes C, Batzer MA. 2015. Sequence analysis and characterization of active human Alu subfamilies based on the 1000 genomes pilot project. Genome Biol Evol 7: 2608–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA. 2012. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet 91: 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper RP, Vissers LE, Venkatachalam R, Bodmer D, Hoenselaar E, Goossens M, Haufe A, Kamping E, Niessen RC, Hogervorst FB, et al. 2011. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat 32: 407–414. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, Carey V. 2009. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25: 1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman MA, Russell DW, Goldstein JL, Brown MS. 1987. Alu-Alu recombination deletes splice acceptor sites and produces secreted low density lipoprotein receptor in a subject with familial hypercholesterolemia. J Biol Chem 262: 3354–3361. [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K. 2003. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4: 712–720. [DOI] [PubMed] [Google Scholar]
- Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. 2006. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet 79: 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Yuan B, Carvalho CM, Wuster A, Walter K, Zhang L, Gambin T, Chong Z, Campbell IM, Coban Akdemir Z, et al. 2017. An organismal CNV mutator phenotype restricted to early human development. Cell 168: 830–842.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. 1998. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 14: 417–422. [DOI] [PubMed] [Google Scholar]
- Lupski JR. 2004. Hotspots of homologous recombination in the human genome: Not all homologous sequences are equal. Genome Biol 5: 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. 2014. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 42: D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G. 2015. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349: 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. 2013. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res 41: W597–W600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Beach A, Haber JE. 2017. Homology requirements and competition between gene conversion and break-induced replication during double-strand break repair. Mol Cell 65: 515–526.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzenberg AB, Wurzer G, Huisman TH, Smithies O. 1991. Homology requirements for unequal crossing over in humans. Genetics 128: 143–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales ME, White TB, Streva VA, DeFreece CB, Hedges DJ, Deininger PL. 2015. The contribution of Alu elements to mutagenic DNA double-strand break repair. PLoS Genet 11: e1005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SR, McCarroll SA. 2006. New insights into the biological basis of genomic disorders. Nat Genet 38: 1363–1364. [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnelly P, McVean G. 2008. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet 40: 1124–1129. [DOI] [PubMed] [Google Scholar]
- Nielsen I, Bentsen IB, Lisby M, Hansen S, Mundbjerg K, Andersen AH, Bjergbaek L. 2009. A Flp-nick system to study repair of a single protein-bound nick in vivo. Nat Methods 6: 753–757. [DOI] [PubMed] [Google Scholar]
- Quemener S, Chen JM, Chuzhanova N, Benech C, Casals T, Macek M Jr, Bienvenu T, McDevitt T, Farrell PM, Loumi O, et al. 2010. Complete ascertainment of intragenic copy number mutations (CNMs) in the CFTR gene and its implications for CNM formation at other autosomal loci. Hum Mutat 31: 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quentin Y. 1992. Fusion of a free left Alu monomer and a free right Alu monomer at the origin of the Alu family in the primate genomes. Nucleic Acids Res 20: 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Reiter LT, Hastings PJ, Nelis E, De Jonghe P, Van Broeckhoven C, Lupski JR. 1998. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am J Hum Genet 62: 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repping S, Skaletsky H, Lange J, Silber S, Van Der Veen F, Oates RD, Page DC, Rozen S. 2002. Recombination between palindromes P5 and P1 on the human Y chromosome causes massive deletions and spermatogenic failure. Am J Hum Genet 71: 906–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudiger NS, Gregersen N, Kielland-Brandt MC. 1995. One short well conserved region of Alu-sequences is involved in human gene rearrangements and has homology with prokaryotic chi. Nucleic Acids Res 23: 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CW, Jelinek WR. 1982. The Alu family of dispersed repetitive sequences. Science 216: 1065–1070. [DOI] [PubMed] [Google Scholar]
- Sen SK, Han K, Wang J, Lee J, Wang H, Callinan PA, Dyer M, Cordaux R, Liang P, Batzer MA. 2006. Human genomic deletions mediated by recombination between Alu elements. Am J Hum Genet 79: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. 2005. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77: 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. 2005. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet 116: 1–7. [DOI] [PubMed] [Google Scholar]
- Shen MR, Batzer MA, Deininger PL. 1991. Evolution of the master Alu gene(s). J Mol Evol 33: 311–320. [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org.
- Startek M, Szafranski P, Gambin T, Campbell IM, Hixson P, Shaw CA, Stankiewicz P, Gambin A. 2015. Genome-wide analyses of LINE–LINE-mediated nonallelic homologous recombination. Nucleic Acids Res 43: 2188–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Skaletsky H, Rozen S, Gromoll J, Nieschlag E, Oates R, Page DC. 2000. Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum Mol Genet 9: 2291–2296. [DOI] [PubMed] [Google Scholar]
- Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, Godiwala N, Yatsenko SA, Sebastian J, Madan-Khetarpal S, et al. 2016. Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Hum Genet 135: 569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL. 2011. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD. 2002. Modern applied statistics with S, 4th ed Springer, New York. [Google Scholar]
- Villarreal DD, Lee K, Deem A, Shim EY, Malkova A, Lee SE. 2012. Microhomology directs diverse DNA break repair pathways and chromosomal translocations. PLoS Genet 8: e1003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, Bhatt SS, Janssen IM, Xia Z, Lalani SR, Pfundt R, Derwinska K, de Vries BB, Gilissen C, Hoischen A, et al. 2009. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum Mol Genet 18: 3579–3593. [DOI] [PubMed] [Google Scholar]
- Waldman AS, Liskay RM. 1988. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol Cell Biol 8: 5350–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AJ, Berg IL, Jeffreys A. 2008. Sperm cross-over activity in regions of the human genome showing extreme breakdown of marker association. Proc Natl Acad Sci 105: 10471–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876. [DOI] [PubMed] [Google Scholar]
- White JJ, Mazzeu JF, Coban-Akdemir Z, Bayram Y, Bahrambeigi V, Hoischen A, van Bon BWM, Gezdirici A, Gulec EY, Ramond F, et al. 2018. WNT signaling perturbations underlie the genetic heterogeneity of Robinow syndrome. Am J Hum Genet 102: 27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.