

Abstract
Contrary to other anticancer targets, topoisomerase I (TOP1) is targeted by only one chemical class of FDA-approved drugs: topotecan and irinotecan, the derivatives of the plant alkaloid, camptothecin. The indenoisoquinolines LMP400, LMP744 and LMP776 are novel non-camptothecin TOP1 inhibitors in clinical trial, which overcome the limitations of camptothecins. To further improve metabolic stability, their methoxy groups have been replaced by a fluorine, as in the fluoroindenoisoquinolines NSC 781517 (LMP517), NSC 779135 (LMP135) and NSC 779134 (LMP134). We tested the induction and stability of TOP1 cleavage complexes (TOP1cc), and the induction and persistence of DNA damage measured by histone H2AX phosphorylation (γH2AX) compared to their parent compounds LMP744 and LMP776 in leukemia CCRF-CEM and colon carcinoma HCT116 cells. The fluoroindenoisoquinolines induced TOP1cc and γH2AX at nanomolar concentrations, and at higher levels than the parent indenoisoquinolines. The fluoroindenoisoquinoline LMP135 showed greater antitumor activity than topotecan in small cell lung cancer cell H82 xenografts. It was also more potent than topotecan in the NCI-60 cancer cell line panel. Bioinformatics tools (http://discover.nci.nih.gov/cellminercdb) were used to investigate: 1/ the correlation of fluoroindenoisoquinolines activity with other drugs, and 2/ genomic determinants of response in the NCI-60. The activity of the fluoroindenoisoquinolines was most correlated with camptothecin derivatives and the parent indenoisoquinolines, consistent with TOP1 targeting. Genomic analyses and activity assays in CCRF-CEM SLFN11-deleted cells showed that SLFN11 expression is a dominant determinant of response to LMP135. This study shows the potential value of the fluoroindenoisoquinolines for further development as novel anticancer agents targeting TOP1.
Keywords: chemotherapy, DNA targeted drugs, DNA repair, topoisomerase inhibitors, TDP1
Introduction
Topoisomerases relax DNA supercoils and resolve DNA knots and intertwining (catenanes) generated during DNA replication, transcription, repair and chromatin remodeling (1). To do so, they induce transient breaks (topoisomerase cleavage complexes; TOPcc) in the DNA backbone that allow one of the strands to rotate around the other (TOP1) or the passage of another DNA segment through the break (TOP2). TOP1 enzymes (TOP1 and TOP1MT) induces single-strand breaks and TOP2 enzymes (TOP2α and TOP2β) induces double-strand breaks (1). Immediately after DNA relaxation or strand passage, topoisomerases ligate the broken DNA, restoring the DNA sequence and ensuring DNA integrity. Among DNA-targeted therapies, topotecan and irinotecan are widely used and effective in a broad range of tumors. They selectively trap TOP1 cleavage complexes (TOP1cc), blocking the religation of the TOP1-linked DNA single-strand breaks. Collisions with DNA replication forks convert the single-strand breaks into double-strand break lesions that ultimately lead to cell death and anticancer activity (2,3).
The only chemical class of FDA-approved TOP1 inhibitors are irinotecan and topotecan, which are water-soluble derivatives of the plant alkaloid, camptothecin (CPT). Irinotecan is widely prescribed for colorectal cancers, and topotecan for ovarian, cervical and small cell lung cancers. Yet, CPT derivatives suffer from well-documented limitations: 1/ chemical instability of their E-ring, leading to the inactive carboxylate derivatives that bind tightly to serum albumin (4-6); 2/rapid elimination due to short plasma half-life; 3/ highly reversible DNA damage due to their rapid dissociation from the TOP1cc (5,7); 4/ active efflux from multidrug-resistant cancer cells expressing ABC transporters (ABCG2 and ABCB1); 5/ gastrointestinal toxicity (including severe diarrheas for irinotecan); and 6/ dose-limiting bone marrow toxicity (6,8,9). Based on phenotypic COMPARE analysis across the 60 cancer cell lines of the National Cancer Institute (NCI-60) (10-13), we identified a novel chemical class of TOP1 inhibitors: the indenoisoquinolines (10,14). Lead optimization led to highly potent and specific TOP1cc-targeted indenoisoquinolines that overcome most of the limitations of the camptothecins. Indeed, the indenoisoquinolines are chemically stable, form less reversible TOP1cc, have long plasma half-life, are not substrates for the ABC drug efflux transporters and do not produce diarrheas (6,9,15).
Three indenoisoquinolines, LMP400 (indotecan, NSC724998), LMP776 (indimitecan, NSC725776) and LMP744 (MJ-III-65, NSC706744) have entered clinical trials. LMP400 recently completed phase 1 with demonstrable target engagement (15), and is now poised for Phase 2 clinical trials. LMP776 is finishing Phase 1 and LMP744 is beginning Phase 1 clinical trials. Because the methoxy groups of LMP400 and LMP776 and LMP744 (Fig. 1) are likely to be metabolized in vivo by O-dealkylation catalyzed chiefly by hepatic cytochrome P450 enzymes (16-18), a common metabolic process that, for example, plays a significant role in human metabolism of etoposide and teniposide (19), the aim of the present study was to determine whether replacing the methoxy groups by a fluorine would generate novel fluoroindenoisoquinolines retaining potent TOP1cc-targeting and antiproliferative activity in cells, thereby making them worthy of development as anticancer agents. Our recent studies have shown that addition of a fluorine in position 3 yields fluoroindenoisoquinolines that retain potent activity on TOP1cc in biochemical assays with recombinant human TOP1 (17,18,20).
Figure 1. Fluoroindenoisoquinolines are potent inhibitors of human TOP1.
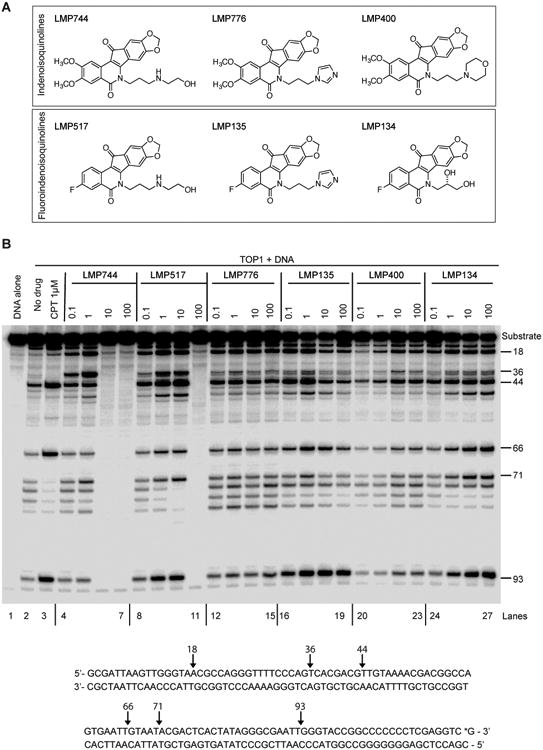
A, Structures of the clinical first generation indenoisoquinolines (LMP744, LMP776 and LMP400) and of the three fluoroindenoisoquinolines (LMP517, LMP135 and LMP134). The methoxy groups of LMP744 and LMP776 are replaced by a fluorine substituent in LMP517 and LMP135, respectively. B, Representative gel showing TOP1cc-associated DNA breaks induced by the indicated compounds. The substrate DNA (3′ end-labeled PvuII/HindIII fragment of pBluescript SK (–) phagemid DNA (pSK), lane 1) was reacted with recombinant TOP1 in the absence of drug (lane 2) or in the presence of the indicated concentrations (micromolar) of CPT, LMP400 (lanes 20 to 23), LMP744 (lanes 4 to 7), LMP776 (lanes 12 to 15), LMP517 (lanes 8 to 11), LMP134 (lanes 24 to 27) and LMP135 (lanes 16 to 19). Reactions were performed at 30°C for 20 min and stopped by adding 0.5% SDS. DNA fragments were separated in 16% denaturing polyacrylamide gels. Cleavage sites are indicated on the right. The sequence of the DNA substrate and TOP1cc sites are shown at the bottom. The asterisk indicates the position of the 32P-end-labelling.
In this study, three fluorinated indenoisoquinolines, NSC 781517 (LMP517), NSC 779135 (LMP135) and NSC 779134 (LMP134) (Fig. 1A) were tested on TOP1cc formation and stability in biochemical and cellular assays. In addition, their abilities to induce stable DNA damage, cell death, antiproliferative activity, and antitumor activity in xenograft model were examined. Two of the fluoroindenoisoquinolines were directly compared with their parent derivatives (LMP517 with LMP744, and LMP135 with LMP776) (see Fig. 1A). The cellular response to LMP135, the most potent fluoroindenoisoquinoline, was found to be dominantly driven by the expression of Schlafen 11 (SLFN11), a putative DNA/RNA helicase, which is a strong determinant of response to TOP1 and PARP inhibitors and widely used anticancer DNA damaging agents (21-28).
Materials and Methods
Cell lines and reagents
Human leukemia CCRF-CEM and colon carcinoma HCT116 cell lines were cultured in RPMI medium (Gibco) supplemented with 10% FBS. HCT116 cell lines were provided by the NCI-Frederick cancer Division of Cancer Treatment and Diagnosis tumor/cell line repository. CCRF-CEM cells are were obtained from the National Cancer Institute Developmental Therapeutics Program (Frederick, MD). The SLFN11-deleted CCRF-CEM cells by CRISPR/Cas9 have been recently described (24,28). H82 cells were obtained from the NCI repository. All cell lines were kept for 45 days maximum after thawing and tested for mycoplasma with MycoAlert™ Mycoplasma Detection Kit (Lonza). CPT, topotecan, LMP400, LMP744 and LMP776 were provided by the NCI Drug Developmental Therapeutics Program (DTP). LMP517, LMP135 and LMP134 were synthesized in the Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University (20,29).
In vitro TOP1cc trapping
A 3′-[32P]-labeled 117-bp DNA substrate oligonucleotide was prepared as previously described (30). Radiolabeled DNA was incubated with recombinant human TOP1 in 20 μL reaction buffer [10 mM Tris-HCl, pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at 25°C for 20 min in the presence of the indicated drug concentrations. Reactions were terminated by adding sodium dodecyl sulfate (SDS) (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of reaction mixtures were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a PhosphorImager and Image Quant software (Molecular Dynamics).
DNA-Protein Cross-Links (DPC)
Alkaline elution was used to assess cellular TOP1cc by measuring DPC as described (4,31-33). Before alkaline elution and drug treatments, genomic DNA of CCRF-CEM cells was radiolabeled with 0.02 μCi/ml [3H]-thymidine for one to two doubling times at 37°C and chased in non-radioactive medium overnight. Cells were treated with the indicated concentrations of LMP compounds or CPT for 1 h before scraping and alkaline elution. For reversal experiments, the cells were cultured in drug-free medium for the indicated times. Radioactivity in all fractions was measured with a liquid scintillation analyzer (PerkinElmer Life and Analytical Sciences), and DPC frequency, which reflects TOP1cc was determined as published (4,31-33).
ICE bioassay
TOP1cc were detected using the immunocomplex of enzyme (ICE) bioassay (34-36). In brief, treated or untreated CCRF-CEM cells were pelleted and immediately lysed in 1% sarkosyl. After homogenization with a Dounce homogenizer, cell lysates were gently layered on CsCl step gradients and centrifuged at 165,000g for 20 h at 20°C. Half-milliliter fractions were collected, diluted with an equal volume of 25 mM sodium phosphate buffer, pH 6.5, and applied to Immobilon-P membranes (Millipore Corporation, Billerica, MA) through a slot-blot vacuum manifold as described (36). TOP1cc were detected using the C21 TOP1 monoclonal antibody (BD Pharmingen cat.556597).
Cellular DNA damage measured by γH2AX staining
HCT116 cells were plated at 50,000 cells per well in 4-well chamber slides, incubated for 24 h and then treated with drugs. Cells were washed with phosphate buffered saline (PBS), fixed and permeabilized with ethanol absolute overnight at 4°C, blocked for 1 h in PBS-BSA 8% at room temperature (RT), and incubated for 2 h at room temperature with primary antibody. After washing with PBS, cells were incubated with secondary antibody for 1 h at room temperature, washed with PBS and mounted with Vectashield® with DAPI (Vector Laboratories). Images were captured with a confocal microscope (Nikon PCM2000). The primary antibody for histone γH2AX was mouse monoclonal (Millipore # 05-636: anti–γH2AX Ser139 antibody, clone JBW301) and the secondary antibody was a donkey anti-mouse conjugated to Alexa Fluor 568 from Molecular Probes. Signals were quantified using ImageJ program. An area was selected (same size) and the signal was measured in each nucleus, giving the intensity in arbitrary unit.
Cellular viability assays
CCR-CEM and CCRF-CEM SLFN11-KO cells were seeded at 5,000 cells per well in 96-well white plates (#6005680 Perkin Elmer Life Sciences), and exposed to the indicated concentrations of topotecan, LMP517, LMP135 or LMP134 for 72 h in triplicate. Cellular viability was determined using ATPlite 1-step kits (PerkinElmer). ATP levels of untreated cells were defined as 100%. Percent viability was defined as: [(ATP in treated cells) / (ATP in untreated cells)] × 100 (28).
In vivo study in nude mice
Athymic nude mice (nu/nu, female, 20-25g, 8-12-week-old) from Charles River, were transplanted with 5 million H82 human small cell lung cancer cells. When the tumor volume reached between 100 and 125 mm3, the animals were randomized into treatment groups based on tumor volume and body weights using the StudyLog software. Ten mice for topotecan and vehicle arm and 6 mice for LMP135 were used. The animals were treated with either topotecan (1.5 mg/kg) administered intraperitoneally (i.p.) or with LMP135 (20 mg/kg) administered by intravenous (i.v.) push via tail vein once a day for 5 consecutive days. LMP135 was dissolved in 10 mM citric acid, 5% dextrose. Topotecan was diluted in sterile water. The three axes (millimeters) of tumors were measured with a caliper to calculate tumor volume. Measurement were made every 3 or 4 days. Maximum allowable weight loss tolerated of 20% was never reached. Mice were euthanized if tumors presented necrosis or exceeded 20 mm in diameter. Animal studies were approved by the Animal Care and Use Committee of the NCI- Frederick, and all animal care was in accordance with institutional guidelines.
Pharmacokinetic study
Blood for pharmacokinetic measurements of each compound were obtained at 5 min, 30 min, 1 hr, 2 hr, and 8 hr post dose via cardiac puncture on euthanized mice and drawn into tubes containing sodium heparin. All animal handling was conducted in accordance with NIH Animal Care and Usage Committee (ACUC) regulations. Plasma was obtained through centrifuging the blood for 10 min at 1500× g, then transferred to cryovials and stored at -80 C until bioanalysis. Each time point was performed in triplicate.
On the day of measurement, 50 μL of thawed plasma was mixed with 3× volume of acetonitrile, mixed to precipitate plasma proteins, and centrifuged. Ten microliters of the resulting supernatant were injected onto a Waters ACQUITY UPLC® BEH C18 column (2.1×50 mm, 1.7 um) and the analytes of interest (LMP134, LMP135, LMP517) were chromatographically separated using a gradient elution. The column eluent was directed into a triple quadrupole mass spectrometer operated in the positive ion mode and the analytes were detected based on their unique mass fragmentation of m/z 384.0 → 309.5 (LMP134), m/z 418.1 → 350.1 (LMP135), and m/z 411.1 → 350.1 (LMP517). The assay had a calibration range of 50-25,000 ng/mL and was validated according to FDA guidelines (https://www.fda.gov/downloads/drugs/guidances/ucm368107.pdf).
Pharmacokinetic parameters were estimated using non-compartmental analysis and the area under the plasma concentration vs time curve (AUC) was estimated using the linear trapezoidal rule with adjustments for destructive sample and the Bailer's method for estimating variance (PMID 3221328; 7724473). The maximum plasma concentration (CMAX) was calculated as the highest average (n=3) plasma concentration. The elimination rate (KEL) was the slope of the line through the terminal natural log-transformed average plasma concentrations; half-life (T1/2) was calculated as natural log 2/KEL. Clearance (CL) was calculated as dose/AUC; volume of distribution was calculated as CL/KEL. All parameter estimates were calculated using Phoenix WinNonlin v7.0 (Certara Pharsight, Cary, NC).
Results
Fluoroindenoisoquinolines efficiently trap TOP1cc in biochemical assays
To determine the potency of the three fluoroindenoisoquinolines (LMP517, LMP135 and LMP134), we performed TOP1cc trapping assays in the presence of recombinant human TOP1 and DNA. All three compounds induced TOP1cc and showed comparable patterns of cleavage sites. Those cleavage patterns were comparable to the first generation indenoisoquinolines LMP400, LMP744 and LMP776 (Fig. 1B). LMP517 and LMP135 were the most potent TOP1cc inducers (Fig. 1B). Like its closest analog LMP744 (14), LMP517 inhibited TOP1cc formation at high concentration of 100 μM, an effect which has been related to the ability of the LMP744 side chain to enable DNA intercalation at high concentration (14). These results demonstrate that derivatives with replacement of the 2,3-dimethoxy groups of LMP744 and LMP776 with a single 3-fluoro substituent, as in LMP135 and LMP517, retain potent TOP1cc trapping activity in vitro.
Fluoroindenoisoquinolines efficiently inhibit cellular TOP1
To assess the trapping of TOP1cc in cell by LMP517, LMP134 and LMP135 in cells, two methods were used: ICE bioassay and alkaline elution (4,31-36). For the ICE bioassay, CCRF-CEM cells and HCT116 cells were treated for 1 h with 1 μM of the three fluoroindenoisoquinolines in parallel with CPT and LMP744. Cell lysates were fractionated by cesium chloride gradient and TOP1cc were measured in the DNA-containing fractions (fraction 5 to 8; Fig. 2A). TOP1cc were detected in the treated but not in the untreated cells, demonstrating that all three fluoroindenoisoquinolines induced cellular TOP1cc.
Figure 2. Cellular TOP1cc induced by the fluoroindenoisoquinolines in human cancer cells.
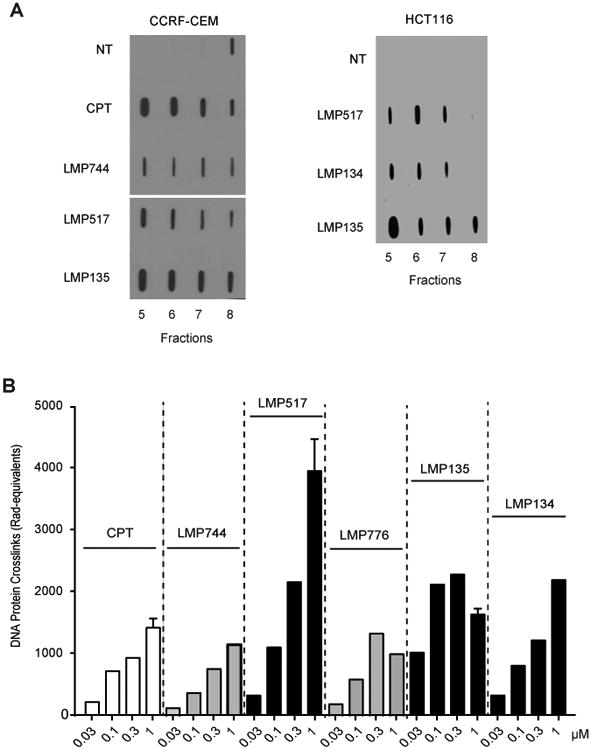
A, TOP1cc detected by ICE bioassay. Human leukemia CCRF-CEM and colon carcinoma HCT116 cells were treated with the indicated drugs (1 μM) for 1 h at 37°C. DNA-containing fractions were blotted and TOP1 was detected using TOP1 C21 monoclonal antibody. B, Quantitative analysis of TOP1cc detected as DNA-protein complexes (DPC) by alkaline elution. Human leukemia CCRF-CEM cells were treated for 1 h at 37°C as indicated.
The alkaline elution assay quantifies TOP1cc as DNA-protein crosslinks (DPC) (4). CCRF-CEM cells were treated for 1 h with a range of drug concentrations of LMP517, LMP134 and LMP135, and in parallel with similar concentrations of CPT, LMP744 (closest analog of LMP517) and LMP776 (closest analog of LMP135). All drugs induced DPC in a concentration-dependent manner (Fig. 2B). LMP517 was the most potent and compared favorably to LMP744. Also, the three fluoroindenoisoquinolines induced 1.5 to 4 times more DPC than CPT, LMP744 and LMP776 at 1 μM (Fig. 2B). Together, the results of the ICE bioassay and alkaline elution experiments demonstrate that the fluoroindenoisoquinolines effectively target TOP1cc at nanomolar concentration in cells.
Fluoroindenoisoquinolines induce cellular DNA damage
Phosphorylation of histone H2AX on serine 139 (γH2AX) is a chromatin modification induced when DNA double-strand breaks are formed following DNA damage (37). Because CPT, topotecan and the indenoisoquinolines LMP400, LMP776 and LMP744 induce this phosphorylation (15,33,38,39), we examined γH2AX signal in cells treated with LMP517, LMP135 or LMP134. Figure 3A shows representative immunofluorescence microscopy images of HCT116 cells treated for 1 h with 1 μM of each of the three fluoroindenoisoquinolines in comparison with topotecan. Time-course experiments with 1 μM drug concentration showed the rapid kinetics of γH2AX signal formation (within 30 min of drug exposure), which indicates the rapid cellular uptake of the fluoroindenoisoquinolines (LMP517, LMP135 and LMP134).
Figure 3. Cellular DNA damage induced by the fluoroindenoisoquinolines.
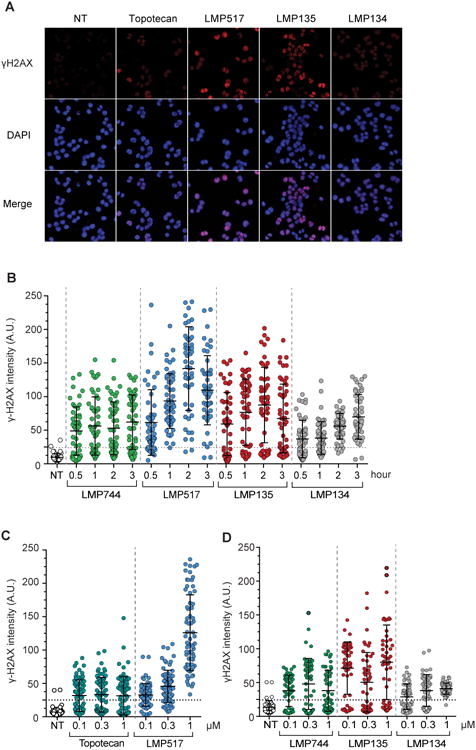
A, Representative immunofluorescence confocal microscopy images. HCT116 cells were treated with the indicated drugs (1 μM for 1 h at 37°C). Following fixation, cells were stained for histone γH2AX and DAPI. B, Quantitative analysis of γH2AX as a function of time. Cells were treated with 1 μM of topotecan, LMP744, LMP517, LMP134 and LMP135 for the indicated times (h). γH2AX signal intensities of 50 cells (each individual cell represented as a dot) were measured by image-J. Same sized area signal was quantified in each nucleus and plotted for each condition. C and D, Quantitative analysis of γH2AX induction as a function of drug concentration. Cells were treated as indicated for 1 h. γH2AX signal intensities of 70 cells were measured by image-J as in panel B. Signals below the horizontal dotted lines are within background signal for untreated cells.
Experiments were also performed to determine drug concentration-dependency. LMP517 was more potent than topotecan at 1 μM (Fig. 3C), and LMP135 was more potent than LMP744 and LMP134 at their lowest concentration (0.1 μM) with respect to γH2AX induction (Fig. 3D). All drugs induce DNA damage at sub-micromolar concentrations.
These results demonstrate that the three fluoroindenoisoquinolines rapidly induce DNA damage response and that LMP517 and LMP135 are superior to the other drugs tested with respect to γH2AX induction.
Persistent TOP1cc and DNA damage in response to the fluoroindenoisoquinolines
Next, we examined the reversal kinetics of the TOP1cc and γH2AX induced by the fluoroindenoisoquinolines using alkaline elution and immunofluorescence microscopy (Fig. 4). CCRF-CEM cells were treated for 1 h with 1 μM CPT, LMP744, LMP776, LMP517, LMP134 or LMP135. After drug removal, cells were grown in fresh drug-free medium for an additional hour. DPC induced by LMP517 and LMP135 remained at 75% and 65% of initial response respectively at 1 h after drug removal (Fig. 4A). By contrast, 2% of the DPC induced by CPT and 15% of the LMP134-induced DPCs remained at 1 h after drug removal (about 50 % for the indenoisoquinolines LMP744 and LMP776).
Figure 4. Persistent DNA damage and TOP1cc in cells treated with the fluoroindenoisoquinolines.
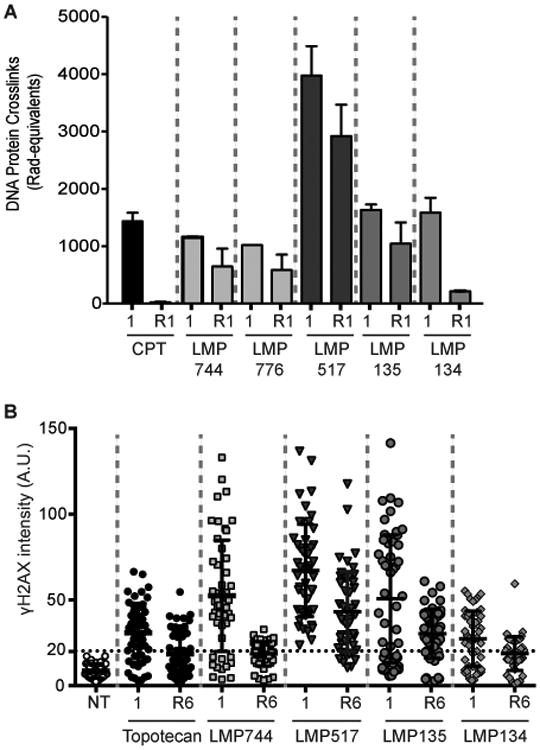
A, Quantitative analysis of TOP1cc after drug removal in CCRF-CEM cells. Cells were treated with 1 μM CPT, LMP744, LMP776, LMP517, LMP134 or LMP135 for 1 h at 37°C. Drugs were removed and cells were grown for another hour in drug-free medium (R1). TOP1cc were determined as DPC by alkaline elution. Percent reversal for each drug is indicated in the text. B, Quantitative analysis of γH2AX persistence 6 h after drug removal. HCT116 were treated for 1 h with 1 μM drug concentration, and either fixed at that point (1 h) or grown for an additional 6 h in drug-free medium (R). γH2AX signal intensities of 50 cells (each individual cell represented as a dot/circle) were measured by image-J. Same sized area signal was quantified in each nucleus. Percent reversal for each drug is indicated in the text. Points below the horizontal dotted lines are γH2AX signals within background signal for untreated cells.
For DNA damage detection using γH2AX, HCT116 cells were treated with 1 μM of drug for 1 h and then grown in fresh drug-free medium for 6 h (Fig. 4B).γH2AX signal was reversible for all drugs. The DNA damage signals elicited by LMP517 and LMP135 were more persistent than with the other drugs tested with approximately 80% of γH2AX signal remaining after drug removal. By contrast, about 35% of the topotecan-, 45% of LMP744- and 40% of LMP134-induced γH2AX signals remained 6 h after drug removal (Fig. 4B). These results demonstrate the persistence of the TOP1cc and DNA damage response (γH2AX) induced by LMP517 and LMP135.
Fluoroindenoisoquinolines behave like TOP1 inhibitors in the NCI-60 panel and LMP135 is the most potent compound
To determine where the new fluoroindenoisoquinolines stand with respect to their antiproliferative activity in comparison to validated TOP1 inhibitors, we used the NCI-60 phenotypic data that monitor drug growth inhibitory 50% concentrations (GI50) (11-13). Figure 5A represents the distribution of GI50 values of LMP517, LMP135 and LMP134 in comparison with topotecan, LMP744 and LMP776 in the NCI-60 cell lines. Figure 5A also shows the average GI50 values for each drug. LMP135 was the most potent drug with an average GI50 of 26 nM while LMP134 was the least potent of the fluoroindenoisoquinolines with an average GI50 of 151 nM (84 nM for LMP517). Yet, all the fluoroindenoisoquinolines showed better potency than topotecan (258 nM for topotecan) and the parent indenoisoquinolines (312 nM for LMP744).
Figure 5. LMP135 is the most potent fluoroindenoisoquinoline in the NCI-60 and shows antitumor activity in small cell lung cancer H82 xenografts model.
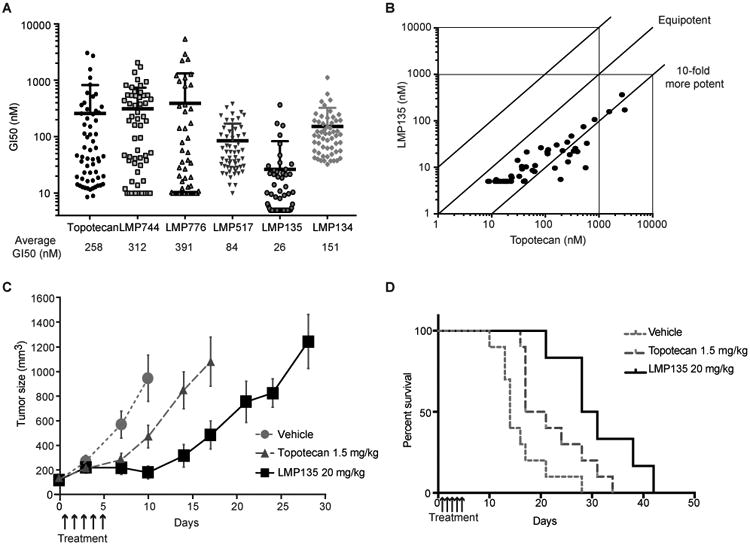
A, Comparison of the NCI-60 responses to the indicated TOP1 inhibitors. Each dot represents the growth inhibitory 50% concentration (GI50) of a given NCI-60 cell line for the indicated drug. Data were obtained from the NCI Developmental Therapeutics Program (DTP). Average GI50 for all 60 cell lines is indicated below the graph. LMP744 and LMP776 were not tested under 10 nM and LMP135 under 5 nM, and in those cell lines in which the GI50 was less than the minimum concentration tested, the GI50 values were recorded as 10 and 5 nM, respectively. The “averages” in these cases therefore represent mean-graph midpoint (MGM) values rather than true GI50 averages. B, Comparison of the sensitivity of the NCI-60 cells to LMP135 vs. topotecan. GI50 of each drug for each cell line is indicated as well as equipotency curve. LMP135 is approximately 10-fold more potent than topotecan. C-D, Antitumor activity of LMP135 (20 mg/kg) compared to topotecan (1.5 mg/kg) represented as tumor volume (C) or as Kaplan Meyer curves (D). Bars represent standard deviations (n = 10 for vehicle and topotecan; n = 6 for LMP135).
Next, the COMPARE analysis (13) of CellMiner (11) was used to test whether the fluoroindenoisoquinolines exhibit a TOP1 inhibitor phenotype across the NCI-60. All fluoroindenoisoquinolines were most highly correlated with camptothecin and its derivatives and other indenoisoquinolines across the NCI-60 drug database, which encompasses over 20,000 publicly available compounds (11) (see Supplementary Table S1 and Table S2 for the complete dataset). Figure 5B shows the high potency of the most potent cellular inhibitor, LMP135, in comparison with topotecan (10-fold lower GI50 than topotecan), and its highly significant correlation with topotecan across the NCI-60 (correlation r=0.88, p<0.001). These analyses demonstrate that the fluoroindenoisoquinolines behave as prototypical TOP1 inhibitors across the NCI-60 cancer cell lines, and that they are more potent than topotecan.
LMP135 shows better tumor response than topotecan in H82 xenografts
As topotecan is FDA-approved for small cell lung cancer (SCLC), we used SCLC H82 cell line as xenograft model to test the antitumor activity and tolerability of LMP135 in comparison with topotecan. Topotecan maximum tolerated dose (MTD) was 1.5 mg/kg and LMP135 was 20 mg/kg. Nude athymic mice were inoculated with H82 cells. After tumors reached 100 mm3, the mice were treated with either topotecan at its MTD (1.5 mg/kg) (38) or with LMP135 at its MTD (20 mg/kg). A single cycle of 5 days of LMP135 was sufficient to inhibit tumor growth with low toxicity (13% body weight loss) (Fig. 5C, D, Fig. S1). LMP135 inhibited tumor growth until day 10, while topotecan inhibited tumor growth only until day 7, leading to a quicker growth. Mouse body weight loss was about 13% for LMP135 and 5% for topotecan (Supplementary Figure S1). These results show that LMP135 is a potentially promising molecule for development as an anticancer drug.
We also determined the pharmacokinetics of the fluoroindenoisoquinolines. The results shown in Supplementary Table S3 show that LMP135 had a half-life of 2.5 h and a volume of distribution of 452 ml, which compares favorably with its parent compound LMP776 (T1/2 = 1.4 h and Vz = 60 ml)(40).
fluoroindenoisoquinolines are selectively active in SLFN11-proficient cells
NCI-60 CellMiner (11,41) (http://discover.nci.nih.gov/cellminercdb)was used to determine the genomic determinants of response to the fluoroindenoisoquinolines. Using the “Multivariate analyses” tool (Fig. 6A), we found that the top genomic determinant was SLFN11 (Schlafen 11) (see also Supplementary Table S4 for the complete dataset), which is a recently established determinant of response to TOP1 inhibitors, PARP inhibitors, hydroxyurea, gemcitabine and other widely used therapeutic agents targeting DNA replication (21-24,26,28,42). Figure 6A also shows the lack of correlation between the activity of LMP135 and two genes encoding most prominent drug efflux transporters, ABCB1 (encoding P-glycoprotein PgP) and ABCG2 [encoding MXR (mitoxantrone resistance protein) and known to confer resistance to topotecan (21)]. The correlation between sensitivity to LMP135 and SLFN11 expression in the NCI-60 cell lines was highly significant (Fig. 6B). Yet there are several outliers that are highly sensitive to LMP135 in spite of low SLFN11 expression, indicating that additional genomic determinants influence the response of cancer cells to LMP135.
Figure 6. SLFN11 is a dominant determinant of response to the fluoroindenoisoquinolines.
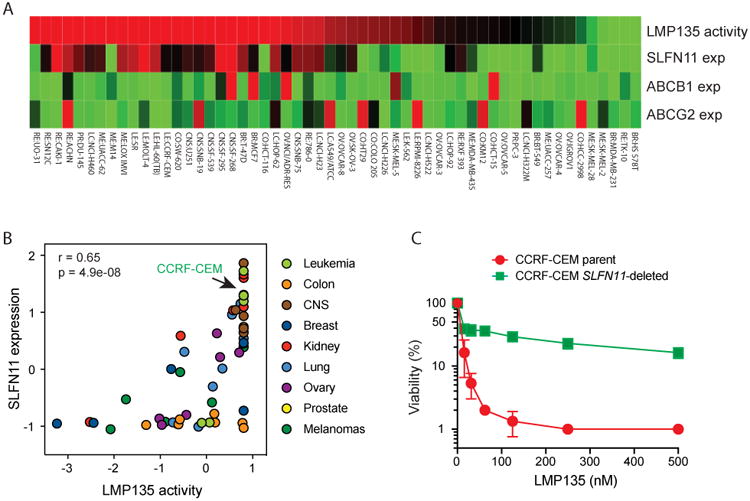
A, Relationship between SLFN11 gene expression and the antiproliferative activity of LMP135 across the NCI-60 cancer cell lines. Gene expression of the drug efflux transporters ABCB1 and ABCG2 is shown for comparison. Cell lines (individual columns) are ranked by drug sensitivity. Color scale: red represents high drug sensitivity and high gene expression. Green is the opposite. B, Correlation between SLFN11 expression and the antiproliferative activity of LMP135 across the NCI-60. C, Causal relationship between SLFN11 expression and sensitivity to LMP135. Growth inhibition of CCRF-CEM parental and SLFN11-deleted cells (24) was measured by ATPlite® assay after treatment with the indicated drugs for 72 h.
To determine the causality between SLFN11 and the activity of the fluoroindenoisoquinolines, we tested the response to the fluoroindenoisoquinolines in SLFN11-proficient CCRF-CEM and isogenic SLFN11-deleted CCRF-CEM cells (24,28) (Fig. 6C and supplementary Fig. S2). Cells were grown in the presence of increasing concentrations of LMP517, LMP134 and LMP135 for 72 h for viability analysis and topotecan was used as a control (21,22). The viability results show that lack of SLFN11 expression confers high resistance to all the drugs. The IC90 of LMP135 was not reached in SLFN11-deleted cells under conditions where it was about 25 nM in CCRF-CEM parental cells (Fig. 6C). The IC90 values of LMP134 and LMP517 in CCRF-CEM parental cells were 120 nM and 55 nM, respectively. In SLFN11-deleted cells, they were much higher (640 nM and 300 nM, respectively, Fig. S2). These results demonstrate that SLFN11 could be a useful determinant of response for the clinical development of the fluoroindenoisoquinolines.
Discussion
Our main conclusions are that: 1/ the fluoroindenoisoquinolines represent a novel chemical class of potent TOP1 inhibitors; 2/ the fluoroindenoisoquinoline LMP135 exerts better antitumor activity compared to topotecan in the small cell lung cancer H82 xenograft model; 3/SLFN11 expression is a prominent determinant of response to the fluoroindenoisoquinolines, and lack of SLFN11 expression predicts resistance; 4/ histone γH2AX can be used as clinical biomarker of response (target engagement) (15,43) for the fluoroindenoisoquinoline.
It is legitimate to develop non-camptothecin TOP1 inhibitors because of the established limitations of camptothecins and because TOP1 is a validated target for a broad range of cancers including colorectal, ovarian, lung, endometrial and pediatric cancers (9,44). The indenoisoquinolines overcome most of the limitations of the camptothecins (see Introduction) and three derivatives are in Phase 1-2 clinical trials (LMP400, indotecan; LMP776, Indimite can and LMP744). The fluoroindenoisoquinoline derivatives (20,29) presented are potent and selective TOP1 inhibitors as they induce TOP1cc at nanomolar concentrations in the presence of recombinant human TOP1 enzymes (Fig. 1) and in cells (Figs. 2 & 4). They also induce DNA damage (γH2AX; Figs. 3-4), and their cellular activity is highly correlated with the established TOP1 inhibitors CPT and topotecan in the NCI-60 (Fig. 5). The potential differences between the fluoroindenoisoquinolines and the current clinical indenoisoquinolines is their enhanced potency (Figs. 1-3 & 5), persistent TOP1cc and γH2AX response (Fig. 4) and lack of O-demethylation (29). The fluorine substituent is likely to affect their distribution and pharmacokinetics. LMP135 showed higher potency than its parent compound LMP776 (see Fig. 1A), and is also more potent than topotecan in the NCI-60 and in tumor model (Fig. 5). Hence, LMP135 might be worthy of consideration for further development as a novel anticancer agent including its use for tumor-targeted delivery using antibody-drug conjugates (ADC), liposomes or PEGylation.
Consistent with previous observation with camptothecins (21-23,26,28,43), SLFN11 expression was the most significant genomic determinant of response to the fluoroindenoisoquinolines (Fig. 6 and Table S3). Lack of SLFN11 was also demonstrated as a causal determinant of resistance to the indenoisoquinolines, as well as topotecan (21,22,42,43). Hence, it should be important to evaluate SLFN11 in clinical samples from patient treated with TOP1 inhibitors to establish the prognostic value of SLFN11 and its role in tumor resistance when tumors suppress its expression (23,42,45,46). Immunohistochemistry studies are ongoing to complement the genomic determination of SLFN11 expression, and it appears logical to use SLFN11 expression as a predictive biomarker for the development of the indenoisoquinolines. In addition, our study suggests that γH2AX could be applied to determine target engagement in response to the fluoroindenoisoquinolines in patient samples (15,37-39).
Supplementary Material
Acknowledgments
Our studies are supported by the Center for Cancer Research, the Intramural Program of the National Cancer Institute (Z01-BC006161, recipient Y. Pommier). We wish to thank the Developmental Therapeutics Branch (DTP) of the NCI for the cellular testing of the indenoisoquinolines in the NCI-60, and the CellMiner Genomics and Pharmacology Facility (CGPF) of the Developmental Therapeutics Branch, CCR, NCI (William Reinhold) for the development of pharmacogenomic tools (http://discover.nci.nih.gov/cellminer) for our analyses of the NCI-60. The xenograft studies were supported and performed by the NCI Drug Development Collaborative (DDC). This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E (recipient Y. Pommier). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Frederick National Laboratory is accredited by AAALAC International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals” (National Research Council; 2011; National Academies Press; Washington, D.C.).
This study has been supported by the Center for Cancer Research, the intramural program of the National Cancer Institute (NCI) (Z01-BC006161); the NCI Drug Development Collaborative (DDC) and federal funds from NCI, National Institute of Health (contract No. HHSN261200800001E)
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Wang JC. DNA topoisomerases. Annu Rev Biochem. 1996;65:635–92. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Ryan AJ, Squires S, Strutt HL, Johnson RT. Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res. 1991;19(12):3295–300. doi: 10.1093/nar/19.12.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol. 2000;20(11):3977–87. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Covey JM, Jaxel C, Kohn KW, Pommier Y. Protein-linked DNA Strand Breaks Induced in Mammalian Cells by Camptothecin, an Inhibitor of Topoisomerase I. Cancer Research. 1989;49(18):5016. [PubMed] [Google Scholar]
- 5.Burke TG, Mi Z. The structural basis of camptothecin interactions with human serum albumin: impact on drug stability. Journal of Medicinal Chemistry. 1994;37(1):40–6. doi: 10.1021/jm00027a005. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 7.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proceedings of the National Academy of Sciences. 2002;99(24):15387–92. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brangi M, Litman T, Ciotti M, Nishiyama K, Kohlhagen G, Takimoto C, et al. Camptothecin resistance: role of the ATP-binding cassette (ABC), mitoxantrone-resistance half-transporter (MXR), and potential for glucuronidation in MXR-expressing cells. Cancer Res. 1999;59(23):5938–46. [PubMed] [Google Scholar]
- 9.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8(1):82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohlhagen G, Paull K, Cushman M, Nagafufuji P, Pommier Y. Protein-linked DNA strand breaks induced by NSC 314622, a non-camptothecin topoisomerase I poison. Mol Pharmacol. 1998;54:50–8. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 11.Reinhold WC, Sunshine M, Varma S, Doroshow JH, Pommier Y. Using CellMiner 16 for Systems Pharmacology and Genomic Analysis of the NCI-60. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(17):3841–52. doi: 10.1158/1078-0432.CCR-15-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol Cancer Ther. 2010;9(5):1451–60. doi: 10.1158/1535-7163.MCT-10-0106. doi:1535-7163.MCT-10-0106[pii]10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, et al. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of a mean graph and COMPARE algorithm. J Natl Cancer Inst. 1989;81:1088–92. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 14.Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, et al. Novel Indenoisoquinolines NSC 725776 and NSC 724998 Produce Persistent Topoisomerase I Cleavage Complexes and Overcome Multidrug Resistance. Cancer Research. 2007;67(21):10397. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 15.Kummar S, Chen A, Gutierrez M, Pfister TD, Wang L, Redon C, et al. Clinical and pharmacologic evaluation of two dosing schedules of indotecan (LMP400), a novel indenoisoquinoline, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;78(1):73–81. doi: 10.1007/s00280-016-2998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cinelli MA, Reddy PV, Lv PC, Liang JH, Chen L, Agama K, et al. Identification, Synthesis, and Biological Evaluation of Metabolites of the Experimental Cancer Treatment Drugs Indotecan (LMP400) and Indimitecan (LMP776) and Investigation of Isomerically Hydroxylated Indenoisoquinoline Analogues as Topoisomerase I Poisons. Journal of medicinal chemistry. 2012;55(24):10844–62. doi: 10.1021/jm300519w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317(5846):1881–6. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 18.Park BK, Kitteringham NR, O'Neill PM. Metabolism of fluorine-containing drugs. Annu Rev Pharmacol Toxicol. 2001;41:443–70. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- 19.Relling MV, Nemec J, Schuetz EG, Schuetz JD, Gonzalez FJ, Korzekwa KR. O-demethylation of epipodophyllotoxins is catalyzed by human cytochrome P450 3A4. Mol Pharmacol. 1994;45(2):352–8. [PubMed] [Google Scholar]
- 20.Beck DE, Lv W, Abdelmalak M, Plescia CB, Agama K, Marchand C, et al. Synthesis and Biological Evaluation of New Fluorinated and Chlorinated Indenoisoquinoline Topoisomerase I Poisons. Bioorganic & medicinal chemistry. 2016;24(7):1469–79. doi: 10.1016/j.bmc.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A. 2012;109(37):15030–5. doi: 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell. 2017;31(2):286–99. doi: 10.1016/j.ccell.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534–50. doi: 10.18632/oncotarget.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO reports. 2016;17(1):94–109. doi: 10.15252/embr.201540964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian L, Song S, Liu X, Wang Y, Xu X, Hu Y, et al. Schlafen-11 sensitizes colorectal carcinoma cells to irinotecan. Anticancer Drugs. 2014;25(10):1175–81. doi: 10.1097/CAD.0000000000000151. [DOI] [PubMed] [Google Scholar]
- 27.Sousa FG, Matuo R, Tang SW, Rajapakse VN, Luna A, Sander C, et al. Alterations of DNA repair genes in the NCI-60 cell lines and their predictive value for anticancer drug activity. DNA repair. 2015;28:107–15. doi: 10.1016/j.dnarep.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol Cell. 2018;69(3):371–84 e6. doi: 10.1016/j.molcel.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elsayed MSA, Su Y, Wang P, Sethi T, Agama K, Ravji A, et al. Design and Synthesis of Chlorinated and Fluorinated 7-Azaindenoisoquinolines as Potent Cytotoxic Anticancer Agents That Inhibit Topoisomerase I. J Med Chem. 2017;60(13):5364–76. doi: 10.1021/acs.jmedchem.6b01870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dexheimer TS, Pommier Y. DNA cleavage assay for the identification of topoisomerase I inhibitors. Nat Protoc. 2008;3(11):1736–50. doi: 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohn KW, Grimek-Ewig RA. Alkaline elution analysis, a new approach to the study of DNA single-strand interruptions in cells. Cancer Res. 1973;33(8):1849–53. [PubMed] [Google Scholar]
- 32.Pommier Y, Kohlhagen G, Kohn KW, Leteurtre F, Wani MC, Wall ME. Interaction of an alkylating camptothecin derivative with a DNA base at topoisomerase I-DNA cleavage sites. Proc Natl Acad Sci U S A. 1995;92(19):8861–5. doi: 10.1073/pnas.92.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, et al. Cellular topoisomerase I inhibition and antiproliferative activity by MJ-III-65 (NSC 706744), an indenoisoquinoline topoisomerase I poison. Mol Pharmacol. 2005;67(2):523–30. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 34.Shaw JL, Blanco J, Mueller GC. Simple procedure for isolation of DNA, RNA and protein fractions from cultured animal cells. Anal Biochem. 1975;65(1-2):125–31. doi: 10.1016/0003-2697(75)90498-4. [DOI] [PubMed] [Google Scholar]
- 35.Subramanian D, Kraut E, Staubus A, Young DC, Muller MT. Analysis of topoisomerase I/DNA complexes in patients administered topotecan. Cancer Res. 1995;55(10):2097–103. [PubMed] [Google Scholar]
- 36.Pourquier P, Takebayashi Y, Urasaki Y, Gioffre C, Kohlhagen G, Pommier Y. Induction of topoisomerase I cleavage complexes by 1-beta -D-arabinofuranosylcytosine (ara-C) in vitro and in ara-C-treated cells. Proc Natl Acad Sci U S A. 2000;97(4):1885–90. doi: 10.1073/pnas.97.4.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. gammaH2AX and cancer. Nat Rev Cancer. 2008;8(12):957–67. doi: 10.1038/nrc2523. doi:nrc2523[pii]10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinders RJ, Hollingshead M, Khin S, Rubinstein L, Tomaszewski JE, Doroshow JH, et al. Preclinical modeling of a phase 0 clinical trial: qualification of a pharmacodynamic assay of poly (ADP-ribose) polymerase in tumor biopsies of mouse xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(21):6877–85. doi: 10.1158/1078-0432.CCR-08-0214. doi:14/21/6877[pii]10.1158/1078-0432.CCR-08-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinders R, Hollingshead MG, Lawrence S, Ji J, Tabb B, Bonner WM, et al. Development of a Validated Immunofluorescence Assay for {gamma}H2AX as a Pharmacodynamic Marker of Topoisomerase I Inhibitor Activity. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010 doi: 10.1158/1078-0432.CCR-09-3076. doi:1078-0432.CCR-09-3076[pii]10.1158/1078-0432.CCR-09-3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirk Culotta JC, Casey Schlutz, Melinda Hollingshead, John Carter, Suzanne Borgel, Carrie Bonomi, Joseph Tomaszewski, Mary Johansen, Timothy Madden. Preclinical pharmacokinetic and comparative biodistribution studies of NSC 706744, NSC 724998, and NSC 725776, three novel indenoisoquinoline topoisomerase I poisons, in CD2F1 mice. AACR Annual Meeting; Apr 12-16, 2008; San Diego, CA. 2008. suppl; abstr 785. [Google Scholar]
- 41.Luna A, Rajapakse VN, Sousa FG, Gao J, Schultz N, Varma S, et al. rcellminer: exploring molecular profiles and drug response of the NCI-60 cell lines in R. Bioinformatics. 2016;32(8):1272–4. doi: 10.1093/bioinformatics/btv701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, et al. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget. 2016;7(3):3084–97. doi: 10.18632/oncotarget.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji J, Zhang Y, Redon CE, Reinhold WC, Chen AP, Fogli LK, et al. Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS One. 2017;12(2):e0171582. doi: 10.1371/journal.pone.0171582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pommier Y, Cushman M. The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol Cancer Ther. 2009;8(5):1008–14. doi: 10.1158/1535-7163.mct-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reinhold WC, Varma S, Sunshine M, Rajapakse V, Luna A, Kohn KW, et al. The NCI-60 Methylome and Its Integration into CellMiner. Cancer Research. 2017;77:601–12. doi: 10.1158/0008-5472.CAN-16-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang SW, Thomas A, Murai J, Trepel J, Bates SE, Rajapakse VN, et al. Overcoming resistance to DNA targeted agents by epigenetic activation of Schlafen 11 (SLFN11) expression with class I histone deacetylase inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018 doi: 10.1158/1078-0432.CCR-17-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.