

In this study, Parsa et al. investigated the mechanisms underlying RNAi-independent heterochromatin assembly by the CTD–RRM protein Seb1 in S. pombe. They show that Seb1 promotes long-lived RNAPII pauses at pericentromeric repeat regions and that their presence correlates with the heterochromatin-triggering activities of the corresponding dg and dh DNA fragments, providing new insight into Seb1-mediated polymerase stalling as a signal necessary for heterochromatin nucleation.
Keywords: heterochromatin, Seb1, polymerase pausing, centromere
Abstract
In Schizosaccharomyces pombe, transcripts derived from the pericentromeric dg and dh repeats promote heterochromatin formation via RNAi as well as an RNAi-independent mechanism involving the RNA polymerase II (RNAPII)-associated RNA-binding protein Seb1 and RNA processing activities. We show that Seb1 promotes long-lived RNAPII pauses at pericentromeric repeat regions and that their presence correlates with the heterochromatin-triggering activities of the corresponding dg and dh DNA fragments. Globally increasing RNAPII stalling by other means induces the formation of novel large ectopic heterochromatin domains. Such ectopic heterochromatin occurs even in cells lacking RNAi. These results uncover Seb1-mediated polymerase stalling as a signal necessary for heterochromatin nucleation.
Packaging of pericentromeric DNA into heterochromatin is crucial for genome stability, development, and health, yet its endogenous triggers remain poorly understood (Allshire and Madhani 2018). A defining feature of pericentromeric heterochromatin is histone H3 Lys9 methylation (H3K9me) (Rea et al. 2000; Bannister et al. 2001; Lachner et al. 2001). Investigations of the fission yeast Schizosaccharomyces pombe have played a major role in our understanding of this type of repressive chromatin. In S. pombe, transcripts derived from the pericentromeric dg and dh repeats during S phase (Volpe et al. 2002; Chen et al. 2008; Kloc and Martienssen 2008) promote heterochromatin formation through two parallel pathways. The first is an RNAi-dependent mechanism involving recruitment of the Clr4 H3K9 methyltransferase complex (CLR-C) via the Argonaute-containing RNA-induced transcriptional silencing (RITS) complex (Motamedi et al. 2004; Noma et al. 2004; Verdel et al. 2004; Sugiyama et al. 2005; Buhler et al. 2006; Bayne et al. 2010).
A second, less-characterized pathway does not require RNAi factors. Components that impact this mechanism include the RNA polymerase II (RNAPII)-associated RNA-binding protein Seb1, the repressor/remodeler complex SHREC, the nuclear 5′–3′ exonuclease Dhp1 (Rat1/Xrn2 in Saccharomyces cerevisiae), the nuclear 3′–5′ exonuclease Rrp6, and the RNA export factor Mlo3/Yra1 (Sadaie et al. 2004; Sugiyama et al. 2007; Reyes-Turcu et al. 2011; Marina et al. 2013; Chalamcharla et al. 2015; Lemay et al. 2016; Tucker et al. 2016; Wittmann et al. 2017). Seb1, Dhp1, and SHREC promote RNAi-independent pericentromeric heterochromatin assembly at pericentromeric regions (Marina et al. 2013; Chalamcharla et al. 2015; Tucker et al. 2016), while Rrp6 and Mlo3 inhibit it (Reyes-Turcu et al. 2011). Other fungi, such as Neurospora crassa and Cryptococcus neoformans, as well as somatic mammalian cells do not require RNAi for heterochromatin assembly (Freitag et al. 2004; Wang et al. 2010; Chan and Wong 2012); thus, poorly understood RNAi-independent mechanisms are important to investigate. In this study, we show that Seb1, whose role in heterochromatin we established in prior work (Marina et al. 2013), displays extensive binding to dg and dh repeat RNAs and promotes long-lived pausing by RNAPII. We show that inducing pausing by other means can trigger the assembly of ectopic heterochromatin domains independently of RNAi. The cleavage polyadenylation (CPA) machinery antagonizes silencing, suggesting a possible mechanism by which heterochromatin is limited at mRNA-coding genes despite the presence of termination-associated pauses. These findings establish a role for Seb1-dependent RNAPII pausing in promoting the formation of repressive chromatin structures.
Results
Nascent elongating transcript sequencing (NET-seq) reveals a role for Seb1 in the pausing of RNAPII
To understand how Seb1 interfaces with the transcription of dg and dh repeats to promote heterochromatin, we used a previously identified viable heterochromatin-defective allele, seb1-1 (Marina et al. 2013). When combined with mutants in the RNAi machinery, seb1-1 eliminates pericentromeric heterochromatin, while the corresponding single mutants decrease H3K9me, indicative of partially redundant pathways (Marina et al. 2013). We examined transcription of heterochromatin at single-nucleotide resolution and tested the impact of the seb1-1 allele using NET-seq (Churchman and Weissman 2011). To analyze the intrinsic transcriptional properties of heterochromatic sequences prior to the establishment of heterochromatin assembly, we used the clr4Δ mutant, which lacks H3K9me and displays full derepression of most silenced chromatin regions. We compared this strain with a clr4Δ seb1-1 double mutant to assess the impact of seb1-1. We first examined the effect of seb1-1 on transcription of nonheterochromatic regions (Fig. 1). Initial inspection revealed numerous genes with a decreased peak density at 5′ regions in the double mutant with increased peak density upstream of annotated CPA sites (often called transcription end sites [TESs]) (see Fig. 1A,B for examples). To analyze these trends genome-wide, travelling ratios were computed on replicate data to assess relative polymerase progression for the 500-base-pair (bp) segment downstream from the transcription start site (TSS; 5′ traveling ratio) and the 500-bp segment upstream of the annotated TES (3′ traveling ratio) (Fig. 1C; see the Materials and Methods). A lower travelling ratio in mutant versus wild type implies lower pausing over the region examined in the mutant and vice versa for higher ratios. Iterative K-means clustering revealed three groups (Fig. 1D), two of which (representing 77% of genes in our data set) are significantly impacted by the seb1-1 allele (Fig. 1D, clusters I and II; Supplemental Fig. S1). The seb1-1 mutation causes a reduced median 5′ traveling ratio and an increased median 3′ traveling ratio for both clusters (Fig. 1D [clusters I and II], E [top and middle panels]), while no significant changes were observed for the third cluster (Fig. 1E, cluster III, bottom panel; see Supplemental Fig. S1 for P-values). These data indicate that the seb1-1 allele leads to decreased RNAPII pausing at gene 5′ ends with an associated increased 3′ signal; the latter may be due to polymerase release from upstream pauses.
Figure 1.

Seb1 controls polymerase progression. (A,B) NET-seq signatures for clr4Δ (black) and clr4Δ seb1-1 (red) strains for two genes (SPAC1B3.12c and SPAC31G5.17c). (C) Traveling ratios at the 5′ and 3′ regions of genes. (D) K-means clustering of NET-seq 5′ and 3′ traveling ratios. Cluster I, n = 373; cluster II, n = 911; cluster III, n = 383. NET-seq replicates are represented for each clr4Δ and clr4Δ seb1-1. (E) Cumulative distribution function (CDF) plots of 5′ (left column) and 3′ (right column) traveling ratios for each cluster from D, comparing clr4Δ (black) with clr4Δ seb1-1 (red) strains in each plot. KS tests were conducted for P-values (Supplemental Fig. S1).
A direct role for Seb1 in RNAPII pausing and heterochromatin assembly in the pericentromeric dh and dg repeats
Our prior RIP-qPCR (RNA immunoprecipitation [RIP] combined with quantitative PCR [qPCR]) analysis indicates that Seb1 functions directly in heterochromatin assembly by binding pericentromeric dg and dh repeat transcripts (Marina et al. 2013). Furthermore, comparison of the transcriptomes of wild-type and seb1-1 using RNA sequencing (RNA-seq) revealed no significant changes [P < 0.01 and |log2(fold change)| > 1] in the transcript levels of known silencing factors (Supplemental Fig. S2; Supplemental Table S1). To assess direct interactions of Seb1 with pericentromeric RNA at single-nucleotide resolution and across the entirety of the dg and dh regions, we conducted PAR-CLIP (photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation) in replicate on a clr4Δ strain. Computational analysis identified statistically significant Seb1 PAR-CLIP read clusters (Corcoran et al. 2011) and confirmed direct binding of Seb1 to pericentromeric transcripts (Fig. 2A,B, top panels; Supplemental Fig. S3A–C) via a motif described previously by others for nonheterochromatic sites bound by Seb1 (Lemay et al. 2016; Wittmann et al. 2017): UGUA (DREME motif analysis; P = 2.9 × 10−9; E = 7.4 × 10−7) (Bailey 2011). Our analysis of Seb1 PAR-CLIP read clusters for coding genes recapitulates published work and is not discussed further here (Lemay et al. 2016; Wittmann et al. 2017).
Figure 2.
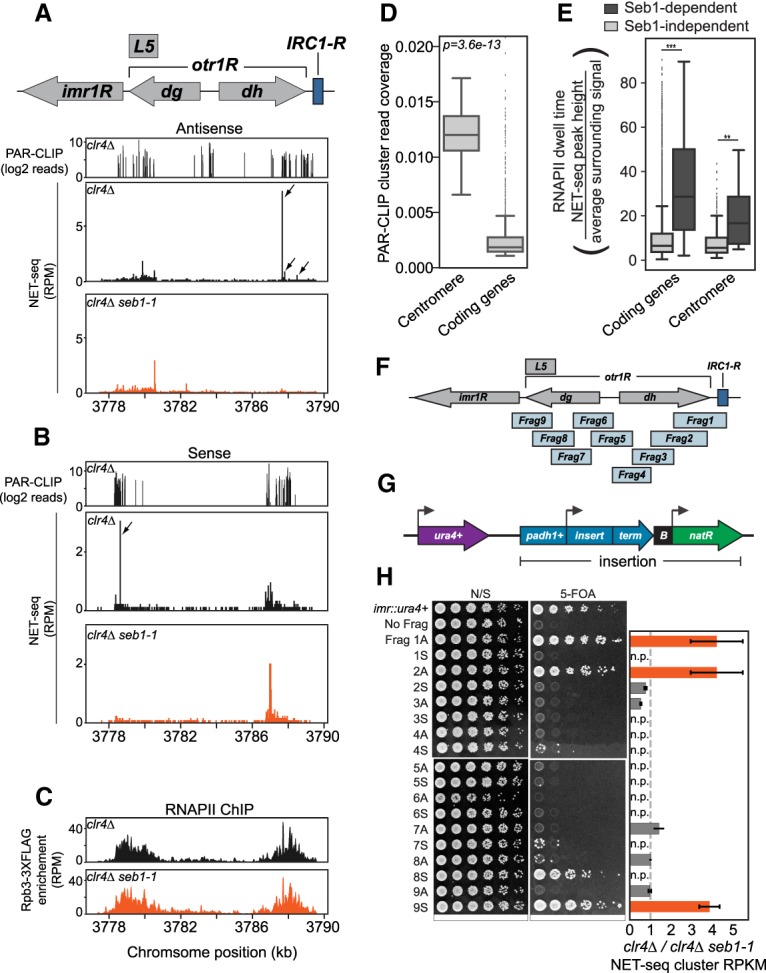
Seb1 directly binds to pericentromeric transcripts and induces RNAPII pausing in silencing-promoting segments. (A,B) Seb1 PAR-CLIP read clusters (log2 reads) and NET-seq peaks (reads per million mapped reads [RPM]) aligned to the right arm of centromere 1 in clr4Δ (PAR-CLIP) or comparing clr4Δ (black) and clr4Δ seb1-1 (red) strains (NET-seq). (A) Read clusters/peaks aligning to the antisense transcript. (B) Read clusters/peaks aligning to the sense transcript. Arrows indicate locations of Seb1-dependent NET-seq peak clusters identified computationally (see the Materials and Methods). (C) ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) data of Rpb3-3xFlag for the right arm of centromere 1, comparing RNAPII enrichment in clr4Δ (black) and clr4Δ seb1-1 (red). (D) Seb1 PAR-CLIP read cluster coverage at the centromeres compared with coding genes. (E) RNAPII dwell time analysis for centromeres and coding genes, comparing Seb1-dependent (dark gray) and Seb1-independent (light gray) pauses. (**) P = 0.0068; (***) P = 3.6 × 10−59. (F) Illustration of the right arm of centromere 1 and nine overlapping fragments analyzed. (G) Illustration of the reporter construct used to determine the silencing capacity of centromere fragments shown in F. (padh1+) adh1+ promoter; (term) a bidirectional terminator; (B) B-boxes boundary element; (natR) nourseothricin-resistance gene. The construct was placed downstream from ura4+. (H) Silencing assays for each fragment in antisense (A) or sense (S) orientations (spotting assay). Cells were plated on nonselective YS medium (N/S) and YS medium supplemented with 5-FOA (5-FOA). Controls for plating were a strain encoding a single functional ura4+ gene placed in the innermost repeats of centromere 1 (imr::ura4+) and a strain with the construct in G containing no fragment (No Frag). Ratios of clr4Δ/clr4Δ seb1-1 for NET-seq clusters in each fragment in antisense and sense transcription units (bar graph). Value of 1 represents no change in NET-seq signal (dotted line), and a value of >1 represents NET-seq clusters in clr4Δ that are reduced in clr4Δ seb1-1. Silencing-competent fragments are orange, and silencing-deficient fragments are dark gray. (n.p.) No peaks.
To compare the binding of Seb1 across transcript classes, we computed the fraction of RNA covered by Seb1 PAR-CLIP read clusters. We observed an ∼12-fold higher PAR-CLIP cluster coverage for pericentromeric repeat intervals than for coding gene intervals (Fig. 2D; Supplemental Fig. S4A). Noncoding RNAs (ncRNAs) display the highest coverage at a mean level ∼100-fold higher than that of coding genes (Supplemental Fig. S4B).
We next examined the NET-seq profiles of pericentromeric heterochromatin sequences of clr4Δ and clr4Δ seb1-1 strains (replicate experiments were conducted). Pericentromeric regions harbor detectable NET-seq signal in clr4Δ cells despite a low level of polymerase engagement at any single nucleotide (Fig. 2A,B, middle panels). The signal overlaps with regions of high Seb1 PAR-CLIP cluster coverage (Fig. 2A,B, top panels). Notably, a handful of discrete peaks indicative of pausing were observed, and the largest were lost in the seb1-1 mutant for both antisense and sense signals (Fig. 2A,B, bottom panels; Supplemental Table S2). RNAPII densities at the pericentromeric regions are comparable between clr4Δ and clr4Δ seb1-1 strains as assessed by ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) analysis of Rpb3-3xFlag (Fig. 2C; Supplemental Fig. S4C), indicating that decreases in NET-seq peak intensities caused by the seb1-1 mutation are not trivially explained by loss of RNAPII recruitment (note that rare pauses may not impact overall RNAPII density as measured by ChIP-seq). Notably, calculation of polymerase dwell times (Larson et al. 2014) at centromeres and across the genome and genotypes revealed that Seb1-dependent pauses are significantly longer-lived on average than Seb1-independent pauses (Fig. 2E; Supplemental Fig. S4D). These data reveal detectable Seb1-dependent RNAPII pauses in pericentromeric sequences.
Previous studies identified two segments of pericentromeric DNA that can trigger heterochromatin: L5 (Partridge et al. 2002; Volpe et al. 2003) and fragment 1 (Frag1) (Marina et al. 2013), respectively. Frag1 defined a segment that requires both RNAi and Seb1 for its activity (Marina et al. 2013). To compare the activity of pericentromeric fragments with their transcription properties described above, we extended this analysis using a system that we used previously (Marina et al. 2013). The cen1R region was divided into nine overlapping fragments (Fig. 2F; Supplemental Table S3). Each fragment was placed downstream from an adh1+ promoter (padh1+) in either forward or reverse orientation and upstream of a transcription terminator. This insert was then placed downstream from ura4+ (Fig. 2G). Silencing of ura4+ was determined using YS-FOA plates, which select for ura4+ repression. The insert of Frag1 displays silencing activity; this construct was used previously to isolate the seb1-1 mutant and was shown to require the padh1+ promoter for silencing activity (Marina et al. 2013). Three additional fragments exhibit strong silencing activity, and each is functional in only one orientation (Frag2A, Frag8S, and Frag9S) (Fig. 2H). Thus, these pericentromeric regions harbor a transcription-dependent orientation-specific signal capable of triggering silencing. To examine the relationship of these regions to those that display detectable Seb1-dependent pauses, we identified clusters of NET-seq peaks (see the Materials and Methods) and computed the total read density of these clusters within each fragment. A comparison of clr4Δ to clr4Δ seb1-1 strains revealed significant correlation (χ2 = 12.6, P < 0.001) (Fig. 2H, Frag1A, Frag2A, and Frag9S; Supplemental Fig. S5A–C). The exception, Frag8S, displays silencing activity but no detectable Seb1-dependent NET-seq peak clusters (although it does display Seb1-dependent NET-seq signal) (Fig. 2H; Supplemental Fig. S5D). The heterochromatin assembly activity of this fragment may be pause-independent, or the relevant RNAPII pauses may be below the sensitivity of NET-seq. Introduction of the seb1-1 allele into these reporter strains (Frag2A, Frag8S, and Frag9S) by homologous recombination leads to reduced silencing of ura4+ (Supplemental Fig. S5E). The residual activity is likely due to the parallel RNAi pathway (Marina et al. 2013). These data indicate that Seb1 directly recognizes dg and dh RNAs, induces detectable pausing in centromere fragments, and promotes the silencing activity directed by these sequences.
Global enhancement of RNAPII pausing can trigger ectopic heterochromatin assembly
The shared defect of the viable seb1-1 allele in both heterochromatin assembly and RNAPII pausing suggests that pausing signals the assembly of heterochromatin. However, given that Seb1 may have other activities impacted by seb1-1, we sought an orthogonal test of the role of pausing. Thus, we pursued an alternative strategy of testing whether increasing RNAPII pausing per se could be sufficient to trigger heterochromatin assembly and, if so, whether such an activity required RNAi. We exploited the conserved elongation factor TFIIS, which binds paused RNAPII complexes and stimulates RNA hydrolysis by RNAPII, enabling restart (Mayer et al. 2017). It is thought that all genes are subject to this type of rescue mechanism, as pausing is a ubiquitous feature of transcription. Two conserved acid residues in domain III of TFIIS are required for catalysis (Jeon et al. 1994). Mutation of these residues to alanine prevents the cleavage of the RNA by RNAPII (Jeon et al. 1994), ultimately resulting in polymerase trapped in a lethal paused/backtracked state (Sigurdsson et al. 2010; Imashimizu et al. 2013). We introduced the corresponding D274A and E275A mutations in the TFIIS gene tfs1+, creating a dominant-negative allele, tfs1DN (Fig. 3A; Lemay et al. 2014). To overcome lethality of this allele (Sigurdsson et al. 2010; Imashimizu et al. 2013), we placed it under control of an nmt1+ thiamine-repressible promoter and inserted it at the leu1+ locus (Matsuyama et al. 2004), enabling concerted expression of tfs1+ and tfs1DN. We conducted replicate NET-seq analysis on clr4Δ and clr4Δ tfs1DN cells under inducing conditions. Genome-wide analysis of 5′ and 3′ traveling ratios indicated TFIISDN-dependent increases in RNAPII pausing at gene 5′ ends and a more modest effect at 3′ ends (Supplemental Fig. S6A,B).
Figure 3.
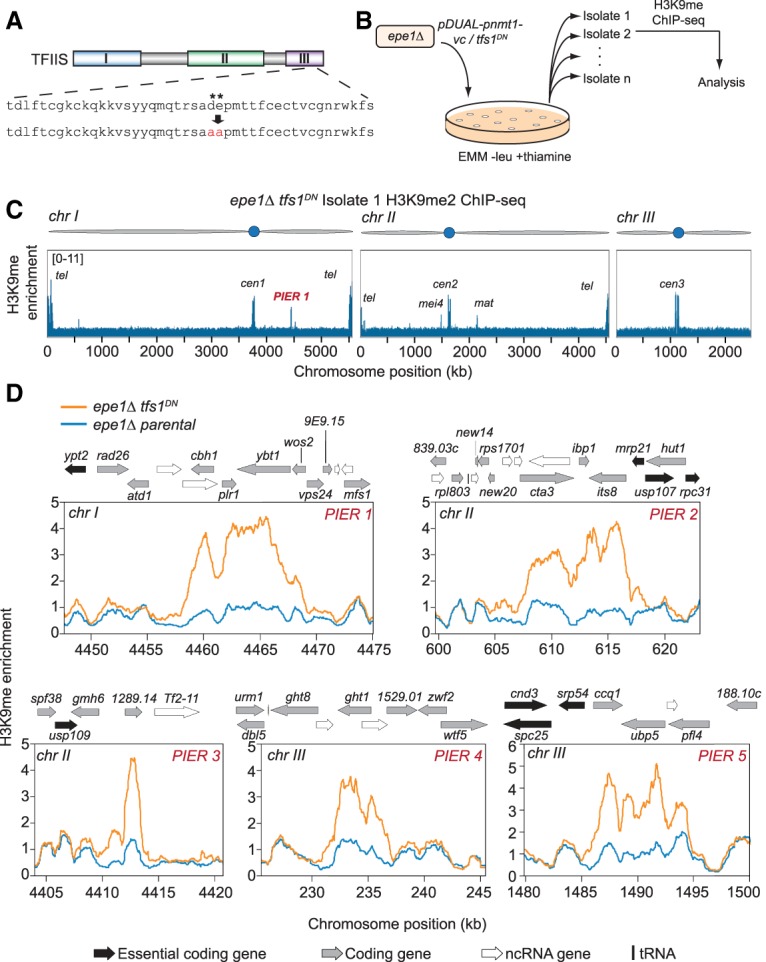
TFIISDN induces ectopic heterochromatin formation. (A) Representation of the domains present in TFIIS. Two acidic residues in domain III (denoted by asterisks) were mutated to alanine residues to produce TFIISDN. (B) Pipeline for isolating TFIISDN-expressing cells and analysis of genome-wide H3K9me2. S. pombe epe1Δ strains were transformed with tfs1DN controlled by the nmt1+ thiamine-repressible promoter (pDUAL-pnmt1+-tfs1DN) or a vector control (pDUAL-pnmt1+-vc) and selected on Edinburgh minimal medium (EMM) −leu +thiamine plates. Thirteen isolates for each epe1Δ tfs1DN and 15 isolates for epe1Δ vector control strains were collected. ChIP-seq for H3K9me2 was conducted and analyzed for each isolate as well as a parental strain for each set of isolates. (C) Genome-wide representation of H3K9me2 ChIP-seq enrichment for epe1Δ tfs1DN isolate 1. (PIER) Pause-induced ectopic heterochromatic region. (D) Genome browser images of PIERs 1–5 that were observed from epe1Δ tfs1DN isolates. Each plot contains the H3K9me2 enrichment of the parental epe1Δ strain (blue) and the epe1Δ tfs1DN strain (orange). Genome features are displayed above each browser image. (Black arrow) Essential coding gene; (gray arrow) coding gene; (white arrow) ncRNA; (black bar) tRNA.
To test whether stabilizing endogenous RNAPII pauses in this manner triggers de novo ectopic heterochromatin, we performed H3K9me2 ChIP-seq analysis on tfs1DN strains; however, we observed no effects in this background (data not shown). Heterochromatin components are limiting and antagonized by anti-silencing factors, particularly Epe1, which actively removes the H3K9me mark (Zofall and Grewal 2006; Aygun et al. 2013; Audergon et al. 2015). Thus, we constructed epe1Δ tfs1DN mutant strains or epe1Δ strains carrying an integrated vector-only control (epe1Δ-vc), collected multiple strain isolates for each, and performed H3K9me2 ChIP-seq on all isolates (Fig. 3B). Because high-level TFIISDN expression is lethal in epe1Δ cells (Supplemental Fig. S6C, middle panel), experiments were performed in the presence of thiamine, enabling viability (Supplemental Fig. S6C, right panel). Consistent with a slight fitness defect under these conditions (Supplemental Fig. S6C, right panel), RNA-seq analysis revealed that 13% of the transcript pool from the tfs1 genes arise from the tfs1DN allele, and 87% arise from the wild-type allele when cells are grown in thiamine (Supplemental Fig. S6D), indicating leaky repression. The ChIP-seq data obtained from strain isolates were examined for H3K9me peaks (see the Materials and Methods). We filtered the results for well-established heterochromatic recruitment sites (including HOODs, islands, meiotic genes, Epe1-bound genes, etc.) (see the Materials and Methods), as these genomic regions have an intrinsic propensity (e.g., via RNAi or the RNA elimination machinery) to nucleate H3K9me (Zofall et al. 2012; Yamanaka et al. 2013; Wang et al. 2015).
Remarkably, of 13 isolates derived from epe1Δ tfs1DN parents analyzed by H3K9me ChIP-seq, five separate isolates harbor a distinct ectopic region of heterochromatin, which we termed the pause-induced ectopic heterochromatic region (PIER) (Fig. 3C,D; Supplemental Fig. S7). No novel ectopic heterochromatic loci were observed by whole-genome H3K9me2 ChIP-seq in the 15 epe1Δ-vc strains (χ2 = 7.02, P = 0.0082). PIERs range in size from ∼3 to ∼15 kb, and each PIER was unique. Three PIERs are bounded by an essential gene on at least one side of the locus, suggesting that selection likely prevents observing PIERs that assemble over essential genes (PIER2, PIER3, and PIER5) (Fig. 3D); this implies that our approach may underestimate the propensity of PIER formation. H3K9me enrichment at known heterochromatin nucleation sites is unrelated to tfs1 genotype and is summarized for all strains in Supplemental Figure S8. These results indicate that TFIISDN-induced RNAPII pausing can be sufficient to nucleate heterochromatin at novel sites.
To determine whether H3K9me at PIERs leads to repression, we conducted RNA-seq analysis on epe1Δ tfs1DN isolate 2 (containing PIER2) (Supplemental Fig. S9A; Supplemental Table S4). We observed a significant decrease (P < 0.01) in two of the three genes present in PIER2: cta3+ and its8+ (Supplemental Fig. S9A; Supplemental Table S4). TFIISDN expression does not alter the expression of known heterochromatin factors [P < 0.01, |log2(fold change)| > 1] (Supplemental Fig. S9B; Supplemental Table S4).
PIER induction is independent of RNAi
Four out of five PIERs contain overlapping convergent genes that have the potential to form dsRNA (PIER1, PIER2, PIER4, and PIER5) (Fig. 3D). As dsRNA can trigger heterochromatin in cis via the RNAi pathway (Simmer et al. 2010), we determined whether the de novo establishment of PIERs requires RNAi. We constructed epe1Δ ago1Δ strains by disrupting ago1+ in the epe1Δ mutant. Initial ChIP-seq analysis of three isolates revealed two that display ectopic heterochromatin at the clr4+ locus and reduced levels of H3K9me at constitutive heterochromatic loci (Supplemental Figs. S10, S11B,C, top panels). Such adaptive silencing of clr4+ has been described previously in epe1Δ mst2Δ strains (Wang et al. 2015) and evidently can occur in strains lacking Epe1 and RNAi. Remarkably, upon integration of tfs1DN into these two epe1Δ ago1Δ strains, H3K9me2 ChIP-seq revealed establishment of heterochromatin at constitutively silenced loci (e.g., centromeres) and its loss at the clr4+ locus (Supplemental Fig. S11B,C, isolates 1, 3, 4, and 5). This observation indicates that pericentromeric repeats harbor the ability to respond to RNAPII pausing and assemble heterochromatin independently of RNAi. Accompanying these changes, six out of the 12 epe1Δ ago1Δ tfs1DN isolates subjected to whole-genome analysis acquired PIERs (Fig. 4; Supplemental Fig. S11). Five of six of these PIERs are bounded by an essential gene on at least one side of the region similar to that seen in the epe1Δ tfs1DN (PIER6–10) (Fig. 4), consistent with the notion that essential genes limit our ability to observe ectopic heterochromatin. Again, each PIER was unique. Thus, PIERs can be triggered by RNAPII pausing even in cells lacking a functional RITS complex.
Figure 4.

TFIISDN-induced ectopic heterochromatin is RNAi-independent. Genome browser images of PIER6–11 that were observed from epe1Δ ago1Δ tfs1DN isolates. epe1Δ ago1Δ strains were transformed with pDUAL-pnmt1+-tfs1DN, and H3K9me2 ChIP-seq was conducted. Genome-wide H3K9me2 enrichment was analyzed, and six PIERs (PIER6–11) were observed from epe1Δ ago1Δ tfs1DN. Each plot contains the H3K9me2 enrichment of the parental epe1Δ ago1Δ strain (blue) and the epe1Δ ago1Δ tfs1DN strain (orange). Genome features are displayed above each browser image. (Black arrow) Essential coding gene; (gray arrow) coding gene; (white arrow) ncRNA; (black bar) tRNA.
The CPA machinery antagonizes heterochromatin assembly
To address the role of Seb1 in the formation of PIERs, we interrogated 15 independent isolates of seb1-1 epe1Δ tfs1DN for ectopic heterochromatin assembly by whole-genome ChIP. A single PIER was observed (Fig. 5A)—a frequency significantly lower than that produced by epe1Δ tfs1DN strains (χ2 = 4.18, P = 0.0409). Two additional regions of ectopic heterochromatin appear evident by visual inspection but were below our cutoffs for H3K9me enrichment (Supplemental Fig. S12A–C). These data suggest that while frequent PIER formation and high levels of H3K9me accumulation are promoted by Seb1, the seb1-1 allele is permissive for some degree of H3K9me triggered by tfs1DN.
Figure 5.

Seb1 promotes PIER formation, and the CPA machinery limits heterochromatin assembly. (A) Graph of the single PIER from seb1-1 epe1Δ tfs1DN isolates. The plot depicts the H3K9me2 enrichment of the parental seb1-1 epe1Δ strain (blue) and the seb1-1 epe1Δ tfs1DN strain (orange). Genome features are displayed above the image. (Black arrow) Essential coding gene; (gray arrow) coding gene. (B–D) Images of H3K9me enrichment at the telomeres and centromere of chromosome I (B), chromosome II (C), and the centromere III (D) in wild-type (blue) and pfs2-11 (orange) mutant backgrounds after temperature shift. (E) Model depicting activation of heterochromatin assembly by Seb1-dependent RNAPII pausing and its repression by the CPA machinery.
Our data demonstrate that Seb1 promotes RNAPII pausing at centromeres, the major site of constitutive heterochromatin assembly, but also at numerous additional sites (Fig. 1). Why, then, is heterochromatin assembly restricted to specific sites under standard growth conditions? A potential clue comes from studies that show that Seb1 promotes CPA termination and copurifies with the CPA machinery (Lemay et al. 2016; Wittmann et al. 2017). One possibility is that recruitment of the CPA by strong polyadenylation signals might suppress heterochromatin assembly, thereby neutralizing the heterochromatin-promoting effects of Seb1. To test this idea, we used a well-characterized temperature-sensitive mutant of a CPA factor, pfs2-11, which produces transcription readthrough past the CPA site under nonpermissive conditions (Wang et al. 2005). Strikingly, under these conditions, we observed increased levels of H3K9me at all centromeres (Fig. 5B–D; Supplemental Fig. S13) as well as increased heterochromatin assembly over a 15-kb subtelomeric region on the right arm of chromosome 1 (Fig. 5B; Supplemental Fig. S13). Notably, the increase in centeromeric H3K9me is not at the expense of its loss elsewhere, as occurs in some mutant backgrounds due to limiting amounts of silencing factors (Allshire and Madhani 2018). These data support a model in which the CPA machinery or its actions suppresses heterochromatin formation at sites of Seb1-promoted CPA (Fig. 5E). They are also consistent with the observation that deletion of sequences that include polyadenylation signal enhance heterochromatin assembly on a reporter gene triggered in trans by expression of an artificial hairpin (Yu et al. 2014).
Discussion
Our results indicate that Seb1, a conserved RNAPII-associated RNA-binding protein that mediates RNAi-independent heterochromatin assembly in S. pombe (Marina et al. 2013), is enriched on pericentromeric ncRNA transcripts relative to coding sequences and promotes long-lived RNAPII pauses. Remarkably, pausing is sufficient to trigger ectopic heterochromatin assembly in an RNAi-independent fashion, indicating that this is a relevant activity of Seb1 in promoting heterochromatin assembly. Binding of Seb1 to euchromatic ncRNAs (e.g., small nuclear RNAs [snRNAs]) is not associated with detectable heterochromatin assembly, which may be due to high levels of transcription, which induces anti-silencing histone marks and histone turnover, both of which antagonize silencing (Allshire and Madhani 2018). At mRNA-coding genes, heterochromatin assembly suppression by the CPA machinery may also play a role in specificity (Fig. 5E). Additionally, the repetitiveness of pericentromeric sequences may also contribute to specificity by producing a threshold density of paused polymerases within a discrete genomic interval. Testing these and other possibilities will require the development of tools that enable the programming of pauses of defined length at defined sites and at defined levels of transcription. Relevant to the issue of heterochromatin assembly specificity is a recent study (Gallagher et al. 2018) that reported that the low-temperature culturing of S. pombe triggers H3K9me heterochromatin islands across the genome independently of RNAi. Thus, the ability of “euchromatic” sites to assemble facultative heterochromatin is evidently higher than might have been assumed.
Our data are germane to the observation that mutations in the Paf1 complex (Paf1-C), a multifunctional elongation complex that binds cooperatively to RNAPII with TFIIS (Xu et al. 2017), enables synthetic hairpin RNAs to trigger heterochromatin in trans and increases heterochromatin spreading in S. pombe (Kowalik et al. 2015; Sadeghi et al. 2015; Verrier et al. 2015). While the elongation-promoting activity of Paf1-C has been suggested to limit heterochromatin by limiting targeting of RITS to the nascent transcript (Kowalik et al. 2015), it may also act via RNAi-independent mechanisms, as we found that increased RNAPII pausing can trigger H3K9me independently of RNAi. Seb1-triggered RNAPII pausing may drive heterochromatin assembly by promoting the heterochromatic stalling of replisomes associated with CLR-C through RNAPII–replisome collisions as proposed (Li et al. 2011; Zaratiegui et al. 2011). Analogous concepts have been put forth in S. cerevisiae, where tight protein–DNA interactions are sufficient to trigger recruitment of the SIR complex (Dubarry et al. 2011). Consistent with this hypothesis, such transcription–replication conflicts are limited by Paf1-C (Poli et al. 2016), which inhibits heterochromatin assembly, while slowing of replisome progression enhances heterochromatin spread (Singh and Klar 2008; Li et al. 2017). It has also been proposed that the 5′ → 3′ RNA exonuclease Dhp1 (related to S. cerevisiae Rat1/Xrn2), which is required for RNAi-independent heterochromatin assembly, recruits the silencing machinery via a physical interaction with CLR-C (Chalamcharla et al. 2015; Tucker et al. 2016). Because RNAPII pausing enhances recruitment of Xrn2 (Wagschal et al. 2012; Contreras et al. 2013), and Seb1 copurifies with the Dhp1 (Lemay et al. 2016; Wittmann et al. 2017), Seb1-induced pausing may promote heterochromatin assembly via this mechanism as well. Our model also readily accommodates genetic observations that null mutants in S. pombe RNAPII elongation factors suppress the H3K9me defect of RNAi mutants (Reyes-Turcu et al. 2011; Sadeghi et al. 2015) as well as analogous observations for mutants in RNA biogenesis factors (Reyes-Turcu et al. 2011), as these factors also promote transcriptional elongation (Luna et al. 2012). Weak CPA signals promote heterochromatin assembly (Yu et al. 2014), which is predicted to result in accumulation of paused RNAPII at S. pombe downstream pause elements (Aranda and Proudfoot 1999). Another key factor recruited to pericentromeric regions by Seb1 is the remodeling/HDAC (histone deactylase) complex SHREC (Marina et al. 2013). Thus, Seb1-paused RNAPII may promote heterochromatin assembly through multiple mechanisms. Given the tight coupling of this heterochromatin signal to RNAPII activity, it is tempting to speculate that Seb1-mediated pausing may have evolved from a surveillance mechanism for silencing foreign DNA. Finally, pathogenic triplet repeat expansions in the Friedreich ataxia gene FXN concomitantly display a block to transcriptional elongation and the appearance of H3K9me on FXN (Punga and Bühler 2010; Li et al. 2015), raising the possibility that pause-induced heterochromatin underlies disease pathogenesis.
Materials and methods
Yeast strains, plasmids, and media
A list of all S. pombe strains and plasmids used in this study is in Supplemental Table S5. Cells were grown at 30°C in synthetic complete (SC) medium with adenine and amino acid supplements with reduced levels of uracil (150 mg/L) for PAR-CLIP or in Edinburgh minimal medium (EMM) supplemented with adenine, uracil, and the appropriate amino acids with or without 15 µM thiamine for NET-seq and ChIP-seq.
NET-seq
NET-seq experiments were conducted as described previously (Churchman and Weissman 2012) with minor alterations for S. pombe. S. pombe cultures were grown in 1 L of EMM without thiamine to an OD600 of 0.7, harvested via filtration, and flash-frozen in liquid nitrogen. Lysis and immunoprecipitation were conducted as described previously (Shetty et al. 2017). Adaptor ligation was performed using random hexamer-barcoded adaptors. All strains were analyzed in duplicate, and sequencing was conducted on a HiSeq 4000 platform.
RNA-seq
Strains were grown in YS medium + 3% glucose overnight to OD600 = 0.7. Cells were harvested by centrifugation, washed twice with ice-cold water, and flash-frozen. Pellets were resuspended in 1 mL of Trizol (Thermo Fisher Scientific, 15596026). Zirconia-silica beads (0.5-mm; BioSpec, 11079105z) were added, and lysis was accomplished by three cycles of bead beating for 90 sec on high (Bead Ruptor 12 homogenizer, OMNI International). Following centrifugation at 14,000 rpm for 10 min at 4°C, the supernatant was transferred to a microcentrifuge tube, extracted once with chloroform, and precipitated with isopropanol. Following resuspension and reprecipitation with isopropanol, pellets were washed with 75% ethanol and air-dried for 30 min. Pellets were resuspended in 150 µL of RNase-free water.
One microgram of total RNA was used to isolate mRNA using PolyATtract systems III and IV (Promega, Z5310) according to the manufacturer's instructions. Input RNA quality and mRNA purity were verified by Bioanalyzer RNA 6000 Pico kit (Agilent, 5067-1513). To address the issue of genomic DNA contamination in RNA samples, we used Zymo RNA Clean and concentrator kit 5 (Zymo Research, 11-326) according to the manufacturer's instruction. A sequencing library was constructed using NEBNext Ultra Directional RNA library preparation kit for Illumina (New England Biolab, E740S). Libraries were analyzed for quality and average size on Bioanalyzer high-sensitivity DNA kit (Agilent, 5067-4626). The sequencing was performed on an Illumina HiSeq 4000 platform.
PAR-CLIP
PAR-CLIP experiments were conducted as a combination of PAR-CLIP and CRAC (cross-linking and analysis of cDNA) protocols (Granneman et al. 2009; Hafner et al. 2010) in two replicates. Cells were grown in 2 L of SC medium to an OD600 of 0.75. 4-thiouracil (Sigma, 440736-1G) was added to a final concentration of 1.3 mM for 15 min in the dark at 30°C. Samples were immediately cross-linked using the UV Power-Shot handheld UV curing system (SPDI UV) at a 365-nm wavelength for 15 min while continuously stirring. Samples were collected by filtration, resuspended in 6 mL of buffer TMN150 (50 mM Tris-HCl at pH 7.8, 150 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 5 mM β-mercaptoethanol), and frozen as “yeast popcorn” by drop-wise addition to liquid nitrogen. This material was lysed by ball mill for five cycles at 15 Hz for 3 min (Mixer Mill MM 301, Retsch). CRAC was then performed from this point on as described previously (Granneman et al. 2009) with the exception of gel extraction, which was conducted by electroelution of the gel piece containing the radioactively labeled RNA sample using D-TubeTM dialyzer midi tubes (EMD Millipore, 71507-3). Electroelution was carried out in 1× MOPS SDS PAGE buffer (5 mM MOPS, 50 mM Tris, 0.1% SDS, 1 mM EDTA) and run for 2 h at 100 V. The isolated sample was transferred to a fresh tube, and 100 µg of Proteinase K (Sigma, P2308) was added to the sample. Procedures after Proteinase K treatment were conducted as described previously (Granneman et al. 2009), and samples were sequenced on the HiSeq 4000 platform.
Spotting assay
Strains were grown overnight to saturation and diluted to an OD600 of 1. Serial dilutions were performed with a dilution factor of 5. For ura4+ silencing assays, cells were grown on nonselective medium and 2 g/L 5-fluoroortic acid (FOA) (unless stated otherwise) YS plates for 3 d at 30°C. For tfs1DN induction assays, cells were grown on YS, EMM −leu −thiamine, or EMM −leu +thiamine for 3 d.
ChIP-seq
ChIP-seq was conducted as described previously (Inada et al. 2016). ChIP-seq immunoprecipitations were performed with 10 μg of anti-H3K9me2 (Abcam, ab1220) and 10 µg of anti-Flag (Sigma, P3165). Samples were sequenced on a HiSeq 4000 platform.
NET-seq analysis: genome alignment
For NET-seq analysis, adapter sequences (using ATCTCGTAT) were removed, and reads were flattened to remove sequence duplicates. Barcoded reads were then mapped to the S. pombe genome (Wood et al. 2012) using Bowtie (Langmead et al. 2009) to align and omit any sequence reads that were misprimed during the reverse transcription step of NET-seq and thus lacked a barcode using the following flags: -M1 --best --strata. Unaligned files were collected for further analysis. Barcodes were removed, and the new unique debarcoded reads were realigned to the genome using the following flags in Bowtie: -M1 --best --strata.
NET-seq analysis: cluster finding
High-density regions of NET-seq signal were defined across centromeres and coding regions to compare NET-seq density between genotypes. First, NET-seq peaks were discovered by calculating robust Z-scores (based on median and median absolute deviation) from the log2 transform of the number of reads starting at each position in the defined region (centromere fragment or transcript). Positions with a robust Z-score of at least 2 and at least 10 unique reads were considered peaks. Next, peaks were clustered together using a sliding window (width = 50, increment = 10). The density of the cluster was calculated as the number of reads in the cluster divided by the size of the cluster in kilobases.
To determine cluster densities for each fragment derived from the right arm of centromere 1, the sum of cluster densities was normalized to the sum of all densities in each sample. Error bars represent the range of two replicates.
NET-seq analysis: traveling ratio
Traveling ratios were calculated for every nonoverlapping annotated transcript at least 1000 nucleotides (nt) in length according to the Pombase annotation (Wood et al. 2012). Transcripts with <50 total reads were excluded from the analysis. The traveling ratio was determined for a 0.5-kb window either immediately after the TSS (5′ traveling ratio) or immediately before the CPA site (3′ traveling ratio). Transcripts <1 kb in length were omitted from this analysis to ensure that the 0.5-kb 5′ and 3′ windows used for each traveling ratio do not overlap. Reads were counted in this window and across the entire transcript and then divided by the size of the window or transcript, respectively. Transcripts were clustered using K-means (sklearn.cluster.Kmeans, three centroids) based on the travelling ratio at each end of the clr4Δ and clr4Δ seb1-1 mutant on one replicate. P-values for each cluster for the difference between the clr4Δ and clr4Δ seb1-1 traveling ratio distributions were determined by KS test for each pair of replicates. Traveling ratio cumulative distribution function (CDF) plots were similar between replicates, and a single replicate is presented.
NET-seq analysis: dwell time
Dwell time was determined by normalizing peak height to the average NET-seq signal density of the surrounding 100 nt. NET-seq peaks with at least a twofold decrease from clr4Δ to clr4Δ seb1-1 in both replicates were considered Seb1-dependent. P-values were determined by KS test.
RNA-seq analysis
Analysis was performed using TopHat (Trapnell et al. 2009) and DESeq2 (Love et al. 2014). Changes in transcript expression levels required a greater than twofold change in mutants compared with wild type to be considered significantly changed enough to have a functional consequence. Data analysis was performed on two replicates per condition.
To determine the fraction of reads derived from the expression of tfs1DN, we divided the total number of reads that specifically aligned to the mutated region of the tfs1DN allele by the total number of reads (both wild-type and mutant alleles) that aligned to this same region.
Seb1 PAR-CLIP data analysis by PARalyzer
For PAR-CLIP analysis, adapter sequences were removed, and reads were mapped to the S. pombe genome (Wood et al. 2012) using Bowtie (Langmead et al. 2009), allowing for three mismatches with the following flags: -M1 -v3 --best --strata. Seb1-binding site read clusters were identified with PARalyzer (Corcoran et al. 2011). Reads of <20 nt were omitted, and read clusters required at least 10 reads and at least two T → C conversions per cluster to be called as a Seb1-binding site. The PARalyzer OUTPUTCLUSTERSFILE file was converted to a genome browser readable file (.bam) for analysis. PAR-CLIP cluster coverage was calculated as the fraction of the interval of interest harboring a PARalyzer-called PAR-CLIP cluster (centromere arm, coding gene, or ncRNA). P-values were determined by KS test.
DREME motif analysis
DREME (Bailey 2011) motif discovery for short ungapped sequences was used to find Seb1-specific binding motifs. PAR-CLIP clusters that were flattened to remove identical recurrent sequence clusters originating from all three centromeric regions were subjected to motif analysis. A shuffled sequence set created from the input sequences was used as a control.
ChIP-seq analysis
ChIP-seq analysis was conducted as described previously (Inada et al. 2016). Briefly, adaptor sequences from ChIP-seq sequencing libraries were removed (using GATCGGAAGA), and reads <20 nt were omitted. Reads were aligned to the S. pombe genome (Wood et al. 2012) using Bowtie (Langmead et al. 2009) with the following flags: -M1 --best --strata. Aligned reads were smoothed over a 1-kb window.
ChIP-seq analysis: PIER discovery
H3K9me ChIP-seq peaks were considered as novel ectopic sites of H3K9me if two criteria were met: (1) H3K9me peaks were threefold or greater than the genome background signal in the isolate, and (2) when normalized to the whole-cell extract (denoted “H3K9me enrichment” in the figures), the H3K9me signal at the peak was threefold or greater higher than the background and parental H3K9me ChIP-seq signals. A curated list of genomic regions previously observed to have a propensity to form heterochromatin in various S. pombe backgrounds (Yamanaka et al. 2012; Zofall et al. 2012; Wang et al. 2015) was generated (Supplemental Table S6). In epe1Δ backgrounds, oscillation and spreading of H3K9me can occur (Trewick et al. 2007); thus, peaks within 10 kb of our curated list of H3K9me nucleation sites or within 10 kb of H3K9me regions present in the parental strain were not counted as novel H3K9me nucleation events.
ChIP-seq analysis: H3K9me levels at HOODs, islands, meiotic genes, and PIERs
For all isolates and whole-cell extracts, RPKMs (reads per kilobase transcript per million mapped reads) for each region in Supplemental Table S6 and all PIERs were normalized to the RPKM of a 10-kb window surrounding each region (5 kb upstream and downstream). The ratio of H3K9me enrichment values from isolates to the whole-cell extracts were plotted as a heat map (Supplemental Fig. S8).
Data sets
All available sequencing data sets are listed in Supplemental Table S7 and were deposited in the Gene Expression Omnibus with the superseries accession number GSE114540.
Supplementary Material
Acknowledgments
We thank the members of the Madhani laboratory for support and scientific discussion, Robin Allshire (The University of Edinburgh) for the gift of the pDUAL-pnmt1+ plasmid, Fred Winston and Ameet Shetty (Harvard Medical School) for sharing their S. pombe NET-seq protocol, Chris Norbury (The University of Oxford) for the gift of the pfs2-11 S. pombe strain, and Jeffrey Hussmann (University of California at San Francisco) for helpful discussions on data analysis. We thank Diana Marina, Smita Shankar, and Ken Finn for their early contributions to this work. We thank Sigurd Braun, Geeta Narlikar, Robin Allshire, Phillip Dumesic, and Bassem Al-Sady for critical review of the manuscript. Research in the Madhani laboratory is supported by grants from the National Institutes of Health. H.D.M. is a Chan-Zuckerberg Biohub Investigator.
Author contributions: J.-Y.P. and H.D.M. designed the study. J.-Y.P. performed all of the experiments reported here except for the RNA-seq work, which was performed by S.B. J.B. performed computational analysis, and C.H. contributed software for the analysis. J.-Y.P. and H.D.M. wrote the manuscript with input from all authors.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.310136.117.
Freely available online through the Genes & Development Open Access option.
References
- Allshire RC, Madhani HD. 2018. Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19: 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda A, Proudfoot NJ. 1999. Definition of transcriptional pause elements in fission yeast. Mol Cell Biol 19: 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audergon PN, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, Allshire RC. 2015. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 348: 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygun O, Mehta S, Grewal SI. 2013. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat Struct Mol Biol 20: 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL. 2011. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124. [DOI] [PubMed] [Google Scholar]
- Bayne EH, White SA, Kagansky A, Bijos DA, Sanchez-Pulido L, Hoe K-L, Kim D-U, Park H-O, Ponting CP, Rappsilber J, et al. 2010. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 140: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler M, Verdel A, Moazed D. 2006. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell 125: 873–886. [DOI] [PubMed] [Google Scholar]
- Chalamcharla VR, Folco HD, Dhakshnamoorthy J, Grewal SI. 2015. Conserved factor Dhp1/Rat1/Xrn2 triggers premature transcription termination and nucleates heterochromatin to promote gene silencing. Proc Natl Acad Sci 112: 15548–15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FL, Wong LH. 2012. Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res 40: 11178–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI. 2008. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451: 734–737. [DOI] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. 2011. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. 2012. Native elongating transcript sequencing (NET-seq). Curr Protoc Mol Biol 98: 14.4.1–14.4.17. [DOI] [PubMed] [Google Scholar]
- Contreras X, Benkirane M, Kiernan R. 2013. Premature termination of transcription by RNAP II. Transcription 4: 72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. 2011. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol 12: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubarry M, Loiodice I, Chen CL, Thermes C, Taddei A. 2011. Tight protein–DNA interactions favor gene silencing. Genes Dev 25: 1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag M, Lee DW, Kothe GO, Pratt RJ, Aramayo R, Selker EU. 2004. DNA methylation is independent of RNA interference in Neurospora. Science 304: 1939. [DOI] [PubMed] [Google Scholar]
- Gallagher PS, Larkin M, Thillainadesan G, Dhakshnamoorthy J, Balachandran V, Xiao H, Wellman C, Chatterjee R, Wheeler D, Grewal SIS. 2018. Iron homeostasis regulates facultative heterochromatin assembly in adaptive genome control. Nat Struct Mol Biol 25: 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D. 2009. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci 106: 9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imashimizu M, Kireeva ML, Lubkowska L, Gotte D, Parks AR, Strathern JN, Kashlev M. 2013. Intrinsic translocation barrier as an initial step in pausing by RNA polymerase II. J Mol Biol 425: 697–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada M, Nichols RJ, Parsa JY, Homer CM, Benn RA, Hoxie RS, Madhani HD, Shuman S, Schwer B, Pleiss JA. 2016. Phospho-site mutants of the RNA polymerase II C-terminal domain alter subtelomeric gene expression and chromatin modification state in fission yeast. Nucleic Acids Res 44: 9180–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon C, Yoon H, Agarwal K. 1994. The transcription factor TFIIS zinc ribbon dipeptide Asp–Glu is critical for stimulation of elongation and RNA cleavage by RNA polymerase II. Proc Natl Acad Sci 91: 9106–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloc A, Martienssen R. 2008. RNAi, heterochromatin and the cell cycle. Trends Genet 24: 511–517. [DOI] [PubMed] [Google Scholar]
- Kowalik KM, Shimada Y, Flury V, Stadler MB, Batki J, Buhler M. 2015. The Paf1 complex represses small-RNA-mediated epigenetic gene silencing. Nature 520: 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MH, Mooney RA, Peters JM, Windgassen T, Nayak D, Gross CA, Block SM, Greenleaf WJ, Landick R, Weissman JS. 2014. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science 344: 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, Larochelle M, Marguerat S, Atkinson S, Bahler J, Bachand F. 2014. The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat Struct Mol Biol 21: 919–926. [DOI] [PubMed] [Google Scholar]
- Lemay JF, Marguerat S, Larochelle M, Liu X, van Nues R, Hunyadkurti J, Hoque M, Tian B, Granneman S, Bahler J, et al. 2016. The Nrd1-like protein Seb1 coordinates cotranscriptional 3′ end processing and polyadenylation site selection. Genes Dev 30: 1558–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Martienssen R, Cande WZ. 2011. Coordination of DNA replication and histone modification by the Rik1–Dos2 complex. Nature 475: 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu Y, Polak U, Lin K, Shen J, Farmer J, Seyer L, Bhalla AD, Rozwadowska N, Lynch DR, et al. 2015. Expanded GAA repeats impede transcription elongation through the FXN gene and induce transcriptional silencing that is restricted to the FXN locus. Hum Mol Genet 24: 6932–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yi J, Agbu P, Zhou Z, Kelley RL, Kallgren S, Jia S, He X. 2017. Replication stress affects the fidelity of nucleosome-mediated epigenetic inheritance. PLoS Genet 13: e1006900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna R, Rondón AG, Aguilera A. 2012. New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim Biophys Acta 1819: 514–520. [DOI] [PubMed] [Google Scholar]
- Marina DB, Shankar S, Natarajan P, Finn KJ, Madhani HD. 2013. A conserved ncRNA-binding protein recruits silencing factors to heterochromatin through an RNAi-independent mechanism. Genes Dev 27: 1851–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama A, Shirai A, Yashiroda Y, Kamata A, Horinouchi S, Yoshida M. 2004. pDUAL, a multipurpose, multicopy vector capable of chromosomal integration in fission yeast. Yeast 21: 1289–1305. [DOI] [PubMed] [Google Scholar]
- Mayer A, Landry HM, Churchman LS. 2017. Pause & go: from the discovery of RNA polymerase pausing to its functional implications. Curr Opin Cell Biol 46: 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamedi MR, Verdel A, Colmenares SU, Gerber SA, Gygi SP, Moazed D. 2004. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell 119: 789–802. [DOI] [PubMed] [Google Scholar]
- Noma K, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S, Moazed D, Grewal SI. 2004. RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet 36: 1174–1180. [DOI] [PubMed] [Google Scholar]
- Partridge JF, Scott KS, Bannister AJ, Kouzarides T, Allshire RC. 2002. Cis-acting DNA from fission yeast centromeres mediates histone H3 methylation and recruitment of silencing factors and cohesin to an ectopic site. Curr Biol 12: 1652–1660. [DOI] [PubMed] [Google Scholar]
- Poli J, Gerhold CB, Tosi A, Hustedt N, Seeber A, Sack R, Herzog F, Pasero P, Shimada K, Hopfner KP, et al. 2016. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev 30: 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punga T, Bühler M. 2010. Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol Med 2: 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599. [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Zhang K, Zofall M, Chen E, Grewal SI. 2011. Defects in RNA quality control factors reveal RNAi-independent nucleation of heterochromatin. Nat Struct Mol Biol 18: 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M, Iida T, Urano T, Nakayama J. 2004. A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J 23: 3825–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi L, Prasad P, Ekwall K, Cohen A, Svensson JP. 2015. The Paf1 complex factors Leo1 and Paf1 promote local histone turnover to modulate chromatin states in fission yeast. EMBO Rep 16: 1673–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A, Kallgren SP, Demel C, Maier KC, Spatt D, Alver BH, Cramer P, Park PJ, Winston F. 2017. Spt5 plays vital roles in the control of sense and Antisense transcription elongation. Mol Cell 66: 77–88.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Dirac-Svejstrup AB, Svejstrup JQ. 2010. Evidence that transcript cleavage is essential for RNA polymerase II transcription and cell viability. Mol Cell 38: 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer F, Buscaino A, Kos-Braun IC, Kagansky A, Boukaba A, Urano T, Kerr ARW, Allshire RC. 2010. Hairpin RNA induces secondary small interfering RNA synthesis and silencing in trans in fission yeast. EMBO Rep 11: 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Klar AJS. 2008. Mutations in deoxyribonucleotide biosynthesis pathway cause spreading of silencing across heterochromatic barriers at the mating-type region of the fission yeast. Yeast 25: 117–128. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Cam H, Verdel A, Moazed D, Grewal SI. 2005. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc Natl Acad Sci 102: 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K, Zofall M, Kobayashi R, Grewal SI. 2007. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128: 491–504. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewick SC, Minc E, Antonelli R, Urano T, Allshire RC. 2007. The JmjC domain protein Epe1 prevents unregulated assembly and disassembly of heterochromatin. EMBO J 26: 4670–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker JF, Ohle C, Schermann G, Bendrin K, Zhang W, Fischer T, Zhang K. 2016. A novel epigenetic silencing pathway involving the highly conserved 5′–3′ exoribonuclease Dhp1/Rat1/Xrn2 in Schizosaccharomyces pombe. PLoS Genet 12: e1005873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D. 2004. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303: 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier L, Taglini F, Barrales RR, Webb S, Urano T, Braun S, Bayne EH. 2015. Global regulation of heterochromatin spreading by Leo1. Open Biol 5: 150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. 2002. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297: 1833–1837. [DOI] [PubMed] [Google Scholar]
- Volpe T, Schramke V, Hamilton GL, White SA, Teng G, Martienssen RA, Allshire RC. 2003. RNA interference is required for normal centromere function in fission yeast. Chromosome Res 11: 137–146. [DOI] [PubMed] [Google Scholar]
- Wagschal A, Rousset E, Basavarajaiah P, Contreras X, Harwig A, Laurent-Chabalier S, Nakamura M, Chen X, Zhang K, Meziane O, et al. 2012. Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell 150: 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SW, Asakawa K, Win TZ, Toda T, Norbury CJ. 2005. Inactivation of the pre-mRNA cleavage and polyadenylation factor Pfs2 in fission yeast causes lethal cell cycle defects. Mol Cell Biol 25: 2288–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hsueh YP, Li W, Floyd A, Skalsky R, Heitman J. 2010. Sex-induced silencing defends the genome of Cryptococcus neoformans via RNAi. Genes Dev 24: 2566–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Reddy BD, Jia S. 2015. Rapid epigenetic adaptation to uncontrolled heterochromatin spreading. Elife 4: e06179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann S, Renner M, Watts BR, Adams O, Huseyin M, Baejen C, El Omari K, Kilchert C, Heo DH, Kecman T, et al. 2017. The conserved protein Seb1 drives transcription termination by binding RNA polymerase II and nascent RNA. Nat Commun 8: 14861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood V, Harris MA, McDowall MD, Rutherford K, Vaughan BW, Staines DM, Aslett M, Lock A, Bahler J, Kersey PJ, et al. 2012. PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res 40: D695–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Bernecky C, Lee CT, Maier KC, Schwalb B, Tegunov D, Plitzko JM, Urlaub H, Cramer P. 2017. Architecture of the RNA polymerase II-Paf1C-TFIIS transcription elongation complex. Nat Commun 8: 15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, Mehta S, Reyes-Turcu FE, Zhuang F, Fuchs RT, Rong Y, Robb GB, Grewal SIS. 2012. RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature 493: 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, Mehta S, Reyes-Turcu FE, Zhuang F, Fuchs RT, Rong Y, Robb GB, Grewal SI. 2013. RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature 493: 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Jih G, Iglesias N, Moazed D. 2014. Determinants of heterochromatic siRNA biogenesis and function. Mol Cell 53: 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, de Castro E, Marin L, Chang AY, Goto D, et al. 2011. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature 479: 135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Grewal SI. 2006. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol Cell 22: 681–692. [DOI] [PubMed] [Google Scholar]
- Zofall M, Yamanaka S, Reyes-Turcu FE, Zhang K, Rubin C, Grewal SI. 2012. RNA elimination machinery targeting meiotic mRNAs promotes facultative heterochromatin formation. Science 335: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.