

Abstract
Galactose oxidase (EC 1.1.3.9) is a monomeric enzyme that contains a single copper ion and catalyses the stereospecific oxidation of primary alcohols to their corresponding aldehydes. The protein contains an unusual covalent thioether bond between a tyrosine, which acts as a radical center during the two-electron reaction, and a cysteine. The enzyme is produced in a precursor form lacking the thioether bond and also possessing an additional 17-aa pro-sequence at the N terminus. Previous work has shown that the aerobic addition of Cu2+ to the precursor is sufficient to generate fully processed mature enzyme. The structure of the precursor protein has been determined to 1.4 Å, revealing the location of the pro-sequence and identifying structural differences between the precursor and the mature protein. Structural alignment of the precursor and mature forms of galactose oxidase shows that five regions of main chain and some key residues of the active site differ significantly between the two forms. The precursor structure provides a starting point for modeling the chemistry of thioether bond formation and pro-sequence cleavage.
Galactose oxidase from Fusarium spp. is a copper-containing enzyme that catalyses the oxidation of primary alcohols to their corresponding aldehydes, a two-electron reaction, but with only a single copper at the active site (1). The second redox active center necessary for the reaction was found to be situated at a tyrosine residue (2). The crystal structure (3) showed the presence of a novel thioether bond, covalently linking Sγ of Cys-228 and Cɛ of Tyr-272, with this tyrosine also acting as a ligand to the copper. The side chains of these two residues form an aromatic plane that stacks with the indole ring of Trp-290. Oxidation of Tyr-272 generates a radical, providing the second redox center in the active state of the enzyme.
The presence of this covalent cross-link places galactose oxidase in a growing class of proteins that contain posttranslationally modified, redox active amino acids (4). Glyoxal oxidase, a copper-containing enzyme involved in lignin degradation, is also believed to have a cysteine-tyrosine bond at the active site (5). Thioether bonds are not the only posttranslational modifications that have been observed. Others include a tyrosine-histidine bridge in cytochrome c oxidase (6), a modified tryptophan-tryptophan bridge in methylamine dehydrogenase (7), a histidine-cysteine bridge in catechol oxidase (8), and 2,4,5- trihydroxyphenylalanine quinone in copper amine oxidase (9). There is currently great interest in such modified amino acids and the mechanisms by which they are formed. Initiation of the autocatalytic processing of tyrosine to trihydroxyphenylalanine quinone in amine oxidase requires only copper and oxygen (10–12). This is also true of thioether bond formation in galactose oxidase (13). Self-processing redox enzymes are likely to play an important role in enzyme evolution by allowing new redox reactions to take place without the requirement for complex biosynthetic pathways to generate cofactors. In addition, a greater understanding of these modifications should allow incorporation of similar centers into designed or adapted enzymes.
Galactose oxidase is of particular interest as the generation of mature enzyme requires at least three processing events: cleavage of a secretion signal sequence, cleavage of a 17-aa N-terminal pro-sequence, and formation of the thioether bond (14). The latter two events have been shown to take place in vitro on the addition of copper and oxygen, even in the presence of protease inhibitors (13). In copper-limited conditions, heterologous expression of galactose oxidase results in three forms of the protein identifiable as distinct bands on SDS/PAGE. The lower band is mature galactose oxidase, which runs anomalously on SDS/PAGE (at 65.5 kDa) (15, 16) owing to the presence of the thioether bond that introduces an intramolecular loop between residues 228 and 272. The middle band (68.5 kDa) migrates with the expected apparent molecular mass of mature galactose oxidase, indicating that this is an incompletely processed form of the protein that lacks the thioether bond. The upper band (70.2 kDa) is a precursor of galactose oxidase, which has been revealed by N-terminal sequencing to have the additional 17 aa at the N terminus. As a first step toward understanding the self-processing events we have crystallized the precursor of the enzyme, containing the pro-sequence, and here describe the structure determined to a resolution of 1.4 Å.
Methods
Galactose oxidase was expressed in Aspergillus nidulans and purified under strict metal-free conditions (13). Crystals of the precursor of galactose oxidase were obtained in Mes ≈pH 6, 18% PEG 8000, and 200 mM calcium acetate at 18°C by using the sitting drop method. To ensure that the enzyme remained as the precursor, crystallization conditions were strictly copper-free. This was achieved by soaking all plastic ware in 0.1 M EDTA before use, treating protein with 50 mM sodium diethyldithiocarbamate, and adding Chelex-100 resin to crystallization solutions. Crystals were obtained within 2 weeks with hexagonal rod-shaped morphology. These orthorhombic crystals are in the space group P212121 with unit cell dimensions a = 69.31, b = 89.52, and c = 93.96 Å (Table 1).
Table 1.
Crystallographic data collection and refinement statistics
| Space group | P212121 |
| Unit cell, Å | a = 69.31, b = 89.52, c = 93.96 |
| Resolution, Å | 64–1.4 |
| Number of observed reflections | 731,202 |
| Number of unique reflections | 115,006 |
| Completeness (%) overall (final shell) | 99.2 (93.7) |
| I/σ(I) overall (final shell) | 72.3 (28.5) |
| Rsym (%)* overall (final shell) | 4.2 (9.2) |
| Wilson B factor, Å2 | 11.6 |
| Rcryst, %† | 17.6 |
| Rfree, %‡ | 19.3 |
| Number of atoms | 5,626 |
| Average overall B factor, Å2 | 12.4 |
| rms bond lengths, Å | 0.005 |
| rms bond angles, ° | 1.4 |
Rsym = Σhkl(ΣI(|Ihkl,I − 〈Ihkl〉|))/Σhkl,IIkhl,I, where Ihkl,I is the intensity of an individual reflection, and 〈Ihkl〉 is the mean intensity of that reflection.
Rcryst = Σhkl(∥Fobshkl| − |Fcalchkl∥)/|Fobshkl|, where |Fobshkl| and |Fcalchkl| are the observed and calculated structure factor amplitudes for reflections used during refinement (working set).
Rfree is equivalent to Rcryst but calculated with reflections omitted from the refinement process (test set).
Initial diffraction data were collected to 1.9-Å resolution at 100 K by using an R-AXIS IIC imaging plate detector and Rigaku RU200 rotating anode x-ray generator and were processed with mosflm (17). Data were scaled by using scala (17) and reduced in truncate (17). The molecular replacement program amore (17, 18) was used to solve the structure with the coordinates for the mature form of galactose oxidase (Protein Data Bank ID code 1GOG) as the search model. A solution was obtained with a correlation coefficient of 63% and an R factor of 35% after rigid body refinement. A free R set was generated by containing 10% of the reflections. The early 2Fo − Fc maps clearly showed the presence of the N-terminal pro-sequence and also revealed regions of main chain that required significant rebuilding. Modeling of the N-terminal pro-sequence and rebuilding was carried out by using the program o (19). A second, higher-resolution data set was subsequently collected to 1.4 Å on station 9.6 at the Daresbury Synchrotron Radiation Source (Cheshire, U.K.) by using an ADSC Quantum IV charge-couple device detector. These data were processed in denzo (20) and reduced in scalepack (20). Refinement was carried out in cns (21–23) by using the simulated annealing, minimization, and individual B factor protocols. Waters were added after examining difference maps and considering hydrogen bonding geometry. They were retained if they maintained sensible B factors.
Once refinement was complete the Cα positions of the precursor and mature structures were aligned in lsqman (24) to identify the most significant differences between the two structures. After an initial alignment, the superposition was improved by using least squares, excluding residues that deviated by more than 2 Å at each round until no further improvement was obtained. After this superposition, the Cαs of residues that deviated by 2 Å or more (Fig. 1 Upper) were excluded from an rms deviation (rmsd) calculation, and the remaining residues were found to have an rmsd of 0.7 Å. In the same alignment, those with a deviation greater that 2 Å had an rmsd of 5.1 Å.
Figure 1.

Structure of the mature form of galactose oxidase (Upper Left) and the precursor form (Upper Right). Domain I is red, domain II is blue, and domain III is purple. In the precursor form the N-terminal pro-sequence is green, and regions that differ from the mature structure by more than 2 Å are yellow. The sequence of galactose oxidase (14) is shown (Lower) and colored as above. The pro-sequence residues are numbered −17 to −1. All figures were produced by using spock (45).
Results
Precursor galactose oxidase, expressed in A. nidulans, was produced, purified (13), and crystallized in copper-free conditions. The structure was solved by molecular replacement using the structure of the mature enzyme as a model. The precursor structure was rebuilt and refined to 1.4 Å (Table 1) to give a final model with good stereochemistry and an Rcryst = 18.0%, Rfree = 19.5%.
Overall Structure.
The mature enzyme comprises three predominately β-sheet domains (3, 25) (Fig. 1 Upper Left). Domain I consists of eight β-strands in a jelly-roll motif with a five-stranded antiparallel β-sheet facing a three-stranded antiparallel β-sheet. Domain II is the largest, with a seven-bladed β-propeller fold surrounding a central cavity. The active site of the mature protein is situated at the surface of this domain, with the copper ion lying close to the central pseudo 7-fold axis. Three of the four protein ligands to the copper, Tyr-272, Tyr-495, and His-496, are provided by domain II. Domain III is a bundle of seven, mostly antiparallel, β-strands surrounding a hydrophobic core. One long antiparallel β-ribbon penetrates into the central cavity of the domain II propeller, and at the tip of this loop is the fourth protein ligand to the copper, His-581. The overall structure of the copper-free precursor is similar to the mature enzyme, but with significant local differences (Fig. 1 Upper Right).
The structure of the precursor also reveals the presence of an additional sugar at the surface of domain II. The electron density suggests at least a disaccharide, with the first ring stacking almost parallel to the ring of Tyr-484, but its identity has not been determined. As the sugar is some distance from the active site its biological relevance is uncertain.
Location and Interactions of the Pro-Sequence.
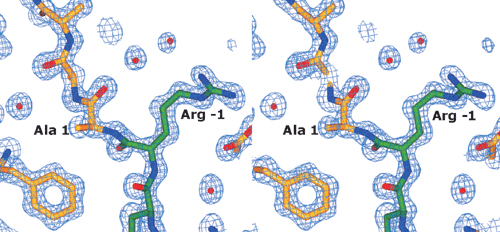
The pro-sequence lies between domains I and II, with 12 of the 17 aa visible in the final electron density maps. These residues form a loop between the domains (Fig. 1 Upper Right), turning back at residues −5/−6, until the N-terminal residues of the pro-sequence turn away from the main body of the protein and become disordered. Eleven of the pro-sequence amino acids (−1 to −11) are clearly defined in the density maps (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org), but the remaining residues (−13 to −17) and the side chain of the twelfth residue (Ile −12), are not visible. The main chain of the pro-sequence makes several hydrogen bonds to the mature sequence (Fig. 2) (Arg −1 O to Asn-521 Nδ2, Arg −1 N to Gly-516 O, Ser −3 O to Tyr-189 Oη and Asn-191 Nδ2, Leu −4 O to Arg Nη1-217, Phe −5 O to Arg Nη2-217) and also with itself, particularly at the turn (Ser −3 N to Ser −8 O, Leu −4 N to Leu −7 O, Phe −5 N to Ser −8 O and Oγ). The side chains also form hydrogen bonds to Asp-108, Thr-111, and Asp-517. There are 14 hydrogen bonds to water molecules.
Figure 2.

Stereoview of the residues of the pro-sequence and the amino acids with which they form hydrogen bonds. The carbon atoms of the pro-sequence are green, whereas those of the mature sequence are yellow.
Structural Differences Between Precursor and Mature Protein.
The Cα positions of the precursor, excluding the pro-sequence, were aligned with those of the mature enzyme (Protein Data Bank ID code 1GOG) with an rmsd of 1.6 Å. Whereas the majority of the two structures are very similar overall (rmsd of 0.7 Å), residues colored yellow in Fig. 1, which include the five regions of the precursor main chain (1–5, 189–200, 216–225, 255–261, and 290–296) show a rmsd of 5.1 Å, with the greatest deviations found in domain II. Some residues of the active site show significant rearrangements, with movements of side chains and adjacent main-chain regions. In addition, the presence of the pro-sequence affects the position of Ala-1 moving it by over 4 Å from its location in the mature protein.
Of the five main-chain regions that differ, the first comprises residues 1–5 of the mature sequence. The second, loop 189–200, is prevented from occupying its position in the mature protein by the presence of the pro-sequence. The Oη group of the Tyr-189 is hydrogen-bonded to the carbonyl oxygen of Ser −3, as is the Nδ2 group of Asn-191. The third region that differs significantly between the two forms is the strand section 216–227, with main-chain positions differing by up to 6.1 Å from the mature form. In the mature form the side chain of Arg-217 is hydrogen-bonded to Asp-258 (see Fig. 6, which is published as supporting information) of the adjacent strand, but in the precursor it is orientated in the opposite direction and is hydrogen-bonded to the main-chain carbonyl of Phe −5. This results in a difference of 4.6 Å in the Cα position. A similar displacement is found in the amino acids 218–221, with these side chains also oppositely orientated. At the end of this strand is Cys-228, the cysteine of the thioether bond. Differences in this strand affect the position of the fourth region corresponding to the adjacent β-strand (residues 254–261) involved in the lost hydrogen bond to Arg-217. None of the residues in this strand are directly interacting with the pro-sequence. The fifth region is the loop (residues 290–296) containing the active site residue Trp-290, which in the mature protein stacks directly on top of Tyr-272 and the thioether bond. The Cα of residue 290 has moved by 6.3 Å from its site in the mature protein, although the Cα of Ser-289 differs by only 1.5 Å. Lys-297 hardly moves between the two forms, but between Trp-290 and Glu-296 the backbone differs by up to 8.0 Å. The density in this region is quite poor, suggesting high mobility or disorder with an average B factor for all atoms of these residues in the precursor structure of 31.2 Å2 (overall average B factor 11.9 Å2).
At the copper binding site of the precursor protein the loop rearrangement that affects the stacking Trp-290 results in a much more open appearance to the active site (Fig. 3). The two residues forming the thioether bond in the mature enzyme, Cys-228 and Tyr-272, are exposed because of the absence of Trp-290. The thioether bond is not present in the precursor and the side chains of both residues have quite different rotamers from their mature form. The ring of Tyr-272 is rotated almost 90° about χ2 and the side chain of Cys-228 is pointing away from its position in the active protein. These two residues sterically hinder Trp-290 from occupying its mature position. Main-chain movements are smaller, with Cα of Tyr-272 and Cys-228 moving by 1.2 and 1.4 Å, respectively, whereas the residues preceding 228 show quite significant deviations from their mature conformations. Phe-227 undergoes a peptide flip with the carbonyl group pointing toward the active site in the precursor and away in the mature enzyme. The side chain is also affected, with χ1 differing by 125°. Met-226 appears to have two possible main-chain conformations, suggesting disorder or flexibility in this region. The remaining three protein ligands to the copper, Tyr-495, His-496, and His-581, have main-chain Cα movements of less than 0.4 Å (0.4, 0.1, and 0.1 Å, respectively). There is little movement in the side chains of Tyr-495 and His-581, but the side chain of His-496 rotates about χ1 by 37°. The electron density at Cys-228 in the precursor shows additional electron density, suggesting oxidation to an S-OH group (Fig. 3 Right). It is uncertain whether this apparent oxidation represents an intermediate in thioether bond formation or is an artifact resulting from crystallization or radiation damage.
Figure 3.

Active site residues in precursor galactose oxidase (Left) and in the mature protein (Center) showing the copper ligands (Tyr-272, Tyr-495, His-496, and His-581), Cys-228 that forms the thioether bond to Tyr-272, the tryptophan that stacks over it (Trp-290), and Phe-227. The Sγ of Cys-228 in the precursor is situated directly above Cɛ2 of Tyr-272. In the precursor the sulfur of the cysteine appears to have been oxidized to a sulfenic group. (Right) Final 2Fo − Fc electron density map for Cys-228 and Tyr-272.
Discussion
It is known that in vitro processing of galactose oxidase results from addition of copper and oxygen (13). Copper is a highly toxic transition metal that is present in vivo as less than one free copper ion per cell (26). Intracellularly, copper is associated with metallochaperone proteins that deliver it, by direct interaction, to target copper-requiring proteins (26–28). Because galactose oxidase can be expressed in heterologous organisms it seems unlikely that these contain a suitable copper chaperone to facilitate intracellular copper delivery to this enzyme. This makes it likely that processing occurs outside the cell where free copper levels will be higher.
Several secreted enzymes are produced in a prepro form in which the pro-sequence acts as an intramolecular chaperone and/or inhibitor of catalytic activity. The best-studied examples are proteases where the pro-sequence is relatively long, comprising some 70 to more than 200 aa (29–33). Processing, commonly by protease activity, leads to removal of the pro-sequence and activation of the enzyme. It has been shown that mutations in the pro-sequence of subtilisin E can influence the folding pathway and final conformation of the active enzyme (34). For secreted enzymes, the pro-sequence can enhance the level of secretion (29, 30). The short length of the galactose oxidase pro-sequence (17 aa) is more similar to that of fungal cutinase (35) than to proteases. This prevents it from acting as a direct steric inhibitor of the active site (Fig. 1) but does not rule out an inhibitory role through the influence on the structure of the active site. Alternatively the pro-sequence may represent an intramolecular chaperone responsible for positioning of active site residues, including copper ligands, to facilitate the chemical processes required for thioether bond formation.
In the latter regard, three of the four copper ligands (Tyr-495, His-496, and His-581) are in very similar positions to their mature conformation, suggesting that copper may be able to bind to the proform. The presence of the pro-sequence directly affects three peptide regions: residues 1–5, 189–200, and 216–227. At the end of the third of these is Cys-228, the cysteine of the thioether bond. The sulfur of Cys-228 is directly over the pz orbital of Cɛ2 of Tyr-272 (Fig. 3 Left) at a distance of 3.6 Å. This geometry is essential for Cys-228 to attack Tyr-272 by a nucleophilic, electrophilic, or radical coupling mechanism. It should be noted, however, that rotational freedom of Tyr-272 and Cys-228 in concert might allow an active site rearrangement to occur before thioether bond formation, if required.
To produce the oxidized, catalytically active enzyme requires a three-electron oxidation of the Cu(II)-containing precursor enzyme. We have previously suggested two potential mechanisms for the Tyr-272/Cys-228 coupling reaction, in which an initial intramolecular Tyr-272→Cu electron transfer is followed by either a radical coupling reaction or nucleophilic attack of Cys-228 at a peroxide-activated tyrosine ring (Fig. 4a) (13). Both the radical and ionic mechanisms plausibly result in a [Cu(II)-Y−] intermediate (A in Fig. 4). The structure and reactivity of A must differ from the semireduced state of the mature enzyme [which also contains Cu(II)-coordinated tyrosine] because the latter is apparently inert to oxidation by O2 (36, 37). There is a precedent for the initial electron-transfer step in Fig. 4a (38), whereas the subsequent radical coupling (39) and peroxidation (ref. 40 and references therein) steps also represent well-known reaction pathways in phenoxyl chemistry. Our observation of apparent oxidation of Cys-228 in the precursor protein, however, suggests a third potential mechanism. This involves the initial oxygenation of Cys-228 to a sulfenate or perhaps a sulfinate derivative, which could then act as an electrophile toward Tyr-272 (Fig. 4b). The oxygenation step might be copper-dependent, because metal-induced oxygenation of thiolates is a well-known reaction (41). Cysteine-sulfenic acid is known to function as a redox center in NADH oxidases and peroxidases, and in some peroxiredoxins and transcription regulators (42). Our suggestion that it might act as an electrophile in galactose oxidase maturation, however, corresponds to a new role for this residue in biochemistry. Generally, sulfenate derivatives require activation (e.g., to a sulfenyl chloride) to enhance their electrophilic reactivity. We suggest in Fig. 4b that Cu(II) coordination could labilize hydroxide; alternatively protonation of the hydroxide would make it an excellent leaving group. A sulfoxide (R2S=O) intermediate, as might result from the reaction of Tyr-272 with a Cys-228 sulfinate, has previously been reported in a thioether bond-forming reaction between Sγ of a cysteine and Cɛ1 of a histidine derivative (43). However, the requirement for reduction of the sulfoxide to the thioether is apparently inconsistent with the previous observation that oxidizing conditions are sufficient for biogenesis to occur.
Figure 4.

Potential mechanisms for thioether bond formation in galactose oxidase. (a) Mechanisms involving initial Tyr→Cu electron transfer. (b) Mechanisms involving initial mono-oxygenation of Cys-228 to a sulfenate group. Species A is a [Cu(II)-Y−] intermediate that is common to both mechanisms.
The order of events for maturation of the protein has not yet been definitively determined. However, results from SDS/PAGE analysis of copper limited preparation (13) suggest that pro-sequence cleavage may occur before thioether bond formation because a band corresponding to a protein with both the pro-sequence and thioether bond has not been identified.
Although the structure reveals the site of pro-sequence cleavage, it does not point to any obvious mechanism. The removal of the pro-sequence requires copper and oxygen (13), but the structure reveals no evidence of an additional binding site for copper near the cleavage point. Site-specific cleavage events have been observed when copper binds close to the site of a disulfide bridge in the amyloid precursor protein that has been implicated in Alzheimer's disease (44). The site of cleavage in galactose oxidase is also near the disulfide formed between Cys-515 and Cys-518.
The high-resolution crystal structures for both precursor and mature forms of galactose oxidase now provide an excellent basis for studies to understand the unusual copper- and oxygen-mediated processing events by trapping of structural and kinetic intermediates.
Supplementary Material
Acknowledgments
We thank Susan Girdwood and the staff of the Daresbury Synchrotron Radiation Source for assistance with data collection. This work was supported by the Biotechnology and Biological Sciences Research Council/Engineering and Physical Sciences Research Council and National Institutes of Health Grant GM 27659. S.J.F is supported by an Engineering and Physical Sciences Research Council studentship, and M.A.H. is a Royal Society (London) Research Fellow. We are also grateful for facilities provided by the Biotechnology and Biological Sciences Research Council-funded North of England Structural Biology Centre.
Abbreviation
- rmsd
rms deviation
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1K3I).
See commentary on page 12863.
References
- 1.Kosman D J, Ettinger M J, Weiner R E, Massaro E J. Arch Biochem Biophys. 1974;165:456–467. doi: 10.1016/0003-9861(74)90271-9. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker M M, Whittaker J W. J Biol Chem. 1990;265:9610–9613. [PubMed] [Google Scholar]
- 3.Ito N, Phillips S E V, Stevens C, Ogel Z B, McPherson M J, Keen J N, Yadav K D S, Knowles P F. Nature (London) 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 4.Okeley N M, Van der Donk W A. Chem Biol. 2000;7:R159–R171. doi: 10.1016/s1074-5521(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 5.Whittaker M M, Kerstern P J, Nakamura N, Sanders-Loehr J, Schweizer E S, Whittaker J W. J Biol Chem. 1996;271:681–687. doi: 10.1074/jbc.271.2.681. [DOI] [PubMed] [Google Scholar]
- 6.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L Y, Doi N, Durley R C E, Chistoserdov A Y, Lidstrom M E, Davidson V L, Mathews F S. J Mol Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 8.Klabunde T, Eicken C, Sacchettini J C, Krebs B. Nat Struct Biol. 1998;5:1084–1090. doi: 10.1038/4193. [DOI] [PubMed] [Google Scholar]
- 9.Janes S M, Mu D, Wemmer D, Smith A J, Kaur S, Maltby D, Burlingame A L, Klinman J P. Science. 1990;248:981–987. doi: 10.1126/science.2111581. [DOI] [PubMed] [Google Scholar]
- 10.Cai D, Klinman J P. Biochemistry. 1994;33:7647–7653. doi: 10.1021/bi00190a019. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzaki R, Fukui T, Sato H, Osaki Y, Tanizawa K. FEBS Lett. 1994;351:360–364. doi: 10.1016/0014-5793(94)00884-1. [DOI] [PubMed] [Google Scholar]
- 12.Ruggiero C E, Smith J A, Tanizawa K, Dooley D M. Biochemistry. 1997;36:1953–1959. doi: 10.1021/bi9628836. [DOI] [PubMed] [Google Scholar]
- 13.Rogers M S, Baron A J, McPherson M J, Knowles P F, Dooley D M. J Am Chem Soc. 2000;122:990–991. [Google Scholar]
- 14.McPherson M J, Ogel Z B, Stevens C, Yadav K D S, Keen J N, Knowles P F. J Biol Chem. 1992;267:8146–8152. [PubMed] [Google Scholar]
- 15.Baron A J, Stevens C, Wilmot C, Seneviratne K D, Blakeley V, Dooley D M, Phillips S E V, Knowles P F, McPherson M J. J Biol Chem. 1994;269:25095–25105. [PubMed] [Google Scholar]
- 16.McPherson M J, Stevens C, Baron A J, Ogel Z B, Seneviratne K, Wilmot C M, Ito N, Brocklebank L, B, Phillips S E V, Knowles P F. Biochem Soc Trans. 1993;21:752–756. doi: 10.1042/bst0210752. [DOI] [PubMed] [Google Scholar]
- 17.Collaborative Computational Project Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 18.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 19.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–325. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Adams P D, Pannu N S, Read R J, Brünger A T. Proc Natl Acad Sci USA. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Pannu N S, Read R J. Acta Crystallogr A. 1996;52:659–668. [Google Scholar]
- 24.Kleywegt G J. Acta Crystallogr D. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 25.Ito N, Phillips S E V, Yadav K D S, Knowles P F. J Mol Biol. 1994;238:794–814. doi: 10.1006/jmbi.1994.1335. [DOI] [PubMed] [Google Scholar]
- 26.Rae T D, Schmidt P J, Pufahl R A, Culotta V C, O'Halloran T V. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 27.Harrison M D, Jones C E, Solioz M, Dameron C T. Trends Biochem Sci. 2000;25:29–32. doi: 10.1016/s0968-0004(99)01492-9. [DOI] [PubMed] [Google Scholar]
- 28.Rosenzweig A C, O'Halloran T V. Curr Opin Chem Biol. 2000;4:140–147. doi: 10.1016/s1367-5931(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 29.Eder J, Fersht A R. Mol Microbiol. 1995;16:609–614. doi: 10.1111/j.1365-2958.1995.tb02423.x. [DOI] [PubMed] [Google Scholar]
- 30.Baardsnes J, Sidhu S, MacLeod A, Elliott J, Morden D, Watson J, Borgford T. J Bacteriol. 1998;180:3241–3244. doi: 10.1128/jb.180.12.3241-3244.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nirasawa S, Nakajima Y, Zhang Z Z, Yoshida M, Hayashi K. Biochem J. 1999;341:25–31. [PMC free article] [PubMed] [Google Scholar]
- 32.Marie-Claire C, Ruffet E, Beaumont A, Roques B P. J Mol Biol. 1999;285:1911–1915. doi: 10.1006/jmbi.1998.2449. [DOI] [PubMed] [Google Scholar]
- 33.Sellman B R, Tweten R K. Mol Microbiol. 1997;25:429–440. doi: 10.1046/j.1365-2958.1997.4541820.x. [DOI] [PubMed] [Google Scholar]
- 34.Shinde U, Fu X, Inouye M. J Biol Chem. 1999;274:15615–15621. doi: 10.1074/jbc.274.22.15615. [DOI] [PubMed] [Google Scholar]
- 35.Longhi S, Cambillau C. Biochim Biophys Acta. 1999;1441:185–196. doi: 10.1016/s1388-1981(99)00159-6. [DOI] [PubMed] [Google Scholar]
- 36.Hamilton G A, Adolf P K, DeJersey J, Dubois G C, Dryacz G R, Libby R D. J Am Chem Soc. 1978;100:1899–1912. [Google Scholar]
- 37.Kosman D. In: Copper Proteins and Copper Enzymes. Lontie R, editor. Vol. 2. Boca Raton, FL: CRC; 1984. pp. 1–26. [Google Scholar]
- 38.Fujisawa K, Iwata Y, Kitajima N, Higashimura H, Kubota M, Miyashita Y, Yamada Y, Okamoto K, Moro-oka Y. Chem. Lett. 1998. 739–740. [Google Scholar]
- 39.Nonhebel D C, Walton J C. Free-Radical Chemistry. Cambridge, U.K.: Cambridge Univ. Press; 1974. pp. 326–345. [Google Scholar]
- 40.d'Alessandro N, Bianchi G, Fang X, Jin F, Schuchmann H P, von Sonntag C. J Chem Soc Perkin Trans. 2000;2:1862–1867. [Google Scholar]
- 41.Grapperhaus C A, Darensbourg M Y. Acc Chem Res. 1998;31:451–459. [Google Scholar]
- 42.Claiborne A, Yeh J I, Mallet T C, Luba J, Crane E J, III, Charrier V, Parsonage D. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 43.Ishikawa Y, Israel S E, Melville D B. J Biol Chem. 1974;249:4420–4427. [PubMed] [Google Scholar]
- 44.Multhaup G, Ruppert T, Schlicksupp A, Hesse L, Bill E, Pipkorn R, Masters C L, Beyreuther K. Biochemistry. 1998;37:7224–7230. doi: 10.1021/bi980022m. [DOI] [PubMed] [Google Scholar]
- 45.Christopher J A. SPOCK: The Structural Properties Observation and Calculation Kit. Texas A&M University, College Station: The Center for Macromolecular Design; 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}