

Abstract
Objective
Pancreatic ductal adenocarcinoma (PDA) is a highly metastatic disease with limited therapeutic options. Genome and transcriptome analyses have identified signaling pathways and cancer driver genes with implications in patient stratification and targeted therapy. However, these analyses were performed in bulk samples and focused on coding genes, which represent a small fraction of the genome.
Design
We developed a computational framework to reconstruct the non-coding transcriptome from cross-sectional RNA-Seq, integrating somatic copy number alterations (SCNA), common germline variants associated to PDA risk, and clinical outcome. We validated the results in an independent cohort of paired epithelial and stromal RNA-Seq derived from laser capture microdissected human pancreatic tumors, allowing us to annotate the compartment-specificity of their expression. We employed systems and experimental biology approaches to interrogate the function of epithelial lncRNAs associated with genetic traits and clinical outcome in PDA.
Results
We generated a catalogue of PDA-associated lncRNAs. We showed that lncRNAs define molecular subtypes with biological and clinical significance. We identified lncRNAs in genomic regions with SCNA and SNPs associated with lifetime risk of PDA and associated with clinical outcome using genomic and clinical data in PDA. Systems biology and experimental functional analysis of two epithelial lncRNAs (LINC00673 and FAM83H AS1) suggest they regulate the transcriptional profile of pancreatic tumor samples and PDA cell lines.
Conclusions
Our findings indicate that lncRNAs are associated with genetic marks of pancreatic cancer risk, contribute to the transcriptional regulation of neoplastic cells and provide an important resource to design functional studies of lncRNAs in PDA.
Keywords: Pancreatic cancer, Gene Regulation, Epithelial cells, Cancer genetics, RNA expression
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is the third–leading cause of cancer mortality in western countries, and is projected to become the second-leading cause by 2030, surpassed only by lung cancer.(1) The median overall survival of PDA patients is less than six months, and only 8% of patients survive more than five years.(2) PDA is characterized by high penetrance mutations in four genes (KRAS, TP53, CDKN2A, and SMAD4). Large–scale sequencing efforts also identified a host of low–frequency alterations in coding genes, with an average of 60 total per patient. However, while these efforts led to novel molecular classification approaches for the disease, it was learned that only a low fraction of PDAs harbor “actionable” mutations.(3-7)
The introduction of unbiased expression profiling technologies like RNA-Seq has resulted in a greater appreciation for the extent of transcription arising from non-coding regions.(8) In particular, transcriptomic analyses across several human tissues and cell lines have identified roughly ten thousand long non-coding RNAs (lncRNAs), which are defined as transcripts longer than 200 nucleotides that lack coding potential.(9, 10) To date, only a few cancer-associated lncRNAs have been extensively characterized;(11) however, there is increasing evidence suggesting that these transcripts play a role in tumorigenesis: 1) genome–wide expression analysis showed that lncRNAs are deregulated in cancer and associated with tumor progression;(10) 2) analysis of copy number somatic variations identified lncRNAs drivers of tumorigenesis;(12, 13) and 3) 93% of the germline variants associated with risk susceptibility identified by genome wide association studies (GWAS) map to chromatin-modified non-coding regions with the features of lncRNA loci.(14) Indeed GWAS in PDA identified risk alleles near lncRNAs.(15) In light of these data, we hypothesized that lncRNAs are an integral part of the signaling pathways that regulate the initiation and progression of PDA, and we sought to develop a means to identify lncRNAs that play a role in this challenging disease.
Here, we present NORI (Non-coding RNA Identification), an open source computational tool to identify lncRNAs using next generation sequencing. We applied NORI to human PDA and identified lncRNAs that we further annotated with information pertinent to somatic recurrent genomic alterations, cancer-enriched germline variants, and clinical prognosis. In addition, we determined epithelial or stromal expression by integrating a cohort of RNA-Seq samples obtained from a large collection of laser captured microdissected (LCM) PDA samples. Functional analysis of two epithelial–enriched PDA–associated lncRNAs validate the significance of the integrative analysis.
RESULTS
Identification of lncRNAs in PDA
We developed NORI, a computational approach to annotate and characterize the non-coding transcriptome using next-generation sequencing data (Fig 1a). We applied NORI to reconstruct the non-coding transcriptome of 109 PDA human samples deposited at The Cancer Genome Atlas (TCGA). We identified 3433 lncRNAs including several described as drivers of tumor progression or as functional regulators of organ development such as UCA1,(16) DEANR1,(17) PVT1,(13) NBR2,(18) MALAT1,(19) and NEAT1 (Sup Table 1).(20) These data confirm the capability of NORI to reconstruct non-coding transcriptomes, and constitute an initial set of PDA-associated lncRNAs as the basis for further analysis.
Figure 1. Identification of lncRNAs and molecular subtyping of PDA.

(a) Schematic representation of the computational analysis. NORI identified 3433 lncRNAs expressed in PDA using RNAseq from a cohort of 109 tumors from TCGA. The output of NORI was subset into abundant lncRNAs (RPKM>1) prioritized for experimental validation, and lncRNAs whose expression correlates (q<0.001) with the allele frequency of PDA driver genes for the identification of molecular subtypes in PDA by non-negative matrix factorization (NMF). Abundant lncRNAs were annotated with the genomic distance to recurrent SCNA and/or SNPs associated with PDA risk and with the expression correlation with clinical outcome. In addition, an independent cohort of LCM PDA samples (n=66 epithelium, 65 stroma) were analyzed to validate expression of lncRNAs in PDA and to select epithelial lncRNAs for functional analysis. (b) NMF using the expression of lncRNAs identified three molecular subtypes in the TCGA cohort (n=147). (c) Kaplan-Meier disease free survival estimations for the individual subtypes. (d) Differential gene expression analysis between molecular subtypes. Relevant genes are shown (see sup table 3 for full list). Each TCGA sample is color coded according to the molecular subtype. KRASmut allele frequency (AF) is depicted as an independent estimation of tumor cellularity of each sample.
Molecular subtyping defined by the expression of lncRNAs
Global analyses of coding transcripts in PDA have been used to dissect molecular subtypes of the disease.(3, 6, 7) We investigated whether lncRNAs might prove useful for the classification of molecular subtypes independent of coding genes. Because expression profiles from TCGA samples represent a mixture of both malignant epithelial cells and stromal cells, we used a computational approach to focus our classification effort on lncRNAs whose expression correlates with the allele frequency of the major drivers in PDA (KRAS, TP53, CDKN2A, and SMAD4), which are mutated in 97% of the TCGA samples (Material and Methods, Sup Table 2). We used the resulting 652 genes to define molecular subtypes, applying non-negative matrix factorization to 147 PDA samples from the TCGA. This analysis revealed the presence of three molecular subtypes, with a cophenetic coefficient of 0.9931 (Fig 1b, Sup Fig 1). To determine whether molecular subtyping was an indirect effect of the transcription of neighbor coding genes, we removed lncRNAs that were within 10kb of coding genes and still obtained very similar clustering (p=7.01 10−50 chi-square test, Sup Fig 2). Clusters 1 and 2 were associated with elevated mutant KRAS allele frequencies, while tumors in Cluster 3 were associated with low frequencies, perhaps indicating that this group emerged because of the inclusion of higher amounts of stroma and/or infiltrated normal tissues. Analysis of outcomes data available through the TCGA indicated an association of tumors from Cluster 2 with reduced disease-free survival relative to those in Cluster 1 and 3 (log-rank test, p=0.0028) (Fig 1c).
To understand the biological significance, we performed differential gene expression analysis (Fig 1d, Sup Table 3). In Cluster 1, we observed enrichment of transcription factors necessary in pancreas development, including FOXA2, FOXA3, GATA6, GATA4, PDX1, MNX1, HNF1b, HNF4g, HNF4a.(3) Many of these genes, as well as others involved in lineage specification, are enriched in the previously identified “Classical” molecular subtype, which was associated with improved overall survival relative to the “Basal-like” subtype. Notably, Cluster 2 included several genes found in the Basal-like subtype and associated with EMT, including TP63, CAV1, SNAI2, MET, HMGA2 and TGFβ. Finally, in Cluster 3, we observed genes related to digestive processes (PLA2G1B, PRSS1, PRSS3), endocrine function (INS, GCG, SST), and the immune system (CD48, CCR2, GIMAP proteins), suggesting that this subtype is defined by the contributions of non-neoplastic cell types. Together these findings demonstrate that lncRNAs reflect the heterogeneity of biological processes in PDA with relevance to clinical outcomes.
Annotation of lncRNAs in PDA
We next sought to prioritize the 3433 lncRNAs expressed in PDA. First, we filtered out low-abundance transcripts, yielding 453 lncRNAs with a mean expression >1 RPKM. Next, we investigated the association of lncRNAs with relevant genomic and clinical features of the disease, including: i) proximity to PDA-associated coding genes;(21) ii) location within SCNA in PDA; iii) proximity to germline variants identified in GWAS; and iv) correlation with clinical outcome data (Fig 2a, Sup Table 1). As expected, analysis of chromatin modifications at these loci found significant enrichment of accessible chromatin regions related to transcriptional activity (Fig 2b, Sup Fig 3; p<0.001, permutation test). Nonetheless, this analysis allowed us to discriminate between bona fide lncRNAs and spurious transcripts or sequencing artifacts.
Figure 2. Annotation of lncRNA with genomic threats of pancreatic cancer and identification of epithelial or stromal expression.
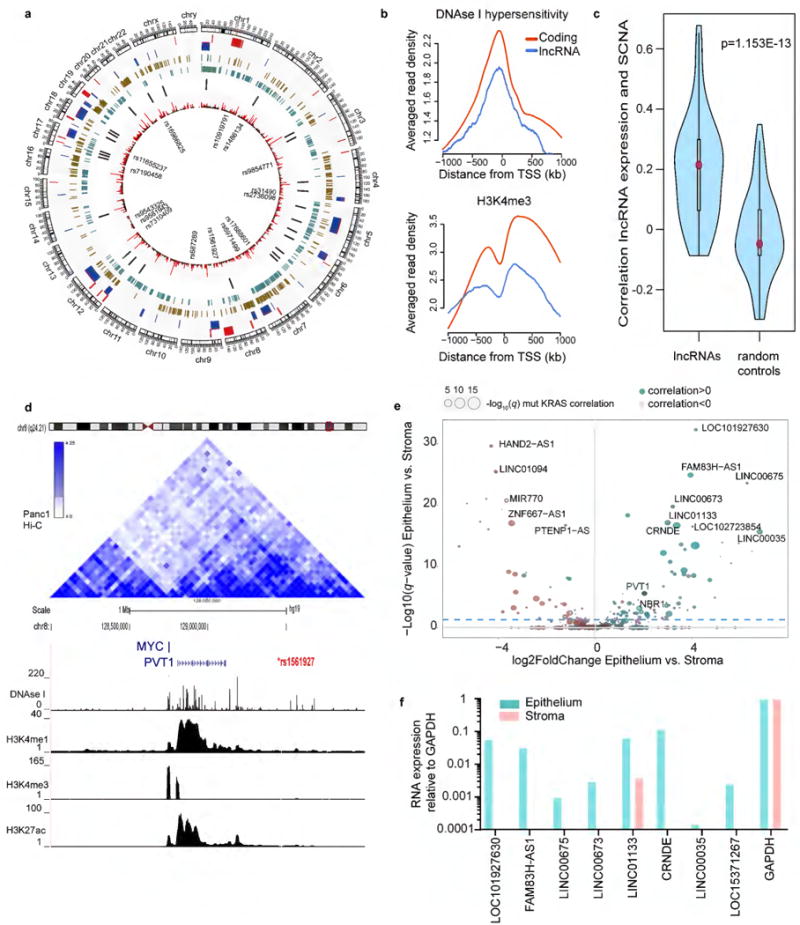
(a) Circos plot depicting location of lncRNAs respective to genomic marks associated with PDA. From inner to outer: SNPs associated to lifetime risk of PDA; lncRNAs identified by NORI (Red: expression > 1 RPKM); Location of PDA associated cancer genes described in supplementary table 1; DNase I hypersensitivity and H3K4me3 in PANC1 cells; recurrent SCNA in the TCGA cohort, amplifications (red) and deletions (blue). The outermost ring shows the chromosomes in clockwise order with sex chromosome at the end. Full annotation of lncRNAs is provided in supplementary table 1. (b) Averaged reads density of DNAseI signal (upper) and H3K4me3 (lower) along the TSS region of ±1kb, summarized for lncRNA and coding genes respectively. Reads depth are log transformed and averaged on each base. (c) Comparison of expression-SCNA correlations on 85 lncRNAs with random controls. P-value is calculated from Wilcox rank sum test.(d) UCSC snapshot of the PVT1 locus, location of the SNP associated with lifetime risk of PDA (red) and TADs in PANC1 cells indicative of higher order of genome organization. The genomic regions are overlap with DNAseI hypersensitivity and epigenetic marks of active transcription in PANC1 cells (ENCODE data). For clarity, only PVT1 and MYC are shown. (e) Scatter plot showing distribution of lncRNAs according to epithelial and stromal expression as determined by LCM RNAseq data (n=131). In addition, as an independent metric for expression in neoplastic epithelium, the size of each circle represents the correlation of lncRNA expression with the allele frequency of KRAS mutation. (f) Validation of epithelial enrichment for the top epithelial lncRNAs. Analysis performed in a pool of epithelial and stromal samples from the CUMC cohort. N=3 technical replicates. Only the eight out of ten candidates that were validated are shown. Expression relative to GAPDH.
Somatic copy number alterations
SCNA are frequently the subject of clonal selection during tumor progression. However, many SCNA lack known coding tumor-drivers,(22, 23) perhaps suggesting that unknown non-coding drivers may be located within these genomic regions. Analysis of SNP data by GISTIC2 in the TCGA cohort revealed 56 recurrent SCNA, 23 amplifications and 33 deletions, including known events in PDA such as amplification of GATA6, KRAS and MYC; and deletions of CDKN2A and SMAD4. We examined the overlap of SCNA genomic locations in PDA with our candidate lncRNAs and observed that 85 of 453 lncRNAs were located within the 56 SCNA identified in the same cohort of patients (Sup Table 1). Significantly higher lncRNA expression-SCNA correlations were found in the 85-paired lncRNAs than in random controls (Fig 2c). Among those lncRNAs, we detected expression of PVT1(13); LINC-PINT;(24) and the antisense lncRNA for GATA6 Given the paucity of established cancer-associated lncRNAs, the identification of several such genes in PDA-associated SCNAs serves as conceptual validation for this approach.
Germline variants associated with PDA
GWAS studies have identified 14 genetic loci associated with increased or reduced lifetime risk of PDA.(15) Interestingly, four of these loci map to genomic regions known to contain functional lncRNAs (PVT1, LINC-PINT, PDX1-AS1 and LINC00673) while several others mapped to unannotated non-coding regions.(26) We performed a similar analysis and identified five candidate lncRNAs within loci that harbored somatic SNP variants associated with increased risk of PDA (Sup Table 1). We detected expression of PVT1, LINC-PINT and LINC00673. In addition, we identified a novel lncRNA located on chromosome 9q34.2 near the ABO gene that is associated with PDA risk ; and LINC01829, located on chromosome 2p14 upstream of ETAA1, a recently characterized protein involved in DNA damage signaling.(27) Furthermore, analysis of chromatin features and topological associated domains (TADs) in PANC1 cells showed chromatin interactions with distant SNPs, suggesting a cis-regulation (Fig 2d, Sup Fig 4). Overall, we identified expression of five PDA lncRNAs that are near SNPs associated with increased risk of pancreatic cancer.
Association with clinical outcome
We also examined the association between the expression of each lncRNA and overall or disease–free survival in the TCGA cohort. We observed that 23 (5.3%) and 36 (7.9%) of 453 lncRNAs were significantly (p<0.05) associated with overall survival and disease free survival respectively (Sup Table 1). This is similar to the fraction of coding genes that are associated with survival and progression differences in PDA (5.7% and 4.1%, respectively), further supporting a possibility of a functional role for lncRNAs in driving PDA.
In summary, we have generated a large-scale resource describing lncRNAs expressed in PDA annotated with information on their expression level, association with genomic landmarks, and association with clinical outcomes. We expect this resource will serve as an aid for functional studies on the contribution of lncRNAs to the progression of pancreatic cancer.
Independent validation of PDA lncRNAs using compartment–specific expression data
Previous studies using both correlative and functional analyses have established that lncRNAs can act as regulators of oncogenic pathways in malignant cells.(28, 29) However, bulk tumor RNA-Seq contains a mixture of transcripts originating from neoplastic epithelial cells and non-neoplastic stromal cells. To explore the cellular origins of the candidate lncRNAs, we performed RNA-Seq on LCM samples from 65 human PDA patients who underwent resection with the Pancreas Center at the Columbia University Medical Center (CUMC) (He et al, in revision). Notably, we detected 80% of the 453 candidate PDA lncRNAs in the CUMC cohort, providing an independent validation set for our earlier analysis. We annotated each lncRNA with its relative enrichment in the stroma or epithelium of PDA samples (Sup Table 1). We identified 138 compartment-enriched lncRNAs (q<0.05, DESeq2) of which 94 are enriched in the epithelium and 44 in the stroma (Fig 2e, Sup table 4). The expression of the top 10 epithelial and stromal candidates as detected by RNA-Seq is depicted in Sup Fig 5. We validated the expression of eight of the 10 epithelial candidates by orthogonal methods in a pool of six random epithelial and stromal samples from the CUMC cohort (Fig 2f). Consistent with previous clustering results, we identified two molecular subtypes in the CUMC cohort that were functionally characterized by gene sets associated with differentiation state (Sup table 5). Overall, these analyses validate the expression of selected lncRNAs in two independent PDA cohorts and confirms our non-coding transcriptome reconstruction approach. Together, these data provide a rich resource of validated PDA-associated lncRNAs annotated with information on their compartment of origin.
Functional validation of epithelial lncRNAs
Next, we sought to study the functional roles of top candidate PDA-associated lncRNAs. We focused our analysis on lncRNAs enriched in the epithelium as potential regulators of molecular pathways altered in malignant cells. In addition, we selected lncRNAs located in genomic regions that have been associated with focal amplification or deletions as potential drivers of tumor progression. We became interested in FAM83HAS1 and LINC00673.
FAM83H-AS1 is the antisense of FAM83H, a recently described coding-gene required for the organization of the keratin cytoskeleton in epithelial cells.(30) The two genes share a promoter region but transcribed in opposite directions (Fig 3a). FAM83H-AS1 is located on chromosome 8 on a genomic region frequently amplified in PDA (8q23.3-8q24.3). We found that FAM83H-AS1 expression correlates significantly with amplification (r=0.67, p=6.5×10-21, Sup Fig 6). RefSeq gene annotation and ENCODE data indicate that it has four exons and that it is located in an actively transcribed chromatin region in PANC1 (Fig 3a, Sup Fig 7). Importantly, high expression of FAM83H-AS1 showed a borderline association with poor clinical outcome (p=0.056, Fig 3b) and FAM83H-AS1 expression across a panel of cancer cell lines showed elevated expression in PDA lines (Fig 3c). We explored the function of FAM83H-AS1 by targeting the gene with two siRNAs in Aspc1 cells, each of which resulted in >60% knockdown efficiency (Fig. 3d). RNA-Seq analysis with transient knockdown of FAM83H-AS1 identified a 1,309 and 2,721 differentially expressed genes with siRNA1 and siRNA2 respectively, and principal component analysis (PCA) clustered samples according to the siRNA (Fig 3e). There was significant overlap in the sets of genes differentially expressed in response to the two siRNAs (Fisher’s exact test, p<2.2×10−16), as expected (Fig 3f). Gene set enrichment analysis (GSEA) of common dysregulated gene sets suggest a less aggressive phenotype when FAM83H-AS1 is downregulated, consistent with our observation of a worse prognosis for pancreatic tumors expressing high levels of FAM83H-AS1 (Sup Fig 8a, Sup Table 6).
Figure 3.
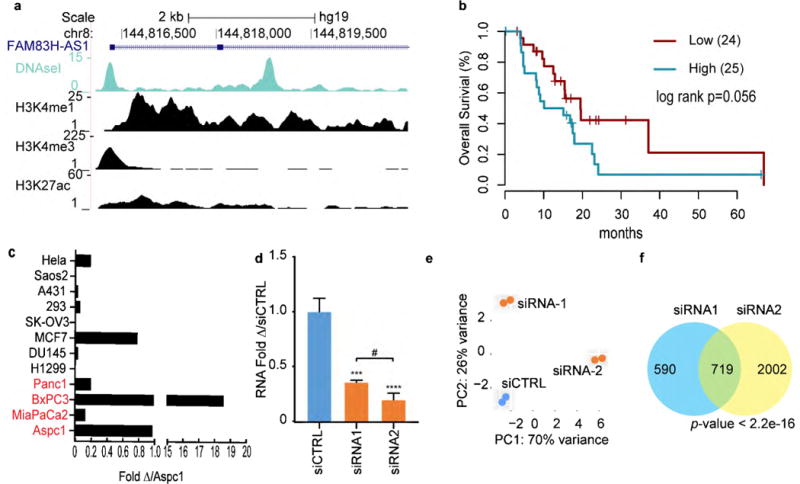
FAM83H-AS1 regulates the transcriptome profile of Aspc1 cells. (a) UCSC snapshot of the FAM83H-AS1 transcriptional start site (TSS) depicting DNase I hypersensitivity and chromatin modifications in PANC1 cells (ENCODE). (b) Kaplan-Meier overall survival estimations for samples with high and low expression of FAM83H AS1. Only samples with KRASmut AF>0.2 were considered. The two groups were defined by partitioning the samples into two equal-sized sets using the median value of FAM83HAS1 expression. (c) FAM83H-AS1 expression across a panel of cell lines. Normalized with GAPDH and relative to the expression in Aapc1. Pancreatic cancer cell lines depicted in red. (d) FAM83H-AS1 RNA expression after transient transfection of Aspc1 cells with two different siRNAs. (e) Cluster of RNAseq samples by principle component analysis. (f) Overlap of dysregulated genes (padj<0.05) with individual siRNAs. Fisher exact test.
As a complementary approach to understand the functions of this lncRNA, we used an information theory–based systems biology technique called regulatory network analysis. Briefly, a regulatory network delineates interactions between regulatory genes (that is, genes whose activity alters RNA transcript abundance) and their target genes. We used the ARACNe algorithm (31) to reconstruct de novo a regulatory network from TCGA expression data in an unbiased manner (Sup Table 7), providing a list of inferred target genes for known transcription factors as well as the 453 lncRNAs defined above. The network comprised over 300,000 total interactions between 1,813 total regulatory genes and 453 lncRNAs. In particular, ARACNe inferred 146 potential target genes for FAM83H-AS1, including 78 positive (activated) targets and 68 negative (inhibited) targets. Computed overlap with gene sets (MSigDB v6.1) found that genes inferred to be negatively regulated by FAM83H-AS1 were associated with more benign processes while positive inferred targets of FAM83H-AS1 were associated with more malignant processes. (Sup Fig 8b, Sup Table 7). Together these data are consistent with a role for FAM83H-AS1 in promoting tumor progression.
Another top candidate identified in our analysis is LINC00673, a transcript that is among the most epithelial-enriched PDA-associated lncRNAs. LINC00673 is located in a recurrent, focally amplified region in PDA and is linked to a PDA–associated SNP. It is located approximately 275 kb telomeric of SOX9 (Sup Fig 7). Notably, SOX9 is a well-known transcription factor expressed in multipotent pancreatic progenitor cells and is required for the neoplastic transformation of PanIN lesions in a mouse model of PDA.(32) ChIP-Seq analysis from ENCODE in PANC1 cells showed enrichment of chromatin modifications associated with active transcription at the promoter of LINC00673 in PANC1 cells (Fig 4a). In addition, we detected that LINC00673 expression correlates with SCNA (r=0.39, p=1.3×10−6, Sup Fig 6) and high expression of LINC00673 in TCGA PDA samples is significantly associated (p=0.050) with better survival (Fig 4b). Expression analysis across various cell lines showed widespread LINC00673 expression in PDA cells (Fig 4c).
Figure 4.
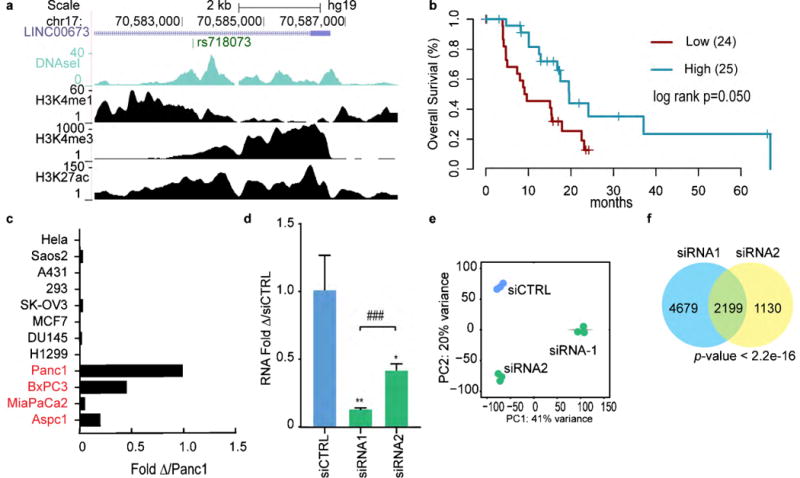
LINC00673 regulates the transcription profile of pancreatic cancer cells and is necessary to maintain epithelial features. (a) UCSC snapshot of the LINC00673 locus as described for FAM83H-AS1 in figure 3. (b) Kaplan-Meier overall survival estimations for tumor samples with high and low expression of LINC00673. Only samples with KRASmut AF>0.2 were considered. The two groups were defined by partitioning the samples into two equal-sized sets using the median value of LINC00673 expression. (c) LINC00673 expression across a panel of PDA cell lines. Normalized with GAPDH and relative to the expression in PANC1. PDA cell lines depicted in red. (d) LINC00673 RNA expression after transient transfection of PANC1 cells with two different siRNAs. (e-g) RNAseq was performed in PANC1 cells transiently transfected with two different siRNAs and a non-targeting control. (e) Principal component analysis. (f) Overlap of dysregulated genes (padj<0.05) with both siRNAs. Fisher exact test.
To test the function of LINC00673, we silenced its expression in PANC1 cells through transient transfection with two different siRNAs. Each siRNA efficiently downregulated the expression of LINC00673, albeit to different levels (Fig 4d). To gain insight into the mechanism of action of LINC00673 we profiled gene expression in PANC1 cells following LINC00673 silencing. The resulting RNA-Seq profiles clustered by treatment group by PCA (Fig 4e). Notably, there was significant overlap in the sets of genes dysregulated by the two siRNAs (Fisher’s exact test, p<2.2×10−16) with a larger total number of differentially expressed in cells treated with the more effective siRNA (Fig 4f), consistent with a dose-dependent effect. Critically, GSEA performed on the differentially expressed genes from each siRNA revealed many overlapping processes, particularly pathways related to EMT in downregulated genes (Sup Fig 9a, Sup Table 8). Next, we applied regulatory network analysis to identify candidate target genes for LINC00673. We inferred 123 potential targets of LINC00673, including 91 positive and 32 negative targets. Overlap of candidate targets with gene sets from MsigDB demonstrated enrichment for gene sets related to maintenance of epithelial properties and downregulation of metastasis (positive) and downregulation of cell to cell communication (negative) (Sup Fig 9b, Sup Table 7), consistent with the results from cell lines.
Functional analysis of cells transiently depleted of LINC00673 by siRNA show impaired colony formation, and this effect cannot be explained by defects in cell cycle (Sup Fig 10). Interestingly, the less efficient siRNA2 produced an intermediate phenotype, again supporting a dose dependency in the function of LINC00673 (Fig 5a). These results were reproduced in two additional human PDA cell lines, MiaPaca2 and BxPC3 (Sup Fig 11). Furthermore, we observed that knockdown of LINC00673 resulted in increased cell motility in several PDA cell lines (Fig 5b, Sup Fig 12), consistent with the effects of EMT. In addition, we assessed the metastatic potential of PDA cells after LINC00673 downregulation by injecting PANC1/Luc cells pretreated with siCTRL or siRNA1 in the spleen of nude mice. We determined that PANC1 cells were more efficient in producing metastatic lesions when LINC00673 was downregulated (Fig 5c). Overall, these data suggest that LINC00673 regulates in vitro and in vivo the metastatic potential of PDA cell lines.
Figure 5. Transient inhibition of LINC00673 leads to loss of epithelial markers and EMT.

(a) PANC1 colony formation assay performed with the indicated siRNA and visualized with crystal violet. N=3 independent experiments with two different siRNAs. Student t-test. (b) PANC1 migration assay in five um transwell membranes. N=4 independent experiments with two different siRNAs. Student t-test. (c) Higher metastatic burden in nude mice after splenic injections of PANC1/Luc cells transfected with siRNA1 targeting LINC00673 for 48 hours prior to surgery. p=0.017 Mann Whitney test (d) MET, FOXA1 and CDH1 mRNA expression in PANC1 cells treated with two different siRNAs against LINC00673. N=3. Student t-test. (e) Western blot of MET and vimentin after transient knockdown of LINC00673. HGF treatment (20ng/ul) included as positive control. Representative blot of at least three independent experiments. (f) Immunofluorescene analysis of vimentin expression in PANC1 cells transfected with siRNA1. Representative images of at least three independent experiments. (g) Molecular subtyping using Bailey and Collisson classifiers of PANC1 cells before and after LINC00673 knockdown. (h) RNA expression after knockdown of LINC00673 of genes containing SOX9 binding sites at the promoter. (i) FOXA1 mRNA expression in PANC1 cells overexpressing SOX9 and LINC00673 knockdown. Errors bars represent ± SD
Examining the genes dysregulated by LINC00673 silencing, we found that the most strongly upregulated gene was MET (Fig 5d), a receptor tyrosine kinase involved in motility, migration and invasion in PDA cell lines.(33) We confirmed this finding by qRT-PCR and observed that upregulation of MET was associated with downregulation of epithelial markers such as FOXA1 and CDH1 (Fig 5d). In addition, loss of LINC00673 induced a mesenchymal phenotype evidenced by gain of vimentin expression (Fig 5e–f, Sup Fig 13). We were unable to confirm a role of LINC00673 in the stimulation of ERK signaling pathway in PDA cell lines, as previously suggested (Sup Fig 14).(34) Genetic downregulation of MET did not prevent the increased migratory capacity of PANC1 cells suggesting that a global transcriptome switch or loss of differentiation status mediates this aggressive behavior (Sup Fig 15). Consistently, we observed enrichment of squamous (log2 fold change 0.86, padj=0.03) and quasimesenchymal subtype (QM, log2 fold change 0.70, padj=0.07) molecular classifiers after downregulation of LINC00673 with siRNA1 in PANC1 cells (Fig 5g). No significant enrichment was found with the less efficient siRNA2. Overall suggesting that LINC00673 is required to maintain epithelial differentiation and prevent expression of mesenchymal markers.
Since SOX9 is located in close proximity to LINC00673, we hypothesized that SOX9 could influence LINC00673 function. We did not detect changes in SOX9 protein following LINC00673 knockdown (Sup Fig 14), however, we observed significant dysregulation of SOX9 target genes, particularly the classical target and known mediator of pancreas differentiation, FOXA1 (Fig 5h).(33) Overexpression of SOX9 resulted in a significant upregulation of FOXA1 that was partially abrogated by LINC00673 downregulation (Fig 5i, Sup Fig 16). These data suggest that LINC00673 participates in the functional regulation of SOX9. Supporting this hypothesis, analysis of the potential SOX9 target genes in PDA identified overlap with gene sets related to the maintenance of epithelial features such as cell-cell junction, apical junction complex and epithelium development (Sup Table 7), consistent with the downregulation of FOXA1 and CDH1 mediated by LINC00673.
Overall, we provided experimental and clinical evidence suggesting that loss of LINC00673 induced a loss of epithelial differentiation in PDA cells, and this is reflected in poor clinical outcome in low LINC00673 tumors, increased migratory capacity in vitro and in vivo and loss of epithelial and gain of mesenchymal markers in vitro and in tumor samples.
DISCUSSION
Our understanding of the role of lncRNAs is rapidly evolving from spurious expression of “junk DNA” to the current understanding that many such genes play a direct functional role in biology. As a whole, a consensus is beginning to emerge that lncRNAs act as critical modulators of cellular regulatory states. In particular, many lncRNAs contribute to the process of lineage specification, a critical need in the evolution of complex organisms with hundreds or thousands of discrete cell types.
Cancer is also intimately linked to cellular differentiation; indeed, loss of differentiation is one of the cardinal features of the disease, both at the point of initiation and during tumor progression. Individual examples of lncRNAs with functional roles, such as XIST (35) and HOTAIR (36) serve as important proofs of principle for the potential contributions of lncRNAs. Likewise, analysis of recurrent SCNA identified FAL1, an oncogene frequently amplified in ovarian cancer;(37) PVT1, co-amplified with MYC ;(13) SAMMSON, frequently amplified in melanoma and required for mitochondrial function.(12) However, overall such efforts have been limited to small numbers of lncRNAs,(38) to small numbers of samples,(39) or by the constraints of array-based technologies.(40) In general, global analysis of lncRNAs has been hindered by a paucity of tools for their identification, annotation, and prioritization. We believe the open-source NORI tool and other computational approaches used here provide a useful framework for investigating lncRNAs in cancer using publically available RNA-Seq data such as that available through TCGA.
In this study, we presented an analysis of lncRNAs expressed in PDA that we validated in two independent cohorts of PDA samples. We used NORI to reconstruct the global expression of lncRNAs and then applied a series of computational criteria to probe their association with features of pancreatic malignancy. For example, localization of candidate PDA lncRNAs to SCNA is of interest because many such regions selected during tumor evolution are devoid of known coding oncogenes or tumor suppressor.(22, 23) Likewise, germline variants associated with risk susceptibility identified by GWAS very frequently map to non-coding regions. Global gene expression analysis shows that lncRNAs in trait-associated loci are expressed in cell types relevant to the trait, again suggesting a role of lncRNAs in disease.(41) By applying multiple such criteria, we prioritized those lncRNA candidates with the highest likelihood of playing a functional role in PDA.
It was therefore notable to us that many of the resulting PDA-associated lncRNAs identified were related at some level to pancreatic lineage specification. In particular, one of our top candidates, LINC00673, is located next to SOX9 and regulates the expression of several SOX9 target genes. Although, we did not detect changes in SOX9 expression mediated by LINC00673, lncRNAs have been found to interact with related protein SOX2 to regulate downstream targets.(42) We also detected lncRNAs located in proximity to GATA6 and FOXA2, both important transcription factors involved in pancreas development. These findings are consistent with the paradigm that loss of developmental genes affecting the terminal differentiation of epithelial cells contributes to tumor progression, both generally (25, 43, 44) and specifically in the pancreas.(45) Furthermore, our data are consistent with previous reports showing that lineage determinants, such as GATA6 and PDX1, are amplified or reactivated at the initiation of PDA (and thus behave as oncogenes); however their expression may be lost during tumor progression, perhaps contributing to the subsequent loss of epithelial character (25, 46). Together with early evidence suggesting that reprogramming of chromatin domains is associated with the acquisition of metastatic clones,(47) our data support a model in which lncRNAs play a role in the metastatic progression of PDA.
Our focus on lncRNAs expressed in the neoplastic epithelium was greatly facilitated by a unique RNA-Seq dataset derived from laser capture microdissected human pancreatic tumors, which also served as a means for independent validation of candidate lncRNA expression. However, we do expect that lncRNAs will also play an important role in the biology of stromal cells. For instance, among the top enriched stromal lncRNAs, we identified HAND2-AS1, a promoter-associated lncRNA, that has been recently shown in a knockout mouse model to regulate the expression of HAND2,(48) which is involved in paracrine stroma-to-epithelium signaling in the uterus.(49) Further investigation of these stroma-enriched lncRNAs is warranted.
LncRNAs are emerging as essential players in the biology and progression of cancer as active regulators of coding gene expression. Critically, the recent FDA approval of the first antisense therapy provides a viable, practical approach for leveraging this new understanding of cancer biology. Nusinersin (Spinraza®, Biogen) is an antisense oligonucleotide directed against the splice junction of SMN2, a paralogue of the SMN1 that is mutated in spinomuscular atrophy. This and other clever strategies may be employed to modulate lncRNA function in vivo, providing a whole new tool set to control and reverse the regulatory states that drive malignancy.
MATERIAL AND METHODS
Patients and Samples
This study used RNAseq data from 147 samples deposited at TCGA annotated as Pancreas-Adenocarcinoma Ductal Type, based on their Neoplasm Histologic Type. BAM files were downloaded from Cancer Genomics Hub (https://cghub.ucsc.edu/) on February 2016. Detailed clinical information of the samples was downloaded from Cbioportal (http://www.cbioportal.org/study?id=paad_tcga#clinical).
LCM-RNA-Seq profiles were acquired from PDA specimens obtained from patients who underwent surgical resection at the Pancreas Center at Columbia University Medical Center. Prior to surgery, all patients had given surgical informed consent, which was approved by the Columbia University Institutional Review Board. Immediately after surgical removal, the specimens were cryopreserved, sectioned and microscopically evaluated by the Columbia University Tumor Bank (IRB protocol AAAB2667). Suitable samples were transferred into OCT medium (Tissue Tek) and snap frozen in a 2-methylbutane dry ice slurry. The tissue blocks were stored at −80°C until further processing. H&E stained sections of frozen PDA samples from the Tumor Bank were initially screened to confirm diagnosis and overall sample RNA quality was assessed by the Pancreas Center supported Next Generation Tumor Banking program using gel electrophoresis, with samples exhibiting high RNA quality utilized for subsequent analyses.
Non Coding RNA identification (NORI)
We developed NORI to identify long non-coding RNAs from RNAseq data. We first used cufflinks (50) to reconstruct the transcriptome profiles on 109 TCGA PAAD cohort. Then, individual transcriptomes from each sample within the TCGA was merged into a single Gene Transfer Format (GTF) file. Our NORI pipeline extracts a list of long non-coding RNAs from the GTF file by removing transcripts if any of the following criteria were met: a) they were overlapped with genes annotated in Ensembl and not annotated as "lincRNA", "non_coding", "antisense", "3prime_overlapping_ncrna", "processed_transcript", "miRNA", "misc_RNA", "polymorphic_pseudogene", "processed_pseudogene", or "pseudogene"; b) overlapped with RefSeq genes (h19) annotated as protein coding, where the RefSeq ID began with "NM"; c) overlapped with pseudogenes from Pseudogene.org; d) monoexonic or less than 200 bp in CDS length; e) predicted to have protein-coding potential by the Coding Potential Assessment Tool (CPAT;(51) coding probability > 0.364); f) gene-level maximum reads per kilobase of transcript per million mapped reads (RPKM) was less than 0.05 * n, where n is the number of samples. NORI is available for download at https://github.com/RabadanLab and accepts a number of optional arguments; see the vignette (browseVignettes(‘NORI’)) for more details.
Expression dataset preparation
For a given molecular feature list, e.g. known coding genes/identified lncRNAs from NORI, we used featureCounts from package ‘Subread’ to call read counts from .bam files (TCGA/CUMC). Genes with low read depths across the cohort are removed. Then, read counts are transformed into RPKM values, followed by log2 transformation, and quantile normalized on sample level.
Molecular subtyping
To identify molecular subtypes with biological relevance, we select a subset of lncRNAs generated by NORI, based on expression correlation with PDA driver mutations. Mutation allele frequency of KRAS, TP53, CDKN2A and SMAD4 were downloaded from Cbioportal (http://www.cbioportal.org/study?id=paad_tcga#mutations). The association between lncRNA expression and mutation allele frequency was assessed by spearman correlation. We only kept the transcripts from NORI with spearman q-value < 0.001 on at least one of the four drivers, resulting in 652 lncRNAs as input features for clustering. Then, Non-negative matrix factorization consensus clustering from GenePattern (52) was employed to identify stable sample clusters on 147 Pancreas-Adenocarcinoma Ductal samples. Detailed clustering parameters are: predefined clusters k from 2 to 7, num clusterings 20, max num iterations 2000, error function euclidean, stop convergence 40 and stop frequency 10. The final number of clusters k was selected with the highest cophenetic coefficient.
Additionally, we determine whether the molecular subtyping of lncRNA transcription is an indirect reflection of the transcription of neighbor coding genes. We removed antisense lncRNAs and run-offs of coding genes by filtering lncRNAs that share an overlap with 10kb up/downstream of a coding gene. NMF was performed as described above for the resulting 354 lncRNAs.
Differential expression analysis
DESeq2 was applied to call differential expressed genes between pre-defined groups with interests.(53) To generate the input matrix required by DESeq2, featureCounts was used to call raw read counts for both lncRNA and coding genes (GRCh37.75.gtf) on the TCGA cohort.(54)
To investigate the biological content for each subtype defined by NMF clustering, one vs. rest comparisons was performed using DESeq2 to identify subtype-specific expressed genes. We also made an additional analysis by comparing lncRNA cluster 1 and 2.
DESeq2 was used to distinguish lncRNAs enriched in the epithelium (n=66) and in the stroma (n=65) using the RNAseq from the CUMC cohort.
Overlap of lncRNAs with chromatin annotations of lncRNAs
For chromatin modifications we selected DNAse1 hypersensitivity, H3K27ac, H3K4me1 and H3K4me3 profiles tested on PANC-1 cell line (ENCODE). MACS2 outputted narrowPeak file were downloaded from UCSC Genome Browser for H3K27ac, H3K04me1 and H3K04me3. Dnase1 narrowPeak file was downloaded from GEO (GSM736519). Overlapping of chromatin modifications or DNAse1 hypersensitivity with lncRNAs was assessed by calculating the ratio of overlapped lncRNA sequence with respect to the total length that we defined as Percentage of Overlapped Region (POR). Controls were generated by keeping the same length and chromosome distributions as the tested lncRNAs. Significance was calculated by performing 10,000-permutation test. P-value is given by n/10000, where n is the number of permutations that the control gave a larger POR than lncRNAs.
Next, we analyzed the chromatin modifications at the transcriptional start site (TSS) of lncRNAs and coding genes. For each lncRNA/coding genes, the reads depth on each base at the TSS±1kb were count from the Chip-seq datasets (DNAse1, H3K27ac, H3K4me1 and H3K4me3) from PANC1 cells available at ENCODE. Raw counts are log transformed and averaged for total number of lncRNAs and coding genes respectively.
Concordant lncRNAs expression and SCNA status
SCNA segment scores from TCGA PDA samples were downloaded from cBioportal (http://www.cbioportal.org/). The correlation between segment scores and lncRNA expression was determined by spearman correlation on patient level. Next, to determine the significance, we compared the lncRNA expression-SCNA correlations with random controls. To generate the controls, we randomly selected 85 lncRNAs and arbitrarily matched their expression with the original SCNA segment scores.
Survival analysis
Overall survival and disease specific survival records of TCGA TAAD cohort were downloaded from Cbioportal (http://www.cbioportal.org/study?id=paad_tcga#clinical). Survival analysis was conducted using the R package ‘survival’ and ‘survcomp’. To demonstrate the clinical relevance of NMF clusters, patient survival in different subtypes were compared using Kaplan-Meier survival curves together with the log-rank test. To evaluate the prognostic power of an lncRNA on a specific sample set, we first split the cohort into two equal-sized subsets based on the median expressing value of the lncRNA, and then compare the survival differences between the two using the log-rank test. The analysis on Figure 3 and 4 was performed in a subset of PDA samples wilt KRAS mutated allele frequency higher than 0.2.
Regulatory network
The pancreatic cancer regulatory network was reverse engineered by ARACNe-AP (55) from the TCGA PAAD cohort. The RNA-Seq level 3 data were downloaded from TCGA data portal, raw counts were normalized to account for different library sizes after filtering out genes with less than 1 fragment per million mapped fragments (FPM) in at least 20% of the samples, and the variance was stabilized by fitting the dispersion to a negative-binomial distribution as implemented in the DESeq2 R package (Bioconductor). ARACNe was run with standard settings (using data processing inequality, with 100 bootstrap iterations using all gene symbols mapping to a set of 1,813 transcription factors that includes genes annotated in the Gene Ontology molecular function database (GO) as GO:0003700, (‘transcription factor activity’), GO:0004677, (‘DNA binding’), GO:0030528 (‘transcription regulator activity’), or as GO:0004677/GO: 0045449, (‘regulation of transcription’). In addition to these coding gene products, we added the 453 lncRNAs to the list of transcriptional regulators. Thresholds for the tolerated data processing inequality (DPI) and mutual information (MI) p-value were set to 0 and 10−8, respectively.
Cell lines and transfection
PANC1, BxPC3, MiaPaCa2 and Aspc1 were passaged and maintained following standard techniques in 5% CO2 and 95% air cultured following manufacturer instructions (ATCC). PANC 1 cells that constitutively express the firefly luciferase gene (PANC1/Luc1) were generated by lentiviral transduction of a bi-cistronic expression vector with dtomato and luciferase (Addgene plasmid # 48688).
Cells were transfected with 5nM siRNA targeting FAM83H-AS1 or LINC00673 and a non-targeting control (Silencer select, Ambion) using Lipofectamine 3000 following manufacturer instructions (Life Technologies). siRNAs sequences are listed in supplemental table 9.
For SOX9 overexpression, SOX9 cDNA sequence (Origene, NM_000346) was cloned into pcDNA3. Cells were transfected with SOX9 and with an empty pcDNA3 vector as control. Transfection was done using Lipofectamine 3000 following manufacturer instructions (Life Technologies). Cell lines were purchased and verified by ATCC, maintained at low passage and tested for mycoplasma.
Splenic Injection and live imaging
PANC1/Luc were transiently transfected 48 hours prior surgery with either control or targeted siRNA by using Lipofectamine 3000 (Invitrogen). Cells were harvested by trypsinization and a single cell suspension of 2 × 106 cells in 100 μl was prepared in phosphate buffered saline and kept on ice before injection. Cells were injected into the spleen of 6-7 weeks of age of NU/J male mice (002019, The Jackson laboratories), keeping syringe inside spleen for 5 minutes post-injection to allow tumor cells to drain. Tumor growth was monitored by bioluminescence imaging using the IVIS whole body imaging system. Luciferin substrate was given by intra-peritoneal injection ten minutes before imaging. Bioluminescence flux was quantified using live imaging software (Perkin Elmer). All procedures were approved by the ethics committee of Columbia University.
Cell Cycle Analysis
PANC1 cells were fixed in 70% ethanol 48hr after transfection with the indicated siRNA were stained with propidium iodide (final concentration 1ug/ml). Cells were sorted by FACS and cell cycle distribution analyzed with FlowJo.
Western Blot
Cell were lysed and proteins extracted following the whole-cell extract from adherent cells protocol provided with the Nuclear Extract Kit (Active Motif). Primary antibodies are listed in supplemental table 9.
RNA extraction and qRT-PCR analysis
Total RNA was isolated from cultured cell lines using the RNeasy Mini Kit (Qiagen). RLT buffer was supplemented with 2-mercaptoethanol (Sigma-Aldrich) and DNase treatment was performed for 20 min using the RNase-Free DNase set (Qiagen). 1 ug of total RNA was reverse transcribed into cDNA using random hexamers with SuperScript III First-Strand Synthesis kit (Life Technologies). 20 ng of cDNA were used in the qRT-PCR reaction with iQ SYBR® Green supermix (Bio-Rad) and custom designed primers. All experiments were calculated as a function of gene expression relative to either control TBP expression or GAPDH. qPCR data were expressed as mean fold change (2−ΔΔCT). Primers are listed in supplemental table 9.
RNA-seq in PANC1 and ASPC1
200 ng of total RNA from culture cell lines was subjected to transcriptome analysis. Total RNA was converted into cDNA libraries (TruSeq RNA Sample Prep Kit v2, Illumina) using poly-A pull down for mRNA enrichment. Sequencing was performed to a depth of 30 million pairs. Differential expression between replicates was assayed using DESeq2 (R package). All samples had RIN values higher than 9.0 as determined with Agilent Bioanalyzer 2100. Complete RNA-Seq data is available through GEO Express GSE96931.
Gene Set Enrichment Analysis
We used GSEA software with defined genesets (MSigDB, v6.1) to investigate molecular profiles enriched before and after targeting the expression of LINC00673 and FAM83HAS1. Overlap with SOX9 target genes was performed using the gene set CATTGTYY_SOX9_B1 from the Broad Institute. Phenotype labels were created for siCTRL and siRNA. siRNA1 and siRNA2 vs siCTRL were assessed independently. Genes with FPKM=0 across all samples studied were removed from the gct file for GSEA analysis. Parameters used were Collapse data: false, Permutation type: gene_set, 1000 permutations, Chip platform: gene symbol.
Gene set variation analysis (GSVA)
We used the R implementation of single sample Gene Set Enrichment analysis: GSVA (gene set variation analysis) with default parameters 1. The input expression matrix was filtered for the most variable 75% of the genes. For annotation of epithelial subtypes, we tested a set of gene sets shown previously to be discriminating between molecular subtypes of pancreatic ductal adenocarcinoma (3, 7). Differential enrichment analysis of these gene sets between the different siRNA treatments was carried out using the limma R package 4 and a FDR < 0.1 was considered significant.
Clonogenic Assay
Human pancreatic cancer cell lines were transfected with targeting siRNAs and siCTRL in triplicates. 48 hr after transfection cells were detached and 5-10× 103 cells were seeded in triplicates in 6 well plates and incubated to allow colony formation for 10-14 days. Colonies were visualized by staining with crystal violet and quantified with image J (http://rsb.info.nih.gov/ij/)
Migration Assay
Cells pre-transfected 48 hr before with siCTRL or siRNA-1 were seeded (50× 103) onto Transwell membrane inserts in 2% FBS culture media (5 µm pore, Corning). Regular media was added to the lower chamber and cells were incubated for 12 hr at 37 °C. After incubation, cells that migrated across the membrane were fixed and stained with crystal violet. For each membrane, the same 9 randomly distributed fields were counted. The data represent the mean of three independent experiments performed in triplicate.
Immunofluorescence
Cells were platted onto coverslips and fixed with 4% paraformaldehyde for 5 min and 100% methanol for additional 5 min. After fixation, coverslips were kept at 4°C submerged in PBS. For immunofluorescence, coverslips containing fixed cells were blocked in 5% serum in 1×PBS +0.5% Triton-X for 30 minutes. Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. The primary antibody used was rabbit α-Vimentin (1:500; ab92547, abcam). Coverslips were incubated with appropriate secondary antibodies conjugated to DyLight-488 (Jackson Immunoresearch). DAPI (1∶1000; Invitrogen) was applied for 30 minutes following secondary antibody incubation.
Code availability
The NORI framework is fully available for academic use on Github (https://github.com/RabadanLab/NORI).
Supplementary Material
Significance of this study.
What is already known in this subject?
Pancreatic ductal adenocarcinoma is one of the most aggressive malignancies, exhibiting only limited and transient responses to current treatments. “Targetable” alterations in protein coding genes are uncommon in PDA.
A large fraction of recurrent somatic copy number alterations and single nucleotide polymorphism associated with lifetime risk of cancer are devoid of coding genes drivers of tumorigenesis.
Long non-coding RNAs (lncRNAs) are emerging as essential players in the biology and progression of a variety of tumors as active regulators of gene expression (non-coding oncogenes or tumor suppressors) and/or passive readouts of tumor progression or clinical prognosis (biomarkers).
What are the new findings?
We used an integrative analysis of genomic and clinical data from PDA tumor samples to define a catalogue of lncRNAs associated with genetic traits of pancreatic cancer and associated with clinical outcome.
The identified set of lncRNAs were independently validated in a cohort of paired epithelial and stromal RNA-Seq profiles derived from laser capture microdissected human pancreatic tumors, allowing us to annotate the compartment-specificity of their expression.
LncRNAs segregate tumor samples into subgroups distinguished by differentiation status and associated with clinical prognosis in PDA.
Using this approach, we identified FAM83H-AS1 and LINC00673 in recurrent amplified genomic regions and associated with clinical outcome in PDA.
We found that loss of LINC00673 regulates the epithelial differentiation state in PDA cells, increases migratory capacity in vitro and in vivo, and results in loss of epithelial and gain of mesenchymal markers, both in vitro and in tumor samples. This finding is further reflected in poor clinical outcome in low LINC00673 tumors.
How might it impact on clinical practice in the foreseeable future?
We expect that the collection of PDA-associated long non-coding RNAs will aid in the design of targeted therapies and may contribute to the development of improved diagnostic tools for PDA. The recent clinical approval of the first antisense therapy for human disease provides a viable, practical approach for leveraging this new understanding of cancer biology.
Acknowledgments
We thank Steve Sastra and Christopher Damocci for excellent assistance with the xenograft model. This work was funded by the IRIS (LA) and CaST (LA) programs at Columbia University, the Juvenile Diabetes Research Foundation (LA) and R21CA188059 (LS). These studies used the resources of the Herbert Irving Comprehensive Cancer Center (Center Grant P30CA013696) and the Diabetes and Endocrinology Research Center (Center Grant 5P30DK063608). R.R. and Z.L. were funded by NIH U54 CA193313. NB was funded by an NIH - NHLBI T35 training grant. LA was funded by the Juvenile Diabetes Research Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, L.A.; Computational analysis, Z.L and J.W.; Software, N.B.; Investigation, L.A., Z.L., H.C.M., I.S., M.S.M., D.C.G. and D.A.B.; Resources, L.A., K.P.O., R.R.; Writing, LA wrote the manuscript with feedback from, K.P.O. and R.R.; Visualization, L.A. and Z.L; Funding Acquisition, L.A; Project Oversight and Management, L.A, K.P.O, R.R. All authors discussed the results and commented on the manuscript.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer research. 2014;74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 4.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature communications. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature genetics. 2015;47(10):1168–78. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature medicine. 2011;17(4):500–3. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Consortium EP. Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, et al. The landscape of long noncoding RNAs in the human transcriptome. Nature genetics. 2015;47(3):199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer cell. 2016;29(4):452–63. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J, et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016;531(7595):518–22. doi: 10.1038/nature17161. [DOI] [PubMed] [Google Scholar]
- 13.Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512(7512):82–6. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nature genetics. 2015;47(8):911–6. doi: 10.1038/ng.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Zhou N, Watabe K, Lu Z, Wu F, Xu M, et al. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1) Cell Death Dis. 2014;5:e1008. doi: 10.1038/cddis.2013.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang W, Liu Y, Liu R, Zhang K, Zhang Y. The lncRNA DEANR1 facilitates human endoderm differentiation by activating FOXA2 expression. Cell Rep. 2015;11(1):137–48. doi: 10.1016/j.celrep.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Xiao ZD, Han L, Zhang J, Lee SW, Wang W, et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol. 2016;18(4):431–42. doi: 10.1038/ncb3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031–41. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 20.Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P, et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nature medicine. 2016;22(8):861–8. doi: 10.1038/nm.4135. [DOI] [PubMed] [Google Scholar]
- 21.Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nature genetics. 2017 doi: 10.1038/ng.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nature genetics. 2013;45(10):1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marin-Bejar O, Marchese FP, Athie A, Sanchez Y, Gonzalez J, Segura V, et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome biology. 2013;14(9):R104. doi: 10.1186/gb-2013-14-9-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinelli P, Carrillo-de Santa Pau E, Cox T, Sainz B, Jr, Dusetti N, Greenhalf W, et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut. 2016 doi: 10.1136/gutjnl-2015-311256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amundadottir LT. Pancreatic Cancer Genetics. Int J Biol Sci. 2016;12(3):314–25. doi: 10.7150/ijbs.15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YC, Zhou Q, Chen J, Yuan J. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr Biol. 2016;26(24):3257–68. doi: 10.1016/j.cub.2016.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huarte M. The emerging role of lncRNAs in cancer. Nature medicine. 2015;21(11):1253–61. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 29.Peng QL, Zhang YM, Yang HB, Shu XM, Lu X, Wang GC. Transcriptomic profiling of long non-coding RNAs in dermatomyositis by microarray analysis. Scientific reports. 2016;6:32818. doi: 10.1038/srep32818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuga T, Sasaki M, Mikami T, Miake Y, Adachi J, Shimizu M, et al. FAM83H and casein kinase I regulate the organization of the keratin cytoskeleton and formation of desmosomes. Scientific reports. 2016;6:26557. doi: 10.1038/srep26557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nature genetics. 2005;37(4):382–90. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 32.Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPt, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer cell. 2012;22(6):737–50. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y, Washington MK, Crawford HC. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer research. 2010;70(5):2115–25. doi: 10.1158/0008-5472.CAN-09-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng J, Huang X, Tan W, Yu D, Du Z, Chang J, et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nature genetics. 2016 doi: 10.1038/ng.3568. [DOI] [PubMed] [Google Scholar]
- 35.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322(5902):750–6. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu X, Feng Y, Zhang D, Zhao SD, Hu Z, Greshock J, et al. A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer cell. 2014;26(3):344–57. doi: 10.1016/j.ccr.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tahira AC, Kubrusly MS, Faria MF, Dazzani B, Fonseca RS, Maracaja-Coutinho V, et al. Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol Cancer. 2011;10:141. doi: 10.1186/1476-4598-10-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Li Z, Zheng S, Zhou Y, Zhao L, Ye H, et al. Expression profile of long non-coding RNAs in pancreatic cancer and their clinical significance as biomarkers. Oncotarget. 2015;6(34):35684–98. doi: 10.18632/oncotarget.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu XL, Liu DJ, Yan TT, Yang JY, Yang MW, Li J, et al. Analysis of long non-coding RNA expression profiles in pancreatic ductal adenocarcinoma. Scientific reports. 2016;6:33535. doi: 10.1038/srep33535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017 doi: 10.1038/nature21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng SY, Bogu GK, Soh BS, Stanton LW. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Molecular cell. 2013;51(3):349–59. doi: 10.1016/j.molcel.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 43.Krah NM, De La OJ, Swift GH, Hoang CQ, Willet SG, Chen Pan F, et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. Elife. 2015;4 doi: 10.7554/eLife.07125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Figura G, Morris JPt, Wright CV, Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2014;63(4):656–64. doi: 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris JPt, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nature reviews Cancer. 2010;10(10):683–95. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roy N, Takeuchi KK, Ruggeri JM, Bailey P, Chang D, Li J, et al. PDX1 dynamically regulates pancreatic ductal adenocarcinoma initiation and maintenance. Genes & development. 2016;30(24):2669–83. doi: 10.1101/gad.291021.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nature genetics. 2017 doi: 10.1038/ng.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson KM, Anderson DM, McAnally JR, Shelton JM, Bassel-Duby R, Olson EN. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature. 2016;539(7629):433–6. doi: 10.1038/nature20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q, Kannan A, DeMayo FJ, Lydon JP, Cooke PS, Yamagishi H, et al. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science. 2011;331(6019):912–6. doi: 10.1126/science.1197454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28(5):511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41(6):e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nature genetics. 2006;38(5):500–1. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 53.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41(10):e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lachmann A, Giorgi FM, Lopez G, Califano A. ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics. 2016;32(14):2233–5. doi: 10.1093/bioinformatics/btw216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.