

Abstract
The innate immunity is critically important in protection against virus infections, and in the case of RNA viral infections, the signaling mechanisms that initiate robust protective innate immunity without triggering autoimmune inflammation remain incompletely defined. Here, we found the E3 ligase TRIM29 was specifically expressed in poly I:C stimulated human myeloid dendritic cells (mDCs). The induced TRIM29 played a negative role in type I interferon (IFN) production in response to poly I:C or dsRNA virus reovirus infection. Importantly, the challenge of wildtype mice with reovirus led to lethal infection. In contrast, deletion of TRIM29 protected the mice from this developing lethality. Additionally, TRIM29−/− mice have lower titers of reovirus in the heart, intestine, spleen, liver, and brain due to elevated production of type I IFN. Mechanistically, TRIM29 was shown to interact with MAVS and subsequently induce its K11-linked ubiquitination and degradation. Taken together, TRIM29 regulates negatively the host innate immune response to RNA virus, which could be employed by RNA viruses for viral pathogenesis.
Introduction
Innate immunity provides the first line of defense against invading pathogens. Activation of innate immunity requires the recognition of pathogen-associated molecular patterns by pattern-recognition receptors (PRRs)(1). RIG-I-like receptors (RLRs), including retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA-5), function as cytoplasmic RNA sensors that recognize viral nucleic acids double strand RNA released during virus replication(2). Once recognizing the viral RNA, these RLRs could be activated and initiate a series of signaling events that lead to the production of type I interferons (IFN-α and IFN-β)(3). IFN-α/β further activate downstream signaling pathways that lead to the transcriptional induction of a wide range of genes encoding antiviral products, and those antiviral products act together to elicit a cellular antiviral response through various mechanisms(4).
The RLR signaling pathway triggers innate immune responses against RNA viruses. Although RIG-I and MDA5 sense distinct types of viruses(5-7), they share the common adaptor protein MAVS (also known as Cardif, IPS-1 or VISA) to transmit signals(8-11), which leads to IFN-β production for initiation of the antiviral innate immune response(12). MAVS is critical in innate antiviral immunity as the sole adaptor for RLRs. MAVS regulation is essential for the prevention of excessive harmful immune responses. Given that MAVS coordinates signals from two independent central sensors RIG-I and MDA5, various mechanisms are employed to modulate MAVS, including protein-protein interactions, post-translational modifications and changes of mitochondrial dynamics(13). Recently, several negative regulators for MAVS have been reported (14-16). However, the detailed molecular regulation mechanisms of MAVS activation are not clear.
Tripartite motif proteins (TRIM) consist of a large family of proteins containing a RING-Bbox-Coiled Coil motif followed by different C-terminal domains. TRIM family proteins control important cellular processes such as intracellular signaling in innate immunity and viral infection, transcriptional regulation, development, autophagy, and carcinogenesis(17). TRIM29 is structurally a member of the TRIM protein family and is a unique multifunctional protein for DNA damage responses(18), molecular cancer biomarker(19), antiviral innate immunity(20), cell adhesion/invasion in tumor(21-23), cell differentiation and signal regulation for the canonical Wnt pathway (24). Previously, we have found that E3 ubiquitin ligase TRIM29 is highly and constitutively expressed in alveolar macrophages and functions to control the respiratory infections by influenza virus and Haemophilus influenza through regulating negatively the innate immunity in the respiratory tract (20). Also, we have shown that TRIM29 is constitutively expressed in human airway epithelial cells and induced by dsDNA in human myeloid dendritic cells (mDCs) and promotes DNA virus infections by inhibiting innate immune response (25). Given that TRIM29 has E3 ligase activity and is a multifunctional protein in cellular processes, we continued to investigate its role in innate immunity and viral infections. In this study, we identify the E3 ligase TRIM29, specifically expressed in poly I:C induced human mDCs, as a critical negative regulator of the innate immune responses to RNA virus through targeting MAVS for degradation. Thus, our report reveals a previously unrecognized regulatory function of TRIM29 in the context of RNA virus infection in human mDCs.
Materials and Methods
Mice
Trim29 knockout (Trim29−/−) mice were from the European Mouse Mutant Archive (EMMA, EM: 07120). Primary bone marrow was collected from wild-type and Trim29−/− mice for inducing mature BMDCs and BMDMs(20). All animals were maintained in the specific pathogen-free facility at Houston Methodist Research Institute in Houston, Texas. Animal use and care were approved by the Houston Methodist Animal Care Committee, in accordance with institutional animal care and use committee guidelines.
Reagents
The high molecular weight dsRNA (long poly I:C, LPIC, Catalog: tlrl-pic-5), low molecular weight dsRNA (short poly I:C, SPIC, Catalog: tlrl-picw), R848 (TLR7/8 ligand, Catalog: tlrl-r848), TSLP (Thymic stromal lymphopoietin), α-CD40 (Neutralizing monoclonal antibody against human CD40L, Catalog: mabg-h40l-3), LPS (TLR4 ligand, Catalog: tlrl-pglps), CpG A (TLR9 agonist, Catalog: tlrl-2216) and CpG B (TLR9 agonist, Catalog: tlrl-2006) were from Invivogen. Lipofectamine 3000 was from Invitrogen. The proteasome inhibitor MG132 was from Sigma. The following antibodies were used for immunoprecipitation: anti-MAVS (1:100) (4983S; Cell Signaling Technology) and anti-TRIM29 (1:100) (A301-210A; Bethyl). The following antibodies were used for immunoblot analysis: anti-MAVS (1:1000) (4983S; Cell Signaling Technology), anti-TRIM29 (1:1,000) (A301-210A; Bethyl), anti-TRIM29 (1:1,000) (sc-33151; H-300; Santa Cruz), anti-ubiquitin (1:1,000) (sc-8017; Santa Cruz), K11-specific anti-ubiquitin (1:1,000) (MABS107-I; Millipore), anti-GAPDH (1:10,000) (clone GAPDH-71.1, G9295; Sigma), anti-HA (1:5,000) (clone HA-7, H6533; Sigma), anti-β-actin (1:20,000) (clone AC-15, A3854; Sigma) and anti-Myc (1:5,000) (ab1326; Abcam). Anti-HA and anti-Myc beads were from Sigma. Lentiviral vectors for shRNA were as follows (all from Open Biosystems): human TRIM29 (clone TRCN0000016348 (TRIM29-#1)) and clone TRCN0000016352 (TRIM29-#2)). The IFN-β and IFN-α ELISA kits were from PBL InterferonSource. The Dual-Luciferase Reporter Assay System (E1910) was from Promega.
Cells culture and lentiviral infection
Human myeloid dendritic cells (mDCs) were isolated from buffy coats of individual healthy donors and maintained in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin (Invitrogen-Gibco) as previously described (26, 27). HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin (Invitrogen-Gibco). All cell lines were tested without mycoplasma contamination. The mDCs were infected with a pLKO.1 lentiviral vector carrying a scrambled shRNA (RHS6848, Open Biosystems) or target gene sequences (Open Biosystems) as described in our previous studies (20, 25, 28, 29). After 24 h of culture, cells were selected by the addition of puromycin (2 ng/ml) to the medium. Cells were stimulated for 16 h with long poly I:C (20 μg/ml) delivered by Lipofectamine 3000. The knockdown efficiency was detected with immunoblot analysis.
Virus infection
For reovirus infection in mice, four-to five-week-old mice were inoculated intraperitoneally with 2×107 pfu of Reovirus (Reovirus type 3 strain Dearing, T3D) in PBS. Reovirus T3D strain was purchased from ATCC (ATCC® VR-824™). Mice were euthanized at various time points following infection and tissues collected for analysis.
For analysis of reovirus replication, mice were euthanized at defined intervals post-inoculation, and organs (heart, liver, spleen, brain, and intestine) were excised into 1ml of PBS and homogenized by freezing, thawing, and sonication. Intestines were transected proximally at the gastroduodenal junction and distally at the rectum before homogenization in 1mL of PBS.
Virus plaque titration
For reovirus infection in mice, viral titers in organ homogenates from infected mice were determined by plaque assay on L929 cells (30). Weights of organs were measured before the assay, and PFU were calculated per mg of tissue. Briefly, tissue was homogenized in 800 μl of PBS. The homogenates were treated with chloroform (10% final concentration), centrifuged briefly and serial dilutions of the aqueous supernatants were incubated on L929 cells at room temperature. After 1 h, the inoculum was removed and cells were covered with 2% agar solution with amphotericin-B. After six days, 2% agar solution containing 2% neutral red solution was added and plaques were visualized with neutral red on the second day.
In vitro pull-down and immunoblot analysis
For the preparation of purified MAVS and TRIM29, HEK293T cells were transfected with expression plasmids encoding full-length or truncated versions of HA- or Myc-tagged MAVS or TRIM29. Lysates were prepared from the transfected cells, followed by incubation with anti-HA or anti-Myc beads. Proteins were eluted from the beads after beads were washed six times with PBS. For precipitation with anti-HA or anti-Myc beads, purified Myc-tagged wild-type MAVS or truncations of MAVS were incubated for 2 h with purified HA-tagged TRIM29 or purified HA-tagged TRIM29 or truncations of TRIM29 were incubated for 2 h with purified Myc-tagged MAVS. Beads were added, after 2 h of incubation, the bound complexes were pelleted by centrifugation. Proteins and beads were analyzed by immunoblot analysis with anti-HA or anti-Myc Abs (20, 26, 27, 31-33).
Ubiquitination
HEK293T cells were transfected with an expression plasmid encoding Myc-tagged full-length MAVS and with or without coexpression of HA-tagged full-length TRIM29 and its mutant T29-N. At 24 h after transfection, cells were treated with 25 μM MG132 for 3h and then were collected for analysis (20, 25, 29, 34). Briefly, cells were lysed and the cell lysis was heated to 65°C for 5 mins. After spinning down, the supernatants were collected and were immunoprecipitated with anti-Myc beads and then analyzed by immunoblot.
Confocal microscopy
BMDCs isolated from the WT mice were infected with reovirus for 4h and were then fixed in 4% paraformaldehyde and permeabilized with 0.1% triton-100, then blocked for 30 min with 5% BSA, incubated with Rabbit anti-MAVS polyclonal antibody (Cat: 14341-1-AP, proteintech) and mouse anti-TRIM29 monoclonal antibody (sc-166707, Santa Cruz) for 2 h, followed by Alexa Fluor 594 goat anti-rabbit secondary antibody and Alexa Fluor 488 goat anti-mouse secondary antibody for 1h and then examined with confocal microscopy. Images of ‘zoomed’ single cells were quantified with Nikon Confocal Software.
Quantitative RT-PCR
RNA was isolated using the RNeasy Kit (Qiagen) according to the manufacturer’s instructions. The isolated RNA was used to synthesize cDNA with the iScript cDNA Synthesis Kit (Bio-Rad). iTaq SYBR Green Supermix with ROX (Bio-Rad) was used for quantitative RT-PCR. PCRs were performed in triplicate. The following primers were used: IFN-β1 forward, 5′-CCCTATGGAGATGACGGAGA-3′ and reverse, 5′-TCCCACGTCAATCTTTCCTC-3′; IFN-α5 forward, 5′- AGTGAGCTGACCCAGCAGAT-3′ and reverse, 5′-CAGGGGCTGTGTTTCTTCTC-3′; IL-6 forward, 5′-CTGATGCTGGTGACAACCAC-3′ and reverse, 5′-TTCTGCAAGTGCATCATCGT-3′; GAPDH forward, 5′-AACTTTGGCATTGTGGAAGG-3′ and reverse, 5′-ACACATTGGGGGTAGGAACA-3′.
Luciferase reporter gene assay
Human HEK293T cells were seeded on 48-well plates (1 × 105 cells per well), then transfected with reporter vectors for Ifnb-firefly luciferase (100 ng) or nfκb-firefly luciferase and renilla luciferase (1 ng) plus expression vector for wild-type MAVS or MAVS mutants (100 ng) with or without expression vector for TRIM29 (300 ng). Empty control vector was added so that a total of 550 ng of vector DNA was transfected into each well of cells. At 24 h after transfection, cells were collected for luciferase activity analysis. Luciferase activity in total cell lysates was detected by Dual-Luciferase Reporter Assay(20).
Statistical analysis
A two-tailed unpaired Student’s t test was used for statistical analysis with Microsoft Excel and GraphPad Prism Software. P values of less than 0.05 were considered significant unless specifically indicated.
Results
TRIM29 is involved in type I IFN production by human mDCs in response to poly I:C
To investigate the function of TRIM29 in humans, we started with analyses of TRIM29 expression in human immune cells. Microarray gene expression analysis showed that TRIM29 was specifically expressed in poly I:C induced human myeloid dendritic cells (mDCs) (data not shown). To further confirm the unique expression of TRIM29 in mDCs, human primary mDCs was isolated from PBMCs of one random donor and stimulated with R848 (TLR7/8 ligand, tlrl-r848, invivogen), poly I:C (high molecular weight poly I:C (LPIC), RLR ligand), TSLP (Thymic stromal lymphopoietin), α-CD40 (Neutralizing monoclonal antibody against human CD40L, mabg-h40l-3, invivogen), LPS (TLR4 ligand), CpG A (TLR9 agonist) and CpG B (TLR9 agonist). The real time PCR analysis showed that TRIM29 was indeed uniquely expressed in human mDCs stimulated by poly I:C, but not by other stimulants (Fig. 1A). To further investigate the biological function of poly I:C highly induced TRIM29 in mDCs, we first established stable knockdown of TRIM29 mDC cell lines through the use of short hairpin RNA (shRNA). Two distinct TRIM29-targeting shRNAs (Trim29#1 and Trim29#2) produced efficient knockdown of TRIM29 expression (Fig. 1B). Additionally, MAVS-targeting shRNA efficiently knocked down the MAVS expression (Fig. 1B). These cells were then stimulated by poly I:C and the production of IFN-β by the cultured cells was measured by ELISA. Consistent with published data, knockdown of MAVS abrogated the production of IFN-β in mDCs that had been induced by cytosolic poly I:C. In contrast, knockdown of TRIM29 enhanced the IFN-β production in mDCs up to threefold in response to intracellular dsRNA (Fig. 1C). These data suggested that TRIM29 negatively regulated type I IFN production in dendritic cells in response to dsRNA.
Figure 1. The induced TRIM29 plays an important role in producing type I IFN by human mDCs in response to poly I:C.
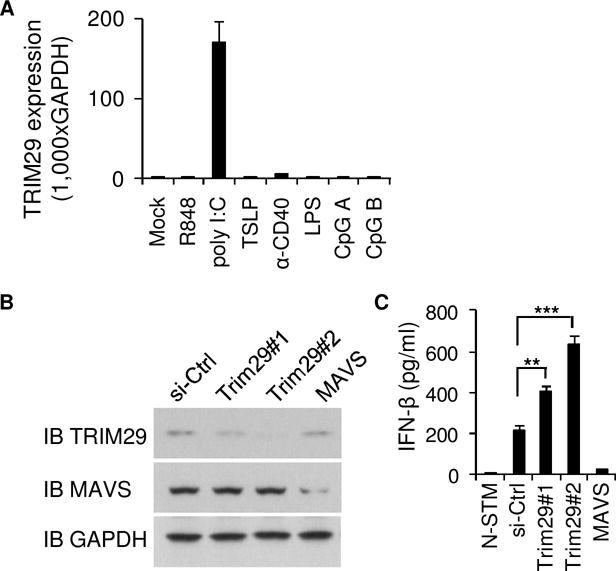
(A) Human myeloid dendritic cells (mDCs) were purified from PBMCs using a cell sorter. Total RNA was isolated from these primary cells induced or not to realtime PCR. The profile of TRIM29 expression in different cells is indicated. The relative expression of TRIM29 was compared by plotting the values extracted from the gene expression database. A value < 1 indicated the absence of gene expression. (B) Immunoblot analysis of TRIM29 and MABVS in human mDCs treated with control shRNA with a scrambled sequence (sh-Ctrl), shRNA targeting mRNA encoding TRIM29 (two shRNAs: Trim29#1 and Trim29#2) or shRNA targeting mRNA encoding MAVS. GAPDH serves as a loading control throughout. (C) ELISA of IFN-β in human primary mDCs treated with scrambled shRNA (sh-Ctrl) and left unstimulated (Mock) or treated with shRNA as above and then stimulated for 16h with long poly I:C (LPIC, 20 μg/ml) delivered by Lipofectamine 3000. *P<0.05, **P<0.01, ***P<0.001 (unpaired t test). N-STM, cells without stimulation. Data are representative of three independent experiments with similar results (mean + s.d.).
TRIM29 knockout enhanced the production of type I IFN and IL-6 in BMDCs and BMDMs in response to poly I:C
To further determine the function of TRIM29 in primary mice cells, we prepared bone marrow-derived dendritic cells (BMDCs) and bone marrow-derived macrophages (BMDMs) from wild-type mice and Trim29-deficient mice. We then stimulated those BMDCs or BMDMs for 16 h with poly I:C. As results, Trim29-deficient BMDCs produced 2- to 3-fold more type I IFN including IFN-α (Fig. 2A), IFN-β (Fig. 2B) and IL-6 (Fig. 2C) in response to poly I:C than did wild-type BMDCs. Similarly, compared to wild-type BMDMs, Trim29-knockout BMDMs produced up to 3-fold more IFN-α (Fig. 2D), IFN-β (Fig. 2E) and IL-6 (Fig. 2F) in response to poly I:C. Additionally, the q-PCR data confirmed that Trim29-deficient BMDCs or BMDMs produced more IFN-α5, IFN-β1 and IL-6 in mRNA levels in response to poly I:C than did wild-type BMDCs (Supplemental Fig. 1A) or BMDMs (Supplemental Fig. 1B). These data indicated a negative role of TRIM29 in murine BMDCs and BMDMs in response to poly I:C.
Figure 2. Knockout-TRIM29 enhanced the production of type I IFN and IL-6 in BMDCs and BMDMs in response to poly I:C.
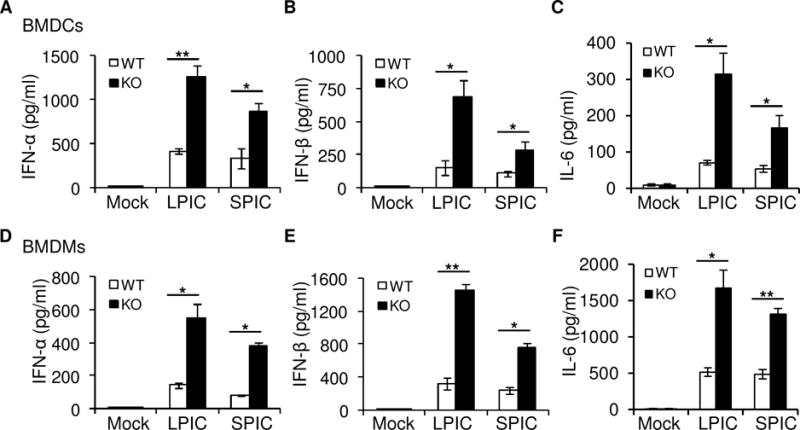
(A-F) ELISA of IFN-α (A, D), IFN-β (B, E) and IL-6 (C, F) in BMDCs (A-C) or BMDMs (D-F) from wildtype (WT) and Trim29-knockout (KO) mice after 16 h of stimulation with long poly I:C (LPIC, 20 μg/ml) or short poly I:C (SPIC, 20 μg/ml) delivered by Lipofectamine 3000. Mock, cells without stimulation. *P<0.01 and **P<0.001 (unpaired t test). Data are representative of three independent experiments with similar results (mean + s.d.).
TRIM29 negatively regulates type I IFN production in BMDCs and BMDMs upon RNA virus infection
The RIG-I-like receptors (RLRs) are cytosolic pattern recognition receptors (PRRs) proteins recognizing viral RNA species including double strand RNA (dsRNA) and 5′-triphosphate RNA (35). In the case of RNA viruses, viral RNA replicase generates 5′-triphosphate RNA and/or dsRNA in ample amounts during replication and transcription of viral RNA genomes. The dsRNA is produced during infection by dsRNA virus reovirus. So we employed the dsRNA virus, Reovirus T3D strain, to investigate the role of TRIM29 in sensing RNA virus. We then prepared BMDCs and BMDMs from wild-type mice and Trim29-deficient mice and infected those BMDCs and BMDMs for 12 or 20 h with reovirus infection. Trim29-deficient BMDCs or BMDMs produced 2- to 4-fold more IFN-β and IFN-α in response to reovirus than did wild-type BMDCs or BMDMs (Fig. 3A and 3B). The q-PCR data also confirmed that Trim29-deficient BMDCs or BMDMs produced more IFN-α5 and IFN-β1 in mRNA levels in response to reovirus infection than did wild-type BMDCs (Supplemental Fig. 2A) or BMDMs (Supplemental Fig. 2B). Additionally, there are much less reovirus production in Trim29-deficient BMDCs (Fig. 3C) or BMDMs (Fig. 3D) than that in wild-type BMDCs or BMDMs after reovirus infection. These data demonstrated that TRIM29 played a negative role in murine BMDCs and BMDMs in sensing RNA virus.
Figure 3. TRIM29 negatively regulates type I IFN production in BMDCs and BMDMs upon RNA virus infection.
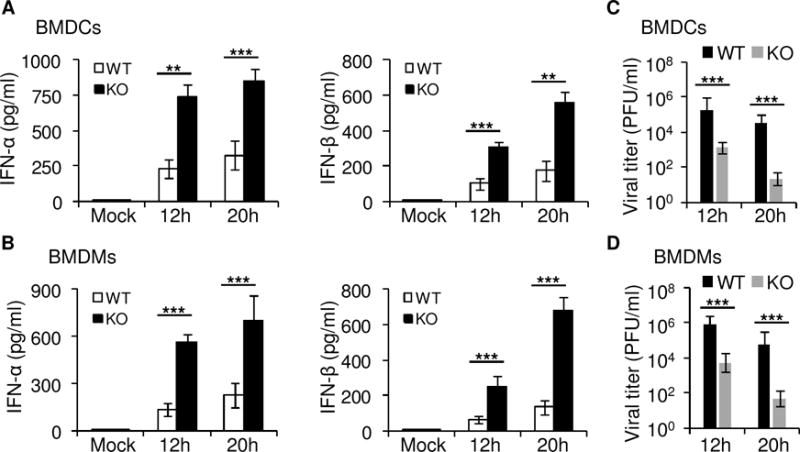
(A, B) ELISA of IFN-α and IFN-β in BMDCs (A) or BMDMs (B) from wildtype (WT) and Trim29-knockout (KO) mice mock infected or infected with reovirus at a multiplicity of infection (MOI) of 5 for 12h or 20h. (C, D) The viral titers of the supernatants from BMDCs (C) or BMDMs (D) from wildtype (WT) and Trim29-knockout (KO) mice mock infected or infected with reovirus at a MOI of 5 by plaque assay. Mock, cells without infection. *P<0.05, **P<0.01 and ***P<0.001 (unpaired t test). Data are representative of three independent experiments with similar results (mean + s.d.).
TRIM29 plays an important role in host defense in vivo
We next evaluated the importance of TRIM29 in host defense against RNA viral infection in vivo. We firstly challenged wildtype mice and Trim29-knockout mice intraperitoneally with T3D strain reovirus and monitored survival over time. The challenge of wildtype mice with reovirus led to lethal infection (Fig. 4A). In contrast, Trim29-knockout mice had significantly higher survival rates after reovirus infection (Fig. 4A). Next, we infected wild-type mice and Trim29-deficient mice intraperitoneally with reovirus and then measured IFN-β in heart homogenates from infected mice. As expected, Trim29-deficient mice produced 2- to 3-fold more IFN-β than did wild-type mice, in response to reovirus (Fig. 4B). Additionally, we harvested intestine, spleen, liver, heart, and brain at days 2 and 4 postinfection, and determined viral titers of reovirus in those organs by plaque assay. We detected significantly less reovirus in Trim29-deficient mice than in wild-type mice, especially at day 4 postinfection (Fig. 4C). These data indicated that RNA virus hijacked TRIM29 to shut down the innate immune responses to immune evasion and viral pathogenesis.
Figure 4. TRIM29 plays an important role in host defense against RNA virus infection in vivo.
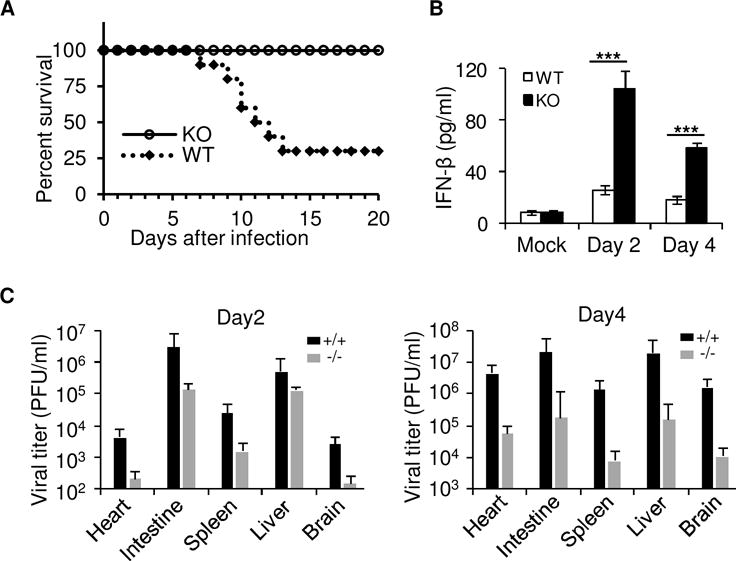
(A) Survival of wildtype (WT) and Trim29-knockout (KO) mice (n=8 per strain) after intraperitoneal injection of reovirus (2×107 plaque-forming units (PFU) per mouse). (B) The wild-type (TRIM29+/+) and Trim29-deficient (TRIM29−/−) mice were inoculated intraperitoneally with 1×107 PFU of reovirus (Reovirus type 3 strain Dearing, T3D). At days 2 (D2) and 4 (D4) post-inoculation, mice were euthanized, and hearts were excised, frozen at −80°C, thawed, and homogenized in PBS. Levels of IFN-β in heart homogenates was quantified by ELISA. (C) The wild-type (WT, +/+) and Trim29-deficient (KO, −/−) mice were inoculated intraperitoneally with 1×107 PFU of reovirus. At day 2 (D2) and day 4 (D4) post-inoculation, mice were euthanized, intestine, spleen, liver, heart, and brain were excised, and viral titers in organ homogenates were determined by plaque assay. Results are expressed as mean viral titers for 3 animals for each time point. Error bars indicate standard errors of the mean. *P<0.05, **P<0.01 and ***P<0.001 (unpaired t test). Mock, mice without infection. Data are representative of three independent experiments with similar results (mean + s.d.).
TRIM29 binds directly to MAVS in RNA sensing signaling pathway
To further investigate how TRIM29 regulates in RNA virus-induced signaling events, we identified TRIM29-interacting proteins by immunoprecipitation with an antibody against TRIM29 (anti-TRIM29) in the mouse mDC line D2SC, followed by protein sequencing by liquid chromatography-mass spectrometry. We identified the MAVS among the group of TRIM29-interacting proteins. We next determined the possible interaction between TRIM29 and MAVS in BMDCs at the endogenous protein level. Anti-MAVS antibody or anti-TRIM29 antibody, but not control IgG, could pull down TRIM29 and MAVS, separately, in BMDCs induced by dsRNA stimulation. By contrast, Anti-MAVS antibody or anti-TRIM29 antibody could not pull down TRIM29 or MAVS without dsRNA stimulation, suggesting there was indeed interaction between TRIM29 and MAVS in BMDCs under the condition of cytosolic dsRNA stimulation (Fig. 5A). To further map the binding sites between MAVS and TRIM29, we analyzed the interactions between hemagglutinin (HA)-tagged recombinant TRIM29 and Myc-tagged recombinant full-length MAVS and truncation mutants of MAVS. Both recombinant full-length and the C-terminal transmembrane (TM) domain of MAVS, but not its N-terminal CARD and Proline domains, bound TRIM29 (Fig. 5B), suggesting that the C-terminal TM domain of MAVS interacted with TRIM29 (Fig. 5B). Next, we analyzed the interactions between Myc-tagged MAVS together with HA-tagged full-length TRIM29 and truncation mutants of TRIM29. As results, the full-length TRIM29 and all mutants except its N terminal domain could interact with MAVS (Fig. 5C), suggesting that the C-terminal domain of TRIM29 bound MAVS (Fig. 5C). By using a luciferase reporter assay of the IFN-β promoter established in human HEK293T cells, we found that overexpression of MAVS activated the IFN-β promoter activity (Fig. 5D). However, overexpression of wild-type TRIM29 (T29-a) and its truncate containing the interaction domain with MAVS (T29-b) inhibited the activation of the IFN-β promoter induced by MAVS (Fig. 5D), while overexpression of TRIM29 truncate without the interaction domain with MAVS lost its inhibitory effect (Fig. 5D). Furthermore, immunofluorescence of TRIM29 and MAVS showed that endogenous TRIM29 could colocalize with endogenous MAVS in the cytosol in BMDCs after reovirus infection (Fig. 5E). Collectively, these data suggested that TRIM29 could bind directly to key adaptor MAVS in the RNA virus sensing signaling pathway.
Figure 5. TRIM29 binds directly to MAVS.
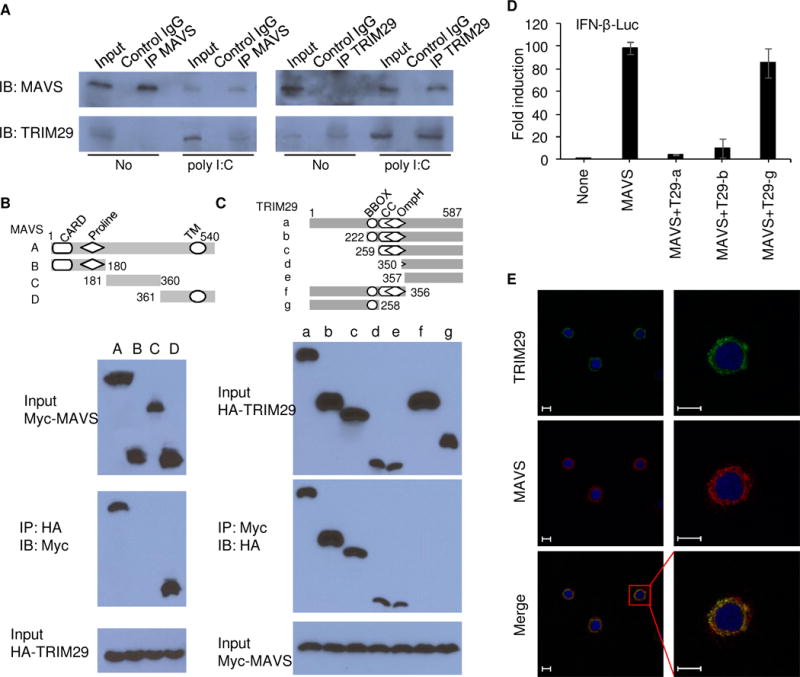
(A) Immunoblot analysis of endogenous proteins of TRIM29 and MAVS precipitated with anti-MAVS, anti-TRIM29 or control immunoglobulin G (control IgG) from whole-cell lysates of BMDCs from wild type mice stimulated without (No) or with poly I:C (25 μg/ml) for 8h, and then with MG132 treatment for 3h. Input, 20% of the BMDCs lysate. (B) Full-length MAVS and serial truncations of MAVS with deletion of various domains (top). Below, immunoblot analysis of purified HA-tagged TRIM29 with anti-HA (bottom blot), and immunoblot analysis of purified Myc-tagged full-length MAVS and MAVS truncation mutants alone with anti-Myc (top blot) or after incubation with HA-tagged full-length TRIM29 and immunoprecipitation with anti-HA (middle blot). (C) Full-length TRIM29 and serial truncations of TRIM29 with deletion (Δ) of various domains (left margin); numbers at ends indicate amino acid positions (top). Below, immunoblot analysis of purified Myc-tagged MAVS with anti-Myc (bottom blot), and immunoblot analysis (with anti-HA) of purified HA-tagged full-length TRIM29 and TRIM29 truncation mutants alone (top blot) or after incubation with Myc-tagged MAVS and immunoprecipitation with anti-Myc (middle blot). (D) Activation of the IFN-β promoter in human HEK293T cells transfected with an IFN-β promoter luciferase reporter, plus expression vector (each 100 ng) for wild-type MAVS or expression vector for wild-type TRIM29 (T29-a) or TRIM29 mutants T29-b and T29-g; results are presented relative to those of renilla luciferase (cotransfected as an internal control). (E) Colocalization of endogenous TRIM29 and MAVS in BMDCs. Confocal microscopy of BMDCs infected with reovirus for 4h. MAVS was stained with Rabbit anti-MAVS polyclonal antibody (Cat: 14341-1-AP, proteintech), followed by Alexa Fluor 594 goat anti-rabbit secondary antibody (green), while TRIM29 was stained with mouse anti-TRIM29 monoclonal antibody (sc-166707, Santa Cruz), followed by Alexa Fluor 488 goat anti-mouse secondary antibody (red). DAPI served as the nuclei marker (blue). Scale bars represent 20 μm. Data are representative of three independent experiments.
TRIM29 ubiquitinates and degrades MAVS
Because TRIM29 is an E3 Ub ligase, we next investigated whether MAVS is the ubiquitination target of TRIM29. We then co-expressed HA-TRIM29 with Myc-MAVS or vector control in HEK293T cells with or without treatment of MG132 and analyzed the expression of MAVS and TRIM29. As results, TRIM29 could significantly degrade the target protein MAVS, compared with the vector control (Fig. 6A). Additionally, the treatment of MG132 could rescue the expression of MAVS (Fig. 6A), suggesting that MAVS is indeed the ubiquitination target of TRIM29. To determine whether the expression of TRIM29 was regulated by poly I:C stimulation, we stimulated the WT and TRIM29 KO BMDCs with poly I:C and then measured the expression of TRIM29 and MAVS in BMDCs after poly I:C stimulation. TRIM29 was upregulated in BMDCs after poly I:C stimulation, while MAVS was downregulated after poly I:C stimulation due to the degradation by TRIM29 (Fig. 6B). Furthermore, the IRF3 and p65 were activated much stronger in TRIM29 KO BMDCs than those in WT BMDCs after poly I:C stimulation (Fig. 6B). These data indicated that TRIM29 was highly inducible and degraded MAVS much strongly after poly I:C stimulation.
Figure 6. TRIM29 induces ubiquitination and degradation of MAVS by K11-linkage.
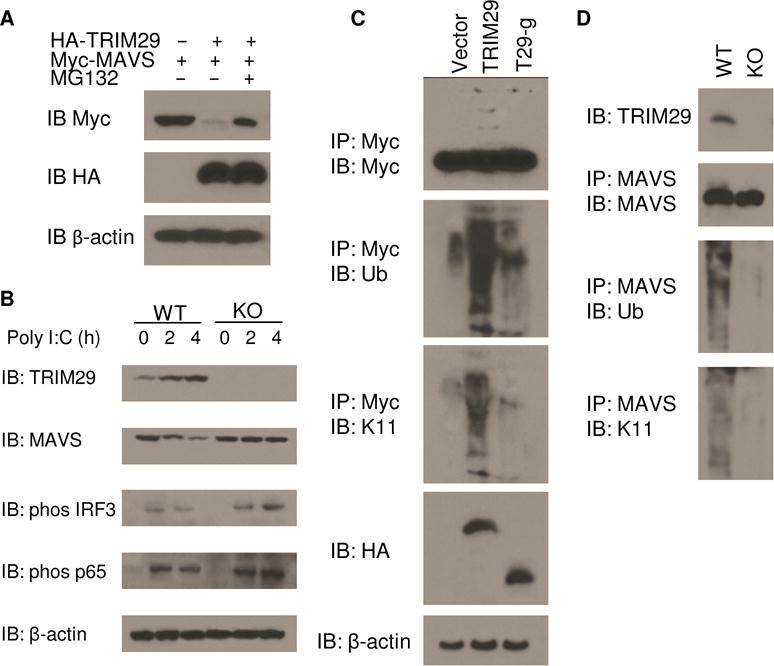
(A) Immunoblot analysis of Myc-tagged MAVS (top blot), HA-tagged TRIM29 (middle blot) and β-actin (bottom blot) in HEK293T cells cotransfected with an expression vector for Myc-tagged MAVS and empty vector or expression vector for HA-tagged TRIM29, with or without treatment of 5 μM MG132. (B) Immunoblot analysis of TRIM29 (top blot), MAVS (second blot), phosphorylated IRF3 (third blot), phosphorylated p65 (fourth blot) and β-actin (bottom blot) in WT and TRIM29 KO BMDCs stimulated for various times (above lanes) with poly I:C (10 μg/ml). (C) Immunoblot analysis (with anti-Myc) of the abundance (top), total ubiquitination (second blot) and K11-linked ubiquitination (third blot) of Myc-tagged MAVS in HEK293T cells transfected with empty vector or expression vector for HA-tagged TRIM29, truncation T29-g (losing binding site of MAVS), and stimulated for 4 h with poly I:C (20 μg/ml), assessed after immunoprecipitation with anti-Myc; and immunoblot analysis whole-cell lysates with anti-HA (fifth blot) and anti-β-actin (bottom). (D) Immunoblot analysis of TRIM29 in WT and KO BMDCs (top), and of the abundance (second blot), total ubiquitination (third blot) and K11-mediated ubiquitination (bottom blot) of MAVS in those cells, stimulated for 4 h with poly I:C (10 μg/ml), assessed after immunoprecipitation with anti-MAVS. Data are representative of three experiments.
To investigate whether the ubiquitination of MAVS was dependent on the binding site of TRIM29 with MAVS, we transfected the HEK293T cells to coexpress Myc-tagged MAVS and HA-tagged full-length TRIM29, truncated TRIM29 lacking the binding site of MAVS (T29-g). We stimulated the cells for 6 h with dsRNA and treated the cells with MG132 for 6h, then prepared cell lysates and incubated them for 5 min at 90 °C with 1% SDS to disrupt protein-protein interactions, followed by immunoprecipitation of Myc-tagged MAVS. Immunoblot analysis of hemagglutinin or ubiquitin demonstrated that the ubiquitination of MAVS was strongly enhanced by overexpression of TRIM29 but not by overexpression of T29-g (Fig. 6C). Immunoblot analysis of K11-linked ubiquitin further demonstrated that TRIM29 induced ubiquitination of MAVS by K11-mediated linkage (Fig. 6C). To determine whether TRIM29 was responsible for the ubiquitination of MAVS ex vivo, we stimulated BMDCs from wildtype mice or Trim29-knockout mice for 4 h with poly I:C. Cell lysates were prepared and analyzed the ubiquitination of MAVS. MAVS was modified via K11-mediated ubiquitination in BMDCs from wildtype mice but not Trim29-knockout mice (Fig. 6D). Together these data indicated that TRIM29 targeted MAVS and induced its ubiquitination for protein degradation by K11-linkage.
Lys371, Lys420, and Lys500 of MAVS are necessary for TRIM29-mediated signaling
To determine the MAVS-ubiquitination sites, we replaced each of these lysine residues individually with arginine (K371R, K420R, K461R, K500R and triple mutation K371RK420RK500R). We coexpressed HA-tagged TRIM29 with Myc-tagged wild-type MAVS and its mutants noted above in HEK293T cells and detected their expressions. As results, TRIM29 were expressed similarly. Moreover, TRIM29 could degrade strongly the wild type MAVS and its mutants (K461R), whereas the MAVS mutants K371R, K420R or K500R could be partially degraded by TRIM29. In contrast, TRIM29 could not degrade the triple mutant MAVS (K371RK420RK500R) (Fig. 7A), suggesting the Lys371, Lys420, and Lys500 of MAVS are the ubiquitination sites mediated by TRIM29. To further determine whether the ubiquitination of MAVS by TRIM29 was affected by the mutation of MAVS, we transfected the HEK293T cells to express Myc-tagged MAVS or its mutants noted above and HA-tagged full-length TRIM29 with the treatment of MG132. Cells were stimulated for 4 h with dsRNA. Cell lysates were prepared and followed by immunoprecipitation of Myc-tagged MAVS. Immunoblot analysis demonstrated that both Myc-tagged MAVS or its mutants and HA-tagged full-length TRIM29 were expressed similarly. As expected, the ubiquitination of MAVS or its mutant K461R was strongly enhanced by overexpression of TRIM29 (Fig. 7B). However, the ubiquitination of MAVS mutant K371R, K420R or K500R by TRIM29 was sharply reduced (Fig. 7B). Especially, ubiquitination of MAVS triple mutant (K371RK420RK500R) was completely lost (Fig. 7B).
Figure 7. Lys371, Lys420, and Lys500 of MAVS are necessary for TRIM29-mediated signaling.
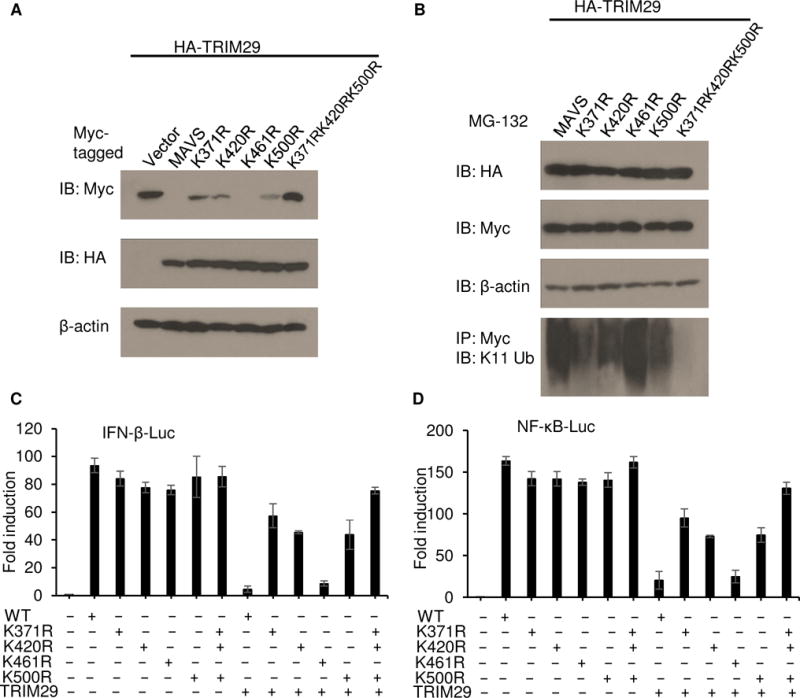
(A) Immunoblot analysis of Myc-tagged MAVS and its mutations (top blot), HA-tagged TRIM29 (middle blot) and β-actin (bottom blot) in HEK293T cells cotransfected with an expression vector for HA-tagged TRIM29 and expression vectors for Myc-tagged wild-type full-length MAVS and its mutations. (B) Immunoblot analysis of HA-tagged TRIM29 (top blot), Myc-tagged MAVS and its mutations (second blot), β-actin (third blot) and K11-linked ubiquitination (bottom blot) in HEK293T cells cotransfected with expression vector for Myc-tagged MAVS or its mutations (above lanes) and expression vector for HA-tagged TRIM29, with treatment of 5 μM MG132 (above lanes). (C,D) Activation of the IFN-β promoter (C) or NF-κB promoter (D) in human HEK293T cells transfected with an IFN-β promoter luciferase reporter (C) or NF-κB promoter luciferase reporter (D), plus expression vector (each 100 ng) for wild-type MAVS or various MAVS mutants alone or expression vector for TRIM29 plus wild-type MAVS or MAVS mutants; results are presented relative to those of renilla luciferase (cotransfected as an internal control). Data are representative of three independent experiments with similar results (mean + s.d.)
Through the use of a luciferase reporter assay of the IFN-β promoter and NF-κB promoter, established in the human HEK293T cells, we found that overexpression of wild-type MAVS enhanced the activity of the IFN-β promoter (Fig. 7C) and NF-κB promoter (Fig. 7D), Overexpression of mutant MAVS with the K371R, K420R, K461R, K500R and K371RK420RK500R substitution could still induce the activation of the IFN-β promoter (Fig. 7C) and NF-κB promoter (Fig. 7D). In addition, overexpression of TRIM29 inhibited the activation of the IFN-β promoter and NF-κB promoter in HEK293T cells expressing wild-type MAVS or its mutant with the K461R substitution (Fig. 7C and 7D). However, overexpression of TRIM29 did not inhibit the activity of the IFN-β promoter and NF-κB promoter in HEK293T cells expressing mutant MAVS with K371R, K420R, K500R or K371RK420RK500R substitution (Fig. 7C and 7D). These data indicated that Lys371, Lys420, and Lys500 were essential for TRIM29-mediated ubiquitination and regulation of MAVS.
DISCUSSION
Immune protection against viral infections relies on activation of the innate immune response to restrict viral replications. Activation of innate immune cells is tightly regulated in that robust anti-viral immunity is achieved without triggering excessive autoimmune inflammation. Previously, we found that TRIM29 was constitutively highly expressed in alveolar macrophages and controlled the local innate immunity in the respiratory tract(20). Recently, we also found that TRIM29 promoted DNA virus infections by inhibiting the innate immune response through degrading STING (25). However, the precise mechanisms of how antiviral signaling activation is strictly controlled during RNA virus infections still remain incompletely defined. Here, we found the E3 ligase TRIM29 was specifically expressed in poly I:C activated human myeloid dendritic cells (mDCs). TRIM29 was highly induced in human mDCs in response to poly I:C and played a negative role in type I interferon (IFN) production. The deficiency in TRIM29 resulted in enhanced type I interferon responses to poly I:C and RNA virus infection. Importantly, the challenge of wildtype mice with reovirus led to lethal infection. In contrast, deletion of TRIM29 protected the mice from developing lethal complications. Mechanistically, TRIM29 was shown to interact with MAVS, the key adaptor in the RNA sensing pathway. Importantly, TRIM29 induced ubiquitination and degradation of MAVS through K11-linked polyubiquitination. These data suggest that RNA virus hijacks the E3 ligase TRIM29 to shut down the host innate immune response for immune evasion.
Similar to many pathways, RLR signaling is tightly regulated to achieve an orchestrated response aimed at maximizing antiviral immunity and minimizing immune- or nonimmune-mediated collateral damage. To achieve an appropriately balanced response, downregulation of antiviral signaling is equally important to its activation. Polyubiquitination has been reported to regulate the activation of MAVS(36). Several E3 ligases have been identified that modulate the poly-ubiquitination of MAVS, including AIP4 (16, 37), RNF5(38), RNF125(39), Smurf1 and Smurf2 (40, 41), TRIM25 (42) and MARCH 5(43). However, all of these E3 ligases promote K48-linked ubiquitination of MAVS. Recently, TRIM31(44) and kinases IKK (45) were identified to mediate positive modification of MAVS through the K63-linked polyubiquitination and phosphorylation, respectively. In our study, we identified firstly TRIM29 as an E3 ligase for the K11-linked polyubiquitination of MAVS. To our knowledge, TRIM29 is the first E3 ligase identified in negatively regulating the activation of MAVS through its ubiquitination in the K11 linkage.
So far, a large number of molecules were proposed and identified as important regulators in the antiviral signaling pathway of type I IFN production. But, only very few concepts were investigated and verified in vivo using knockout mice. In the present study, our observations based on the study of Trim29−/− mice solidify a role for TRIM29 as an in vivo checkpoint of type I IFN response. We have extended these findings to the RNA virus and identified TRIM29 as a key negative regulator in the host defense against the RNA virus.
Type I IFNs play a critical role in not only eliminating the invading pathogens, but also many autoimmune diseases via various immune modulatory actions(46). Recent onset of type 1 diabetes is strongly correlated with infection by RNA viruses such as enteroviruses (47, 48). In this context, the identification of TRIM29 as a negative regulator of key adaptor MAVS in RNA sensing signaling pathway will have important implications for the understanding of not only antiviral innate immunity but also the pathogenesis of human autoimmune diseases.
Supplementary Material
Acknowledgments
We thank the Wellcome Trust Sanger Institute Mouse Genetics Project (Sanger MGP) and its funders for providing the mutant mouse line Trim29, and the European Mouse Mutant Archive (www.emmanet.org) partner from which the mouse line was received.
This work was supported by Lupus Research Alliance grant 519418 (Z.Z.) and National Institutes of Health grant R01AI080779 (X.C.L.).
References
- 1.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 2.Kato H, Takahasi K, Fujita T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev. 2011;243:91–98. doi: 10.1111/j.1600-065X.2011.01052.x. [DOI] [PubMed] [Google Scholar]
- 3.Chan YK, Gack MU. RIG-I-like receptor regulation in virus infection and immunity. Curr Opin Virol. 2015;12:7–14. doi: 10.1016/j.coviro.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 5.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 6.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 9.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 12.Yoneyama M, Fujita T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity. 2008;29:178–181. doi: 10.1016/j.immuni.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Jacobs JL, Coyne CB. Mechanisms of MAVS regulation at the mitochondrial membrane. J Mol Biol. 2013;425:5009–5019. doi: 10.1016/j.jmb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, Ting JP. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 15.Yasukawa K, Oshiumi H, Takeda M, Ishihara N, Yanagi Y, Seya T, Kawabata S, Koshiba T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal. 2009;2:ra47. doi: 10.1126/scisignal.2000287. [DOI] [PubMed] [Google Scholar]
- 16.You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, Jiang Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. 2009;10:1300–1308. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- 17.Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 18.Masuda Y, Takahashi H, Sato S, Tomomori-Sato C, Saraf A, Washburn MP, Florens L, Conaway RC, Conaway JW, Hatakeyama S. TRIM29 regulates the assembly of DNA repair proteins into damaged chromatin. Nat Commun. 2015;6:7299. doi: 10.1038/ncomms8299. [DOI] [PubMed] [Google Scholar]
- 19.Kanno Y, Watanabe M, Kimura T, Nonomura K, Tanaka S, Hatakeyama S. TRIM29 as a novel prostate basal cell marker for diagnosis of prostate cancer. Acta Histochem. 2014;116:708–712. doi: 10.1016/j.acthis.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, Lu H, Li XC, Liu YJ, Zhang Z. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nature immunology. 2016;17:1373–1380. doi: 10.1038/ni.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Yang H, Abel EV, Ney GM, Palmbos PL, Bednar F, Zhang Y, Leflein J, Waghray M, Owens S, Wilkinson JE, Prasad J, Ljungman M, Rhim AD, Pasca di Magliano M, Simeone DM. ATDC induces an invasive switch in KRAS-induced pancreatic tumorigenesis. Genes Dev. 2015;29:171–183. doi: 10.1101/gad.253591.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmbos PL, Wang L, Yang H, Wang Y, Leflein J, Ahmet ML, Wilkinson JE, Kumar-Sinha C, Ney GM, Tomlins SA, Daignault S, Kunju LP, Wu XR, Lotan Y, Liebert M, Ljungman ME, Simeone DM. ATDC/TRIM29 Drives Invasive Bladder Cancer Formation through miRNA-Mediated and Epigenetic Mechanisms. Cancer Res. 2015;75:5155–5166. doi: 10.1158/0008-5472.CAN-15-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ai L, Kim WJ, Alpay M, Tang M, Pardo CE, Hatakeyama S, May WS, Kladde MP, Heldermon CD, Siegel EM, Brown KD. TRIM29 suppresses TWIST1 and invasive breast cancer behavior. Cancer Res. 2014;74:4875–4887. doi: 10.1158/0008-5472.CAN-13-3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Heidt DG, Lee CJ, Yang H, Logsdon CD, Zhang L, Fearon ER, Ljungman M, Simeone DM. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and beta-catenin stabilization. Cancer Cell. 2009;15:207–219. doi: 10.1016/j.ccr.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, Zhang Z. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. 2017;8:945. doi: 10.1038/s41467-017-00101-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xing J, Ly H, Liang Y. The Z proteins of pathogenic but not nonpathogenic arenaviruses inhibit RIG-I-like receptor-dependent interferon production. J Virol. 2015;89:2944–2955. doi: 10.1128/JVI.03349-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing J, Chai Z, Ly H, Liang Y. Differential Inhibition of Macrophage Activation by Lymphocytic Choriomeningitis Virus and Pichinde Virus Is Mediated by the Z Protein N-Terminal Domain. J Virol. 2015;89:12513–12517. doi: 10.1128/JVI.01674-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol. 2013;14:172–178. doi: 10.1038/ni.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Virgin HWT, Mann MA, Fields BN, Tyler KL. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol. 1991;65:6772–6781. doi: 10.1128/jvi.65.12.6772-6781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xing J, Wang S, Lin F, Pan W, Hu CD, Zheng C. Comprehensive characterization of interaction complexes of herpes simplex virus type 1 ICP22, UL3, UL4, and UL20.5. J Virol. 2011;85:1881–1886. doi: 10.1128/JVI.01730-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xing J, Wang S, Lin R, Mossman KL, Zheng C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol. 2012;86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol. 2013;87:9788–9801. doi: 10.1128/JVI.01440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weng L, Mitoma H, Trichot C, Bao M, Liu Y, Zhang Z, Liu YJ. The E3 ubiquitin ligase tripartite motif 33 is essential for cytosolic RNA-induced NLRP3 inflammasome activation. J Immunol. 2014;193:3676–3682. doi: 10.4049/jimmunol.1401448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol. 2015;32:48–53. doi: 10.1016/j.coi.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Heaton SM, Borg NA, Dixit VM. Ubiquitin in the activation and attenuation of innate antiviral immunity. J Exp Med. 2016;213:1–13. doi: 10.1084/jem.20151531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou X, You F, Chen H, Jiang Z. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS) Cell Res. 2012;22:717–727. doi: 10.1038/cr.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong B, Zhang Y, Tan B, Liu TT, Wang YY, Shu HB. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J Immunol. 2010;184:6249–6255. doi: 10.4049/jimmunol.0903748. [DOI] [PubMed] [Google Scholar]
- 39.Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104:7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Tong X, Ye X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J Immunol. 2012;189:5304–5313. doi: 10.4049/jimmunol.1201445. [DOI] [PubMed] [Google Scholar]
- 41.Pan Y, Li R, Meng JL, Mao HT, Zhang Y, Zhang J. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J Immunol. 2014;192:4758–4764. doi: 10.4049/jimmunol.1302632. [DOI] [PubMed] [Google Scholar]
- 42.Castanier C, Zemirli N, Portier A, Garcin D, Bidère N, Vazquez A, Arnoult D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoo YS, Park YY, Kim JH, Cho H, Kim SH, Lee HS, Kim TH, Sun Kim Y, Lee Y, Kim CJ, Jung JU, Lee JS. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat Commun. 2015;6:7910. doi: 10.1038/ncomms8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, Wang P, Zhao K, Hou J, Wang X, Zhang L, Gao C. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. 2017;18:214–224. doi: 10.1038/ni.3641. [DOI] [PubMed] [Google Scholar]
- 45.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 46.González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dotta F, Sebastiani G. Enteroviral infections and development of type 1 diabetes: The Brothers Karamazov within the CVBs. Diabetes. 2014;63:384–386. doi: 10.2337/db13-1441. [DOI] [PubMed] [Google Scholar]
- 48.Richardson SJ, Horwitz MS. Is type 1 diabetes “going viral”? Diabetes. 2014;63:2203–2205. doi: 10.2337/db14-0510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.