

Abstract
Influenza A virus (IAV) remains a major worldwide health threat, especially to high-risk populations, including the young and elderly. There is an unmet clinical need for therapy that will protect the lungs from damage caused by lower respiratory infection. Here, we analyzed the role of EMAPII, a stress- and virus-induced pro-inflammatory and pro-apoptotic factor, in IAV-induced lung injury. First, we demonstrated that IAV induces EMAPII surface translocation, release, and apoptosis in cultured endothelial and epithelial cells. Next, we showed that IAV induces EMAPII surface translocation and release to bronchoalveolar lavage fluid (BALF) in mouse lungs, concomitant with increases in caspase 3 activity. Injection of monoclonal antibody (mAb) against EMAPII attenuated IAV-induced EMAPII levels, weight loss, reduction of blood oxygenation, lung edema, and increase of the pro-inflammatory cytokine TNF alpha. In accordance with the pro-apoptotic properties of EMAPII, levels of caspase 3 activity in BALF were also decreased by mAb treatment. Moreover, we detected EMAPII mAb-induced increase in lung levels of M2-like macrophage markers YM1 and CD206. All together, these data strongly suggest that EMAPII mAb ameliorates IAV-induced lung injury by limiting lung cell apoptosis and shifting the host inflammatory setting toward resolution of inflammation.
Keywords: EMAPII, IAV, lung injury, apoptosis, barrier dysfunction
Graphical Abstract
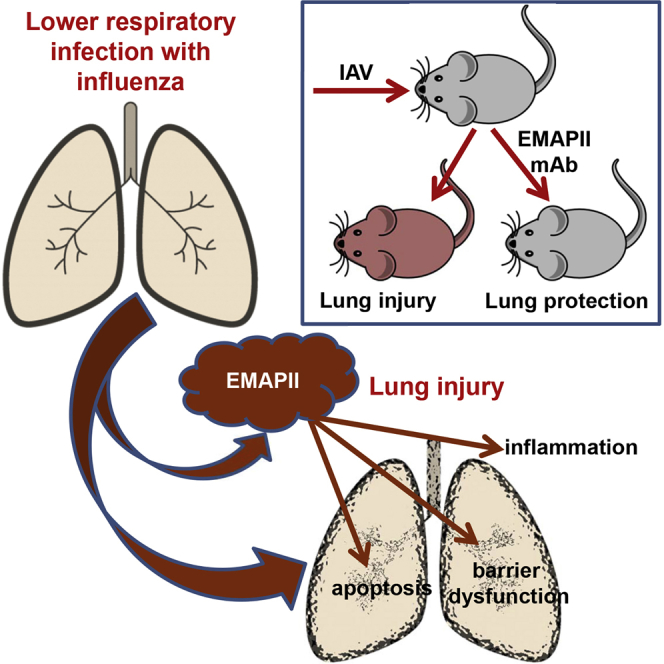
Infection with influenza A virus is followed by the membrane translocation and release of pro-apoptotic and pro-inflammatory mediator EMAPII. Here, Lu and colleagues show that EMAPII neutralization with monoclonal antibody attenuates epithelial apoptosis and barrier dysfunction in vitro and mitigates influenza A virus-induced lung injury in vivo.
Introduction
Influenza infections are associated with the risk of development of lower respiratory tract infections, pneumonia, and lung injury.1, 2 Current treatment of influenza infection relies primarily on neuraminidase inhibitors, which are most effective when given within the first 48 hr,3 a narrow window missed by the majority of patients admitted to ERs with pulmonary complications. Importantly, the latest meta-analysis shows that even early administration of antivirals does not decrease the likelihood of influenza-related pneumonia, although it lowers mortality and the need for ventilator support.4 Moreover, presently used antivirals may lose their therapeutic potency in the future due to mutations of the virus.5, 6 Given the current paucity of therapeutic choices, there is an unmet medical need for a therapy that will not be virus specific, but rather protect the infected lung by modulating detrimental host responses regardless of the type of viral infection.
In the past decade, suppression of many pro-inflammatory responses was successfully attained by a new class of therapeutic agents, monoclonal antibodies targeting host pro-inflammatory molecules.7 Whereas this approach is now widely used for the treatment of diseases such as rheumatoid arthritis and multiple sclerosis, only limited therapeutic monoclonal antibodies are available to treat lung diseases.8 Here, we study the ability of monoclonal antibody toward AIMP1/EMAPII to treat lung injury in mice. AIMP1 encodes non-catalytic component of tRNA synthetase complex, which, upon stress stimuli and apoptosis, can be translocated to the membrane and released as pro-apoptotic and pro-inflammatory product EMAPII.9 EMAPII emerged as a novel target for treating lung diseases due to the fact that it is released by lung endothelial cells.10, 11, 12, 13 EMAPII levels increase in chronic lung inflammatory conditions such as chronic obstructive pulmonary disease (COPD)14 and developmental lung disorders such as bronchopulmonary dysplasia.15 EMAPII monoclonal antibody (mAb) was shown to abrogate progression of emphysema in mice exposed to cigarette smoke.14
Here, we analyze whether EMAPII mAb can limit an acute pathological condition of the lung, namely influenza A virus (IAV)-induced lung injury. We show that EMAPII is translocated to the surface and released in response to IAV in vitro and in vivo, and EMAPII translocation and/or release potentiates IAV-induced endothelial and epithelial apoptosis and barrier dysfunction. Importantly, we demonstrate that EMAPII ablation with mAb attenuates IAV-induced lung injury in mice and promotes a shift toward resolution of inflammation.
Results
IAV Induces EMAPII-Dependent Apoptosis in Pulmonary Endothelium and Epithelium
We have previously demonstrated that EMAPII mediates HIV protein gp120-induced apoptosis in endothelial cells.11 To elucidate whether EMAPII plays a similar role in IAV-infected cells, we first tested whether IAV evokes caspase 3 cleavage in pulmonary endothelium and epithelium. Caspase 3 was chosen due to the convergence of both extrinsic and intrinsic apoptotic pathways on this important player in the terminal execution pathway.16 Our results showed that normal human bronchial epithelial cells (NHBECs) and alveolar epithelial cells (A549), as well as human pulmonary artery endothelial cells (HPAECs) and human lung microvascular endothelial cells (HLMVECs) responded to IAV with caspase 3 cleavage observed as early as 4 hr after infection (Figures 1A and 1B). Accordingly, surface annexin V staining, an alternative readout for apoptosis, was increased in IAV-exposed epithelium and endothelium (Figure 1C). IAV-induced apoptosis was accompanied by a release of extracellular EMAPII (detected as early as 2–6 hr after infection; Figure 1D) and increase in EMAPII cell surface expression (Figure 1E). These events were not accompanied by the increase in AIMP1/EMAPII intracellular expression (Figure 1B) or AIMP1/EMAPII mRNA levels (data not shown).
Figure 1.

IAV Induces Apoptosis and EMAPII Release in Pulmonary Endothelium and Epithelium
(A–C and E) Human pulmonary artery endothelial cells (HPAEC), normal human bronchial epithelial cells (NHBEC), human lung microvascular endothelial cells (HLMVEC), and human alveolar epithelial line A549 were stimulated with 1 pfu/cell IAV and then analyzed for (A and B) caspase 3 cleavage, caspase 3 (A) and EMAPII (B) expression, (C) surface annexin V staining, (D) released EMAPII levels, (E) surface EMAPII staining, or (F) concomitant caspase 3 cleavage and surface EMAPII staining. *p < 0.05 by t test when compared to control values. n = 3–6, data are shown as mean ± SEM.
To assess the role of EMAPII surface translocation, we evaluated surface EMAPII fluorescence levels in cleaved caspase 3-positive and -negative populations. In both endothelial and epithelial cells, surface EMAPII mean fluorescence levels were significantly higher in populations positive for cleaved caspase 3 (Figure 1F), linking IAV-induced EMAPII surface translocation to the activation of pro-apoptotic pathways.
To show that EMAPII release potentiates IAV-induced apoptosis, we compared caspase 3 cleavage in cells treated with IAV in the presence and absence of recombinant EMAPII. EMAPII enhanced IAV-induced caspase 3 cleavage in endothelial (Figure 2A) and epithelial (data not shown) cells, potentiating IAV effect. To demonstrate that IAV-induced apoptosis is mediated by EMAPII, we applied EMAPII neutralizing antibody to IAV-stimulated cells. The mAb dose previously described to limit EMAPII-induced apoptosis in endothelial cells17 was also able to limit IAV-induced apoptosis in A549 bronchial epithelium (Figure 2B).
Figure 2.

IAV-Induced Apoptosis Is Potentiated by EMAPII and Suppressed by EMAPII mAb
HLMVECs (A) and A549 (B) were stimulated with 1 pfu/cell IAV (A) in the presence of 30 μg/mL recombinant EMAPII (E) or (B) 10 μg/mL control IgG or EMAPII mAb, then analyzed for cleaved and total caspase 3 levels. n = 3–5, data are presented as mean ± SEM; *p < 0.05 by one-way ANOVA with Tukey post-hoc.
IAV and EMAPII Induce Barrier Dysfunction in Pulmonary Endothelium and Epithelium
To further analyze the role of EMAPII in IAV-induced lung injury, we assessed the effects of IAV and EMAPII on transendothelial permeability. Endothelial monolayers challenged with 0.2–1 MOI of IAV responded with transient decreases of transendothelial resistance (Figure 3A). Control treatment with heat-inactivated virus did not induce endothelial hyperpermeability, suggesting that active virus is required for barrier dysfunction. However, EMAPII mAb failed to attenuate this short-term IAV-induced barrier dysfunction in endothelial cells (Figure 3B), suggesting EMAPII-independent mechanism of the observed immediate loss of barrier in response to IAV. On the contrary, alveolar epithelium responded to IAV with delayed barrier disruption, evident 72 hr–96 hr after initial challenge (Figures 3C and 3E). Notably, recombinant EMAPII was also able to induce delayed barrier dysfunction and enhance IAV-induced effect, potentiating hyperpermeability (Figure 3C). In concert with pro-apoptotic effect of EMAPII, EMAPII-induced decrease in transepithelial resistance (TER) was attenuated with cell-permeable pan-caspase inhibitor QVD-OPh hydrate (Figure 3D). To demonstrate that IAV-induced barrier dysfunction is mediated by EMAPII, we applied EMAPII-neutralizing antibody to IAV-stimulated cells. EMAPII mAb significantly attenuated IAV-induced decrease in TER, indicative of EMAPII role in barrier-disruptive effects of IAV.
Figure 3.

IAV and EMAPII Induce Hyperpermeability in Endothelial and/or Epithelial Monolayers
(A) HLMVECs grown to confluence were stimulated with 0.2 and 1 pfu/cell of IAV or 1 pfu/cell of heat-inactivated IAV. (B) HPAECs were stimulated with 0.75 pfu/cell IAV in the presence of 10 μg/mL control IgG or EMAPII mAb. (C) A549 were stimulated with 2 pfu/cell IAV, 40 μg/mL recombinant EMAPII, or their combination. (D) A549 were stimulated with 40 μg/mL recombinant EMAPII (E) in the presence/absence of 9 μM QVD (Q). (E) A549 were stimulated with 2 pfu/cell IAV in the presence of 15 μg/mL control IgG or EMAPII mAb. Shown are mean ± SE of three parallel recordings; resistance is normalized to the moment of stimulation with IAV/EMAPII (shown with an arrow in A and B). *p < 0.05 by t test.
IAV Lung Injury Is Associated with EMAPII Translocation/Release and Apoptosis in Lung
To assess the kinetics of IAV-induced lung injury development and to identify the optimal time window for therapeutic intervention with EMAPII-neutralizing antibody, we analyzed lung infection in mice following oropharyngeal aspiration of IAV. Delivery of 750 pfu (particle forming units) of IAV to mouse lung caused a self-limiting infection in mice, characterized by an initial decrease in body weight reaching 78% of the original weight at day 7 and followed by a spontaneous restoration to 95% of the original weight at day 10–14 (Figure 4A). Consistent with the course of malaise, conscious blood oxygenation decreased from 95% at day 0 to 82% at day 7, recovering to 90% by day 14 (Figure 4B). Assessment of lung injury indices showed that lung edema peaked at day 7 (Figure 4C), whereas inflammatory cell and protein extravasation in bronchoalveolar lavage fluid (BALF) were greater at day 10 (Figures 4D and 4E). Majority of inflammatory cells recruited to lung in this model of IAV infection were macrophages (Figure 4E).
Figure 4.

Self-Limiting IAV Infection in Mice Is Accompanied by Lung Injury
Mice were administered 750 pfu/mouse IAV to lung and analyzed for (A) weight loss, (B) conscious blood oxygenation, (C) lung edema, (D) BALF protein extravasation, and (E) BALF white blood cell (WBC) count including total WBC (t), macrophages (m), lymphocytes (l), and neutrophils (n). (A and B) n = 5, (C and D) n = 7 for control group and 3 or 4 for all other groups; data are presented as mean ± SEM. *p < 0.05 by ANOVA with Tukey post-hoc when compared to control values.
Analysis of EMAPII levels in IAV-infected lungs has shown that EMAPII release to BALF occurs relatively late in the course of lung injury and is concomitant with an increase in EMAPII expression in total lung (Figures 5A and 5C). No increase in EMAPII plasma concentration was noted (data not shown). Consistently, IAV-induced increase in caspase 3 activity in BALF and total lung tissue was not significant until day 10 post-infection (Figures 5B and 5D). To increase sensitivity of detection and further analyze cellular subsets affected by apoptosis in IAV-damaged lung, we performed flow cytometric assessment of endothelial (CD31+), epithelial (CD326+), and hematopoietic (CD45+) populations in lung digests.18 We observed significant increases in the amount of cleaved caspase 3-positive cells in endothelial and hematopoietic lineages as early as days 5–7 post-infection (Figures 6B and 6D). Increases in the amount of surface EMAPII+ cells were observed at days 5–7 in the same populations (Figures 6A and 6C).
Figure 5.

IAV-Induced Lung Injury Is Accompanied by EMAPII Release and Induction of Pulmonary Apoptosis
Mice were administered 750 pfu/mouse IAV to lung and analyzed for (A) BALF level of EMAPII, (B) BALF level of caspase 3/7 activity, (C) lung levels of EMAPII, (D) lung levels of cleaved caspase 3. n = 7 for control group and 3–4 for all other groups; data are presented as mean ± SEM. *p < 0.05 by ANOVA with Tukey post-hoc when compared to control values.
Figure 6.

IAV-Induced Lung Injury Is Accompanied by Surface EMAPII Translocation and Caspase 3 Cleavage in Cells of Hematopoietic and Endothelial Lineage
Mice infected with 750 pfu/mouse IAV were sacrificed at day 5 (A and B) or day 7 (C and D). Lungs were digested and subjected to concomitant staining for surface CD31/CD326/CD45 and EMAPII (A and C) or permeabilized and subjected to concomitant staining for CD31/CD326/CD45 and cleaved caspase 3 (B and D). n = 3–5; data are presented as mean ± SEM. *p < 0.05 by t test with Welch correction when compared to control values.
EMAPII mAb Attenuates EMAPII Release, Lung Injury, and Apoptosis
To increase the clinical relevance of the tested treatment protocol, we chose to start administration of EMAPII mAb 4 days after infection (Figure 7A). Day 9 post-infection was chosen for terminal analyses. Repeated subcutaneous injections of EMAPII mAb attenuated the levels of EMAPII released to BALF (Figure 7B). Importantly, EMAPII mAb ameliorated IAV-induced loss of body weight (Figure 7C), decreased blood oxygenation (Figure 6D), lung edema, and protein extravasation into BALF (Figure 7E) in mice. Furthermore, EMAPII ablation with mAb significantly attenuated IAV-induced caspase 3 activity in BALF (Figure 7F). However, EMAPII mAb therapy did not reduce the total amount of inflammatory cell in BALF or corresponding amounts of macrophages, lymphocytes, or neutrophils (Figure 8A).
Figure 7.

EMAPII mAb Subcutaneous Injections Reduce Levels of Released EMAPII, Weight Loss, and Indices of Lung Injury in IAV-Infected Mice
(A) Mice received 750 pfu/mouse IAV or equal volume of saline (cntr). Half of IAV-infected mice were treated with EMAPII mAb (2.5 mg/kg) on days 4, 6, and 8 post-infection. Mice were analyzed for (B) EMAPII levels in BALF, (C) IAV-induced weight loss, (D) conscious blood oxygenation, (E) lung edema and protein in BALF (day 9), and (F) BALF caspase 3/7 activity (day 9). n = 5 for all groups; data are presented as mean ± SEM. *p < 0.05 by ANOVA with Tukey post-hoc (B and D–F) or repeated-measurements ANOVA (C).
Figure 8.

EMAPII mAb Subcutaneous Injections Reduce BALF Levels of TNF-α and Increase Levels of M2 Markers in the Lung of IAV-Infected Mice
At day 9, mice from Figure 6 were analyzed for (A) WBC count in BALF including total WBC (t), macrophages (m), lymphocytes (l), and neutrophils (n), (B) TNF-α levels in BALF, (D) YM1 levels in lung, (E) CD206 levels in lung. n = 5 for all groups; data are presented as mean ± SEM. *p < 0.05 by ANOVA with Tukey post-hoc; shown are differences between IAV and control, and IAV and IAV + ab groups. (C) Mice from Figures 4 and 5 were analyzed for YM1, and CD206 levels in lung; vinculin was used as a loading control.
EMAPII mAb Shifts the Balance between Pro- and Anti-inflammtory Markers in IAV-Infected Lungs
To assess the balance of pro-inflammatory and anti-inflammatory players in mice responding to therapy with EMAPII neutralizing antibody, we analyzed markers of M1/M2 macrophages.
Analysis of BALF level of tumor necrosis factor alpha (TNF-α) revealed significant attenuation of this M1 macrophage marker in EMAPII mAb-treated mice (Figure 8B). Next, we analyzed lung levels of M2 markers YM1 and CD206. Western blot analysis revealed that both YM1 and CD206 levels were increased at days 7–10 post-infection with IAV (Figure 8C), suggesting natural resolution of inflammation in our model of infection. Interestingly, EMAPII neutralizing antibody further increased lung levels of YM1 and CD206, reaching significant differences with YM1 and CD206 levels detected in control mice (Figures 8D and 8E).
Discussion
With the realization that influenza complications are often associated with excessive host response,19 several host-directed therapies were tested recently in an attempt to mitigate IAV-induced lung injury. Some of them, such as the nuclear factor κB (NF-κB) inhibiting derivative of acetylsalicylic acid, already reached clinical trials (www.clinicalregister.eu, EudraCT number: 2012-004072-19); others, such as epithelium-restoring granulocyte-macrophage colony-stimulating factor (GM-CSF), showed promising preliminary results.20 Surprisingly, immunomodulatory and lung-regenerative bone marrow-derived mesenchymal stromal cells failed to attenuate lung injury in animal models of seasonal influenza,21, 22 although mitigation of avian influenza-induced lung injury was demonstrated.23, 24 Interestingly, the key mechanism proposed was the protection of alveolar epithelium from avian influenza-induced damage.23 Our study investigates the ability of EMAPII-neutralizing antibody to protect endothelium and epithelium from IAV H1N1-induced apoptosis and is first to show that this antibody can ameliorate IAV-induced lung injury in mice. Previously, pro-apoptotic activity of EMAPII was proven to be a key pathological factor in chronic lung inflammatory condition, such as COPD.14 Here, we show that pro-apoptotic EMAPII contributes to the pathogenesis of acute inflammatory condition such as viral lung injury.
IAV is known to induce host cell apoptosis by increasing activity of pro-apoptotic factors Bax and Bad via interruption of clusterin and Bax association25 or Bad phosphorylation and cleavage.26 In addition, accumulation of pro-apoptotic proteins such as TRAIL, death receptor Fas, and its ligand FasL in response to IAV was also shown.27 Here, we show that IAV causes surface translocation and release of pro-apoptotic EMAPII from endothelial and epithelial cells. Our data indicate that both surface translocation and challenge with extracellular EMAPII increase caspase 3 cleavage in endothelium and epithelium, inferring both autocrine and paracrine roles of translocated and/or released EMAPII. Earlier, epithelial-released interferon α (IFNα) was shown to relay epithelial damage from cells infected with IAV H1N1 to the adjacent non-infected cells.28 Similarly, epithelial damage by avian influenza was attributed to the release of soluble mediators rather than direct cytopathic effects of virus.23 Our data suggest that EMAPII is likely to play a similar role as the propagator of the initial injury caused by IAV in endothelium and epithelium.
We have shown here for the first time that EMAPII directly induces barrier dysfunction in epithelial cells, and this dysfunction is apoptosis dependent. Acting as an edemagenic agent, translocated and/or released EMAPII is likely to contribute to the pathogenesis of IAV-induced lung edema, prompting the investigation of EMAPII ablation effects in in vivo lung injury model.
To assess the effect of EMAPII neutralization in vivo and establish the basis for possible therapeutic intervention, we have tested the effect of repeated subcutaneous administration of EMAPII mAb on IAV-induced lung injury. As of now, therapeutic monoclonal antibodies are administered via parenteral route in the form of injections;29 no technological platform is currently available for inhaled administration of this type of biologic. Subcutaneous route of administration was chosen here since this route gives a patient an easy opportunity for self-administration as opposed to intravenous administration in the acute care setting. We have shown that subcutaneously delivered EMAPII mAb, first administered at day 4 post-infection, effectively limits IAV-induced body weight loss and lung injury indices including decreased blood oxygenation, lung edema, and levels of the pro-inflammatory cytokine TNF-α. Importantly, this therapeutic window exceeds 48 hr post-infection period currently recommended for the standard-of-care antivirals such as oseltamivir. This advantage can be strongly appreciated by the at-risk patients who tested positive for influenza outside of the 48-hr window of the first symptom manifestation.
Interestingly, although significant increases in surface EMAPII-positive cells in lung were observed at day 7 post-infection, increases in levels of released EMAPII were not detected in BALF until day 9 or 10 of IAV infection. These data suggest that the time course of EMAPII release to BALF is delayed when compared to the changes in lung injury indices, making changes in surface EMAPII expression a better clinical indicator of lung injury. Furthermore, data of literature suggest that EMAPII-mediated apoptosis is induced more efficiently via contact with EMAPII-presenting cells rather than via soluble mediator signaling.30 The fact that administration of EMAPII mAb on day 4 allowed us to see improvement in body weight as early as day 6 post-infection strongly suggests that early EMAPII surface translocation and rise in local EMAPII levels (in vicinity of leukocyte-endothelial interface) is of clinical significance.
In accordance with the pro-apoptotic role of EMAPII,13, 14 we detected strong attenuation of caspase 3/7 activity in BALF of EMAPII mAb-treated mice. While our in vitro data strongly suggested that the reduction of EMAPII-induced lung cell apoptosis and barrier hyperpermeability are the main mechanisms leading to lung edema attenuation by EMAPII mAb, we also explored modulation of inflammation as an additional mechanism of lung injury mitigation. To our surprise, initial assessment revealed no suppression of IAV-induced extravasation of inflammatory cell by EMAPII mAb. However, a reduced level of the M1 macrophage marker TNF-α in BALF prompted further investigation of the shift between the pro-inflammatory M1 and anti-inflammatory M2-like macrophage phenotype in EMAPII mAb-treated animals. Analysis of the M2-like macrophage markers CD206 and YM1 revealed that their expression is increased between day 7 and day 10 in the self-limiting model of IAV infection analyzed here. Importantly, CD206 and YM1 levels were further increased in mice receiving EMAPII mAb therapy. These results are in concert with the recent findings23 demonstrating that therapy with mesenchymal stem cells, protecting epithelium, also shifts M1/M2 balance of infiltrating macrophages without changing the total amount of inflammatory cells in BALF. These data suggest that attenuation of lung cell damage is likely to affect macrophage machinery via paracrine factor cross-talk facilitating resolution of inflammation. With EMAPII mAb therapy, EMAPII ablation can also have a direct effect on macrophage polarization, since macrophages express EMAPII receptor CXCR3,31 and CXCR3 deficiency is known to promote M2 phenotype.32 M2-like macrophages were recently shown to play important roles in the resolution of lung injury.33 All together, our data demonstrate that EMAPII mAb therapy exerts its beneficial effects via several mechanisms involving suppression of lung cell apoptosis and/or barrier dysfunction as well as promotion of the resolution of inflammation.
In conclusion, this study has clearly shown that EMAPII mAb effectively attenuates IAV-induced lung injury in mice. This therapy targets a novel component of endothelial and/or epithelial injury34, 35 along with more conventional components of host inflammatory milieu. In contrast to existing anti-viral therapies19 and prospective therapies with virus-specific monoclonal antibodies,36 this therapy will not be compromised by development of resistance in viruses and can be possibly extended to treat lung injury from other viruses frequently causing pulmonary complications, such as adenovirus. Another important conclusion from our study is that careful investigation of the EMAPII role in clinical disorders where endothelium and/or epithelium are exposed to noxious stimuli may reveal yet-undiscovered involvement of EMAPII in pathogenesis. For these disorders, EMAPII mAb therapy may be of clinical relevance.
Materials and Methods
Cell Culture-Based Assays
HPAECs, HLMVECs, NHBECs (Lonza, Walkerville, MD), and A549 (ATCC, Manassas, VA) were stimulated with H1N1 A/PR/8/34 (ATCC, #VR-1469) in the absence or presence of rat immunoglobulin G (IgG) (Abcam, Cambridge, UK, ab#37361) or rat anti-human EMAPII M7/1 mAb.14 Media was analyzed with human AIMP1 competitive ELISA kit (MyBioSource, San Diego, CA). Cells were stained for surface proteins with anti-EMAPII mAb14 or annexin V antibody (Abcam, #ab14085) and subjected to flow cytometry using a fluorescence-activated cell sorting (FACS) Calibur flow cytometer and Cell-Quest Pro software (BD Biosciences, San Jose CA) or analyzed for cell fluorescence using FlexStation II (Molecular Devices, Sunnyvale, CA). Mean fluorescence was assessed for EMAPII; percentage of positive cells was assessed for annexin V. For EMAPII mRNA level analysis, qRT-PCR was performed with EMAPII primers from Sino Biological (Beijing, China), using β-actin as the housekeeping gene. Alternatively, cells were digested with 1% SDS-containing PBS and analyzed by western blot with anti-cleaved caspase 3, anti-caspase 3 (Cell Signaling, Danvers, MA, #49662 and #96625, respectively), rabbit anti-EMAPII17 and anti-β-actin antibodies (Sigma, St. Louis, MO, #A5441).
Measurement of Transendothelial Permeability
Transendothelial electrical resistance (TER) was measured using the highly sensitive biophysical assay with an electrical cell-substrate impedance sensor as described previously.37 In brief, HPAECs, HLMVECs, and A549 grown to confluence in recommended growth media were stimulated with IAV, EMAPII, or their combination. When indicated, EMAPII mAb M7/1 mAb,14 control rat IgG (#400533, Biolegend, San Diego CA), or pan-caspase inhibitor QVD-OPh hydrate (ApexBio, Houston, TX) was applied.
Infection of Mice with IAV
All animal procedures were approved by Indiana University Institutional Animal Care and Use Committee and conformed to the requirements of Animal Welfare Act.
To induce lung injury, 750 pfu/mouse of IAV were delivered to 12-week-old female C57BL/6 mice by oropharyngeal aspiration.38 Two and a half milligrams per kilogram rat anti-human EMAPII mAb14 were administered subcutaneously on days 4, 6, and 8 post-infection. Blood oxygenation levels were measured in alert animals using MouseOx Plus neck sensor (Starr Life Sciences, Oakmont, PA). At sacrifice, lungs were collected and/or used to extract BALF.
BALF and Lung Collection and Analyses
BALF was obtained from anesthetized animals by flushing the right lung with three portions of ice-cold PBS (0.8 mL). The left lung was excised and used for wet to dry weight ratio analysis; the right lung was snap-frozen and used for western blot analyses. BALF was centrifuged at 600 × g to sediment cells; the pellet was subjected to red blood cell lysis; the supernatant was frozen for future analyses. BALF supernatants were analyzed with Apo-one caspase 3/7 activity assay (Promega, Madison, WI), mouse AIMP1 competitive ELISA (myBioSource) and TNF-α ELISA (R&D Systems, Minneapolis, MN). For western blotting, lung tissue was digested with 1% SDS (in PBS containing anti-protease cocktail) using beads homogenizer (1 mm, Biospec products, Bartlesville, OK). Extracts were analyzed with anti-YM1 (R&D Systems #AF2446), CD206 (Abcam #ab64693), and vinculin (Abcam, #ab18058) antibodies. For flow cytometry, lung tissue was digested with collagenase and DNase (1 mg/mL and 0.12 mg/mL, respectively) mixture for 60 min at 37°C. Homogenates were stained with CD31/CD326/CD45/EMAPII antibody mixture, or permeabilized (FoxP3 staining kit, eBioscience, San Diego, CA) and stained with CD31/CD326/CD45/cleaved caspase 3 mixture. Fluorescent CD31 and CD326 antibodies were from Biolegend (#102409 and #118219, respectively); CD4 and CD45 antibodies were from BD Biosciences (#553729 and #553080, respectively). Fluorescent cleaved caspase 3 antibodies were purchased from both BD Biosciences (#560901) and R&D systems (IC835G025). Cells were FACS sorted using BD Fortessa; 5 × 105 events were obtained per sample and analyzed using FlowJo V10 software. For each panel of staining, fluorescence minus one (FMO) was done using corresponding IgG. We adapted the following gating strategy:18 epithelial cells were defined as CD326+CD31−CD45−, endothelial as CD326−CD31+CD45−, and hematopoietic lineage as CD326−CD31−CD45+ (Figure S1).
Statistical Analysis
Quantitative data are presented as mean ± SEM. Statistical analysis was performed by t test, t test with Welch correction (unequal variance), one-way ANOVA with Tukey post-hoc, or repeated-measurements ANOVA using Origin 8.0 or GraphPad Prism6 software. A p value of <0.05 was considered statistically significant.
Author Contributions
H.L. carried out the majority of experiments. S.C. performed flow cytometry analyses. C.P. and J.S. performed in vivo experiments. K.L.M. and M.C. assisted in experimental design and critically reviewed the manuscript. N.V.B. conceived and carried out experiments, analyzed data, and wrote the manuscript. All authors were involved in manuscript revision and granted final approval of the submitted version.
Conflicts of Interest
N.V.B., M.C., and K.L.M. have a provisional patent for the treatment of influenza-induced lung injury with EMAPII antibody. M.C. is a co-founder of Allinaire Therapeutics, which aims to find a cure for pulmonary diseases with EMAPII being a major target.
Acknowledgments
This publication was supported by an Indiana Clinical and Translational Sciences Institute grant (UL1TR001108) CECARE (to N.V.B.), the Project Development Team (to N.V.B.), a Fisch-Knoebel Cardiovascular Research Award (to N.V.B.), NIH/NHLBI funding (1R01HL1 29843-01 to M.C.), the Vascular and Cardiac Center for Adult Stem Cell Therapy, and the Cryptic Masons Medical Research Foundation. The work was done with the use of facilities of Roudebush VA Medical Center. The contents do not represent the views of the US Department of Veterans Affairs or the United States government. The authors are grateful to Dr. Purvi Mehrotra for assistance with analysis of flow cytometry results, Todd Cook for assistance with animal experiments, and Noelle Dahl for help with manuscript preparation.
Footnotes
Supplemental Information includes one figure and can be found with this article online at https://doi.org/10.1016/j.ymthe.2018.05.017.
Supplemental Information
References
- 1.Nair H., Brooks W.A., Katz M., Roca A., Berkley J.A., Madhi S.A., Simmerman J.M., Gordon A., Sato M., Howie S. Global burden of respiratory infections due to seasonal influenza in young children: a systematic review and meta-analysis. Lancet. 2011;378:1917–1930. doi: 10.1016/S0140-6736(11)61051-9. [DOI] [PubMed] [Google Scholar]
- 2.Mertz D., Kim T.H., Johnstone J., Lam P.P., Science M., Kuster S.P., Fadel S.A., Tran D., Fernandez E., Bhatnagar N., Loeb M. Populations at risk for severe or complicated influenza illness: systematic review and meta-analysis. BMJ. 2013;347:f5061. doi: 10.1136/bmj.f5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu J., Santesso N., Mustafa R., Brozek J., Chen Y.L., Hopkins J.P., Cheung A., Hovhannisyan G., Ivanova L., Flottorp S.A. Antivirals for treatment of influenza: a systematic review and meta-analysis of observational studies. Ann. Intern. Med. 2012;156:512–524. doi: 10.7326/0003-4819-156-7-201204030-00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muthuri S.G., Venkatesan S., Myles P.R., Leonardi-Bee J., Lim W.S., Al Mamun A., Anovadiya A.P., Araújo W.N., Azziz-Baumgartner E., Báez C., PRIDE Consortium Investigators Impact of neuraminidase inhibitors on influenza A(H1N1)pdm09-related pneumonia: an individual participant data meta-analysis. Influenza Other Respir. Viruses. 2016;10:192–204. doi: 10.1111/irv.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barik S. New treatments for influenza. BMC Med. 2012;10:104. doi: 10.1186/1741-7015-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayden F.G., de Jong M.D. Emerging influenza antiviral resistance threats. J. Infect. Dis. 2011;203:6–10. doi: 10.1093/infdis/jiq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ecker D.M., Jones S.D., Levine H.L. The therapeutic monoclonal antibody market. MAbs. 2015;7:9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cockle S.M., Stynes G., Gunsoy N.B., Parks D., Alfonso-Cristancho R., Wex J., Bradford E.S., Albers F.C., Willson J. Comparative effectiveness of mepolizumab and omalizumab in severe asthma: An indirect treatment comparison. Respir. Med. 2017;123:140–148. doi: 10.1016/j.rmed.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Mirando A.C., Francklyn C.S., Lounsbury K.M. Regulation of angiogenesis by aminoacyl-tRNA synthetases. Int. J. Mol. Sci. 2014;15:23725–23748. doi: 10.3390/ijms151223725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnett G., Jakobsen A.M., Tas M., Rice K., Carmichael J., Murray J.C. Prostate adenocarcinoma cells release the novel proinflammatory polypeptide EMAP-II in response to stress. Cancer Res. 2000;60:2850–2857. [PubMed] [Google Scholar]
- 11.Green L.A., Yi R., Petrusca D., Wang T., Elghouche A., Gupta S.K., Petrache I., Clauss M. HIV envelope protein gp120-induced apoptosis in lung microvascular endothelial cells by concerted upregulation of EMAP II and its receptor, CXCR3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;306:L372–L382. doi: 10.1152/ajplung.00193.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knies U.E., Behrensdorf H.A., Mitchell C.A., Deutsch U., Risau W., Drexler H.C., Clauss M. Regulation of endothelial monocyte-activating polypeptide II release by apoptosis. Proc. Natl. Acad. Sci. USA. 1998;95:12322–12327. doi: 10.1073/pnas.95.21.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarz M.A., Kandel J., Brett J., Li J., Hayward J., Schwarz R.E., Chappey O., Wautier J.L., Chabot J., Lo Gerfo P., Stern D. Endothelial-monocyte activating polypeptide II, a novel antitumor cytokine that suppresses primary and metastatic tumor growth and induces apoptosis in growing endothelial cells. J. Exp. Med. 1999;190:341–354. doi: 10.1084/jem.190.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clauss M., Voswinckel R., Rajashekhar G., Sigua N.L., Fehrenbach H., Rush N.I., Schweitzer K.S., Yildirim A.Ö., Kamocki K., Fisher A.J. Lung endothelial monocyte-activating protein 2 is a mediator of cigarette smoke-induced emphysema in mice. J. Clin. Invest. 2011;121:2470–2479. doi: 10.1172/JCI43881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quintos-Alagheband M.L., White C.W., Schwarz M.A. Potential role for antiangiogenic proteins in the evolution of bronchopulmonary dysplasia. Antioxid. Redox Signal. 2004;6:137–145. doi: 10.1089/152308604771978444. [DOI] [PubMed] [Google Scholar]
- 16.Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajashekhar G., Mitnacht-Kraus R., Ispe U., Garrison J., Hou Y., Taylor B., Petrache I., Vestweber D., Clauss M. A monoclonal rat anti-mouse EMAP II antibody that functionally neutralizes pro- and mature-EMAP II in vitro. J. Immunol. Methods. 2009;350:22–28. doi: 10.1016/j.jim.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singer B.D., Mock J.R., D’Alessio F.R., Aggarwal N.R., Mandke P., Johnston L., Damarla M. Flow-cytometric method for simultaneous analysis of mouse lung epithelial, endothelial, and hematopoietic lineage cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016;310:L796–L801. doi: 10.1152/ajplung.00334.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herold S., Becker C., Ridge K.M., Budinger G.R. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur. Respir. J. 2015;45:1463–1478. doi: 10.1183/09031936.00186214. [DOI] [PubMed] [Google Scholar]
- 20.Herold S., Hoegner K., Vadász I., Gessler T., Wilhelm J., Mayer K., Morty R.E., Walmrath H.D., Seeger W., Lohmeyer J. Inhaled granulocyte/macrophage colony-stimulating factor as treatment of pneumonia-associated acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2014;189:609–611. doi: 10.1164/rccm.201311-2041LE. [DOI] [PubMed] [Google Scholar]
- 21.Darwish I., Banner D., Mubareka S., Kim H., Besla R., Kelvin D.J., Kain K.C., Liles W.C. Mesenchymal stromal (stem) cell therapy fails to improve outcomes in experimental severe influenza. PLoS ONE. 2013;8:e71761. doi: 10.1371/journal.pone.0071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gotts J.E., Abbott J., Matthay M.A. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;307:L395–L406. doi: 10.1152/ajplung.00110.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan M.C., Kuok D.I., Leung C.Y., Hui K.P., Valkenburg S.A., Lau E.H., Nicholls J.M., Fang X., Guan Y., Lee J.W. Human mesenchymal stromal cells reduce influenza A H5N1-associated acute lung injury in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2016;113:3621–3626. doi: 10.1073/pnas.1601911113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y., Xu J., Shi W., Chen C., Shao Y., Zhu L., Lu W., Han X. Mesenchymal stromal cell treatment prevents H9N2 avian influenza virus-induced acute lung injury in mice. Stem Cell Res. Ther. 2016;7:159. doi: 10.1186/s13287-016-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tripathi S., Batra J., Cao W., Sharma K., Patel J.R., Ranjan P., Kumar A., Katz J.M., Cox N.J., Lal R.B. Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis. 2013;4:e562. doi: 10.1038/cddis.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran A.T., Cortens J.P., Du Q., Wilkins J.A., Coombs K.M. Influenza virus induces apoptosis via BAD-mediated mitochondrial dysregulation. J. Virol. 2013;87:1049–1060. doi: 10.1128/JVI.02017-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wurzer W.J., Ehrhardt C., Pleschka S., Berberich-Siebelt F., Wolff T., Walczak H., Planz O., Ludwig S. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J. Biol. Chem. 2004;279:30931–30937. doi: 10.1074/jbc.M403258200. [DOI] [PubMed] [Google Scholar]
- 28.Peteranderl C., Morales-Nebreda L., Selvakumar B., Lecuona E., Vadász I., Morty R.E., Schmoldt C., Bespalowa J., Wolff T., Pleschka S. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J. Clin. Invest. 2016;126:1566–1580. doi: 10.1172/JCI83931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skalko-Basnet N. Biologics: the role of delivery systems in improved therapy. Biologics. 2014;8:107–114. doi: 10.2147/BTT.S38387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Youssef M.M., Symonds P., Ellis I.O., Murray J.C. EMAP-II-dependent lymphocyte killing is associated with hypoxia in colorectal cancer. Br. J. Cancer. 2006;95:735–743. doi: 10.1038/sj.bjc.6603299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green L.A., Petrusca D., Rajashekhar G., Gianaris T., Schweitzer K.S., Wang L., Justice M.J., Petrache I., Clauss M. Cigarette smoke-induced CXCR3 receptor up-regulation mediates endothelial apoptosis. Am. J. Respir. Cell Mol. Biol. 2012;47:807–814. doi: 10.1165/rcmb.2012-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oghumu S., Varikuti S., Terrazas C., Kotov D., Nasser M.W., Powell C.A., Ganju R.K., Satoskar A.R. CXCR3 deficiency enhances tumor progression by promoting macrophage M2 polarization in a murine breast cancer model. Immunology. 2014;143:109–119. doi: 10.1111/imm.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Alessio F.R., Craig J.M., Singer B.D., Files D.C., Mock J.R., Garibaldi B.T., Fallica J., Tripathi A., Mandke P., Gans J.H. Enhanced resolution of experimental ARDS through IL-4-mediated lung macrophage reprogramming. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016;310:L733–L746. doi: 10.1152/ajplung.00419.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herold S., Steinmueller M., von Wulffen W., Cakarova L., Pinto R., Pleschka S., Mack M., Kuziel W.A., Corazza N., Brunner T. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008;205:3065–3077. doi: 10.1084/jem.20080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teijaro J.R., Walsh K.B., Cahalan S., Fremgen D.M., Roberts E., Scott F., Martinborough E., Peach R., Oldstone M.B., Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shriver Z., Trevejo J.M., Sasisekharan R. Antibody-based strategies to prevent and treat influenza. Front. Immunol. 2015;6:315. doi: 10.3389/fimmu.2015.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu H., Poirier C., Cook T., Traktuev D.O., Merfeld-Clauss S., Lease B., Petrache I., March K.L., Bogatcheva N.V. Conditioned media from adipose stromal cells limit lipopolysaccharide-induced lung injury, endothelial hyperpermeability and apoptosis. J. Transl. Med. 2015;13:67. doi: 10.1186/s12967-015-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang S., Danchuk S.D., Imhof K.M., Semon J.A., Scruggs B.A., Bonvillain R.W., Strong A.L., Gimble J.M., Betancourt A.M., Sullivan D.E., Bunnell B.A. Comparison of the therapeutic effects of human and mouse adipose-derived stem cells in a murine model of lipopolysaccharide-induced acute lung injury. Stem Cell Res. Ther. 2013;4:13. doi: 10.1186/scrt161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.