

Abstract
No disease-modifying osteoarthritis (OA) drugs are available to prevent posttraumatic osteoarthritis (PTOA). Mitochondria (MT) mediate the pathogenesis of many degenerative diseases, and recent evidence indicates that MT dysfunction is a peracute (within minutes to hours) response of cartilage to mechanical injury. The goal of this study was to investigate cardiolipin-targeted mitoprotection as a new strategy to prevent chondrocyte death and cartilage degeneration after injury. Cartilage was harvested from bovine knee joints and subjected to a single, rapid impact injury (24.0 ±1.4 MPa, 53.8 ±5.3 GPa/s). Explants were then treated with a mitoprotective peptide, SS-31 (1μM), immediately post-impact, or at 1, 6, or 12 hours after injury, and then cultured for up to 7 days. Chondrocyte viability and apoptosis were quantified in situ using confocal microscopy. Cell membrane damage (lactate dehydrogenase activity) and cartilage matrix degradation (glycosaminoglycan loss) were quantified in cartilage-conditioned media. SS-31 treatment at all time points after impact resulted in chondrocyte viability similar to that of un-injured controls. This effect was sustained for up to a week in culture. Further, SS-31 prevented impact-induced chondrocyte apoptosis, cell membrane damage, and cartilage matrix degeneration.
Clinical Significance
This study is the first investigation of cardiolipin-targeted mitoprotective therapy in cartilage. These results suggest that even when treatment is delayed by up to 12 hours after injury, mitoprotection may be a useful strategy in the prevention of PTOA.
Keywords: Cartilage, disease-modifying, osteoarthritis, therapy, mitochondria
Graphical Abstract
The goal of this study was to investigate cardiolipin-targeted mitoprotection as a new strategy to prevent chondrocyte death and cartilage degeneration after injury. Cartilage explants were subjected to a single, rapid impact then treated with a mitoprotective peptide (SS-31) immediately post-impact or at 1, 6, or 12 hours after injury. Treatment resulted in chondrocyte viability similar to that of un-injured controls and this effect was sustained for up to a week in culture. Further, SS-31 prevented impact-induced chondrocyte apoptosis, cell membrane damage, and cartilage matrix degeneration.
INTRODUCTION
Currently, no disease-modifying osteoarthritis (OA) drugs are available to prevent or slow the progression of posttraumatic OA (PTOA).1,2 Evidence suggests that therapies targeting acute biological responses after articular injury may provide benefit by preventing ongoing chondrocyte death and progressive extracellular matrix degradation.3,4 Mitochondrial dysfunction mediates the pathogenesis of many degenerative diseases, including syndromes that develop secondary to traumatic injury.5,6 Some of the earliest known pathologic changes in OA, including oxidative stress and chondrocyte apoptosis are mediated by mitochondria (MT).3,4,7–11
Mitochondrial dysfunction is well documented in the later stages of OA.7,8 For example, chondrocytes isolated from patients with end-stage OA exhibit MT respiratory dysfunction, with lower spare respiratory capacity and higher proton leak compared to healthy chondrocytes, as well as down-regulation of the MT-specific antioxidant superoxide dismutase-2.12 OA is also associated with decreased numbers of MT, and deficiencies in the metabolic biosensors AMPK and SIRT1 that regulate MT biogenesis.13 MT-associated disease pathways are linked to chronic stages of the disease, including decreased synthesis of collagen and proteoglycans and pathologic calcification of cartilage.7,8,14–17 Furthermore, mutations of mtDNA affecting MT function are associated with an increased incidence of knee OA.18
Several in vivo models have assessed MT function in the sub-acute to early chronic stages after cartilage insult. Chondrocyte respiratory function was impaired 4 weeks after surgical destabilization of the medial meniscus (DMM) in rabbits.19 Similarly, in a mouse DMM model, MT superoxide overproduction and SOD2 down regulation occurred in knee cartilage two weeks postoperatively.20 Chondrocytes isolated from SOD-2 deficient cartilage displayed MT depolarization, decreased MT respiratory function, and swollen MT with disrupted cristae structure. Furthermore, proteoglycan content was decreased and extracellular matrix metabolism was impaired by down regulation of anabolic genes and upregulation of catabolic genes, including MMP-13.20
Increasing experimental evidence also indicates that MT dysfunction plays a central role in the initiation and very early pathogenesis of PTOA, although the mechanisms are not fully characterized. Evidence from ex vivo chondral injury models suggests that in cartilage, as in other tissues, MT may act as intracellular mechanotransducers via strain-activated release of reactive oxygen species (ROS).21–24 This theory is supported by the findings that chondrocyte compression distorts the MT network, MT-derived reactive oxygen species induced chondrocyte death, and chondrocyte cytoskeleton dissolution prevents elevated ROS and cell death in injured cartilage.22,24–26 Increases in intracellular calcium lead to MT depolarization within 3–6 hours of cartilage injury, activation of the caspase cascade and chondrocyte apoptosis.27 Recently, MT respiratory dysfunction was identified in the acute phase (within 2 hours) after cartilage impact injury.28
Evidence for the role of MT dysfunction in OA has led to interest in developing clinical therapies that target MT-associated pathways. To date, investigational therapies have generally targeted downstream consequences of MT dysfunction, namely redox imbalance and activation of the caspase cascade,4 however no specific mitoprotective therapies have been evaluated as potential disease modifying OA drugs. In contrast to drugs that are simply targeted to the MT, or participate in MT-associated disease pathways, mitoprotective drugs can be defined as agents that directly protect MT structure and function, and restore MT plasticity.29
The Szeto-Schiller peptides (SS-31) represent a novel class of mitoprotective agents that repair mitochondrial cristae structure, restore mitochondrial function, improve cellular bioenergetics, and prevent cell death.29–31 SS-31 (Elamipretide; Stealth Peptides, Newton, MA) exerts these effects through its specific interaction with cardiolipin, a phospholipid that is uniquely expressed on the inner mitochondrial membrane.32 Cardiolipin promotes folding of cristae membranes and is important for the formation of respiratory supercomplexes that facilitate electron transfer and ATP synthesis. However, cardiolipin is highly susceptible to oxidative damage, and cardiolipin peroxidation results in loss of cristae membranes, a decline in ATP production, increase in ROS production, release of cytochrome c and initiation of apoptosis.33 By interacting with cardiolipin, SS-31 promotes cristae curvature, improves mitochondrial coupling, ATP production, reduces ROS production, inhibits cardiolipin peroxidation, and prevents apoptosis.33,34
SS-31 is currently in multiple Phase II and III trials for both rare and common MT diseases, demonstrating its safe use in humans.35,36 SS-31 has not previously been studied in cartilage, therefore the goals of this study were to test if SS-31 could prevent impact-induced chondrocyte death and cartilage degeneration, and if mitoprotective efficacy would be affected by the time of administration after impact.
METHODS
Tissue Harvest
Full thickness cartilage explants were harvested from the medial femoral condyles of healthy bovids (n = 8 animals, 1–3 days of age) within 48 hours of euthanasia using an 8mm biopsy punch, as previously described.28 Specimens were rinsed in phosphate-buffered saline (PBS), trimmed to a uniform thickness of 3mm from the articular surface (leaving the articular surface intact) using a custom jig, and placed in media (phenol free DMEM containing 1% FBS, HEPES 0.025 ml/ml, penicillin 100 U/mL, streptomycin 100 U/mL and 2.5mM glucose). Experiments were approved by the University Institutional Animal Care and Use Committee.
Rapid Impact Injury Model
Explants were subjected to injury using a validated rapid-impact model, or served as un-impacted controls, as previously described.28,37 Briefly, explants were positioned in a well containing PBS under the plane-ended tip of a spring-loaded impacting device, with the articular surface facing up (Figure 1).38,39 The impactor was used to deliver a single, rapid cycle of unconfined axial compression (24.0 ±1.4 MPa peak stress; 53.8 ±5.3 GPa/s peak stress rate). Impact force was measured at 50 kHz by a load cell (PCBPiezotronics, Depew, NY) attached to the impactor tip. Voltage from the load cell was recorded with a custom LabVIEW program (NI, Austin TX). The impact magnitude was adjusted by setting the deflection of the impactor’s internal spring and mechanical parameters for each impact were calculated as previously described.37
Figure 1. Experimental Methods. A) Study Design.

Half the cartilage explants were impacted (X) at time 0. Injured groups (I; red bars) and non-injured groups (C; grey bars) were then treated (hatched line) with SS-31 (1μM) at time zero (T0), 1 hour (T1), 6 hours (T6), or 12 hours (T12) after injury, or left untreated (NT). SS-31 treatment was withdrawn at 24 hours post-injury. Explants were imaged on day 1 or 7 for cell death or apoptosis, and cartilage conditioned medium was collected at 1, 6, and 12 hours, and 1, 3, 5, and 7 days after injury to assess glycosaminoglycan (GAG) loss and cell membrane damage.
B) Impactor setup and impact geometry. Modified from Bonnevie, et al., 2015. Reused with permission from SAGE Publications (pending).
Study Design
Following injury, explants were kept moist and cut perpendicular to the articular surface into 2 hemicylinders using a custom jig, then placed into a 24-well plate containing 1.5 ml of media. Cartilage hemicylinders were randomly assigned to one of 10 treatment groups (n = 6/group, Figure 1). Injured (I) and uninjured control (C) explants in the non-treated (NT) groups (INT, CNT) were placed into wells containing only media. Other explants were treated with SS-31 (1μM) at time zero (IT0, CT0), 1 hour (IT1, CT1), 6 hours (IT6, CT6), or 12 hours (IT12, CT12) after impact (Figure 1). Explants were maintained under standard tissue culture conditions (37 °C, 21% O2, and 5%CO2) for up to 7 days. After 24 hours, culture media were replaced with media containing no SS-31, then replenished every other day for the duration of the experiment. Medium was sampled (200 μL/well) at 1, 6, 12, and 24 hours, and at 3, 5, and 7 days after impact, and stored at −80°C until biochemical assays were performed. After imaging was complete, cartilage explants were lyophilized and weighed.
Chondrocyte Viability
Chondrocyte viability was assessed in situ using a live/dead imaging assay. At 1 or 7 days post-impact, cartilage was rinsed and placed in PBS containing calcein AM (2μM) and ethidium homodimer (1μM) for 30 minutes at 37°C, to stain live and dead cells, respectively. Explants were then imaged on a Leica SP5 confocal microscope. Digital images were acquired in two channel sequential scans (green; 488/498–544 and red; 514/563–663 nm excitation/emission, respectively) using a modified 3D scanning protocol; A minimum of three z-stacks (ten images, 512×512 pixel/387.5μm ×387.5μm, spaced 10um apart in the z plane) per explant were acquired at 10× magnification. All imaging parameters were optimized during preliminary studies, then the same settings were used throughout the study.
The number of live, dead, and total cells in each image was quantified using custom ImageJ macros (Mac OS X version 10.2, Wayen Rasband, U.S. National Institutes of Health, Bethesda, MD) as previously described.28 Briefly, Each image was thresholded based on mean pixel intensity of that image, then individual particles were identified and counted based on particle size. For each image, the % of dead cells was calculated as the number of dead cells counted in the red channel divided by the total number of cells (live + dead) counted in both channels. The final reported values for cell death are the mean of a minimum of 3 z-stacks obtained for each explant.
Apoptosis (Activated Caspase Staining)
At 1 or 7 days post-impact, cartilage was placed in PBS containing CellEvent Caspase 3/7 (Molecular Probes, Eugene, OR) to detect activated caspase activity. Explants were imaged on a Leica SP5 confocal microscope and digital z-stacked images were acquired as described above in two channel sequential scans; 488/498–544 excitation/emission to image apoptotic cells and reflectance to highlight collagen in the extracellular matrix. The number of caspase-positive cells per field were counted using a custom ImageJ macro and expressed as the number of apoptotic cells per mm2.
Cell Membrane Damage
Imaging studies were validated with biochemical assays performed on cartilage-conditioned media. As a measure of cell membrane damage, the release of lactate dehydrogenase (LDH) from cartilage explants was quantified using a commercial kit (Sigma-Aldrich, St. Louis, MO). NADH absorbance was measured at 450nm in 5-minute intervals by a spectrophotometric microplate reader (Tecan Safire; Männedorf, Switzerland). LDH activity was calculated following subtraction of background media values and expressed as milliunits of LDH per ml of cartilage conditioned media.
Cartilage Matrix Degeneration
To determine if SS-31 could inhibit cartilage matrix degradation after impact injury, loss of glycosaminoglycan (GAG) into media was determined by routine 1,9-dimethyl-methylene blue dye binding (DMMB) assay, as previously described.40 Briefly, media samples were digested using papain (Sigma Aldrich) 0.25 mg/ml at 65°C for 4 hours. A standard curve was prepared using chondroitin-4-sulfate (Sigma Aldrich). Equal volumes of sample and DMMB dye (Sigma Aldrich) were mixed in a 96-well plate. Total GAG content was read fluorometrically, and expressed as the total GAG released into cartilage conditioned media over the culture period per μg dry weight of cartilage.
Distribution and Localization of SS-31 in Cartilage
A concern with any chondrocyte-targeted drug is the question of diffusion through the avascular, highly charged extracellular matrix of cartilage. To investigate the distribution of SS-31, 6mm diameter cartilage explants were harvested as described above and incubated in PBS with biotinylated SS-31 (SPI-72; Biotin-D-Arg-Dmt-Lys-Phe-NH2) 2nM and Mitotracker Green (200nM; Molecular Probes, Eugene, OR) for 1 hour at 25°C. Cartilage explants were then bisected, and hemicylinders were fixed for 24 hours in 10% neutral buffered formalin. After fixation, bisected halves were exposed to streptavadin-AlexaFluor633 (5ug/ml; Molecular Probes, Eugene OR) for 4 hours, rinsed for 15 minutes in PBS, then imaged using confocal microscopy as previously described.32
To confirm that SS-31 can localize to chondrocyte mitochondria in vivo, aladanated SS-31 (SPI-70; D-Arg-Dmt-Lys-ALD-NH2; 30mls of 10uM)41 was injected into the femeropatellar joints of two adult horses (age unknown) immediately following euthanasia. Horses were euthanized for reasons other than this study. The limbs were manipulated through a passive range of motion intermittently for 60 minutes, then cartilage explants were harvested from the medial femoral condyles, tibial plateau and patellofemoral grooves and placed in PBS containing tetramethylrhodamine methyl ester perchlorate (TMRM;10nM, Molecular Probes) for 40 minutes, then rinsed in PBS. Cartilage was imaged using a Zeiss LSM880 confocal/multiphoton microscope (2 photon excitation at 720nm/510–540nm excitation and 561 nm emission/569–611nm excitation) and colocalization analysis was performed using ZEN Imaging software (Carl Zeiss AG, Oberkochen, Germany).
Effect of SS-31 in Adult tissue
To investigate the effects of mitoprotection in adult tissue, and to validate findings obtained in immature cartilage, impact experiments were performed on cartilage harvested from the medial femoral condyles of adult bovine stifle joints (n = 2 animals, >18 months of age), and chondrocyte viability was assessed as described above.
Statistical Analysis
Data were analyzed using a linear mixed effects model, with a random effect of trial/animal and fixed effects of injury (I, C), treatment time (T0, T1, T6, T12, NT), and response time (day 1, day 7), including all interactions. Comparisons between groups were performed using Tukey’s HSD method. Residual analyses were performed to ensure the assumptions of normality and homogeneous variance were met. Differences were considered statistically significant at p ≤ 0.05. Statistical analyses were performed using JMP Pro Version 11.0 (SAS Inc.) software.
RESULTS
SS peptides Diffuse Throughout Cartilage and Localize to Chondrocyte Mitochondria
After incubation of explants with biotinylated SS peptide for 1 hour, fluorescent imaging revealed mitochondrial probe (MitoTracker Green) and the streptavidin-conjugated fluorophore (AlexaFluor633) in chondrocytes throughout the depth of the cartilage (Figure 2). This confirms that SS-31 readily diffuses through the extracellular matrix. After incubation of cartilage with fluorescent SS-31 ([ald]SS-31)32 and a mitochondrial probe (TMRM), multiphoton imaging of individual chondrocytes and colocalization analysis revealed localization of SS-31 to chondrocyte mitochondria.
Figure 2. SS-31 distributes throughout the thickness of cartilage and localizes to chondrocyte mitochondria after intra-articular administration.
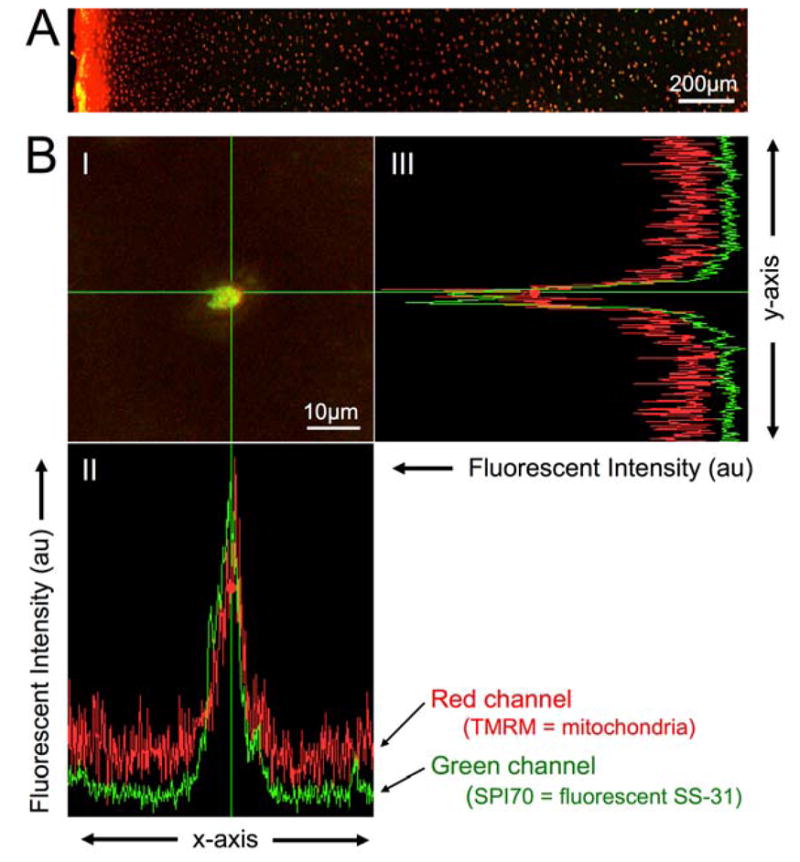
A) Cartilage was incubated with biotinylated SS peptide and MitoTracker (green) for 1 hour, fixed, sectioned, and exposed to streptavadin-AlexaFluor633 (red), then imaged using confocal microscopy. The articular surface is oriented to the left. Note distribution of red (peptide) signal in chondrocytes throughout the depth of the cartilage. B) One hour after intraarticular injection of aladanated (fluorescent) SS-31 (SPI-70; green), cartilage was harvested and incubated with TMRM (red). I) Multiphoton image of a single chondrocyte. II & III) Colocalization analysis depicting the fluorescent intensity (in arbitrary units) in the red and green channels, along the x and y axes, respectively. Note overlap of the red (mitochondria) and green (peptide) signals in both axes, indicating colocalization.
Mitoprotection Prevents Impact-induced Chondrocyte Death and Apoptosis
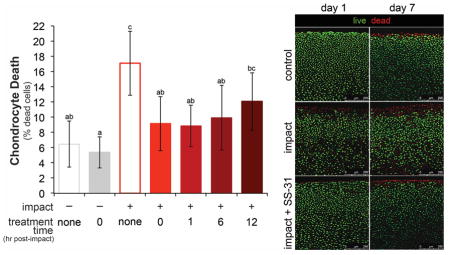
To test if mitoprotection could prevent impact-induced chondrocyte death, cartilage was injured and immediately treated with SS-31, then chondrocyte viability was assessed on days 1 and 7 post-impact by live/dead staining. Cartilage injury resulted in a 2.5-fold increase in cell death (Figure 3). SS-31 treatment prevented impact-induced chondrocyte death at 1 day post-injury (p < 0.001), resulting in chondrocyte viability similar to un-injured controls. When chondrocyte viability was assessed 7 days post-injury, findings were similar (Figure 3). This indicates that although SS-31 was withdrawn from culture media after 24 hours, its cytoprotective effects were sustained for a week in culture. Cell death was not significantly different on days 1 and 7 in uninjured, non-treated (CNT) explants (p = 0.98), indicating that chondrocyte viability was maintained for the 7-day culture period. SS-31 had no effect on chondrocyte viability in uninjured explants. SS-31 also provided cytoprotection in adult cartilage, decreasing the amount of impact induced chondrocyte death by half (Figure S1).
Figure 3. Mitoprotection prevents impact-induced chondrocyte death up to 1 week after injury.
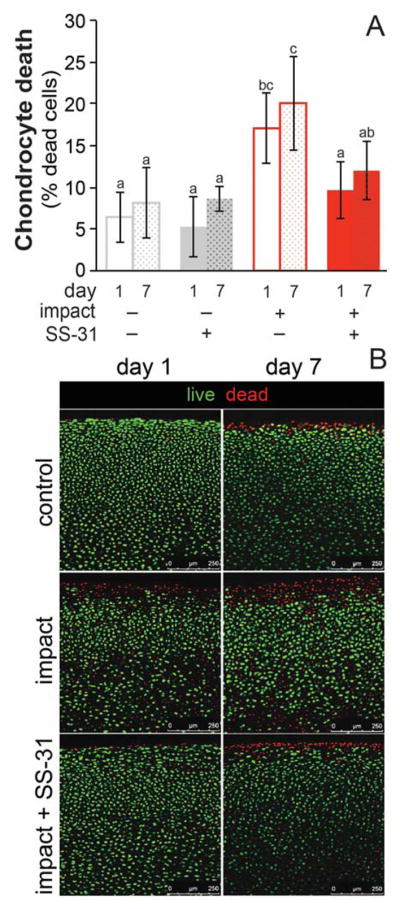
SS-31 treatment at 1 hour after injury was effective at preventing chondrocyte death on both days 1 and 7 post-impact, with chondrocyte death not different after one week of culture compared 1 day in any group. Groups that do not share a letter are significantly different at p ≤ 0.05, error bars = ±s.d. B) Representative confocal images of uninjured (control), injured (impact) and injured, treated (impact + SS-31) cartilage on days 1 and 7 post-injury. Explants were stained for live and dead cells with calcein AM (green) and ethidium homodimer (red), respectively.
Treatment with SS-31 also prevented impact-induced caspase 3/7 activation, a marker of apoptosis. Imaging of explants one day after injury revealed an increase in the number of caspase-positive cells throughout the depth of the cartilage (Figure 4). SS-31 prevented impact-induced apoptosis on days 1 (p = 0.007) and 7 (p = 0.04) after injury. There was a trend toward fewer apoptotic cells on day 7 than day 1 in all groups, most notably in injured, treated explants but this difference did not reach significance (p = 0.07).
Figure 4. SS-31 prevents impact-induced apoptosis.
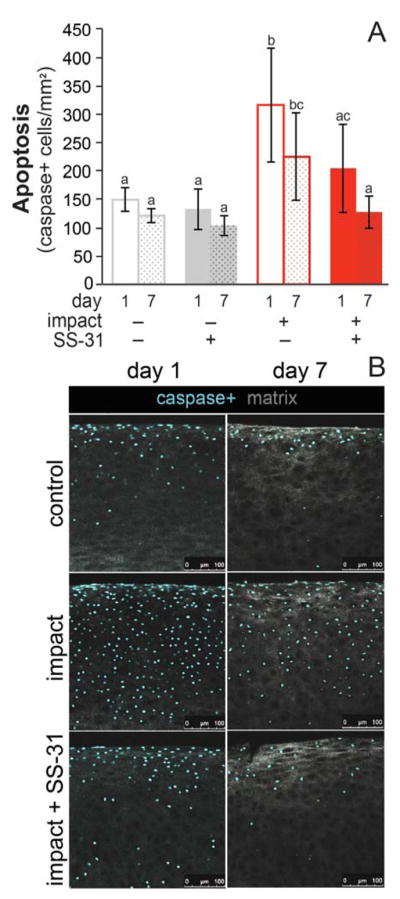
A) Caspase-positive staining in injured explants treated with SS-31 was equivalent to uninjured controls at day 1 and 7. Groups that do not share a letter are significantly different at p ≤ 0.05. Error bars = ±s.d. B) Representative confocal images of uninjured (control), injured (impact) and injured, treated (impact + SS-31) cartilage on day 1 and 7. Explants were imaged for activated caspase 3 and 7 (caspase+) and confocal reflectance of collagen.
Mitoprotection Prevents Cell Membrane Damage and Cartilage GAG Loss
Cumulative cell membrane damage, quantified by LDH activity in cartilage conditioned media for 7 days following injury, was twofold lower after injury in treated versus untreated cartilage (p = 0.0005, Figure 5a). SS-31 also had a protective effect against cell membrane damage in uninjured controls; uninjured, treated samples had a lower cumulative LDH than untreated controls (p = 0.05).
Figure 5. SS-31 prevents impact-induced chondrocyte membrane damage and cartilage matrix degradation.

A) LDH activity in the media of injured, treated groups was lower than injured, untreated explants, and similar to uninjured controls. SS-31 also had a protective effect against cell membrane damage in treated controls (p = 0.05). B) Cumulative GAG loss into the media was increased in injured, untreated explants compared to uninjured controls. GAG loss was similar in injured, treated and control groups. Groups that do not share a letter are significantly different at p ≤ 0.05. Error bars = ±s.d.
SS-31 treatment protected against cartilage matrix degradation after injury. GAG loss into cartilage conditioned media was significantly (p= 0.03) increased in injured, untreated (INT) explants (4.6 s.d. 1.0 μg GAG/ml media) starting on day 3 post-injury compared to uninjured controls (3.7 s.d. 0.8 μg GAG/ml media). Cumulative GAG loss over the 7 day culture period was ~30% greater in injured, untreated explants (INT) compared to uninjured controls (Figure 5b), indicating impact-induced cartilage matrix degeneration. SS-31 prevented impact-induced GAG loss (p = 0.002), resulting in values similar to non-impacted controls (CNT, CT0).
The Effect of Treatment Time on Mitoprotective Efficacy
To investigate the therapeutic window for mitoprotection after acute cartilage injury, explants were injured and treatment was delayed for up to 12 hours. SS-31 initiated at 0, 1 or 6 hours after impact significantly reduced cell death in injured groups versus the INT group (IT0; p = 0.0007, IT1; p < 0.0001, IT6; p = 0.0003) resulting in chondrocyte viability similar to uninjured controls (CNT; p = 0.16). This represents a 37–48% reduction in impact-induced chondrocyte death in IT0, IT1 and IT6 groups (Figure 6a). The cytoprotective effect of SS-31 was less pronounced when treatment was delayed 12 hours after injury. Cell death in the IT12 group was significantly greater than the treated control group (CT0), and not different than the injured, untreated group (INT).
Figure 6. Mitoprotective efficacy depends on treatment time.

A) Chondrocyte death (% dead cells) in injured explants (n=6/ group), treated with SS-31 at 0, 1, or 6 hours was equivalent to uninjured controls. B) Cartilage matrix degeneration, measured by glycosaminoglycan (GAG) loss into the media was equivalent to uninjured controls in all treatment groups. Groups that do not share a letter are significantly different at p ≤ 0.05. Error bars = ±s.d.
The efficacy of SS-31 to provide structural protection after cartilage injury was similarly time-sensitive (Figure 6b). Injured groups treated up to 6 hours post-impact (IT0, IT1 and IT6 ) had GAG loss similar to un-impacted groups (CNT and CT0), but when treatment was delayed 12 hours, GAG loss was not different than injured, untreated samples (INT).
DISCUSSION
The aim of this work was to investigate mitoprotection as a new strategy to prevent posttraumatic osteoarthritis, and our data indicate that SS-31 prevents injury-induced chondrocyte death, caspase activation, cell membrane damage, and matrix degradation. The concept of targeting MT-associated disease pathways in the treatment and prevention of OA is not novel; several groups have targeted events downstream of MT dysfunction42–46 (Figure 7). Despite promising preclinical results, traditional antioxidants such as N-acetyl cysteine and vitamin C have not provided clinical benefit in OA patients.1,2,4 Caspase inhibitors, including synthetic inhibitors of caspase 3 and 9, prevent MT-mediated cell death by preventing activation of the caspase cascade and apoptosis in vitro, but have not progressed to clinical testing.4,46 Ideally, to prevent the initiation and progression of PTOA, interventions would target upstream events and preserve cartilage homeostasis.3 However, until recently no specific mitoprotective therapies (i.e. agents that directly protect MT structure and function) were available.
Figure 7. Basic mitochondria-associated pathways linking cartilage injury to posttraumatic osteoarthritis.
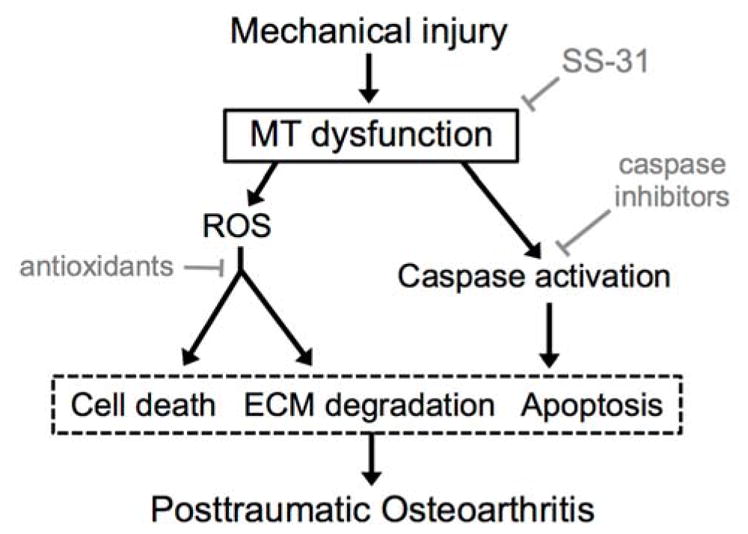
Potential therapeutics to prevent PTOA (grey) and their likely sites of action. Note traditional antioxidants and caspase inhibitors act downstream of mitochondria (MT).
Most modern drugs target single proteins in signaling pathways involved in specific disease processes. However, MT are complex, multi-compartment organelles that rely on a vast network of proteins and lipids to carry out and regulate ATP synthesis. Therefore, previous attempts to identify a single molecular target to improve MT function have been unrewarding.29 SS peptides offer a new therapeutic strategy; by specifically protecting cardiolipin, these novel peptides preserve MT structure and promote MT function. Cardiolipin is a phospholipid dimer expressed exclusively on the inner MT membrane, with small acidic head groups and four diverging hydrophobic acyl chain tails resulting a conical shape.47 Incorporation of cardiolipin into the lipid bilayer causes the inner MT membrane to bend, forming the characteristic folded structure of MT cristae.47 Cardiolipin rafts organize electron transport chain proteins into supercomplexes, shortening the distance between redox partners and increasing efficiency of electron transfer. This optimizes ATP production, and reduces ROS generation. Cardiolipin also anchors cytochrome C to the inner MT membrane, where it executes the rate-limiting step of oxidative phosphorylation. During MT stress, cardiolipin is easily oxidized, which distorts cristae structure. This leads to decreased ATP production, increased electron leak in the electron transport chain with increased ROS generation, and dissociation of cytochrome C from the inner MT membrane, setting the stage for apoptosis.20,33 Therefore, by protecting cardiolipin, SS-31 can optimize the function of all integral proteins on the inner mitochondrial membrane to promote oxidative phosphorylation, reduce excess ROS production, and prevent MT-induced apoptosis and autophagy.48
Mitotoxicity is a major concern with any MT-targeted compound, and many candidate drugs are not effectively delivered to MT. Recently, several MT-targeted antioxidants have been developed. In order to target drugs to MT, lipophilic compounds are often conjugated to cationic moieties such as TPP+ (triphenylphosphonium ion), which enter the MT matrix due to the potential gradient across the inner MT membrane, which can lead to MT depolarization. Unlike these TPP+-conjugated antioxidants, SS-31 does not enter the MT matrix, and has no pro-oxidant activity.29 It is clinically important to note that extensive toxicology studies have shown SS-31 to be safe for in vivo use; SS-31 has no effect on normal cells, and no cytotoxic or mitotoxic effects have been observed in studies at concentrations exceeding 100uM ex vivo, up to 50mg/kg in vivo animal models, and up to 0.25 mg kg−1 h−1 over 4 hours in human safety studies.30
A concern with any chondrocyte-targeted osteoarthritic drug is the question of diffusion through the avascular, highly charged cartilage matrix. Owing to their unique chemical structure, SS peptides are highly water soluble, while able to freely diffuse (translocate) across lipid membranes by transcellular transport.49 This results in a volume of distribution similar to blood volume, and no accumulation in lipid, further limiting concerns for toxicity. Our ex vivo and in vivo imaging studies confirm that SS-31 distributes throughout the thickness of cartilage and localizes to chondrocyte mitochondria after intra-articular injection.
To investigate the possible clinical relevance of SS-31 as a “point-of-injury” therapy in the acute stages following cartilage trauma, a time course experiment was performed. Our findings suggest that even when treatment was delayed by 12 hours after injury, mitoprotective therapy may be a useful strategy to prevent ongoing chondrocyte death and cartilage degeneration. Previous in vitro work indicates that impact-induced cell death peaks at 2–3 hours following injury.28,50 However, MT-mediated cell death evolves over a slower time scale than necrotic cell death (days versus hours, respectively).50–54 Time course studies of progressive apoptosis in cartilage have been investigated by several methods, and indicate chondrocyte apoptosis may be initiated around 6 hours and sustained for up to 7 days post-injury.51–54 In a recent in vitro study, chondrocyte MT respiratory dysfunction occurred within 2 hours of cartilage impact.28 These findings suggest that following injury, a subset of chondrocytes experience acute MT-dysfunction, but remain viable for an indeterminate amount of time. SS-31 may act to stabilize this subpopulation, and rescue chondrocytes experiencing MT-dysfunction but not yet committed to programmed cell death.
The absolute time course presented in this study should not be over-interpreted, however. It is important to note that SS-31 was withdrawn at 24 hours in all treatment groups. Therefore, it is possible that the inferior mitoprotective efficacy observed in the IT12 group may be due to shorter drug exposure, and equal protection may be observed if treatment is continued for similar lengths of time. Furthermore, this ex vivo model utilizes a single-impact followed by static tissue culture. In vivo, MT dysfunction likely continues following acute cartilage trauma. For example, recent work suggests that articular injury inhibits cartilage lubrication mechanisms, and that increased frictional coefficients due to inadequate lubrication are associated with MT depolarization and chondrocyte apoptosis.55,56 Taken together, this suggests that in a clinical scenario, the effective window for mitoprotective intervention after joint injury is likely not limited to hours, but may continue throughout the course of ongoing cartilage degeneration. Recent evidence also indicates that beyond protecting MT from damage, SS-31 promotes MT repair after injury and can restore MT function. Once repaired, mitochondria were protected for at least 6 months after termination of drug treatment. Preclinical and clinical studies would be required to determine the most effective dosing regimens in patients after joint trauma.
Our findings indicate that, in addition to providing cytoprotection, SS-31 can prevent impact-induced cartilage matrix degradation. GAG release into cartilage-conditioned media is a well-established indicator of cartilage injury. In the present study, GAG loss was significantly increased in injury groups versus controls starting on day 3 post-injury. Conversely, previous studies have documented immediate injury-induced GAG loss; i.e. between days 1 and 3 post-injury, but not after day 3.57 The reason for this difference is likely related to the distinct loading regimens employed between studies. Immediate and non-sustained GAG loss observed in other models may be due to mechanical disruption of matrix rather than cell-mediated enzymatic degradation.57 A lag time in injury-induced GAG release in the present study, and inhibition of GAG release by SS-31 suggests that mitoprotection prevents injury-induced cartilage matrix degradation. Although the mechanism for cartilage matrix protection by SS-31 was not directly investigated in the present study, the current findings are consistent with known mechanisms in other cell types. When oxidized, cardiolipin can translocate to the outer MT membrane and trigger multiple cell stress responses, including inflammasome activation and induction of IL-1β.58 Recent evidence supports a role for MT damage in inflammasome activation in OA, and treatment with SS-31 one month after injury reversed upregulation of the inflammatory markers IL-1β, IL-18 and TNF-α in a model of chronic inflammation and fibrosis.59–61 Furthermore, previous work has demonstrated that MT-derived reactive oxygen species are important regulators of matrix degrading enzymes, matrix metalloproteinases (MMPs).16,62–64 In human cartilage explants, MT respiratory chain dysfunction led to upregulation of MMP-1, -3 and -13 and loss of proteoglycan staining.16 Mitochondrial redox imbalance also increased MMP-1 and 13 levels, and a MT-targeted superoxide scavenger reduced MMP levels in primary chondrocytes.63
A limitation of the current study is that experiments were performed at 21% O2 concentration, which is considered relative hyperoxia for normal cartilage.65 There is disagreement in the literature regarding the effect of O2 concentration on chondrocyte metabolism, and most studies have investigated this question in isolated chondrocytes.66–69 One group assessed the level of oxidative phosphorylation in freshly harvested 1mm cubes of cartilage from young mature bovids. They found that at O2 concentrations between 5 and 21%, oxygen consumption rate (OCR) was relatively constant at ~10 nM/106 cells/hour,67 similar to OCR values measured in recent work using the same techniques described in the present study.28 OCR in cultured chondrocytes was also found to be independent of O2 and glucose concentration in short term (48 hour) culture.66 Therefore, the finding of mitoprotection at 24 hours post-injury is unlikely to be affected by oxygen concentration. Furthermore, although 21% O2 is considered physiologic for normal cartilage, it is unclear what oxygen concentration chondrocytes experience after joint trauma. The current study was conducted at 21% O2 to be consistent with recent work linking cartilage trauma to mitochondrial dysfunction in the same experimental system.28 Additional studies are warranted to investigate the effect of mitoprotection at various oxygen concentrations in situ.
In summary, this is the first study to investigate targeted mitoprotection in cartilage. Our findings demonstrate that SS-31 prevents chondrocyte death and cartilage matrix degradation, even when treatment is delayed up to 6 hours after cartilage injury. The unique properties of this class of drugs confer their ability to provide structural protection to MT cristae by specifically interacting with cardiolipin, thereby preventing MT dysfunction.30,32 Our data suggest that SS-31 has the potential to be an effective disease modifying osteoarthritis drug, and preclinical testing is warranted.
Supplementary Material
Acknowledgments
The authors thank Lynn Johnson for statistical consulting, and Alexis Gale, Meg Goodale and Becky Hicks for help executing assays and technical support. Multiphoton imaging data was acquired with the help of Johanna Dela Cruz at the Cornell University Biotechnology Resource Center, with NIH S10OD018516 funding for the shared Zeiss LSM880 confocal/multiphoton microscope. This work was supported by Weill Cornell Medical College Clinical & Translational Science Center Award/National Center for Advancing Translational Sciences (5 UL1 TR000457-09) and The Harry M. Zweig Memorial Fund for Equine Research. MD was supported by NIH 5T32OD011000-20 and NIH 1K08AR068470. EB was supported by the NSF GRFP.
Footnotes
Author Contributions:
MD and LF are responsible for conception and design of the study. MD developed the methodologies, performed the assays and drafted the article. MD and EB were responsible for data acquisition. All authors contributed intellectual content, participated in analysis and interpretation of the data, provided critical revision and approve the final submitted version of the manuscript.
References
- 1.Cheng DS, Visco CJ. Pharmaceutical therapy for osteoarthritis. PM&R. 2012;4(5):S82–S88. doi: 10.1016/j.pmrj.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Gallagher B, Tjoumakaris FP, Harwood MI, et al. Chondroprotection and the prevention of osteoarthritis progression of the knee: a systematic review of treatment agents. The American Journal of Sports Medicine. 2015;43(3):734–744. doi: 10.1177/0363546514533777. [DOI] [PubMed] [Google Scholar]
- 3.Anderson DD, Chubinskaya S, Guilak F, et al. Post-traumatic osteoarthritis: improved understanding and opportunities for early intervention. J Orthop Res. 2011;29(6):802–809. doi: 10.1002/jor.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chubinskaya S, Wimmer MA. Key Pathways to Prevent Posttraumatic Arthritis for Future Molecule-Based Therapy. Cartilage. 2013;4(3 Suppl):13S–21S. doi: 10.1177/1947603513487457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pagano G, Talamanca AA, Castello G, et al. Oxidative stress and mitochondrial dysfunction across broad-ranging pathologies: toward mitochondria-targeted clinical strategies. Oxid Med Cell Longev. 2014;2014:541230. doi: 10.1155/2014/541230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazzeo AT, Beat A, Singh A, Bullock MR. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Experimental Neurology. 2009;218(2):363–370. doi: 10.1016/j.expneurol.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 7.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7(3):161–169. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 8.Terkeltaub R, Johnson K, Murphy A, Ghosh S. Invited review: the mitochondrion in osteoarthritis. Mitochondrion. 2002;1(4):301–319. doi: 10.1016/s1567-7249(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 9.Johnson EO, Charchandi A, Babis GC, Soucacos PN. Apoptosis in osteoarthritis: morphology, mechanisms, and potential means for therapeutic intervention. J Surg Orthop Adv. 2008;17(3):147–152. [PubMed] [Google Scholar]
- 10.Del Carlo M, Loeser R. Cell death in osteoarthritis. Curr Rheumatol Rep. 2008;10(1):37–42. doi: 10.1007/s11926-008-0007-8. [DOI] [PubMed] [Google Scholar]
- 11.Lotz M, Hashimoto S, Kühn K. Mechanisms of chondrocyte apoptosis. Osteoarthr Cartil. 1999;7(4):389–391. doi: 10.1053/joca.1998.0220. [DOI] [PubMed] [Google Scholar]
- 12.Gavriilidis C, Miwa S, Zglinicki von T, et al. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis & Rheumatism. 2013;65(2):378–387. doi: 10.1002/art.37782. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Zhao X, Lotz M, et al. Mitochondrial biogenesis is impaired in osteoarthritic chondrocytes but reversible via peroxisome proliferator-activated receptor-γ coactivator 1α. Arthritis & Rheumatology. 2015;67(8):2141–2153. doi: 10.1002/art.39182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz-Romero C, Calamia V, Mateos J, et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. 2009;8(1):172–189. doi: 10.1074/mcp.M800292-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maneiro E, Martín MA, de Andres MC, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis & Rheumatism. 2003;48(3):700–708. doi: 10.1002/art.10837. [DOI] [PubMed] [Google Scholar]
- 16.Cillero-Pastor B, Rego-Pérez I, Oreiro N, et al. Mitochondrial respiratory chain dysfunction modulates metalloproteases -1, -3 and -13 in human normal chondrocytes in culture. BMC Musculoskelet Disord. 2013;14:235–235. doi: 10.1186/1471-2474-14-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson K, Jung A, Murphy A, et al. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis & Rheumatism. 2000;43(7):1560–1570. doi: 10.1002/1529-0131(200007)43:7<1560::AID-ANR21>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 18.Chang M-C, Hung S-C, Chen WY-K, et al. Accumulation of mitochondrial DNA with 4977-bp deletion in knee cartilage – an association with idiopathic osteoarthritis. Osteoarthritis and Cartilage. 2014;13(11):1004–1011. doi: 10.1016/j.joca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 19.Goetz JE, Coleman MC, Fredericks DC, et al. Time-dependent loss of mitochondrial function precedes progressive histologic cartilage degeneration in a rabbit meniscal destabilization model. J Orthop Res. 2016 doi: 10.1002/jor.23327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koike M, Nojiri H, Ozawa Y, et al. Mechanical overloading causesmitochondrial superoxide andSOD2 imbalance in chondrocytesresulting in cartilage degeneration. Nature Publishing Group; 2015. pp. 1–16. [Google Scholar]
- 21.Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287(3):L486–L496. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- 22.Szafranski JD, Grodzinsky AJ, Burger E, et al. Chondrocyte mechanotransduction: effects of compression on deformation of intracellular organelles and relevance to cellular biosynthesis. Osteoarthr Cartil. 2004;12(12):937–946. doi: 10.1016/j.joca.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Li D, Xie G, Wang W. Reactive oxygen species: the 2-edged sword of osteoarthritis. Am J Med Sci. 2012;344(6):486–490. doi: 10.1097/MAJ.0b013e3182579dc6. [DOI] [PubMed] [Google Scholar]
- 24.Brouillette MJ, Ramakrishnan PS, Wagner VM, et al. Strain-dependent oxidant release in articular cartilage originates from mitochondria. Biomech Model Mechanobiol. 2014;13(3):565–572. doi: 10.1007/s10237-013-0518-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knight MM, Bomzon Z, Kimmel E, et al. Chondrocyte deformation induces mitochondrial distortion and heterogeneous intracellular strain fields. Biomech Model Mechanobiol. 2006;5(2–3):180–191. doi: 10.1007/s10237-006-0020-7. [DOI] [PubMed] [Google Scholar]
- 26.Sauter E, Buckwalter JA, McKinley TO, Martin JA. Cytoskeletal dissolution blocks oxidant release and cell death in injured cartilage. J Orthop Res. 2012;30(4):593–598. doi: 10.1002/jor.21552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huser CAM, Davies ME. Calcium signaling leads to mitochondrial depolarization in impact-induced chondrocyte death in equine articular cartilage explants. Arthritis & Rheumatism. 2007;56(7):2322–2334. doi: 10.1002/art.22717. [DOI] [PubMed] [Google Scholar]
- 28.Delco ML, Bonnevie ED, Bonassar LJ, Fortier LA. Mitochondrial dysfunction is an acute response of articular chondrocytes to mechanical injury. J Orthop Res. 2017 doi: 10.1002/jor.23651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szeto HH, Birk AV. Serendipity and the Discovery of Novel Compounds That Restore Mitochondrial Plasticity. Clin Pharmacol Ther. 2014 doi: 10.1038/clpt.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171(8):2029–2050. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane--from discovery to clinical development. Pharm Res. 2011;28(11):2669–2679. doi: 10.1007/s11095-011-0476-8. [DOI] [PubMed] [Google Scholar]
- 32.Birk AV, Liu S, Soong Y, et al. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. Journal of the American Society of Nephrology. 2013;24(8):1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birk AV, Chao WM, Liu S, et al. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochim Biophys Acta. 2015;1847(10):1075–1084. doi: 10.1016/j.bbabio.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birk AV, Chao WM, Bracken C, et al. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171(8):2017–2028. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibson CM, Giugliano RP, Kloner RA, et al. EMBRACE STEMI study: a Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur Heart J. 2016;37(16):1296–1303. doi: 10.1093/eurheartj/ehv597. [DOI] [PubMed] [Google Scholar]
- 36.Saad A, Herrmann SMS, Eirin A, et al. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ Cardiovasc Interv. 2017;10(9) doi: 10.1161/CIRCINTERVENTIONS.117.005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonnevie ED, Delco ML, Fortier LA, et al. Characterization of Tissue Response to Impact Loads Delivered Using a Hand-Held Instrument for Studying Articular Cartilage Injury. Cartilage. 2015;6(4):226–232. doi: 10.1177/1947603515595071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alexander PG, Song Y, Taboas JM, et al. Development of a Spring-Loaded Impact Device to Deliver Injurious Mechanical Impacts to the Articular Cartilage Surface. Cartilage. 2012;4(1):52–62. doi: 10.1177/1947603512455195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexander PG, McCarron JA, Levine MJ, et al. An In Vivo Lapine Model for Impact-Induced Injury and Osteoarthritic Degeneration of Articular Cartilage. Cartilage. 2012;3(4):323–333. doi: 10.1177/1947603512447301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fortier LA, Motta T, Greenwald RA, et al. Synoviocytes are more sensitive than cartilage to the effects of minocycline and doxycycline on IL-1alpha and MMP-13-induced catabolic gene responses. J Orthop Res. 2010;28(4):522–528. doi: 10.1002/jor.21006. [DOI] [PubMed] [Google Scholar]
- 41.Cohen BE, McAnaney TB, Park ES, et al. Probing protein electrostatics with a synthetic fluorescent amino acid. Science. 2002;296(5573):1700–1703. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]
- 42.D’Lima D, Hermida J, Hashimoto S, et al. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis & Rheumatism. 2006;54(6):1814–1821. doi: 10.1002/art.21874. [DOI] [PubMed] [Google Scholar]
- 43.Huser CAM, Peacock M, Davies ME. Inhibition of caspase-9 reduces chondrocyte apoptosis and proteoglycan loss following mechanical trauma. Osteoarthritis and Cartilage. 2006;14(10):1002–1010. doi: 10.1016/j.joca.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 44.Kurz B, Lemke A, Kehn M, et al. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis & Rheumatism. 2004;50(1):123–130. doi: 10.1002/art.11438. [DOI] [PubMed] [Google Scholar]
- 45.Martin JA, McCabe D, Walter M, et al. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. J Bone Joint Surg Am. 2009;91(8):1890–1897. doi: 10.2106/JBJS.H.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garrido CP, Hakimiyan AA, Rappoport L, et al. Anti-apoptotic treatments prevent cartilage degradation after acute trauma to human ankle cartilage. Osteoarthritis and Cartilage. 2009;17(9):1244–1251. doi: 10.1016/j.joca.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. The Journal of Cell Biology. 2011;192(1):7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao K, Zhao G-M, Wu D, et al. Cell-permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. Journal of Biological Chemistry. 2004;279(33):34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 49.Zhao K, Zhao GM, Wu D, et al. Cell-permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. Journal of Biological Chemistry. 2004;279(33):34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 50.Bartell LR, Fortier LA, Bonassar LJ, Cohen I. Measuring microscale strain fields in articular cartilage during rapid impact reveals thresholds for chondrocyte death and a protective role for the superficial layer. Journal of Biomechanics. 2015;48(12):3440–3446. doi: 10.1016/j.jbiomech.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kühn K, D’Lima DD, Hashimoto S, Lotz M. Cell death in cartilage. Osteoarthritis and Cartilage. 2004;12(1):1–16. doi: 10.1016/j.joca.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 52.Grogan SP, Aklin B, Frenz M, et al. In vitro model for the study of necrosis and apoptosis in native cartilage. J Pathol. 2002;198(1):5–13. doi: 10.1002/path.1169. [DOI] [PubMed] [Google Scholar]
- 53.Green DM, Noble PC, Ahuero JS, Birdsall HH. Cellular events leading to chondrocyte death after cartilage impact injury. Arthritis & Rheumatism. 2006;54(5):1509–1517. doi: 10.1002/art.21812. [DOI] [PubMed] [Google Scholar]
- 54.D’lima D. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthritis and Cartilage. 2001;9(8):712–719. doi: 10.1053/joca.2001.0468. [DOI] [PubMed] [Google Scholar]
- 55.Bonnevie ED, Delco ML, Galesso D, et al. Journal of Biomechanics. 2017:1–7. doi: 10.1016/j.jbiomech.2016.12.034. [DOI] [PubMed] [Google Scholar]
- 56.Bonnevie ED, Delco ML, Jasty N, et al. Chondrocyte death and mitochondrial dysfunction are mediated by cartilage friction and shear strain. Osteoarthr Cartil. 2016;24(Supplement 1 IS):S46EP. [Google Scholar]
- 57.Patwari P, Cook MN, DiMicco MA, et al. Proteoglycan degradation after injurious compression of bovine and human articular cartilage in vitro: interaction with exogenous cytokines. Arthritis & Rheumatism. 2003;48(5):1292–1301. doi: 10.1002/art.10892. [DOI] [PubMed] [Google Scholar]
- 58.Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11(1):35–44. doi: 10.1038/nrrheum.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szeto HH, Liu S, Soong Y, et al. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1β and IL-18 and Arrests CKD. Journal of the American Society of Nephrology. 2017;28(5):1437–1449. doi: 10.1681/ASN.2016070761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Svineng G, Ravuri C, Rikardsen O, et al. The Role of Reactive Oxygen Species in Integrin and Matrix Metalloproteinase Expression and Function. Connective Tissue Research. 2009;49(3–4):197–202. doi: 10.1080/03008200802143166. [DOI] [PubMed] [Google Scholar]
- 63.Reed KN, Wilson G, Pearsall A, Grishko VI. The role of mitochondrial reactive oxygen species in cartilage matrix destruction. Mol Cell Biochem. 2014;397(1–2):195–201. doi: 10.1007/s11010-014-2187-z. [DOI] [PubMed] [Google Scholar]
- 64.Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radic Biol Med. 2004;37(6):768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 65.Rajpurohit R, Koch CJ, Tao Z, et al. Adaptation of chondrocytes to low oxygen tension: relationship between hypoxia and cellular metabolism. J Cell Physiol. 1996;168(2):424–432. doi: 10.1002/(SICI)1097-4652(199608)168:2<424::AID-JCP21>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 66.Ströbel S, Loparic M, Wendt D, et al. Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Research & Therapy. 2010;12(2):R34–R34. doi: 10.1186/ar2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schneider N, Mouithys-Mickalad A, Lejeune J-P, et al. Oxygen consumption of equine articular chondrocytes: Influence of applied oxygen tension and glucose concentration during culture. Cell Biol Int. 2007;31(9):878–886. doi: 10.1016/j.cellbi.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 68.Zhou S, Cui Z, Urban JPG. Factors influencing the oxygen concentration gradient from the synovial surface of articular cartilage to the cartilage-bone interface: A modeling study. Arthritis & Rheumatism. 2004;50(12):3915–3924. doi: 10.1002/art.20675. [DOI] [PubMed] [Google Scholar]
- 69.Grimshaw MJ, Mason RM. Bovine articular chondrocyte function in vitro depends upon oxygen tension. Osteoarthr Cartil. 2000;8(5):386–392. doi: 10.1053/joca.1999.0314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.