

Abstract
Trans-eQTLs have been implicated in complex traits and common diseases, but many were initially identified on the basis of having an effect in cis, and there has been no assessment of the significance of the overlap in relation to chance expectations. Here, we investigated whether trans-expression quantitative trait loci (eQTL) associations identified in whole blood contribute to variance in complex traits by determining (1) whether genome-wide significant (GWS) single-nucleotide polymorphisms (SNPs) were enriched for trans-eQTL (including trans-only eQTL), and (2) whether the genomic regions surrounding associated trans-genes were enriched for statistical associations in the relevant GWAS. On average for a given phenotype, we identify 4.8% of GWS SNPs overlapping with trans-eQTL present in blood, and show that for the majority of these phenotypes, this observation does not exceed that expected by chance. Likewise, we observe no enrichment for genetic associations with the GWAS phenotype in the regions surrounding the linked trans-genes, with the exception of rheumatoid arthritis. Interestingly, the GWS SNPs for each phenotype were consistently more enriched for unique trans-eQTL SNPs than trans-eQTL SNP-probe pairs (p = 4 × 10−7), with schizophrenia the only exception. This relative enrichment for trans-eQTL SNPs over trans-eQTL SNP-probe pairs implies that trait-associated trans-eQTL SNPs in whole blood are less likely to be 'master regulators' than random trans-eQTL SNPs. Taken together, these results suggest little evidence for the role of blood-based trans-eQTL in complex traits and disease, although this may reflect the finite size of currently available data sets and our findings may not hold for trans-eQTLs in more trait-relevant tissues. All software is publically available at https://github.com/IMB-Computational-Genomics-Lab/eqtlOverlapper.
Introduction
Genome-wide association studies (GWAS) have identified thousands of single-nucleotide polymorphisms (SNPs) that are associated with common diseases and complex traits [1]. The majority of these variants are in non-coding regions of the genome and are likely to act by altering gene regulation [2]. Expression quantitative trait loci (eQTL) are polymorphisms whose genotype is associated with an additive effect on gene expression levels. They are classified as either cis (local) or trans (distal) depending on the location of the associated gene relative to the polymorphism. Numerous studies have identified an enrichment of cis-eQTL among genome-wide significant (GWS) (p < 5 × 10−8) SNPs identified by GWAS [3–11], suggesting that the functional basis of many genetic associations involves dysregulation of transcript levels of disease-related genes [12]. Owing to its accessibility, the majority of large-scale eQTL studies have used whole blood, or subsets of blood cell types [13–16]. As such, blood eQTLs are frequently interrogated for disease loci overlap [13, 17].
Biologically, trans-eQTL tend to alter the structure, function, or expression of diffusible factors such as transcription, signalling or splicing factors [12], though there is also evidence that they may participate in transcription factories [18, 19]. In contrast, cis-eQTL are thought to predominantly influence gene expression through transcription factor binding sites [12], DNA methylation [20], chromatin accessibility [21] and regulatory RNAs [22].
Locally acting cis-eQTL are more readily identified [23] than distally acting trans-eQTL. This is because the average effect size of cis-eQTL is larger, and the multiple-testing burden is smaller relative to analysis of trans-eQTL [13, 24]. However, the majority of genetic variance underlying gene expression is located in trans-regions of the genome [24], and thus it follows that most eQTL are likely to exist in trans [18, 25]. Recently, Westra et al. [13] identified 233 trans-eQTL SNPs by conducting the largest trans-eQTL study to date: a meta-analysis of 5311 peripheral blood samples from seven studies, with replication in a further 2775 samples. Critically, they only searched for trans-eQTL using SNPs that had already been shown to be associated with GWAS, with many of the identified effects also having cis-eQTL effects.
There is strong evidence that disease and trait-associated SNPs are enriched for cis-eQTL [10, 26, 27]. However, independent trans effects, i.e., those that are not also cis-eQTL, have not been closely studied, and may help to identify functional pathways and gene networks. These may also be more interesting than those also associated with cis-eQTL, as the trans association may be mediated by the cis association.
Here, we performed analyses using data from two whole blood eQTL studies and GWAS summary statistics to determine whether trans-eQTL contribute to 11 complex traits (Table 1). We defined a trans-eQTL as the sentinel SNP of a locus associated with the gene expression of a transcript on a different chromosome, and cis-eQTL as loci associated with the expression of a gene on the same chromosome. We further defined trans-only-eQTL as the subset of trans-eQTL without (detectable) effects in cis. For a given complex trait, we hypothesised (i) that GWS SNPs would be enriched for trans-eQTL, including trans-only-eQTL, and (ii) that trans-probe regions (Fig. 1) would be enriched for statistical associations in the relevant disease GWAS.
Table 1.
Summary of GWAS used in this study
| Phenotype | Abbreviation | Publication | EUR GWS SNPs (in BSGS database) |
|---|---|---|---|
| Body mass index | BMI | Speliotes et al., 2010 | 31 |
| Diastolic blood pressure | BPD | ICBP, 2011 | 25 |
| Systolic blood pressure | BPS | ICBP, 2011 | 24 |
| Coronary artery disease | CAD | CARDIoGRAMplusC4D., 2013 | 44 |
| Crohn’s disease | CD | Jostins et al., 2012 | 28 |
| Type II diabetes | DB | Morris et al., 2012 | 63 |
| Height | Height | Wood et al., 2014 | 651 |
| Inflammatory bowel disease | IBD | Jostins et al., 2012 | 107 |
| Rheumatoid arthritis | RA | Okada et al., 2014 | 44 |
| Ulcerative colitis | UC | Jostins et al., 2012 | 23 |
| Schizophrenia | SCZ | SWGPGC., 2014 | 128 |
Fig. 1.
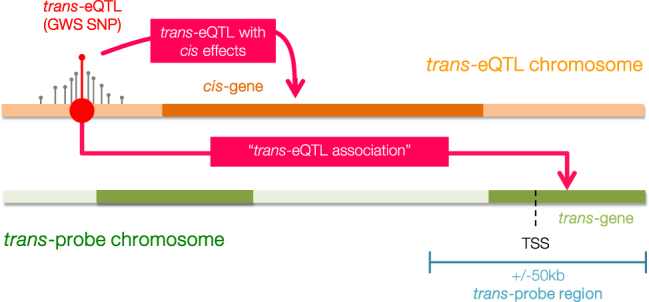
Graphical summary of nomenclature used throughout this study. Orange represents the chromosome with the trans-acting regulatory locus. Red circles correspond to SNPs. Trans-eQTLs are loci that regulate gene expression from another chromosome. We defined trans-eQTL as the sentinel SNP with regulatory trans effects. The trans-eQTL in our study were also GWS SNPs from one of 11 studied phenotypes. Some trans-eQTL also had cis effects on genes located on the same chromosome. Green represents the trans-probe chromosome. The trans-probe region refers to the genetic locus ± 50 kb around the transcription start site (TSS). This region often includes SNPs outside the UCSC-defined genes. Each trans-probe tagged a trans-gene.
Methods
GWAS summary statistics
GWAS summary statistics were downloaded for the following 11 complex traits and common diseases: body mass index (BMI) [4], diastolic blood pressure (DBP) [28], systolic blood pressure [28], coronary artery disease (CAD) [5], Crohn’s disease [7], type 2 diabetes (DB) [6], height [3], inflammatory bowel disease (IBD) [7], rheumatoid arthritis (RA) [29], ulcerative colitis [7] and schizophrenia (SCZ) [8]. Details are given in Table 1 and the Supplementary Note.
eQTL data
For an initial discovery, we used eQTL data (see URL below) generated from the analysis of expression levels of transcripts measured in whole blood from the Brisbane Systems Genetics Study (BSGS) [30]. BSGS comprises 862 individuals of European descent, with expression levels measured on Illumina HT12 arrays and genotype data imputed to 1000 Genomes (phase 1v3). eQTL summary statistics from an additive model are available for each combination of 17994 expression probes and 6.2 million autosomal SNPs. At a 1 × 10−10 significance threshold, the BSGS data set contains 2512 eQTL: 1953 with cis effects, and 602 trans-eQTL, of which 559 are trans-only [30]. For replication of trans-eQTL, we used eQTL data generated by the Consortium for the Architecture of Gene Expression (CAGE). CAGE is comprised of whole blood expression data, generated using Illumina HT12 arrays for 1852 unrelated individuals of European ancestry [31]. The full CAGE data set includes the BSGS results, so we excluded the BSGS data set to perform replication using an independent cohort. Probe and SNP positional information was annotated to the hg19 genome build; for SNPs, we used the FDb.UCSC.snp137common.hg19 package [32] (available from BioConductor).
Trans-eQTL—GWAS overlap
For each of the 11 complex traits and common diseases, we identified GWS SNPs that overlapped with independent trans-eQTL in BSGS (Table 1). We considered only the most significantly associated (hereafter referred to as 'sentinel') SNP in each GWAS locus. We applied the following stringent criteria to determine overlap: (1) the trans-eQTL sentinel SNP was the sentinel GWS SNP for the phenotype, (2) the eQTL association p value was < 1 × 10−6 (corresponding to a false discovery rate of < 0.1), and 3) the GWAS SNP and the BSGS transcript probe were located on different chromosomes. We collected data for the number of trans-eQTL SNP-probe associations, unique trans-eQTL SNPs, and trans-eQTL SNPs that also had local-acting cis effect(s) (i.e., on the same chromosome), also at a 1 × 10−6 significance threshold (corresponding to a false discovery rate of < 0.05). We applied a more stringent FDR for cis-eQTL because effect sizes tend to be larger than for trans-eQTL.
Trans-probe regional enrichment
We investigated whether there was enrichment of GWAS p values within the trans-probe region(s) involving sentinel GWAS SNPs for a given phenotype. Such enrichment would suggest that genetic variation in the vicinity of a trans-gene was more strongly associated with the phenotype than expected by chance. For each trans-probe, we extracted SNPs from the original GWAS study within a ± 50 kb window of the probe start site. We accounted for genome build by cross-referencing trans-probes to a Bioconductor GenomicRanges file containing probe positional coordinates in hg19. By using the flank() and findOverlaps() function from the Bioconductor GenomicRanges package [33], we extracted hg19 SNPs from the FDb.UCSC.snp137common.hg19 package located within ± 50 kb of the trans-probe start site. SNPs within each ± 50 kb trans-probe window were aggregated for each phenotype. We calculated median lambda statistics from SNP p values aggregated across the trans-probe regions (Supplementary Figure 2–4). The median lambda was given by the median chi-squared statistic divided by 0.456, the expected median chi-squared statistic for a 1-degree of freedom test.
Comparison of trans-eQTL counts to a null distribution produced by SNP resampling
We determined an empirical null distribution for comparison with the observed data. This was achieved by resampling 1000 × n SNPs without replacement from the GWAS summary statistics, where n was the number of GWS SNPs in European ancestry populations for the focal phenotype. We re-sampled SNPs matched to the minor allele frequency (MAF) distribution (10 bins) of the observed SNPs. Where the GWAS summary statistics did not provide allele frequencies, we used frequencies taken from the 1000 Genomes Phase 3 EUR reference panel. For each resampling of n SNPs, we performed the same procedures to determine trans-eQTL overlap. The SNP resampling process provided empirical null distributions for the following: (1) number of trans-eQTL associations, (2) number of unique, independent trans-eQTL and (3) the number of trans-eQTL that also had cis effect(s) at a 1 × 10−6 significance threshold (Table 2). We considered results to be statistically significant if ≤ 4/1000 re-sampled SNP sets exceeded the observed result (i.e., a rank of ≥ 996/1000 corresponding to significance level < 0.05 after Bonferroni correction for 11 phenotypes).
Table 2.
Summary of trans-eQTL in BSGS
| EUR GWS SNPs (BSGS database) |
trans-eQTL detection | trans-probe enrichment | ||||
|---|---|---|---|---|---|---|
| Phenotype | Associations | SNPs | % SNPs | cis-eQTLs | Median lambda | |
| BMI | 31 | 2 (846/1000) | 2 (945/1000) | 6.45% | 1 (976/1000)~ | 615/1000 |
| BPD | 25 | 1 (800/1000) | 1 (871/1000) | 4.00% | 0 | 519/1000 |
| BPS | 24 | 1 (795/1000) | 1 (873/1000) | 4.17% | 0 | 563/1000 |
| CAD | 44 | 7*(908/1000) | 4^(977/1000)~ | 9.09% | 2 (943.1000) | 326/1000 |
| CD | 28 | 0 | 0 | 0.00% | 0 | – |
| DB | 63 | 3 (437/1000) | 3 (641/1000) | 4.76% | 0 | 702/1000 |
| Height | 651 | 21 (307/1000) | 15 (415/1000) | 2.15% | 3 (783/1000) | 236/1000 |
| IBD | 107 | 6*(714/1000) | 6^(945/1000) | 5.61% | 2 (955/1000)~ | 665/1000 |
| RA | 44 | 1 (625/1000) | 1 (713/1000) | 2.27% | 0 | 999/1000+ |
| UC | 23 | 0 | 0 | 0.00% | 0 | – |
| SCZ | 128 | 8 (808/1000) | 4 (773/1000) | 3.13% | 0 | 428/1000 |
The number of trans effects found per phenotype is shown in 'Associations'. When these values were ranked against a distribution from 1000 SNP re-samplings (in brackets after the number of associations), no phenotype had more associations than expected by chance. * denotes phenotypes where there was one trans-eQTL association with a probe on a sex chromosome; these associations were not displayed on circle or QQ-plots. 'SNPs' gives the number of unique trans-eQTL SNPs involved in trans-eQTL associations. '%SNPs' refers to the number of ttrans-eQTL SNPs as a percentage of the total number of EUR GWS SNPs. 'cis-eQTL' refers to the number of trans-eQTL that also had cis effects (p < 1 × 10−6). The 'trans-probe enrichment' column shows evidence for GWAS p value enrichment around the aggregated trans-probes. It shows the median lambda rank for the trans-probe SNPs identified in the discovery analysis, compared with the probe resampling null distribution evidence. + denotes ranks that passed a Bonferroni significance threshold. ~ denote ranks that passed a nominal significance threshold.
Comparison of enrichment signal to a null distribution produced by probe resampling
We created an additional empirical null distribution to evaluate enrichment of GWAS associations in trans-probe regions by performing 1000 × n probe re-samplings from the BSGS database, where n was the number of trans-probes found in the initial analysis. For some GWAS summary statistics, the genetic markers provided were quite sparse, and there were instances where there were no statistics available for SNPs within a randomly sampled trans-probe region. Hence, we ensured that the aggregated trans-probe region always contained one or more GWS SNPs, and if it did not, we generated another sample of aggregated trans-probe regions that did. We considered results to be statistically significant if ≤ 4/1000 re-samplings exceeded the observed result (i.e., a rank of ≥ 996/1000 corresponding to significance level < 0.05 after Bonferroni correction for 11 phenotypes; Table 2, Supplementary Figures 5-6).
Results
For nine of the 11 phenotypes, a proportion of GWS SNPs were also trans-eQTL identified from whole blood gene expression in BSGS samples (Table 2). Across all phenotypes, we identified a total of 50 trans-eQTL SNP-probe associations involving 37 unique SNPs. A trans-eQTL can act as a master regulator by controlling multiple transcripts, and this was true for eight of the 37 trans-eQTL (22%). However, when compared to the results from 1000 re-samplings of SNPs matched by MAF, none of the phenotypes had more trans-eQTL associations or trans-eQTL than expected by chance (rank threshold of ≥ 996/1000) in the BSGS data set (Table 2). Thus, although we observe some overlap between GWS SNPs and trans-eQTL, the overlap for these 11 phenotypes is consistent with that expected by randomly resampling SNPs. The phenotype that most closely approached significance was CAD (trans-eQTL associations rank 908/1000; trans-eQTL rank 977/1000) (Table 2).
Overall, ranks for the number of sentinel trans-eQTL tended to be higher than ranks for the number of trans-eQTL SNP-probe associations for all of the phenotypes except SCZ (Supplementary Figure 1). A two-sided Pearson’s Χ2-test comparing the trans-eQTL ranks to the trans-eQTL association ranks yielded p = 2.3 × 10−7, suggesting that this is not due to chance. This implied that trait-associated trans-eQTL accounted for fewer trans-eQTL associations than random trans-eQTL SNPs. The percentage of European GWAS SNPs with trans effects correlated with the rank of the number of trans-eQTL (Pearson’s correlation coefficient p = 0.032). That is, phenotypes with a greater percentage of trans-eQTL had higher trans-eQTL ranks. This is reassuring as it suggests that the null distributions scaled well between phenotypes with regard to trans-eQTL SNP percentage.
A proportion of trans-eQTL were also found to be cis-eQTL [34] (Table 2), as has been reported in previous studies. However, we did not observe evidence for enrichment in comparison with the null distribution based on SNP resampling. The observed numbers of trans-eQTL with cis effects was nominally significant for BMI (rank of 976/1000) and IBD (955/1000) (Table 2).
We investigated whether the trans-eQTL identified in BSGS replicated in the CAGE (minus BSGS) data set. In BSGS, a total of 602 trans-eQTL were identified at a significance threshold of 1 × 10−10. Of these, 526 (87%) replicated in CAGE at a significance level p < 0.05/602, with a matched allelic direction for 94% of the 526 (p = 5.5 × 10−16). These results support the reliability of trans-eQTL identification in the BSGS cohort. The summary statistics from the CAGE cohort for 50 overlapping GWAS/trans-eQTL are provided in Supplementary Table 1. The full summary statistics for the CAGE and BSGS cohorts are available at: http://cnsgenomics.com/shiny/CAGE.
We next investigated if there was an enrichment of GWAS associations in trans-regions of the 48 trans-eQTL associations (representing 36 trans-eQTL SNPs) involving autosomal genes. RA was the only phenotype that showed enrichment in the probe resampling analysis at either the nominal and Bonferroni-corrected rank significance thresholds (median lambda rank 999/1000) (Table 2, Fig. 2). Only one trans-eQTL was found for RA, between rs13330176 and ILMN_1772218 (Table 2, Supplementary Figure 1). There was no trans-probe enrichment of GWAS p values for BMI, BPD, BPS, CAD, DB, IBD or SCZ when compared with the null distribution generated by probe resampling (Table 2, Supplementary Figures 5–6). However, it is important to note that the trans-probe enrichment results for CAD were difficult to interpret, as the SNPs within the GWAS summary statistics were sparse.
Fig. 2.

Analysis workflow, with results from the rheumatoid arthritis (RA) analysis. a Flow diagram of trans-eQTL analysis. To find trans-eQTL associations, three filters were applied: (i) eQTL matched to GWS SNPs, (ii) eQTL association p value of p < 1 × 10−6 (corresponding to an FDR < 0.1), and (iii) eQTL where the SNP and probe were on separate chromosomes. The resultant trans-eQTL associations were summarised using a circle plot b. To test for enrichment of GWAS p values around the gene, SNPs within ± 50 kb of the probe start site were extracted to produce a QQ plot c. Resampling analyses were performed to test for enrichment d–g. d, e A SNP resampling of 1000 × n GWS SNPs provided a null distribution to test enrichment of the number of trans-eQTL associations d, trans-eQTL SNPs e and trans-eQTL that were also cis-eQTL (results not shown as RA did not have any of these associations). f, g A probe resampling of 1000 × n GWAS SNPs provided a null distribution for enrichment (determined using median lambda) of GWAS associations in trans-probe regions. f QQ plot showing trans-probe region enrichment for RA against the probe resampling distribution (orange). The black line represents the discovery analysis results. Ranks were calculated to quantify enrichment (Bonferroni-corrected rank threshold ≥ 996/1000). g Histograms of median lambda distributions from probe resampling analyses. The red line represents the median lambda rank for RA.
In the BSGS enrichment analysis, height qualitatively showed a prominent enrichment signal (Supplementary Figure 3). However, the lambda rank compared with the null distribution based on probe resampling yielded a median lambda rank of 236/1000 (Supplementary Figure 3), suggesting that a few, strongly enriched trans-probe regions drove the enrichment signal. Interestingly, one of the trans-eQTL associations that replicated in both BSGS and CAGE at a p < 1 × 10−10 threshold contributed to one of these highly-enriched trans-probe regions. This was the height trans-eQTL association between rs926438 and the probe ILMN_1752758 (tagging the BTN2A2 gene) (Supplementary Figure 3–4).
Discussion
Here we used GWAS summary statistics and independent whole blood eQTL data to investigate whether trans-only-eQTL contribute to variation in 11 common diseases and complex traits. For most diseases and traits, a proportion of GWS SNPs exhibited trans effects (on average, 4.8%) for gene expression, including trans-only effects, but the observations did not exceed that expected by chance. A previous study had reported that trait-associated SNPs are enriched for trans-eQTL [34]. However, the analyses differ in that Pierce et al. [34]. investigated aggregated trait-associated SNPs generally, rather than for specific phenotypes, and they matched SNPs by distance to transcription start site in addition to MAF, and thus include trans-eQTL with a cis- effect also. Our analysis is novel because we sought to identify trans-eQTL irrespective of whether they had an effect in cis, whereas previous studies examined cis-eQTL for trans effects to identify trans-eQTL [13, 34]. The majority of the trans-eQTL associations in our study were trans-only (Table 2), insomuch as the trans-eQTL did not also exhibit cis effects (p < 1 × 10−6) in the BSGS database.
If a trans-eQTL controlled more than one gene (that is, if it was involved in multiple trans-eQTL associations), we considered it to be a master regulator. The proportion of trans-eQTL that were master regulators in our study (22%) was similar to that reported by Grundberg et al. [24], who found that master regulators for expression levels measured in skin, Lymphoblastoid Cell Lines (LCL) and adipose accounted for 21–32% of trans-eQTL associations. In our study, the GWS SNPs for each phenotype were consistently more enriched for unique sentinel trans-eQTL than trans-eQTL associations (p = 2 × 10−7) (Supplementary Figure 1), with SCZ the only exception to this rule. This relative enrichment for trans-eQTL over trans-eQTL associations implies that trait-associated trans-eQTL have fewer trans effects than random trans-eQTL in whole blood; that is, they are less likely to be master regulators. This concurs with prior studies that have found that trans-eQTL influence relatively few genes in peripheral blood [15]. We speculate that this could be due to the polygenic nature of complex traits; a variant of a trans-master regulator would have an amplified effect on the phenotype, lending itself to a less complex aetiology.
We compared our results with two null distributions based on resampling by SNP and by probe, allowing us to evaluate statistical confidence in observed overlapping GWS loci and trans-eQTL. The SNP resampling allowed us to test for enrichment of trans-eQTL associations, trans-eQTL, and trans-eQTL that also had a cis-eQTL effect; the probe resampling enabled us to test for trans-region enrichment of GWAS associations (median lambda) (Fig. 2). We note that the SNP-resampling enrichment p values may be 'inflated' as we calculated the rank as the total number of random samples minus the number of samples with counts greater than that was observed. This inflation was particularly prominent for our analysis of trans-eQTL that also had a cis-eQTL effect; for example, BMI only had one trans-eQTL with a cis-eQTL effect, yet had a rank of 976/1000. For the trans-region enrichment analysis, we decided to use the median lambda statistic rather than the mean lambda, as the latter could demonstrate inflated results if the trans-region contained a few GWS SNPs with extremely low p values. This was evident in the height analysis, for example (Supplementary Figure 3). Although it was possible to generate another trans-region distribution by using trans-eQTLs identified in each SNP resampling, we elected not to do so. (However, we provide example code in the accompanying package regardless). This was because SNP resampling occasionally identified zero trans-eQTLs, and assigning a 'no-result' p value (i.e., p = 0.5) deflated the lambda statistic.
RA was the only phenotype with significant trans-region enrichment of GWAS association (Fig. 2). The contributing trans-eQTL association was between the chromosome 16 SNP rs13330176 (allele frequency of 0.781) and ILMN_1772218 (tagging the chromosome 6 gene HLA-DPA1). This association may be biologically relevant because RA is an autoimmune disorder and the HLA-DPA1 gene is located within the major histocompatibility complex (MHC). However, there are two caveats: (1) linkage disequilibrium is high in this region so the strong enrichment signal may in fact tag other MHC loci [35], and (2) the association did not replicate in our independent eQTL data set, CAGE.
Of the 11 phenotypes, the analysis for height yielded some noteworthy associations. We identified a trans-eQTL association between SNP rs926438, in the intronic region of the RHCE gene (which encodes the Rh blood antigen), and probe ILMN_1752758 (tagging the BTN2A2 gene). This association, which has been previously reported in monocytes [36], replicated in CAGE at p = 1.1 × 10−43. There is little evidence of transcription factor binding to site rs926438 according to RegulomeDB [37]. BTN2A2 belongs to the butyrophilin family and immunoglobulin superfamily. It is a glycoprotein that participates in lipid, sterol and fatty-acid metabolism, and is associated with fat droplets in milk. Around the ILMN_1752758/BTN2A2 trans-probe region, the SNP with the lowest height GWAS p value was rs10456328 (p = 3.6 × 10−18) (Supplementary Figure 3). The GWAS height SNP rs806794 (p = 4.6 × 10−74) is ~1.5 Mb away, but is not in LD with rs10456328 (r2 = 0.0088, D’ = 0.31) (Supplementary Figure 4). Hence, the trans-probe enrichment appears to be relevant to the BTN2A2 gene, or neighbouring butyrophilin family genes.
The trans-probe region contributing to the strongest enrichment of GWAS signal occurred in the height analysis and corresponded to the trans-eQTL association between rs1325596 and ILMN_1745152 (tagging the gene UQCC). Related functional annotations suggest that there is an interesting link. The GWAS SNP with the lowest p value in the ILMN_1745152/UQCC trans-probe region was rs6060369. Of the SNPs within the trans-probe region, rs6060369 was also the top cis-eQTL (p = 5 × 10−6) for ILMN_1745152/UQCC in the BSGS database. rs6060369 forms a haplotype with rs143384—a height GWS SNP (p = 1.2 × 10−121) [3]. This haplotype has been associated with hip osteoarthritis, a condition associated with adult height by altering posture [38]. The trans-eQTL SNP rs1325596 is an intron variant within the PAPPA2 gene, which has been associated with hip dysplasia [39], which is one of two major hip osteoarthritis risk factors, along with age [40]. Taken together, the rs1325596/UQCC trans-eQTL association may represent a functional mechanism underlying variation in height.
We interpreted trans-region enrichment of GWAS associations as supporting evidence that the trans-eQTL corresponded with a region that contributed to variation in the phenotype. The ± 50 kb region should capture regulatory variants within that gene’s enhancer and promoter regions, as well as coding variants. Intuitively, if variation in the gene code contributes to variation in the phenotype, then nearby regulatory or coding variants should contribute to genetic risk for that trait and manifest as low GWAS p values. In enriched trans-probe regions, it could be interesting to examine how the trans-probe SNPs relate to the gene; for example, whether they are cis-eQTL for that probe, like rs6060369 (in the trans-probe region corresponding to the rs1325596/UQCC trans-eQTL association in height) is for UQCC. However for RA, the trans-probe region was within the MHC and high LD likely led to an inflation of the enrichment signal.
One of the strengths of our analysis was its use of stringent thresholds. First, for the eQTL detection filters, we applied false discovery rate thresholds of 0.1 and 0.05 for trans- and cis-eQTLs, respectively, corresponding in each case to a p value threshold of 1 × 10−6. In addition, we defined trans-eQTL as loci that act on separate chromosomes to the gene. This avoided potential confounds from long-range linkage disequilibrium, such as within the MHC. Secondly, for the trans-probe enrichment analyses, we calculated the median lambda statistic. A phenotype was only considered to show enrichment if the median lambda had a rank ≥ 996/1000 (Bonferroni-adjusted) (Table 2).
Some aspects of this analysis warrant further investigation. The primary limitation of this study was that eQTL are tissue-specific. Although we found little evidence of trans-eQTL contributing to complex traits in whole blood, Wright et al. [15] showed that whole blood has fewer significant trans-eQTL and trans-master regulators than other tissues. Hence, it is still important to study trans-eQTL, especially in different tissues. More broadly, eQTL vary between tissues: 69–80% of regulatory variants are tissue-specific [41], and gene expression between whole blood and LCL eQTL studies differ [42]. Intuitively, blood is more likely to be a biologically relevant tissue for CAD, RA, BMI and BP than height or SCZ, for example; this may help to explain why there was more evidence of enrichment among the former phenotypes for trans-eQTL and trans-probe region GWAS p values. Therefore, it would be interesting to study how the number of trans-eQTL associations, and the trans-probe enrichment signal changes in more biologically relevant tissues. Second, eQTL can be temporally specific, and the BSGS data set used expression data from adolescents (aged 12–16), and their parents. Hence, studies focussing on specific developmental stages should be informative for traits such as height and adolescent-onset diseases such as SCZ. Third, precise trans-eQTL mechanisms and pathways were not investigated here; the rs926438/ILMN_1752758 (tagging the BTN2A2 gene) trans-eQTL association is a particularly promising candidate for functional follow-up. The accompanying software package created here may be used to conduct these analyses, as tissue- and age-specific data sets become available.
In conclusion, trans-eQTL found in adult peripheral whole blood did not exert a significant influence on the eleven complex traits and diseases we examined, although this may reflect the finite size of currently available data sets and, as noted above, our findings may not hold for trans-eQTLs in more trait-relevant tissues and developmental stages.
URLs
BSGS eQTL results: https://bsgseqtlbrowser.qbi.uq.edu.au
CAGE eQTL results: http://cnsgenomics.com/shiny/CAGE/
Electronic supplementary material
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41431-018-0174-7) contains supplementary material, which is available to authorised users.
References
- 1.Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–86. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Consortium CAD, Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards AL, Jones L, Moskvina V, et al. Schizophrenia susceptibility alleles are enriched for alleles that affect gene expression in adult human brain. Mol Psychiatry. 2012;17:193–201. doi: 10.1038/mp.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolae DL, Gamazon E, Zhang W, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fehrmann RS, Jansen RC, Veldink JH, et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16:197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- 13.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–43. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powell JE, Henders AK, McRae AF, et al. Congruence of additive and non-additive effects on gene expression estimated from pedigree and SNP data. PLoS Genet. 2013;9:e1003502. doi: 10.1371/journal.pgen.1003502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright FA, Sullivan PF, Brooks AI, et al. Heritability and genomics of gene expression in peripheral blood. Nat Genet. 2014;46:430–7. doi: 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirsten H, Al-Hasani H, Holdt L, et al. Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding locidagger. Hum Mol Genet. 2015;24:4746–63. doi: 10.1093/hmg/ddv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 18.Cheung VG, Nayak RR, Wang IX, et al. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol. 2010;8:pii: e1000480.. doi: 10.1371/journal.pbio.1000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–7. doi: 10.1038/nature05916. [DOI] [PubMed] [Google Scholar]
- 20.Banovich NE, Lan X, McVicker G, et al. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 2014;10:e1004663. doi: 10.1371/journal.pgen.1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Degner JF, Pai AA, Pique-Regi R, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–4. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pai AA, Cain CE, Mizrahi-Man O, et al. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 2012;8:e1003000. doi: 10.1371/journal.pgen.1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westra HJ, Franke L. From genome to function by studying eQTLs. Biochim Et Biophys Acta. 2014;1842:1896–902. doi: 10.1016/j.bbadis.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Grundberg E, Small KS, Hedman AK, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–9. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smirnov DA, Morley M, Shin E, et al. Genetic analysis of radiation-induced changes in human gene expression. Nature. 2009;459:587–91. doi: 10.1038/nature07940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–8. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 27.Ding J, Gudjonsson JE, Liang L, et al. Gene expression in skin and lymphoblastoid cells: refined statistical method reveals extensive overlap in cis-eQTL signals. Am J Hum Genet. 2010;87:779–89. doi: 10.1016/j.ajhg.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International Consortium for Blood Pressure Genome-Wide Association S. Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–81.. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powell JE, Henders AK, McRae AF, et al. The Brisbane Systems Genetics Study: genetical genomics meets complex trait genetics. PLoS One. 2012;7:e35430. doi: 10.1371/journal.pone.0035430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lloyd-Jones LR, Holloway A, McRae A, et al. The genetic architecture of gene expression in peripheral blood. Am J Hum Genet. 2017;100:371. doi: 10.1016/j.ajhg.2017.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karolchik D, Barber GP, Casper j, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:764–770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence M, Huber W, Pages H, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierce BL, Tong L, Chen LS, et al. Mediation analysis demonstrates that trans-eQTLs are often explained by cis-mediation: a genome-wide analysis among 1,800 South Asians. PLoS Genet. 2014;10:e1004818. doi: 10.1371/journal.pgen.1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raychaudhuri S, Sandor C, Stahl EA, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291–6. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeller T, Wild P, Szymczak S, et al. Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PLoS ONE. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanna S, Jackson AU, Nagaraja R, et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet. 2008;40:198–203. doi: 10.1038/ng.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi D, Sun W, Xu X, et al. A replication study for the association of rs726252 in PAPPA2 with developmental dysplasia of the hip in Chinese Han population. Biomed Res Int. 2014;2014:979520. doi: 10.1155/2014/979520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobsen S, Sonne-Holm S. Hip dysplasia: a significant risk factor for the development of hip osteoarthritis. A cross-sectional survey. Rheumatology. 2005;44:211–8. doi: 10.1093/rheumatology/keh436. [DOI] [PubMed] [Google Scholar]
- 41.Dimas AS, Deutsch S, Stranger BE, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–50. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powell JE, Henders AK, McRae AF, et al. Genetic control of gene expression in whole blood and lymphoblastoid cell lines is largely independent. Genome Res. 2012;22:456–66. doi: 10.1101/gr.126540.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.