

Abstract
During evolution, cells have developed a wide spectrum of stress response modules to ensure homeostasis. The genome and proteome damage response pathways constitute the pillars of this interwoven ‘defensive’ network. Consequently, the deregulation of these pathways correlates with ageing and various pathophysiological states, including cancer. In the present review, we highlight: (1) the structure of the genome and proteome damage response pathways; (2) their functional crosstalk; and (3) the conditions under which they predispose to cancer. Within this context, we emphasize the role of oncogene‐induced DNA damage as a driving force that shapes the cellular landscape for the emergence of the various hallmarks of cancer. We also discuss potential means to exploit key cancer‐related alterations of the genome and proteome damage response pathways in order to develop novel efficient therapeutic modalities. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: homeostasis, stress response, cancer, DNA damage response, proteome damage response, oncogenes, tumor suppressors, replication stress
Introduction: homeostasis and stress response
The human body is continually exposed to a variety of stressors, a generic/collective term describing exogenous and/or endogenous noxious events/factors that disrupt the steady state of the cell 1. The cell's tendency to resist changes in order to maintain the status quo is called homeostasis [derived from the Greek words homeo (same) and histimi (standing up)] 2. From a thermodynamic point of view, living organisms are open systems that are constantly pushed away from their equilibrium. This measure of disorder or ataxia is termed entropy, and represents ‘the general trend of the universe towards death and disorder’, as stated by Newman 3. When a stressor ‘enters the scene’, this delicate balance between order and disorder tilts towards pathophysiological states, if not dealt with efficiently (Figure 1A). To preserve homeostasis and counteract such challenges, cells have developed the capacity to mount a multiplicity of stress responses (SRs) 4. Remarkably, reconstruction of the phylogeny of SRs makes it apparent that many of the key players participating in SRs are not only evolutionarily conserved, but still serve related functions.
Figure 1.
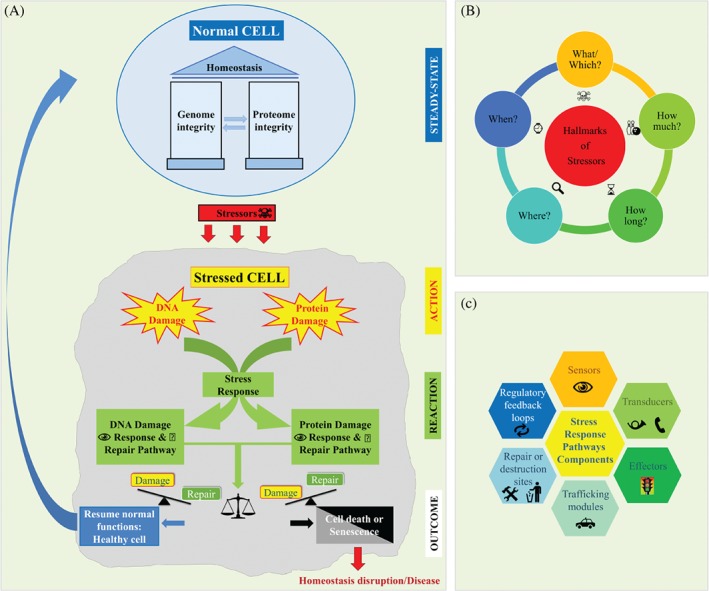
Overview of homeostatic mechanisms. (A) DDRs and PDRs. (B) Hallmarks of stressors. (C) SRP components. See supplementary material for a detailed legend.
Both the nature and the end result of a SR are determined by the type (what/which), magnitude (how much) and duration (how long) of the deleterious stimulus. Additionally, the spatiotemporal parameters of where (tissue organ/cellular/subcellular site) and when (young, old, or stage of a pathophysiogical process – early or late) the stressogenic event takes place constitute the parameters that affect the final outcome in a particular cellular context (Figure 1B). Although all biomolecules are susceptible to damage, dysfunction of the genome and protein machineries will mainly affect the cell's fate. Highly sophisticated and complex protein networks curate and maintain the integrity of the genome and proteome, comprising the so‐called DNA damage response (DDR) and protein damage response (PDR) pathways 5, 6. These damage/stress response pathways (SRPs) are hierarchically constructed and composed of sensors, signalling cascades (transducers and effectors), trafficking and repair modules, or destruction sites (Figure 1C). Each step/process is regulated by regulatory feedback loops that ensure an efficient and fine‐tuned response, which is gradually turned off once the damage is repaired, or proceeds to cell death if the damage is irreversible. However, things are not always so clear. Depending on the SR parameters (Figure 1B), there are variations in the way in which the cell orchestrates its reaction (supplementary material, Figure S1). For instance, a high level of damage in a low bioenergetic state (deprivation of ATP resources) will trigger necrosis instead of apoptosis, which is energy‐based 7. Necrosis predisposes to inflammation, whereas apoptosis is a ‘benign and inflammation‐free’ process 8. Then again, moderate DNA damage persisting because of repair difficulties, in an energy‐proficient environment, can trigger a prolonged cell cycle arrest called senescence* (see the Glossary in the supplementary material for further information on items marked with an asterisk). Senescence has a bright side and a dark side; it can operate as a tumor barrier, but can also foster a protumorigenic environment by resulting in the secretion of a broad spectrum of growth factors and cytokines (SASP*) 9, 10. As senescent cells are resistant to apoptosis, clearance by the immune system will neutralize long‐term harmful effects induced by SASP 11. Concurrently, as energy supplies are consumed, autophagy* is induced to secure cellular fitness, and restore metabolism and homeostasis 12, 13. In contrast, low levels (how much) of persistent (how long) DNA damage may have a long‐term beneficial effect, termed hormesis 14, recalling Nietzsche's quote ‘That which does not kill us, makes us stronger.’
Moreover, RNAs have an active role in SRs. They are considered to be the ancestral life‐encoding molecules 15, and are heterogeneous and versatile molecules that regulate many cellular processes 16. They include long and short RNAs, as well as coding and non‐coding RNAs, with the last group being in the majority 17. RNAs are critical in the genome reaction to SRs. They are involved in development and normal cell physiology, as well as in disease states, including cancer (supplementary material, Table S1), and other SRs 16. From the pathologist's point of view, the cellular reaction to SRs at the RNA level is illustrated by the generation of stress granules, which are dense cytosolic aggregations lacking a membrane, composed of RNAs and proteins 18. These granules store and protect RNAs in stressed cells, and are mainly identified by use of the TIA‐1* and G3BP1* factors 19. Detailed information on this field is provided in other excellent reviews (see supplementary material, Table S1).
Notably, in metazoans, such as humans, the response to stress is not limited to the cellular (local) level, but, importantly, is also elicited intercellularly/systematically (global). This intercellular response is orchestrated mainly via the immune system. It is a paracrine and circulation‐based response that attracts the diverse immune cell types at the sites of ‘damage’ in an attempt to ‘repair and clear it’. The immune cells execute specific and highly contextualized transcriptional programmes promoting immunity, while maintaining homeostasis. Remarkably, >1600 genes are involved in innate and adaptive immune responses 20, 21. The immune system is a constantly evolving functional entity via mechanisms of adaptation, expansion, maturation, and, ultimately, immunological memory, and represents the cornerstone of stewardship at the organismal level 22, 23, 24.
Consequently, the outcome of this ‘stressor versus SR confrontation’ will be determined by the cellular, microenvironmental and organismal context, leading either to healing or to various pathophysiological conditions. In most pathological situations, quantitative and/or qualitative defects in the DDR and PDR pathways are almost always observed (supplementary material, Figure S1). In the case of DDR, the significance of such alterations is underscored by the wide spectrum of clinical manifestations that occur when its components are defective (supplementary material, Table S2). Among the most unfavourable consequences of DDR and PDR deregulation is predisposition to cancer. Cancer is a complex, heterogeneous disease with an adverse prognosis if not diagnosed early. It evolves in a multistep process, and each phenotypic stage reflects a particular set of molecular alterations that provides a selective advantage to the tumor cells carrying them. Depending on cancer type, diverse molecular defects may occur, although key events, such as p53 mutations or pRb pathway inactivation, are common among malignancies 25, 26. Deciphering these molecular dynamics will help us to better understand the trails of carcinogenesis, and will aid in the development of efficient therapeutic strategies. Therefore, mutational signatures* identified by analysing the genomes of various tumors represent the ‘DNA repair traits’, and can help to unveil the repair routes engaged during carcinogenesis, thus suggesting potential therapeutic targets 27, 28. These ‘DNA repair traits’ form part of a broader process termed genomic instability.
DDR–PDR: an integrated genome–proteome maintenance network
GI* is a feature that nearly all tumors share, and is considered to be a hallmark of cancer 29. By the term GI, we refer to the high frequency of genetic alterations within the genome of a cellular lineage. Genetic alterations can range from single‐nucleotide substitutions (SNSs) (point mutations) to more complex quantitative and/or qualitative changes such as chromosome losses, gains, and/or rearrangements. GI can result from malfunctions at various steps of the DNA cycle, from replication to chromosome segregation 30, 31. The corollary of GI is that the transcriptome and proteome landscapes will progressively change, affecting the functionality of the cell. To comprehend the nature of this abnormal, albeit dynamic, process, we need to go through the essential operational characteristics of the DDR and PDR pathways and how they are physiologically wired and rewired during cancer development (Figures 2, 3, 4).
Figure 2.
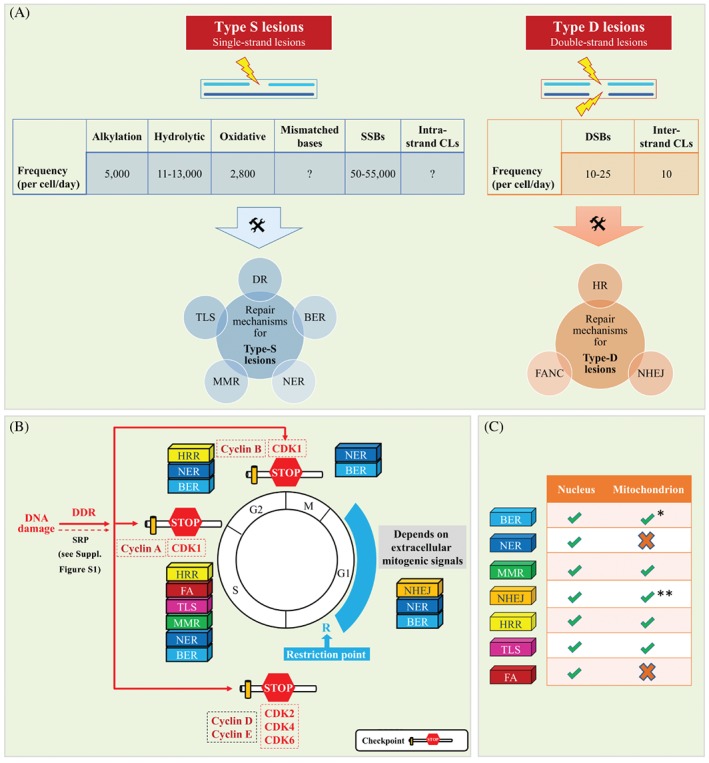
Synopsis of DNA damage type frequency and repair (A), DDR signalling cascades that activate the various checkpoints (B), and DNA repair mechanisms (C). See supplementary material for a detailed legend.
Figure 3.
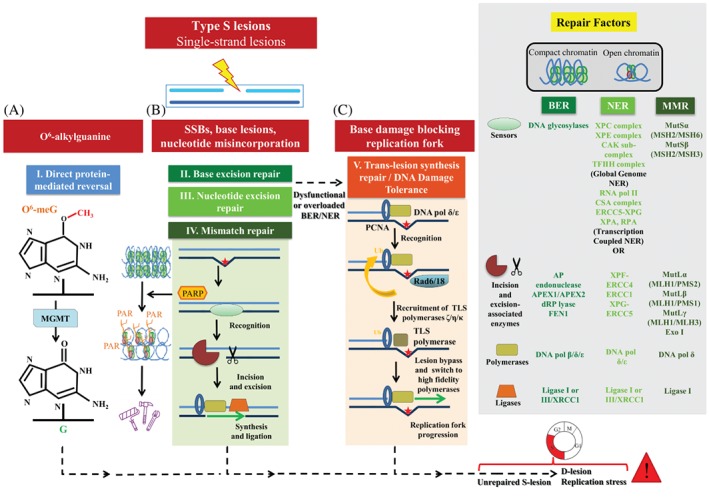
Repair routes for category‐S lesions (DDR surveillance). (A) Direct protein‐mediated reversal. (B) BER and NER. (C) TLS repair. See supplementary material for a detailed legend.
Figure 4.
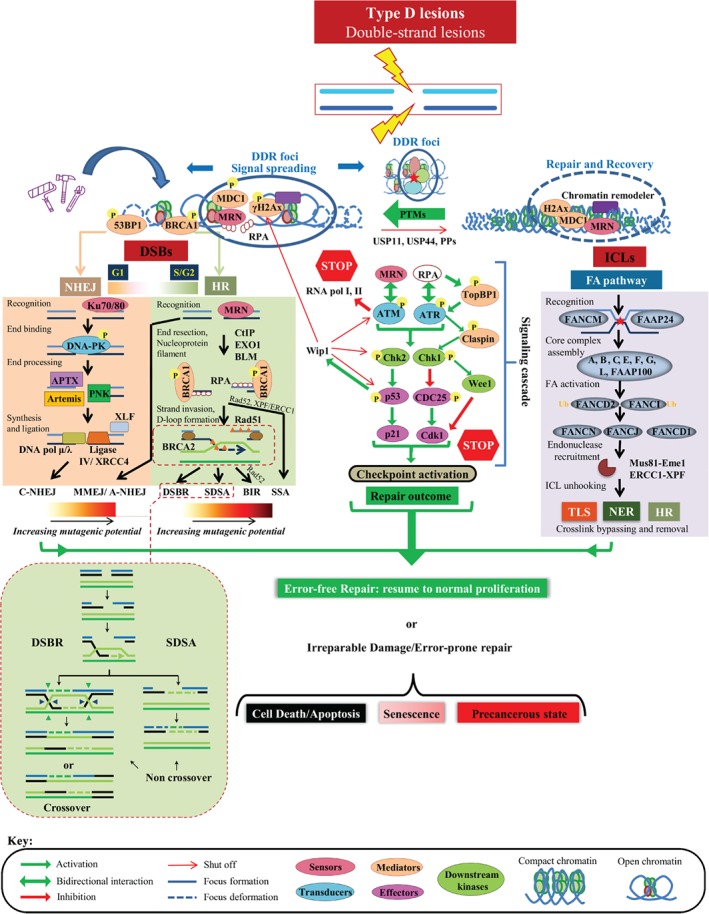
Repairing category‐D lesions (DDR surveillance). See supplementary material for a detailed legend.
Overview of the DNA damage response and repair (DDR/R) network
As DNA is the repository of genetic information, the ultimate goal of the DDR/R network is to preserve its integrity. However, because of the large number of DNA lesions induced in a cell every day, this task is not always achieved without cost (Figure 2) 32, 33. DNA lesions can be divided in two broad categories: those occurring on one strand of the double helix (category‐S lesions), such as modified bases, abasic sites, helix‐distorting base lesions, and single‐strand breaks; and those involving both strands [category‐D lesions: interstrand crosslinks and double‐strand breaks (DSBs)]. In certain cases, such as exposure to ionizing radiation, category‐S and/or category‐D lesions coexist, and, when they are in close vicinity, they are termed clustered DNA lesions* 34. As shown in Figure 2, the type of DNA lesion and the cell‐cycle phase largely dictate the repair programme to be engaged. Whereas some types of damage, such as O 6‐methylguanine, are subjected to direct protein‐mediated reversal (Figure 3A), most are repaired by a series of catalytic events entailing multiple proteins and generally including two steps: (1) damage recognition by sensors; and (2) processing and repair of the lesion (Figures 3B and 4). Depending on which damage category is to be repaired, these steps encompass unique characteristics.
Repairing category‐S lesions
For category‐S lesions, subsequent to recognition, the following are required for processing and repair: (1) incisions flanking the damage; (2) excision of the damaged area; (3) filling of the gap by nucleotide polymerization; and (4) ‘sealing’ the gap with ligation (Figure 3). More specifically, the high‐fidelity (error‐free) pathways base excision repair (BER) 35, 36 and nucleotide excision repair (NER) 37 deal with single‐base DNA defects and helix‐disorting base lesions, respectively, whereas repair of nucleotide misincorporation is mediated by mismatch repair (MMR) (Figure 3B) 38. In the case of NER, the global release of RNAPII waves from promoter proximal pausing sites maximizes sensing and accelerates the repair of category‐S lesions equally well in genes with low and high expression via transcription‐coupled (TC) NER, guaranteeing unbiased transcriptome surveillance 39. However, if BER and NER malfunction or are overloaded by ‘fixing’ demands, then the translesion synthesis (TLS) pathway, which is a low‐fidelity repair module (error‐prone) pathway known as DNA‐damage tolerance (DDT), takes over (Figure 3C) 40. To avoid replication of damaged DNA that could lead to fork collapse, DSB production, and genome destabilization, cells opt to recruit TLS/DDT to bypass encountered lesions and repair them at a later time 40. Thus, TLS/DDT is considered to be responsible for the majority of mutagenic events, playing a central role in carcinogenesis. Although the latter is an undesired event, from a broader perspective it is a ‘cost’ that the cell has to pay to avoid DSBs, thus preserving double helix continuity 41, 42. In line with this, inhibition of factors involved in category‐S defect repair processes has the potential to induce DSB formation during replication, triggering RS* and death if the cell is also deficient in components implicated in DSB repair (Figure 3). One of the best examples supporting this scenario is the enzyme poly(ADP‐ribose) polymerase (PARP) 43. PARP is a vital repair protein involved in single‐strand break (SSB) repair and BER. PARP catalyses the synthesis of negatively charged poly(ADP‐ribose) chains by utilizing the respiratory coenzyme NAD+, with release of nicotinamide. The negative charge of the ADP‐ribose polymers around SSBs repels the positively charged histones of the nucleosomes, opening chromatin and thus allowing access to the repair machinery 8. Targeting of PARP has been shown to kill cancer cells deficient in the homologous recombination factors breast cancer susceptibility gene 1 (BRCA1) and breast cancer susceptibility gene 2 (BRCA2) (see below), paving the way for the use of PARP inhibitors in clinical practice 44, 45.
Repairing category‐D lesions
DSBs are category‐D defects, and are considered to be the most deleterious types of DNA damage. From their frequency (Figure 2), it is apparent that the cell cannot tolerate them, as repair of a single DSB requires >105 ATP molecules 46. Such an energy investment calls for a staggering level of cellular reorganization once DSBs occur. With the exception of immune receptor diversity [V(D)J and class switch (CS) recombination] and chromosomal crossover during meiosis II of gamete production, in which DSBs form part of physiological programmes 47, 48, the cellular reaction to DSBs epitomizes an integrated cellular SR to ‘imminent danger’. Once a DSB is formed, a cascade of biochemical events take place within minutes in an effort to efficiently ‘access, repair, and restore’ 49, 50. These biochemical processes are characterized by extensive PTMs* of the involved DDR proteins and chromatin structure of the damaged area; these processes: (1) are much faster chemical reactions than transcription; and (2) form docking sites for the recruitment of repair factors 51.
Two classes of DDR proteins are recruited at damaged sites: those that present directly at DSBs (called sensors* and mediators*), and those associated with the DSB‐flanking chromatin, altogether constituting so‐called DDR foci (Figure 4) 52. Over time, the DDR foci spread away from the DSB to distances up to megabases in mammals, forming an amplification mechanism recruiting signal transduction factors that further amplify the signal with effectors that set the cell in an ‘alarm’ state (Figure 4; DDR cascade). This mechanism has, on the one hand, a local effect by relaxing the chromatin and increasing the concentration of repair factors at the damaged site, and, on the other hand, a systemic effect, termed checkpoint activation*, that reduces the activity of CDKs* (Figure 2B) 5. Notably, in certain cases and depending on the cellular context, checkpoint activation, apart from the DDR signalling cascade, also involves the parallel action of other SR signalling routes (Figure 2), like the p38 mitogen‐activated protein kinase (p38MAPK) pathway, which coordinates several cellular functions 53. The endpoint of the SR signalling cascade is always the cyclin–CDK complex* (Figure 2B). The cyclin–CDK complexes represent drivers of the cell cycle and, when they are suppressed, the cell enters a state of arrest, providing time for repair.
One of the earliest features that mark these DDR foci is histone variant H2AX phosphorylation at serine 139, (also referred to as γH2AX), by ataxia telangiectasia mutated (ATM) backed up by ataxia telangiectasia and Rad3‐related (ATR) and DNA‐dependent protein kinase, catalytic subunit (DNA‐PK); all three kinases are members of the phosphatidylinositol 3‐kinase (PI3K) family and key DDR signalling components (transducers) 51, 54, 55, 56, 57. Subsequently, the DNA damage mediator called mediator of DNA‐damage checkpoint 1 (MDC1) attaches to γH2AX, acting as a platform for the meiotic recombination 11 (MRE11)–Rad50–Nijmegen breakage syndrome 1 (NBS1) (MRN) sensor complex that activates ATM, thus forming an amplification loop 58, 59. Concurrently, MRN complexes bind the DSB avidly, playing a pivotal role in the initial processing of the break, generating single‐strand (ssDNA) DNA 3′‐overhangs that are recognized by replication protein A (RPA). This event brings into play the ATR transducer kinase, which, in cooperation with ATM, turns on the downstream transducer* kinases checkpoint kinase 1 (Chk1) and checkpoint kinase 2 (Chk2) (Figure 4) 60. In concert, these kinases activate a key effector* of the DDR pathway, namely p53 61, 62. p53 is a transcription factor that governs a complex SR programme covering a bewildering range of biological functions, explaining why p53 is frequently mutated in cancer 63. Activation of p53, mainly via PTMs, induces the expression of numerous downstream effectors, including the universal CDK inhibitor p21WAF1/Cip1, leading to cell‐cycle arrest. Concurrently, ATM imposes a transcriptional silencing programme by shutting down both RNA polymerase II* and RNA polymerase I*, thus saving the energy that transcription demands and preventing collision between transcription and repair (analysed below in ‘Malfunction of endogenous operations as a cause of DNA damage’) 64, 65, 66. In parallel with this, the cell gains time and reshuffles energy resources for the repair machinery to ‘seal’ the DSBs, or it otherwise proceeds to apoptosis or senescence, depending on the extent of damage (Figure 4; supplementary material, Figure S1). Concomitantly with these global effects, repair is facilitated by extensive chromatin modifications and remodelling* at the site of the DSB 67. In brief, SWI/SNF‐dependent histone H2A.Z exchange for histone H2A destabilizes the nucleosomes surrounding the DSB. This nucleosome remodelling event exposes the N‐terminal tail of histone H4, which, in turn, is acetylated by TIP60 histone acetyltransferase, further relaxing chromatin and enabling access to downstream repair factors 68.
The actual repair of the DSB lesion is carried out by homologous recombination repair (HRR) and non‐homologous end joining (NHEJ) (Figure 4; supplementary material, Figure S1) 69, 70. HRR is considered to be an error‐free repair system occurring during the S and G2 cell‐cycle phases, whereas NHEJ is an error‐prone repair pathway dealing mainly with non‐replication‐associated DSBs, and represents the predominant repair route that functions irrespective of the cell cycle (Figure 2). HRR is initiated by the binding of BRCA1 to the ubiquitin chain added by the E3 ligases ring finger protein 8 (RNF8) and ring finger protein 168 (RNF168) to the remodelled nucleosome 49, 71, 72, 73. In this way the BASC* connects sensing and signalling with the repair component BRCA2, which controls the Rad51 recombinase that replaces RPA. The BRCA2–Rad51 complex then invades the homologous template and primes DNA synthesis, copying and restoring the genetic information 74, 75. When the homologue donor strand is the sister chromatid, HRR is accurate. However, recombination may take place across different genome regions, challenging previous notions concerning the error‐free nature of HRR (Figure 5) 76. Hence, to secure sealing of DSBs, various routes of HRR exist that may favour inappropriate pairing. Alternatively, three critical histone modifications, namely histone H4 lysine 20 dimethylation (H4K20me2) [catalysed sequentially by methyltransferases SU(VAR)3‐9H1 and SETD8], ubiquitylation of histone H2A on lysine 15 (H2AK15ub) (induced by the E3 ligase RNF168), and histone H3 lysine 79 methylation (H3K79me), are recognized by the signalling mediator p53‐binding protein 1 (53BP1) at DSBs, promoting NHEJ 77, 78, 79, 80, 81, 82. Importantly, 53BP1 accumulation antagonizes BRCA1‐mediated HRR in favour of NHEJ (Figure 4) 83, 84, 85. In NHEJ, the DSB is sensed by the lupus Ku autoantigen protein p80 (Ku80)–lupus Ku autoantigen protein p70 (Ku70) heterodimer, which recruits and assembles the DNA–PK complex, which, in turn, processes the DNA ends and increases the recruitment of ligase IV/X‐ray repair complementing defective repair in Chinese hamster cells 4 (XRCC4), which carries out the rejoining reaction 74. Although HRR is the favoured pathway to deal with a DSB, ensuring DNA sequence fidelity, in the event that HRR is non‐functional the cell ‘prefers’ to follow inappropriate repair routes to secure cell viability 86. In this case, the faster kinetics of the Ku heterodimer for DSBs compared to those of the HRR factors 87 make the error‐prone NHEJ (the repair pathway of choice) operate even during S phase, with potential unfavourable effects for the cell 86, 88, 89, 90.
Figure 5.
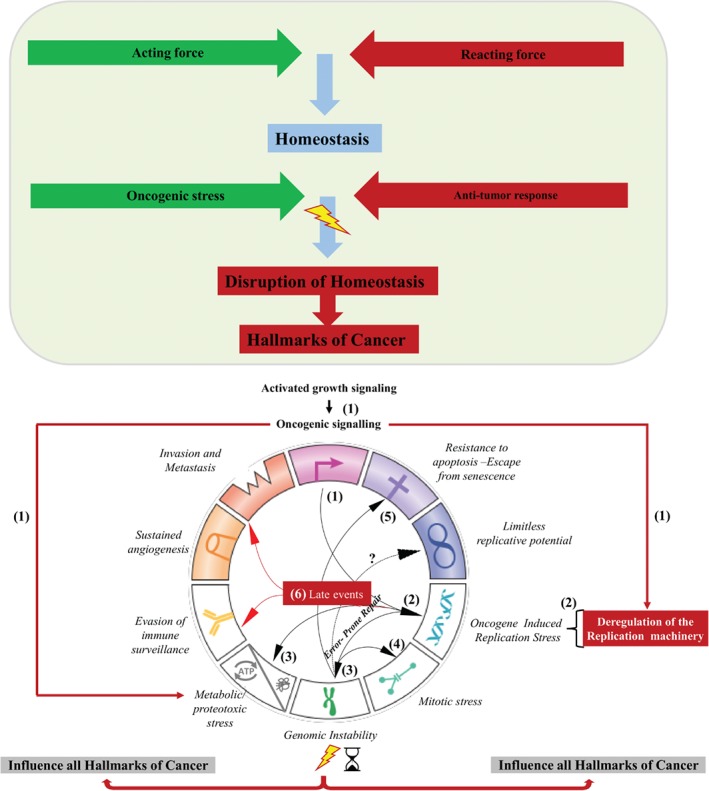
A model depicting how oncogene‐induced RS aids the progressive formation of certain hallmarks of cancer (early events: steps 1–5), while paving the way for angiogenesis, evasion from immune surveillance, invasion, and metastasis (late events: step 6). See supplementary material for a detailed legend.
The second type of category‐D defects comprises DNA interstrand crosslinks (ICLs) that are generated by a class of agents such as mitomycin C (MMC) and diepoxybutane (DEB), or circulating metabolites such as formaldehyde. ICLs are toxic, because the covalent links that they form prevent DNA from unwinding, thereby blocking replication and transcription, causing replication and transcription stress, respectively. These lesions are fixed by the Fanconi anaemia (FA) pathway, which is a replication‐dependent repair mechanism that appeared relatively late in evolution (Figure 4) 91. It is considered to be the most sophisticated repair route, enlisting modules of three classic repair pathways, i.e. TLS, NER, and HRR. The central components of the FA pathway are the 13 complementation groups identified so far, i.e. Fanconi anaemia complementation group (FANC) A, FANCB, FANCC, and FANCD1 (better known as BRCA2), FANCD2, FANCE, FANCF, FANCG, FAQNCI, and FANCJ [BRCA1‐interacting protein C‐terminal helicase 1 (BRIP1) or BTB domain and CNC homologue 1 (BACH1)], and FANCL, FANCM, and FANCN [also termed partner and localizer of BRCA2 (PALB2)], which are mutated with various frequencies in FA* 92. FANCA–FANCG, FANCC–FANCE–FANCF and FANCB–FANCL–FAAP100 form the core complex that is recruited at the damage site by FANCM and FA‐associated protein of 24 kDa (FAAP24), which sense the stalled replication fork. The FA core complex* possesses ubiquitin ligase properties; it mono‐ubiquitinates FANCD2 and FANCI, which is regarded as the essential step for FA activation, in analogy to proliferating cell nuclear antigen (PCNA) ubiquitination‐dependent recruitment of TLS polymerases. Subsequently, DNA repair is organized by engaging ubiquitinated FANCD2 and FANCI with the downstream factors FANCD1 (BRCA2), the helicase FANCJ (BRIP1 or BACH1) and BRCA2 (FANCD1)‐interacting partner FANCN, also termed PALB2 93. The subsequent repair steps require coordinated and sequential activity of TLS, NER and HR enzymes. Once the replication fork moving from both directions stalls at the ICL, a dual incision by endonucleases methyl methanesulphonate and ultraviolet (UV)‐sensitive clone 81 (MUS81) and essential meiotic structure‐specific endonuclease 1 (EME1) at the 3′‐end of the lesion, and excision repair cross‐complementation group 1 (ERCC1) (XPF) at the 5′‐end unhooks the ICL, which is then bypassed by a TLS polymerase, probably Rev1 (polymerase ζ). NER removes the bypassed crosslink and HRR repairs the broken chromatid by using the newly repaired by TLS sister as a template (Figure 4) 37, 94.
Shutting off the DDR pathway
In a normal setting, DDR/R activation is coupled with inactivation to allow a cell to complete its cycle. Feedback control of DDR fine‐tunes the magnitude and duration of the response, limiting aberrant DNA repair. Turning off DDR/R is a more arcane process than it was initially considered to be. Timely termination of DDR is controlled in a spatiotemporal manner, by different mechanisms acting in an overlapping manner 95. As activation of DDR/R signalling encompasses a series of PTMs, shutting it off calls for the reverse procedure. To date, several protein phosphatases (PPs) and deubiquitinating enzymes (DUBs) have been recognized as critical players in DDR/R termination (Figure 4). PP2A is among the best studied PPs; it catalyses dephosphorylation of γH2AX 96. PP2 catalytic subunit‐α (PP2A) inhibition results in persistent γH2AX foci, compromising DDR/R and rendering cells hypersensitive to DNA damage. Another DDR/R regulator is Wip1, which is involved in the dephosphorylation of multiple DDR components 97, 98. Notably, Wip1 is a transcriptional target of p53, and is stimulated after genotoxic stress 99. Activation of Wip1 100 facilitates p53 degradation by murine double minute 2 (MDM2), forming a negative feedback loop that allows termination of DDR. Conversely, overexpression of Wip1 has an oncogenic effect, signifying the importance of fine‐tuning for proper DDR/R operation (Figure 4) 98, 101. Recruitment of ubiquitin‐specific peptidase (USP) 44 and USP11 at DSBs antagonizing RNF8/RNF168 mono‐ubiquitination of histone H2A 102, 103 represents an example of how DUBs regulate DDR/R. Likewise, the activity of the deubiquitinating complex USP1–UAF1 over FANCD2 keeps the FA pathway in check 104, 105.
An additional way to terminate the activity of phosphorylated checkpoint proteins is through proteolytic degradation 106. Phosphorylation marks targets for proteolysis via the ubiquitin–proteasome (UPP) pathway, pinpointing the interdependence of the proteostasis and DDR/R networks 107. This mechanism is exemplified by SCFβTrcP, which acts as a switch between checkpoint initiation and recovery 6. Upon DNA damage, phosphorylated CDC25A is recognized by SCFβTrcP, promoting G2 arrest, whereas, when the DNA damage is repaired, the same complex mediates claspin* and Wee1* degradation, favouring checkpoint recovery and progression to mitosis.
Malfunction of endogenous operations as a cause of DNA damage
Further to various exogenous mutagenic agents (clastogens*), deregulation of endogenous processes can create a genetic landscape prone to DSBs, adding an extra level of pathophysiological complexity. As with exogenous stimuli, a key aspect related to endogenous operations is keeping CDK activity in check. In mammals, several CDK–cyclin complexes are essential for normal proliferation 108. The significance of these complexes is underscored by the fact that aberrant CDK activity is a universal feature of human tumors. Deranged function of cyclin–CDK complexes 108 is counteracted by checkpoint activation, which is aimed at preserving genome integrity (Figure 2) 5, 109. Distinct checkpoints halt the cell cycle at specific phases and activate the DDR/R pathway, allowing time for DNA repair completion before entry into mitosis (Figure 4) 6. If there is extensive DNA damage, and depending on its severity, checkpoints activate apoptosis or senescence instead of cell‐cycle resumption. Thus, aberrant CDK activity exerts selective pressure on the checkpoints, eventually breaching them, allowing DNA damage accumulation and the emergence of GI with pathological consequences 29, 109, making CDKs attractive targets for inhibition 108. As another example, RT* is a highly regulated programme giving rise to early, middle and late time zones, and is strictly coordinated with transcription to avoid replication–transcription collision 110, 111. If this spatiotemporal process is altered by misregulation of an RT‐related gene 32 or a replication process step, e.g. replication licensing* 112, 113, the propensity to DSB formation due to replication–transcription collision increases 114, 115, 116. In this regard, low‐density replication–initiation may lead to unfinished replication, owing to the long distance between replication forks, making breakage a probable event 117. Common fragile sites (CFSs), which are late‐replicating areas of the genome with a paucity of replication origins, are particularly vulnerable to RS, and are at high risk of breaking 118. Conversely, ill‐timed replication–initiation can cause re‐replication, a form of RS that leads to DSBs and GI, and, as discussed later, is frequently observed in cancer from its earliest stages. Overproduction of replication and/or transcription intermediates that are normally formed in low amounts can also pose a threat to genome integrity (supplementary material, Figure S3A,B) 119, 120. A particular DNA:RNA hybrid termed the R‐loop* (supplementary material, Figure S3B), identified 42 years ago 121, has drawn attention as a source of GI. R‐loops usually have short lifespans (∼20 min), as they are efficiently removed under normal circumstances, and play an important role in various processes such as immunoglobulin class switches, and transcriptional regulation and termination 122. However, production of R‐loops resulting from various defects, such as lack of RBPs*, which coat and protect nascent RNA from illicit DNA hybridization 123, make these structures ‘hotspots’ for damage 124. The thermodynamically stable DNA:RNA hybrid structure of the R‐loop can impede proper completion of replication, leading to replication fork stalling and collapse*, and DSB formation, whereas the relatively unstable non‐template ssDNA of the R‐loop can act as a substrate for deaminases. Another endogenous cause of DNA damage is an inability of nucleotide biogenesis to cope with hyperproliferative states, resulting in replication fork deceleration, which can lead to fork stalling and, if not reversed, to fork collapse 125. Exogenous supplementation with nucleosides reverses these adverse effects 126.
A delicate operation that the cell has to carry out following stalling or collapse replication forks is repair and replication restart (supplementary material, Figure S3C) 127. Depending on the duration (how long) of the replication block, there are two options. (1) In short replication blocks (2–4 h), restart is mediated by fork remodelling of replication intermediate structures (fork reversal: ‘chicken foot’). Replication fork reversal is a mechanism in which a three‐way junction at a replication fork is converted to a four‐way junction by the annealing of the two newly replicated strands into a regressed arm at the forks 128. Regressed fork restart requires restoration of the typical replication fork structure, and involves helicase (RECQ‐1*, WRN*, BLM* and DNA2 nuclease*) 128, 129 and translocase (SMARCAL1*, ZRANB3* and HLTF*) activity in the resolution of these intermediates 129. Whether these helicases recognize different structures or act on the same substrates is a matter of investigation 130, 131, 132. RAD51 recombinase has been described as an essential factor in replication fork restart 127, as RAD51‐mediated strand invasion rapidly and effectively leads to replication fork remodelling 133. As the regressed arm of a reversed replication fork resembles a one‐ended DSB, it has to be protected from cleavage. RAD51‐coated nucleofilaments protect nascent DNA from MRE11*‐mediated nucleolytic attack 134, 135. BRCA2 has been shown to be dispensable for RAD51‐mediated fork reversal 136, but is crucial for the assembly of stable protective RAD51 nucleofilaments on regressed arms 134, 135, 137, 138. MRE11‐mediated degradation has recently been shown to be one of the leading causes of sensitivity to DNA‐damaging agents in BRCA‐deficient cancers 139. Recent studies have shed light on MRE11 recruitment at regressed forks by highlighting the role of PTIP*, MLL4*, and RAD52* 136, 139. Recently, a combination of electron microscopy with DNA fibre analysis further defined the events in reversed fork resection in BRCA‐deficient tumors 140. It was shown that CtIP* initiates MRE‐mediated degradation, which is then extended by EXO1* 140. How cells cope with extensive resection at the forks and how different remodellers collaborate to catalyse fork reversal is still poorly understood. (2) In long replication blocks (>24 h) resulting in fork collapse, new origins are fired in an attempt to compensate, and repair takes place by remodelling, utilizing structure‐specific endonuclease complexes* 30, 115 that generate DSBs, promoting Rad51‐dependent homologous recombination (Figure 4). The significance of the above factors is underscored by the severity of the clinical manifestations presented in disorders such as Bloom* and Werner* syndromes, in which corresponding helicases are mutated 141, 142.
Chromatin structure, DNA damage, and DDR/R
Remarkably, susceptibility to DNA damage is not the same across the genome (where), as chromatin compaction seems to provide a ‘shield’ against DNA insults, with heterochromatin (compact chromatin) providing significant protection against breaks, but euchromatin (open chromatin), which is a relaxed and transcriptionally active conformation, being vulnerable to damage 143. A feature of heterochromatin is its high level of enrichment in repetitive DNA sequences* and its concentration in pericentromeric and telomeric genomic regions. Given that repetitive sequences represent recombination ‘hotspots’, and are thus prone to DSBs, heterochromatin formation at these sites has evolved as a protective mechanism preventing illegitimate recombination events 144. A specialized type of compact chromatin is present at telomeres* 145, 146. Telomeres resemble staggered DSBs, and, if not protected by the shelterin complex*, will be recognized as a damaged area, eliciting a DDR with detrimental effects (see ‘Monitoring mitosis: the last checkpoint’ below) 147, 148.
However, a disadvantage of compaction is that it is refractory to repair, as demonstrated by the inability to expand γH2AX 149. Studies in yeast and mammals showed fewer γH2AX foci at DSBs in heterochromatin than in euchromatin 150. When a break occurs in euchromatin, a complex of heterochromatin‐associated proteins*, including KRAB‐associated protein 1 (KAP1), heterochromatin protein 1 (HP1), and SU(VAR)3‐9H1, assist in trimethylation of H3K9, which is a repressive histone PTM. This reaction facilitates an amplification process that spreads over many kilobases from the damage 116. Activated TIP60 (see above) binds to H3K9me3, leading to ATM activation and H4 acetylation, promoting chromatin relaxation. In turn, ATM phosphorylates KAP1, releasing it from chromatin and enhancing access to the damaged site. It appears, that following DNA damage, there is a transient shift in euchromatin, like the ‘squeezing phase of an accordion’, to increase compaction to recruit TIP60 and ATM 68. Compaction can also function as a regulatory mechanism for uncontrolled DSB‐end resection and HR, as we recently showed, via retention of KAP1 and HP1 at the damaged site by the histone chaperone SET/TAF‐Ib/I2PP2A/INHAT 151. In contrast, KAP1 undergoes localized phosphorylation by ATM in a 53BP1‐dependent manner in heterochromatin, dispersing the nucleosome remodeller* CHD3.1, leading to focal relaxation, unlike the diffused relaxation observed in euchromatin 152, 153, 154. The difference in DDR initiation between euchromatin and heterochromatin influences the choice of repair, with transcriptionally active euchromatin regions preferring HRR, and compacted chromatin favouring NHEJ 143. Another repetitive area that is influenced by chromatin compaction, and that is of immense importance for genome and proteome integrity, is composed of the ribosomal DNA (rDNA) clusters, located in the nucleolus.
The nucleolus at the crossroad of stress response
The nucleolus, which is the largest structure in the nucleus, is responsible for rRNA production and ribosomal assembly, as reflected by its characteristic tripartite spatial organization* (supplementary material, Figure S4A) 155, 156. To cope with increased protein production, rDNA is organized into clusters of gene repeats around areas termed NORs* 157. Depending on the demands of protein synthesis, an additional regulation step occurs, which involves adjustment of the number of ‘active’ repeats and alteration of their transcriptional rates 158. RNA polymerase I transcribes rDNA into rRNA, forming the framework of the ribosome. rRNA constitutes 80% of the total cellular RNA, making rDNA the highest‐transcribed locus of the genome. Obviously, the propensity for replication–transcription collisions and R‐loop formation is increased in rDNA loci, especially under conditions of oncogene‐induced RS 159. As a ‘precautionary measure’, the RFB* (supplementary material, Figure S4A), which is an intergenic rDNA site, is recognized as a site coordinating replication and transcription 160, whereas heterochromatin associated with the rDNA clusters acts as a ‘buffer zone’ against genotoxic conditions 161, 162, 163. Regardless of cellular demands, not all rDNA sequences are transcriptionally active, and a fraction of nucleolar rDNA is always silent (supplementary material, Figure S4A). The stability of the repetitive and recombinogenic‐prone rDNA sequences requires chromatin silencing complexes 162, 164, 165. The fraction of ‘active’ versus ‘silent’ rDNA is regulated by the rDNA remodelling complexes NoRC*, eNoSC*, and NuRD*, which limit accessibility to the recombinogenic machinery 166, 167, 168, 169. Disruption of rDNA‐associated silencing proteins within the inner nuclear membrane* disturbs the nucleolus–nucleoplasm boundary, induces the formation of recombination foci, and destabilizes the repeats 170, 171. Finally, almost 70% of nucleolar proteins have functions unrelated to ribosome biogenesis, including triggering SR pathways. A characteristic example is p53 activation following nucleolar segregation (ribosomal stress*) upon DNA damage or transcription inhibition (supplementary material, Figure S4A) 172, 173, 174.
Surveillance and maintenance of mitochondrial genome integrity
Mitochondria are the ‘energy factories’ of cells, and they also regulate other vital functions, including apoptosis. The symbiotic relationship between mitochondria and eukaryotic cells started more than a billion years ago, leading to synchronized action between the two genomes, with most proteins involved in mitochondrial DNA (mtDNA) metabolism being encoded by nuclear DNA 175. Conversely, proteins participating in oxidative phosphorylation are encoded by mtDNA, making mtDNA integrity of paramount importance for cell homeostasis 176. mtDNA is more prone to damage than nuclear DNA, with a 10–20‐fold higher mutation rate, possibly because of proximity to reactive oxygen species (ROS) production (supplementary material, Figure S4B).
mtDNA is an approximately 16‐kb closed circular DNA containing a specific regulatory region, termed the D‐loop*, harbouring initiation sites for both replication and transcription. The mitochondrion‐specific polymerase‐γ (pol‐γ) holoenzyme is responsible for replication, with >200 mutations in POLG having been linked to mitochondrial diseases 177. In contrast to the nuclear genome, which is organized into nucleosomes, mtDNA lacks nucleosomes and is arranged as protein–DNA complexes known as nucleoids. Interestingly, super‐resolution microscopy has revealed that the mtDNA packaging density is higher than that of nuclear chromatin, forming a ‘shield’ against mutagenesis 178. Interestingly, Twinkle helicase is essential for mtDNA maintenance and a key regulator of mtDNA copy number by linking the mitochondrial replication machinery with the cytoplasmic dNTP pool 179. As mitochondria are the largest consumers of dNTPs in the cell, controlling mtDNA copy number is apparently essential for meeting cellular energy requirements 180.
Oxidative damage is a common mechanism of mitochondrial injury. Byproducts of oxidative phosphorylation are ROS, because, during the series of redox reactions, a small percentage of electrons leak directly to oxygen. Thus, 1–2% of the oxygen consumed within the cell is released from mitochondria as ROS. Under normal conditions, the amount of ROS produced is relatively low and essential for proper intracellular signalling, metabolism, and responses to pathogens; however, during periods of increased and/or prolonged ROS production, extensive and persistent mtDNA damage may occur (supplementary material, Figure S4B) 181. BER is the predominant DNA repair pathway for category‐S lesions in mitochondria, whereas they lack effective MMR and are deficient in NER. Adducts requiring NER for their removal will accumulate, resulting in mtDNA mutations and, ultimately, mtDNA degradation 182. Importantly, pol‐γ can bypass some lesions by TLS. Regarding TLS, primase‐polymerase (PrimPol), which is an archaic enzyme with dual primase and polymerase activities, identified in human mitochondria, has the unique feature of de novo DNA synthesis, and the ability to tolerate lesions such as 8‐oxoguanine (8‐oxoG) and apurinic/apyrimidinic or abasic (AP) sites 183. Along the same line, Pif1, a 5′–3′ DNA helicase that is essential for mtDNA replication 184, is involved in the repair of many types of mtDNA damage, including the unwinding of genotoxic G‐quadruplex DNA* 183. For category‐D lesions, the prevailing view is that both HR and NHEJ are active in mitochondria, although recent evidence suggests that NHEJ is replaced by microhomology‐mediated end‐joining (MMEJ), also known as alternative non‐homologous end joining (alt‐NHEJ) (Figure 4) 185.
Depending on the type and amount of damage, inefficient mtDNA repair activates either mitochondrial fusion* or fission* 186. Fusion rearranges the matrix content of a damaged mitochondrion with a healthy one; this event results in diluting the damage that relates e.g. to unfolded proteome or to mutated DNA. On the other hand, fission partitions damaged material to daughter organelles. If the above fail, mitophagy* or apoptosis will take place, determined by the extent of damage (supplementary material, Figure S4B) 187. Molecular players dictating the outcome include ATM, p53, and Sirt1* 188. Sirt1 is a master regulator inhibiting p53‐mediated apoptosis, and, by interacting with AMPK*, directs the SR to mitophagy. Concurrently, by deacetylating PGC1‐a*, it stimulates mitochondrial biogenesis to compensate for losses.
Monitoring mitosis: the last checkpoint
The cell's final level of surveillance occurs during mitosis, as its genetic material must be accurately and equally transferred to offspring. Mitosis is an orchestrated process leading to aneuploidy, CIN* or death if deregulated (supplementary material, Figure S5) 189, 190, 191. Owing to its complexity and short duration (∼1 h), multiple control checkpoints ensure the fidelity of genome inheritance 192, 193. Identified checkpoints occur: (1) between prophase and prometaphase, controlled by checkpoint with forkhead and ring finger domain (CHFR), which is implicated in sensing microtubule poisons 194, 195; (2) during metaphase, governed by the SAC* 196; and (3) in telophase, regulated by the cytokinesis or ‘abscission’ checkpoint* (AC), which is Aurora‐B*‐dependent (supplementary material, Figure S5) 197, 198, 199.
The SAC is the most important surveillance mechanism, monitoring kinetochore–microtubule attachment, and ensuring correct alignment of chromosome pairs before segregation 200, 201, 202. During metaphase, the unattached kinetochores recruit the SAC machinery*, which sequesters CDC20* to block activation of the APC/C* 203. Proper chromosome alignment results in SAC silencing, allowing activation of APC/C, which, in turn, targets securin* and cyclin‐B* for degradation 204. Destruction of securin releases separase*, which cleaves the cohesin rings*, promoting sister chromatid separation (supplementary material, Figure S5). The significance of SAC was demonstrated in mouse models showing that complete loss of these genes results in early embryonic lethality, whereas heterozygous and hypomorphic* mice are viable and fertile despite showing increased levels of aneuploidy. Although aneuploidy is evident in these mice, malignant transformation is a rare and late event 189, questioning whether aneuploidy as such is causative for cancer development 205. Differences in cancer incidence among individuals with identical aneuploidy, as well as between common genetic disorders such as trisomies 13 206, 18 207 and 21 208, suggest that additional hits are required for cancer to evolve. Moreover, extensive analysis in common sporadic cancers with aneuploidy demonstrated the low frequency of mutations in caretaker genes*, including SAC genes, arguing against their role in promoting cancer 29. In this regard, the role of centrosome* aberrations in aneuploidy and CIN 189, 209, 210 requires more clarification, especially in view of emerging concepts such as ‘centrosome inactivation checkpoint’ 211 that link components of the DDR/R machinery with centrosome ‘status’ and ‘mitotic catastrophe’* (supplementary material, Figure S5) 211, 212. Likewise, how delayed abscission affects chromosome homeostasis requires further investigation 199. Unexpectedly, overexpression of SAC genes is a more frequent event in human cancer 29. Conditional Mad2 upregulation predisposes mice to a wide range of early‐onset, aneuploid malignant tumors 213. The main difference from haplosufficienct Mad2 mice, which develop only benign tumors 189, 214, is the occurrence of extensive structural aberrations, implying that the structural branch of CIN, involving DSBs, lies behind cancer progression 109, 215, 216 (see the next section). Notably, p53 and pRb inactivation lead to Mad2 overexpression, CIN, and a malignant phenotype encompassing highly aggressive features 217, 218, 219. These data corroborate previous reports linking p53/pRb expression aberrations with aneuploidy and CIN in common human malignancies 220, 221. Overall, these observations support the idea that deregulation of critical checkpoints impinges on mitotic surveillance and fidelity, determining the fate of daughter cells.
The significance of engaging the repair machinery in the correct context (where and/or when) is vital. During interphase, DNA repair is essential to maintain genome stability, whereas in mitosis it may be deleterious 222. Induction of DSBs in mitosis leads to a ‘muted’, non‐classic DDR (supplementary material, Figure S5). Mitotic DSBs are marked by MRN, MDC1 and γH2AX foci in a PI3K‐dependent manner, whereas RNF8 and RNF168 localization with mitotic γH2AX does not take place, excluding the recruitment of 53BP1 and BRCA1 223, thus blocking NHEJ and HRR, respectively 224. By the suppression of DRR/R in mitosis, telomere–telomere fusion is avoided. In metaphase, the shelterin complex aquires a loose configuration, termed telomere dispersion, promoting condensin loading and chromosome segregation 225, that concurrently renders chromosomes prone to fusions favouring structural CIN 222. Notably, restoration of RNF8 and 53BP1 accumulation at mitotic DSB sites results in telomere–telomere end‐to‐end fusion and aneuploidy, especially in the presence of exogenous genotoxic stress 226. Likewise, mitotic DDR/R activation can lead to deleterious chromosomal alterations 224. In particular, instead of suppressing Plk1*, as it does in G2 phase 227, 228, 229, it upregulates Plk1 activity and, along with Aurora‐A*, it increases kinetochore*–microtubule stability, favouring merotelic attachments*, lagging chromosomes*, micronucleus formation*, and chromothripsis* (supplementary material, Figure S5) 230, 231, 232, 233.
The outcome of mitotic DDR/R activation depends on the underlying stress parameters (Figure 1B). For instance, cells with DNA damage but intact p53 (cellular context) escape or slip out from mitotic arrest and succumb to G1 arrest, whereas p53‐deficient cells continue to cycle and become aneuploid 234, 235. Likewise, in cells with low DNA damage levels (how much), mitotic DDR/R marks DSBs (memory signals) for repair in the subsequent G1 phase 236, 237, 238, resulting in a similar fate to that of cells with DNA damage and signalling effects occurring in earlier cell‐cycle phases 239, 240, 241. In contrast, excessive DNA damage (how much) activates the SAC, leading to mitotic arrest 237. Arrested cells may either die by mitotic catastrophe* or may exit mitosis prematurely without proper chromosome segregation and cytokinesis, through a process termed mitotic slippage* (supplementary material, Figure S5) 242. The destiny of these cells is not clear. They can follow the road to cancer, acquire a senescent phenotype, or die (supplementary material, Figure S5). Altogether, rewiring DSB repair to a ‘repressive mode’ during mitosis has advantages and disadvantages. Specifically, as execution of classic DDR/R is an ATP‐consuming process, it possibly saves energy for the demanding process of mitosis 243; also, it favours rapid execution of mitosis at the expense of increased sensitivity to DNA‐damaging stressors; and it protects against structural alterations. On the other hand it is prone to causing numerical CIN.
Overview of the proteostasis network (PN): connecting the PDR with DDR
Downstream of the critical, but ‘lifeless’, genetic information, there is a world of immense complexity, namely the proteome. The entry point to this world occurs via protein synthesis, which takes place in the cytosol and endoplasmic reticulum (ER) by a complicated machine (i.e. the ribosome) that (1) decodes the information stored in nucleic acids, and (2) shifts the chemistry from nucleic acids to amino acids.
Overview of the PN: links with DDR/R
Human cells express millions of polypeptides of >10 000 different species 244 that fold into well‐defined three‐dimensional structures that form parts of protein machines. Because the average protein and proteome sizes have increased significantly during evolution 245, and the consequences of an unstable proteome can be catastrophic 246, 247, cells have evolved a modular, but integrated, system that ensures general proteome quality control, called the PN 248. The PN performs the daunting task of curating polypeptide synthesis, folding, conformational maintenance, sorting–trafficking, and degradation; it also responds to conditions of proteotoxic stress by addressing the triage decision of fold, hold, or degrade (supplementary material, Figure S6A). The PN comprises ∼2000 chaperones, folding enzymes, trafficking modules, and degradation components, and it is not surprising that proteome stability maintenance consumes the majority of cellular ATP 249, 250. Because of the wiring and interdependence of various PN branches, defects in one module trigger a breakdown of the entire network; these effects are evident during ageing and in age‐related diseases such as cancer 251.
The ‘chaperome’ consists of hundreds of cytosolic and organelle‐specific chaperones that, along with their associated factors, bind to a wide range of distinct substrates 252, 253. Chaperones* are involved in proper polypeptide folding, unfolding, and remodelling, and in the assembly of protein machines or the delivery of damaged polypeptides to degradation machineries (supplementary material, Figure S6). Nuclear chaperones provide examples demonstrating how components of the PN assist the DDR/R machinery, unveiling a new role of proteome quality control in preserving genomic stability (supplementary material, Figure S6A). Specifically, nuclear chaperones orchestrate the delivery of newly produced histones to DNA, and also facilitate histone turnover 68, 254. Hence, reassembly of chromatin upon DNA repair is highly dependent on chaperones 51, 255.
When folding of a mutated or a post‐translationally irreversibly modified polypeptide fails, cellular proteases take over. The two main branches of this part of the PN are the UPP and autophagy–lysosome (ALP) pathways, comprising ∼850 and ∼500 different components, respectively (supplementary material, Figure S6A) 256, 257, 258, 259, 260. The UPP pathway degrades short‐lived poly‐ubiquitinated normal proteins and non‐functional or misfolded polypeptides and is composed of ubiquitin‐conjugating enzymes and the 26S (or 20S) proteasome* 261, 262. The ubiquitin ligase family confers substrate specificity and comprises almost 600 genes in humans 257, 263. The UPP pathway is central to protein synthesis quality control, as non‐functional newly synthesized polypeptides are targeted for degradation to cytosolic or ER‐bound proteasomes [ER‐associated protein degradation (ERAD)] 264. Proteasomes are also located in the outer mitochondrial membrane, and perform outer mitochondrial membrane‐associated degradation (OMMAD) during activation of the mitochondrial unfolded protein response (UPRMT) 265. The UPP pathway is also involved in the degradation of mitochondrial fusion/fission proteins, and thus, apart from genome and proteome stability, PN modules are also critical for mitostasis* 186. It is not thus surprising that ubiquitin ligases are frequently deregulated in cancer 262.
The ALP pathway is an intracellular self‐catabolic process that comprises three forms in mammalian cells: (1) chaperone‐mediated autophagy (CMA); (2) microautophagy; and (3) macroautophagy 12, 266, 267, 268. In CMA, damaged polypeptides are degraded in lysosomes after being recognized by chaperones, which bind to the lysosomal LAMP2A receptor 269; CMA offers an alternative to the UPP pathway for degradation of misfolded proteins. Microautophagy is a less well understood process involving engulfment of small cytosolic regions by the lysosomal membrane. Finally, in macroautophagy, the Atg* proteins form autophagosomes that capture lipids, proteins, or even organelles, transferring them to lysosomes for degradation 270. The ALP pathway also degrades ubiquitinated substrates (including protein aggregates) via the action of microtubule‐associated histone deacetylase 6 (HDAC6) and sequestrome‐1 (p62/SQSTM1) 271.
Genome damage results in the activation of proteostatic modules, and crosstalk between the DDR/R machinery and autophagy has been established (supplementary material, Figure S6A) 272, 273, 274. Prominent connections include those between ATM and PARP1 with AMPK*, one of the central metabolic regulators in eukaryotes. AMPK is activated when the AMP/ATP ratio is high, setting in motion autophagic flux 275, 276, 277. Likewise, p53 has been shown to upregulate components of autophagy, thus forming an amplification loop 278. Autophagy modulates DNA repair by degrading (among others) KAP1, HP1, and sequestrome‐1 (p62/SQSTM1), which hinder BRCA1 and Rad51 accessibility to DSB sites, promoting successful completion of HRR 279. Similarly, the UPP pathway and ubiquitylation are integral parts of the DDR pathway, as ubiquitylation of DDR factors has emerged as a switch that initiates signalling cascades and also as a proteolytic signal coordinating recruitment and disassembly of these proteins 280. Furthermore, the proteasome is involved in the degradation of DNA repair proteins following completion of the process (see above). Given the extensive ongoing damage and genome remodelling during cancer, it is not surprising that proteostatic machineries are deregulated during oncogenic transformation (supplementary material, Figure S6B) 251, 262, 281. However, the link between DDR/R and the PN or the PDR warrants further investigation, especially during carcinogenesis (supplementary material, Figure S6C), as this interrelationship is not straightforward. Specifically, recent in vivo data indicate an inverse relationship in precancerous lesions, whereas, at the cancerous stage, PN modules and the DDR operate in parallel 272, 274, 281.
PDR signalling
Polypeptides are post‐translationally modified either by PTMs, which are normal regulatory processes and do not increase proteome instability, or by non‐enzymatic protein modifications (NEPMs), which are stochastic and disrupt their structure and function 282. Unfolded or damaged proteome components impinge on the PN, triggering a response pathway called PDR (see Introduction) (supplementary material, Figure S6A). The PDR branches are coordinated by a series of complementary homeostatic mechanisms, which sense and respond to imbalances in proteostasis and/or to increased amounts of stressors. These signalling cascades, namely the heat shock response, hypoxia response, oxidative stress response, unfolded protein response in the ER (UPRER)*, and UPRMT*, are modulated by transcription factors that sense stress and mobilize genomic–cytoprotective responses 274, 281, 283. These responses are also coupled with a decrease in protein synthesis, thereby reducing the influx of newly synthesized proteins and/or allowing preferential translation of stress‐responsive mRNAs until proteome stability restoration 284, 285, 286.
Proteome instability is mainly counteracted by heat shock factor 1 (Hsf1), which is maintained in an inert state by chaperone binding 287. Upon heat or other types of stress that destabilize the proteome, these chaperones are titrated away from Hsf1 by binding to denatured proteins, and Hsf1 thus translocates to the nucleus, inducing transcription of a wide range of proteostatic modules 288, 289. Similarly, chaperones that are localized in the ER guide the folding of membrane or secreted proteins. Furthermore, the ER has its own stress response pathway (UPRER) that is activated in cases of increased flux or heavy secretory loads, or after heat shock that increases protein misfolding 290. The UPRER attenuates de novo protein synthesis and induces the expression of chaperones to aid proper polypeptide folding, or, if organelle proteostasis cannot be restored, triggers apoptosis. Likewise, the UPRMT maintains mitochondrial functional integrity by increasing the rates of polypeptide folding and degradation through the transcriptional activation of specific mitochondrial chaperones and proteases 291, 292, 293. Additionally, the UPRMT induces OMMAD, mitophagy, or apoptosis if disruption of mitostasis is irreversible 293, 294. An SR of paramount importance for cellular survival is triggered by hypoxia and is regulated by the Hif‐1 transcription factor 295. Under normoxic conditions, Hif‐1 is targeted for proteosomal degradation by the VHL E3‐ubiquitin ligase. In hypoxic conditions, Hif‐1 is protected, activating a transcriptional programme that includes (among other things) the upregulation of chaperones 296. Finally, the Nrf2 signalling pathway plays a crucial role in defence against oxidative and/or xenobiotic damage by activating [after binding to antioxidant response elements (AREs)] a broad range of detoxification enzymes 297, 298 and by inducing UPP and ALP pathway genes 299, 300.
The PDR and DDR/R: an intermingled fate
Unmitigated stress will eventually exceed the buffering capacity of the PN, leading to proteome instability (supplementary material, Figure S6B) 301, which is a daunting prospect. As the DDR/R machinery includes complex protein machines, the faithful execution of the triage ‘access–repair–restore’ is obviously dependent on proteome stability. Failure of the PDR pathway to cope with proteotoxic stress poses a risk for genomic integrity. Conversely, constitutive DDR/R activation will eventually wear out the proteome because of defective transcription and reduced polypeptide quality (supplementary material, Figure S6B). Suppressed ribosomal biogenesis during DNA damage is an example of how activation of DDR/R may affect proteostasis 302. Furthermore, sustained DNA damage can compromise DDR/R efficiency, increasing the production of mutated polypeptides and proteome instability. Subsequently, a vicious cycle of low‐fidelity DDR/R–PN activity develops that will trigger pathophysiological states, including carcinogenesis 303. The most characteristic paradigm of a ‘saturated’ PDR linked with aberrant DDR activation is accumulation of lipofuscin during oncogene‐induced senescence, with detrimental effects for the cell (see the next section; supplementary material, Figure S6).
Oncogene‐induced DNA damage and cancer development; ‘a model to rule them all?’
In a seminal paper in the early 1990s, Fearon and Vogelstein 304 proposed a genetic model for cancer development, using colorectal cancer as the basis of their study. They suggested that salient molecular alterations characterize each morphological stage of colorectal cancer progression, from the early small adenomas (benign phase) to the large metastatic carcinomas (malignant phase). Molecular changes increase from the benign to the malignant phase, and include mutational activation of oncogenes* coupled with inactivation of tumor suppressors* 305. Since then, the general features of this model have been applied to other epithelial neoplasms 306. The idea of analysing the molecular traits of each developmental stage of cancer has revolutionized the field and played a major role in the emergence of molecular pathology. Twenty years later, findings largely based on the above models led Hanahan and Weinberg 307 to propose six hallmarks of cancer. These include sustained proliferative signals, inactivation of tumor suppressors, resistance to apoptosis, replicative immortality, increased angiogenesis, and invasion/metastasis. Conceptual advances in the last decade provided two additional hallmarks: escape from immune surveillance and deregulated metabolism 308.
In spite of the value that the above model provided by setting the timeline of molecular events, it does not offer an explanation of how one molecular alteration leads to the next. In other words, it is a static model lacking the dynamic parameter of the driving force. Some years ago, we put forward a model for cancer development postulating that oncogene‐induced DNA damage followed by error‐prone repair could be the link between the hallmarks of cancer 109. The concept was based on the simple finding that carcinomas show DDR foci from their earliest stages of development, whereas adjacent normal epithelium is ‘clear’ 309, 310. This observation suggested that an endogenous source within the incipient cancerous environment caused DNA damage, triggering constitutive activation of the DDR pathway. We hypothesized that a hyperproliferative state could be the origin. Hyperproliferative signals, in most cases, represent parts of SR pathways that deviate from their adaptive/protective role, particularly when SR parameters stray beyond certain limits. For example, squamous metaplasia of the bronchial epithelium is an adaptive response to toxic injury caused by cigarette smoke stimulated by the epidermal growth factor receptor (EGFR) pathway 311. Depending on the cellular and environmental context, squamous metaplasia can shift progressively to dysplasia and to full neoplastic transformation in which EGFR becomes frequently amplified, fulfilling the first hallmark of cancer [Figure 5(1), lower panel] 307, 312, 313.
To functionally recapitulate the above scenario, we utilized various types of normal cells and precancerous models, and showed that hyperproliferative stimuli, including activated oncogenes, caused DSBs 159, 309, 310, 314. Interestingly, the lesions occurred predominantly at CFSs* 159, 309, 314, 315. As mentioned above, CFSs are late‐replicating regions of the genome with a sequence composition that makes them vulnerable to RS [Figure 5(2,3); lower panel] 118, 316, 317. As a result, they show breaks, losses and rearrangements, which are genetic alterations collectively termed CFS expression [Figure 5(3); lower panel] 118, 318, 319, 320. Their expression is not just a ‘passive’ event, but an incident with severe repercussions, as CFSs constitute highly ‘functional’ entities harbouring cancer‐related genes, often extending over large genomic regions (long genes), microRNAs (miRNAs), and regulatory sequences, with a much higher density than in non‐fragile sites 118, 321. For instance, two of the long genes located within CFSs are FHIT in FRA3B, and WWOX in FRA16D, both of which are potent tumor suppressors that are frequently inactivated in cancer 322, 323, 324, 325. Consistently, sequencing of colon adenomas revealed that SNSs mapped more often to very large genes 326. Likewise, in a precancerous cellular model, we noticed that, apart from CFSs, the repetitive and damage‐prone rDNA (see ‘The nucleolus at the crossroad of stress response’ above) was an additional ‘hotspot’ for oncogene‐mediated damage [Figure 5(3); lower panel] 159.
The above observations led us to suggest that activated oncogenes compromise DNA replication, provoking RS and, in turn, activating the DDR/R pathway in an effort to ‘fix’ the generated lesions [Figure 5(2); lower panel]. Concurrently, to impede the transition of mutated genetic material to offspring, the DDR sets in motion the antitumor barriers of apoptosis and senescence 109, 159, 309, 310, 314, 327, 328, 329, 330. However, sustained RS would lead to accumulation of damage that, at some point, would overwhelm the capacity of the high‐fidelity DNA repair routes, shifting error‐free to error‐prone repair [Figure 5(2,3)]. This switch will, in due course, modify the genome landscape, exhausting the integrity of antitumor responses and paving the way for cancer progression [Figure 5(2–5); lower panel].
A prediction of this model whereby oncogene‐induced RS (OIRS) acts as a driving force for cancer progression is that the replication machinery and its regulatory network* should play a vital role in cancer initiation and progression [Figure 5(2); lower panel] 331, 332. Genes that either positively or negatively regulate growth would be primary targets for activating or inactivating mutations, respectively, whereas DNA repair genes will be spared to ‘fix’, as otherwise the incipient cancer cell will die. Recent high‐throughput sequencing studies in sporadic cancers support this notion, showing: (1) a high frequency of inactivating mutations in checkpoint genes such as RB (retinoblastoma), TP53, and ATM; (2) a high incidence of activating events (e.g. mutations and gene amplifications) in growth‐promoting genes such as RAS and EGFR; and (3) a paucity of mutations in DNA repair genes 29. Particular attention should be given to the p16INK4A–Rb–E2F pathway, which is a major ‘molecular crossroad’ that most mitogenic/oncogenic signals converge to. Its phosphorylation status determines whether the cell will bypass the ‘restriction point’ of G1 phase, committing the cell to progress to S phase without the requirement for extracellular stimulants 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179, 180, 181, 182, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192, 193, 194, 195, 196, 197, 198, 199, 200, 201, 202, 203, 204, 205, 206, 207, 208, 209, 210, 211, 212, 213, 214, 215, 216, 217, 218, 219, 220, 221, 222, 223, 224, 225, 226, 227, 228, 229, 230, 231, 232, 233, 234, 235, 236, 237, 238, 239, 240, 241, 242, 243, 244, 245, 246, 247, 248, 249, 250, 251, 252, 253, 254, 255, 256, 257, 258, 259, 260, 261, 262, 263, 264, 265, 266, 267, 268, 269, 270, 271, 272, 273, 274, 275, 276, 277, 278, 279, 280, 281, 282, 283, 284, 285, 286, 287, 288, 289, 290, 291, 292, 293, 294, 295, 296, 297, 298, 299, 300, 301, 302, 303, 304, 305, 306, 307, 308, 309, 310, 311, 312, 313, 314, 315, 316, 317, 318, 319, 320, 321, 322, 323, 324, 325, 326, 327, 328, 329, 330, 331, 332, 333. Hence, inactivation of p16INK4A–Rb signalling is not a surprising finding in cancer 221, 334, 335, 336, 337, as it releases E2F transcription factor 1 (E2F1) from its inhibitory control 329, 333, 338, 339, stimulating the sustained expression of cell‐cycle drivers such as cyclin E, Cdc6, and Cdt1, converting them into ‘oncogenic stimuli’ [Figure 5(1); lower panel] 112, 210, 314, 327, 338, 340. In support of this, we observed that E2F1, Cdc6 and Cdt1 are aberrantly expressed in most cancer types examined, and, importantly, from their earliest stages 112, 314, 329, 338, 339, 340. Deregulated production of E2F1, Cdc6 and Cdt1 was not a mere reflection of an increased proliferation rate, as their forced expression (utilizing various inducible cellular systems covering the whole spectrum of carcinogenesis) triggered RS, DNA damage and DDR activation with induction of apoptosis and/or senescence 159, 314, 327, 329, 340, 341, 342. In accordance with the main prediction of our model, p53 function was gradually attenuated, apoptosis was reduced, and a fraction of senescent cells escaped, showing aggressive traits [Figure 5(4–6); lower panel] 159, 314, 327, 342, such as increased invasiveness, aneuploidy,* and features reminiscent of EMT* 159, 314, 343, which is an embryonic developmental programme exploited by cancer cells to invade and metastasize 344, 345. The above sequence of events explains why p53 needs to be inactived in tumors for oncogenes to exert their adverse effects 221, 335, 338, 339, 346, 347, 348, 349, 350.
The functional coupling between oncogene activation and inactivation of tumor barriers provides experimental evidence that OIRS acts as a driving force exerting selective pressure that eventually shapes the stage for cancer progression (Figure 5). The mechanism behind OIRS and cancer development depends on the availability of ‘quantitative and qualitative repair resources’. When the latter condition is not met, continuous rewiring of DNA repair networks occurs, favouring the survival of the ‘fittest incipient cancer cell’. As a consequence, chronic activation of the DDR/R network results in the exhaustion of essential short‐lived repair factors controlling high‐fidelity repair, such as Rad51 or 53BP1, thus forcing repair to follow less accurate routes 342, 351, 352. Within this framework, we noticed that, in a p53‐deficient environment, p21WAF1/Cip1 (a traditional tumor suppressor) revealed a ‘dark side’ of promoting GI by deregulating the replication licensing machinery and rewiring the DNA repair network to favour Rad52‐dependent error‐prone break‐induced replication (BIR) and single‐strand annealing (SSA) [Figure 5(2,3); lower panel] 342, 353, 354. This observation challenges the conventional view of dividing cancer‐related genes into ‘oncogenes’ and ‘tumor suppressors’, stressing the significance of cellular context in protein function.
As the activities of the DDR/R network go well beyond the boundaries of just ‘sensing, signalling and repairing DNA lesions’, by crosstalking with crucial SR pathways, OIRS‐mediated DNA damage makes the situation more complex 23, 274, 355. It is thus apparent that, when DDR components such as p53 and ATM are targeted, essential intercellular and intracellular surveillance operations will be modified, favouring, in the end, cellular transformation. There are two additional DDR interactions worth mentioning: (1) the immune system; and (2) metabolic pathways [Figure 5(3); lower panel]. Regarding the first, the DDR pathway communicates with nuclear factor‐κB (NF‐κB), the central hub of the immune response. Two of the best‐characterized connections involve ATM‐dependent activation of NEMO, a regulatory subunit of the IKK complex, stimulating NF‐κB activity 356, 357, 358, 359, 360, and of NKG2D and DNAM‐1/CD226 ligands, two key players in innate imunity 361, 362, 363. Another paradigm of DDR and immune response crosstalk is that between p53 and ICAM1 364, 365, 366, showing a direct role of p53 in immunosurveillance. As a result, tumor‐promoting inflammation, which is an enabling characteristic of cancer 308, can be attributed, to a certain degree, to a sustained DDR 23, 109, 367, 368, 369. Considering the link with metabolism, the most prominent example is that of p53 inhibiting glycolysis by inducing TIGAR 355, 370. Within this context, cells deficient in ATM or p53 will create, over time, a permissive environment favouring immune evasion and ‘aerobic glycolysis’, both of which are hallmarks of cancer cells (Figure 5).
Collectively, the above data further support the role of GI in carcinogenesis, making it a hallmark of cancer [Figure 5(3); lower panel] 29, 308. Nevertheless, although our model provides a unified mechanistic explanation of how the hallmarks of cancer are formulated during cancer development, several questions remain to be answered (Figure 5). For instance, how does OIRS lead to replicative immortality? Surprisingly, reactivation of telomerase, which occurs in almost 90% of human cancers, is attributable to two nucleotide substitutions, C228T and C250T, located within the TERT promoter, implying that error‐prone repair was involved in the selection of these mutations (Figure 5) 371. Another issue is how angiogenesis is induced within the context that we propose. A potential mechanism could be via ROS production, which prevents Hif‐prolyl hydroxylase from activating Hif, which, in turn, stimulates neovascularization 372. Escaping senescence is an emerging concept in cancer progression. OIRS seems to play a role in shaping the genetic landscape for ‘escape’ to occur 159, 342, 354, 373. However, apart from genetic alterations, epigenetic modifications, extensive chromatin remodelling, increased proteome instability and metabolic reprogramming should also take place for such a dramatic shift in cellular behaviour to occur 68, 251, 374, 375, 376, 377. Addressing the mechanistic details linking OIRS with these molecular adjustments would help immensely in finding the ‘Achilles’ heel' and designing analogous therapeutic strategies.
Therapeutic strategies, novel tools, and future perspectives
If OIRS and error‐prone repair drives cancer, targeting this process will kill cancer cells 303, 378, 379. This hypothesis has been tested by designing various relevant therapeutic approaches over the last few years. The most characteristic example is PARP inhibition. In 2014, olaparib was the first PARP inhibitor approved for the treatment of advanced BRCA1/2 mutant ovarian cancer 380, 381. ATR or Chk1 inhibitors are not particularly toxic for normal cells, but cancer cells harbouring DNA lesions rely on ATR for survival. Two highly selective and potent ATR inhibitors, AZD6738 and VX‐970, are in early‐phase clinical trials, either as monotherapies or in combination with a variety of genotoxic chemotherapies 382. In addition to inhibitors of RS, cell‐cycle checkpoint inhibitors against the Wee1 kinase and several CDKs are also under development as stand‐alone or combination therapies 383, 384. A parameter to be taken into consideration in future therapeutic interventions is the circadian rhythm, as increasing evidence has demonstrated its impact on key biochemical functions, including the DDR/R 385, 386, 387, 388. This constraint should be into consideration when novel therapeutic modalities are designed, to augment the therapeutic effect.
The dramatic changes that occur in cellular metabolism during cancer development have drawn attention to novel therapeutic approaches. Specifically, the increased oxidative stress that characterizes most cancer cells has been an object of intense investigation. Targeting enzymes that hydrolyse and remove oxidized nucleotides, such as mutT homologue 1 (MTH1), has shown promising results 389, 390, 391, 392. An additional relationship that can be exploited therapeutically is that between ATM and alternative reading frame (ARF). ARF is encoded together with p16INK4A by the CDKN2 locus. Historically, it was the first tumor suppressor reported to sense and react to oncogenic stimuli 393. It is interesting that, for many years, it was considered to act in a DDR‐independent manner, showing multiple functions such as regulating p53 stability and ribosome biogenesis 394, 395. We recently showed that ATM keeps ARF in check, as a ‘second line of defence’ 174. Moreover, ARF shows a higher activation threshold to ‘oncogenic load’ than the DDR pathway, thus forming together the DDR with a hierarchically organized ‘antitumor barrier’ 330. Consequently, targeting ATM, particularly in p53‐deficient tumors, will probably set in motion the antitumor properties of ARF, simultaneously crippling DDR signalling and cumulatively inhibiting tumor growth 174.
As the functionality of proteostatic modules 261 and anti‐stress responses 298 decline during ageing, these events also fuel ageing and age‐related diseases, including cancer (supplementary material, Figure S6B,C) 248, 301, 396. However, during the late phases of carcinogenesis and because of accumulating stressors, there should be a selective pressure for upregulation of the cytoprotective PN, and advanced and/or metastatic tumors may thus become ‘addicted’ to the higher expression levels and/or activities of proteostatic modules [Figure 5(3); lower part]. This hallmark of advanced tumors can be exploited therapeutically, because, apart from the increased load of mutated polypeptides, the high replication rates of cancer cells requires upregulated protein synthesis and maintenance that are out of balance relative to differentiated cells. Indeed, cancer cells are highly sensitive to Hsp90 inhibitors 397, and proteasome inhibitors have shown clinical efficacy in haematological cancers 398. Likewise, the ‘stress phenotype’ of cancer cells offers novel therapeutic strategies, as cancer cells have probably exhausted their capacity to survive under conditions of increased stress. In support of this, it was found recently that piperlongumine (a compound that upregulates ROS in both cancer and normal cells) had selective antitumor effects with no apparent toxicity in physiological tissues 399.
However, one thing that cancer has taught us is its resilience, adaptation to therapy, and evolution 400. One of the most intriguing features of cancer evolution, aside from loss or enhancement of function, is gain of novel functions, further increasing the level of ‘plasticity’. Evasion from senescence, circumvention of immune surveillance and p53 ‘gain‐of‐function’ mutations are representative examples 159, 342, 349, 373, 401, 402, 403, 404, 405, 406, 407. A decade ago, the weapons in our arsenal with which to investigate such complicated phenomena were still limited. The era of ‐omics provides us with tremendous amounts of data, and molecular resolution capabilities have reached the single‐cell level 408. Nevertheless, the rapid increase in the amount of information can lead to erroneous or misleading conclusions in certain cases, challenging established knowledge. At this point, advanced bioinformatics tools combined with sophisticated molecular methods take centre stage, unveiling hidden patterns and providing accurate mechanistic insights into disease and particularly cancer development 409, 410, 411. For instance, whole genome sequencing, even at the single‐cell level, can identify mutational signatures that actually represent the repair history of a cancer cell, thus highlighting potential therapeutic targets 27, 28, 39, 412, 413. Other revolutionary methods include chromosome conformation capture techniques that facilitate a three‐dimensional view of genome–proteome interactions, providing, for the first time, unique opportunities to monitor ‘holistically’ the regulation and deregulation of homeostatic mechanisms 414, 415, 416. Nevertheless, conventional methods still play a role, not only at the diagnostic level but also at the front line of research. The best example is the emergence of lipofuscin, a substance identified 175 years ago by Adolf Hannover, as a key hallmark of cellular senescence (supplementary material, Figure S6D) 417, 418. On the basis of this feature, we recently developed a novel reagent that is able to monitor senescence in any biological setting, including archival material, providing a solution to a challenge that haunted the field for almost 50 years 418. The latter is of vast importance now that senolytic drugs are entering the scene. From all of the above, it is evident that we are moving from the ‘era of ‐omics’ to the ‘era of quantum bioinformatics*’ 419; thus, for the first time, the prospects for precision medicine are bright.
Author contributions statement
VGG and IPT conducted writing and manuscript preparation. DEP and ISP performed the literature search, and figure and manuscript preparation. VGG supervised manuscript preparation.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary figure legends
Full legends for main figures
Glossary and list of abbreviations
Figure S1. The cellular fate following genotoxic insults
Figure S2. Other pathways that contribute to DDR signaling
Figure S3. Replication‐transcription intermediates and replication fork restart
Figure S4. Nucleolus and rDNA organization and maintaining mitochondrial DNA integrity
Figure S5. Monitoring Mitosis (DDR surveillance)
Figure S6. Functional interplay and interdependence of genome and proteome maintenance modules (DDR and PDR surveillance)
Table S1. Representative categories and types of human RNAs involved in physiological processes and diseases, including cancer
Table S2. The implications of defective key components of the DNA damage response mechanisms in the pathogenesis of specific clinical syndromes in humans
Supporting information
Supplementary figure legends
Full legends for main figures
Appendix S1. Glossary and list of abbreviations
Figure S1. The cellular fate following genotoxic insults. The magnitude of the genotoxic insult (low, moderate, excessive) determines cells fate (effective repair, senescence or cell‐death, respectively). Under certain conditions, determined by the stress response parameters (Figure 1B), senescence can present a “dark side”. Likewise, necrosis and/or resistance to apoptosis can build up a pro‐tumorigenic environment (see text for details and references). SASP: senescence‐associated secretory phenotype.
Figure S2. Other pathways that contribute to DDR signaling. Accumulating data demonstrate that the DDR function is complemented and/or it cross‐talks with other signaling routes, which also respond to DNA damage 420, 421, 422. To what extent these signaling pathways modulate the DDR function is a subject that has not been fully elucidated. Nevertheless, the implementation of multiple signaling cascades in a DDR network highlights the need for the DNA damage machinery to detect and respond to a wide range of stimuli in various cellular scenarios, underscoring the highly modular organization of the DDR 422. (i) One such signaling pathway involved in DDR is the p38 MAPK. It is one of the three main groups of mitogen‐activated protein kinases (MAPK). It contributes in the G2/M checkpoint, to facilitate DNA repair, via three possible routes: a) the direct phosphorylation of p53, which results in the dissociation of p53 from Mdm2 thus preventing p53 ubiquitination and degradation, b) the association with Gadd45α, which interacts with p53 and increases its stability, and c) the phosphorylation and inhibition of the phosphatase Cdc25B which is responsible for driving the cell cycle through activation of the Cyclin B/Cdc2 complex 53, 423. In addition, p38 MAPK activation can induce G1/S checkpoint in response to a variety of cellular stresses such as osmotic shock or cellular senescence 53, 423. (ii) Hippo signaling pathway is also implicated in the DDR. Further to a wide spectrum of cellular roles, components of the Hippo pathway cooperate with central orchestrators of the DDR, namely the ATR‐Chk1 and ATM‐Chk2 signaling nodes 424, 425. (iii) Wnt/ β catenin pathway, which has important functions in controlling gene expression, cell polarity and adhesion, is also involved in the repair of DNA damage specifically due to oxidative stress, through interaction with DDR at different levels 426, 427, 428. (iv) NOTCH pathway is a highly conserved signaling system that functions in developmental processes related to cell‐fate determination, particularly in stem cells. In mammalian cells, activation of human Notch1 results in reduced ATM signaling in a manner independent of Notch 1 transcriptional activity 429. Notch1 binds directly to the regulatory FATC domain of ATM, thus inhibiting ATM kinase activity 429. (v) An additional paradigm of interaction between DDR and other signaling routes is that of the Hedgehog (Hh) pathway on the DNA repair mechanism. Inhibition of Hh signaling can repress almost all of the DNA repair mechanisms (i.e. BER, NER, MMR and DSB repair including HR and NHEJ) 430. (vi) Immune responses upon DNA damage are supported by a growing body of evidence 24. DNA‐PK, Ku70 and MRE11 are all capable of sensing cytosolic DNA and activating the cGAS‐STING pathway promoting type I and type III interferon‐signaling. Additionally, PARP‐1 and ATM interact with subunits of IκB kinase triggering NF‐κB‐dependent gene expression. ATM and ATR activation is also involved in the upregulation of ligands for the NKG2D receptor upon stalled DNA replication forks. Conversely, key immune system players like the classical cytokine IL‐1α can act as intracellular DNA damage sensors and signal the presence of genotoxic stress 23, 431.
Figure S3. Replication‐transcription intermediates and replication fork restart. (A) Replication intermediate lesions harboring single stranded DNA (ssDNA). (i) Uncoupling of the replicative helicase and polymerases results in generation of ssDNA due to excessive unwinding of the template (stalled fork). (L: leading strand; l: lagging strand) (ii) A stalled replication fork may undergo remodeling by creating an intermediate reverse fork also known as “chicken foot” structure: (ii‐1) Direct CtIP processing of the reversed fork may lead to nascent strand ssDNA formation. (ii‐2) Cleavage by SLX4‐docking nucleases generates DNA double strand break that is subsequently followed by resection resulting into nascent strand ssDNA generation. (iii) Unequal branch migration or resection (by CtIP) of a reversed fork can also lead to generation of template ssDNA. (iv) Deregulated firing of clustered origins leads to replication stress and accumulation of gaps in the nascent strands, leaving template ssDNA (see text for details and references) (B) Transcription intermediates. R loops are the predominant transcription generated intermediates and represent a three‐stranded nucleic acid structure that comprises two branches, an RNA–DNA hybrid and an ssDNA. The former can impede completion of replication leading to replication fork stalling, collapse and DSBs formation, while the latter can serve as a substrate to DNA damaging agents and cellular enzymes [APOBEC deaminases (Table 1)] resulting in DNA lesions and/or nicks (see text for details and references). (C) Restart of stalled or collapsed replication forks. Depending on the duration (how long) of the replication block, forks can stall or collapse. Restart of stalled forks is promoted by fork remodeling factors, while collapsed forks rely on DSB mediated restart through homologous recombination repair, whereas new origins are concurrently fired (see text for details and references).
Figure S4. (A) Nucleolus and rDNA organization. Schematic representation of the rDNA repeats, an organization that renders them susceptible to replication‐transcription collisions (see text for details). PHC: Perinuclear Heterochromatin, FC: Fibrillar Centre, DFC: Dense Fibrillar Component and CC: Granular Component (see text for details and references). (B) Maintaining mitochondrial DNA integrity: Nuclear and mitochondrial DNAs are interdependent. Cartoon of the mitochondrial DNA: D (Displacement)‐loop: a short nucleotide segment complementary to the light (L)‐strand that displaces the heavy (H)‐strand of the mitochondrial DNA. It contains promoters (LSP and HSP) for the RNA transcription from the two strands (heavy and light, respectively) of mitochondrial DNA, possibly involved in the organization of the mitochondrial nucleoid (see text for details and references); LSP: Light strand promoter. The promoter is responsible for gene transcription from the light strand (lower molecular mass) of mitochondrial DNA; HSP: Heavy strand promoter. The promoter is responsible for gene transcription from the heavy strand (higher molecular mass) of mitochondrial DNA. Depending on the magnitude of the mitochondrial DNA damage three levels of repair may take place (see text for details and references).
Figure S5. Monitoring Mitosis (DDR surveillance). During M phase checkpoints monitor the proper alignment, segregation and cytokinesis (see lower left panel). In response to a mitotic defect, such as misalignment and/or DSBs, cell fate depends on the context (i.e. p53 status) and the extent of the damage (how much): i) low mitotic damage is marked, and repaired in the subsequent cell cycle in the daughter cells (continuous green line corresponds to G1 phase where the majority of DNA lesions are repaired, however, mitotic DNA lesions can also be repaired in S and G2 phase depicted by the dashed green line‐see lower left cell cycle panel), ii) high DNA damage or mitotic spindle defects may lead to mitotic catastrophe or mitotic slippage, which in turn generates aneuploidy and/or CIN. The later can lead to cell death, senescence or development of precancerous lesions. Upon induction of DSBs during mitosis, MRN, and phosphorylated MDC1 and Η2ΑΧ are recruited to the damaged site forming the mitotic DDR foci (see right panel). Notably, 53BP1 and BRCA1 are not recruited to the site of damage blocking NHEJ and HR activation, respectively, preventing telomere fusion (mute DDR). An adverse outcome of mitotic DDR activation is kinetochore‐microtubule stabilization mediated by activation of PLK1 and Aurora kinase A that in turn promotes merotelic attachment and the formation of lagging chromosomes resulting in numerical CIN. However, it is not yet clear under what circumstances activation of mitotic DDR leads to this unfavorable outcome, instead of marking the DNA damage and proceeding to repair in the following cell cycle (marked with a question mark; right lower panel). “P” within red colored circles depicts the two phosphorylation sites of 53BP1 at Threonine‐1609 and Threonine‐1618 that prevent it from recruitment to DDR foci. CIN: chromosomal instability
Figure S6. Functional interplay and interdependence of genome and proteome maintenance modules (DDR and PDR surveillance). (A) The PN along with PDR are actively involved in DDR efficiency since by assuring proteome integrity they maintain the functionality of the protein machines that safeguard genome stability. On the other hand, DDR induces a number of proteostatic and/or metabolic adaptations, including suppression of transcription and ribosomal biogenesis, indicating the functional interdependence of the two pathways. These pathways are fully active in young organisms. (B‐C) The age‐related collapse of proteostatic modules functionality and/or expression levels (B) results in the gradual accumulation of non‐functional polypeptides, protein aggregates or lipofuscin, compromising proteome integrity and leading to genomic instability (and thus increased chances for carcinogenesis) as a result of ineffective DNA maintenance and/or repair. Eventually, a vicious cycle may form where a mildly unstable genome accelerates proteome instability due to synthesis of mutated polypeptides that progressively increase the attrition of protein machines resulting in an increasingly stressful cellular landscape that favors the appearance (C) of cellular senescence, cell death or age‐related diseases (e.g. cancer). (D) In normal cells, production of ROS or RNS is neutralized by anti‐oxidant responses while intact PN ensures normal protein turnover. During stress induced premature senescence (Glossary) or in aged tissues the levels of ROS/RNS increase leading to lipid and protein oxidation in the cytoplasm. As this process evolves, oxidized proteins become unfolded and intra‐ and/or inter‐molecular cross links occur, forming non‐degradable oxidized protein aggregates; the latter along with oxidized lipids/lipoproteins, carbohydrate residues and metals form undegradable lipofuscin which accumulates mainly in lysosomes, while only a minor amount is found free in the cytosol. Cytosolic lipofuscin occurs either due to impaired uptake by stalled autophagy or following autophagosome/phagophore rapture. Lipofuscin also inhibits proteasome activity further boosting lipid/lipoprotein oxidation in the cytoplasm.
Table S1. Representative categories and types of human RNAs involved in physiological processes and diseases, including cancer
Table S2. The implications of defective key components of the DNA damage response mechanisms in the pathogenesis of specific clinical syndromes in humans
Acknowledgements
We would like to thank Dr Athanasios Kotsinas, Dr Konstantinos Evangelou, Dr Panagiotis Vasileiou and Aggelos Margetis for their assistance with the preparation of the manuscript. Financial support was from a Greek National Scholarships Foundation–Siemens Aristeia Fellowship and from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grants agreement No. 722729, Welfare Foundation for Social & Cultural Sciences (KIKPE), Greece. We would like to apologise for not being able to include all relevant works in the field.
No conflicts of interest were declared.
References
*Cited only in supplementary material.
- 1. Report on Carcinogens . 2016. Available from: https://ntp.niehs.nih.gov/pubhealth/roc/index-1.html
- 2. Davies KJ. Adaptive homeostasis. Mol Aspects Med 2016; 49: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolfe J. Cellular Thermodynamics: the Molecular and Macroscopic Views. eLS: Wiley, 2001. [Google Scholar]
- 4. Nesse RM, Bhatnagar S, Young EA. Evolutionary origins and functions of the stress response In Encyclopedia of Stress. Elsevier: San Diego, 2010; 965–970. [Google Scholar]
- 5. Jackson SP, Bartek J. The DNA‐damage response in human biology and disease. Nature 2009; 461: 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19: 238–245. [DOI] [PubMed] [Google Scholar]
- 7. Galluzzi L, Vitale I, Aaronson SA, et al Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 2018; 25: 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernstein C, Bernstein H, Payne CM, et al DNA repair/pro‐apoptotic dual‐role proteins in five major DNA repair pathways: fail‐safe protection against carcinogenesis. Mutat Res 2002; 511: 145–178. [DOI] [PubMed] [Google Scholar]
- 9. Gorgoulis VG, Halazonetis TD. Oncogene‐induced senescence: the bright and dark side of the response. Curr Opin Cell Biol 2010; 22: 816–827. [DOI] [PubMed] [Google Scholar]
- 10. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–740. [DOI] [PubMed] [Google Scholar]
- 11. Munoz‐Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15: 482–496. [DOI] [PubMed] [Google Scholar]
- 12. Galluzzi L, Baehrecke EH, Ballabio A, et al Molecular definitions of autophagy and related processes. EMBO J 2017; 36: 1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simon HU, Friis R, Tait SW, et al Retrograde signaling from autophagy modulates stress responses. Sci Signal 2017; 10: eagg2791. [DOI] [PubMed] [Google Scholar]
- 14. Schumacher B. Transcription‐blocking DNA damage in aging: a mechanism for hormesis. Bioessays 2009; 31: 1347–1356. [DOI] [PubMed] [Google Scholar]
- 15. Robertson MP, Joyce GF. The origins of the RNA world. Cold Spring Harb Perspect Biol 2012; 4: a005608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amaral PP, Dinger ME, Mattick JS. Non‐coding RNAs in homeostasis, disease and stress responses: an evolutionary perspective. Brief Funct Genomics 2013; 12: 254–278. [DOI] [PubMed] [Google Scholar]
- 17. Ng KW, Anderson C, Marshall EA, et al Piwi‐interacting RNAs in cancer: emerging functions and clinical utility. Mol Cancer 2016; 15: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Protter DS, Parker R. Principles and properties of stress granules. Trends Cell Biol 2016; 26: 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson P, Kedersha N. RNA granules. J Cell Biol 2006; 172: 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bardoel BW, Strijp JA. Molecular battle between host and bacterium: recognition in innate immunity. J Mol Recognit 2011; 24: 1077–1086. [DOI] [PubMed] [Google Scholar]
- 21. Cao X. Self‐regulation and cross‐regulation of pattern‐recognition receptor signalling in health and disease. Nat Rev Immunol 2016; 16: 35–50. [DOI] [PubMed] [Google Scholar]
- 22. Liston A, Masters SL. Homeostasis‐altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol 2017; 17: 208–214. [DOI] [PubMed] [Google Scholar]
- 23. Pateras IS, Havaki S, Nikitopoulou X, et al The DNA damage response and immune signaling alliance: is it good or bad? Nature decides when and where. Pharmacol Ther 2015; 154: 36–56. [DOI] [PubMed] [Google Scholar]
- 24. Nakad R, Schumacher B. DNA damage response and immune defense: links and mechanisms. Front Genet 2016; 7: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81: 323–330. [DOI] [PubMed] [Google Scholar]
- 27. Helleday T, Eshtad S, Nik‐Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 2014; 15: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability – an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11: 220–228. [DOI] [PubMed] [Google Scholar]
- 30. Aguilera A, Garcia‐Muse T. Causes of genome instability. Annu Rev Genet 2013; 47: 1–32. [DOI] [PubMed] [Google Scholar]
- 31. Abbas T, Keaton MA, Dutta A. Genomic instability in cancer. Cold Spring Harb Perspect Biol 2013; 5: a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017; 168: 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Georgakilas AG, O'Neill P, Stewart RD. Induction and repair of clustered DNA lesions: what do we know so far? Radiat Res 2013; 180: 100–109. [DOI] [PubMed] [Google Scholar]
- 35. Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol 2013; 5: a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single‐strand interruption repair pathway in mammalian cells. Cell Res 2008; 18: 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liakos A, Lavigne MD, Fousteri M. Nucleotide excision repair: from neurodegeneration to cancer. Adv Exp Med Biol 2017; 1007: 17–39. [DOI] [PubMed] [Google Scholar]
- 38. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer – the stable evidence. Nat Rev Clin Oncol 2010; 7: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lavigne MD, Konstantopoulos D, Ntakou‐Zamplara KZ, et al Global unleashing of transcription elongation waves in response to genotoxic stress restricts somatic mutation rate. Nat Commun 2017; 8: 2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghosal G, Chen J. DNA damage tolerance: a double‐edged sword guarding the genome. Transl Cancer Res 2013; 2: 107–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Livneh Z, Cohen IS, Paz‐Elizur T, et al High‐resolution genomic assays provide insight into the division of labor between TLS and HDR in mammalian replication of damaged DNA. DNA Repair (Amst) 2016; 44: 59–67. [DOI] [PubMed] [Google Scholar]
- 42. Hedglin M, Benkovic SJ. Eukaryotic translesion DNA synthesis on the leading and lagging strands: unique detours around the same obstacle. Chem Rev 2017; 117: 7857–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rouleau M, Patel A, Hendzel MJ, et al PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010; 10: 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Farmer H, McCabe N, Lord CJ, et al Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–921. [DOI] [PubMed] [Google Scholar]
- 45. Bryant HE, Schultz N, Thomas HD, et al Specific killing of BRCA2‐deficient tumors with inhibitors of poly(ADP‐ribose) polymerase. Nature 2005; 434: 913–917. [DOI] [PubMed] [Google Scholar]
- 46. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009; 361: 1475–1485. [DOI] [PubMed] [Google Scholar]
- 47. Keeney S, Neale MJ. Initiation of meiotic recombination by formation of DNA double‐strand breaks: mechanism and regulation. Biochem Soc Trans 2006; 34: 523–525. [DOI] [PubMed] [Google Scholar]
- 48. Dudley DD, Chaudhuri J, Bassing CH, et al Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol 2005; 86: 43–112. [DOI] [PubMed] [Google Scholar]
- 49. Mladenov E, Magin S, Soni A, et al DNA double‐strand‐break repair in higher eukaryotes and its role in genomic instability and cancer: cell cycle and proliferation‐dependent regulation. Semin Cancer Biol 2016; 37–38: 51–64. [DOI] [PubMed] [Google Scholar]
- 50. Thompson LH. Recognition, signaling, and repair of DNA double‐strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res 2012; 751: 158–246. [DOI] [PubMed] [Google Scholar]
- 51. Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25: 409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clouaire T, Marnef A, Legube G. Taming tricky DSBs: ATM on duty. DNA Repair (Amst) 2017; 56: 84–91. [DOI] [PubMed] [Google Scholar]
- 53. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J 2010; 429: 403–417. [DOI] [PubMed] [Google Scholar]
- 54. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14: 197–210. [PubMed] [Google Scholar]
- 55. Lopez‐Contreras AJ, Fernandez‐Capetillo O. The ATR barrier to replication‐born DNA damage. DNA Repair (Amst) 2010; 9: 1249–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumor progression. Oncogene 2007; 26: 7773–7779. [DOI] [PubMed] [Google Scholar]
- 57. Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J 2009; 28: 3067–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Uziel T, Lerenthal Y, Moyal L, et al Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003; 22: 5612–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee JH, Paull TT. ATM activation by DNA double‐strand breaks through the Mre11–Rad50–Nbs1 complex. Science 2005; 308: 551–554. [DOI] [PubMed] [Google Scholar]
- 60. Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013; 5: a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 2009; 137: 413–431. [DOI] [PubMed] [Google Scholar]
- 62. Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358: 15–16. [DOI] [PubMed] [Google Scholar]
- 63. Greenblatt MS, Bennett WP, Hollstein M, et al Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54: 4855–4878. [PubMed] [Google Scholar]
- 64. Shanbhag NM, Rafalska‐Metcalf IU, Balane‐Bolivar C, et al ATM‐dependent chromatin changes silence transcription in cis to DNA double‐strand breaks. Cell 2010; 141: 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kruhlak M, Crouch EE, Orlov M, et al The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007; 447: 730–734. [DOI] [PubMed] [Google Scholar]
- 66. Larsen DH, Hari F, Clapperton JA, et al The NBS1–Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol 2014; 16: 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem 2009; 78: 273–304. [DOI] [PubMed] [Google Scholar]
- 68. Hauer MH, Gasser SM. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 2017; 31: 2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jasin M, Haber JE. The democratization of gene editing: insights from site‐specific cleavage and double‐strand break repair. DNA Repair (Amst) 2016; 44: 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mehta A, Haber JE. Sources of DNA double‐strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 2014; 6: a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Doil C, Mailand N, Bekker‐Jensen S, et al RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136: 435–446. [DOI] [PubMed] [Google Scholar]
- 72. Ranjha L, Howard SM, Cejka P. Main steps in DNA double‐strand break repair: an introduction to homologous recombination and related processes. Chromosoma 2018; 127: 187–214. [DOI] [PubMed] [Google Scholar]
- 73. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double‐strand break repair pathway choice. Mol Cell 2012; 47: 497–510. [DOI] [PubMed] [Google Scholar]
- 74. Wyman C, Kanaar R. DNA double‐strand break repair: all's well that ends well. Annu Rev Genet 2006; 40: 363–383. [DOI] [PubMed] [Google Scholar]
- 75. Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol 2009; 4: 461–487. [DOI] [PubMed] [Google Scholar]
- 76. Rodgers K, McVey M. Error‐prone repair of DNA double‐strand breaks. J Cell Physiol 2016; 231: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fradet‐Turcotte A, Canny MD, Escribano‐Diaz C, et al 53BP1 is a reader of the DNA‐damage‐induced H2A Lys 15 ubiquitin mark. Nature 2013; 499: 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zgheib O, Pataky K, Brugger J, et al An oligomerized 53BP1 tudor domain suffices for recognition of DNA double‐strand breaks. Mol Cell Biol 2009; 29: 1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schotta G, Sengupta R, Kubicek S, et al A chromatin‐wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev 2008; 22: 2048–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yang H, Pesavento JJ, Starnes TW, et al Preferential dimethylation of histone H4 lysine 20 by Suv4‐20. J Biol Chem 2008; 283: 12085–12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Botuyan MV, Lee J, Ward IM, et al Structural basis for the methylation state‐specific recognition of histone H4‐K20 by 53BP1 and Crb2 in DNA repair. Cell 2006; 127: 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Huyen Y, Zgheib O, Ditullio RA, Jr , et al Methylated lysine 79 of histone H3 targets 53BP1 to DNA double‐strand breaks. Nature 2004; 432: 406–411. [DOI] [PubMed] [Google Scholar]
- 83. Aparicio T, Baer R, Gautier J. DNA double‐strand break repair pathway choice and cancer. DNA Repair (Amst) 2014; 19: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zimmermann M, de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol 2014; 24: 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol 2010; 17: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9: 297–308. [DOI] [PubMed] [Google Scholar]
- 87. Kim JS, Krasieva TB, Kurumizaka H, et al Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol 2005; 170: 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Snouwaert JN, Gowen LC, Latour AM, et al BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non‐homologous recombination that is corrected by expression of a brca1 transgene. Oncogene 1999; 18: 7900–7907. [DOI] [PubMed] [Google Scholar]
- 89. Tutt A, Bertwistle D, Valentine J, et al Mutation in Brca2 stimulates error‐prone homology‐directed repair of DNA double‐strand breaks occurring between repeated sequences. EMBO J 2001; 20: 4704–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xia F, Taghian DG, DeFrank JS, et al Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc Natl Acad Sci U S A 2001; 98: 8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 2016; 17: 337–349. [DOI] [PubMed] [Google Scholar]
- 92. Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer 2018; 18: 168–185. [DOI] [PubMed] [Google Scholar]
- 93. Che R, Zhang J, Nepal M, et al Multifaceted Fanconi anemia signaling. Trends Genet 2018; 34: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bukowska B, Karwowski BT. Actual state of knowledge in the field of diseases related with defective nucleotide excision repair. Life Sci 2018; 195: 6–18. [DOI] [PubMed] [Google Scholar]
- 95. Panier S, Durocher D. Push back to respond better: regulatory inhibition of the DNA double‐strand break response. Nat Rev Mol Cell Biol 2013; 14: 661–672. [DOI] [PubMed] [Google Scholar]
- 96. Chowdhury D, Keogh MC, Ishii H, et al gamma‐H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double‐strand break repair. Mol Cell 2005; 20: 801–809. [DOI] [PubMed] [Google Scholar]
- 97. Pechackova S, Burdova K, Macurek L. WIP1 phosphatase as pharmacological target in cancer therapy. J Mol Med (Berl) 2017; 95: 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lowe J, Cha H, Lee MO, et al Regulation of the Wip1 phosphatase and its effects on the stress response. Front Biosci (Landmark Ed) 2012; 17: 1480–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fiscella M, Zhang H, Fan S, et al Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53‐dependent manner. Proc Natl Acad Sci U S A 1997; 94: 6048–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lu X, Ma O, Nguyen TA, et al The Wip1 phosphatase acts as a gatekeeper in the p53–Mdm2 autoregulatory loop. Cancer Cell 2007; 12: 342–354. [DOI] [PubMed] [Google Scholar]
- 101. Goloudina AR, Kochetkova EY, Pospelova TV, et al Wip1 phosphatase: between p53 and MAPK kinases pathways. Oncotarget 2016; 7: 31563–31571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mosbech A, Lukas C, Bekker‐Jensen S, et al The deubiquitylating enzyme USP44 counteracts the DNA double‐strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem 2013; 288: 16579–16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yu M, Liu K, Mao Z, et al USP11 is a negative regulator to gammaH2AX ubiquitylation by RNF8/RNF168. J Biol Chem 2016; 291: 959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nijman SM, Huang TT, Dirac AM, et al The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 2005; 17: 331–339. [DOI] [PubMed] [Google Scholar]
- 105. Smogorzewska A, Matsuoka S, Vinciguerra P, et al Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007; 129: 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Calonge TM, O'Connell MJ. Turning off the G2 DNA damage checkpoint. DNA Repair (Amst) 2008; 7: 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Maniatis T. A ubiquitin ligase complex essential for the NF‐kappaB, Wnt/Wingless, and Hedgehog signaling pathways. Genes Dev 1999; 13: 505–510. [DOI] [PubMed] [Google Scholar]
- 108. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9: 153–166. [DOI] [PubMed] [Google Scholar]
- 109. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene‐induced DNA damage model for cancer development. Science 2008; 319: 1352–1355. [DOI] [PubMed] [Google Scholar]
- 110. Blumenfeld B, Ben‐Zimra M, Simon I. Perturbations in the replication program contribute to genomic instability in cancer. Int J Mol Sci 2017; 18: E1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rivera‐Mulia JC, Gilbert DM. Replication timing and transcriptional control: beyond cause and effect – part III. Curr Opin Cell Biol 2016; 40: 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Petrakis TG, Komseli ES, Papaioannou M, et al Exploring and exploiting the systemic effects of deregulated replication licensing. Semin Cancer Biol 2016; 37‐38: 3–15. [DOI] [PubMed] [Google Scholar]
- 113. Blow JJ, Gillespie PJ. Replication licensing and cancer – a fatal entanglement? Nat Rev Cancer 2008; 8: 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vitelli V, Galbiati A, Iannelli F, et al Recent advancements in DNA damage‐transcription crosstalk and high‐resolution mapping of DNA breaks. Annu Rev Genomics Hum Genet 2017; 18: 87–113. [DOI] [PubMed] [Google Scholar]
- 115. Gaillard H, Garcia‐Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer 2015; 15: 276–289. [DOI] [PubMed] [Google Scholar]
- 116. Helmrich A, Ballarino M, Nudler E, et al Transcription–replication encounters, consequences and genomic instability. Nat Struct Mol Biol 2013; 20: 412–418. [DOI] [PubMed] [Google Scholar]
- 117. Techer H, Koundrioukoff S, Nicolas A, et al The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet 2017; 18: 535–550. [DOI] [PubMed] [Google Scholar]
- 118. Georgakilas AG, Tsantoulis P, Kotsinas A, et al Are common fragile sites merely structural domains or highly organized ‘functional’ units susceptible to oncogenic stress? Cell Mol Life Sci 2014; 71: 4519–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Neelsen KJ, Zanini IM, Mijic S, et al Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev 2013; 27: 2537–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Couch FB, Bansbach CE, Driscoll R, et al ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 2013; 27: 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Thomas M, White RL, Davis RW. Hybridization of RNA to double‐stranded DNA: formation of R‐loops. Proc Natl Acad Sci U S A 1976; 73: 2294–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sebastian R, Oberdoerffer P. Transcription‐associated events affecting genomic integrity. Phil Trans R Soc Lond B Biol Sci 2017; 372: 20160288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Nishida K, Kuwano Y, Nishikawa T, et al RNA binding proteins and genome integrity. Int J Mol Sci 2017; 18: E1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Freudenreich CH. R‐loops: targets for nuclease cleavage and repeat instability. Curr Genet 2018; 10.1007/s00294-018-0806-z [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hills SA, Diffley JF. DNA replication and oncogene‐induced replicative stress. Curr Biol 2014; 24: R435–R444. [DOI] [PubMed] [Google Scholar]
- 126. Giannattasio M, Branzei D. S‐phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell Mol Life Sci 2017; 74: 2361–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 2010; 11: 683–687. [DOI] [PubMed] [Google Scholar]
- 128. Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 2015; 16: 207–220. [DOI] [PubMed] [Google Scholar]
- 129. Quinet A, Lemacon D, Vindigni A. Replication fork reversal: players and guardians. Mol Cell 2017; 68: 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Betous R, Couch FB, Mason AC, et al Substrate‐selective repair and restart of replication forks by DNA translocases. Cell Rep 2013; 3: 1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hishiki A, Hara K, Ikegaya Y, et al Structure of a novel DNA‐binding domain of helicase‐like transcription factor (HLTF) and its functional implication in DNA damage tolerance. J Biol Chem 2015; 290: 13215–13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kile AC, Chavez DA, Bacal J, et al HLTF's ancient HIRAN domain binds 3′ DNA ends to drive replication fork reversal. Mol Cell 2015; 58: 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zellweger R, Dalcher D, Mutreja K, et al Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 2015; 208: 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hashimoto Y, Ray Chaudhuri A, Lopes M, et al Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 2010; 17: 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Schlacher K, Christ N, Siaud N, et al Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mijic S, Zellweger R, Chappidi N, et al Replication fork reversal triggers fork degradation in BRCA2‐defective cells. Nat Commun 2017; 8: 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ying S, Hamdy FC, Helleday T. Mre11‐dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 2012; 72: 2814–2821. [DOI] [PubMed] [Google Scholar]
- 138. Pefani DE, Latusek R, Pires I, et al RASSF1A–LATS1 signalling stabilizes replication forks by restricting CDK2‐mediated phosphorylation of BRCA2. Nat Cell Biol 2014; 16: 962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chaudhuri AR, Callen E, Ding X, et al Erratum: Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature 2016; 539: 456. [DOI] [PubMed] [Google Scholar]
- 140. Lemacon D, Jackson J, Quinet A, et al MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81‐dependent fork rescue in BRCA2‐deficient cells. Nat Commun 2017; 8: 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Cunniff C, Bassetti JA, Ellis NA. Bloom's syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol Syndromol 2017; 8: 4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lebel M, Monnat RJ, Jr . Werner syndrome (WRN) gene variants and their association with altered function and age‐associated diseases. Ageing Res Rev 2017; 41: 82–97. [DOI] [PubMed] [Google Scholar]
- 143. Nair N, Shoaib M, Sorensen CS. Chromatin dynamics in genome stability: roles in suppressing endogenous DNA damage and facilitating DNA repair. Int J Mol Sci 2017; 18: E1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Nishibuchi G, Dejardin J. The molecular basis of the organization of repetitive DNA‐containing constitutive heterochromatin in mammals. Chromosome Res 2017; 25: 77–87. [DOI] [PubMed] [Google Scholar]
- 145. Maciejowski J, de Lange T. Telomeres in cancer: tumor suppression and genome instability. Nat Rev Mol Cell Biol 2017; 18: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Moyzis RK, Buckingham JM, Cram LS, et al A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A 1988; 85: 6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Schoeftner S, Blasco MA. A ‘higher order’ of telomere regulation: telomere heterochromatin and telomeric RNAs. EMBO J 2009; 28: 2323–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Arnoult N, Karlseder J. Complex interactions between the DNA‐damage response and mammalian telomeres. Nat Struct Mol Biol 2015; 22: 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Cann KL, Dellaire G. Heterochromatin and the DNA damage response: the need to relax. Biochem Cell Biol 2011; 89: 45–60. [DOI] [PubMed] [Google Scholar]
- 150. Kalousi A, Soutoglou E. Nuclear compartmentalization of DNA repair. Curr Opin Genet Dev 2016; 37: 148–157. [DOI] [PubMed] [Google Scholar]
- 151. Kalousi A, Hoffbeck AS, Selemenakis PN, et al The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep 2015; 11: 149–163. [DOI] [PubMed] [Google Scholar]
- 152. Klement K, Luijsterburg MS, Pinder JB, et al Opposing ISWI‐ and CHD‐class chromatin remodeling activities orchestrate heterochromatic DNA repair. J Cell Biol 2014; 207: 717–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Huen MS, Huang J, Leung JW, et al Regulation of chromatin architecture by the PWWP domain‐containing DNA damage‐responsive factor EXPAND1/MUM1. Mol Cell 2010; 37: 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Noon AT, Shibata A, Rief N, et al 53BP1‐dependent robust localized KAP‐1 phosphorylation is essential for heterochromatic DNA double‐strand break repair. Nat Cell Biol 2010; 12: 177–184. [DOI] [PubMed] [Google Scholar]
- 155. Orsolic I, Jurada D, Pullen N, et al The relationship between the nucleolus and cancer: current evidence and emerging paradigms. Semin Cancer Biol 2016; 37–38: 36–50. [DOI] [PubMed] [Google Scholar]
- 156. Boisvert FM, van Koningsbruggen S, Navascues J, et al The multifunctional nucleolus. Nat Rev Mol Cell Biol 2007; 8: 574–585. [DOI] [PubMed] [Google Scholar]
- 157. Montanaro L, Trere D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol 2008; 173: 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Russell J, Zomerdijk JC. RNA‐polymerase‐I‐directed rDNA transcription, life and works. Trends Biochem Sci 2005; 30: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Komseli ES, Pateras IS, Krejsgaard T, et al A prototypical non‐malignant epithelial model to study genome dynamics and concurrently monitor micro‐RNAs and proteins in situ during oncogene‐induced senescence. BMC Genomics 2018; 19: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Akamatsu Y, Kobayashi T. The human RNA polymerase I transcription terminator complex acts as a replication fork barrier that coordinates the progress of replication with rRNA transcription activity. Mol Cell Biol 2015; 35: 1871–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Deregowska A, Adamczyk J, Kwiatkowska A, et al Shifts in rDNA levels act as a genome buffer promoting chromosome homeostasis. Cell Cycle 2015; 14: 3475–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Grummt I. The nucleolus – guardian of cellular homeostasis and genome integrity. Chromosoma 2013; 122: 487–497. [DOI] [PubMed] [Google Scholar]
- 163. Kobayashi T. A new role of the rDNA and nucleolus in the nucleus – rDNA instability maintains genome integrity. Bioessays 2008; 30: 267–272. [DOI] [PubMed] [Google Scholar]
- 164. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999; 13: 2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gottlieb S, Esposito RE. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell 1989; 56: 771–776. [DOI] [PubMed] [Google Scholar]
- 166. Santoro R, Li J, Grummt I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 2002; 32: 393–396. [DOI] [PubMed] [Google Scholar]
- 167. Murayama A, Ohmori K, Fujimura A, et al Epigenetic control of rDNA loci in response to intracellular energy status. Cell 2008; 133: 627–639. [DOI] [PubMed] [Google Scholar]
- 168. Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 2011; 11: 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Srivastava R, Srivastava R, Ahn SH. The epigenetic pathways to ribosomal DNA silencing. Microbiol Mol Biol Rev 2016; 80: 545–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Mekhail K, Seebacher J, Gygi SP, et al Role for perinuclear chromosome tethering in maintenance of genome stability. Nature 2008; 456: 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Nemeth A, Langst G. Genome organization in and around the nucleolus. Trends Genet 2011; 27: 149–156. [DOI] [PubMed] [Google Scholar]
- 172. Pelletier J, Thomas G, Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer 2018; 18: 51–63. [DOI] [PubMed] [Google Scholar]
- 173. Boulon S, Westman BJ, Hutten S, et al The nucleolus under stress. Mol Cell 2010; 40: 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Velimezi G, Liontos M, Vougas K, et al Functional interplay between the DNA‐damage‐response kinase ATM and ARF tumor suppressor protein in human cancer. Nat Cell Biol 2013; 15: 967–977. [DOI] [PubMed] [Google Scholar]
- 175. Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem 2007; 76: 781–821. [DOI] [PubMed] [Google Scholar]
- 176. Vasileiou PVS, Mourouzis I, Pantos C. Principal aspects regarding the maintenance of mammalian mitochondrial genome integrity. Int J Mol Sci 2017; 18: E1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Stumpf JD, Copeland WC. Mitochondrial DNA replication and disease: insights from DNA polymerase gamma mutations. Cell Mol Life Sci 2011; 68: 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Bogenhagen DF. Mitochondrial DNA nucleoid structure. Biochim Biophys Acta 2012; 1819: 914–920. [DOI] [PubMed] [Google Scholar]
- 179. Tyynismaa H, Sembongi H, Bokori‐Brown M, et al Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet 2004; 13: 3219–3227. [DOI] [PubMed] [Google Scholar]
- 180. Nikkanen J, Forsstrom S, Euro L, et al Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one‐carbon metabolism. Cell Metab 2016; 23: 635–648. [DOI] [PubMed] [Google Scholar]
- 181. Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer 2014; 14: 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 2012; 13: 659–671. [DOI] [PubMed] [Google Scholar]
- 183. Garcia‐Gomez S, Reyes A, Martinez‐Jimenez MI, et al PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 2013; 52: 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Futami K, Shimamoto A, Furuichi Y. Mitochondrial and nuclear localization of human Pif1 helicase. Biol Pharm Bull 2007; 30: 1685–1692. [DOI] [PubMed] [Google Scholar]
- 185. Tadi SK, Sebastian R, Dahal S, et al Microhomology‐mediated end joining is the principal mediator of double‐strand break repair during mitochondrial DNA lesions. Mol Biol Cell 2016; 27: 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Gumeni S, Trougakos IP. Cross talk of proteostasis and mitostasis in cellular homeodynamics, ageing, and disease. Oxid Med Cell Longev 2016; 2016: 4587691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta 2008; 1777: 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Fang EF, Scheibye‐Knudsen M, Chua KF, et al Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 2016; 17: 308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009; 10: 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Potapova TA, Zhu J, Li R. Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev 2013; 32: 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 2008; 180: 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Funk LC, Zasadil LM, Weaver BA. Living in CIN: mitotic infidelity and its consequences for tumor promotion and suppression. Dev Cell 2016; 39: 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kastan MB, Bartek J. Cell‐cycle checkpoints and cancer. Nature 2004; 432: 316–323. [DOI] [PubMed] [Google Scholar]
- 194. Scolnick DM, Halazonetis TD. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature 2000; 406: 430–435. [DOI] [PubMed] [Google Scholar]
- 195. Mariatos G, Bothos J, Zacharatos P, et al Inactivating mutations targeting the chfr mitotic checkpoint gene in human lung cancer. Cancer Res 2003; 63: 7185–7189. [PubMed] [Google Scholar]
- 196. Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol 2015; 25: R1002–R1018. [DOI] [PubMed] [Google Scholar]
- 197. Steigemann P, Wurzenberger C, Schmitz MH, et al Aurora B‐mediated abscission checkpoint protects against tetraploidization. Cell 2009; 136: 473–484. [DOI] [PubMed] [Google Scholar]
- 198. Norden C, Mendoza M, Dobbelaere J, et al The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell 2006; 125: 85–98. [DOI] [PubMed] [Google Scholar]
- 199. Nahse V, Christ L, Stenmark H, et al The abscission checkpoint: making it to the final cut. Trends Cell Biol 2017; 27: 1–11. [DOI] [PubMed] [Google Scholar]
- 200. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 2005; 5: 773–785. [DOI] [PubMed] [Google Scholar]
- 201. Lara‐Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol 2012; 22: R966–R980. [DOI] [PubMed] [Google Scholar]
- 202. Bakhoum SF, Thompson SL, Manning AL, et al Genome stability is ensured by temporal control of kinetochore–microtubule dynamics. Nat Cell Biol 2009; 11: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Benanti JA, Toczyski DP. Cdc20, an activator at last. Mol Cell 2008; 32: 460–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Liu ST, Zhang H. The mitotic checkpoint complex (MCC): looking back and forth after 15 years. AIMS Mol Sci 2016; 3: 597–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Brinkley BR, Goepfert TM. Supernumerary centrosomes and cancer: Boveri's hypothesis resurrected. Cell Motil Cytoskeleton 1998; 41: 281–288. [DOI] [PubMed] [Google Scholar]
- 206. Satge D, Nishi M, Sirvent N, et al A tumor profile in Patau syndrome (trisomy 13). Am J Med Genet A 2017; 173: 2088–2096. [DOI] [PubMed] [Google Scholar]
- 207. Satge D, Nishi M, Sirvent N, et al A tumor profile in Edwards syndrome (trisomy 18). Am J Med Genet C Semin Med Genet 2016; 172: 296–306. [DOI] [PubMed] [Google Scholar]
- 208. Xavier AC, Ge Y, Taub JW. Down syndrome and malignancies: a unique clinical relationship: a paper from the 2008 William Beaumont hospital symposium on molecular pathology. J Mol Diagn 2009; 11: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2002; 2: 815–825. [DOI] [PubMed] [Google Scholar]
- 210. Koutsami MK, Tsantoulis PK, Kouloukoussa M, et al Centrosome abnormalities are frequently observed in non‐small‐cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J Pathol 2006; 209: 512–521. [DOI] [PubMed] [Google Scholar]
- 211. Loffler H, Lukas J, Bartek J, et al Structure meets function – centrosomes, genome maintenance and the DNA damage response. Exp Cell Res 2006; 312: 2633–2640. [DOI] [PubMed] [Google Scholar]
- 212. Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ 2008; 15: 1153–1162. [DOI] [PubMed] [Google Scholar]
- 213. Sotillo R, Hernando E, Diaz‐Rodriguez E, et al Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007; 11: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Michel LS, Liberal V, Chatterjee A, et al MAD2 haplo‐insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001; 409: 355–359. [DOI] [PubMed] [Google Scholar]
- 215. Pihan G, Doxsey SJ. Mutations and aneuploidy: co‐conspirators in cancer? Cancer Cell 2003; 4: 89–94. [DOI] [PubMed] [Google Scholar]
- 216. Sarni D, Kerem B. Oncogene‐induced replication stress drives genome instability and tumorigenesis. Int J Mol Sci 2017; 18: 1339. [Google Scholar]
- 217. Schvartzman JM, Duijf PH, Sotillo R, et al Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011; 19: 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Malumbres M. Oncogene‐induced mitotic stress: p53 and pRb get mad too. Cancer Cell 2011; 19: 691–692. [DOI] [PubMed] [Google Scholar]
- 219. Hernando E, Nahle Z, Juan G, et al Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 2004; 430: 797–802. [DOI] [PubMed] [Google Scholar]
- 220. Manning AL, Benes C, Dyson NJ. Whole chromosome instability resulting from the synergistic effects of pRB and p53 inactivation. Oncogene 2014; 33: 2487–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Gorgoulis VG, Zacharatos P, Kotsinas A, et al Altered expression of the cell cycle regulatory molecules pRb, p53 and MDM2 exert a synergetic effect on tumor growth and chromosomal instability in non‐small cell lung carcinomas (NSCLCs). Mol Med 2000; 6: 208–237. [PMC free article] [PubMed] [Google Scholar]
- 222. Cesare AJ. Mitosis, double strand break repair, and telomeres: a view from the end: how telomeres and the DNA damage response cooperate during mitosis to maintain genome stability. Bioessays 2014; 36: 1054–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double‐strand breaks during mitosis. J Cell Biol 2010; 190: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Bakhoum SF, Kabeche L, Compton DA, et al Mitotic DNA damage response: at the crossroads of structural and numerical cancer chromosome instabilities. Trends Cancer 2017; 3: 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Reyes C, Serrurier C, Gauthier T, et al Aurora B prevents chromosome arm separation defects by promoting telomere dispersion and disjunction. J Cell Biol 2015; 208: 713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Orthwein A, Fradet‐Turcotte A, Noordermeer SM, et al Mitosis inhibits DNA double‐strand break repair to guard against telomere fusions. Science 2014; 344: 189–193. [DOI] [PubMed] [Google Scholar]
- 227. Macurek L, Lindqvist A, Lim D, et al Polo‐like kinase‐1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455: 119–123. [DOI] [PubMed] [Google Scholar]
- 228. Seki A, Coppinger JA, Jang CY, et al Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320: 1655–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Smits VA, Klompmaker R, Arnaud L, et al Polo‐like kinase‐1 is a target of the DNA damage checkpoint. Nat Cell Biol 2000; 2: 672–676. [DOI] [PubMed] [Google Scholar]
- 230. Bakhoum SF, Kabeche L, Murnane JP, et al DNA‐damage response during mitosis induces whole‐chromosome missegregation. Cancer Discov 2014; 4: 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Liu D, Davydenko O, Lampson MA. Polo‐like kinase‐1 regulates kinetochore–microtubule dynamics and spindle checkpoint silencing. J Cell Biol 2012; 198: 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Stolz A, Ertych N, Kienitz A, et al The CHK2–BRCA1 tumor suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol 2010; 12: 492–499. [DOI] [PubMed] [Google Scholar]
- 233. Zhang CZ, Spektor A, Cornils H, et al Chromothripsis from DNA damage in micronuclei. Nature 2015; 522: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Hayashi MT, Cesare AJ, Rivera T, et al Cell death during crisis is mediated by mitotic telomere deprotection. Nature 2015; 522: 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Hayashi MT, Cesare AJ, Fitzpatrick JA, et al A telomere‐dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol 2012; 19: 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Terasawa M, Shinohara A, Shinohara M. Double‐strand break repair‐adox: restoration of suppressed double‐strand break repair during mitosis induces genomic instability. Cancer Sci 2014; 105: 1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Heijink AM, Krajewska M, van Vugt MA. The DNA damage response during mitosis. Mutat Res 2013; 750: 45–55. [DOI] [PubMed] [Google Scholar]
- 238. Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol 2010; 20: 1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Lukas C, Savic V, Bekker‐Jensen S, et al 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13: 243–253. [DOI] [PubMed] [Google Scholar]
- 240. Moreno A, Carrington JT, Albergante L, et al Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc Natl Acad Sci U S A 2016; 113: E5757–E5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Yang HW, Chung M, Kudo T, et al Competing memories of mitogen and p53 signalling control cell‐cycle entry. Nature 2017; 549: 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Topham CH, Taylor SS. Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol 2013; 25: 780–785. [DOI] [PubMed] [Google Scholar]
- 243. Salazar‐Roa M, Malumbres M. Fueling the cell division cycle. Trends Cell Biol 2017; 27: 69–81. [DOI] [PubMed] [Google Scholar]
- 244. Kulak NA, Geyer PE, Mann M. Loss‐less nano‐fractionator for high sensitivity, high coverage proteomics. Mol Cell Proteomics 2017; 16: 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Balchin D, Hayer‐Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science 2016; 353: aac4354. [DOI] [PubMed] [Google Scholar]
- 246. Gidalevitz T, Ben‐Zvi A, Ho KH, et al Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006; 311: 1471–1474. [DOI] [PubMed] [Google Scholar]
- 247. Gidalevitz T, Krupinski T, Garcia S, et al Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet 2009; 5: e1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol 2018; 217: 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Balch WE, Morimoto RI, Dillin A, et al Adapting proteostasis for disease intervention. Science 2008; 319: 916–919. [DOI] [PubMed] [Google Scholar]
- 250. Powers ET, Morimoto RI, Dillin A, et al Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009; 78: 959–991. [DOI] [PubMed] [Google Scholar]
- 251. Trougakos IP, Sesti F, Tsakiri E, et al Non‐enzymatic post‐translational protein modifications and proteostasis network deregulation in carcinogenesis. J Proteomics 2013; 92: 274–298. [DOI] [PubMed] [Google Scholar]
- 252. Brehme M, Voisine C, Rolland T, et al A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 2014; 9: 1135–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Sala AJ, Bott LC, Morimoto RI. Shaping proteostasis at the cellular, tissue, and organismal level. J Cell Biol 2017; 216: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Hammond CM, Stromme CB, Huang H, et al Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol 2017; 18: 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Li X, Tyler JK. Nucleosome disassembly during human non‐homologous end joining followed by concerted HIRA‐ and CAF‐1‐dependent reassembly. Elife 2016; 5: e15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Nijman SM, Luna‐Vargas MP, Velds A, et al A genomic and functional inventory of deubiquitinating enzymes. Cell 2005; 123: 773–786. [DOI] [PubMed] [Google Scholar]
- 257. Li W, Bengtson MH, Ulbrich A, et al Genome‐wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One 2008; 3: e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Sowa ME, Bennett EJ, Gygi SP, et al Defining the human deubiquitinating enzyme interaction landscape. Cell 2009; 138: 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Varshavsky A. The ubiquitin system, an immense realm. Annu Rev Biochem 2012; 81: 167–176. [DOI] [PubMed] [Google Scholar]
- 260. Garcia‐Prat L, Martinez‐Vicente M, Perdiguero E, et al Autophagy maintains stemness by preventing senescence. Nature 2016; 529: 37–42. [DOI] [PubMed] [Google Scholar]
- 261. Tsakiri EN, Trougakos IP. The amazing ubiquitin–proteasome system: structural components and implication in aging. Int Rev Cell Mol Biol 2015; 314: 171–237. [DOI] [PubMed] [Google Scholar]
- 262. Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer 2018; 18: 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Hohn TJ, Grune T. The proteasome and the degradation of oxidized proteins: part III – Redox regulation of the proteasomal system. Redox Biol 2014; 2: 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum‐associated degradation. Nat Rev Mol Cell Biol 2008; 9: 944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Neutzner A, Youle RJ, Karbowski M. Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found Symp 2007; 287: 4–14; discussion 14–20. [PubMed] [Google Scholar]
- 266. Cuervo AM. Chaperone‐mediated autophagy: selectivity pays off. Trends Endocrinol Metab 2010; 21: 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Mizushima N, Levine B, Cuervo AM, et al Autophagy fights disease through cellular self‐digestion. Nature 2008; 451: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Klionsky DJ, Abdelmohsen K, Abe A, et al Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12: 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Cuervo AM, Wong E. Chaperone‐mediated autophagy: roles in disease and aging. Cell Res 2014; 24: 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13: 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Kirkin V, Dikic I. Ubiquitin networks in cancer. Curr Opin Genet Dev 2011; 21: 21–28. [DOI] [PubMed] [Google Scholar]
- 272. Havaki S, Vlachou V, Zampetidis CP, et al Monitoring autophagy immunohistochemically and ultrastructurally during human head and neck carcinogenesis. Relationship with the DNA damage response pathway. Int J Mol Sci 2017; 18: E1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Eliopoulos AG, Havaki S, Gorgoulis VG. DNA damage response and autophagy: a meaningful partnership. Front Genet 2016; 7: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Gumeni S, Evangelakou Z, Gorgoulis VG, et al Proteome stability as a key factor of genome integrity. Int J Mol Sci 2017; 18: E2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Alexander A, Cai SL, Kim J, et al ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A 2010; 107: 4153–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010; 6: 672–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Rodriguez‐Vargas JM, Ruiz‐Magana MJ, Ruiz‐Ruiz C, et al ROS‐induced DNA damage and PARP‐1 are required for optimal induction of starvation‐induced autophagy. Cell Res 2012; 22: 1181–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Pietrocola F, Izzo V, Niso‐Santano M, et al Regulation of autophagy by stress‐responsive transcription factors. Semin Cancer Biol 2013; 23: 310–322. [DOI] [PubMed] [Google Scholar]
- 279. Gomes LR, Menck CFM, Leandro GS. Autophagy roles in the modulation of DNA repair pathways. Int J Mol Sci 2017; 18: E2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Kouranti I, Peyroche A. Protein degradation in DNA damage response. Semin Cell Dev Biol 2012; 23: 538–545. [DOI] [PubMed] [Google Scholar]
- 281. Sklirou A, Papanagnou ED, Fokialakis N, et al Cancer chemoprevention via activation of proteostatic modules. Cancer Lett 2018; 413: 110–121. [DOI] [PubMed] [Google Scholar]
- 282. Nedic O, Rattan SI, Grune T, et al Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic Res 2013; 47(suppl 1): 28–38. [DOI] [PubMed] [Google Scholar]
- 283. Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem 2015; 84: 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Harding HP, Novoa I, Zhang Y, et al Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 2000; 6: 1099–1108. [DOI] [PubMed] [Google Scholar]
- 285. Biamonti G, Caceres JF. Cellular stress and RNA splicing. Trends Biochem Sci 2009; 34: 146–153. [DOI] [PubMed] [Google Scholar]
- 286. Shalgi R, Hurt JA, Krykbaeva I, et al Widespread regulation of translation by elongation pausing in heat shock. Mol Cell 2013; 49: 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Niforou K, Cheimonidou C, Trougakos IP. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol 2014; 2: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Zou J, Guo Y, Guettouche T, et al Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress‐sensitive complex with HSF1. Cell 1998; 94: 471–480. [DOI] [PubMed] [Google Scholar]
- 289. Zheng X, Krakowiak J, Patel N, et al Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. Elife 2016; 5: e18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 2005; 74: 739–789. [DOI] [PubMed] [Google Scholar]
- 291. Haynes CM, Petrova K, Benedetti C, et al ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 2007; 13: 467–480. [DOI] [PubMed] [Google Scholar]
- 292. Nargund AM, Pellegrino MW, Fiorese CJ, et al Mitochondrial import efficiency of ATFS‐1 regulates mitochondrial UPR activation. Science 2012; 337: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol 2018; 19: 109–120. [DOI] [PubMed] [Google Scholar]
- 294. Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 2011; 36: 254–261. [DOI] [PubMed] [Google Scholar]
- 295. Lendahl U, Lee KL, Yang H, et al Generating specificity and diversity in the transcriptional response to hypoxia. Nat Rev Genet 2009; 10: 821–832. [DOI] [PubMed] [Google Scholar]
- 296. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 2004; 5: 343–354. [DOI] [PubMed] [Google Scholar]
- 297. Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis 2010; 31: 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Sykiotis GP, Bohmann D. Stress‐activated cap'n'collar transcription factors in aging and human disease. Sci Signal 2010; 3: re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Tsakiri EN, Sykiotis GP, Papassideri IS, et al Proteasome dysfunction in Drosophila signals to an Nrf2‐dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 2013; 12: 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Li X, Matilainen O, Jin C, et al Specific SKN‐1/Nrf stress responses to perturbations in translation elongation and proteasome activity. PLoS Genet 2011; 7: e1002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Lopez‐Otin C, Blasco MA, Partridge L, et al The hallmarks of aging. Cell 2013; 153: 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. van Sluis M, McStay B. Ribosome biogenesis: Achilles heel of cancer? Genes Cancer 2014; 5: 152–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non‐oncogene addiction. Cell 2009; 136: 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767. [DOI] [PubMed] [Google Scholar]
- 305. Jass JR. Colorectal cancer: a multipathway disease. Crit Rev Oncog 2006; 12: 273–287. [DOI] [PubMed] [Google Scholar]
- 306. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789–799. [DOI] [PubMed] [Google Scholar]
- 307. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 308. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 309. Gorgoulis VG, Vassiliou LV, Karakaidos P, et al Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434: 907–913. [DOI] [PubMed] [Google Scholar]
- 310. Bartkova J, Horejsi Z, Koed K, et al DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 2005; 434: 864–870. [DOI] [PubMed] [Google Scholar]
- 311. Franklin WA, Veve R, Hirsch FR, et al Epidermal growth factor receptor family in lung cancer and premalignancy. Semin Oncol 2002; 29: 3–14. [DOI] [PubMed] [Google Scholar]
- 312. Roskoski R, Jr . The ErbB/HER family of protein‐tyrosine kinases and cancer. Pharmacol Res 2014; 79: 34–74. [DOI] [PubMed] [Google Scholar]
- 313. Gorgoulis V, Giatromanolaki A, Karameris A, et al Epidermal growth factor receptor expression in squamous cell lung carcinomas: an immunohistochemical and gene analysis in formalin‐fixed, paraffin‐embedded material. Virchows Arch A Pathol Anat Histopathol 1993; 423: 295–302. [DOI] [PubMed] [Google Scholar]
- 314. Liontos M, Koutsami M, Sideridou M, et al Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res 2007; 67: 10899–10909. [DOI] [PubMed] [Google Scholar]
- 315. Tsantoulis PK, Kotsinas A, Sfikakis PP, et al Oncogene‐induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome‐wide study. Oncogene 2008; 27: 3256–3264. [DOI] [PubMed] [Google Scholar]
- 316. Harrigan JA, Belotserkovskaya R, Coates J, et al Replication stress induces 53BP1‐containing OPT domains in G1 cells. J Cell Biol 2011; 193: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Chan KL, Palmai‐Pallag T, Ying S, et al Replication stress induces sister‐chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 2009; 11: 7537–7560. [DOI] [PubMed] [Google Scholar]
- 318. Glover TW, Wilson TE, Arlt MF. Fragile sites in cancer: more than meets the eye. Nat Rev Cancer 2017; 17: 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Sarni D, Kerem B. The complex nature of fragile site plasticity and its importance in cancer. Curr Opin Cell Biol 2016; 40: 131–136. [DOI] [PubMed] [Google Scholar]
- 320. Debatisse M, Le Tallec B, Letessier A, et al Common fragile sites: mechanisms of instability revisited. Trends Genet 2012; 28: 22–32. [DOI] [PubMed] [Google Scholar]
- 321. Karras JR, Schrock MS, Batar B, et al Fragile genes that are frequently altered in cancer: players not passengers. Cytogenet Genome Res 2016; 150: 208–216. [DOI] [PubMed] [Google Scholar]
- 322. Del Mare S, Husanie H, Iancu O, et al WWOX and p53 dysregulation synergize to drive the development of osteosarcoma. Cancer Res 2016; 76: 6107–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Waters CE, Saldivar JC, Hosseini SA, et al The FHIT gene product: tumor suppressor and genome ‘caretaker’. Cell Mol Life Sci 2014; 71: 4577–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Gardenswartz A, Aqeilan RI. WW domain‐containing oxidoreductase's role in myriad cancers: clinical significance and future implications. Exp Biol Med (Maywood) 2014; 239: 253–263. [DOI] [PubMed] [Google Scholar]
- 325. Paige AJ, Taylor KJ, Taylor C, et al WWOX: a candidate tumor suppressor gene involved in multiple tumor types. Proc Natl Acad Sci U S A 2001; 98: 11417–11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Nikolaev SI, Sotiriou SK, Pateras IS, et al A single‐nucleotide substitution mutator phenotype revealed by exome sequencing of human colon adenomas. Cancer Res 2012; 72: 6279–6289. [DOI] [PubMed] [Google Scholar]
- 327. Bartkova J, Rezaei N, Liontos M, et al Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444: 633–637. [DOI] [PubMed] [Google Scholar]
- 328. Di Micco R, Fumagalli M, Cicalese A, et al Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature 2006; 444: 638–642. [DOI] [PubMed] [Google Scholar]
- 329. Liontos M, Niforou K, Velimezi G, et al Modulation of the E2F1‐driven cancer cell fate by the DNA damage response machinery and potential novel E2F1 targets in osteosarcomas. Am J Pathol 2009; 175: 376–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Evangelou K, Bartkova J, Kotsinas A, et al The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ 2013; 20: 1485–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol 2015; 10: 425–448. [DOI] [PubMed] [Google Scholar]
- 332. O'Driscoll M. The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery. J Pathol 2017; 241: 192–207. [DOI] [PubMed] [Google Scholar]
- 333. Tsantoulis PK, Gorgoulis VG. Involvement of E2F transcription factor family in cancer. Eur J Cancer 2005; 41: 2403–2414. [DOI] [PubMed] [Google Scholar]
- 334. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell 2002; 2: 103–112. [DOI] [PubMed] [Google Scholar]
- 335. Gorgoulis VG, Zacharatos P, Kotsinas A, et al Alterations of the p16–pRb pathway and the chromosome locus 9p21–22 in non‐small‐cell lung carcinomas: relationship with p53 and MDM2 protein expression. Am J Pathol 1998; 153: 1749–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Gorgoulis VG, Koutroumbi EN, Kotsinas A, et al Alterations of p16–pRb pathway and chromosome locus 9p21–22 in sporadic invasive breast carcinomas. Mol Med 1998; 4: 807–822. [PMC free article] [PubMed] [Google Scholar]
- 337. Mariatos G, Gorgoulis VG, Zacharatos P, et al Expression of p16(INK4A) and alterations of the 9p21–23 chromosome region in non‐small‐cell lung carcinomas: relationship with tumor growth parameters and ploidy status. Int J Cancer 2000; 89: 133–141. [DOI] [PubMed] [Google Scholar]
- 338. Gorgoulis VG, Zacharatos P, Mariatos G, et al Transcription factor E2F‐1 acts as a growth‐promoting factor and is associated with adverse prognosis in non‐small cell lung carcinomas. J Pathol 2002; 198: 142–156. [DOI] [PubMed] [Google Scholar]
- 339. Zacharatos P, Kotsinas A, Evangelou K, et al Distinct expression patterns of the transcription factor E2F‐1 in relation to tumor growth parameters in common human carcinomas. J Pathol 2004; 203: 744–753. [DOI] [PubMed] [Google Scholar]
- 340. Karakaidos P, Taraviras S, Vassiliou LV, et al Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non‐small‐cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability – evidence of E2F‐1 transcriptional control over hCdt1. Am J Pathol 2004; 165: 1351–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Walter D, Hoffmann S, Komseli ES, et al SCF(Cyclin F)‐dependent degradation of CDC6 suppresses DNA re‐replication. Nat Commun 2016; 7: 10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Galanos P, Vougas K, Walter D, et al Chronic p53‐independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016; 18: 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Sideridou M, Zakopoulou R, Evangelou K, et al Cdc6 expression represses E‐cadherin transcription and activates adjacent replication origins. J Cell Biol 2011; 195: 1123–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Nieto MA, Huang RY, Jackson RA, et al Emt: 2016. Cell 2016; 166: 21–45. [DOI] [PubMed] [Google Scholar]
- 345. Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest 2009; 119: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 2005; 6: 44–55. [DOI] [PubMed] [Google Scholar]
- 347. Garinis GA, Gorgoulis VG, Mariatos G, et al Association of allelic loss at the FHIT locus and p53 alterations with tumor kinetics and chromosomal instability in non‐small cell lung carcinomas (NSCLCs). J Pathol 2001; 193: 55–65. [DOI] [PubMed] [Google Scholar]
- 348. Gorgoulis VG, Zacharatos P, Mariatos G, et al Deregulated expression of c‐mos in non‐small cell lung carcinomas: relationship with p53 status, genomic instability, and tumor kinetics. Cancer Res 2001; 61: 538–549. [PubMed] [Google Scholar]
- 349. Kotsinas A, Evangelou K, Zacharatos P, et al Proliferation, but not apoptosis, is associated with distinct beta‐catenin expression patterns in non‐small‐cell lung carcinomas: relationship with adenomatous polyposis coli and G(1)‐to S‐phase cell‐cycle regulators. Am J Pathol 2002; 161: 1619–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Apostolopoulou K, Pateras IS, Evangelou K, et al Gene amplification is a relatively frequent event leading to ZBTB7A (Pokemon) overexpression in non‐small cell lung cancer. J Pathol 2007; 213: 294–302. [DOI] [PubMed] [Google Scholar]
- 351. Ochs F, Somyajit K, Altmeyer M, et al 53BP1 fosters fidelity of homology‐directed DNA repair. Nat Struct Mol Biol 2016; 23: 714–721. [DOI] [PubMed] [Google Scholar]
- 352. Ottaviani D, LeCain M, Sheer D. The role of microhomology in genomic structural variation. Trends Genet 2014; 30: 85–94. [DOI] [PubMed] [Google Scholar]
- 353. Sotiriou SK, Kamileri I, Lugli N, et al Mammalian RAD52 functions in break‐induced replication repair of collapsed DNA replication forks. Mol Cell 2016; 64: 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Galanos P, Pappas G, Polyzos A, et al Mutational signatures reveal the role of RAD52 in p53‐independent p21‐driven genomic instability. GenomeBiol 2018; 19: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol 2010; 2: a001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Miyamoto S. Nuclear initiated NF‐kappaB signaling: NEMO and ATM take center stage. Cell Res 2011; 21: 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Pasparakis M. Regulation of tissue homeostasis by NF‐kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol 2009; 9: 778–788. [DOI] [PubMed] [Google Scholar]
- 358. Bredemeyer AL, Helmink BA, Innes CL, et al DNA double‐strand breaks activate a multi‐functional genetic program in developing lymphocytes. Nature 2008; 456: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Wu ZH, Shi Y, Tibbetts RS, et al Molecular linkage between the kinase ATM and NF‐kappaB signaling in response to genotoxic stimuli. Science 2006; 311: 1141–1146. [DOI] [PubMed] [Google Scholar]
- 360. Li N, Banin S, Ouyang H, et al ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem 2001; 276: 8898–8903. [DOI] [PubMed] [Google Scholar]
- 361. Soriani A, Zingoni A, Cerboni C, et al ATM‐ATR‐dependent up‐regulation of DNAM‐1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK‐cell susceptibility and is associated with a senescent phenotype. Blood 2009; 113: 3503–3511. [DOI] [PubMed] [Google Scholar]
- 362. Gasser S, Raulet DH. The DNA damage response arouses the immune system. Cancer Res 2006; 66: 3959–3962. [DOI] [PubMed] [Google Scholar]
- 363. Gasser S, Orsulic S, Brown EJ, et al The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005; 436: 1186–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Xue W, Zender L, Miething C, et al Senescence and tumor clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445: 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Gorgoulis VG, Pratsinis H, Zacharatos P, et al p53‐dependent ICAM‐1 overexpression in senescent human cells identified in atherosclerotic lesions. Lab Invest 2005; 85: 502–511. [DOI] [PubMed] [Google Scholar]
- 366. Gorgoulis VG, Zacharatos P, Kotsinas A, et al p53 activates ICAM‐1 (CD54) expression in an NF‐kappaB‐independent manner. EMBO J 2003; 22: 1567–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Lasry A, Ben‐Neriah Y. Senescence‐associated inflammatory responses: aging and cancer perspectives. Trends Immunol 2015; 36: 217–228. [DOI] [PubMed] [Google Scholar]
- 368. Ermolaeva MA, Schumacher B. Systemic DNA damage responses: organismal adaptations to genome instability. Trends Genet 2014; 30: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Chatzinikolaou G, Karakasilioti I, Garinis GA. DNA damage and innate immunity: links and trade‐offs. Trends Immunol 2014; 35: 429–435. [DOI] [PubMed] [Google Scholar]
- 370. Wang SJ, Gu W. To be, or not to be: functional dilemma of p53 metabolic regulation. Curr Opin Oncol 2014; 26: 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Li Y, Tergaonkar V. Telomerase reactivation in cancers: mechanisms that govern transcriptional activation of the wild‐type vs. mutant TERT promoters. Transcription 2016; 7: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Boulahbel H, Duran RV, Gottlieb E. Prolyl hydroxylases as regulators of cell metabolism. Biochem Soc Trans 2009; 37: 291–294. [DOI] [PubMed] [Google Scholar]
- 373. Milanovic M, Fan DNY, Belenki D, et al Senescence‐associated reprogramming promotes cancer stemness. Nature 2018; 553: 96–100. [DOI] [PubMed] [Google Scholar]
- 374. Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 2012; 22: 50–55. [DOI] [PubMed] [Google Scholar]
- 375. Daskalos A, Nikolaidis G, Xinarianos G, et al Hypomethylation of retrotransposable elements correlates with genomic instability in non‐small cell lung cancer. Int J Cancer 2009; 124: 81–87. [DOI] [PubMed] [Google Scholar]
- 376. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 2012; 21: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Ogrunc M, Di Micco R, Liontos M, et al Oncogene‐induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ 2014; 21: 998–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Dobbelstein M, Sorensen CS. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 2015; 14: 405–423. [DOI] [PubMed] [Google Scholar]
- 379. Aziz K, Nowsheen S, Pantelias G, et al Targeting DNA damage and repair: embracing the pharmacological era for successful cancer therapy. Pharmacol Ther 2012; 133: 334–350. [DOI] [PubMed] [Google Scholar]
- 380. Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 2017; 18: 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481: 287–294. [DOI] [PubMed] [Google Scholar]
- 382. Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res 2015; 21: 4780–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013; 12: 3159–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Asghar U, Witkiewicz AK, Turner NC, et al The history and future of targeting cyclin‐dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14: 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Miki T, Matsumoto T, Zhao Z, et al p53 regulates Period2 expression and the circadian clock. Nat Commun 2013; 4: 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Gaddameedhi S, Selby CP, Kaufmann WK, et al Control of skin cancer by the circadian rhythm. Proc Natl Acad Sci U S A 2011; 108: 18790–18795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Wang H, van Spyk E, Liu Q, et al Time‐restricted feeding shifts the skin circadian clock and alters UVB‐induced DNA damage. Cell Rep 2017; 20: 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Manzella N, Bracci M, Strafella E, et al Circadian modulation of 8‐oxoguanine DNA damage repair. Sci Rep 2015; 5: 13752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Puigvert JC, Sanjiv K, Helleday T. Targeting DNA repair, DNA metabolism and replication stress as anti‐cancer strategies. FEBS J 2016; 283: 232–245. [DOI] [PubMed] [Google Scholar]
- 390. Warpman Berglund U, Sanjiv K, Gad H, et al Validation and development of MTH1 inhibitors for treatment of cancer. Ann Oncol 2016; 27: 2275–2283. [DOI] [PubMed] [Google Scholar]
- 391. Gad H, Koolmeister T, Jemth AS, et al MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014; 508: 215–221. [DOI] [PubMed] [Google Scholar]
- 392. Giribaldi MG, Munoz A, Halvorsen K, et al MTH1 expression is required for effective transformation by oncogenic HRAS. Oncotarget 2015; 6: 11519–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res 2000; 60: 3689–3695. [PubMed] [Google Scholar]
- 394. Sherr CJ, Bertwistle D, Den Besten W, et al p53‐Dependent and ‐independent functions of the Arf tumor suppressor. Cold Spring Harb Symp Quant Biol 2005; 70: 129–137. [DOI] [PubMed] [Google Scholar]
- 395. Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer 2006; 6: 663–673. [DOI] [PubMed] [Google Scholar]
- 396. Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med 2015; 21: 1406–1415. [DOI] [PubMed] [Google Scholar]
- 397. Trepel J, Mollapour M, Giaccone G, et al Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 2010; 10: 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Dimopoulos MA, Moreau P, Palumbo A, et al Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open‐label, multicentre study. Lancet Oncol 2016; 17: 27–38. [DOI] [PubMed] [Google Scholar]
- 399. Raj L, Ide T, Gurkar AU, et al Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011; 475: 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 400. Koutsami M, Velimezi G, Kotsinas A, et al Is exclusive Skp2 targeting always beneficial in cancer therapy? Blood 2008; 112: 4777–4779. [DOI] [PubMed] [Google Scholar]
- 401. Solomon H, Dinowitz N, Pateras IS, et al Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 2018; 37: 1669–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Oren M, Tal P, Rotter V. Targeting mutant p53 for cancer therapy. Aging (Albany NY) 2016; 8: 1159–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Cooks T, Pateras IS, Tarcic O, et al Mutant p53 prolongs NF‐kappaB activation and promotes chronic inflammation and inflammation‐associated colorectal cancer. Cancer Cell 2013; 23: 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Gorgoulis VG, Zacharatos PV, Manolis E, et al Effects of p53 mutants derived from lung carcinomas on the p53‐responsive element (p53RE) of the MDM2 gene. Br J Cancer 1998; 77: 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Gorgoulis VG, Zoumpourlis V, Rassidakis GZ, et al A molecular and immunohistochemical study of the MDM2 protein isoforms and p53 gene product in bronchogenic carcinoma. J Pathol 1996; 180: 129–137. [DOI] [PubMed] [Google Scholar]
- 406. Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumor immune responses. Nat Rev Cancer 2018; 18: 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Cooks T, Pateras IS, Jenkins LM, et al Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR‐1246. Nat Commun 2018; 9: 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Edifizi D, Schumacher B. Omics approaches for identifying physiological adaptations to genome instability in aging. Int J Mol Sci 2017; 18: E2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Langmead B, Nellore A. Cloud computing for genomic data analysis and collaboration. Nat Rev Genet 2018; 19: 325. [DOI] [PubMed] [Google Scholar]
- 410. Cooper LA, Demicco EG, Saltz JH, et al Pan‐cancer insights from The Cancer Genome Atlas: the pathologist's perspective. J Pathol 2018; 244: 512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Angermueller C, Parnamaa T, Parts L, et al Deep learning for computational biology. Mol Syst Biol 2016; 12: 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Campbell BB, Light N, Fabrizio D, et al Comprehensive analysis of hypermutation in human cancer. Cell 2017; 171: 1042–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Martincorena I, Raine KM, Gerstung M, et al Universal patterns of selection in cancer and somatic tissues. Cell 2017; 171: 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Schmitt AD, Hu M, Ren B. Genome‐wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol 2016; 17: 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Mumbach MR, Rubin AJ, Flynn RA, et al HiChIP: efficient and sensitive analysis of protein‐directed genome architecture. Nat Methods 2016; 13: 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Rao SS, Huntley MH, Durand NC, et al A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014; 159: 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Georgakopoulou EA, Tsimaratou K, Evangelou K, et al Specific lipofuscin staining as a novel biomarker to detect replicative and stress‐induced senescence. A method applicable in cryo‐preserved and archival tissues. Aging (Albany NY) 2013; 5: 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Evangelou K, Lougiakis N, Rizou SV, et al Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017; 16: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Asano M, Basieva I, Khrennikov A, et al A model of differentiation in quantum bioinformatics. Prog Biophys Mol Biol 2017; 130: 88–98. [DOI] [PubMed] [Google Scholar]
- 420. Kotsinas A, Papanagnou P, Galanos P, et al MKK7 and ARF: new players in the DNA damage response scenery. Cell Cycle 2014; 13: 1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Kotsinas A, Papanagnou P, Evangelou K, et al ARF: a versatile DNA damage response ally at the crossroads of development and tumorigenesis. Front Genet 2014; 5: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Burgess RC, Misteli T. Not all DDR are created equal: non‐canonical DNA damage responses. Cell 2015; 162: 944–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9: 537–549. [DOI] [PubMed] [Google Scholar]
- 424. Park JH, Shin JE, Park HW. The role of Hippo pathway in cancer stem cell biology. Mol Cells 2018; 41: 83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Pefani DE, O'Neil E. Hippo pathway and protection of genome stability in response to DNA damage. FEBS J 2016; 283: 1392–1403. [DOI] [PubMed] [Google Scholar]
- 426. Karimaian A, Majidinia M, Bannazadeh Baghi H, et al The crosstalk between Wnt/β‐catenin signaling pathway with DNA damage response and oxidative stress: implications in cancer therapy. DNA Repair (Amst) 2017; 51: 14–19. [DOI] [PubMed] [Google Scholar]
- 427. Essers MA, de Vries‐Smits LM, Barker N, et al Functional interaction between beta‐catenin and FOXO in oxidative stress signaling. Science 2005; 308: 1181–1184. [DOI] [PubMed] [Google Scholar]
- 428. Funato Y, Michiue T, Asashima M, et al The thioredoxin‐related redox‐regulating protein nucleoredoxin inhibits Wnt–beta‐catenin signalling through dishevelled. Nat Cell Biol 2006; 8: 501–508. [DOI] [PubMed] [Google Scholar]
- 429. Vermezovic J, Adamowicz M, Santarpia L, et al Notch is a direct negative regulator of the DNA‐damage response. Nature 2015; 22: 417–424. [DOI] [PubMed] [Google Scholar]
- 430. Meng E, Hanna A, Samant RS, et al The impact of Hedgehog signaling pathway on DNA repair mechanisms in human cancer. Cancers 2015; 7: 1333–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Cohen I, Rider P, Vornov E, et al IL‐1α is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci Rep 2015; 5: 14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Engreitz JM, Pandya‐Jones A, McDonel P, et al The Xist lncRNA exploits three‐dimensional genome architecture to spread across the X chromosome. Science 2013; 341: 1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Rao AKDM, Rajkumar T, Mani S. Perspectives of long non‐coding RNAs in cancer. Mol Biol Rep 2017; 44: 203–218. [DOI] [PubMed] [Google Scholar]
- 434. Pateras IS, Apostolopoulou K, Koutsami M, et al Downregulation of the KIP family members p27(KIP1) and p57(KIP2) by SKP2 and the role of methylation in p57(KIP2) inactivation in nonsmall cell lung cancer. Int J Cancer 2006; 119: 2546–2556. [DOI] [PubMed] [Google Scholar]
- 435. Pateras IS, Apostolopoulou K, Niforou K, et al p57KIP2: ‘Kip'ing the cell under control. Mol Cancer Res 2009; 7: 1902–1919. [DOI] [PubMed] [Google Scholar]
- 436. Mowel WK, Kotzin JJ, McCright SJ, et al Control of immune cell homeostasis and function by lncRNAs. Trends Immunol 2018; 39: 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol 2009; 4: 199–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiol Rev 2011; 91: 827–887. [DOI] [PubMed] [Google Scholar]
- 439. Stepanov GA, Filippova JA, Komissarov AB, et al Regulatory role of small nucleolar RNAs in human diseases. Biomed Res Int 2015; 2015: 206849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Mannoor K, Liao J, Jiang F. Small nucleolar RNAs in cancer. Biochim Biophys Acta 2012; 1826: 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. McMahon M, Contreras A, Ruggero D. Small RNAs with big implications: new insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip Rev RNA 2015; 6: 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Greene J, Baird AM, Brady L, et al Circular RNAs: biogenesis, function and role in human diseases. Front Mol Biosci 2017; 4: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Benitez‐Buelga C, Vaclová T, Ferreira S, et al Molecular insights into the OGG1 gene, a cancer risk modifier in BRCA1 and BRCA2 mutations carriers. Oncotarget 2016; 7: 25815–25825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Weren RD, Ligtenberg MJ, Geurts van Kessel A, et al NTHL1 and MUTYH polyposis syndromes: two sides of the same coin? J Pathol 2018; 244: 135–142. [DOI] [PubMed] [Google Scholar]
- 445. Shen J, Gilmore EC, Marshall CA, et al Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet 2010; 42: 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Pachlopnik Schmid J, Lemoine R, Nehme N, et al Polymerase ϵ1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (‘FILS syndrome’). J Exp Med 2012; 209: 2323–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer 2005; 5: 564‐573. [DOI] [PubMed] [Google Scholar]
- 448. Berneburg M, Lehmann AR. Xeroderma pigmentosum and related disorders: defects in DNA repair and transcription. Adv Genet 2001; 43: 71–102. [DOI] [PubMed] [Google Scholar]
- 449. Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech Ageing Dev 2013; 134: 161–170. [DOI] [PubMed] [Google Scholar]
- 450. Stefanini M, Botta E, Lanzafame M, et al Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst) 2010; 9: 2–10. [DOI] [PubMed] [Google Scholar]
- 451. Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: the growing complex landscape of hereditary colon cancer. World J Gastroenterol 2015; 21: 9253–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Ponti G, Manfredini M, Tomasi A, et al Muir–Torre syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene 2016; 589: 127–132. [DOI] [PubMed] [Google Scholar]
- 453. Schmidt MH, Pearson CE. Disease‐associated repeat instability and mismatch repair. DNA Repair (Amst) 2016; 38: 117–126. [DOI] [PubMed] [Google Scholar]
- 454. Kobayashi H, Ohno S, Sasaki Y, et al Hereditary breast and ovarian cancer susceptibility genes. Oncol Rep 2013; 30: 1019–1029. [DOI] [PubMed] [Google Scholar]
- 455. Salewsky B, Wessendorf P, Hirsch D, et al Nijmegen breakage syndrome: the clearance pathway for mutant nibrin protein is allele specific. Gene 2013; 519: 217–221. [DOI] [PubMed] [Google Scholar]
- 456. Suhasini AN, Brosh RM Jr. Disease‐causing missense mutations in human DNA helicase disorders. Mutat Res 2013; 752: 138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Taylor AM, Groom A, Byrd PJ. Ataxia‐telangiectasia‐like disorder (ATLD) – its clinical presentation and molecular basis. DNA Repair (Amst) 2004; 3: 1219–1225. [DOI] [PubMed] [Google Scholar]
- 458. Pichierri P, Ammazzalorso F, Bignami M, et al The Werner syndrome protein: linking the replication checkpoint response to genome stability. Aging (Albany NY) 2011; 3: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurobiol 2011; 94: 166–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Belizna C, Henrion D, Beucher A, et al Anti‐Ku antibodies: clinical, genetic and diagnostic insights. Autoimmun Rev 2010; 9: 691–694. [DOI] [PubMed] [Google Scholar]
- 461. O'Driscoll M, Gennery AR, Seidel J, et al An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS‐SCID and ATR‐Seckel syndrome. DNA Repair (Amst) 2004; 3: 1227–1235. [DOI] [PubMed] [Google Scholar]
- 462. Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non‐homologous end‐joining. DNA Repair (Amst) 2014; 16: 84–96. [DOI] [PubMed] [Google Scholar]
- 463. Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev 2015; 33: 32–40. [DOI] [PubMed] [Google Scholar]
- 464. Su X, Huang J. The Fanconi anemia pathway and DNA interstrand cross‐link repair. Protein Cell 2011; 2: 704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Taylor AM, Lam Z, Last JI, Byrd PJ. Ataxia telangiectasia: more variation at clinical and cellular levels. Clin Genet 2015; 87: 199–208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure legends
Full legends for main figures
Appendix S1. Glossary and list of abbreviations
Figure S1. The cellular fate following genotoxic insults. The magnitude of the genotoxic insult (low, moderate, excessive) determines cells fate (effective repair, senescence or cell‐death, respectively). Under certain conditions, determined by the stress response parameters (Figure 1B), senescence can present a “dark side”. Likewise, necrosis and/or resistance to apoptosis can build up a pro‐tumorigenic environment (see text for details and references). SASP: senescence‐associated secretory phenotype.
Figure S2. Other pathways that contribute to DDR signaling. Accumulating data demonstrate that the DDR function is complemented and/or it cross‐talks with other signaling routes, which also respond to DNA damage 420, 421, 422. To what extent these signaling pathways modulate the DDR function is a subject that has not been fully elucidated. Nevertheless, the implementation of multiple signaling cascades in a DDR network highlights the need for the DNA damage machinery to detect and respond to a wide range of stimuli in various cellular scenarios, underscoring the highly modular organization of the DDR 422. (i) One such signaling pathway involved in DDR is the p38 MAPK. It is one of the three main groups of mitogen‐activated protein kinases (MAPK). It contributes in the G2/M checkpoint, to facilitate DNA repair, via three possible routes: a) the direct phosphorylation of p53, which results in the dissociation of p53 from Mdm2 thus preventing p53 ubiquitination and degradation, b) the association with Gadd45α, which interacts with p53 and increases its stability, and c) the phosphorylation and inhibition of the phosphatase Cdc25B which is responsible for driving the cell cycle through activation of the Cyclin B/Cdc2 complex 53, 423. In addition, p38 MAPK activation can induce G1/S checkpoint in response to a variety of cellular stresses such as osmotic shock or cellular senescence 53, 423. (ii) Hippo signaling pathway is also implicated in the DDR. Further to a wide spectrum of cellular roles, components of the Hippo pathway cooperate with central orchestrators of the DDR, namely the ATR‐Chk1 and ATM‐Chk2 signaling nodes 424, 425. (iii) Wnt/ β catenin pathway, which has important functions in controlling gene expression, cell polarity and adhesion, is also involved in the repair of DNA damage specifically due to oxidative stress, through interaction with DDR at different levels 426, 427, 428. (iv) NOTCH pathway is a highly conserved signaling system that functions in developmental processes related to cell‐fate determination, particularly in stem cells. In mammalian cells, activation of human Notch1 results in reduced ATM signaling in a manner independent of Notch 1 transcriptional activity 429. Notch1 binds directly to the regulatory FATC domain of ATM, thus inhibiting ATM kinase activity 429. (v) An additional paradigm of interaction between DDR and other signaling routes is that of the Hedgehog (Hh) pathway on the DNA repair mechanism. Inhibition of Hh signaling can repress almost all of the DNA repair mechanisms (i.e. BER, NER, MMR and DSB repair including HR and NHEJ) 430. (vi) Immune responses upon DNA damage are supported by a growing body of evidence 24. DNA‐PK, Ku70 and MRE11 are all capable of sensing cytosolic DNA and activating the cGAS‐STING pathway promoting type I and type III interferon‐signaling. Additionally, PARP‐1 and ATM interact with subunits of IκB kinase triggering NF‐κB‐dependent gene expression. ATM and ATR activation is also involved in the upregulation of ligands for the NKG2D receptor upon stalled DNA replication forks. Conversely, key immune system players like the classical cytokine IL‐1α can act as intracellular DNA damage sensors and signal the presence of genotoxic stress 23, 431.
Figure S3. Replication‐transcription intermediates and replication fork restart. (A) Replication intermediate lesions harboring single stranded DNA (ssDNA). (i) Uncoupling of the replicative helicase and polymerases results in generation of ssDNA due to excessive unwinding of the template (stalled fork). (L: leading strand; l: lagging strand) (ii) A stalled replication fork may undergo remodeling by creating an intermediate reverse fork also known as “chicken foot” structure: (ii‐1) Direct CtIP processing of the reversed fork may lead to nascent strand ssDNA formation. (ii‐2) Cleavage by SLX4‐docking nucleases generates DNA double strand break that is subsequently followed by resection resulting into nascent strand ssDNA generation. (iii) Unequal branch migration or resection (by CtIP) of a reversed fork can also lead to generation of template ssDNA. (iv) Deregulated firing of clustered origins leads to replication stress and accumulation of gaps in the nascent strands, leaving template ssDNA (see text for details and references) (B) Transcription intermediates. R loops are the predominant transcription generated intermediates and represent a three‐stranded nucleic acid structure that comprises two branches, an RNA–DNA hybrid and an ssDNA. The former can impede completion of replication leading to replication fork stalling, collapse and DSBs formation, while the latter can serve as a substrate to DNA damaging agents and cellular enzymes [APOBEC deaminases (Table 1)] resulting in DNA lesions and/or nicks (see text for details and references). (C) Restart of stalled or collapsed replication forks. Depending on the duration (how long) of the replication block, forks can stall or collapse. Restart of stalled forks is promoted by fork remodeling factors, while collapsed forks rely on DSB mediated restart through homologous recombination repair, whereas new origins are concurrently fired (see text for details and references).
Figure S4. (A) Nucleolus and rDNA organization. Schematic representation of the rDNA repeats, an organization that renders them susceptible to replication‐transcription collisions (see text for details). PHC: Perinuclear Heterochromatin, FC: Fibrillar Centre, DFC: Dense Fibrillar Component and CC: Granular Component (see text for details and references). (B) Maintaining mitochondrial DNA integrity: Nuclear and mitochondrial DNAs are interdependent. Cartoon of the mitochondrial DNA: D (Displacement)‐loop: a short nucleotide segment complementary to the light (L)‐strand that displaces the heavy (H)‐strand of the mitochondrial DNA. It contains promoters (LSP and HSP) for the RNA transcription from the two strands (heavy and light, respectively) of mitochondrial DNA, possibly involved in the organization of the mitochondrial nucleoid (see text for details and references); LSP: Light strand promoter. The promoter is responsible for gene transcription from the light strand (lower molecular mass) of mitochondrial DNA; HSP: Heavy strand promoter. The promoter is responsible for gene transcription from the heavy strand (higher molecular mass) of mitochondrial DNA. Depending on the magnitude of the mitochondrial DNA damage three levels of repair may take place (see text for details and references).
Figure S5. Monitoring Mitosis (DDR surveillance). During M phase checkpoints monitor the proper alignment, segregation and cytokinesis (see lower left panel). In response to a mitotic defect, such as misalignment and/or DSBs, cell fate depends on the context (i.e. p53 status) and the extent of the damage (how much): i) low mitotic damage is marked, and repaired in the subsequent cell cycle in the daughter cells (continuous green line corresponds to G1 phase where the majority of DNA lesions are repaired, however, mitotic DNA lesions can also be repaired in S and G2 phase depicted by the dashed green line‐see lower left cell cycle panel), ii) high DNA damage or mitotic spindle defects may lead to mitotic catastrophe or mitotic slippage, which in turn generates aneuploidy and/or CIN. The later can lead to cell death, senescence or development of precancerous lesions. Upon induction of DSBs during mitosis, MRN, and phosphorylated MDC1 and Η2ΑΧ are recruited to the damaged site forming the mitotic DDR foci (see right panel). Notably, 53BP1 and BRCA1 are not recruited to the site of damage blocking NHEJ and HR activation, respectively, preventing telomere fusion (mute DDR). An adverse outcome of mitotic DDR activation is kinetochore‐microtubule stabilization mediated by activation of PLK1 and Aurora kinase A that in turn promotes merotelic attachment and the formation of lagging chromosomes resulting in numerical CIN. However, it is not yet clear under what circumstances activation of mitotic DDR leads to this unfavorable outcome, instead of marking the DNA damage and proceeding to repair in the following cell cycle (marked with a question mark; right lower panel). “P” within red colored circles depicts the two phosphorylation sites of 53BP1 at Threonine‐1609 and Threonine‐1618 that prevent it from recruitment to DDR foci. CIN: chromosomal instability
Figure S6. Functional interplay and interdependence of genome and proteome maintenance modules (DDR and PDR surveillance). (A) The PN along with PDR are actively involved in DDR efficiency since by assuring proteome integrity they maintain the functionality of the protein machines that safeguard genome stability. On the other hand, DDR induces a number of proteostatic and/or metabolic adaptations, including suppression of transcription and ribosomal biogenesis, indicating the functional interdependence of the two pathways. These pathways are fully active in young organisms. (B‐C) The age‐related collapse of proteostatic modules functionality and/or expression levels (B) results in the gradual accumulation of non‐functional polypeptides, protein aggregates or lipofuscin, compromising proteome integrity and leading to genomic instability (and thus increased chances for carcinogenesis) as a result of ineffective DNA maintenance and/or repair. Eventually, a vicious cycle may form where a mildly unstable genome accelerates proteome instability due to synthesis of mutated polypeptides that progressively increase the attrition of protein machines resulting in an increasingly stressful cellular landscape that favors the appearance (C) of cellular senescence, cell death or age‐related diseases (e.g. cancer). (D) In normal cells, production of ROS or RNS is neutralized by anti‐oxidant responses while intact PN ensures normal protein turnover. During stress induced premature senescence (Glossary) or in aged tissues the levels of ROS/RNS increase leading to lipid and protein oxidation in the cytoplasm. As this process evolves, oxidized proteins become unfolded and intra‐ and/or inter‐molecular cross links occur, forming non‐degradable oxidized protein aggregates; the latter along with oxidized lipids/lipoproteins, carbohydrate residues and metals form undegradable lipofuscin which accumulates mainly in lysosomes, while only a minor amount is found free in the cytosol. Cytosolic lipofuscin occurs either due to impaired uptake by stalled autophagy or following autophagosome/phagophore rapture. Lipofuscin also inhibits proteasome activity further boosting lipid/lipoprotein oxidation in the cytoplasm.
Table S1. Representative categories and types of human RNAs involved in physiological processes and diseases, including cancer
Table S2. The implications of defective key components of the DNA damage response mechanisms in the pathogenesis of specific clinical syndromes in humans