

Abstract
Increasing evidence suggests that α-synuclein (αS) aggregates in brains of individuals with Parkinson's disease and dementia with Lewy bodies can spread in a prion-like manner. Although the initial αS nuclei are pivotal in determining αS fibril polymorphs and resulting phenotypes, it is not clear how the initial fibril seeds are generated. Previous studies have shown that αS truncation might have an important role in αS aggregation. However, little is known about how this truncation influences αS's propagation properties. In the present study, we generated αS fibrils from a series of truncated human αS constructs, characterized their structures and conformational stabilities, and investigated their ability to convert the conformation of full-length αS in vitro, in cultured cells, and in WT mice. We show that both C- and N-terminal truncations of human αS induce fibril polymorphs and exhibit different cross-seeding activities. N-terminally 10- or 30-residue–truncated human αS fibrils induced more abundant αS pathologies than WT fibrils in mice, whereas other truncated fibrils induced less abundant pathologies. Biochemical analyses of these truncated fibrils revealed that N-terminal 10- or 30-residue truncations of human αS change the fibril conformation in a manner that increases their structural compatibility with WT mouse αS fibrils and reduces their stability. C-terminally 20-residue–truncated fibrils displayed enhanced seeding activity in vitro. Our findings imply that truncation of αS can influence its prion-like pathogenicity, resulting in phenotypic diversity of α-synucleinopathies.
Keywords: protein aggregation, alpha-synuclein (a-synuclein), amyloid, electron microscopy (EM), fibril, immunohistochemistry, neurodegenerative disease, Parkinson's disease, Western blot, neurodegeneration, polymorphs, prion-like, truncation, protein truncation, protein structure, prion, neurodegeneration
Introduction
Misfolding and aggregation of normally soluble proteins are common pathological features of many neurodegenerative diseases (1). α-Synucleinopathies, including Parkinson's disease (PD)3, dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) are associated with formation of abnormal aggregates of α-synuclein (αS), a natively unfolded 14-kDa protein consisting of 140 amino acids. These diseases are neuropathologically characterized by the deposition of filamentous αS aggregates with cross-β structure (2), which are abnormally phosphorylated (3) and partially ubiquitinated (4). αS and various other neurodegenerative disease–related proteins, including β-amyloid (Aβ), tau, and TAR-DNA–binding protein-43 (TDP-43), can propagate through neural networks in a similar manner to abnormal prion proteins in prion diseases, in which abnormal insoluble protein acts as a seed or template for converting soluble protein to abnormal form (5, 6). Although in vitro (7, 8) and in vivo (9–11) experiments using detergent-insoluble fraction of diseased brains and aggregated recombinant proteins as seeds have provided support for the “prion-like propagation hypothesis,” it is not yet clear how αS aggregates propagate through the diseased brain.
In MSA, αS is mainly deposited in glial cells, whereas αS aggregates are found in neuronal cells in PD and dementia with Lewy bodies (DLB) (12). Such phenotypic differences suggest the existence of different “strains” of pathogenic αS, whose aggregate structures determine clinical phenotypes, as in prion diseases (13, 14). Although the differences in cross-seeding activity between MSA and PD brain extracts in heterozygous mice transgenic for A53T human αS suggest that structural differences exist (11, 15), the mechanisms that cause such structural variations among sporadic α-synucleinopathies remain unclear.
In Lewy bodies, not only full-length (FL), but also truncated αS is accumulated (16). Truncation is a common posttranslational modification that is not specific to the diseased brain, and about 15% of αS is truncated even in the brain of healthy individuals (17). In the diseased brain, both N-terminally and C-terminally truncated αS species are present (18). Overexpression of truncated αS or coexistence of truncated and FL αS can enhance aggregation and neurodegeneration both in vitro (19, 20) and in vivo (21, 22). Although it has been suggested that truncation has an important influence on αS aggregation (19, 23, 24), very few studies have focused on the effect of truncation on prion-like propagation.
In the present study, we systematically investigated the structural polymorphism of truncated αS fibrils and examined whether or not these fibrils can act as templates for FL αS aggregation, using in vitro and in vivo experimental models. We found that differently truncated human αS fibrils exhibit a variety of cross-seeding activities in vitro and in vivo. Our results imply that truncation of αS in the initial nucleation phase might not only influence aggregation, but also result in phenotypic diversity.
Results
Fibril formation from truncated αS proteins
To investigate the effects of C- and N-terminal truncations on αS aggregation, we prepared a series of wildtype (WT) and truncated human αS (Fig. 1A). The constructs were expressed in Escherichia coli, and the purified soluble αS proteins were analyzed by SDS-PAGE (Fig. 1B). After agitation in the presence of 150 mm KCl, the formation of fibrillar aggregates was confirmed by negative-staining transmission electron microscopy (TEM) (Fig. 1C). The fibrils were 6–10 nm in diameter and did not branch. In agreement with a previous report (25), human C-terminally truncated αS fibrils (ΔC20 and ΔC30) tended to be shorter than WT fibrils and were bundled laterally, whereas human N-terminally truncated αS formed long fibrils similar to WT fibrils (26). These fibrils bound to thioflavin T (ThT), which is commonly used to detect amyloid formation, and showed enhanced fluorescence (Fig. S1).
Figure 1.

Human WT and truncated αS fibril seeds. A, schematic representation of the human WT and truncated αS variants used in this study. The shaded portion indicates the reported core region (residues 31–109) of WT αS fibrils. B, CBB staining of purified αS proteins. C, negative staining TEM images of WT and truncated αS fibrils. Scale bars, 100 nm. D, aggregation kinetics of human FL αS monomers under quiescent conditions in the absence or presence of 1 mol % human WT or truncated αS fibril seeds, monitored by ThT fluorescence. Curves are averages of three samples. The αS concentration was 150 μm in all experiments.
To examine whether these truncated αS fibrils could act as seeds for FL αS aggregation, seeding assay against human WT αS monomer was performed by incubation under quiescent conditions in the presence or absence of 1 mol % of preformed human αS fibril seeds in vitro (Fig. 1D). WT and all truncated αS fibrils induced the aggregation of FL αS protein, but the ThT fluorescence was not increased in the absence of αS fibril seeds. We found that ΔC20 seeds drastically increased the rate of fibril assembly (t½ = 2 ± 0 h), compared with WT seeds (t½ = 26 ± 6 h). ΔN10 seeds and ΔN30 also induced fibril assembly, but more slowly (t½ = 76 ± 3 and 58 ± 1 h, respectively).
Effect of truncation on induction of phospho-αS pathology in WT mice
To examine the cross-seeding activity of human truncated αS fibril seeds in WT mouse brain, we injected 750 pmol human WT and truncated αS fibril seeds into the right striatum of WT mice. Three months after injection, paraffin-embedded brain sections were prepared and immunostained with anti–phospho-αS antibody (pSer-129/EP1536Y) (Fig. 2, A and B). In mice injected with WT seeds, Lewy body/Lewy neurite-like phospho-αS aggregates were distributed bilaterally through the brain, including striatum (Str), substantia nigra (SN), amygdala (Amy), and anterior cingulate cortex (ACC). Only sparse phospho-αS–positive structures were observed in mice injected with ΔC10 seeds, ΔC20 seeds, and ΔC30 seeds, and these were restricted to Str, SN, and Amy in the injected hemisphere. On the contrary, abundant phospho-αS aggregates were observed throughout the bilateral brain, including Str, SN, Amy, and ACC, in mice injected with ΔN10 seeds and ΔN30 seeds. To evaluate the accumulated αS biochemically, we analyzed sarkosyl-insoluble fractions (Sar-ppt) from whole mouse brain (Fig. 2C). The amounts of insoluble phospho-αS in mice injected with ΔN10 seeds and ΔN30 seeds were larger than in mice injected with WT seeds, whereas only very faint bands were detected in mice injected with ΔC10 seeds, ΔC20 seeds, ΔC30 seeds, and ΔN20 seeds (Fig. 2D). These phospho-αS bands were stained with anti-mouse αS-specific antibody (D37A6), but not with anti-human αS-specific antibody (LB509). We also compared the amount of insoluble mouse αS but did not find any difference among the mice because of the high basal signal detected in mock-treated mice. These results indicate that N-terminal 10- or 30-residue truncation of human αS enhances the cross-seeding activity with WT mouse αS in vivo, despite the low seeding activity in vitro (Fig. 1D). On the other hand, the cross-seeding activities are reduced by C-terminal truncations and N-terminal 20-residue truncation.
Figure 2.

Comparison of αS pathologies in WT mouse brains by inoculation of human WT and truncated αS fibril seeds. A, distributions of phospho-αS pathology in WT mouse brains. WT mice injected with human αS fibrils into the striatum were assessed at 3 months after injection. αS pathology was evaluated by immunohistochemistry with anti–pSer-129 antibody (EP1536Y). Sections were counterstained with hematoxylin. Representative images of ipsilateral striatum (Str), substantia nigra (SN), amygdala (Amy), and anterior cingulate cortex (ACC) of inoculated mice are shown. Scale bars, 25 μm. B, quantification of phospho-αS pathology of each area (red, injection side; blue, contralateral side). Data are shown as mean percentage area occupied by pSer-129–positive structures ± S.E. (n = 3 per group). N.D., not detected. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, Western blot analysis of sarkosyl-soluble (sup) and sarkosyl-insoluble (ppt) fractions of injected brain extracts. The blots were probed using anti-pSer-129 (pSyn#64), anti-mouse αS specific (D37A6), anti-human αS specific (LB509), and anti-tubulin α antibodies. LB509, which recognizes 115–122 of human αS, cannot bind ΔC20 and ΔC30. Data are representative of three independent experiments. D, quantification of relative insoluble phospho-αS (upper) and mouse-αS (lower). Data presented are the means ± S.E. (n = 3 per group). ***, p < 0.001.
Effect of truncation on seed-dependent aggregation of αS in cultured cells
To further confirm these differences, we compared the cross-seeding activities using a cultured cell model. SH-SY5Y cells transiently expressing mouse WT αS were transfected with human WT and truncated αS fibril seeds: ΔC20, ΔN10, ΔN20, and ΔN30 seeds. Two days after transfection, we checked the presence of insoluble αS aggregates in cells by biochemical fractionation and Western blot analysis (Fig. 3A). The levels of mouse αS in sarkosyl-insoluble fraction were increased in cells treated with αS fibril seeds, and the insoluble mouse αS bands were positive to anti–phospho-αS antibody (pSer-129/pSyn#64). The amount of insoluble phosphorylated αS in cells treated with ΔN30 seeds was larger than those in cells treated with WT seeds, whereas the amounts in cells treated with ΔN20 seeds and ΔC20 seeds were relatively small (Fig. 3B). We also compared the amount of insoluble mouse αS and found the amount of insoluble mouse αS in cells treated with ΔN30 seeds was larger than those in cells treated with WT seeds. Similar results were observed when SH-SY5Y cells overexpressing FLAG-human αS were treated with truncated fibril seeds (Fig. S2). These results indicate that human truncated αS fibrils can convert soluble mouse and human FL αS into insoluble form, and some N-terminal truncations have more potent cross-seeding activities in cultured cells.
Figure 3.

Cross-seeding activity of human WT and truncated αS fibrils in cultured cells. SH-SY5Y cells expressing WT mouse-αS were transfected with the αS fibril seeds. After 2 days, cells were collected, lysed with A68 buffer containing 1% sarkosyl, and fractionated. The supernatant (sup) and the pellet (ppt) fractions were subjected to Western blot analysis. A, Western blot analysis using anti-pSer-129 (pSyn#64), anti-mouse αS (D37A6), and anti-tubulin α antibodies. B, quantification of relative insoluble phospho-αS (upper) and mouse-αS (lower). Data presented are the means ± S.E. (n = 3 per group). *, p < 0.05.
Truncation induces fibril polymorphs of αS
As described above, N-terminal 10- or 30-residue truncation of human αS results in increased cross-seeding activity in vivo, suggesting that these truncations induce different fibrillar polymorphs. In this context, the protease digestion pattern reflect the structure of the cross-β sheet–rich fibril core region, and is commonly used to characterize the conformations of amyloid fibrils (27–29). Therefore, we compared the proteinase K (PK)–resistant fractions of truncated human αS fibrils with those of WT mouse αS fibrils. WT mouse αS fibrils (WTMo seed) showed ∼10 and ∼8 kDa PK-resistant fractions after PK digestion, whereas WT human αS fibrils (WTHu seed) yielded ∼12 and ∼8 kDa fractions (Fig. 4A). The N-terminally truncated human αS fibrils showed a WT mouse-like digestion pattern, suggesting that these fibrils share a common fibril core structure with WT mouse fibrils. In contrast, the ΔC20 fibrils showed a WT human-like digestion pattern. Next, we compared the digestion patterns of FL mouse αS aggregates (FLAMo) seeded with the αS fibrils. FL mouse αS aggregates obtained with WTMo seed (FLAMo–WTMo) showed ∼10 and ∼8 kDa resistant fractions after PK digestion, whereas aggregates with WTHu seed (FLAMo–WTHu) were digested into low-molecular fractions (Fig. 4B). FL mouse αS aggregates obtained with N-terminal truncated fibril seeds (FLAMo–ΔN10, –ΔN20, and –ΔN30) showed FLAMo–WTMo–like digestion patterns, whereas FLAMo–ΔC20 showed a FLAMo–WTHu–like pattern. These results imply that the N-terminal truncations of human αS have similar conformations to WT mouse αS fibrils, and can work as seeds for mouse αS monomer to form αS fibrils with structural properties similar to those of WT mouse αS fibrils.
Figure 4.
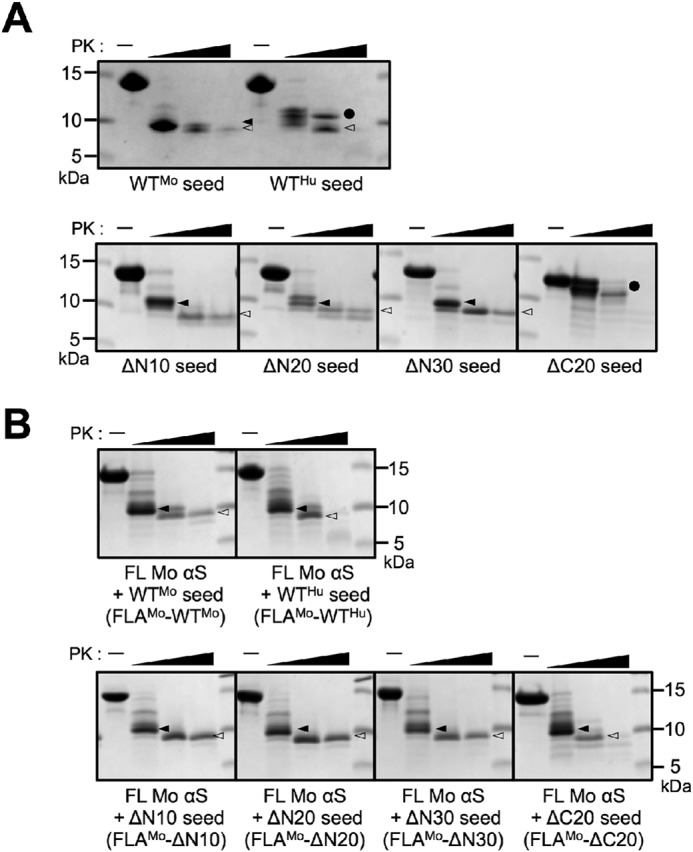
Concentration-dependent proteinase K digestion assays of αS aggregates. αS fibrils were treated with PK (0, 1, 10, 100 μg/ml) for 30 min at 37 °C. After treatment, the samples were resolved using SDS-PAGE and stained by CBB. A, FL and truncated human αS fibril seeds. B, FL mouse αS aggregates seeded with mouse WT αS fibril seeds (WTMo), human WT αS fibril seeds (WTHu), and human truncated αS fibril seeds. A filled circle indicates the band at ∼12 kDa; a filled arrowhead indicates the band at ∼10 kDa; an open arrowhead indicates the band at ∼8 kDa. Three independent experiments were done and representative data are shown.
N-terminal 10- and 30-residue–truncated human αS fibrils have decreased conformational stability
As described above, the N-terminal truncations of human αS form mouse αS–like fibrils, but sequential N-terminal truncations show biphasic seeding effects in mouse brain: Fibrils of ΔN10 and ΔN30 have greater effects than fibrils of ΔN20 (Fig. 2). In the prion field, there is a general consensus that seeding capacity is inversely related to fibril stability (30). Here, we examined the stability of the fibrils by treating WT and N-terminally truncated human αS fibrils with a chemical denaturant, sarkosyl, and separating the products into supernatant and pellet fractions by ultracentrifugation (Fig. 5). In the case of WT human αS, ∼36% of the total protein was fractionated into the pellet (36 ± 4% of total protein). The ΔN10 and ΔN30 fibrils, which could propagate efficiently in mouse brain, showed decreased conformational stability (15 ± 5% and 19 ± 6% of total protein in the pellet, respectively). On the other hand, the ΔN20 fibrils, which exhibited inefficient seeding in vivo, showed increased stability (58 ± 3% of total protein in the pellet). These results indicate that N-terminal 10- or 30-residue truncation enhances the cross-seeding activity in vivo as a result of decreased stability, implying that αS fibrils are propagated through a prion-like molecular mechanism.
Figure 5.
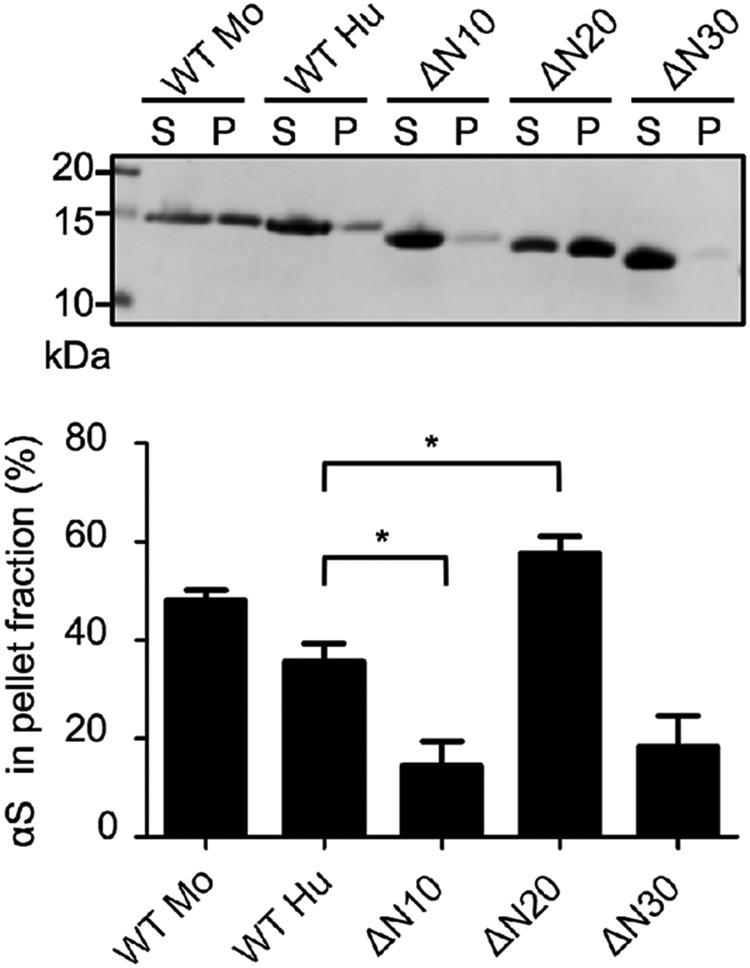
Comparison of the stability of WT and N-terminal truncated αS fibrils. WT and N-terminally truncated αS fibrils were treated with 1% sarkosyl for 5 min at room temperature. The samples were centrifuged and the supernatant (S) and the pellet (P) fractions were analyzed by SDS-PAGE. The image of a gel stained with CBB (upper) and quantification data of αS protein (lower) are shown. Data presented are the means ± S.E. percentage of αS protein in the pellet from five independent experiments. *, p < 0.05 compared with WTHu.
C-terminal 20-residue truncation induces distinctive prion-like properties
Although ΔC20 fibrils showed drastically enhanced seeding activity in vitro, they induced only sparse αS pathology in vivo (Figs. 1D and 2). To investigate whether the apparent inconsistency is because of their structural characteristics, we analyzed the structures of the seeded FL αS aggregates (FLA)–ΔC20 in vitro and in cultured cells. TEM images showed that FLA seeded with ΔC20 seeds (FLA–ΔC20) had a twisted morphology that differed from the others, which showed straight, unbranched fibrillar structures with a diameter of ∼10 nm (Fig. 6A; Fig. S3A). To explore the difference of the underlying structures, we quantified the ThT-binding properties of the seeded FL αS aggregates. FLA-ΔC20 showed higher binding affinity (211 ± 8% of FLA–WT) (Fig. S3B). The rate of FL WT αS aggregation in the presence of FLA–ΔC20 was faster (t½ = 5 ± 0 h) than that of FLA–WTHu (t½ = 28 ± 2 h) (Fig. 6B). We also examined the seeding activities of the FL αS aggregates in cultured cells. FLA–ΔC20 showed poor seeding activity in cultured cells (Fig. 6, C and D). These results indicate that ΔC20 fibrils induce distinct structures of FL αS aggregates and these aggregates have similar but different seeding properties with ΔC20 fibrils in vitro and in cells (Figs. 1 and 3).
Figure 6.
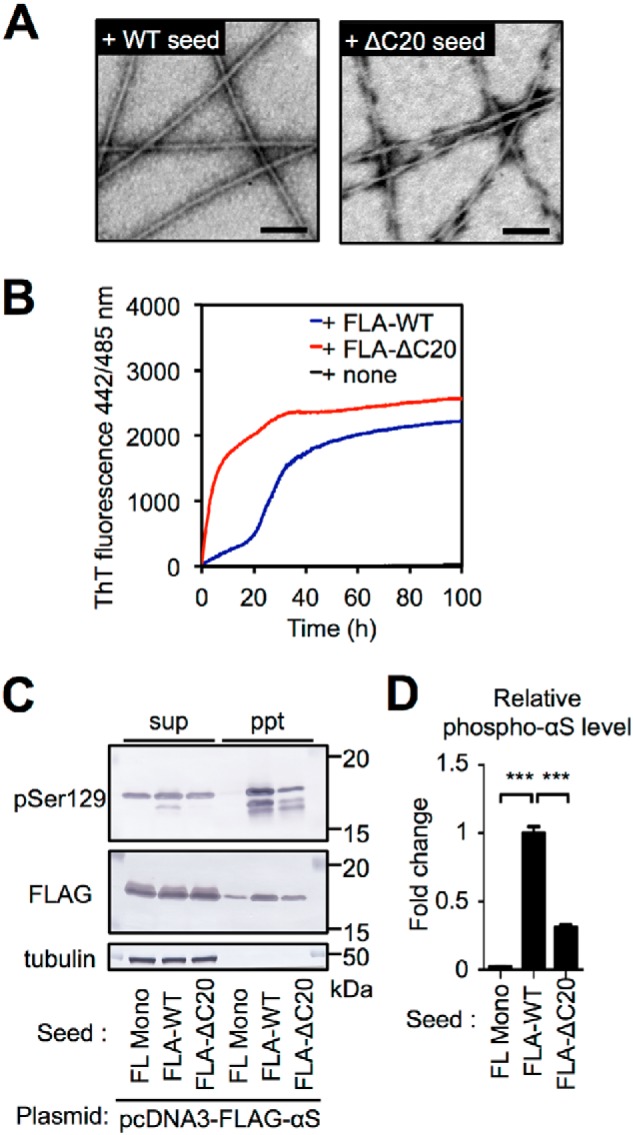
C-terminal 20-residue truncation of human αS also produces distinctive prion-like properties. A, negative staining TEM imaging of human FL αS aggregates seeded with FL (left) or ΔC20 (right) seeds. Scale bars, 100 nm. Human FL αS aggregates seeded with ΔC20 seeds showed twisted fibrillar structure. B, aggregation kinetics of human FL αS monomers under quiescent conditions in the absence or presence of 1 mol % human FL αS aggregates seeded with WT or ΔC20 seeds, monitored by measuring ThT fluorescence. Data are averages of three samples. C, seed-dependent aggregation of αS induced by human FL αS aggregates seeded with WT or ΔC20 seeds in the cultured cell model. SH-SY5Y cells expressing FLAG-αS were transfected with αS monomer (FL Mono), human FL αS aggregates seeded with WT (FLA-WT), or ΔC20 (FLA-ΔC20). Cell extracts were prepared with A68 buffer containing 1% sarkosyl and centrifuged. The supernatant (sup) and the pellet (ppt) fractions were subjected to Western blot analysis using anti-pSer-129 (pSyn#64), anti-FLAG, and anti-tubulin antibodies. D, the intensities of phosphorylated αS in (C) are quantified. ***, p < 0.001.
Discussion
Aggregation of αS can be influenced by mutations of SNCA gene (31–38), phosphorylation (39), and various other external conditions, including salt concentration (40, 41) and pH (42), during in vitro assembly. Human αS can be cleaved either at the N or C terminus by calpain 1 (16), plasmin (43), neurosin (44), 20S proteasome (19, 23), cathepsin D (45), caspase-1 (46), matrix metalloproteinase-3 (47), and asparagine endopeptidase (48), resulting in the production of a range of aggregation-prone truncated species (20, 49, 50). Our results show that C-terminal and N-terminal truncations not involved in the reported human αS fibril core region (residues 31–109) (28) induced fibril polymorphism characterized by different in vitro and in vivo propagation properties (Fig. 7).
Figure 7.

Schematic representation of the effect of truncation on prion-like properties of αS. The N-terminal truncation of human αS changes the structure of the fibrils to a form similar to that of WT mouse αS fibrils. Furthermore, the N-terminally 10- or 30-residue–truncated human αS fibrils have lower conformational stability than WT human αS fibrils. As a result, these fibrils propagate effectively in WT mice. The C-terminally 20-residue–truncated fibrils show enhanced seeding activity in vitro, but reduced seeding activity in cell model and mouse model. There is no direct correlation between in vitro and in vivo propagation activities.
The C-terminal region (residues 96–140) of αS is highly enriched in negatively charged residues (aspartate and glutamate) (51) and proline residues (52). Long-range interactions of the C-terminal region (residues 110–130) with the central hydrophobic NAC region (residues 85–95) (53, 54) are important to stabilize the natively unfolded structure (55–57). When the interaction between the C-terminal and the NAC region is weakened by low pH, addition of polycations, or changes in ionic strength, exposure of the NAC region increases, and more tightly packed αS fibrils are formed (50, 58). Tyrosine residues located at positions 125, 133, and 136 in the C-terminal region are crucial to maintain the N- and C-terminal intermolecular interactions and are related to aggregation (59). In addition to deletion of charged residues, complete removal of these tyrosine residues can result in structural polymorphisms. In the present study, the C-terminal 20-residue truncated fibrils showed enhanced seeding activity in vitro, and FLA seeded with ΔC20 seeds (FLA–ΔC20) exhibited twisted morphologies distinct from that of the original ΔC20 seeds and others, which showed straight structure. In our opinion, such twisted morphologies were likely caused by the negatively charged residues, which exist in FL monomers, but not in ΔC20 seeds. These residues might interact with the core region of ΔC20 to form the twisted morphologies. As a result, C-terminal 20-residue truncated fibrils induced full-length αS polymorphisms.
The N-terminal region (residues 1–60) of human αS contains four imperfect 11-residue repeats with a conserved motif (XKTKEGVXXXX), which interacts with the lipid membrane, with conformational changes to the α-helical structure (26, 56). Although the N-terminal region directly interacts with the C-terminal region, but not the central NAC region, (57), N-terminal truncation also increases the ratio of β-sheet structure within αS fibrils (56). Recent studies have shown that familial PD-related mutations located in the N-terminal region influence the relative stability of the fibrils and their conformational preferences, rather than the global fibril structures (60, 61).
Truncation of the N-terminal 10 or 30 residues markedly enhanced cross-seeding activity in mouse brain. Structural compatibility between the seed and the host protein is important for cross-seeding activity in vivo (62). We found that N-terminal truncation switches the conformational preference of human αS fibrils, making them with structural properties similar to those of WT mouse αS fibrils, whereas C-terminal truncation does not have such an effect. Furthermore, we also found that N-terminal 10- or 30-residue truncation reduces conformational stability in a manner that is inversely correlated with in vivo propagation efficacy. Propagation of prion proteins depends on the stability of the pathogenic aggregates; less-stable prions replicate faster than more-stable prions (30). This would be another reason why these N-terminally truncated fibrils reduce the species barrier in WT mice. These results indicate that N-terminal truncation influences the prion-like seeding activity of αS.
As previously reported (62), we also found that propagation activities in vitro, in cells, and in vivo were not always consistent. In cultured cells, seeding reaction occurs in a more complex environment, such as molecular crowding, effects of chaperones and protein degradation machineries, other proteins interacting with seeds or monomers, and so on, compared with the simple reaction buffers used for in vitro assay (63). Moreover, seeds should be taken from outside of cells and escaped from endosomal vesicles if taken by endosome system in cultured cells. In mice, the cell to cell transmissibility might be influenced by secretion and uptake of seeds (64). In this study, we found both ΔC20 seeds and FLA–ΔC20 seeds had markedly different seeding activities in vitro versus in cultured cells and mice. These seeds induced rapid aggregation in vitro, but not in cells and in mice. Contrary to these seeds, N-terminally truncated seeds such as ΔN30 induced severe pathology in cells and mice but had reduced seeding activity in vitro. These differences in propagation characteristics may result from different susceptibility of fibrils to external factors described above. It was reported that even αS fibrils that could not propagate effectively in WT mice might trigger αS pathology and neurodegeneration when proteolytic systems become less effective with aging (65) or in the presence of a genetic factor, such as heterozygous mutation in the lysosomal glucocerebrosidase gene (GBA1) (66, 67). Therefore, there is a possibility that ΔC20 seeds induce severe αS pathology in aged mice or lysosomal mutation mice. Consequently, it is important to evaluate the propagation properties of αS fibrils using multiple models.
In summary, our findings indicate that differently truncated human αS can induce various polymorphisms of FL αS fibrils via a seeding mechanism. The properties of N-terminal truncations of human αS imply that the pathogenicity of αS involves a prion-like molecular mechanism. We believe these findings provide new insights into both the pathogenicity and phenotypic diversity of sporadic α-synucleinopathies.
Experimental procedures
Antibodies
Antibodies used in the present study were anti–phospho-αS antibodies to pSer-129, including pSyn#64 (3) and EP1536Y (Abcam), and other anti-αS antibodies, including LB509 (a gift from Dr. Iwatsubo) and D37A6 (Cell Signaling Technology). Biotin-labeled secondary antibodies were purchased from Vector Laboratories for use in the avidin–biotin complex method.
Expression and purification of recombinant αS proteins
C-terminally truncated αS (ΔC10: residues 1–130, ΔC20: residues 1–120, and ΔC30: residues 1–110) and N-terminally truncated αS (ΔN10: residues 11–140, ΔN20: residues 21–140, and ΔN30: residues 31–140) were amplified by PCR with the following primers and subcloned into pRK172 (68, 69): ΔC10, forward, 5′-aggagatatacatatggatgtattcatgaa-3′ and reverse, 5′-atcggaattcaagcttttactcataaggcatttcat-3′; ΔC20, forward, 5′-aggagatatacatatggatgtattcatgaa-3′ and reverse, 5′-atcggaattcaagcttttactcataaggcatttcat-3′; ΔC30, forward, 5′-aggagatatacatatggatgtattcatgaa-3′ and reverse, 5′-atcggaattcaagcttttactcataaggcatttcat-3′; ΔN10, forward, 5′-aggagatatacatatggccaaggagggagttgt-3′ and reverse, 5′-atcggaattcaagcttttaggcttcaggttcgt-3′;ΔN20, forward, 5′-aggagatatacatatgaaaaccaaacagggtgt-3′ and reverse, 5′-atcggaattcaagcttttaggcttcaggttcgt-3′; ΔN30, forward, 5′-aggagatatacatatgggaaagacaaaagaggg-3′ and reverse, 5′-atcggaattcaagcttttaggcttcaggttcgt-3′.
All constructs were verified by DNA sequencing. Recombinant proteins were expressed in E. coli BL21 (DE3) as described previously (70). Bacterial pellets were sonicated in extraction buffer A (50 mm Tris-HCl, pH 7.5, 1 mm EGTA, 1 mm DTT) on ice. The homogenates were centrifuged at 16,000 × g for 15 min, and the supernatants were boiled for 5 min. Then, the supernatants (heat-stable fraction) of 15-min centrifugation at 16,000 × g were subjected to anion-exchange (for WT, ΔC10, ΔC20, ΔN10, Δ20, and ΔN30) or cation-exchange (for ΔC30) chromatography on columns (Q Sepharose or SP Sepharose Fast Flow, respectively; GE Healthcare Japan) equilibrated with extraction buffer A. The columns were washed with extraction buffer A and eluted with a 100–500 mm NaCl gradient. The eluates were concentrated by precipitation with 50% saturated ammonium sulfate. The pellets were re-suspended in and dialyzed against 30 mm Tris-HCl, pH 7.5. The protein concentrations were determined by reversed phase-HPLC (RP-HPLC), using an Aquapore RP300 column (71). Purified αS samples were analyzed by 13.5% SDS-PAGE and stained with CBB. Gel images were recorded with a Gel DocTM EZ Imager (Bio-Rad).
Preparation of αS fibril seeds
αS fibrils were prepared as follows. Purified recombinant αS proteins were dissolved in 30 mm Tris-HCl, pH 7.5, containing 150 mm KCl and 0.1% NaN3, to a final concentration of 150 μm protein. The proteins were incubated at 37 °C under linear shaking at 200 rpm for 7 days. The assembled αS was collected by ultracentrifugation at 135,000 × g for 20 min at 25 °C. The resultant pellets were re-suspended and sonicated in 30 mm, Tris-HCl, pH 7.5, on ice with an ultrasonic homogenizer (VP-5S, TAITEC) and ultracentrifuged again. The pellet was re-suspended and sonicated again in 30 mm Tris-HCl, pH 7.5. The proteins were dissolved in 6 m guanidine hydrochloride and their concentrations were determined by HPLC as described above.
Transmission EM
The αS fibrils were diluted in 30 mm Tris-HCl pH 7.5 to 15 μm, plated on carbon-coated 300-mesh copper grids (Nissin EM), and stained with 2% (v/v) phosphotungstate. Micrographs were recorded on a JEM-1400Plus electron microscope (JEOL).
Thioflavin T-binding assay
The degree of fibrillation was measured in terms of thioflavin T fluorescence intensity, which is increased when ThT binds to amyloid-like fibrils. The αS proteins (7.5 μm) were incubated with 20 μm ThT in 30 mm Tris-HCl buffer, pH 7.5, for 30 min at 37 °C. Fluorometry was performed using a microplate reader (Varioskan® Flash, excitation 442 nm, emission 485 nm; Thermo Scientific) and normalized to the ThT intensity of WT αS fibrils.
αS aggregation assay
Full-length αS aggregation experiments were performed using a microplate reader (Varioskan® Flash, excitation 442 nm, emission 485 nm; Thermo Scientific) and monitored by measuring ThT fluorescence in the absence or presence of 1 mol % sonicated αS fibril seeds. All experiments were performed at 37 °C, under quiescent conditions in flat-bottomed 96-well black plates (Sumitomo Bakelite) sealed with MicroAmp Optical Adhesive Film (Applied Biosystems) to prevent evaporation. The reaction mixture consisted of 30 mm Tris-HCl (pH 7.5) containing 150 mm KCl, 0.1% NaN3, and 20 μm ThT. Protein concentration was 150 μm unless otherwise indicated. During experiments under quiescent conditions, ThT fluorescence change was read every 6 min.
Proteinase K digestion of αS fibrils
αS fibrils (1 mg/ml) in 30 mm Tris-HCl, pH 7.5, were incubated for 30 min at 37 °C in the presence of various concentrations of proteinase K (1, 10, and 100 μg/ml; Promega). The reaction was stopped by boiling the mixture for 10 min. The samples were analyzed by 15–20% Tris–Tricine–SDS-PAGE; the gels were stained with CBB (28). Gel images were recorded with a Gel DocTM EZ Imager (Bio-Rad).
Conformational stability assay of αS fibrils
αS fibrils (1 mg/ml) were incubated in 30 mm Tris-HCl, pH 7.5, containing 1% sarkosyl (w/w) for 5 min at room temperature. After ultracentrifugation at 135,000 × g for 20 min at 25 °C, the supernatant was collected as sarkosyl-soluble fraction, and the pellet was re-suspended in 1× SDS-PAGE buffer. The samples were analyzed by 15% Tris–Tricine–SDS-PAGE; the gels were stained with CBB (28). Gel images were recorded with a Gel DocTM EZ Imager (Bio-Rad). The band intensities were quantified using ImageJ software. Conformational stability was determined as the ratio of αS in the pellet to total αS (supernatant + pellet).
Cell culture, transfection of plasmids, and treatment of αS proteins
Human neuroblastoma SH-SY5Y cells were maintained at 37 °C in 5% CO2 in DMEM/F12 medium (Sigma-Aldrich) with 10% fetal calf serum, penicillin-streptomycin-glutamine (Gibco), and Minimum Eagle's Medium Nonessential Amino Acid Solution (Gibco). Cells at a concentration of 1.5 × 105 cells/ml were cultured to ∼30–50% confluence in collagen-coated 6-well plates and transfected with pcDNA3-WT mouse αS or pcDNA3-FLAG-human αS plasmids using X-tremeGENE 9 (Roche), followed by culture for 16 h. The cells were treated with final 150 nm αS proteins using MultiFectam (Promega), as previously described (72).
Immunocytochemistry
SH-SY5Y cells grown on collagen-coated coverslips were fixed with 4% paraformaldehyde and stained with the indicated primary antibodies at 1:1,000 dilution. After incubation for 1 h, cells were washed and treated with secondary antibodies (anti-rabbit IgG-conjugated Alexa Fluor 568 and anti-mouse IgG-conjugated Alexa Fluor 488; Invitrogen) and Hoechst 33324 (Sigma-Aldrich) to counterstain nuclear DNA. The cells were mounted and analyzed using a BZ-X710 (Keyence) and BZ-X analyzer (Keyence).
Biochemical analysis of cultured cells
After incubation for 2 days, cells were harvested by centrifugation (1,800 × g, 5 min) and washed with PBS. The cellular proteins were extracted with 300 μl of buffer A68 (10 mm Tris-HCl, pH 7.5, 1 mm EGTA, 10% sucrose, 0.8 m NaCl) containing final 1% sarkosyl (w/w) by sonication. After ultracentrifugation at 135,000 × g for 20 min at 25 °C, the supernatant was collected as sarkosyl-soluble fraction, and the protein concentration was determined by BCA assay. The pellet was washed once with 300 μl of buffer A68 containing 1% sarkosyl, and solubilized in 100 μl of SDS-sample buffer. Both sarkosyl-soluble and -insoluble fractions were analyzed by immunoblotting with appropriate antibodies as indicated (73). The band intensities of insoluble phospho-αS and mouse-αS were quantified using ImageJ software. The relative amount of precipitate mouse-αS was calculated by insoluble mouse-αS divided by insoluble plus soluble mouse-αS. Data are expressed as -fold induction relative to that in the WT.
αS inoculation into mice
Ten-week-old male C57BL/6J mice were purchased from CLEA Japan, Inc. All experimental protocols were performed according to the recommendations of the Animal Care and Use Committee of Tokyo Metropolitan Institute of Medical Science. αS proteins (150 μm, 5 μl) were injected into the right striatum (anterior-posterior, 0.5 mm; medial-lateral, −2.0 mm; dorsal-ventral, −3.5 mm). Inoculation into mouse brain was performed as described previously (10).
Immunohistochemistry of mouse brains
Three months after inoculation, mice were deeply anesthetized with pentobarbital injection and sacrificed. The brain was perfused with 0.1 m PBS, followed by 10% formalin neutral buffer solution. After fixation, the dissected brain was embedded in paraffin and 5 μm–thick serial sections were prepared. For antigen retrieval, sections were treated with 100% formic acid (Wako) for 5 min and washed under running tap water, then brought to a boil in 0.1 m sodium citrate buffer (pH 6.0) for 10 min, followed by cooling for 1 h. Immunohistochemistry with mAb EP1536Y (1:2,000) directed against αS phosphorylated at Ser-129 was performed as described previously (10). Sections were counterstained with hematoxylin. αS pathologies were analyzed with a BZ-X710 (Keyence) and quantified using a BZ-X analyzer (Keyence).
Biochemical analysis of mouse brains
Frozen whole mouse brains were homogenized in 20 volumes of buffer A68 containing final 2% sarkosyl (w/v), and incubated at 37 °C for 30 min. The brain homogenates were centrifuged at 9,460 × g for 20 min at 25 °C, and then the supernatants were collected and ultracentrifuged at 135,000 × g for 20 min at 25 °C. The pellets were washed with 30 mm Tris-HCl (pH 7.5) and ultracentrifuged as before. The pellet was once washed with 300 μl of buffer A68 containing 1% sarkosyl, and solubilized in 100 μl of SDS–sample buffer. Both sarkosyl-soluble and -insoluble fractions were analyzed by Western blotting with appropriate antibodies as indicated (10). The band intensities of insoluble phospho-αS and mouse-αS were quantified using ImageJ software. The relative amount of precipitate mouse-αS was calculated by insoluble mouse-αS divided by insoluble plus soluble mouse-αS. Data are expressed as -fold induction relative to that in the WT.
Statistical analysis
Graphs were created, and statistics were calculated in Prism ver. 5 (GraphPad Software, San Diego, CA). Multiple group comparisons were made by one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. Data are presented as mean ± S.E. unless otherwise noted. Significance was determined at *, p < 0.05, **, p < 0.01, and ***, p < 0.001.
Author contributions
M. T., G. S., and M. H. conceptualization; M. T., G. S., T. N., F. K., and M. H. resources; M. T. and M. H. data curation; M. T., G. S., T. N., F. K., A. T., and M. H. formal analysis; M. T., G. S., T. N., F. K., and M. H. validation; M. T., G. S., and M. H. investigation; M. T., G. S., and M. H. visualization; M. T., G. S., T. N., F. K., and M. H. methodology; M. T., G. S., and M. H. writing-original draft; M. T., G. S., T. N., F. K., A. T., and M. H. writing-review and editing; G. S., T. N., F. K., A. T., and M. H. supervision; M. H. funding acquisition; M. H. project administration.
Supplementary Material
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology Grants-in-Aid for Scientific Research (KAKENHI) Grant JP26117005 (to M. H.), Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (KAKENHI) Grant JP23228004 (to M. H.), and Japan Agency for Medical Research and Development (AMED) Grant-in-Aid for research on Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) Grant JP14533254 (to M. H.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
- PD
- Parkinson's disease
- αS
- α-synuclein
- MSA
- multiple system atrophy
- Hu
- human
- Mo
- mouse
- FL
- full-length
- FLA
- seeded full-length αS aggregates
- TEM
- transmission electron microscopy
- ThT
- thioflavin T
- PK
- proteinase K
- CBB
- Coomassie Brilliant Blue
- Str
- striatum
- SN
- substantia nigra
- Amy
- amygdala
- ACC
- anterior cingulate cortex
- buffer A
- 50 mm Tris-HCl, pH 7.5, 1 mm EGTA, 1 mm DTT
- NAC
- non-amyloid-β component.
References
- 1. Ross C. A., and Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, (suppl.) S10–S17 10.1038/nm1066 [DOI] [PubMed] [Google Scholar]
- 2. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., and Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- 3. Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M. S., Shen J., Takio K., and Iwatsubo T. (2002) α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164 10.1038/ncb748 [DOI] [PubMed] [Google Scholar]
- 4. Hasegawa M., Fujiwara H., Nonaka T., Wakabayashi K., Takahashi H., Lee V. M., Trojanowski J. Q., Mann D., and Iwatsubo T. (2002) Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076 10.1074/jbc.M208046200 [DOI] [PubMed] [Google Scholar]
- 5. Jucker M., and Walker L. C. (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51 10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frost B., and Diamond M. I. (2010) Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159 10.1038/nrn2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Conway K. A., Harper J. D., and Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early onset Parkinson disease. Nat. Med. 4, 1318–1320 10.1038/3311 [DOI] [PubMed] [Google Scholar]
- 8. Volpicelli-Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q., and Lee V. M. (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luk K. C., Song C., O'Brien P., Stieber A., Branch J. R., Brunden K. R., Trojanowski J. Q., and Lee V. M. (2009) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056 10.1073/pnas.0908005106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Masuda-Suzukake M., Nonaka T., Hosokawa M., Oikawa T., Arai T., Akiyama H., Mann D. M., and Hasegawa M. (2013) Prion-like spreading of pathological α-synuclein in brain. Brain 136, 1128–1138 10.1093/brain/awt037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watts J. C., Giles K., Oehler A., Middleton L., Dexter D. T., Gentleman S. M., DeArmond S. J., and Prusiner S. B. (2013) Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 110, 19555–19560 10.1073/pnas.1318268110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tu P. H., Galvin J. E., Baba M., Giasson B., Tomita T., Leight S., Nakajo S., Iwatsubo T., Trojanowski J. Q., and Lee V. M. (1998) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann. Neurol. 44, 415–422 10.1002/ana.410440324 [DOI] [PubMed] [Google Scholar]
- 13. Peelaerts W., Bousset L., Van der Perren A., Moskalyuk A., Pulizzi R., Giugliano M., Van den Haute C., Melki R., and Baekelandt V. (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- 14. Lee S. J., and Masliah E. (2015) Neurodegeneration: Aggregates feel the strain. Nature 522, 296–297 10.1038/nature14526 [DOI] [PubMed] [Google Scholar]
- 15. Prusiner S. B., Woerman A. L., Mordes D. A., Watts J. C., Rampersaud R., Berry D. B., Patel S., Oehler A., Lowe J. K., Kravitz S. N., Geschwind D. H., Glidden D. V., Halliday G. M., Middleton L. T., Gentleman S. M., Grinberg L. T., and Giles K. (2015) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 112, E5308–E5317 10.1073/pnas.1514475112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q., and Iwatsubo T. (1998) Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884 [PMC free article] [PubMed] [Google Scholar]
- 17. Muntané G., Ferrer I., and Martinez-Vicente M. (2012) α-Synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience 200, 106–119 10.1016/j.neuroscience.2011.10.042 [DOI] [PubMed] [Google Scholar]
- 18. Kellie J. F., Higgs R. E., Ryder J. W., Major A., Beach T. G., Adler C. H., Merchant K., and Knierman M. D. (2014) Quantitative measurement of intact α-synuclein proteoforms from post-mortem control and Parkinson's disease brain tissue by intact protein mass spectrometry. Sci. Rep. 4, 5797 10.1038/srep05797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu C. W., Giasson B. I., Lewis K. A., Lee V. M., Demartino G. N., and Thomas P. J. (2005) A precipitating role for truncated α-synuclein and the proteasome in α-synuclein aggregation: Implications for pathogenesis of Parkinson disease. J. Biol. Chem. 280, 22670–22678 10.1074/jbc.M501508200 [DOI] [PubMed] [Google Scholar]
- 20. Murray I. V., Giasson B. I., Quinn S. M., Koppaka V., Axelsen P. H., Ischiropoulos H., Trojanowski J. Q., and Lee V. M. (2003) Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42, 8530–8540 10.1021/bi027363r [DOI] [PubMed] [Google Scholar]
- 21. Periquet M., Fulga T., Myllykangas L., Schlossmacher M. G., and Feany M. B. (2007) Aggregated α-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 27, 3338–3346 10.1523/JNEUROSCI.0285-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Michell A. W., Tofaris G. K., Gossage H., Tyers P., Spillantini M. G., and Barker R. A. (2007) The effect of truncated human α-synuclein (1–120) on dopaminergic cells in a transgenic mouse model of Parkinson's disease. Cell Transplant. 16, 461–474 10.3727/000000007783464911 [DOI] [PubMed] [Google Scholar]
- 23. Li W., West N., Colla E., Pletnikova O., Troncoso J. C., Marsh L., Dawson T. M., Jäkälä P., Hartmann T., Price D. L., and Lee M. K. (2005) Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 2162–2167 10.1073/pnas.0406976102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prasad K., Beach T. G., Hedreen J., and Richfield E. K. (2012) Critical role of truncated α-synuclein and aggregates in Parkinson's disease and incidental Lewy body disease. Brain Pathol. 22, 811–825 10.1111/j.1750-3639.2012.00597.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Serpell L. C., Berriman J., Jakes R., Goedert M., and Crowther R. A. (2000) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc. Natl. Acad. Sci. U.S.A. 97, 4897–4902 10.1073/pnas.97.9.4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qin Z., Hu D., Han S., Hong D. P., and Fink A. L. (2007) Role of different regions of α-synuclein in the assembly of fibrils. Biochemistry 46, 13322–13330 10.1021/bi7014053 [DOI] [PubMed] [Google Scholar]
- 27. Yonetani M., Nonaka T., Masuda M., Inukai Y., Oikawa T., Hisanaga S., and Hasegawa M. (2009) Conversion of wild-type α-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J. Biol. Chem. 284, 7940–7950 10.1074/jbc.M807482200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miake H., Mizusawa H., Iwatsubo T., and Hasegawa M. (2002) Biochemical characterization of the core structure of α-synuclein filaments. J. Biol. Chem. 277, 19213–19219 10.1074/jbc.M110551200 [DOI] [PubMed] [Google Scholar]
- 29. Tsuji H., Arai T., Kametani F., Nonaka T., Yamashita M., Suzukake M., Hosokawa M., Yoshida M., Hatsuta H., Takao M., Saito Y., Murayama S., Akiyama H., Hasegawa M., Mann D. M., and Tamaoka A. (2012) Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 135, 3380–3391 10.1093/brain/aws230 [DOI] [PubMed] [Google Scholar]
- 30. Legname G., Nguyen H. O., Peretz D., Cohen F. E., DeArmond S. J., and Prusiner S. B. (2006) Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. U.S.A. 103, 19105–19110 10.1073/pnas.0608970103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li J., Uversky V. N., and Fink A. L. (2001) Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 40, 11604–11613 10.1021/bi010616g [DOI] [PubMed] [Google Scholar]
- 32. Fredenburg R. A., Rospigliosi C., Meray R. K., Kessler J. C., Lashuel H. A., Eliezer D., and Lansbury P. T. Jr. (2007) The impact of the E46K mutation on the properties of α-synuclein in its monomeric and oligomeric states. Biochemistry 46, 7107–7118 10.1021/bi7000246 [DOI] [PubMed] [Google Scholar]
- 33. Ghosh D., Sahay S., Ranjan P., Salot S., Mohite G. M., Singh P. K., Dwivedi S., Carvalho E., Banerjee R., Kumar A., and Maji S. K. (2014) The newly discovered Parkinson's disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry 53, 6419–6421 10.1021/bi5010365 [DOI] [PubMed] [Google Scholar]
- 34. Giasson B. I., Uryu K., Trojanowski J. Q., and Lee V. M. (1999) Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 274, 7619–7622 10.1074/jbc.274.12.7619 [DOI] [PubMed] [Google Scholar]
- 35. Porcari R., Proukakis C., Waudby C. A., Bolognesi B., Mangione P. P., Paton J. F., Mullin S., Cabrita L. D., Penco A., Relini A., Verona G., Vendruscolo M., Stoppini M., Tartaglia G. G., Camilloni C., Christodoulou J., Schapira A. H., and Bellotti V. (2015) The H50Q mutation induces a 10-fold decrease in the solubility of α-synuclein. J. Biol. Chem. 290, 2395–2404 10.1074/jbc.M114.610527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fares M. B., Ait-Bouziad N., Dikiy I., Mbefo M. K., Jovičić A., Kiely A., Holton J. L., Lee S. J., Gitler A. D., Eliezer D., and Lashuel H. A. (2014) The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 23, 4491–4509 10.1093/hmg/ddu165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rospigliosi C. C., McClendon S., Schmid A. W., Ramlall T. F., Barré P., Lashuel H. A., and Eliezer D. (2009) E46K Parkinson's-linked mutation enhances C-terminal-to-N-terminal contacts in α-synuclein. J. Mol. Biol. 388, 1022–1032 10.1016/j.jmb.2009.03.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bussell R. Jr., and Eliezer D. (2001) Residual structure and dynamics in Parkinson's disease-associated mutants of α-synuclein. J. Biol. Chem. 276, 45996–46003 10.1074/jbc.M106777200 [DOI] [PubMed] [Google Scholar]
- 39. Ma M. R., Hu Z. W., Zhao Y. F., Chen Y. X., and Li Y. M. (2016) Phosphorylation induces distinct α-synuclein strain formation. Sci. Rep. 6, 37130 10.1038/srep37130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bousset L., Pieri L., Ruiz-Arlandis G., Gath J., Jensen P. H., Habenstein B., Madiona K., Olieric V., Böckmann A., Meier B. H., and Melki R. (2013) Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4, 2575 10.1038/ncomms3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peelaerts W., and Baekelandt V. (2016) α-Synuclein strains and the variable pathologies of synucleinopathies. J. Neurochem. 139, Suppl. 1, 256–274 10.1111/jnc.13595 [DOI] [PubMed] [Google Scholar]
- 42. Buell A. K., Galvagnion C., Gaspar R., Sparr E., Vendruscolo M., Knowles T. P., Linse S., and Dobson C. M. (2014) Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 111, 7671–7676 10.1073/pnas.1315346111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim K. S., Choi Y. R., Park J. Y., Lee J. H., Kim D. K., Lee S. J., Paik S. R., Jou I., and Park S. M. (2012) Proteolytic cleavage of extracellular α-synuclein by plasmin: Implications for Parkinson disease. J. Biol. Chem. 287, 24862–24872 10.1074/jbc.M112.348128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iwata A., Maruyama M., Akagi T., Hashikawa T., Kanazawa I., Tsuji S., and Nukina N. (2003) Alpha-synuclein degradation by serine protease neurosin: Implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 12, 2625–2635 10.1093/hmg/ddg283 [DOI] [PubMed] [Google Scholar]
- 45. Sevlever D., Jiang P., and Yen S. H. (2008) Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687 10.1021/bi800699v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang W., Nguyen L. T., Burlak C., Chegini F., Guo F., Chataway T., Ju S., Fisher O. S., Miller D. W., Datta D., Wu F., Wu C. X., Landeru A., Wells J. A., Cookson M. R., et al. (2016) Caspase-1 causes truncation and aggregation of the Parkinson's disease-associated protein α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 113, 9587–9592 10.1073/pnas.1610099113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sung J. Y., Park S. M., Lee C. H., Um J. W., Lee H. J., Kim J., Oh Y. J., Lee S. T., Paik S. R., and Chung K. C. (2005) Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J. Biol. Chem. 280, 25216–25224 10.1074/jbc.M503341200 [DOI] [PubMed] [Google Scholar]
- 48. Zhang Z., Kang S. S., Liu X., Ahn E. H., Zhang Z., He L., Iuvone P. M., Duong D. M., Seyfried N. T., Benskey M. J., Manfredsson F. P., Jin L., Sun Y. E., Wang J. Z., and Ye K. (2017) Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson's disease. Nat. Struct. Mol. Biol. 24, 632–642 10.1038/nsmb.3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crowther R. A., Jakes R., Spillantini M. G., and Goedert M. (1998) Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 436, 309–312 10.1016/S0014-5793(98)01146-6 [DOI] [PubMed] [Google Scholar]
- 50. Hoyer W., Cherny D., Subramaniam V., and Jovin T. M. (2004) Impact of the acidic C-terminal region comprising amino acids 109–140 on α-synuclein aggregation in vitro. Biochemistry 43, 16233–16242 10.1021/bi048453u [DOI] [PubMed] [Google Scholar]
- 51. Levitan K., Chereau D., Cohen S. I., Knowles T. P., Dobson C. M., Fink A. L., Anderson J. P., Goldstein J. M., and Millhauser G. L. (2011) Conserved C-terminal charge exerts a profound influence on the aggregation rate of α-synuclein. J. Mol. Biol. 411, 329–333 10.1016/j.jmb.2011.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meuvis J., Gerard M., Desender L., Baekelandt V., and Engelborghs Y. (2010) The conformation and the aggregation kinetics of α-synuclein depend on the proline residues in its C-terminal region. Biochemistry 49, 9345–9352 10.1021/bi1010927 [DOI] [PubMed] [Google Scholar]
- 53. Lee H. J., and Lee S. J. (2002) Characterization of cytoplasmic α-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J. Biol. Chem. 277, 48976–48983 10.1074/jbc.M208192200 [DOI] [PubMed] [Google Scholar]
- 54. Zibaee S., Jakes R., Fraser G., Serpell L. C., Crowther R. A., and Goedert M. (2007) Sequence determinants for amyloid fibrillogenesis of human α-synuclein. J. Mol. Biol. 374, 454–464 10.1016/j.jmb.2007.09.039 [DOI] [PubMed] [Google Scholar]
- 55. McLean P. J., and Hyman B. T. (2002) An alternatively spliced form of rodent α-synuclein forms intracellular inclusions in vitro: Role of the carboxy-terminus in α-synuclein aggregation. Neurosci. Lett. 323, 219–223 10.1016/S0304-3940(02)00154-4 [DOI] [PubMed] [Google Scholar]
- 56. Kessler J. C., Rochet J. C., and Lansbury P. T. Jr. (2003) The N-terminal repeat domain of α-synuclein inhibits β-sheet and amyloid fibril formation. Biochemistry 42, 672–678 10.1021/bi020429y [DOI] [PubMed] [Google Scholar]
- 57. Bertoncini C. W., Jung Y. S., Fernandez C. O., Hoyer W., Griesinger C., Jovin T. M., and Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435 10.1073/pnas.0407146102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fernández C. O., Hoyer W., Zweckstetter M., Jares-Erijman E. A., Subramaniam V., Griesinger C., and Jovin T. M. (2004) NMR of α-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 23, 2039–2046 10.1038/sj.emboj.7600211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ulrih N. P., Barry C. H., and Fink A. L. (2008) Impact of Tyr to Ala mutations on α-synuclein fibrillation and structural properties. Biochim. Biophys. Acta 1782, 581–585 10.1016/j.bbadis.2008.07.004 [DOI] [PubMed] [Google Scholar]
- 60. Xu L., Ma B., Nussinov R., and Thompson D. (2017) Familial mutations may switch conformational preferences in α-synuclein fibrils. ACS Chem. Neurosci. 8, 837–849 10.1021/acschemneuro.6b00406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Uversky V. N. (2017) Looking at the recent advances in understanding α-synuclein and its aggregation through the proteoform prism. F1000Res 6, 525 10.12688/f1000research.10536.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Luk K. C., Covell D. J., Kehm V. M., Zhang B., Song I. Y., Byrne M. D., Pitkin R. M., Decker S. C., Trojanowski J. Q., and Lee V. M. (2016) Molecular and biological compatibility with host α-synuclein influences fibril pathogenicity. Cell Rep. 16, 3373–3387 10.1016/j.celrep.2016.08.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Oueslati A., Ximerakis M., and Vekrellis K. (2014) Protein transmission, seeding and degradation: Key steps for α-synuclein prion-like propagation. Exp. Neurobiol. 23, 324–336 10.5607/en.2014.23.4.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sacino A. N., Brooks M. M., Chakrabarty P., Saha K., Khoshbouei H., Golde T. E., and Giasson B. I. (2017) Proteolysis of α-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J. Neurochem. 140, 662–678 10.1111/jnc.13743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vilchez D., Saez I., and Dillin A. (2014) The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5, 5659 10.1038/ncomms6659 [DOI] [PubMed] [Google Scholar]
- 66. Dehay B., Martinez-Vicente M., Caldwell G. A., Caldwell K. A., Yue Z., Cookson M. R., Klein C., Vila M., and Bezard E. (2013) Lysosomal impairment in Parkinson's disease. Movement Disorders 28, 725–732 10.1002/mds.25462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bae E. J., Yang N. Y., Lee C., Lee H. J., Kim S., Sardi S. P., and Lee S. J. (2015) Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 47, e188 10.1038/emm.2015.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Masuda M., Dohmae N., Nonaka T., Oikawa T., Hisanaga S., Goedert M., and Hasegawa M. (2006) Cysteine misincorporation in bacterially expressed human α-synuclein. FEBS Lett. 580, 1775–1779 10.1016/j.febslet.2006.02.032 [DOI] [PubMed] [Google Scholar]
- 69. Jakes R., Spillantini M. G., and Goedert M. (1994) Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32 10.1016/0014-5793(94)00395-5 [DOI] [PubMed] [Google Scholar]
- 70. Masuda M., Suzuki N., Taniguchi S., Oikawa T., Nonaka T., Iwatsubo T., Hisanaga S., Goedert M., and Hasegawa M. (2006) Small molecule inhibitors of α-synuclein filament assembly. Biochemistry 45, 6085–6094 10.1021/bi0600749 [DOI] [PubMed] [Google Scholar]
- 71. Nonaka T., Iwatsubo T., and Hasegawa M. (2005) Ubiquitination of α-synuclein. Biochemistry 44, 361–368 10.1021/bi0485528 [DOI] [PubMed] [Google Scholar]
- 72. Oikawa T., Nonaka T., Terada M., Tamaoka A., Hisanaga S., and Hasegawa M. (2016) α-Synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J. Biol. Chem. 291, 15046–15056 10.1074/jbc.M116.736355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nonaka T., Watanabe S. T., Iwatsubo T., and Hasegawa M. (2010) Seeded aggregation and toxicity of α-synuclein and tau: Cellular models of neurodegenerative diseases. J. Biol. Chem. 285, 34885–34898 10.1074/jbc.M110.148460 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.