

Abstract
Osteogenesis imperfecta, or brittle bone disease, is a congenital disease that primarily causes low bone mass and bone fractures but it can negatively affect other organs. It is usually inherited in an autosomal dominant fashion, although rarer recessive and X-chromosome-linked forms of the disease have been identified. In addition to type I collagen, mutations in a number of other genes, often involved in type I collagen synthesis or in the differentiation and function of osteoblasts, have been identified in the last several years. Seldom, the study of a rare disease has delivered such a wealth of new information that have helped our understanding of multiple processes involved in collagen synthesis and bone formation. In this short review I will describe the clinical features and the molecular genetics of the disease, but then focus on how OI dysregulates all aspects of extracellular matrix biology. I will conclude with a discussion about OI therapeutics.
Keywords: Osteogenesis imperfecta, collagen, bone, fragility, mineralization, proteoglycans, genetics
Introduction
Osteogenesis imperfecta (OI or brittle bone disease) is an inherited, generalized, connective-tissue disease that primarily affects the skeleton by lowering bone mass and causing fractures. It is most often an autosomal dominant condition, although rarer recessive and X-chromosome-linked forms of the disease also have been identified. Describing a newborn with numerous fractures in the 1840s, Willem Vrolik coined the term osteogenesis imperfecta, which in Latin means imperfect bone formation [1, 2]. Several subsequent studies reported on clinical, hereditary, epidemiological, and social aspects of OI [3–7]. These studies, along with research progress on collagen biology in the 1970s [8–11], laid the groundwork for a series of seminal papers that finally associated type I collagen mutations with autosomal dominant OI in the early 1980s [12–18].
While research on OI continued in the following years, the emphasis on type I collagen partially shadowed the interest in those OI cases that had a clear pattern of recessive inheritance and lacked an association with collagen genes [19–21]. In the 21st century, progress in genetic technology fueled another round of seminal discoveries in the OI field. Starting in 2006, the identification of several genes associated with rarer forms of OI has significantly revitalized the field, taught us many new notions about bone formation, and triggered discussions about what OI nomenclature to adopt.
Regardless of the underlying genetic mutation, OI is a disease characterized by significant alterations in quantity, quality, mineralization and homeostasis of the extracellular matrix (ECM). This mini-review will describe the clinical features and genetic heterogeneity of the disease, followed by the role of the ECM in the pathogenesis of OI and recent progress in OI therapeutics.
Clinical features of the disease
OI is the most common bone-fragility disorder with an incidence of about 1 in 15,000 and an estimated prevalence in the US of between 25,000 and 50,000 affected individuals [22]. Both genetically and clinically, it is a highly heterogeneous disease [7]. For instance, the same type I collagen mutation may express very different disease symptoms among members of a family or of different families [23]. The clinical severity of the disease is evaluated along a spectrum, ranging from perinatal lethal (OI type II), to severe (OI type III), to moderate (OI type IV), to mild (OI type I), as originally classified by Sillence and Rimoin [5]. An example of this wide range of severity is shown in Figure 1.
Figure 1.
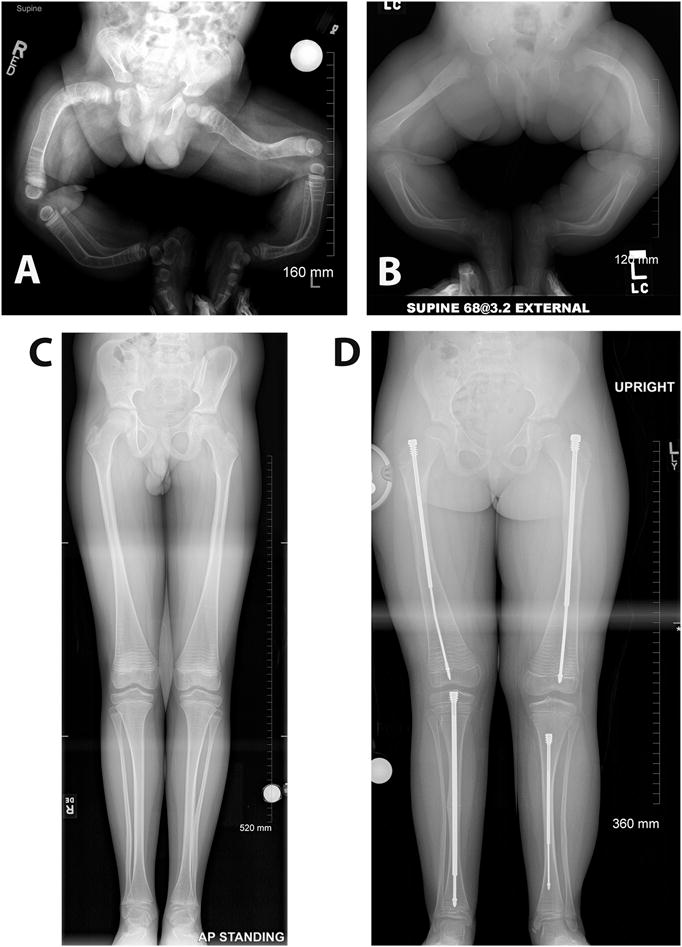
Radiographic images of lower extremities of patients affected with different types of OI, demonstrating the wide range of severity that affects the skeleton in this disease. A. One-year-old infant diagnosed with severe type III OI. Note the severe bowing of the legs and the lack of bone modeling in both femurs and tibiae. B. A nine-month-old infant with moderately severe type IV OI. C. A 12-year-old patient with type I OI. Note the relative constriction of the femur diaphysis and the flaring of the distal metaphysis which is known as Erlenmeyer flask deformity. Metaphyseal bands due to cyclical bisphosphonate treatment are also visible. D. Same patient shown in B at 7 years of age and after years of medical and surgical management. Note the telescoping rods implanted in both femurs and tibiae, a common surgical procedure in OI patients that have significant long bone deformities. All the radiographic images are a courtesy of Dr. Paul W. Esposito and Maegen Wallace (University of Nebraska Medical Center).
The disease primarily affects the skeletal system, and the main clinical manifestations are generalized low bone mass, recurrent fractures following minor trauma (these can be detected in utero in severe OI), bowing of the long bones, vertebral compression, scoliosis, bone pain, stunted growth, and ligamentous and joint laxity [24, 25]. A number of additional clinical features can be present or develop during growth or in adult life, sometimes based on the severity or the underlying genetic mutation. These additional features may include dentinogenesis imperfecta with poorly mineralized and translucent dentin, dental malocclusion, basilar invagination of the base of the cranium, blue sclerae, conductory or sensory hearing impairment/loss, thinning and easily bruised skin, susceptibility to bleeding, joint contractures, restrictive or obstructive pulmonary disease and cardiovascular manifestations [25].
With such varied symptoms, the medical management of OI patients requires a multidisciplinary team approach at all ages (including adulthood) to provide coordinated and comprehensive assessments and interventions aimed at maximizing function, independence, well-being, and, ultimately, quality of life [26]. A recent study on 687 cases of OI patients who were followed for more than 35 years in Denmark reported that patients with OI were at increased risk of death due to respiratory disease, gastrointestinal disease, and trauma; compared with the general population, OI patients had a higher mortality rate throughout their life [27].
Genetics of OI: an evolving story
As mentioned earlier, OI was associated with type I collagen alterations in the early 1980s. It is now estimated that about 85% of OI cases are caused by autosomal dominant mutations in either the COL1A1 or COL1A2 gene, encoding the α1(I) or α 2(I) chain of type I collagen, respectively. The remainder of OI cases are associated with dominant, recessive or X-linked mutations in a number of genes involved in different aspects of collagen synthesis, processing or crosslinking, or in osteoblast differentiation and function [25]. Heterozygous COL1A1 mutations causing a null allele result in haploinsufficiency and are the most common type of mutations that result in dominant OI [28]. They cause a quantitative loss of α 1(I) chain and, with few exceptions, cause a mild form of OI (type I) because the other allele produces collagen of normal quality and structure [28]. OI type I patients have increased susceptibility to fractures, few to no deformities, and short to normal stature [29].
Conversely, other mutations in COL1A1 or COL1A2, most often glycine substitutions in the Gly-X-Y collagen repeat, cause structural defects of the collagen triple helix that have a dominant negative action on the normal collagen chains, and result in moderate, severe or lethal OI [30]. These structural mutations often cause protracted collagen folding with over-modification of lysine residues in the endoplasmic reticulum (ER), enlargement of the ER due to accumulation of misfolded chains, abnormal collagen trafficking, and delayed secretion in the ECM [29]. A database on osteogenesis imperfecta variants indicates that more than 1,500 unique COL1A1 and COL1A2 (combined) DNA variants were reported as of October 2017 (www.le.ac.uk/ge/collagen/). Genotype-phenotype correlations among the different structural mutations and the moderate to lethal phenotype spectrum of severity have been challenging to assess, but some general observations on mutations affecting the triple helical domain can be taken and are reported elsewhere [23].
The tremendous progress in genetic technologies of the 1990s and the beginning of the 21st century dramatically accelerated the pace of new medical discoveries, including in the OI field. Gene-targeting techniques in rodents, for instance, promoted reverse genetic approaches to studying the function of poorly understood genes and generating new mouse models for human diseases [31]. The sequencing of the human genome and the development of high-throughput DNA sequencing technologies also led to an explosion of forward genetic studies and made the identification of new genes involved in rare diseases, including OI, an easier task [32]. Beginning in 2004, this climate has allowed scientists to identify at least one new gene associated with a rare form of OI every year. Although the exact count of such genes may vary depending on the classification, these number no less than 15 genes (in addition to COL1A1 and COL1A2), most of which cause recessive OI; this count also includes one gene that causes dominant OI and one gene that causes an X-linked form of OI [25]. The newly identified genes contribute to about 15% of all OI cases.
The identification of mutations in PLOD2 (encodes Lysyl hydroxylase 2) and CRTAP (encodes Cartilage-associated protein) enabled a major breakthrough: the discovery that alterations in collagen post-translational modifications can cause OI [33–35]. This advancement quickly led to the identification of mutations in P3H1 (encodes Prolyl 3-hydroxylase 1) [36] and PPIB (encodes Cyclophilin B) [37, 38], which, together with CRTAP, form the collagen prolyl 3-hydroxylation complex that also acts as collagen chaperone in the ER [39]. Mutations in two other collagen chaperones, SERPINH1 (encodes HSP47) [40] and FKBP10 (encodes FKBP65) [41], that participate in type I collagen folding, downstream of the prolyl 3-hydroxylation complex, were also identified in recessively inherited OI. Interestingly, mutations in the cation channel TMEM38B (encodes TRIC-B) expressed on the ER membrane, cause recessive OI by dysregulating collagen post-translational modification and causing ER stress and reduced collagen secretion [42, 43]. The identification of mutations in CREB3L1 (which encodes OASIS) and MBTPS2 (which encodes S2P) further highlighted the importance of collagen misfolding in the ER, the ensuing ER stress, and the role of regulated intramembrane proteolysis (RIP) in the complex cross-talk between the ER and Golgi. CREB3L1 codes for a putative ER stress transducer that regulates procollagen I expression [44]. S2P is a site-2 metalloprotease involved in intramembrane proteolysis that somehow affects type I collagen lysyl hydroxylation and secretion, and causes the only X-linked form of OI described so far [45].
The significance of proper collagen processing is also evident in the ECM; mutations in BMP1 (encodes bone morphogenetic protein 1), the main extracellular protease involved in procollagen 1 C-propeptide (PICP) excision, cause recessive OI [46]. Recessive OI has been associated with mutations in two genes encoding secreted proteins that bind to type I collagen in the ECM, SPARC (which encodes SPARC/osteonectin) [47, 48] and SERPINF1 (which encodes PEDF – pigment epithelium-derived factor) [49–51]. The latter is the gene responsible for what was earlier named OI type VI; it causes an atypical bone mineralization defect based on histological observations [20]. PEDF appears to be involved in a poorly characterized signaling pathway that includes the plasma membrane receptor BRIL, encoded by IFITM5. IFITM5 mutations cause the only other dominant form of OI (OI type V) through a putative gain of function or neomorphic activity of the most common identified mutation, which adds 5 amino acid (MALEP) at the N-terminal end of the protein [19, 52–54].
One of the best-studied anabolic signaling pathways for bone is the canonical WNT pathway [55]. Thus, it is not surprising that its dysregulation may also cause OI. In fact, loss of function mutations in LRP5 (which encodes LDL receptor-related protein 5), a WNT co-receptor, cause osteoporosis-pseudoglioma syndrome (formerly known as the ocular form of OI) [56]. More recently, mutations in WNT1 (which encodes proto-oncogene WNT1) were also identified in early-onset osteoporosis and OI [57, 58]. Finally, a mutation in SP7 (encodes SP7/Osterix), an essential transcription factor for murine osteoblast differentiation [59], can also cause recessive OI, though the mechanism is still poorly studied [60].
The classification of OI has been debated in recent years in light of the discovery of several genes, including non-collagen genes, that can cause the disease. While a strictly genetic classification has been utilized, with an increasing number of distinct OI types based on the underlying genetic mutation [25], the most recent recommendations on the nosology and classification of skeletal dysplasias is to continue to use the Sillence classification (types I-IV) for osteogenesis imperfecta. This recommendation is based on the phenotypical features of the disease and not on its molecular genetics. An OI type V is also added since this type is radiologically distinguishable from the other ones [61].
It is important to realize that the study of a rare bone disease such as OI has led to more than the identification of the genes that cause it. The functional studies of the proteins those genes encode are generating a significant new body of knowledge and increased understanding of collagen processing, bone mineralization, osteoblast differentiation and matrix homeostasis. Table 1 provides a list of the genes associated with OI, classified based on the localization of the protein they encode.
TABLE 1.
Classification of osteogenesis imperfecta (OI) based on the cellular localization (color-coded) of the defective protein. It is important to note that mutations in proteins located in the ER and Golgi ultimately cause type I collagen defects in the bone matrix. AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive.
| Localization | Protein | Function | Gene | Inheritance | Clinical severity |
|---|---|---|---|---|---|
| Matrix | α1(I) | Matrix structural component | COL1A1 | AD | Mild to lethal OI depending on themutation |
| Matrix | α 2(I) | Matrix structural component | COL1A2 | AD | Moderate tolethal depending on the mutation |
| Matrix | PEDF | Collagen-binding protein/pigment epithelium derived factor | SERPINF1 | AR | Moderate to severe OI – postnatal onset |
| Matrix | BMP1 | Extracellular protease | BMP1 | AR | Moderate to severe OI |
| Matrix | WNT1 | Secreted signaling molecule | WNT1 | AR | Moderate tosevere OI – CNS defects |
| Matrix | SPARC | Matrix-associated protein | SPARC | AR | Moderate to severe OI |
| Plasma membrane | BRIL | Bone-restricted interferon-induced transmembrane-like protein | IFITM5 | AD | Variable severity– hyperplastic callus formation, calcification of interosseous membrane |
| Plasma membrane | LRP5 | WNT signaling co-receptor | LRP5 | AR | Osteoporosis-pseudoglioma syndrome – ocular defects |
| ER-Golgi | TRIC-B | Cation channel | TMEM38B | AR | Moderate to severe OI |
| ER-Golgi | S2P | Membrane-bound transcription factorsite-2 protease | MBTPS2 | XLR | Moderate to severe OI – chest deformities |
| ER-Golgi | OASIS | Cyclic AMP responsive element binding protein 3-like protein 1 | CREB3L1 | AR | Severe OI |
| ER-Golgi | HSP47 | Type I procollagen chaperone | SERPINH1 | AR | Severe OI |
| ER | FKBP65 | Type I procollagen chaperone | FKBP10 | AR | Moderate to severe OI |
| ER | LH2 | Collagen telo-peptide lysyl hydroxylase | PLOD2 | AR | Moderate to severe OI – joints contractures |
| ER | CYCLOPHILIN B | Peptidylprolyl Isomerase B | PPIB | AR | Moderate to severe OI |
| ER | P3H1 | Prolyl 3-hydroxylase | P3H1 | AR | Severe to lethal OI |
| ER | CRTAP | Adapter protein??? | CRTAP | AR | Severe to lethal OI |
| Nucleus | SP7/Osterix | Transcription factor | SP7 | AR | Mild to moderate OI |
Bone extracellular matrix in the pathogenesis of OI
Engineers teach us that the solidity and strength of a building depend on its architectural design, the amount of building materials used, and the quality of the building materials. The same is true for our skeleton: the ultimate strength of our bones and their resistance to fracture depend on how the bones are built (architecture), how much bone is there (bone mass), and the quality of the bone components (material properties). Furthermore, bones have a hierarchical structural organization across several length-scales, from macro to micro to nano, which ensure great energy dissipation within such arrangement [62]. Changes in any of the above parameters affect bone strength, and OI is known to negatively impact all of them and thus cause the severest form of brittle bone disease [63].
While bone mass and architecture can be intrinsically related, and usually an increase in bone mass results in improved bone architecture, the bone material properties, derived from the ordered assembly of individual bone matrix components and the mineralization of the matrix, are usually independent variables. Among the three parameters, the study of OI has clearly highlighted the essential role of material properties in bone strength [63]. In fact, genetic mutations that cause a quantitative reduction of type I collagen essentially reduce bone mass and affect bone architecture but cause milder forms of OI disease. This is contrary to structural mutations of type I collagen, which have a dominant negative action on the product of the healthy allele, significantly impact the quality of the bone matrix and result in moderate to lethal forms of OI. Therefore, at least for what concerns type I collagen, it can be argued that a poor quality of a main matrix component is less well-tolerated compared with variations in its amount. Nevertheless, OI affects the ECM in multiple ways.
OI and matrix organic components
The extracellular matrix proteins that are found in bone can be divided into collagen and non-collagenous proteins. Collagen, primarily type I with smaller amounts of type III and V collagen, constitutes about 85-90% of the total bone protein content; the rest is represented by proteoglycans, SIBLING proteins (small integrin-binding ligand, N-linked glycoproteins), glycosylated proteins, γ-carboxylated proteins, and other serum-derived proteins [64, 65]. Type I collagen not only plays a major structural role in bone but also contributes to tissue organization and, given that it is essential for mineralization, to its mechanical properties [66, 67]. As such, it is not surprising that defects in type I collagen have dramatic effects on the skeleton. In most OI cases, whether dominant or recessive, there is a significant reduction of type I collagen in the matrix produced by osteoblasts and skin fibroblasts (Fig. 2) [25, 68–70]. This reduction can be due to reduced synthesis (COL1A1 null alleles) or to a number of alterations in the collagen synthetic pathway that range from defective procollagen chain association, slowing of the triple helix winding, over-modification of lysine and proline residues, regional misfolding of the triple helical region, defective procollagen chaperone activity, or dysregulated intracellular trafficking [29, 34, 71]. These processes can increase the retention of the procollagen in the ER, cause enlargement of the ER, trigger the ER-associated degradation pathway [72] or autophagy [73–75], and result in both delayed and decreased collagen secretion. Moreover, the collagen that is secreted in the matrix can have altered post-translational modification [76] and glycosylation patterns, which can lead to defective/altered cross-linking and disruption of proper collagen fibrillogenesis [34]. The latter is likely also a consequence of the reduced ability of the abnormal collagen to correctly bind to other matrix molecules, including proteoglycans such as Decorin, which is known to regulate the assembly of collagen fibrils [77–79]. Notably, type I collagen mutations that localize along triple helical regions identified as proteoglycan binding sites, are often perinatal lethal [23, 80].
Figure 2.
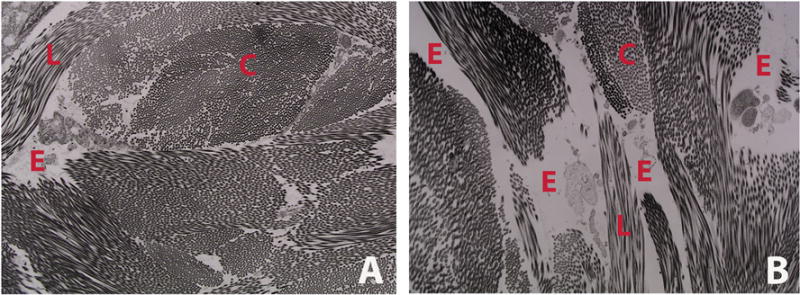
Representative transmission electron micrograph of dermal collagen from WT (A) and CrtapKO (B) mouse, showing decreased amount of collagen and increased space among collagen bundles in this murine model of recessive OI. This translates into skin laxity and reduced dermal thickness. C = cross section of collagen fibrils; L = longitudinal section of collagen fibrils; E = empty space.
A study comparing steady-state levels of specific matrix proteins secreted by human bone cells from OI patients and age-matched controls showed that in addition to reduced collagen, biglycan (a large chondroitin-sulfate proteoglycan), decorin, and osteonectin were present in reduced levels in the matrix of OI cells. Conversely, some other matrix components such as thrombospondin, fibronectin, and hyaluronan were present in higher amount in the OI matrix compared to controls [81]. This is a clear indication that the extracellular matrix stoichiometry is altered in OI [81]. Consistent with that study, biallelic mutations in the gene coding for osteonectin (also known as SPARC, a secreted acidic cysteine-rich glycoprotein) that affect its binding to type I collagen were shown to cause OI [47]. Interestingly, it was suggested that SPARC might be a collagen chaperone and that its loss may lead to defects in collagen deposition [82]. Therefore, OI-causing mutations, whether in type I collagen, in genes affecting its processing or in genes encoding other matrix components, result in both quantitative and qualitative changes of the bone matrix and can lead to changes in its material properties [81].
OI and matrix inorganic components
Mineral in the form of hydroxyapatite, a crystalline complex of calcium and phosphate, is the most abundant component of bone [64]. Active osteoblasts lay down new ECM on the bone surface, which is termed osteoid before it mineralizes. The mineralization of osteoid takes several days [83] and is a process which is actively investigated, yet not completely understood. Two proteins appear essential in bone mineralization: the first is type I collagen and the fibrils it forms which constitute the protein scaffold upon which mineral is deposited; the second is alkaline phosphatase which acts on the surface of osteoblasts to eliminate pyrophosphate, a strong inhibitor of mineralization, and to modify the phosphorylation status of osteopontin [67, 84]. Other bone matrix proteins are also known to regulate the mineralization process such as proteoglycans [85, 86], matrix Gla-protein [87] and various phosphate-regulating proteins including MEPE (matrix extracellular phosphoglycoprotein), DMP1 (dentin matrix protein 1) and PHEX (phosphate-regulating endopeptidase homolog X-linked) [88], among others. Calcium and phosphate ions are actively concentrated within matrix vesicles, which are 100nm particles secreted by osteoblasts in a polarized fashion onto the bone surface, and are where initial formation of mineral crystals takes place [89]. These vesicles then release the preformed hydroxyapatite crystals, and these serve as templates for further propagation of the mineralization process, in the presence of sufficient extracellular Ca2+ and PO4 3− [89]. The calcification of osteoid is thus the result of a combination of inorganic and organic components, and is regulated by factors that may promote or inhibit this process.
Because type I collagen is so important in bone mineralization, any changes in its quantity and/or quality such as those seen in OI, have negative consequences on this process. Studies of iliac crest biopsy cores taken from patients affected with different types of OI have shown higher average mineralization densities compared to age-matched healthy controls, as assessed by quantitative backscattered electron imaging (qBEI) [90]. One interpretation is that the abnormal ECM assembly in OI results in an increased water fraction that is available for mineral deposition [90]. Additional studies have demonstrated a higher degree of mineralization in OI bones and confirmed the finding of increased bone mineral density distribution (BMDD) in all types of OI, including the dominant and some of the recessive forms of the disease [91–93]. Analyses using small-angle X-ray scattering (SAXS) and wide-angle X-ray diffraction (WAXD) showed that the high bone matrix mineralization observed in children with OI type I is not due to larger mineral particles; rather, the size of mineral particles was the same or smaller but their packing density is increased compared to controls [94]. Several other studies utilizing both Raman and Fourier-transform infrared (FTIR) spectroscopy were conducted on various animal models of OI, especially in oim (osteogenesis imperfecta mouse), to investigate mineral composition, crystal size and alignment, and mineral-to-matrix ratio. These are summarized elsewhere [95]. Most of these studies agree that the hydroxyapatite crystals are reduced in size and are less-well aligned along collagen fibrils compared to controls. Conversely, reports of mineral-to-matrix ratio were less consistent and varied between decreased or increased depending on the OI mouse model, the instrumentation used or the group that performed the study [95, 96].
IFITM5 and SERPINF1 are two recently identified genes associated with dominant OI type V and recessive OI type VI, respectively, and encode for proteins that directly affect the bone mineralization process [54]. Histological studies on iliac crest biopsy specimens from OI type V described lamellae with irregular organization and a mesh-like appearance [19]. Similar studies on iliac crest biopsies from OI type VI patients revealed an absence of the birefringent pattern of normal lamellar bone under polarized light, accompanied with a “fish-scale” pattern [20]. Although the defective proteins, BRIL and PEDF (respectively), apparently are not involved in the synthesis of type I collagen, there is evidence for reduced type I collagen production and increased mineralization in primary osteoblast cultures [97] and hyper-mineralization of bone tissue [98] similar to other forms of OI.
Collectively, multiple studies on OI bones have shown decreased bone mass with increased bone mineralization, accompanied by hydroxyapatite crystals that are reduced in size, more densely packed and less-well organized along collagen fibrils. These features lead to changes in intrinsic bone material properties, including a reduction in bone toughness, elastic modulus and ultimate strength and in increase in bone brittleness which are typical distinguishing features of OI brittle bone compared to other bone fragility conditions [63, 99].
OI and matrix-to-cell signaling
The ECM used to be considered a rather static structure providing support to various cells and tissues. It is now well accepted that the ordered assembly of the various matrix components is a highly dynamic process [100] and that the final ECM array serves multiple additional functions, including a reservoir of several growth factors and cytokines, and the presentation of a number of binding and adhesion sites to receptors expressed on the cell surface [101]. The overall organization of the matrix can affect cell proliferation, differentiation, migration, survival, and behavior via the integration of a number of signals that constitute the matrix-to-cell signaling [101].
The importance of locally retained and local-acting growth factors and cytokines versus their counterpart detected in serum has been highlighted in elegant studies, including those on important molecules that regulate osteoclastogenesis and bone resorption [102, 103]. It is therefore reasonable to think that the pathogenesis of a disease such as OI, most often caused by alterations in the most abundant bone matrix component, i.e. type I collagen, implicates the signaling dysregulation of local-acting growth factors. Indeed, such is the case for transforming growth factor-beta (TGF-beta) signaling. TGF-beta is secreted as part of a latent complex that includes the latency-associated propeptide and the latent TGF-beta binding protein (LTBP) [104]. This complex is targeted to the ECM, where it binds in an inactive form to small interstitial proteoglycans, such as biglycan, decorin, and fibromodulin, and other proteins such as fibrillins [105–107]. The activation and release of active TGF-beta can be mediated by multiple mechanisms, including bone resorption but also in an integrin-mediated fashion [108, 109].
One study that used two distinct mouse models of OI showed that excessive TGF-beta signaling is an important disease mechanism that contributes to the OI phenotype [110]. Although the precise process that leads to TGF-beta over-activity hasn’t been worked out yet, it is likely that type I collagen and consequent proteoglycan alterations in OI cause inefficient retention of TGF-beta in the matrix [110]. Another pathway, the WNT/beta-catenin signaling pathway, is strongly anabolic for bone and is mediated by WNT proteins, which are lipid-modified, hydrophobic and considered short-range acting molecules [111]. As discussed above, the loss of WNT1 also causes OI [57, 58] and could be interpreted as another example of defective matrix-to-cell or cell-to-cell signaling.
An important class of cell-surface proteins that mediate cell adhesion to the ECM is represented by integrins, which function as αβ heterodimers and have different binding affinity for various ECM components [101]. Similar to what was observed for proteoglycans, type I collagen triple-helical regions that are rich in lethal OI mutations also contain binding sites for integrins [23]. The importance of integrin signaling following their interaction with type I collagen for the survival of mesenchymal stem cells has also been shown [112]. Interestingly, mutations in ITGB6 (encodes integrin beta 6) are associated with amelogenesis imperfecta, indicating that tooth enamel formation, a process that is similar to bone mineralization, appears closely dependent upon cell-matrix interactions [113]. Furthermore, the relevance of integrin-mediated signaling in bone formation by cytoskeletal organization and its role in modulating BMP and WNT signaling in osteoprogenitors has also been recently demonstrated [114]. Finally, bone histomorphometry studies showed that there is increased bone turnover and decreased bone formation in pediatric OI patients compared to healthy controls [70, 115]. While the mechanisms that trigger high bone turnover in OI are still unknown it could be hypothesized that osteocytes, the long lived cells of the skeleton, receive altered matrix feedback regulation or biochemical stimulation; this could dysregulate their behavior and trigger the production and release of cytokines, e.g. RANKL, that stimulate osteoclastogenesis and affect bone resorption. Evidence of dysregulated matrix-to-osteocyte signaling in mouse models of OI is beginning to emerge (Roy Morello, unpublished results).
In summary, a reduced quantity of type I collagen and alterations in multiple processes that contribute to its synthesis, secretion, processing, assembly and interaction with other matrix components, as shown in Figure 3, result in a poor quality extracellular matrix and various degrees of bone brittleness and material property changes. These aspects represent major pathogenetic mechanisms in OI and the most difficult to treat, as discussed below.
Figure 3.
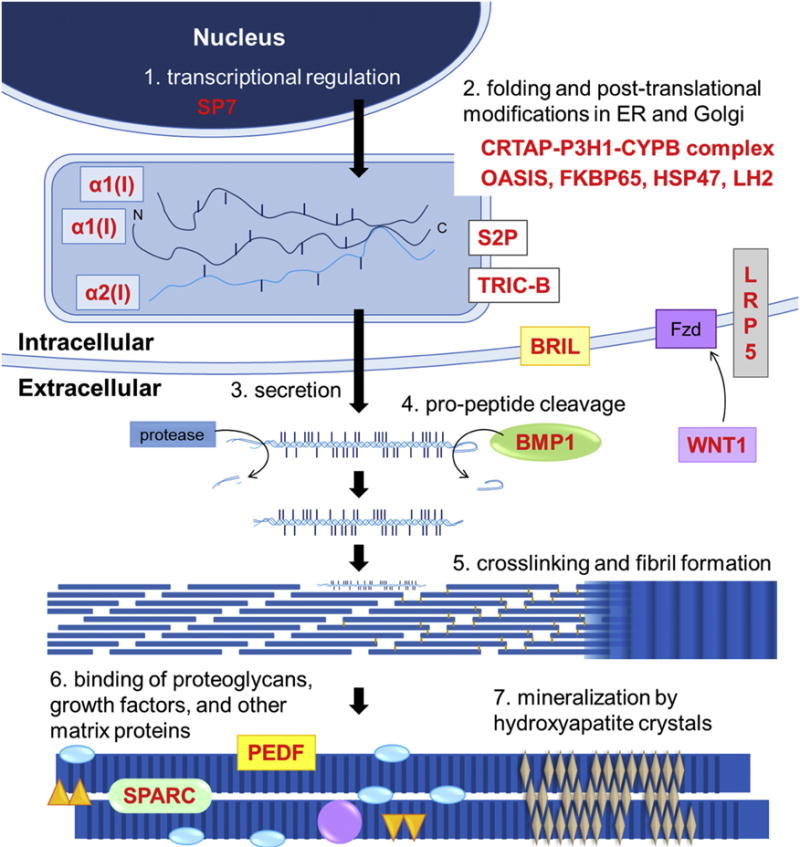
Schematic diagram illustrating essential processes of type I collagen synthesis, its assembly in the matrix, interaction with other matrix components, and mineralization. Some of the proteins that regulate these processes and that are mutated in various types of OI are also indicated. Alterations of one or more of these key steps in the formation of ECM represent major pathogenetic mechanisms in OI.
OI therapeutics
Unfortunately, a cure for osteogenesis imperfecta does not exist yet. Most therapeutic approaches for OI are borrowed from those developed and utilized to treat osteoporosis or other conditions such as osteolytic bone metastases and their complications. As such, their goal is to increase bone mass and architecture via a bone anti-resorptive or anabolic activity, but they do not address the underlying matrix defect. It is hypothesized that in OI, an increase in bone mass provides a clinical benefit of reducing fracture incidence regardless of any defective qualities in the bone matrix.
Bone anti-resorptive treatments
The standard of care for newborns with moderate to severe OI is either oral or intravenous administration of nitrogen-containing bisphosphonates, such as pamidronate, alendronate, ibandronate, risedronate, or zoledronic acid. These compounds are analogues of pyrophosphate, have high-binding affinity for hydroxyapatite and accumulate at sites of active bone remodeling (for a review see [116, 117]). Bisphosphonates inhibit the bone resorptive activity of osteoclasts and induce their apoptosis by interfering with the mevalonate pathway and the synthesis of cholesterol and other isoprenoid lipids [118]. As a result, it is believed that certain GTP-binding proteins that are critical for osteoclast activity are not properly post-translationally modified, become misplaced/dysregulated and ultimately lead to osteoclast cell death [119]. Early bisphosphonates were first tested in a few cases of OI in the late 1980s [120, 121], but the effects of the use of intravenous pamidronate on the treatment of children with severe OI were reported about ten years later [122–124]. In most cases there were no adverse effects, and the treatment significantly increased bone mineral density. Some children had improved mobility, reduced pain and an overall improvement in their quality of life [122, 124].
Dr. Glorieux (Shriners Hospital, Montreal, CA) and collaborators developed a treatment protocol of cyclical pamidronate infusions in children with severe OI. In these cases, post-treatment iliac bone histomorphometry studies showed increased cortical width (+88%), increased cancellous bone (+46%) due to higher trabecular number, reduced bone surface-based indicators of bone remodeling (−26-75%), and a positive impact on several functional parameters [125, 126]. Numerous studies testing the use of bisphosphonates in children and adults with OI have been published; due to space constraints it is impossible to cite them all, but excellent reviews on this topic are available [127–130]. A critical issue on the use of bisphosphonates in OI, i.e. the reduction of fracture incidence, is still debated, with several clinical trials showing reduced fractures at certain skeletal sites [122, 124, 131–134] and others that have not reached similar conclusions [128, 135, 136]. Because bisphosphonates suppress bone turnover and can persist in the skeleton for years [116, 137], to this day several questions on their use remain a matter of discussion, including the optimal length of therapy in children with OI, the long-term consequences on skeletal remodeling, the potential delayed healing of osteotomies, and other concerns such, as osteonecrosis of the jaw or atypical femoral fractures [138].
Two additional approaches were undertaken by the pharmacological industry to inhibit bone resorption for the treatment of osteoporosis. One of these approaches, the inhibition of receptor activator of nuclear factor kappa-B ligand (RANKL), is currently being tested for the treatment of OI. Denosumab is a human monoclonal antibody developed by Amgen that selectively blocks RANKL, an essential cytokine for osteoclastogenesis [139, 140]. The results of a large, phase III clinical trial (FREEDOM trial) in postmenopausal women with osteoporosis after 10 years of treatment with denosumab were recently published [141] and showed low rates of adverse events, low fracture incidence, and sustained elevation of BMD [141]. RANKL inhibition in the osteogenesis imperfecta mouse (oim) model showed some improvement in bone biomechanical parameters but not a reduction in number of fractures compared to placebo-treated controls [142]. The first use of denosumab in children was reported for the treatment of two boys with OI due to mutations in SERPINF1 (encodes PEDF) [143]. The results of a first prospective trial on the safety and efficacy of denosumab in children with OI indicated a significant increase in lumbar spine aBMD after 48 weeks of treatment, with no changes in mobility parameters and no serious adverse events [144]. Therefore, in children who do not respond to bisphosphonates therapy, the use of denosumab instead appears promising so far. However, given the role of RANKL in the maturation of the immune system in rodents, potential alterations in the response to certain infections may be a concern [145].
The second approach consists of the inhibition of cathepsin K, a lysosomal enzyme that osteoclasts secrete for the breakdown of demineralized collagen fibrils [146]. Because osteoclastogenesis is not affected and mature osteoclasts are still able to adhere to the bone surface and dissolve hydroxyapatite by creating an acid environment, this strategy had the advantage of retaining the ability to release factors that couple bone resorption to bone formation [147], unlike denosumab. Very promising results were reported in phase II clinical trials of Odanacatib, an inhibitor of cathepsin K developed by Merck for the treatment of postmenopausal women with low BMD [148, 149]. Unfortunately, further development of this drug was discontinued due to an increased risk of cardiovascular problems [150].
Bone anabolic treatments
The study of sclerosteosis, a recessive bone dysplasia that causes high bone mass and skeletal overgrowth, led to the discovery of a new gene named SOST and encoding for the secreted cystine knot-containing protein Sclerostin [151]. Sclerostin is mainly produced by osteocytes, and its expression decreases following mechanical stimulation of bone [152]. It binds to the low-density lipoprotein receptor-related proteins 5 and 6 which are co-receptor for WNT ligands and inhibits canonical WNT signaling and thus bone formation [153, 154]. Importantly, the use of antibodies against sclerostin in animal models showed increased bone formation but also decreased bone resorption at both modeling and remodeling skeletal sites [155–157]. Human monoclonal antibodies against sclerostin were developed by multiple pharmacological companies and tested in clinical trials on postmenopausal women with osteoporosis [158, 159]. The results demonstrated significant increases in BMD and reduction in fracture risk compared to the placebo group after only 12 months [158, 159]. Sclerostin-neutralizing antibodies have been tested in multiple mouse models of OI and have shown a good anabolic bone response, although this was always less than that of control mice. In most cases, improvements in bone mass and bone architecture were seen at all ages tested, with consequent amelioration of certain bone biomechanical parameters but no effect on bone brittleness [160–163]. Collectively, sclerostin inhibition therapy appears very promising for osteoporosis and perhaps also OI, and a new study in adult patients with type I, III or IV OI treated with BPS804 (another Sclerostin antibody) sponsored by Mereo BioPharma (https://clinicaltrials.gov/ct2/show/NCT03118570) has recently been announced.
The old observation that a fragment of the parathyroid hormone (PTH) has anabolic effects on bone when administered intermittently [164–168], led to the development of recombinant human PTH aa1-34 which is known as Teriparatide and commercialized as Forteo (Eli Lilly & Co.). Its effects, as once-daily injections, on postmenopausal women with osteoporosis were well-tolerated and demonstrated a decreased risk of vertebral and non-vertebral fractures and an increase in vertebral, femoral, and total-body bone-mineral density [169]. The mechanisms that lead to increased bone formation by intermittent PTH administration were shown to involve a reduction of osteoblast apoptosis in cancellous bone [170] and a pro-differentiation/pro-survival effect on pre-osteoblasts located on the periosteum [171]. Additional mechanisms, though, are known to contribute to the anabolic effects of PTH on bone. These mechanisms include, among others, the suppression of sclerostin production with a consequent increase in WNT signaling [172, 173]. The studies on the use of Teriparatide in OI are few and are limited to adults, since it is not approved for use in the pediatric population. In patients with mild OI (OI type I), osteoblasts were shown to have a preserved anabolic response, although this was somewhat lower compared to that observed in postmenopausal osteoporotic women treated for the same length of time [174]. In mild OI, Teriparatide treatment increased serum levels of markers of bone formation, and improved hip and spine areal bone-mineral density and estimated vertebral strength [174–176]. Patients affected with more severe forms of OI (type III and IV), however, showed no benefits after 18 months of treatment with Teriparatide, indicating that its clinical use in OI may be useful only to patients with mild disease [175].
The effects of TGF-beta signaling on bone homeostasis are complex and have been studied for several years [177, 178]. It is known that TGF-beta is secreted by osteoblasts and efficiently stored in an inactive latent complex in the bone matrix, from which it can be released via multiple mechanisms. Osteoclast activity releases active TGF-beta from bone, which represents a coupling factor between bone resorption and formation [109]. In cells of the osteoblast lineage, TGF-beta can repress Runx2 and osteocalcin expression [179, 180], thus inhibiting osteoblast differentiation while, at the same time, favoring the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis [181, 182]. As discussed above, a recent study on the pathogenesis of OI identified excessive TGF-beta signaling in both dominant (Col1a2tm1.1Mcbr) and recessive (CrtapKO) mouse models of OI [110]. Importantly, the administration of a pan-TGF-beta antibody for 8 weeks to CrtapKO or Col1a2tm1.1Mcbr mice resulted in almost complete rescue of bone mass, although the bone remained brittle [110]. Nonetheless, given the promising results in pre-clinical models, a phase I clinical trial to assess the safety of TGF-beta inhibition in the treatment of adults with OI is now recruiting subjects (https://clinicaltrials.gov/ct2/show/NCT03064074).
Because a single bone anti-resorptive or anabolic treatment may have efficacious but quickly reversible effects (i.e. rapid bone loss), the latest trend in the treatment of osteoporosis is a combination therapy in which an anabolic agent (e.g. Teriparatide) is given for several months and then followed by an anti-resorptive agent (e.g. denosumab or bisphosphonates) [183–185]. As such, the bone mass that is gained during the treatment with the anabolic drug should be retained longer because of the suppression of bone turnover caused by the anti-resorptive drug. This approach likely also will be adopted in the treatment of OI.
Cell therapy, gene targeting and gene editing
The best treatment for OI would be a therapy to administer as early as possible during growth (ideally in utero, to allow healthy bone formation and prevent progressive damage to the skeleton) and to enable directly repairing the underlying gene defect to yield a normal bone matrix. One approach has been the allogeneic transplantation of bone marrow, bone marrow stromal cells, or marrow-derived mesenchymal stem cells (MSCs) to test whether osteo-progenitor cells that normally reside in the marrow would successfully graft into the OI patient marrow and contribute to healthy bone formation. This strategy was supported by the observation that mosaicism for a type I collagen mutation results in a mild phenotype [186] and by promising results in a mouse model of OI [187]. Three toddlers (age range 13-32 months) afflicted with severe OI received bone marrow transplantation from compatible siblings and showed small percentage of donor-derived osteoblast cells on bone biopsies but improved parameters of bone formation, increased total body bone mineral content, and increased growth velocity about 7 months after the procedure [188]. Follow-up studies treating mouse models of OI, including intra-uterine transplantation, with either human or murine MSCs also showed very positive effects on the skeleton [189–191]. The most recent studies have performed pre- and post-natal transplantation of fetal allogeneic MSCs in two fetuses with severe OI (type III and IV) and resulted in improved linear growth, mobility, and fracture incidence for several months. The second, post-natal transplantation resumed these positive effects without any toxicity or alloreactivity toward the donor hMSCs [192, 193]. Although too few patients have been treated with this protocol and therefore more experience needs to be accumulated, cell therapy in OI holds great promise for the future.
Two different “gene targeting” attempts were also tried ex-vivo to correct the collagen defect that causes OI [194–196]. The rationale is that the inactivation of the mutated allele or its transcriptional silencing (and hence the elimination of its dominant negative action) would translate to production of only good collagen from the healthy allele and hence in a mild form of OI. Chamberlain et al. partially achieved this result by transducing MSCs from OI patients with a known COL1A1 mutation with an adeno-associated virus carrying a construct that would target and disrupt the mutant allele [194]. The other two studies obtained type I collagen allele-specific silencing using small interfering RNAs (siRNAs) in either human bone-derived cells or mouse fibroblasts from the Brtl mouse [195, 196]. Although partially successful, the clinical translation of these proof-of-concept studies carries many uncertainties, including safety of the vectors utilized for the delivery of the construct, possible insertional mutagenesis, duration of effects, and others.
Finally, while gene editing has not yet been attempted in OI, it clearly holds the most promise for the future treatment of OI. The ability to precisely edit any mutation, in an allele-specific fashion, using the CRISPRs/Cas9 system is likely to revolutionize the way we think about treating human Mendelian disorders [197, 198]. The treatment of autosomal dominant conditions utilizing novel in vivo delivery methods of gene editing Cas9-guide RNA complexes in animal models has already been shown [199].
Future directions
The study of osteogenesis imperfecta has led to a dramatic progress in understanding its complex natural history, genetics, and pathogenesis. Although there is no definite cure, future therapeutic interventions will likely be based on a precision medicine approach based on individual patients’ mutation and pathway affected. Although good instrumentation tools exist to study changes in bone architecture and bone mass and these allow some prediction on their consequences and the inherent susceptibility to fractures, it has been difficult to analyze how changes in bone matrix components affect the overall ECM composition, contribute to alter its material properties and ultimately cause bone fragility. More modern technologies, such as semi-quantitative and quantitative proteomics will be helpful for a better characterization of the cellular and matrix composition changes caused by single gene mutations, including in OI. The recent use of OMICS approaches has already provided interesting clues about potentially new pathogenetic processes in OI [200, 201] and may eventually identify new therapeutic targets.
HIGHLIGHTS.
-
-
OI primarily causes low bone mass and skeletal fragility but may also exhibit a number of extra-skeletal manifestations
-
-
OI is genetically and clinically heterogeneous. Its study significantly increased our knowledge of skeletal biology
-
-
Extracellular matrix alterations, affecting organic, inorganic and matrix-to-cell signaling components are key features of OI
-
-
OI therapeutic approaches have improved but have not addressed the underlying matrix defect
Acknowledgments
This work was supported in part by the National Institute of Arthritis and Musculoskeletal and Skin Diseases grant 1 R01 AR060823. I would like to thank Sarah M. Zimmerman, a graduate student in my laboratory, for help in generating Figure 3, and the Science Communication Office of the University of Arkansas for Medical Sciences for editorial assistance during the preparation of this manuscript. I would also like to thank Dr. Paul W. Esposito and Maegen Wallace (University of Nebraska Medical Center) for the radiographic images shown in Figure 1. Finally, I would like to thank the Osteogenesis Imperfecta Foundation (OIF) for past support and continuous inclusion in their annual research meeting.
Abbreviations
- OI
osteogenesis imperfect
- Brtl
Brittle IV mouse
- hMSCs
human mesenchymal stem cells
- aBMD
areal bone mineral density
- PEDF
pigment epithelium-derived factor
- BRIL
bone restricted ifitm-like protein
- ER
endoplasmic reticulum
- ECM
extra-cellular matrix
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vrolik W. Tabulae ad illustrandam embryogenesin hominis et mammalium, naturalem tam abnormem 1849 [Google Scholar]
- 2.Baljet B. Aspects of the history of osteogenesis imperfecta (Vrolik’s syndrome) Ann Anat. 2002;184:1–7. doi: 10.1016/S0940-9602(02)80023-1. [DOI] [PubMed] [Google Scholar]
- 3.Seedorf K. Osteogenesis imperfecta: a study of clinical features and heredity based on 55 Danish families comprising 180 affected members. Copenhagen: UniversitetsforlagetI Aarhus; 1949. [Google Scholar]
- 4.Smars G. Clinical, Genetic, Epidemiological and Socio-medical Aspects. Stockholm: Scandinavian University Books; 1961. Osteogenesis imperfecta in Sweden. [Google Scholar]
- 5.Sillence DO, Rimoin DL. Classification of osteogenesis imperfect. Lancet. 1978;1:1041–1042. doi: 10.1016/s0140-6736(78)90763-8. [DOI] [PubMed] [Google Scholar]
- 6.Sillence DO, Rimoin DL, Danks DM. Clinical variability in osteogenesis imperfecta-variable expressivity or genetic heterogeneity. Birth Defects Orig Artic Ser. 1979;15:113–129. [PubMed] [Google Scholar]
- 7.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bornstein P, Traub W, editors. The chemistry and biology of collagen. New York: Academic Press, Inc.; 1979. [Google Scholar]
- 9.Prockop DJ, Kivirikko KI, Tuderman L, Guzman NA. The biosynthesis of collagen and its disorders (first of two parts) New Engl J Med. 1979;301:13–23. doi: 10.1056/NEJM197907053010104. [DOI] [PubMed] [Google Scholar]
- 10.Prockop DJ, Kivirikko KI, Tuderman L, Guzman NA. The biosynthesis of collagen and its disorders (second of two parts) New Engl J Med. 1979;301:77–85. doi: 10.1056/NEJM197907123010204. [DOI] [PubMed] [Google Scholar]
- 11.Bellamy G, Bornstein P. Evidence for procollagen, a biosynthetic precursors of collagen. P Natl Acad Sci USA. 1971;68:1138–1142. doi: 10.1073/pnas.68.6.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trelstad RL, Rubin D, Gross J. Osteogenesis imperfecta congenita: evidence for a generalized molecular disorder of collagen. Lab Invest. 1977;36:501–508. [PubMed] [Google Scholar]
- 13.Barsh GS, Byers PH. Reduced secretion of structurally abnormal type I procollagen in a form of osteogenesis imperfecta. P Natl Acad Sci USA. 1981;78:5142–5146. doi: 10.1073/pnas.78.8.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu ML, Williams CJ, Pepe G, Hirsch JL, Prockop DJ, Ramirez F. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nature. 1983;304:78–80. doi: 10.1038/304078a0. [DOI] [PubMed] [Google Scholar]
- 15.Pihlajaniemi T, Dickson LA, Pope FM, Korhonen VR, Nicholls A, Prockop DJ, Myers JC. Osteogenesis imperfecta: cloning of a pro-alpha 2(I) collagen gene with a frameshift mutation. J Biol Chem. 1984;259:12941–12944. [PubMed] [Google Scholar]
- 16.Chu ML, Gargiulo V, Williams CJ, Ramirez F. Multiexon deletion in an osteogenesis imperfecta variant with increased type III collagen mRNA. J Biol Chem. 1985;260:691–694. [PubMed] [Google Scholar]
- 17.Barsh GS, Roush CL, Bonadio J, Byers PH, Gelinas RE. Intron-mediated recombination may cause a deletion in an alpha 1 type I collagen chain in a lethal form of osteogenesis imperfecta. P Natl Acad Sci USA. 1985;82:2870–2874. doi: 10.1073/pnas.82.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deak SB, van der Rest M, Prockop DJ. Altered helical structure of a homotrimer of alpha 1(I)chains synthesized by fibroblasts from a variant of osteogenesis imperfecta. Coll Relat Res. 1985;5:305–313. doi: 10.1016/s0174-173x(85)80020-0. [DOI] [PubMed] [Google Scholar]
- 19.Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F, Bishop NJ. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000;15:1650–1658. doi: 10.1359/jbmr.2000.15.9.1650. [DOI] [PubMed] [Google Scholar]
- 20.Glorieux FH, Ward LM, Rauch F, Lalic L, Roughley PJ, Travers R. Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Miner Res. 2002;17:30–38. doi: 10.1359/jbmr.2002.17.1.30. [DOI] [PubMed] [Google Scholar]
- 21.Ward LM, Rauch F, Travers R, Chabot G, Azouz EM, Lalic L, Roughley PJ, Glorieux FH. Osteogenesis imperfecta type VII: an autosomal recessive form of brittle bone disease. Bone. 2002;31:12–18. doi: 10.1016/s8756-3282(02)00790-1. [DOI] [PubMed] [Google Scholar]
- 22.Martin E, Shapiro JR. Osteogenesis imperfecta:epidemiology and pathophysiology. Curr Osteoporos Rep. 2007;5:91–97. doi: 10.1007/s11914-007-0023-z. [DOI] [PubMed] [Google Scholar]
- 23.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 25.Marini JC, Forlino A, Bachinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM, Krakow D, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. doi: 10.1038/nrdp.2017.52. [DOI] [PubMed] [Google Scholar]
- 26.Marr C, Seasman A, Bishop N. Managing the patient with osteogenesis imperfecta: a multidisciplinary approach. J Multidiscip Healthc. 2017;10:145–155. doi: 10.2147/JMDH.S113483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folkestad L, Hald JD, Canudas-Romo V, Gram J, Hermann AP, Langdahl B, Abrahamsen B, Brixen K. Mortality and Causes of Death in Patients With Osteogenesis Imperfecta: A Register-Based Nationwide Cohort Study. J Bone Miner Res. 2016;31:2159–2166. doi: 10.1002/jbmr.2895. [DOI] [PubMed] [Google Scholar]
- 28.Byers PH. Haploinsufficiency for Mutations in Type I Collagen Genes: Mechanisms and Clinical Effects. In: Shapiro JR, editor. Osteogenesis Imperfecta: A Translational Approach to Brittle Bone Disease. First. Elsevier Inc.; 2013. pp. 125–127. [Google Scholar]
- 29.Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brodsky B, Persikov A. Structural Consequences of Glycine Missense Mutations in Osteogenesis Imperfecta. In: Shapiro JR, editor. Osteogenesis Imperfecta: A Translational Approach to Brittle Bone Disease. Elsevier Inc.; 2013. pp. 115–124. [Google Scholar]
- 31.Capecchi M. The first transgenic mice: an interview with Mario Capecchi. Interview by Kristin Kain. Dis Model Mech. 2008;1:197–201. doi: 10.1242/dmm.001966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sule G, Campeau PM, Zhang VW, Nagamani SC, Dawson BC, Grover M, Bacino CA, Sutton VR, Brunetti-Pierri N, Lu JT, et al. Next-generation sequencing for disorders of low and high bone mineral density. Osteoporosis Int. 2013;24:2253–2259. doi: 10.1007/s00198-013-2290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ha-Vinh R, Alanay Y, Bank RA, Campos-Xavier AB, Zankl A, Superti-Furga A, Bonafe L. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet. 2004;131:115–120. doi: 10.1002/ajmg.a.30231. [DOI] [PubMed] [Google Scholar]
- 34.Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, et al. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 35.Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. New Engl J Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnes AM, Carter EM, Cabral WA, Weis M, Chang W, Makareeva E, Leikin S, Rotimi CN, Eyre DR, Raggio CL, et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. New Engl J Med. 2010;362:521–528. doi: 10.1056/NEJMoa0907705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, et al. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishikawa Y, Wirz J, Vranka JA, Nagata K, Bachinger HP. Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein.cyclophilin B complex. J Biol Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volodarsky M, Markus B, Cohen I, Staretz-Chacham O, Flusser H, Landau D, Shelef I, Langer Y, Birk OS. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum Mutat. 2013;34:582–586. doi: 10.1002/humu.22274. [DOI] [PubMed] [Google Scholar]
- 43.Cabral WA, Ishikawa M, Garten M, Makareeva EN, Sargent BM, Weis M, Barnes AM, Webb EA, Shaw NJ, Ala-Kokko L, et al. Absence of the ER Cation Channel TMEM38B/TRIC-B Disrupts Intracellular Calcium Homeostasis and Dysregulates Collagen Synthesis in Recessive Osteogenesis Imperfecta. PLoS Genet. 2016;12:e1006156. doi: 10.1371/journal.pgen.1006156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Symoens S, Malfait F, D’Hondt S, Callewaert B, Dheedene A, Steyaert W, Bachinger HP, De Paepe A, Kayserili H, Coucke PJ. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis. 2013;8:154. doi: 10.1186/1750-1172-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindert U, Cabral WA, Ausavarat S, Tongkobpetch S, Ludin K, Barnes AM, Yeetong P, Weis M, Krabichler B, Srichomthong C, et al. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun. 2016;7:11920. doi: 10.1038/ncomms11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Glez V, Valencia M, Caparros-Martin JA, Aglan M, Temtamy S, Tenorio J, Pulido V, Lindert U, Rohrbach M, Eyre D, et al. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012;33:343–350. doi: 10.1002/humu.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mendoza-Londono R, Fahiminiya S, Majewski J, Tetreault M, Nadaf J, Kannu P, Sochett E, Howard A, Stimec J, Dupuis L, et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet. 2015;96:979–985. doi: 10.1016/j.ajhg.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosset EM, Bradshaw AD. SPARC/osteonectin in mineralized tissue. Matrix Biol. 2016;52–54:78–87. doi: 10.1016/j.matbio.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Becker J, Semler O, Gilissen C, Li Y, Bolz HJ, Giunta C, Bergmann C, Rohrbach M, Koerber F, Zimmermann K, et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Homan EP, Rauch F, Grafe I, Lietman C, Doll JA, Dawson B, Bertin T, Napierala D, Morello R, Gibbs R, et al. Mutations in SERPINF1 cause Osteogenesis imperfecta Type VI. J Bone Miner Res. 2011;12:2798–803. doi: 10.1002/jbmr.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Venturi G, Gandini A, Monti E, Dalle Carbonare L, Corradi M, Vincenzi M, Valenti MT, Valli M, Pelilli E, Boner A, et al. Lack of expression of SERPINF1, the gene coding for pigment epithelium-derived factor causes progressively deforming osteogenesis imperfecta with normal type I collagen. J Bone Miner Res. 2011;3:723–8. doi: 10.1002/jbmr.1480. [DOI] [PubMed] [Google Scholar]
- 52.Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, et al. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012;91:349–357. doi: 10.1016/j.ajhg.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, et al. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012;91:343–348. doi: 10.1016/j.ajhg.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farber CR, Reich A, Barnes AM, Becerra P, Rauch F, Cabral WA, Bae A, Quinlan A, Glorieux FH, Clemens TL, et al. A novel IFITM5 mutation in severe atypical osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor. J Bone Miner Res. 2014;29:1402–1411. doi: 10.1002/jbmr.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 56.Ai M, Heeger S, Bartels CF, Schelling DK. Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Genet. 2005;77:741–753. doi: 10.1086/497706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, Lu JT, Pekkinen M, Wessman M, Heino TJ, et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368:1809–1816. doi: 10.1056/NEJMoa1215458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013;50:345–348. doi: 10.1136/jmedgenet-2013-101567. [DOI] [PubMed] [Google Scholar]
- 59.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 60.Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA, Valencia M, Leton R, Martinez-Glez V, Elhossini R, Amr K, Vilaboa N, et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet Part A. 2015;167A:2869–2892. doi: 10.1002/ajmg.a.37365. [DOI] [PubMed] [Google Scholar]
- 62.Nair AK, Gautieri A, Chang SW, Buehler MJ. Molecular mechanics of mineralized collagen fibrils in bone. Nat Commun. 2013;4:1724. doi: 10.1038/ncomms2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bishop N. Bone Material Properties in Osteogenesis Imperfecta. J Bone Miner Res. 2016;31:699–708. doi: 10.1002/jbmr.2835. [DOI] [PubMed] [Google Scholar]
- 64.Boskey AL. Bone composition: relationship to bone fragility and antiosteoporotic drug effects. Bonekey Rep. 2013;2:447. doi: 10.1038/bonekey.2013.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iozzo RV, Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. 2011;3:a004978. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Gene Dev. 2005;19:1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valli M, Barnes AM, Gallanti A, Cabral WA, Viglio S, Weis MA, Makareeva E, Eyre D, Leikin S, Antoniazzi F, et al. Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix. Clin Genet. 2012;82:453–459. doi: 10.1111/j.1399-0004.2011.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uveges TE, Collin-Osdoby P, Cabral WA, Ledgard F, Goldberg L, Bergwitz C, Forlino A, Osdoby P, Gronowicz GA, Marini JC. Cellular mechanism of decreased bone in Brtl mouse model of OI: imbalance of decreased osteoblast function and increased osteoclasts and their precursors. J Bone Miner Res. 2008;23:1983–1994. doi: 10.1359/JBMR.080804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rauch F, Travers R, Parfitt AM, Glorieux FH. Static and dynamic bone histomorphometry in children with osteogenesis imperfecta. Bone. 2000;26:581–589. doi: 10.1016/s8756-3282(00)00269-6. [DOI] [PubMed] [Google Scholar]
- 71.Chessler SD, Wallis GA, Byers PH. Mutations in the carboxyl-terminal propeptide of the pro alpha 1(I) chain of type I collagen result in defective chain association and produce lethal osteogenesis imperfecta. J Biol Chem. 1993;268:18218–18225. [PubMed] [Google Scholar]
- 72.Lisse TS, Thiele F, Fuchs H, Hans W, Przemeck GK, Abe K, Rathkolb B, Quintanilla-Martinez L, Hoelzlwimmer G, Helfrich M, et al. ER stress-mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet. 2008;4:e7. doi: 10.1371/journal.pgen.0040007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ishida Y, Yamamoto A, Kitamura A, Lamande SR, Yoshimori T, Bateman JF, Kubota H, Nagata K. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell. 2009;20:2744–2754. doi: 10.1091/mbc.E08-11-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bateman JF, Boot-Handford RP, Lamande SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet. 2009;10:173–183. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- 75.Makareeva E, Aviles NA, Leikin S. Chaperoning osteogenesis: new protein-folding disease paradigms. Trends Cell Biol. 2011;21:168–176. doi: 10.1016/j.tcb.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chang W, Barnes AM, Cabral WA, Bodurtha JN, Marini JC. Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex. Hum Mol Genet. 2010;19:223–234. doi: 10.1093/hmg/ddp481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang G, Ezura Y, Chervoneva I, Robinson PS, Beason DP, Carine ET, Soslowsky LJ, Iozzo RV, Birk DE. Decorin regulates assembly of collagen fibrils and acquisition of biomechanical properties during tendon development. J Cell Biochem. 2006;98:1436–1449. doi: 10.1002/jcb.20776. [DOI] [PubMed] [Google Scholar]
- 78.Robinson KA, Sun M, Barnum CE, Weiss SN, Huegel J, Shetye SS, Lin L, Saez D, Adams SM, Iozzo RV, et al. Decorin and biglycan are necessary for maintaining collagen fibril structure, fiber realignment, and mechanical properties of mature tendons. Matrix Biol. 2017;64:81–93. doi: 10.1016/j.matbio.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gubbiotti MA, Vallet SD, Ricard-Blum S, Iozzo RV. Decorin interacting network: A comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol. 2016;55:7–21. doi: 10.1016/j.matbio.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Di Lullo GA, Sweeney SM, Korkko J, Ala-Kokko L, San Antonio JD. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J Biol Chem. 2002;277:4223–4231. doi: 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]
- 81.Fedarko NS, Robey PG, Vetter UK. Extracellular matrix stoichiometry in osteoblasts from patients with osteogenesis imperfecta. J Bone Miner Res. 1995;10:1122–1129. doi: 10.1002/jbmr.5650100718. [DOI] [PubMed] [Google Scholar]
- 82.Martinek N, Shahab J, Sodek J, Ringuette M. Is SPARC an evolutionarily conserved collagen chaperone? J Dent Res. 2007;86:296–305. doi: 10.1177/154405910708600402. [DOI] [PubMed] [Google Scholar]
- 83.Melsen F, Mosekilde L. Trabecular bone mineralization lag time determined by tetracycline double-labeling in normal and certain pathological conditions. Acta Pathol Microbiol Scand A. 1980;88:83–88. doi: 10.1111/j.1699-0463.1980.tb02470.x. [DOI] [PubMed] [Google Scholar]
- 84.Narisawa S, Yadav MC, Millan JL. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J Bone Miner Res. 2013;28:1587–1598. doi: 10.1002/jbmr.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dziewiatkowski DD, Majznerski LL. Role of proteoglycans in endochondral ossification: inhibition of calcification. Calcified Tissue Int. 1985;37:560–564. doi: 10.1007/BF02557842. [DOI] [PubMed] [Google Scholar]
- 86.Nikitovic D, Aggelidakis J, Young MF, Iozzo RV, Karamanos NK, Tzanakakis GN. The biology of small leucine-rich proteoglycans in bone pathophysiology. J Biol Chem. 2012;287:33926–33933. doi: 10.1074/jbc.R112.379602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 88.Martin A, David V, Laurence JS, Schwarz PM, Lafer EM, Hedge AM, Rowe PS. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology. 2008;149:1757–1772. doi: 10.1210/en.2007-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222–226. doi: 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- 90.Boyde A, Travers R, Glorieux FH, Jones SJ. The mineralization density of iliac crest bone from children with osteogenesis imperfecta. Calcified Tissue Int. 1999;64:185–190. doi: 10.1007/s002239900600. [DOI] [PubMed] [Google Scholar]
- 91.Roschger P, Fratzl-Zelman N, Misof BM, Glorieux FH, Klaushofer K, Rauch F. Evidence that abnormal high bone mineralization in growing children with osteogenesis imperfecta is not associated with specific collagen mutations. Calcified Tissue Int. 2008;82:263–270. doi: 10.1007/s00223-008-9113-x. [DOI] [PubMed] [Google Scholar]
- 92.Fratzl-Zelman N, Morello R, Lee B, Rauch F, Glorieux FH, Misof BM, Klaushofer K, Roschger P. CRTAP deficiency leads to abnormally high bone matrix mineralization in a murine model and in children with osteogenesis imperfecta type VII. Bone. 2010;46:820–826. doi: 10.1016/j.bone.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blouin S, Fratzl-Zelman N, Glorieux FH, Roschger P, Klaushofer K, Marini JC, Rauch F. Hypermineralization and High Osteocyte Lacunar Density in Osteogenesis Imperfecta Type V Bone Indicate Exuberant Primary Bone Formation. J Bone Miner Res. 2017;32:1884–1892. doi: 10.1002/jbmr.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fratzl-Zelman N, Schmidt I, Roschger P, Glorieux FH, Klaushofer K, Fratzl P, Rauch F, Wagermaier W. Mineral particle size in children with osteogenesis imperfecta type I is not increased independently of specific collagen mutations. Bone. 2014;60:122–128. doi: 10.1016/j.bone.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 95.Boskey A, Doty SB. Mineralized Tissue: Histology, Biology and Biochemistry. In: Shapiro J, editor. Osteogenesis Imperfecta: A Translational Approach to Brittle Bone Disease. First. Elsevier; 2013. pp. 31–43. [Google Scholar]
- 96.Lietman CD, Lim J, Grafe I, Chen Y, Ding H, Bi X, Ambrose CG, Fratzl-Zelman N, Roschger P, Klaushofer K, et al. Fkbp10 Deletion in Osteoblasts Leads to Qualitative Defects in Bone. J Bone Miner Res. 2017;32:1354–1367. doi: 10.1002/jbmr.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reich A, Bae AS, Barnes AM, Cabral WA, Hinek A, Stimec J, Hill SC, Chitayat D, Marini JC. Type V OI primary osteoblasts display increased mineralization despite decreased COL1A1 expression. J Clin Endocr Metab. 2015;100:E325–332. doi: 10.1210/jc.2014-3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fratzl-Zelman N, Schmidt I, Roschger P, Roschger A, Glorieux FH, Klaushofer K, Wagermaier W, Rauch F, Fratzl P. Unique micro- and nano-scale mineralization pattern of human osteogenesis imperfecta type VI bone. Bone. 2015;73:233–241. doi: 10.1016/j.bone.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 99.Vanleene M, Porter A, Guillot PV, Boyde A, Oyen M, Shefelbine S. Ultra-structural defects cause low bone matrix stiffness despite high mineralization in osteogenesis imperfecta mice. Bone. 2012;50:1317–1323. doi: 10.1016/j.bone.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sivakumar P, Czirok A, Rongish BJ, Divakara VP, Wang YP, Dallas SL. New insights into extracellular matrix assembly and reorganization from dynamic imaging of extracellular matrix proteins in living osteoblasts. J Cell Sci. 2006;119:1350–1360. doi: 10.1242/jcs.02830. [DOI] [PubMed] [Google Scholar]
- 101.Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. 2011;209:139–151. doi: 10.1530/JOE-10-0377. [DOI] [PubMed] [Google Scholar]
- 102.Nandi S, Akhter MP, Seifert MF, Dai XM, Stanley ER. Developmental and functional significance of the CSF-1 proteoglycan chondroitin sulfate chain. Blood. 2006;107:786–795. doi: 10.1182/blood-2005-05-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xiong J, Cawley K, Piemontese M, Fujiwara Y, Macleod R, Goellner J, Zhao H, O’Brien CA. The soluble form of RANKL contributes to cancellous bone remodeling in adult mice but is dispensable for ovariectomy-induced bone loss. ASBMR. 2017 Volume Available at: http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=51d4e88b-f79d-47e2-a15b-134f0c57b52e. (Denver (CO): J Bone Miner Res 32 (Suppl 1))
- 104.Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41:233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 105.Hildebrand A, Romaris M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, Ruoslahti E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 1994;302(Pt 2):527–534. doi: 10.1042/bj3020527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaartinen V, Warburton D. Fibrillin controls TGF-beta activation. Nat Genet. 2003;33:331–332. doi: 10.1038/ng0303-331. [DOI] [PubMed] [Google Scholar]
- 107.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 108.Henderson NC, Sheppard D. Integrin-mediated regulation of TGFbeta in fibrosis. Biochim Biophys Acta. 2013;1832:891–896. doi: 10.1016/j.bbadis.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Crane JL, Xian L, Cao X. Role of TGF-beta Signaling in Coupling Bone Remodeling. Methods Mol Biol. 2016;1344:287–300. doi: 10.1007/978-1-4939-2966-5_18. [DOI] [PubMed] [Google Scholar]
- 110.Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, Bertin T, Munivez E, Chen Y, Dawson B, et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20:670–675. doi: 10.1038/nm.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nusse R, Clevers H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 112.Popov C, Radic T, Haasters F, Prall WC, Aszodi A, Gullberg D, Schieker M, Docheva D. Integrins alpha2beta1 and alpha11beta1 regulate the survival of mesenchymal stem cells on collagen I. Cell Death Dis. 2011;2:e186. doi: 10.1038/cddis.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang SK, Choi M, Richardson AS, Reid BM, Lin BP, Wang SJ, Kim JW, Simmer JP, Hu JC. ITGB6 loss-of-function mutations cause autosomal recessive amelogenesis imperfecta. Hum Mol Genet. 2014;23:2157–2163. doi: 10.1093/hmg/ddt611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dejaeger M, Bohm AM, Dirckx N, Devriese J, Nefyodova E, Cardoen R, St-Arnaud R, Tournoy J, Luyten FP, Maes C. Integrin-Linked Kinase Regulates Bone Formation by Controlling Cytoskeletal Organization and Modulating BMP and Wnt Signaling in Osteoprogenitors. J Bone Miner Res. 2017;32:2087–2102. doi: 10.1002/jbmr.3190. [DOI] [PubMed] [Google Scholar]
- 115.Baron R, Gertner JM, Lang R, Vignery A. Increased bone turnover with decreased bone formation by osteoblasts in children with osteogenesis imperfecta tarda. Pediatr Res. 1983;17:204–207. doi: 10.1203/00006450-198303000-00007. [DOI] [PubMed] [Google Scholar]
- 116.Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032–1045. doi: 10.4065/83.9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Soares AP, do Espirito Santo RF, Line SR, Pinto M, Santos Pde M, Toralles MB, do Espirito Santo AR. Bisphosphonates: Pharmacokinetics, bioavailability, mechanisms of action, clinical applications in children, and effects on tooth development. Environ Toxicol Pharmacol. 2016;42:212–217. doi: 10.1016/j.etap.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 118.Luckman SP, Hughes DE, Coxon FP, Graham R, Russell G, Rogers MJ. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res. 1998;13:581–589. doi: 10.1359/jbmr.1998.13.4.581. [DOI] [PubMed] [Google Scholar]
- 119.Dunford JE, Thompson K, Coxon FP, Luckman SP, Hahn FM, Poulter CD, Ebetino FH, Rogers MJ. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J Pharmacol Exp Ther. 2001;296:235–242. [PubMed] [Google Scholar]
- 120.Devogelaer JP, Malghem J, Maldague B, Nagant de Deuxchaisnes C. Radiological manifestations of bisphosphonate treatment with APD in a child suffering from osteogenesis imperfecta. Skeletal Radiol. 1987;16:360–363. doi: 10.1007/BF00350961. [DOI] [PubMed] [Google Scholar]
- 121.Huaux JP, Lokietek W. Is APD a promising drug in the treatment of severe osteogenesis imperfecta? J Pediatr Orthop. 1988;8:71–72. doi: 10.1097/01241398-198801000-00017. [DOI] [PubMed] [Google Scholar]
- 122.Bembi B, Parma A, Bottega M, Ceschel S, Zanatta M, Martini C, Ciana G. Intravenous pamidronate treatment in osteogenesis imperfecta. J Pediatr. 1997;131:622–625. doi: 10.1016/s0022-3476(97)70074-x. [DOI] [PubMed] [Google Scholar]
- 123.Fujiwara I, Ogawa E, Igarashi Y, Ohba M, Asanuma A. Intravenous pamidronate treatment in osteogenesis imperfecta. Eur J Pediatr. 1998;157:261–262. [PubMed] [Google Scholar]
- 124.Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339:947–952. doi: 10.1056/NEJM199810013391402. [DOI] [PubMed] [Google Scholar]
- 125.Glorieux FH. Experience with bisphosphonates in osteogenesis imperfecta. Pediatrics. 2007;119(Suppl 2):S163–165. doi: 10.1542/peds.2006-2023I. [DOI] [PubMed] [Google Scholar]
- 126.Rauch F, Travers R, Plotkin H, Glorieux FH. The effects of intravenous pamidronate on the bone tissue of children and adolescents with osteogenesis imperfecta. J Clin Invest. 2002;110:1293–1299. doi: 10.1172/JCI15952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rijks EB, Bongers BC, Vlemmix MJ, Boot AM, van Dijk AT, Sakkers RJ, van Brussel M. Efficacy and Safety of Bisphosphonate Therapy in Children with Osteogenesis Imperfecta: A Systematic Review. Horm Res Paediatr. 2015;84:26–42. doi: 10.1159/000381713. [DOI] [PubMed] [Google Scholar]
- 128.Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2016;10:CD005088. doi: 10.1002/14651858.CD005088.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Biggin A, Munns CF. Long-Term Bisphosphonate Therapy in Osteogenesis Imperfecta. Curr Osteoporos Rep. 2017;15:412–418. doi: 10.1007/s11914-017-0401-0. [DOI] [PubMed] [Google Scholar]
- 130.Marom R, Lee YC, Grafe I, Lee B. Pharmacological and biological therapeutic strategies for osteogenesis imperfecta. Am J Med Genet C Semin Med Genet. 2016;172:367–383. doi: 10.1002/ajmg.c.31532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gatti D, Antoniazzi F, Prizzi R, Braga V, Rossini M, Tato L, Viapiana O, Adami S. Intravenous neridronate in children with osteogenesis imperfecta: a randomized controlled study. J Bone Miner Res. 2005;20:758–763. doi: 10.1359/JBMR.041232. [DOI] [PubMed] [Google Scholar]
- 132.Letocha AD, Cintas HL, Troendle JF, Reynolds JC, Cann CE, Chernoff EJ, Hill SC, Gerber LH, Marini JC. Controlled trial of pamidronate in children with types III and IV osteogenesis imperfecta confirms vertebral gains but not short-term functional improvement. J Bone Miner Res. 2005;20:977–986. doi: 10.1359/JBMR.050109. [DOI] [PubMed] [Google Scholar]
- 133.Lindahl K, Kindmark A, Rubin CJ, Malmgren B, Grigelioniene G, Soderhall S, Ljunggren O, Astrom E. Decreased fracture rate, pharmacogenetics and BMD response in 79 Swedish children with osteogenesis imperfecta types I, III and IV treated with Pamidronate. Bone. 2016;87:11–18. doi: 10.1016/j.bone.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 134.Shi CG, Zhang Y, Yuan W. Efficacy of Bisphosphonates on Bone Mineral Density and Fracture Rate in Patients With Osteogenesis Imperfecta: A Systematic Review and Meta-analysis. Am J Ther. 2016;23:e894–904. doi: 10.1097/MJT.0000000000000236. [DOI] [PubMed] [Google Scholar]
- 135.Ward LM, Rauch F, Whyte MP, D’Astous J, Gates PE, Grogan D, Lester EL, McCall RE, Pressly TA, Sanders JO, et al. Alendronate for the treatment of pediatric osteogenesis imperfecta: a randomized placebo-controlled study. J Clin Endocr Metab. 2011;96:355–364. doi: 10.1210/jc.2010-0636. [DOI] [PubMed] [Google Scholar]
- 136.Hald JD, Evangelou E, Langdahl BL, Ralston SH. Bisphosphonates for the prevention of fractures in osteogenesis imperfecta: meta-analysis of placebo-controlled trials. J Bone Miner Res. 2015;30:929–933. doi: 10.1002/jbmr.2410. [DOI] [PubMed] [Google Scholar]
- 137.Khan SA, Kanis JA, Vasikaran S, Kline WF, Matuszewski BK, McCloskey EV, Beneton MN, Gertz BJ, Sciberras DG, Holland SD, et al. Elimination and biochemical responses to intravenous alendronate in postmenopausal osteoporosis. J Bone Miner Res. 1997;12:1700–1707. doi: 10.1359/jbmr.1997.12.10.1700. [DOI] [PubMed] [Google Scholar]
- 138.Saita Y, Ishijima M, Kaneko K. Atypical femoral fractures and bisphosphonate use: current evidence and clinical implications. Ther Adv Chronic Dis. 2015;6:185–193. doi: 10.1177/2040622315584114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front Immunol. 2014;5:511. doi: 10.3389/fimmu.2014.00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Feng X, Teitelbaum SL. Osteoclasts: New Insights. Bone Res. 2013;1:11–26. doi: 10.4248/BR201301003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bone HG, Wagman RB, Brandi ML, Brown JP, Chapurlat R, Cummings SR, Czerwinski E, Fahrleitner-Pammer A, Kendler DL, Lippuner K, et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. 2017;5:513–523. doi: 10.1016/S2213-8587(17)30138-9. [DOI] [PubMed] [Google Scholar]
- 142.Bargman R, Huang A, Boskey AL, Raggio C, Pleshko N. RANKL inhibition improves bone properties in a mouse model of osteogenesis imperfecta. Connect Tissue Res. 2010;51:123–131. doi: 10.3109/03008200903108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hoyer-Kuhn H, Netzer C, Koerber F, Schoenau E, Semler O. Two years’ experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J Rare Dis. 2014;9:145. doi: 10.1186/s13023-014-0145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hoyer-Kuhn H, Franklin J, Allo G, Kron M, Netzer C, Eysel P, Hero B, Schoenau E, Semler O. Safety and efficacy of denosumab in children with osteogenesis imperfect–a first prospective trial. J Musculoskelet Neuronal Interact. 2016;16:24–32. [PMC free article] [PubMed] [Google Scholar]
- 145.Ferrari-Lacraz S, Ferrari S. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos Int. 2011;22:435–446. doi: 10.1007/s00198-010-1326-y. [DOI] [PubMed] [Google Scholar]
- 146.Troen BR. The role of cathepsin K in normal bone resorption. Drug News Perspect. 2004;17:19–28. doi: 10.1358/dnp.2004.17.1.829022. [DOI] [PubMed] [Google Scholar]
- 147.Ikeda K, Takeshita S. Factors and mechanisms involved in the coupling from bone resorption to formation: how osteoclasts talk to osteoblasts. J Bone Metab. 2014;21:163–167. doi: 10.11005/jbm.2014.21.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Langdahl B, Binkley N, Bone H, Gilchrist N, Resch H, Rodriguez Portales J, Denker A, Lombardi A, Le Bailly De Tilleghem C, Dasilva C, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase 2 study. J Bone Miner Res. 2012;27:2251–2258. doi: 10.1002/jbmr.1695. [DOI] [PubMed] [Google Scholar]
- 149.Rizzoli R, Benhamou CL, Halse J, Miller PD, Reid IR, Rodriguez Portales JA, DaSilva C, Kroon R, Verbruggen N, Leung AT, et al. Continuous treatment with odanacatib for up to 8 years in postmenopausal women with low bone mineral density: a phase 2 study. Osteoporos Int. 2016;27:2099–2107. doi: 10.1007/s00198-016-3503-0. [DOI] [PubMed] [Google Scholar]
- 150.Mullard A. Merck &Co. drops osteoporosis drug odanacatib. Nat Rev Drug Discov. 2016;15:669. doi: 10.1038/nrd.2016.207. [DOI] [PubMed] [Google Scholar]
- 151.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–5875. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 153.Lewiecki EM. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther Adv Musculoskelet Dis. 2014;6:48–57. doi: 10.1177/1759720X13510479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcified Tissue Int. 2010;87:99–107. doi: 10.1007/s00223-010-9372-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li X, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Grisanti M, Dwyer D, Stouch B, Thway TM, Stolina M, et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res. 2010;25:2647–2656. doi: 10.1002/jbmr.182. [DOI] [PubMed] [Google Scholar]
- 156.Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25:948–959. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 157.Ominsky MS, Niu QT, Li C, Li X, Ke HZ. Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. J Bone Miner Res. 2014;29:1424–1430. doi: 10.1002/jbmr.2152. [DOI] [PubMed] [Google Scholar]
- 158.Recker RR, Benson CT, Matsumoto T, Bolognese MA, Robins DA, Alam J, Chiang AY, Hu L, Krege JH, Sowa H, et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res. 2015;30:216–224. doi: 10.1002/jbmr.2351. [DOI] [PubMed] [Google Scholar]
- 159.Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, Hofbauer LC, Lau E, Lewiecki EM, Miyauchi A, et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N Engl J Med. 2016;375:1532–1543. doi: 10.1056/NEJMoa1607948. [DOI] [PubMed] [Google Scholar]
- 160.Sinder BP, White LE, Salemi JD, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Adult Brtl/+ mouse model of osteogenesis imperfecta demonstrates anabolic response to sclerostin antibody treatment with increased bone mass and strength. Osteoporos Int. 2014;25:2097–2107. doi: 10.1007/s00198-014-2737-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sinder BP, Salemi JD, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Rapidly growing Brtl/+ mouse model of osteogenesis imperfecta improves bone mass and strength with sclerostin antibody treatment. Bone. 2015;71:115–123. doi: 10.1016/j.bone.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roschger A, Roschger P, Keplingter P, Klaushofer K, Abdullah S, Kneissel M, Rauch F. Effect of sclerostin antibody treatment in a mouse model of severe osteogenesis imperfecta. Bone. 2014;66:182–188. doi: 10.1016/j.bone.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 163.Grafe I, Alexander S, Yang T, Lietman C, Homan EP, Munivez E, Chen Y, Jiang MM, Bertin T, Dawson B, et al. Sclerostin Antibody Treatment Improves the Bone Phenotype of Crtap(−/−) Mice, a Model of Recessive Osteogenesis Imperfecta. J Bone Miner Res. 2016;31:1030–1040. doi: 10.1002/jbmr.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Reeve J, Hesp R, Williams D, Hulme P, Klenerman L, Zanelli JM, Darby AJ, Tregear GW, Parsons JA. Anabolic effect of low doses of a fragment of human parathyroid hormone on the skeleton in postmenopausal osteoporosis. Lancet. 1976;1:1035–1038. doi: 10.1016/s0140-6736(76)92216-9. [DOI] [PubMed] [Google Scholar]
- 165.Reeve J, Meunier PJ, Parsons JA, Bernat M, Bijvoet OL, Courpron P, Edouard C, Klenerman L, Neer RM, Renier JC, et al. Anabolic effect of human parathyroid hormone fragment on trabecular bone in involutional osteoporosis: a multicentre trial. Br Med J. 1980;280:1340–1344. doi: 10.1136/bmj.280.6228.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tam CS, Heersche JN, Murray TM, Parsons JA. Parathyroid hormone stimulates the bone apposition rate independently of its resorptive action: differential effects of intermittent and continuous administration. Endocrinology. 1982;110:506–512. doi: 10.1210/endo-110-2-506. [DOI] [PubMed] [Google Scholar]
- 167.Dempster DW, Cosman F, Parisien M, Shen V, Lindsay R. Anabolic actions of parathyroid hormone on bone. Endocr Rev. 1993;14:690–709. doi: 10.1210/edrv-14-6-690. [DOI] [PubMed] [Google Scholar]
- 168.Lane NE, Kimmel DB, Nilsson MH, Cohen FE, Newton S, Nissenson RA, Strewler GJ. Bone-selective analogs of human PTH(1-34) increase bone formation in an ovariectomized rat model. J Bone Miner Res. 1996;11:614–625. doi: 10.1002/jbmr.5650110509. [DOI] [PubMed] [Google Scholar]
- 169.Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 170.Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–446. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jilka RL, O’Brien CA, Ali AA, Roberson PK, Weinstein RS, Manolagas SC. Intermittent PTH stimulates periosteal bone formation by actions on post-mitotic preosteoblasts. Bone. 2009;44:275–286. doi: 10.1016/j.bone.2008.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Aslan D, Andersen MD, Gede LB, de Franca TK, Jorgensen SR, Schwarz P, Jorgensen NR. Mechanisms for the bone anabolic effect of parathyroid hormone treatment in humans. Scand J Clin Lab Invest. 2012;72:14–22. doi: 10.3109/00365513.2011.624631. [DOI] [PubMed] [Google Scholar]
- 173.Drake MT, Srinivasan B, Modder UI, Peterson JM, McCready LK, Riggs BL, Dwyer D, Stolina M, Kostenuik P, Khosla S. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J Clin Endocr Metab. 2010;95:5056–5062. doi: 10.1210/jc.2010-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gatti D, Rossini M, Viapiana O, Povino MR, Liuzza S, Fracassi E, Idolazzi L, Adami S. Teriparatide treatment in adult patients with osteogenesis imperfecta type I. Calcified Tissue Int. 2013;93:448–452. doi: 10.1007/s00223-013-9770-2. [DOI] [PubMed] [Google Scholar]
- 175.Orwoll ES, Shapiro J, Veith S, Wang Y, Lapidus J, Vanek C, Reeder JL, Keaveny TM, Lee DC, Mullins MA, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124:491–498. doi: 10.1172/JCI71101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Leali PT, Balsano M, Maestretti G, Brusoni M, Amorese V, Ciurlia E, Andreozzi M, Caggiari G, Doria C. Efficacy of teriparatide vs neridronate in adults with osteogenesis imperfecta type I: a prospective randomized international clinical study. Clin Cases Miner Bone Metab. 2017;14:153–156. doi: 10.11138/ccmbm/2017.14.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen G, Deng C, Li YP. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. 2012;8:272–288. doi: 10.7150/ijbs.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rys JP, Monteiro DA, Alliston T. Mechanobiology of TGFbeta signaling in the skeleton. Matrix Biol. 2016;52–54:413–425. doi: 10.1016/j.matbio.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kang JS, Alliston T, Delston R, Derynck R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005;24:2543–2555. doi: 10.1038/sj.emboj.7600729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Alliston T, Choy L, Ducy P, Karsenty G, Derynck R. TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. 2001;20:2254–2272. doi: 10.1093/emboj/20.9.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Borton AJ, Frederick JP, Datto MB, Wang XF, Weinstein RS. The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J Bone Miner Res. 2001;16:1754–1764. doi: 10.1359/jbmr.2001.16.10.1754. [DOI] [PubMed] [Google Scholar]
- 182.Karsdal MA, Larsen L, Engsig MT, Lou H, Ferreras M, Lochter A, Delaisse JM, Foged NT. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J Biol Chem. 2002;277:44061–44067. doi: 10.1074/jbc.M207205200. [DOI] [PubMed] [Google Scholar]
- 183.Cosman F. Combination therapy for osteoporosis: a reappraisal. Bonekey Rep. 2014;3:518. doi: 10.1038/bonekey.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Leder BZ, Tsai JN, Uihlein AV, Wallace PM, Lee H, Neer RM, Burnett-Bowie SA. Denosumab and teriparatide transitions in postmenopausal osteoporosis (the DATA-Switch study): extension of a randomised controlled trial. Lancet. 2015;386:1147–1155. doi: 10.1016/S0140-6736(15)61120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kitaguchi K, Kashii M, Ebina K, Kaito T, Okada R, Makino T, Noguchi T, Ishimoto T, Nakano T, Yoshikawa H. Effects of single or combination therapy of teriparatide and anti-RANKL monoclonal antibody on bone defect regeneration in mice. Bone. 2018;106:1–10. doi: 10.1016/j.bone.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 186.Constantinou CD, Pack M, Young SB, Prockop DJ. Phenotypic heterogeneity in osteogenesis imperfecta: the mildly affected mother of a proband with a lethal variant has the same mutation substituting cysteine for alpha 1-glycine 904 in a type I procollagen gene (COL1A1) Am J Hum Genet. 1990;47:670–679. [PMC free article] [PubMed] [Google Scholar]
- 187.Pereira RF, O’Hara MD, Laptev AV, Halford KW, Pollard MD, Class R, Simon D, Livezey K, Prockop DJ. Marrow stromal cells as a source of progenitor cells for nonhematopoietic tissues in transgenic mice with a phenotype of osteogenesis imperfecta. P Natl Acad Sci USA. 1998;95:1142–1147. doi: 10.1073/pnas.95.3.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M, Sussman M, Orchard P, Marx JC, Pyeritz RE, et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–313. doi: 10.1038/6529. [DOI] [PubMed] [Google Scholar]
- 189.Guillot PV, Abass O, Bassett JH, Shefelbine SJ, Bou-Gharios G, Chan J, Kurata H, Williams GR, Polak J, Fisk NM. Intrauterine transplantation of human fetal mesenchymal stem cells from first-trimester blood repairs bone and reduces fractures in osteogenesis imperfecta mice. Blood. 2008;111:1717–1725. doi: 10.1182/blood-2007-08-105809. [DOI] [PubMed] [Google Scholar]
- 190.Otsuru S, Gordon PL, Shimono K, Jethva R, Marino R, Phillips CL, Hofmann TJ, Veronesi E, Dominici M, Iwamoto M, et al. Transplanted bone marrow mononuclear cells and MSCs impart clinical benefit to children with osteogenesis imperfecta through different mechanisms. Blood. 2012;120:1933–1941. doi: 10.1182/blood-2011-12-400085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Panaroni C, Gioia R, Lupi A, Besio R, Goldstein SA, Kreider J, Leikin S, Vera JC, Mertz EL, Perilli E, et al. In utero transplantation of adult bone marrow decreases perinatal lethality and rescues the bone phenotype in the knockin murine model for classical, dominant osteogenesis imperfecta. Blood. 2009;114:459–468. doi: 10.1182/blood-2008-12-195859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Le Blanc K, Gotherstrom C, Ringden O, Hassan M, McMahon R, Horwitz E, Anneren G, Axelsson O, Nunn J, Ewald U, et al. Fetal mesenchymal stem-cell engraftment in bone after in utero transplantation in a patient with severe osteogenesis imperfecta. Transplantation. 2005;79:1607–1614. doi: 10.1097/01.tp.0000159029.48678.93. [DOI] [PubMed] [Google Scholar]
- 193.Gotherstrom C, Westgren M, Shaw SW, Astrom E, Biswas A, Byers PH, Mattar CN, Graham GE, Taslimi J, Ewald U, et al. Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: a two-center experience. Stem Cells Transl Med. 2014;3:255–264. doi: 10.5966/sctm.2013-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chamberlain JR, Schwarze U, Wang PR, Hirata RK, Hankenson KD, Pace JM, Underwood RA, Song KM, Sussman M, Byers PH, et al. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science. 2004;303:1198–1201. doi: 10.1126/science.1088757. [DOI] [PubMed] [Google Scholar]
- 195.Lindahl K, Kindmark A, Laxman N, Astrom E, Rubin CJ, Ljunggren O. Allele dependent silencing of collagen type I using small interfering RNAs targeting 3′UTR Indels – a novel therapeutic approach in osteogenesis imperfecta. Int J Med Sci. 2013;10:1333–1343. doi: 10.7150/ijms.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rousseau J, Gioia R, Layrolle P, Lieubeau B, Heymann D, Rossi A, Marini JC, Trichet V, Forlino A. Allele-specific Col1a1 silencing reduces mutant collagen in fibroblasts from Brtl mouse, a model for classical osteogenesis imperfecta. Eur J Hum Genet. 2014;22:667–674. doi: 10.1038/ejhg.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548:413–419. doi: 10.1038/nature23305. [DOI] [PubMed] [Google Scholar]
- 198.Hess GT, Tycko J, Yao D, Bassik MC. Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol Cell. 2017;68:26–43. doi: 10.1016/j.molcel.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, Hu YJ, Hu JH, Thompson DB, Shu Y, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2017 doi: 10.1038/nature25164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bianchi L, Gagliardi A, Maruelli S, Besio R, Landi C, Gioia R, Kozloff KM, Khoury BM, Coucke PJ, Symoens S, et al. Altered cytoskeletal organization characterized lethal but not surviving Brtl+/− mice: insight on phenotypic variability in osteogenesis imperfecta. Hum Mol Genet. 2015;24:6118–6133. doi: 10.1093/hmg/ddv328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gagliardi A, Besio R, Carnemolla C, Landi C, Armini A, Aglan M, Otaify G, Temtamy SA, Forlino A, Bini L, et al. Cytoskeleton and nuclear lamina affection in recessive osteogenesis imperfecta: A functional proteomics perspective. J Proteomics. 2017;167:46–59. doi: 10.1016/j.jprot.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]