

Abstract
Epidemiologic evidence suggests that air pollution is a risk factor for childhood obesity. Limited experimental data have shown that early-life exposure to ambient particles either increases susceptibility to diet-induced weight gain in adulthood or increases insulin resistance, adiposity, and inflammation. However, no data have directly supported a link between air pollution and non-diet-induced weight increases. In a rodent model, we found that breathing Beijing’s highly polluted air resulted in weight gain and cardiorespiratory and metabolic dysfunction. Compared to those exposed to filtered air, pregnant rats exposed to unfiltered Beijing air were significantly heavier at the end of pregnancy. At 8 wk old, the offspring prenatally and postnatally exposed to unfiltered air were significantly heavier than those exposed to filtered air. In both rat dams and their offspring, after continuous exposure to unfiltered air we observed pronounced histologic evidence for both perivascular and peribronchial inflammation in the lungs, increased tissue and systemic oxidative stress, dyslipidemia, and an enhanced proinflammatory status of epididymal fat. Results suggest that TLR2/4-dependent inflammatory activation and lipid oxidation in the lung can spill over systemically, leading to metabolic dysfunction and weight gain.—Wei, Y., Zhang, J., Li, Z., Gow, A., Chung, K. F., Hu, M., Sun, Z., Zeng, L., Zhu, T., Jia, G., Li, X., Duarte, M., Tang, X. Chronic exposure to air pollution particles increases the risk of obesity and metabolic syndrome: findings from a natural experiment in Beijing.
Keywords: chronic inflammation, metabolic dysfunction, particulate matter
It is increasingly recognized that environmental factors such as air pollution may contribute to the global obesity epidemic. Epidemiologic studies have shown an association between traffic density (a proxy of air pollution exposure) and body mass index levels at age 18 years in subjects living in Southern California (1) and an association between maternal exposure to polycyclic aromatic hydrocarbons in combustion particles during pregnancy with childhood obesity in children living in New York City (2). Limited animal experimental data support these epidemiologic associations. For example, in a mouse model, prenatal exposure to diesel exhaust particles predisposes the offspring to increased susceptibility to diet-induced weight gain and neuroinflammation in adulthood (3). However, this study did not establish a direct link between particle exposure and weight gain under normal dietary conditions. Similarly, another mouse study did not find a significant effect of early-life particle exposure on body weight in adulthood, although the exposure resulted in increased insulin resistance, adiposity, and inflammation (4, 5). A recent study using diesel exhaust exposure of pregnant mice showed increased placental inflammation and fetal resorption, along with increased weight in the surviving offspring at 10 wk of age (6). These experimental studies have focused on prenatal or early-life exposures and used an intermittent exposure protocol either with laboratory-resuspended particles or with artificially concentrated ambient particles (in the absence of gaseous copollutants). These exposure protocols may not represent real-life situations of continuous exposure to the air pollution mixture.
Ambient concentrations of PM2.5 (particulate matter with a diameter of ≤2.5 µm) in Beijing have in recent years been high enough to reach comparable levels in an exposure chamber (5). Without the need for further artificial concentration, natural ambient air in Beijing was used in the present study to examine whether living naturally in such an environment can lead to weight gain, and if so, what pathophysiologic processes are involved at the organ, tissue, and molecular levels. Our control exposure was the same Beijing air with particulate matter partially removed and without changing gaseous pollutants, as we focused on examining the effects of PM2.5 in the context of the real-world air pollution mixture.
MATERIALS AND METHODS
Ethics
All animal experimentations were conducted in compliance with the guidelines of ethical animal research. The study protocol was approved by the Medicine Animal Care and Use Committee at Peking University Health Science Center before commencement of experiments.
Exposure protocol
Two identically sized chambers, each ∼1 m3 in volume (1.2 × 0.8 × 1.2 m), were used in the present study. Each chamber could house a maximum of 9 rat cages (4 on the upper rack and 5 on the lower rack). These chambers were placed side by side for whole body inhalation exposure of rats in an air-conditioned room in Beijing. The only difference between the 2 chambers was the presence or absence of a high-efficiency particulate air (HEPA) filter placed in the inlet duct. The room was about 2 km away from northwestern fourth Ring Road, a major artery of the city with 8 main lanes and 6 auxiliary lanes that carries nearly 220,000 vehicles per day. Particulate matter concentrations were measured using 2 sets of single particle soot photometer and scanning mobility particle sizer and aerodynamic particle size, one for the chamber airstream and the other for ambient atmosphere. We calculated the removal efficiency of every particulate size. The removal efficiency of the HEPA filter was 98.99 ± 0.86% for particles larger than 2.5 μm in diameter and 70.61 ± 19.34% for PM2.5. PM2.5 concentrations inside the chamber equipped with the HEPA filter were estimated using outdoor PM2.5 concentrations and the filter removal efficiency for PM2.5. The temperature inside these chambers was controlled at 24 ± 1°C.
We started our experiments with pregnant Sprague Dawley rats. All the rats were kept in an animal care facility before the experiments commenced. We conducted the first set of experiments using 30 pregnant Sprague Dawley rats that were 12 wk old. On gestation d 4, rats were assigned to 2 groups: 18 entered the unfiltered chamber, and the remaining 12 entered the filtered chamber. The assignment was done by consideration of baseline body weight in such a way that the baseline weights were not significantly different between the two groups. In each chamber, no more than 2 rats were housed per cage to avoid overcrowding. One group was placed inside a chamber with the inlet air directly coming from outdoors; the other group was housed inside a same-size chamber equipped with a HEPA filter at the air inlet. The chambers were placed side by side inside an air-conditioned room in Beijing. Rats lived in these chambers naturally under a 12 h light/12 h dark cycle and were fed a normal chow diet. The animals were weighed when entering the chamber (d 0) and then every 3 d until d 14, when a series of end points was analyzed. Over this 14-d period, (December 28, 2009, to January 10, 2010), PM2.5 concentrations were 73.5 ± 61.3 μg/m3 in the unfiltered chamber and 19.8 ± 9.0 μg/m3 in the filtered chamber (means ± sd of hourly concentrations measured throughout the 14-d period). The sd values indicated large variability in the hourly data. We observed large diurnal and day-to-day fluctuations in ambient PM2.5 concentrations.
We found that 7 of 10 rats in the unfiltered air group and 4 of 6 in the filtered air group had produced pups. Among the rest of the pregnant rats not dissected, we found that 6 in the unfiltered group and 6 in the filtered group had delivered pups normally. The pups prenatally exposed to unfiltered air stayed in the unfiltered chamber, and the pups prenatally exposed to filtered air stayed in the filtered chamber continuously until they were analyzed either at 3 or 8 wk of age.
To allow for monitoring body weight for a longer period and for measuring more endpoints in pregnant rats, we conducted a second set of experiments using 16 pregnant Sprague Dawley rats at 12 wk old (10 in the unfiltered air group and 6 in the filtered air group). The experiments started at gestation d 1 and lasted for 19 d. Pups of these rats were not further studied. All the exposure experiments were carried out continuously 24 h/d and 7 d/wk under a 12 h light/12 h dark cycle throughout the 19 d period. During this set of the experiments (March 12 – 31, 2010), PM2.5 concentrations were 64.6 ± 72.7 and 16.7 ± 9.6 μg/m3 in the unfiltered and filtered chambers, respectively.
Blood and tissue biomarker assays
After euthanasia, the blood was collected in EDTA anticoagulant tubes, then centrifuged at 3000 rpm for 10 min at 4°C. The EDTA plasma was separated and collected. Plasma cholesterol, glucose, oxidative stress biomarkers, and inflammatory cytokines were measured using enzymatic colorimetric assays for malondialdehyde (MDA), glutathione (GSH), total cholesterol (TC), triglyceride (TG), LDL, HDL, glucagon-like peptide 1 (GLP-1), C-X-C motif chemokine 5 (CXCL5), chemokine (C-C motif) ligand 2 (CCL2), TNF-α, IL-10, and IL-6. We ground 30 g each of lung, liver, spleen, and brain in 1 ml PBS on ice and centrifuged the samples at 5000 rpm for 15 min. The clarifying middle layer was carefully aspirated for MDA and 8-isoprostane assay kit analyses.
Lung morphometric analysis
The completely cut-down left lungs were fixed in 10% formalin and embedded in paraffin for making hematoxylin and eosin–stained slides, performing morphometric analysis, and performing further immunohistochemistry using slides. These slides were analyzed by an optical microscope with a digital camera. The right lung tissue samples were immediately frozen in liquid nitrogen for further RNA extraction. Other organs were collected, weighed, and cryopreserved for further analysis.
Second-generation RNA sequencing for gene expression analysis
Second-generation RNA sequencing (RNA-Seq) was used to analyze the genome-wide gene expression profiling in the lungs. RNA-Seq was analyzed by a commercial company (BGI, Beijing, China). Gene expression results are presented as the log2 ratio of data from the unfiltered air group to the data from the filtered air group. Total RNA was extracted using a combination of Trizol reagent and ethanol precipitation according to manufacturer’s instructions (Tiangen Biotech, Beijing, China). The integrity and quantity of RNA in the samples were determined using a spectrophotometer and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Real-time quantitative PCR (qPCR) for verification of selected genes
qPCR was used for verifying the significantly up- or down-regulated genes that were identified by RNA-Seq analysis. On the basis of the RNA-Seq results, we selected 30 genes for further qPCR analysis (Supplemental Table S1). The analysis was performed on an Opticon 2 qPCR machine (Bio-Rad, Hercules, CA, USA) using SYBR Green mix (Tiangen Biotech) with the following cycling program: 95°C for 10 min, 40 cycles of 95°C for 25 s, and 60°C for 1 min. Glyceraldehyde 3-phostaphate dehydrogenase (GAPDH) was used as an internal control for normalization, and each sample was run in triplicate. The aliquot of cDNA was amplified with a pair of primers for each.
Statistical analysis
SAS 9.1.3 for windows (SAS Institute, Cary, NC, USA) was used to perform data analysis. The data are expressed as means ± sd. Pairwise comparison was conducted by Student’s t test (2-tailed) with equal or unequal variance depending on F test results. A value of P < 0.05 was considered statistically significant.
RESULTS
Effects in pregnant rats
At the beginning of the experiments, there was no difference in body weight between pregnant rats to be exposed to unfiltered air and those to be exposed to filtered air. The mean body weight of the unfiltered air group was consistently higher than that of the filtered air group throughout the experiments, reaching statistical significance on d 14 and 19 (Fig. 1A). The unfiltered air group was 7% heavier on d 14 (P = 0.046) and 15% heavier on d 19 (P = 0.0041). On d 19, the lungs of the unfiltered group were heavier (by 25%; 1.47 ± 0.19 vs. 1.18 ± 0.10 g; P = 0.0004) and displayed both perivascular and peribronchial inflammation (Fig. 1B). We observed thickened alveolar septa and the presence of mononuclear cell infiltration in the airway of the rats in the unfiltered group compared to the lung tissues of rats in the filtered group.
Figure 1.
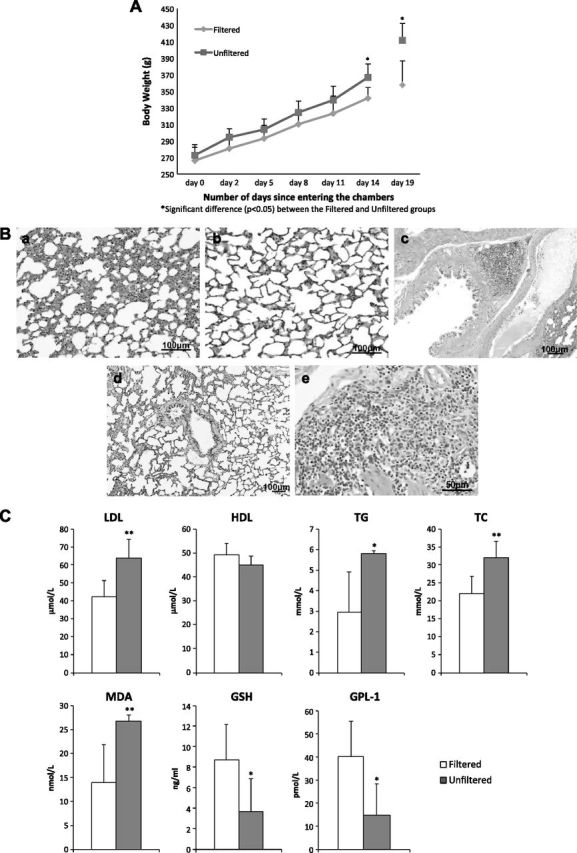
Pregnant rats exposed to unfiltered Beijing air vs. those exposed to filtered Beijing air. A) Body mass measured from d 0, when rats went into chambers, to d 14 (in first set of experiments) and measured at d 19 (in second set of experiments). Error bars represent ±sd. B) Lung histology (hematoxylin and eosin stain) showing inflammation in alveoli of rat after 19 d exposure to unfiltered air (a), but no apparent inflammation in alveolus of rat after 19 d filtered air exposure (b). Similar histologic evidence for inflammation in bronchus of unfiltered air exposed rat (c) in reference to that of filtered air exposed rat (d). Higher-magnification micrograph indicates that inflammatory cells are mainly mononuclear cells (e). C) Blood biomarkers measured in pregnant rats after 19 d exposure protocol. Significant differences between groups are indicated. *P < 0.05, **P < 0.01.
The rats in the unfiltered group also had heavier livers (by 16%; 15.62 ± 1.80 vs. 13.42 ± 0.71 g; P = 0.011). The mass of other organs, including adrenal, thymus, brain, and epididymal fat, was not significantly different. As shown in Fig. 1C, after the 19 d exposure protocol, the unfiltered air group showed a significantly worsened plasma lipid profile compared to the filtered air group: LDL was 51% higher, TC was 97% higher, and TG was 46% higher. Moreover, GLP-1, an incretin hormone that reinforces glucose-dependent secretion of insulin, was significantly lower (by −64%) in the unfiltered air group. GLP-1 is regulated by its degradation, which is accelerated by systemic inflammation and has anti-inflammatory properties within adipose tissue (7). This altered metabolic profile is consistent with a loss of insulin-dependent regulation of circulating glucose levels, which can lead to production of reactive oxygen species. There were accordingly a significant reduction (−62%) in plasma GSH (a major antioxidant) and a significant increase (+91%) in plasma MDA in the unfiltered air group. These indicate that unfiltered Beijing air induced a systemic oxidative stress resulting in an increase in oxidized lipids within the circulation, as MDA is a product of reactions between lipids (e.g., n-6-polyunsaturated acids in cell membranes) and reactive oxygen species such as peroxides (8).
Effects in rat offspring
We continued our experiments with the offspring of the Sprague Dawley rats that had undergone the 14 d exposure protocol. The pups were placed into the same chambers where their mothers had lived and continuously lived there until humanely killed for analyses. At 8 wk old, both female and male rats in the unfiltered air group had significantly greater body mass (by 10 and 18%, respectively), lung mass (by 12 and 13%, respectively), and mass for other organs (liver, spleen, and heart) compared to the filtered air group (Table 1). Blood markers showed significant differences between the 2 groups, indicating that exposure to unfiltered air resulted in a worsened lipid profile (+24% LDL, −21% HDL, +86% TG, +16% TC), reduced incretin levels (−44% GLP-1), reduced antioxidant capacity (−46% GSH), and increased oxidative stress (+90% MDA) (Table 2). Furthermore, we observed significantly higher concentrations of MDA and 8-isoprostane in tissue samples of lung, liver, spleen, and brain in the unfiltered air group (Fig. 2A).
TABLE 1.
Body and organ mass of rats exposed prenatally and continuously for 8 wk to unfiltered or filtered Beijing air
| Site | Male | Female | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Filtered (n = 8) |
Unfiltered (n = 6) |
P | Filtered (n = 8) |
Unfiltered (n = 6) |
P | |||||
| Mean | sd | Mean | sd | Mean | sd | Mean | sd | |||
| Body | 275 | 33.6 | 324 | 27.6 | 0.011 | 194 | 8.54 | 214 | 14.5 | 0.019 |
| Liver | 11.5 | 0.91 | 14.2 | 1.55 | 0.005 | 7.72 | 0.61 | 9.07 | 0.83 | 0.008 |
| Spleen | 0.79 | 0.05 | 0.88 | 0.06 | 0.008 | 0.49 | 0.06 | 0.65 | 0.10 | 0.009 |
| Adrenal | 0.04 | 0.00 | 0.06 | 0.01 | 0.002 | 0.05 | 0.01 | 0.06 | 0.01 | 0.092 |
| Thymus | 0.60 | 0.08 | 0.66 | 0.06 | 0.112 | 0.53 | 0.09 | 0.53 | 0.07 | 0.938 |
| Heart | 1.01 | 0.06 | 1.18 | 0.13 | 0.024 | 0.73 | 0.04 | 0.79 | 0.05 | 0.041 |
| Brain | 1.41 | 0.07 | 1.46 | 0.04 | 0.095 | 1.34 | 0.07 | 1.36 | 0.08 | 0.569 |
| Lung | 0.82 | 0.09 | 0.93 | 0.09 | 0.035 | 0.66 | 0.04 | 0.74 | 0.03 | 0.002 |
| Epididymal fat | 1.25 | 0.23 | 1.48 | 0.19 | 0.064 | |||||
TABLE 2.
Biomarkers of male rats exposed prenatally and continuously for 8 wk to unfiltered or filtered Beijing air
| Biomarker | Filtered (n = 10) | Unfiltered (n = 10) | P | ||
|---|---|---|---|---|---|
| Mean | sd | Mean | sd | ||
| LDL (µM) | 50.7 | 8.02 | 63.0 | 9.18 | 0.005 |
| HDL (µM) | 58.8 | 7.84 | 46.5 | 3.49 | 0.001 |
| TG (mM) | 2.93 | 2.07 | 5.45 | 1.41 | 0.006 |
| TC (mM) | 23.4 | 5.39 | 27.2 | 2.15 | 0.058 |
| MDA (nM) | 13.4 | 6.84 | 25.5 | 6.05 | 0.001 |
| GSH (ng/ml) | 8.69 | 3.51 | 4.69 | 2.66 | 0.011 |
| GLP-1 (pM) | 30.9 | 14.0 | 17.4 | 12.0 | 0.033 |
Figure 2.
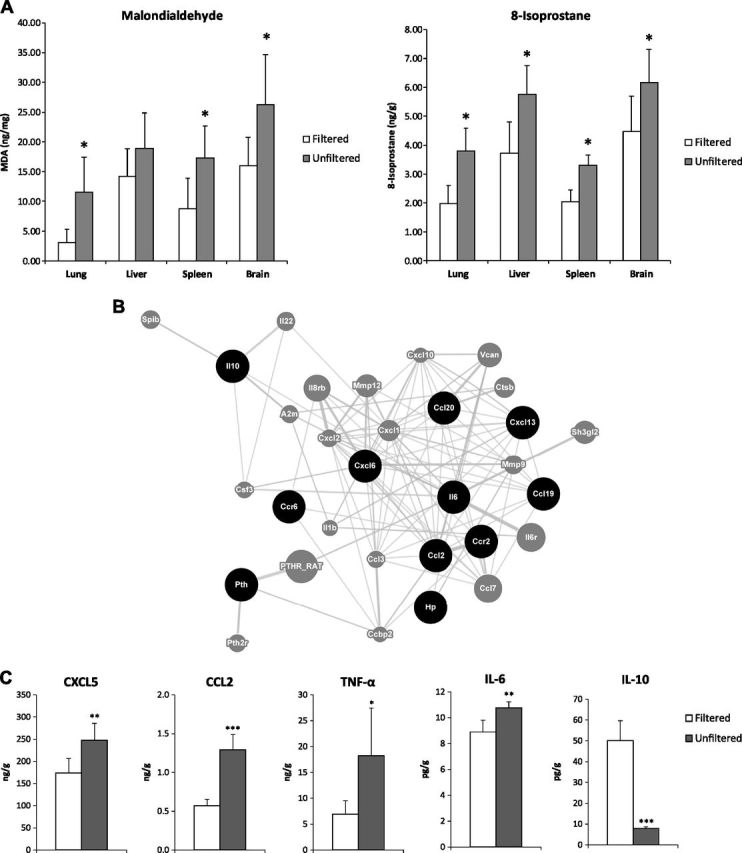
Rats exposed prenatally and continuously for 8 wk to unfiltered Beijing air vs. those exposed to filtered Beijing air. A) Biomarkers of oxidative stress, MDA, and 8-isoprostane, in tissues of lung, liver, spleen, and brain of 8-wk-old male pups. *P < 0.05. B) GeneMania network of up-regulated genes involved in cytokine signaling from lung tissue. Black circles indicate query genes identified as up-regulated between groups by RNA-Seq; gray circles, those connected via prediction or expression network analysis. C) Cytokine/chemokine concentrations in epididymal fat tissue of male rats. *P < 0.05, **P < 0.01, ***P < 0.001.
Similar to that for the rat dams, lung histology revealed both peribronchial and perivascular inflammation in these rat offspring (Fig. 3). When we conducted RNA-Seq analysis of lung tissue from both dams and pups comparing the unfiltered group to the filtered group, we found gene expression changes for over 100 genes, some of which were further confirmed for their expression changes using qPCR assays (Supplemental Table S1). Using the DAVID bioinformatic database revealed a focus of these up-regulated genes upon inflammatory processes and glucocorticoid response elements (9). A further analysis of the up-regulated genes, using the online network tool GeneMANIA (10), revealed a proinflammatory network focused on acute inflammatory signaling and classic macrophage activation, namely IL-6, CCL-2, CXCL-6, and CCL-19 (Fig. 2B). In this context, it is important to note that classic macrophage activation has been suggested to play a role in metabolic responses to inhaled pollutants (11).
Figure 3.

Lung histology (hematoxylin and eosin stain) of 8-wk-old rats, showing inflammation in alveoli of rat exposed to unfiltered air (A) compared to alveoli of rat exposed to filtered air exposure (B). Similar histologic evidence of inflammation in bronchus of unfiltered air exposed rats (C) in reference to that of filtered air exposed rats (D). Higher-magnification micrograph indicates that inflammatory cells are mainly mononuclear cells (E).
In association with lung inflammation, we also found evidence of systemic inflammation in the epididymal fat, as indicated by the significantly raised concentrations of 4 proinflammatory chemokines and cytokines (CXCL5, CCL2, IL-6, and TNF-α) and significantly lower concentrations of the anti-inflammatory cytokine (IL-10) in the unfiltered air group (Fig. 2C). We particularly observed a >6-fold reduction in IL-10 and >2-fold increases in TNF-α and CCL2, respectively. The presence of worsened metabolic profile and adipose inflammation led us to suspect that adipose tissue metabolism may be altered. We found that the mass of epididymal fat was 18% greater in the male rats exposed to unfiltered air than in those exposed to filtered air (Table 1). This indicates that the chronic exposure disrupted the balance between the proinflammatory and anti-inflammatory functions, priming the adipose tissue toward inflammatory status that may be linked to metabolic dysfunction.
DISCUSSION
The explanation of how airborne particles (or an air pollution mixture) may cause metabolic dysfunction is important in understanding the role of air pollution in cardiometabolic diseases and underlying biologic mechanisms of particle health effects. In the present study, we examined whether and how inhaled particles could lead to non-diet-induced weight gain through one or more biologic pathways. We used a real-life air pollution exposure protocol, pathologic assessment, biochemical analysis, and gene expression assays. We also examined molecular processes involved in the pathophysiologic pathways by measuring RNA and protein biomarkers.
In both rat dams and their 8-wk-old offspring, we observed histologic evidence for perivascular and peribronchial inflammation in the lungs, increased concentration of MDA in the tissue, increased MDA and 8-isoprostane, and decreased concentration of GSH in the blood. The findings of the present study are consistent with previous studies reporting that PM2.5 exposure induces oxidative stress and inflammation in organ tissues and the circulatory system, as summarized in a statement of the American Heart Association (12).
The findings on epididymal fat mass and plasma incretin also augment to the emerging evidence that chronic exposure to PM2.5 leads to increased lipid deposition in adipose tissue (13) and increased insulin resistance (14). Because chronic inflammation is increasingly recognized as part of the etiology of obesity, and because metabolic diseases and obesity are closely related (15, 16), our findings consistently provide evidence that chronic exposure to a real-world air pollution mixture containing high-level of PM2.5 increases the risk for developing obesity.
Previous studies have shown that administration of TNF-α led to an increase in serum triglycerides and very-low-density lipoproteins in rats and humans (17) and inhibited insulin-stimulated glucose transport (18). CXCL5 is produced concomitantly with IL-8 in response to stimulation with TNF-α and mediates the effects of TNF-α in insulin resistance (19, 20). Chronically elevated IL-6 levels have been associated with the development of insulin resistance (19). In contrast, levels of IL-10, an anti-inflammatory cytokine that attenuates the inflammatory processes induced by TNF-α, IL-6, and IL-1, have been adversely correlated with body mass index, fat mass, and fasting glucose levels (21, 22). Low levels of IL-10 in adipose tissues have been associated with both metabolic syndrome and type 2 diabetes (23).
In stark contrast to the above findings in the offspring after the 8 wk exposure protocol, we did not observe significant differences in body and organ mass and blood biomarkers (apart from HDL) between the 2 exposure groups in offspring that had only lived in the chamber for 3 wk (Supplemental Table S2). Lung histology results were also similar, showing no significant inflammation in both groups (Supplemental Fig. S1). This suggests that longer-term exposure may be needed to generate continuous inflammatory and metabolic changes that ultimately increase body weight.
Our exposure model used unfiltered and filtered Beijing air. The filtration we used in our study resulted in PM2.5 concentrations comparable to ambient concentrations measured in typical urban/suburban areas in and below the United States. 24 h PM2.5 standard of 35 µg/m3. The filtration only removed the particulate component of the air pollution mixture. Therefore, the health effects we observed, more precisely speaking, reflect particulate effects in the presence of gaseous copollutants. Moreover, it is known that HEPA filters are more efficient in removing larger particles than smaller particles. In the present study, we observed a removal efficiency of 99% for PM larger than 2.5 µm and of 71% for PM2.5. Within the PM2.5, we did not measure ultrafine particles (PM0.1), but we expect even lower removal efficiency for PM0.1. Therefore, this study is limited in terms of further elucidating the role of specific role of ultrafine particles and PM2.5 within the context of the air pollution mixture.
Nevertheless, using the comprehensive data from our experiments in combination with previously postulated mechanisms (12), we developed a mechanistic framework, as shown in Fig. 4, for future hypothesis testing. In a rat model, we found that exposure to ambient air containing high concentrations of PM2.5 induced TLR2/4-dependent inflammatory activation and lipid oxidation in the lung, which then spilled over systemically, leading to metabolic dysfunction and weight gain. The study provides mechanistic evidence that chronic exposure to air pollution increases the risk for developing obesity and metabolic syndrome. If translated to and verified in humans, these findings will add support to the urgent need to reduce air pollution exposure, given the growing burden of obesity and related metabolic abnormalities in today’s highly polluted world.
Figure 4.

Mechanistic framework explaining how inhaled air pollutants disrupt metabolic state. Inhalation of air pollutants (especially particulate matter) can lead to direct activation of alveolar macrophages through TLR2/4-dependent mechanisms and generation of oxidized lipids within lung lining. These oxidized lipids can further activate inflammatory processes through TLR2/4 binding or be released to vasculature, where they will initiate systemic inflammation and oxidative stress responses. TLR2/4-dependent inflammatory activation, through activity of MyD88, will lead to release of proinflammatory cytokines, such as CCL-2 and IL-6, from lung, also generating systemic inflammation. These systemic inflammatory activation processes, along with loss of anti-inflammatory functions from incretins such as GLP-1, will lead to increased recruitment of activated inflammatory cells to tissues and in particular adipose. Recruitment of such cells to adipose will worsen metabolic profile, leading to weight gain and metabolic disease state. Prenatal exposure may enhance cellular and functional responses shown here.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
Acknowledgments
This work was supported in part by grants from the National Natural Science Foundation of China (21190051, 21477119), the Open Fund of the State Key Joint Laboratory of Environment Simulation and Pollution Control (13K03ESPCP), and the China Postdoctoral Science Foundation (201003010).
Glossary
- CCL2
chemokine (C-C motif) ligand 2
- CXCL5
C-X-C motif chemokine 5
- GLP-1
glucagon-like peptide 1
- GSH
glutathione
- HEPA
high-efficiency particulate air
- MDA
malondialdehyde
- PM2.5
particulate matter with a diameter of ≤2.5 µm
- RNA-Seq
second-generation RNA sequencing
- qPCR
quantitative PCR
- TC
total cholesterol
- TG
triglyceride
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Jerrett M., McConnell R., Chang C. C. R., Wolch J., Reynolds K., Lurmann F., Gilliland F., Berhane K. (2010) Automobile traffic around the home and attained body mass index: a longitudinal cohort study of children aged 10–18 years. Prev. Med. (Suppl 1), S50–S58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rundle A., Hoepner L., Hassoun A., Oberfield S., Freyer G., Holmes D., Reyes M., Quinn J., Camann D., Perera F., Whyatt R. (2012) Association of childhood 1obesity with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy. Am. J. Epidemiol. , 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolton J. L., Smith S. H., Huff N. C., Gilmour M. I., Foster W. M., Auten R. L., Bilbo S. D. (2012) Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. , 4743–4754 [DOI] [PubMed] [Google Scholar]
- 4.Sun Q., Yue P., Deiuliis J. A., Lumeng C. N., Kampfrath T., Mikolaj M. B., Cai Y., Ostrowski M. C., Lu B., Parthasarathy S., Brook R. D., Moffatt-Bruce S. D., Chen L. C., Rajagopalan S. (2009) Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation , 538–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu X., Yavar Z., Verdin M., Ying Z., Mihai G., Kampfrath T., Wang A., Zhong M., Lippmann M., Chen L.-C., Rajagopalan S., Sun Q. (2010) Effect of early particulate air pollution exposure on obesity in mice: role of p47phox. Arterioscler. Thromb. Vasc. Biol. , 2518–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weldy C. S., Liu Y., Liggitt H. D., Chin M. T. (2014) In utero exposure to diesel exhaust air pollution promotes adverse intrauterine conditions, resulting in weight gain, altered blood pressure, and increased susceptibility to heart failure in adult mice. PLoS One , e88582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y. S., Park M. S., Choung J. S., Kim S. S., Oh H. H., Choi C. S., Ha S. Y., Kang Y., Kim Y., Jun H. S. (2012) Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia , 2456–2468 [DOI] [PubMed] [Google Scholar]
- 8.Frankel E. N., Neff W. E. (1983) Formation of malonaldehyde from lipid oxidation products. Biochim. Biophys. Acta Lipids Lipid Metab. , 264–270 [Google Scholar]
- 9.Huang W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. , 44–57 [DOI] [PubMed] [Google Scholar]
- 10.Zuberi K., Franz M., Rodriguez H., Montojo J., Lopes C. T., Bader G. D., Morris Q. (2013) GeneMANIA prediction server 2013 update. Nucleic Acids Res. , W115–W122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopalan S., Brook R. D. (2012) Air pollution and type 2 diabetes: mechanistic insights. Diabetes , 3037–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brook R. D., Rajagopalan S., Pope C. A. III, Brook J. R., Bhatnagar A., Diez-Roux A. V., Holguin F., Hong Y., Luepker R. V., Mittleman M. A., Peters A., Siscovick D., Smith S. C. Jr., Whitsel L., Kaufman J. D.; American Heart Association Council on Epidemiology and Prevention, Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism (2010) Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation , 2331–2378 [DOI] [PubMed] [Google Scholar]
- 13.Mendez R., Zheng Z., Fan Z., Rajagopalan S., Sun Q., Zhang K. (2013) Exposure to fine airborne particulate matter induces macrophage infiltration, unfolded protein response, and lipid deposition in white adipose tissue. Am. J. Transl. Res. , 224–234 [PMC free article] [PubMed] [Google Scholar]
- 14.Thiering E., Cyrys J., Kratzsch J., Meisinger C., Hoffmann B., Berdel D., von Berg A., Koletzko S., Bauer C. P., Heinrich J. (2013) Long-term exposure to traffic-related air pollution and insulin resistance in children: results from the GINIplus and LISAplus birth cohorts. Diabetologia , 1696–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrante A. W., Jr (2007) Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J. Intern. Med. , 408–414 [DOI] [PubMed] [Google Scholar]
- 16.Shoelson S. E., Herrero L., Naaz A. (2007) Obesity, inflammation, and insulin resistance. Gastroenterology , 2169–2180 [DOI] [PubMed] [Google Scholar]
- 17.Grunfeld C., Feingold K. R. (1991) The metabolic effects of tumor necrosis factor and other cytokines. Biotherapy , 143–158 [DOI] [PubMed] [Google Scholar]
- 18.Hotamisligil G. S., Murray D. L., Choy L. N., Spiegelman B. M. (1994) Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA , 4854–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chavey C., Fajas L. (2009) CXCL5 drives obesity to diabetes, and further. Aging (Albany, N.Y.) , 674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chavey C., Lazennec G., Lagarrigue S., Clapé C., Iankova I., Teyssier J., Annicotte J.-S., Schmidt J., Mataki C., Yamamoto H., Sanches R., Guma A., Stich V., Vitkova M., Jardin-Watelet B., Renard E., Strieter R., Tuthill A., Hotamisligil G. S., Vidal-Puig A., Zorzano A., Langin D., Fajas L. (2009) CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell Metab. , 339–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blüher M., Fasshauer M., Tönjes A., Kratzsch J., Schön M. R., Paschke R. (2005) Association of interleukin-6, C-reactive protein, interleukin-10 and adiponectin plasma concentrations with measures of obesity, insulin sensitivity and glucose metabolism. Exp. Clin. Endocrinol. Diabetes. , 534–537 [DOI] [PubMed] [Google Scholar]
- 22.Juge-Aubry C. E., Somm E., Pernin A., Alizadeh N., Giusti V., Dayer J.-M., Meier C. A. (2005) Adipose tissue is a regulated source of interleukin-10. Cytokine , 270–274 [DOI] [PubMed] [Google Scholar]
- 23.Charles B. A., Doumatey A., Huang H., Zhou J., Chen G., Shriner D., Adeyemo A., Rotimi C. N. (2011) The roles of IL-6, IL-10, and IL-1RA in obesity and insulin resistance in African-Americans. J. Clin. Endocrinol. Metab. , E2018–E2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.