

Abstract
Survival bias is a long-recognized problem in case-control studies, and many varieties of bias can come under this umbrella term. We focus on one of them, termed Neyman’s bias or “prevalence-incidence bias.” It occurs in case-control studies when exposure affects both disease and disease-induced mortality, and we give a formula for the observed, biased odds ratio under such conditions. We compare our result with previous investigations into this phenomenon and consider models under which this bias may or may not be important. Finally, we propose three hypothesis tests to identify when Neyman’s bias may be present in case-control studies. We apply these tests to three data sets, one of stroke mortality, another of brain tumors, and the last of atrial fibrillation, and find some evidence of Neyman’s bias in the former two cases, but not the last case.
Keywords: odds ratio, survival bias, truncation
1. Introduction.
Survival bias is a frequent source of concern in case-control studies (Sackett, 1979; Rothman et al., 2008). Sackett describes nine types of bias common in case-control studies, and we focus our investigation on one of them, first identified by Jerzy Neyman and now known as Neyman’s bias or “prevalence-incidence bias” (Neyman, 1955). It is a bias that occurs when prevalent cases are sampled and exposure affects disease and disease-associated mortality. Since Neyman’s article was written in the 1950’s when the relationship between smoking and lung cancer was under debate, he uses an example that focuses on that subject. He disregards competing risks and supposes that if, in fact, smoking is protective against lung cancer, but lung cancer mortality is far higher among non-smokers than smokers, then the odds ratio would suggest that smoking is a risk factor for disease as was being observed at the time. In our study of the subject, we focus on three other examples, one coming from a study of brain tumors and chemotherapy, another coming from a GWAS of ischemic stroke, and the last coming from a study of atrial fibrillation in the Framingham Heart Study. Prevalence-incidence bias could arise in the study of brain tumors if certain patients are assessed to have disease too progressed to benefit from chemotherapy and therefore do not undergo treatment. The GWAS could suffer from prevalence-incidence bias if a certain subset of patients die before admission to a hospital and study entry. We use our study of atrial fibrillation in the Framingham Heart study as an example of a prospective design that should therefore not suffer from prevalence-incidence bias. We consider these data as being generated under the null hypothesis of no prevalence-incidence bias in order to substantiate the validity of the test.
Despite Neyman’s early identification of this bias, methodological investigation into it has been limited. Hill (2003) uses a compartment model to show how bias arises when performing case-control studies on prevalent cases if the risk factor impacts both disease and mortality from disease. He also shows that any impact of the risk factor on mortality from other causes does not impact the observed odds ratio, which demonstrates that Neyman was justified in ignoring competing risks. While trying to draw inference on incidence instead of the odds ratio, Fluss et al. (2012), Keiding (1991), and Keiding (2006) all consider the problem of using cross-sectional designs and their resultant sampling biases.
Anderson et al. (2011) performs a computational investigation into Neyman’s bias, recognizing that genome-wide association studies (GWAS) and their use of prevalent cases in case-control study designs were susceptible to it. If an allele is a risk factor for both disease and mortality from disease, then the common practice of calculating an odds ratio from prevalent cases and controls could lead to biased inference. Since the odds ratios in such studies are usually small, differences in disease-associated mortality between the exposed and unexposed would not be required for a risk allele to be observed as protective, or vice versa. Their own investigation is motivated by a locus found to be significantly associated with ischemic stroke in longitudinal studies that did not replicate using a case-control design. As a solution, they simulate data under different disease and mortality risk models and then fit regression models for percent bias of the odds ratio to the disease and mortality risk model parameters. These fitted models give researchers a means to investigate the potential biases of estimated odds ratios in their own studies.
In this paper, we propose a framework for consideration of Neyman’s bias and examine it from a modeling perspective. We suggest three hypothesis tests to assess whether Neyman’s bias is present in a study and then apply these tests to three data sets mentioned: one of brain tumors and chemotherapy, another a GWAS of ischemic stroke, and the last focused on atrial fibrillation in the Framingham Heart Study. We propose hypothesis tests rather than methods to recover the true, unbiased odds ratio, since that quantity is unrecoverable under the framework we consider.
2. Methods.
2.1. Notation and background.
Suppose that we have a setting similar to that described in Anderson et al. (2011), where we have some binary risk SNP or gene, G, that takes on the value 1 with probability p (“exposed”) and 0 with probability 1 - p (“unexposed”). Let D denote age of disease onset, and suppose that G may be associated with D. Let {Ma,j}, j = 1,...,n, denote age at mortality from all other causes not associated with disease. Let {Xi}, i = 1,...,m, denote latent time from disease onset to the ith mortality cause related to disease and let X = min{Xi}. Thus, Ma,j ⫫ (X D)T | G for all j, where W ⫫ Y | Z denotes statistical independence of W and Y conditional on Z (Dawid, 1979). We define Ma ≡ min{Ma,j}, and thus Ma ⫫ (X D)T | G. Let Md,i ≡ D+Xi and Md = min(Md,i) = D+X. If Xi ⫫ D, then Md,i is necessarily associated with D because Md,i ≡ D + Xi (i.e., Md,i denotes the age at disease-associated mortality cause i). In fact, Xi would have to be associated with D in a specific way to have Md,i ⫫ D. We do not assume Xi is a positive random variable so that we can have P(Md,i < D) ≥ 0. While it may seem counterintuitive to allow for disease-associated mortality prior to disease, this flexibility fits into a realistic framework. For example, if the disease of interest is stroke, and there exists an association between death from myocardial infarction and stroke, then indeed mortality associated with disease, though not directly caused by it, can occur before disease and can bias the odds ratio, as we show later.
It is not a limitation of this conceptual framework to assume the existences of the Ma,j’s, ages at causes of mortality unrelated to our disease of interest. They are present to show their lack of effect on the observed odds ratio in the work to follow. Since there is no cap on the possible number of Md,i’s, all causes of mortality can be considered as disease-associated if desired by the analyst.
2.2. Formulae.
Suppose we perform a case-control study of prevalent cases at age t*, and define Ca ≡ I(t* ≤ Ma), Cd ≡ I(t* ≤ Md), where I(·) is the indicator function, and C ≡ Cd × Ca. While Cd and Ca are functions of Md and Ma, mortality causes, we can consider Md and Ma more generally as anything that would render a subject unable to enter the study that is associated and unassociated with disease, respectively. A subject is available to enter the study at age t* if C = 1; i.e., if the subject has not died from any cause by age t*. Denote the cumulative distribution function associated with random variable Y as FY (t). Then the target odds ratio among the population at age t* is
In contrast, the observed odds ratio among prevalent cases at age t* is
Now consider the term P(D ≤ t*,G = 1,Ca = 1,Cd = 1). We can factor the probability as
Since Ma ⫫ (X D)T | G and Md ≡ X +D, Ma ⫫ Md | G, and since Ca and Cd are functions of only Ma and Md (with fixed and known t*), respectively, (D Cd)T ⫫ Ca | G. Using this conditional independence, the probability further simplifies to
which is equal to
Analogous simplifications of the other terms of ORob(t*) yield
| (1) |
Note that when X ⫫ D | G, we observe
This assumption may be reasonable for some exposures that are risk factors for diseases whose course is independent of the age of onset given G.
Returning to the general case (1), we consider ways in which ORob(t*) = ORtr(t*) holds. Recall that Md ≡ D + X, and that X need not be a positive random variable. Suppose that X ≡ A - D, for some positive random variable A independent of D, conditional on G. Then Md ≡ D + X = D + (A - D) = A. So Md = A and is independent of D given G, or in notation, Md ⫫ D | G (Dawid, 1979). Notice that when Md is defined in this way, an association necessarily exists between X and D, conditional on G, since X is itself a function of D. If Md ⫫ D | G holds, then (1) reduces to
| (2) |
where (2) follows from
So when Md ⫫ D | G, Md behaves like Ma in the sense that ORob(t*) is no longer a function of the distribution of Md and ORtr(t*) = ORob(t*). While Md ⫫ D | G is a sufficient condition for ORtr(t*) = ORob(t*), it is not necessary; there exist multivariate distributions (X D G)T such that ORtr(t*) = ORob(t*), but Md
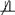 D | G (Md is not independent of D conditional on G). For example, consider the case in which if x ≤ t* or x ≤ t and , otherwise for g ∈ {0,1}; i.e., no disease-related death occurs prior to t* and in this way cannot bias ORob(t*), but in the region D > t*, Md is perfectly correlated with D so that Md
D | G (Md is not independent of D conditional on G). For example, consider the case in which if x ≤ t* or x ≤ t and , otherwise for g ∈ {0,1}; i.e., no disease-related death occurs prior to t* and in this way cannot bias ORob(t*), but in the region D > t*, Md is perfectly correlated with D so that Md
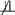 D | G. Nonetheless, in § 5 we propose tests of deviations from Md ⫫ D | G since the cases in which Md
D | G. Nonetheless, in § 5 we propose tests of deviations from Md ⫫ D | G since the cases in which Md
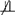 D | G, but ORtr(t*) = ORob(t*) holds are unlikely to occur as is the case in this example.
D | G, but ORtr(t*) = ORob(t*) holds are unlikely to occur as is the case in this example.
3. Scientific hypotheses versus sampling bias hypotheses.
We distinguish between H0S : ORtr(t*) = 1 (at some time t*, the true odds ratio is one), which we term the “scientific null hypothesis” and H0B : ORtr(t*) = ORob(t*) (there is no bias in the odds ratios at time t*), which we term the “sampling bias null hypothesis.” The alternative hypothesis in each case is the complement of the null hypothesis. We describe characteristics of these hypotheses.
Even if mortality from other causes, Ma, depends on G, it does not affect the bias of the observed odds ratio; in other words, ORob(t*) and ORtr(t*) are not a function of the distribution of Ma. Thus, we may assume, as Neyman (1955) does in his original example and Hill (2003) confirms, that mortality from other causes is not present and death can only occur from disease. Similarly, the probability of exposure, p, does not affect ORob(t*). Also, if FMd|G=g(t*) = 0 for g 2 {0,1} (which is the case when no disease-associated mortality occurs prior to t*), then ORob(t*) is unbiased: ORob(t*) = ORtr(t*). This result is expected since it is disease-related mortality that results in the bias-inducing differential selection between the exposed and unexposed.
Under the following four conditions, bias exists (i.e., ORob(t*) ≠ ORtr(t*)):
FX|D=t,G=0(t* − t) = FX|G=0(t* − t) = FX|G=1(t* − t) = FX|D=t, G=1(t* − t) for all t (i.e., the mortality distribution from disease-onset is identical between the exposed and unexposed and not dependent on age at disease-onset).
FX|G=g(t**) > 0 for some g ∈ {0,1}. In other words, either the exposed or unexposed have positive probability of dying from disease by t**, where t** is defined as the time between t* and the first possible presence of disease among the exposed or unexposed (i.e., inf{FD|G=g(t) > 0 : t ∈ [0,∞),g 2 {0,1}}) so that the bias-inducing event will have some chance of occurring prior to study entry at age t*).
P(X > 0) = 1, implying P(D < Md) = 1.
FD|G=0(x) = FD|G=1(x − k) for all x for some k ≠ 0 , and FD|G=0(t*) > 0 or FD|G=1(t*) > 0 (i.e., the disease distributions for the exposed and unexposed are in the same location family, and k ≠ 0 implies ORtr(t*) ≠ 1).
These assumptions seem plausible if some exposure affects the mean age of disease, though the shape of the disease distribution is approximately the same between exposed and unexposed, and after disease occurrence, hazard of mortality is identical among those with and without the exposure and not a function of age at disease onset. The theorem and proof of this result is found in the Appendix (Theorem 1). As becomes evident upon examination of that theorem, we expect greater bias with a higher hazard function of death, holding all else constant. Additionally, in that proof we examine the direction of bias; we find that when ORtr(t*) < 1, then ORob(t*) > ORtr(t*), and when ORtr(t*) > 1, then ORob(t*) < ORtr(t*). Thus, if the degree of bias is relatively small, then it can be viewed as a bias toward an observed odds ratio of 1. However, ORob(t*) is by no means bounded by 1 and so if the amount of bias is great, ORob(t*) and ORtr(t*) can lie on opposite sides of 1, leading to wrongly inferring a truly protective exposure as a risk factor for the outcome or a true risk factor as protective against the outcome.
This result of ORob(t*) ≠ ORtr(t*) will not necessarily hold if conditions 1 − 3 hold, but condition 4 is not satisfied (the distributions of disease of exposed and unexposed are not in the same location family). Under such a scenario, there may not be bias, as Example 1 in the Appendix illustrates. Additionally, if we only assume that conditions 2 − 3 are satisfied, then there may or may not be bias. See Examples 2 and 3 in the Appendix for instances of ORob(t*) = ORtr(t*) and ORob(t*) ≠ ORtr(t*), respectively, when X is associated with G (but is independent of D given G: X ⫫ D | G). It follows that if there exist no conditional independences, one can make no conclusions regarding the relationship between ORtr(t*) and ORob(t*) as there is even greater flexibility in the joint model. Lastly, if only X ⫫ G | D is assumed so that X may depend on D (i.e., time to disease-induced mortality may depend on age at disease-onset), again ORtr(t*) and ORob(t*) may or may not be equal. This result follows from the proof with location families and Example 1 because they are special cases of only assuming X ⫫ G | D.
If we only assume that ORtr(t*) = 1 with no conditions on ORtr(t) for t < t*, and also that X ⫫ D | G and FX|G=0(t) ≠ FX|G=1(t) for some t < t**, one cannot conclude anything regarding the relationship between ORtr(t*) and ORob(t*). Consider Examples 4 and 5 in the Appendix for instances of ORob(t*) = ORtr(t*) = 1 and ORob(t*) ≠ ORtr(t*) = 1, respectively. We also observe that if ORtr(t) = 1 for all t ≤ t* and FX|D,G=0(t) = FX|D,G=1(t) for all t < t**, ORtr(t*) = ORob(t*) = 1.
4. The odds ratio when T* is not fixed.
If the case-control study consists of people of many ages, then t*, previously considered fixed, can be considered random. Let us denote this random variable T*. Under these conditions, the target odds ratio becomes
Making no assumptions about the joint model (D X Ma G)T, the observed odds ratio is
While P(G = 1) = p cancels from ORob(T*) as before with ORob(t*), we see that even if (D X)T ⫫ Ma | G, we cannot factor P(T* < Ma|G = g) out of the expression. So ORob(T*) becomes a function of Ma, causes of mortality unassociated with the disease under investigation. Additionally, regardless of whether P(T* < Ma|G = g) factors out of the expression, D ⫫ Md | G, which we have stated before as being sufficient for ORob(t*) = ORtr(t*), is not sufficient for ORob(T*) = ORtr(T*). This point is relevant as we propose hypothesis tests below. This is also important to remember since ORob(T*) is generally what would be measured in a real-world case-control study where ages of subjects vary. While outside the scope of this investigation, investigators might want to only pool those groups of subjects where FD|G(·) is relatively constant across their age ranges, in which case D ⫫ Md | G would be sufficient for ORob(T*) = ORtr(T*). Also, if the sample size of a case-control study is sufficient, stratifying subjects by age and calculating age-specific odds ratios would be another way to be assured that D ⫫ Md | G is sufficient for those stratum-specific odds ratios.
5. Hypothesis testing.
5.1. Description.
We develop three methods for testing for the presence of Neyman’s bias in a study. Again, the “bias null hypothesis” of these tests is ORtr(t*) = ORob(t*), and the alternative is ORtr(t*) ≠ ORob(t*). While power may vary as a function of ORtr(t*), the tests we propose are valid under all values of ORtr(t*). Each of these three methods makes use of characteristics unique to the data when Neyman’s bias is absent, and each test may be more fitting to use than the other two under certain study designs. So, for example, Tests 1 and 2 require study observations to have some variation in age at study entry, a random variable we denote T*, while Test 3 does not, though Test 3 requires external knowledge of population prevalence of disease and exposure, while neither Test 1 nor Test 2 does.
We have demonstrated above that Md ⫫ D | G is a sufficient condition for ORtr(t*) = ORob(t*). Ideally, we would have data on all of D, Md, and G and could test for conditional independences. However, in practice, it may be unlikely that one would have follow-up data on controls and perhaps even cases, in which case Md would be unknown for one or both groups. Thus, we propose these tests with real-world data limitations in mind.
The first two hypothesis tests we propose attempt to test whether this independence condition holds. Both of these hypothesis tests make use of previous work coming from the truncation methodology literature for tests of “quasi-independence,” which refers to independence of random variables in a certain “observable” region of their joint distribution, which we explain further below (Martin and Betensky, 2005; Tsai, 1990). Tests for quasi-independence are based on U-statistics, a class of statistics with broad application outside of these tests and first described in Hoeffding et al. (1948).
The last hypothesis test we propose assumes P(D < Md) = 1, which may be unreasonable in some settings, but reasonable in others, and depends on whether causes of mortality associated with disease can come before disease onset. The test uses the fact that with data collected under a case-control study design along with population disease prevalence, one can estimate the population exposure proportion. If one has knowledge of the true exposure proportion, any comparison between the true, known value and the calculated quantity can reveal bias in the odds ratio from which it was calculated. Thus, in contrast to the first two tests that detect a sufficient, though not necessary, condition for ORtr(t*) = ORob(t*), rendering the test potentially slightly conservative (though likely not very conservative), this last test achieves its nominal type 1 error rate under the null of ORtr(t*) = ORob(t*) and has power greater than it under the alternative of ORtr(t*) ≠ ORob(t*).
5.2. Test 1: Testing for “quasi-independence” using D and Md.
We are interested in testing independence of D and Md given G, and our observable region is D < T* < Md given G; i.e., realizations of observed (because T* < Md) cases (because D < T*) of a given exposure status. To accomplish this, we modify a U-statistic test of association of Austin et al. (2013), whose null hypothesis assumes in our context mutual independence of D, T*, Md; this is stronger than our null hypothesis. This is a valid approach to testing D ⫫ Md | G, which is sufficient for no Neyman’s bias, because D ⫫ Md | G necessarily implies independence in the region we are defining as observable, D < T* < Md given G. Additionally, we focus on the cases in the study, under the assumption that follow-up data on Md is more likely to be available among them. While the power of this test may suffer in comparison to one that makes use of all observations, the approach makes fewer assumptions on data availability, and in settings in which P(D < Md) is close to 1, power will not suffer significantly.
To implement the hypothesis test, first we categorize all causes of mortality as Md, since if D and Md are associated given G, and D ⫫ Ma | G, then categorizing Ma as Md will maintain that association and avoid the need to censor observations. Doing so is not an approximation nor does it invalidate the test; rather, the test could become invalid if mortality related to disease (Md) are incorrectly categorized as unrelated to disease (Ma). Also, if D ⫫ Md | G and D ⫫ Ma | G, categorizing Ma as Md will maintain D ⫫ Md | G. This approach is also legitimate from the perspective that Md was originally defined as causes of mortality potentially, though not necessarily, associated with disease. Now suppose that we have 1,...,n realizations of (Gi Di Ti* Md,i)T, all cases so that one can assume D < T* and on whom there is follow-up so Md is known, and that (alternatively, ) if G = 0 (alternatively, G = 1) and max{Di,Dj} ≤ min{Md,i,Md,j}, the comparability criterion, is satisfied, and (alternatively, otherwise. Define and .
The test statistic for the stratum G = g, Tg, with g ∈ {0,1}, is
Then Tg ~ N(0,vg), where
and where and , with sgn(x) = 1 for x > 0, −1 for x < 0, and 0 for x = 0.
Since we would reject if either T0 or T1 falls in some predetermined critical region because dependence between D and Md given either G = 0 or G = 1 may mean ORtr(t*) ≠ ORob(t*), in order to achieve a size α test, we can use a p-value threshold of α* for T0 and T1, where α* satisfies the equation α = 1 − (1 − α*)2. So we propose a test that rejects for , where abs(x) denotes the absolute value of x and z1 − α*/2is the (z1 − α*/2)th quantile of a standard normal random variable.
Also, though D ⫫ Md | G characterizes a subset of situations for which ORtr(t*) = ORob(t*), our test is likely not overly conservative. The majority of situations in which ORtr(t*) = ORob(t*) holds result from D ⫫ Md | G being satisfied.
Power curves for Test 1 as a function of the association between D and Md are shown in Figs. 1 and 2. These curves were generated at 11 different values of Kendall’s 𝜏, used as a measure of the association between D and Md. In our case a Kendall’s 𝜏 value of 0 corresponds to independence between D and Md, and the power of the test at that value demonstrates the desired type 1 error rate of 0.05. The power curve in Fig. 1 was generated using 3000 iterations at each value, while that in Fig. 2 was generated using 1000 iterations at each value, and power was estimated by averaging over these iterations. In Fig. 1, at each iteration the test statistic was calculated using 1000 comparable pairs (i.e., those pairs that satisfy the comparability criterion mentioned in the description of Test 1). In Fig. 2, the test statistic was calculated using approximately 670 comparable pairs at each iteration–a subset of the 1000 comparable pairs used for Test 2, described below, that satisfied Test 1’s more stringent comparability criterion. Fig. 2 also demonstrates a type 1 error rate of 0.05. D, T, and Md were all distributed normal with means of 5, 9, and 9, respectively, and standard deviations of 0.7.
Figure 1:

Comparison of power between tests 1 (short dashes) and 2 (long dashes) as a function of the asso-ciation between D and Md, measured by Kendall’s 𝜏, holding the sample size constant.
Figure 2:

Comparison of power for tests 1 (short dashes) and 2 (long dashes) as a function of the D and Md as measured by Kendall’s 𝜏, holding the number of comparable pairs constant.
We now consider realistic settings in which age of entry, T*, is random. In § 4 above, we saw that the distribution for Ma did not factor out of the odds ratio even when (D X) ⫫ Ma | G, and that additionally even under the assumption of D ⫫ Md | G, whether or not the previous assumption held, ORob(T*) could be biased; we needed a fixed t* for these conditional independencies to result in ORtr(t*) = ORob(t*). Thus, it may seem illogical to propose a test that requires variation in T*, which is precisely when the odds ratio will almost certainly be biased as shown in § 4. If we do find that D ⫫ Md | G, sufficient for no Neyman’s bias, we would need to then stratify our sample according to similar values of T* such that, within each stratum, T* can be effectively considered fixed, and then calculate the odds ratio for these different strata. We could then combine these strata into a average odds ratio if desirable or just consider each stratum-specific odds ratio separately.
5.3. Test 2: Testing “quasi-independence” with D and T*.
We now describe a test related to Test 1, which does not require knowledge of Md and again focuses on cases, those observations for whom D < T* is true. Such a test is appropriate if a study did not obtain follow-up on subjects, but did record age at onset of disease for cases. The foundation for the test is based on causal directed acyclic graphs (DAGs), borrowed from the causal inference literature (Hernan and Robins, ress). We use DAGs not for the sake of justifying causal interpretations of ORob, but rather as a convenient means of encoding conditional independencies. If DAGs are unfamiliar with the reader, Hernan and Robins (ress) describes them well.
By definition, the event I(T* < Md) = 1 must be satisfied for any subject in the study and can therefore betreated as a conditioning event. Additionally, by definition of I(T* < Md), there exists an association between it and both T* and Md; to borrow language from the causal inference literature, I(T* < Md) is called a “collider” in this instance because both T* and Md cause it. Thus, we see in Figs. 3 and 4 arrows between these random variables, indicative of a possible association, and a square around I(T* < Md), indicative of a conditioning event. Assuming 0 < P(T* < Md) < 1 so the conditioning event is non-trivial, an association between D and T* given G implies D
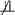 Md | G, and the converse of this statement is also true. Association between D and T* given G therefore serves as a powerful and valid proxy for association between D and Md given G. These associations result from conditioning on the “collider” I(T* < Md); association paths are opened between D and T* given G. Were I(T* < Md) not to be conditioned on, D and T* would be independent given G. The structure of this DAG is identical to that found in classic selection bias (even if the variables are not), where exposure and outcome both cause some indicator that is conditioned on in the analysis, which results in a spurious association between exposure and outcome even under the null.
Md | G, and the converse of this statement is also true. Association between D and T* given G therefore serves as a powerful and valid proxy for association between D and Md given G. These associations result from conditioning on the “collider” I(T* < Md); association paths are opened between D and T* given G. Were I(T* < Md) not to be conditioned on, D and T* would be independent given G. The structure of this DAG is identical to that found in classic selection bias (even if the variables are not), where exposure and outcome both cause some indicator that is conditioned on in the analysis, which results in a spurious association between exposure and outcome even under the null.
Figure 3:

This DAG provides the framework for Test 2. When D is not associated with Md, there is no association between D and T*, despite the conditioning event, using rules of DAGs. This figure represents these random variables within each stratum of G.
Figure 4:

This DAG provides the framework for Test 2. When D is associated with Md, an association between D and T* is induced due to the conditioning event using rules of DAGs. This figure represents these random variables within each stratum of G.
We could assume D and T* are known for all observations in our data set and propose a test of association for these random variables under the framework described above. However, doing so is unrealistic as it assumes follow-up data on age at disease, D, for those observed at T* as controls (i.e., those with D > T*). Thus, we assume D and T* are observed only for cases (i.e., those realizations satisfying D < T*) and propose a test of “quasi-independence” between D and T* given G in the region of D < T* given G. If we assume that the independence which holds on the region D < T* also holds for the entire joint distribution of (D T*)T, then since there is an association between D and T* given G if and only if D and Md are associated given G, this test is valid. We do not feel this assumption is an overly strong one, but is instead reasonable. In general, a joint distribution that exhibits dependence will not have that structure isolated to a certain region–examination of just a single region will reveal it, even if there are pathological counterexamples where this behavior does not hold.
We describe here this proposed test of quasi-independence. As before, let there be n realizations of , and again define (alternatively, ) similarly to how we did with (alternatively, ), where if G = 0 and and = 0 otherwise, and where = 1 if G = 1 and and = 0 otherwise. Also, define and .
Then the test statistic for the stratum G = g, Wg, with g 2 {0,1}, is
Then Wg ~ N(0,ug), where
and where and . As with Test 1, since we would reject if either W0 or W1 falls in some predetermined critical region because dependence between D and Md given either G = 0 or G = 1 may mean ORtr(t*) ≠ ORob(t*), for a size α test, our p-value threshold α* for W0 and W1 satisfies α = 1− (1 – α*)2. Thus, our test rejects for .
Power curves for Test 2 as a function of the association between D and Md are shown in Figs. 1 and 2. As with the simulations for Test 1, these curves were generated at 11 different values of Kendall’s 𝜏, used as a measure of the association between D and Md. In our case a Kendall’s 𝜏 value of 0 corresponds to independence between D and Md. The power curve for Test 2 in Fig. 1 was generated using 3000 iterations at each value, while that in Fig. 2 was generated using 1000 iterations at each value, and power was again estimated by averaging over these iterations. In both Figs. 1 and 2, at each iteration the test statistic was calculated using 1000 comparable pairs. D, T, and Md were distributed multivariate normal with means of 5, 9, and 9, respectively, and standard deviations of 0.7. The correlation between D and Md varied as measured by Kendall’s 𝜏, while T was assumed independent of (T, Md)T.
As mentioned at the end of the description of Test 1 and for reasons given there, if this test does not reject D ⊥ ⊥ T* | G, implying D ⫫ Md | G, we would again need to stratify the data by T* in order for ORob(t*) to be unbiased for ORtr(t*). In other words, one could split the data in subsets based on age at entry, T*, and calculate stratum-specific odds ratios. It is important to note that the reason for doing so applies no more in the context of testing for Neyman’s bias than it would any standard case-control study, where a mixture of ages at study entry results in an odds ratio that is difficult to interpret and possibly biased as shown in § 4.
5.4. Test 3: Estimating population exposure proportion.
With knowledge of disease prevalence, we can construct an estimate of the exposure in the general population from case-control study data that is unbiased in the absence of Neyman’s bias, but biased otherwise. Thus, if the exposure proportion in the population is also known, as might be the case in GWAS where minor allele frequencies (MAFs) are oftentimes known for SNPs in different populations, we can test for the presence of Neyman’s bias by examining their discrepancy. We develop one possible hypothesis test below where, again, H0 is ORtr(t*) = ORob(t*), and Ha is the complement of H0.
If we make an assumption of P(D < Md) = 1, then in comparing ORtr(t*) and ORob(t*), we see that their equivalence depends on
| (3) |
if and only if
So define p2(t*) ≡ P(G = 1|D < t*) = P(G = 1 | Case at t*). Then defining
we have p2(t*) = h(t*)/(1 + h(t*)), and defining
then we have ≡ P(G = 1 | Case at t*,Not censored from disease by t*) = h*(t*)/(1 + h*(t*)). When equation 3 does not hold,
Thus, if bias is present so that ORtr(t*) ≠ ORob(t*), then h(t*) ≠ h*(t*), and it will follow that . This idea can be leveraged in a hypothesis test if there is external knowledge of the population exposure proportion and population prevalence of disease.
By definition of , its estimator, , is the observed exposure proportion among cases where . Let p1(t*) ≡ P(G = 1 | D > t*, Md > t*), and since P(D < Md) = 1 by assumption, p1(t*) = P(G = 1 | D > t*) = P(G = 1 | Control at t*). Then is the observed exposure proportion among controls, and .
We will estimate with . Also, define p*(t*) ≡ P(Case at t*) = P(D < t*), which implies (1 p*(t*b)) = P(Control at t*) =b P(D > t*). So p*(t*) is the population prevalence of disease at a common age t* and is considered fixed and known. Since
if , which indicates that ORob(t*) = ORtr(t*), then pN(t*) = P(G = 1). Since we consider P(G = 1) fixed and known, the discrepancy between and P(G = 1) will inform our test.
Define . Then
So P(G = 1) and pN(t*) differ by δ(t*)p*(t*). The variance associated with our estimate of the exposure proportion is
where n2 is the number of cases and n1 the number of controls. We can estimate v with and and call the quantity . So, using a large sample approximation, we can construct an 𝛼 level hypothesis test for the presence ofb Neyman’s bias by rejecting for
The power becomes
assuming one tail probability negligible. We see that power decreases as p*(t*) decreases and increases with δ(t*), interpreted as the “degree of Neyman’s bias.”
Power curves for Test 3 are shown in Fig. 5. Consistent with our understanding of the test, power increases as p*(t*) increases. The type 1 error rate of the test is 0.05. The x-axis of Fig. 5, relative probability of observation, is defined as ; the x-axis begins at one and values greater than one imply that exposed cases are less likely to be sampled than unexposed cases because exposed subjects get disease earlier. Curves were generated using 300 cases and 300 controls in each simulated study, and the population-level exposure proportion was 0.09. Power was calculated based on 4000 iterations at each of the 11 total relative probabilities seen on the x-axis. This entire procedure was done for disease prevalences, p*(t*), of 0.1, 0.2, and 0.3. We assume that there is no variation in t* so that p*(t*) is also fixed.
Figure 5:

Power for test 3 as a function of p*(t*) and the relative probability of observing the unexposed cases versus exposed cases. As the relative probability increases (i.e., it is more likely to observed unexposed cases than exposed cases) as is the case when there are a greater number of mortality-inducing events among the exposed, there is more bias and power. The solid, dashed, and dotted lines represent population prevalences of disease (p*(t*)) of 0.1, 0.2, and 0.3, respectively.
6. Data analysis.
For illustration we apply our three hypothesis tests to three different data sets. The data sets are used for demonstrative purposes of interpretation and applicability of the proposed tests. Because the tests developed above make diverse assumptions about the nature of the data available, we found data sets with very different characteristics such that we could apply each test.
In general we do not know whether underlying Neyman’s bias exists, which is why we propose our hypothesis tests in the first place. However, in the case of the data set used for Test 1, we know the underlying truth by virtue of the prospective design of the Framingham Heart Study. Neyman’s bias cannot be present in a prospective cohort study because all relevant observations are in fact present in the sample, and bias is driven by the fact that some relevant ones are not. The data set therefore serves as a helpful “negative control” of at least Test 1. Indeed, as described below, the hypothesis test is not significant indicating a lack of Neyman’s bias.
On the other hand, in the cases of tests 2 and 3 applied to two additional data sets, we do not know whether Neyman’s bias is in fact present. The high mortality of both conditions, brain tumor and stroke, respectively, in those data sets makes the presence of Neyman’s bias more likely than if mortality were to occur more slowly after those outcomes. This expectation is a direct result of Theorem 1, as we describe in Section 3 above: holding all else equal, we expect greater bias with a higher hazard function of death.
For these reasons, it is not unexpected for Neyman’s bias to be present in both of these data sets and so they are good ones on which to test. Consistent with our reasoning, we reject both hypothesis tests as written below, indicating that Neyman’s bias is likely present.
6.1. Test 1 applied to an atrial fibrillation data set.
We apply Test 1 to the Framingham Heart study, a longitudinal study where we would not expect to find evidence of Neyman’s bias; subjects would not be lost to high-mortality diseases by nature of the study design. We would hope that the null hypothesis for Test 1 would not be rejected for each exposure stratum. We consider our exposure to be gender and the disease atrial fibrillation. Bear in mind we test not whether there is an association between gender and atrial fibrillation, but whether there would be bias in such an association. Among the cohort, there were 82 males and 188 females who had had atrial fibrillation and died of a related cause. Z-statistics calculated using the methodology of Test 1 for the male and female strata are 0.84 and 0.68, respectively, with associated p-values of 0.20 and 0.25. We do not reject the null hypothesis for either exposure stratum. The interpretation of this result is that, were we to calculate an odds ratio for atrial fibrillation and gender at some set age, we would not expect to encounter bias. Again, however, the test is used solely for illustration in this case because the study is prospective and not subject to the bias.
6.2. Test 2 applied to a brain tumor data set.
We apply Test 2 to a brain tumor data set. Seventy-five subjects with oligodendroglioma, a malignant brain tumor, were enrolled in a study at the London Regional Cancer Centre from 1984–1999 (Betensky et al., 2003; Ino et al., 2001). The data set consisted of patient age at diagnosis of oligodendroglioma (i.e., age at disease, D) and age at start of chemotherapy (i.e., entry into the study, T*) in addition to genetic markers and other covariates. We consider the marker at the 1pLOH locus, thought to be potentially associated with tumor sensitivity to chemotherapy. Applying Test 2 to the data set, first within the exposed stratum of the 1pLOH marker, we obtain a Z-statistic of 6.85, significant at the 0.05 level (p < 0.001). The sample size was insufficient to apply the test to the unexposed stratum. However, since a significant test statistic within any stratum is sufficient for rejection of the null hypothesis, we reject the null hypothesis of D ⫫ Md | G and conclude that there could be an association between D and Md within strata of G. The result of the test suggests that if one were to calculate an odds ratio of oligodendroglioma for the 1pLOH marker for subjects at a fixed age, the result is likely biased.
Consistent with the comparability criterion of Test 1 being more strict than that for Test 2, the sample size of 75 subjects was insufficient to additionally use Test 1. Had it been used, conclusions drawn from it would not have changed a lack of faith put on the odds ratio in this data set due to results from Test 2.
6.3. Test 3 applied to a stroke-mortality data set.
We apply Test 3 to a GWAS data set of ischemic stroke coming from a cohort based at Massachusetts General Hospital consisting of 383 cases and 384 controls. We use a wide interval estimate of ischemic stroke prevalence, ranging from 0.5%−5%, based on a search of the stroke literature (Feigin et al., 2009; Johnston et al., 2009; CDC, 2012). With this range of p*(t*), we reconstruct what would be population exposure proportion, which is unbiased for the true population exposure proportion assuming that Neyman’s bias is not present. We calculate a test statistic based on the difference between the true population exposure proportion and our estimate of it, divided by an estimate of the standard error. Using a 0.0005 Bonferroni-adjusted significance level, we find that 42 of the 99 SNPs in the study suggest that Neyman’s bias may be present. The interpretation of this result is that any one of the odds ratios calculated for these 42 SNPs might be biased. We additionally perform a power calculation for this test using realized minor allele frequencies in the data set and generous estimates of both , δ(t*), and p*(t*). Doing so yields power calculations little above 𝛼, at 0.06, which we discuss below.
7. Discussion.
Test 2 with the brain tumor data suggests that Neyman’s bias may be present because the within exposure stratum association between D and T* suggests a within exposure stratum association of D and Md. However, we should restate that an association within strata does not necessary imply that bias is present; it is only when the D ⫫ Md | G holds that we can conclude that Neyman’s bias is not present. Additionally, the study design may contribute to a within stratum association between D and T* and so the authors suggest that more work is needed to form stronger conclusions regarding the potential presence of Neyman’s bias in this study.
As with the result from Test 2, the rejection of the null hypothesis of no Neyman’s bias in the stroke-mortality data by Test 3 needs confirmatory analyses. A primary concern is that if the population underlying the measurements in dbSNP, the source of our “true” population MAFs against which we compare the estimate, is significantly different than that composing the study subjects, the type 1 error could be inflated. Since, for many of the SNPs in the data set, the MAF among cases and the MAF among controls did not contain the population MAF, which should be the case as the sample size gets large, there is some evidence of different underlying populations. Another assumption that may not be satisfied is P(D < Md) = 1. While P(D < Md) = 1 is unlikely to ever be fully satisfied, ischemic stroke is an event with numerous comorbidities and so violations of the assumption may be too large for a valid test (Ostwald et al., 2006; Bots et al., 1997). Lastly, the description of Test 3 showed that the power for detection of bias goes to 𝛼 as the population prevalence of disease gets small. The implication of this result is that any bias detected when the population prevalence of disease ranges over a relatively small 0.5%−5% is more likely due to unsatisfied assumptions than genuine Neyman’s bias. The generous power calculation of 0.06 confirms this belief–it is unlikely that a large proportion of SNPs would have significant p-values, as we have, when there is littler power to detect the bias. It is more likely that the reference population minor allele frequencies are unreflective of the population in the MGH study, which is an assumption that must be satisfied for a valid test.
We did not use Test 1 on the brain tumor and stroke data sets because of an insufficient sample size and insufficient covariates, respectively. The sample size was insufficient in the brain tumor data set because the comparability criterion for Test 1 is more stringent than that for Test 2, so there are only a limited number of pairs of observations that can contribute to estimation of the necessary parameters, especially when overlap between the multivariate random variables (D T* Md)T is minimal. Thus, while Test 2 might be thought of as somewhat removed from testing D ⫫ Md | G because it tests D ⫫ T* | G as a proxy, one advantage of Test 2 over Test 1 is that there are fewer restrictions imposed by the comparability criterion, allowing for more flexible use of the data.
Supplementary Material
8. Acknowledgements and address.
The authors wish to thank Dr. Deborah Blacker for many helpful comments used in the preparation of this manuscript as well as Drs. Gregory Cairncross and David Louis for use of the brain tumor data. Dr. Guido Falcone provided invaluable support in the preparation of the ischemic stroke dataset. The MGH ischemic stroke dataset was supported by the American Heart Association/Bugher Foundation Centers for Stroke Prevention Research, the National Institute of Neurological Disorders and Stroke, the Deane Institute for Integrative Study of Atrial Fibrillation and Stroke, and the Keane Stroke Genetics Research Fund. Dr. Anderson is supported by a Clinical Research Training Fellowship from the American Brain Foundation. Prof. Rebecca Betensky is supported by the National Institutes of Health grant CA075971. Dr. Swanson was supported by the National Institutes of Health Training Grant T32 NS048005 while the work was completed at Harvard School of Public Health. His current address is Oslo universitetssykehus Ulleval, Oslo, NO 0407. His email address is dms866@mail.harvard.edu.
9. Appendix: direction of bias and examples.
We provide a theorem regarding the direction of Neyman’s bias under certain modeling assumptions and examples of when Neyman’s bias does or does not occur.
Theorem 1
If G is associated with D such that OR(t*) ≠ 1 , the distribution of D | (G = 0) and D | (G = 1) belong to the same location family, P(X > 0) = 1, P(X < t**) > 0 (where t** is defined as the time between t* and the first possible presence of disease among the exposed or unexposed), and X ⫫ (D G)T, then ORob(t*) ≠ ORtr(t*). Specifically, if D | (G = 0) is stochastically greater than D | (G = 1) (alternatively, stochastically less than) so that exposure is a risk factor for disease (alternatively, protective against disease), then ORob(t*) < ORtr(t*) (alternatively, ORob(t*) > ORtr(t*)).
Proof. Define 𝜕FD|G=0(x)/𝜕x = f0(x) and 𝜕FD|G=1(x)/𝜕x = f1(x), and suppose that f1(x) = f0(x − k) for some k positive, without loss of generality. Such a scenario corresponds to exposure being protective against disease, though below we will also consider it a risk factor. f1(x) and f0(x) are in the same location family. Define F(x) as the cumulative distribution function of X evaluated at x and remember F(0) = 0 and F(t*) > 0. Consider the two quantities:
which we call the “percent erosion” of and , respectively. Then
Since F(·) a cumulative distribution function and therefore increasing, we have
| (4) |
because at every “successive” 𝜕x in each integral, 1 − F(t* − (x+k)) ≥ 1 − F(t* − x) and there is some 0 < x < t* for which 1 − F(t* − (x + k)) > 1 F(t* − x). Thus, the “percent erosion” of f0(x) will always be greater than that of f1(x) = f0(x − k), which is intuitive since f1(·) is located to the right of f0(·) and thus subject to the corrosive effects of F(·) for less “time.” Then using the inequality in (4),
which implies that
since P(X > 0) implies P(Control, Exposed, Observed) = P(Control, Exposed) and P(Control, Unexposed, Observed) = P(Control, Unexposed). Again, these inequalities only hold when exposure is protective against disease. When exposure is a risk factor for disease and therefore shifts the mean age of disease onset to the left under the above assumptions,
using analogous results. So we see that the bias is not toward the null, but in a definite direction depending on model assumptions.
Example 1. Consider D | (G = 1) uniform on (0,2), D | (G = 0) uniform on (0,1), and X uniform on (0,3), inde-pendent of G. Clearly the distributions of disease for exposed and unexposed are not in the same location family in this case, and the model for X corresponds to disease-induced mortality necessarily occurring within 3 times units after disease, D. We need only consider cases when investigating the odds ratio since we assume P(X > 0) = 1, implying P(D < Md) = 1. Taking t* = 1,
So we have X independent of exposure status and time of disease-onset, as was the case above, but here ORob = ORtr.
Example 2. Consider again D | (G = 1) uniform on (0,2), and D | (G = 0) uniform on (0,1). However, consider X | (G = 1) uniform on (0,3) and X | (G = 0) with density fX|G=0(x) = 2/3(1 x)2 on [0,1 + (9/2)1/3]. Again, we need only consider cases when investigating potential bias of the odds ratio since we assume P(D < Md) = 1 so that controls are not subject to the bias-inducing mortality event. Taking t* = 1,
and so here we have no bias again.
Example 3. Assume the same models of D conditional on G, and suppose X | (G = 1) is uniform on (0,3) and X | (G = 0) has density fX|G=0(x) = 5/2(1 − x)4 on [0,1 + 21/5]. For the reasons given above, we again only consider cases for investigating the bias of the odds ratio. Taking t* = 1,
and so here we have bias.
Example 4. Take D | (G = 1) with density fD|G=1(x) = x2/4 on [0,121/3], D | (G = 0) with density fD|G=0(x) = x/3 [0,61/2]. Then let X | (G = 1) have density fX|G=1(x) = (2 x)2/4 on [0,2+41/3] and X | (G = 0) be uniform on [0,2]. As before, we need only consider cases when investigating the odds ratio since we assume P(D < Md) = 1 so that controls are not subject to the bias-inducing mortality event. Taking t* = 2,
Remember that P(Case, Exposed)/P(Case, Unexposed) = p/(1 − p) implies ORtr(t*) = 1 when P(D < Md) = 1, which is assumed from condition 3.
Example 5. On the other hand, we can obtain a biased odds ratio using the same conditional disease models as in the previous example and having X | (G = 1) with density fX|G=1(x) = (2 − x)2/4 on [0,2 + 41/3] and X | (G = 0) uniform on [0,2]. We again assume P(D < Md) = 1 from condition 3. Taking t* = 2,
References
- Anderson C, Nalls M, Biffi A, Rost N, Greenberg S, Singleton A, Meschia J, and Rosand J (2011). The effect of survival bias on case-control genetic association studies of highly lethal diseases. Circulation. Cardiovascular genetics, 4(2):188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin MD, Simon DK, and Betensky RA (2013). Computationally simple estimation and improved efficiency for special cases of double truncation. Lifetime data analysis, pages 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betensky R, Louis D, and Cairncross J (2003). Analysis of a molecular genetic neuro-oncology study with partially biased selection. Biostatistics (Oxford, England), 4(2):167–178. [DOI] [PubMed] [Google Scholar]
- Bots M, Hoes A, Koudstaal P, Hofman A, and Grobbee D (1997). Common carotid intima-media thickness and risk of stroke and myocardial infarction: the rotterdam study. Circulation, 96(5):1432–1437. [DOI] [PubMed] [Google Scholar]
- CDC (2012). Prevalence of stroke–united states, 2006–2010. MMWR, 61(20):379–382. [PubMed] [Google Scholar]
- Dawid AP (1979). Conditional independence in statistical theory. Journal of the Royal Statistical Society. Series B (Methodological), pages 1–31. [Google Scholar]
- Feigin V, Lawes C, Bennett D, Barker-Collo S, and Parag V (2009). Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet neurology, 8(4):355–369. [DOI] [PubMed] [Google Scholar]
- Fluss R, Mandel M, Freedman LS, Weiss IS, Zohar AE, Haklai Z, Gordon E-S, and Simchen E (2012). Correction of sampling bias in a cross-sectional study of post-surgical complications. Statistics in Medicine. [DOI] [PubMed] [Google Scholar]
- Hernan M and Robins J (in press). Causal Inference. Chapman and Hall/CRC. [Google Scholar]
- Hill G (2003). Neyman’s bias re-visited. Journal of Clinical Epidemiology, 56. [DOI] [PubMed] [Google Scholar]
- Hoeffding W et al. (1948). A class of statistics with asymptotically normal distribution. The Annals of Mathematical Statistics, 19(3):293–325. [Google Scholar]
- Ino Y, Betensky R, Zlatescu M, Sasaki H, Macdonald D, Stemmer-Rachamimov A, Ramsay D, Cairncross J, and Louis D (2001). Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clinical cancer research : an official journal of the American Association for Cancer Research, 7(4):839–845. [PubMed] [Google Scholar]
- Johnston S, Mendis S, and Mathers C (2009). Global variation in stroke burden and mortality: estimates from monitoring, surveillance, and modelling. Lancet neurology, 8(4):345–354. [DOI] [PubMed] [Google Scholar]
- Keiding N (1991). Age-specific incidence and prevalence: a statistical perspective. Journal of the Royal Statistical Society. Series A (Statistics in Society), pages 371–412. [Google Scholar]
- Keiding N (2006). Event history analysis and the cross-section. Statistics in medicine, 25(14):2343–2364. [DOI] [PubMed] [Google Scholar]
- Martin E and Betensky R (2005). Testing quasi-independence of failure and truncation times via conditional kendall’s tau. Journal of the American Statistical Association, 100. [Google Scholar]
- Neyman J (1955). Statistics; servant of all sciences. Science (New York, N.Y.), 122(3166):401–406. [DOI] [PubMed] [Google Scholar]
- Ostwald S, Wasserman J, and Davis S (2006). Medications, comorbidities, and medical complications in stroke survivors: the cares study. Rehabilitation nursing : the official journal of the Association of Rehabilitation Nurses, 31(1):10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman K, Greenland S, and Lash T (2008). Modern epidemiology. Lippincott Williams and Wilkins. [Google Scholar]
- Sackett D (1979). Bias in analytic research. Journal of chronic diseases, 32(1–2):51–63. [DOI] [PubMed] [Google Scholar]
- Tsai W (1990). Testing the assumption of independence of truncation time and failure time. Biometrika, 77(1):169–177. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.