

IncFIIK plasmids are associated with the acquisition and dissemination of multiple-antimicrobial resistance in Klebsiella pneumoniae and often encountered in clinical isolates of this species. Since the phylogeny and evolution of IncFIIK plasmids remain unclear, here we performed large-scale in silico typing and comparative analysis of these plasmids in publicly available bacterial/plasmid genomes.
KEYWORDS: plasmid, replicon, conjugation, evolution
ABSTRACT
IncFIIK plasmids are associated with the acquisition and dissemination of multiple-antimicrobial resistance in Klebsiella pneumoniae and often encountered in clinical isolates of this species. Since the phylogeny and evolution of IncFIIK plasmids remain unclear, here we performed large-scale in silico typing and comparative analysis of these plasmids in publicly available bacterial/plasmid genomes. IncFIIK plasmids are prevalent in K. pneumoniae, being found in 69% of sequenced genomes, covering 66% of sequenced STs (sequence types), but sparse in other Enterobacteriaceae. IncFIIK replicons have three lineages. One IncFIIK allele could be found in distinct K. pneumoniae STs, highlighting the lateral genetic flow of IncFIIK plasmids. A set of 77 IncFIIK plasmids with full sequences were further analyzed. A pool of 327 antibiotic resistance genes or remnants were annotated in 75.3% of these plasmids. Plasmid genome comparison reiterated that they often contain other replicons belonging to IncFIA, IncFIB, IncFIIYp, IncFIIpCRY, IncR, IncL, and IncN groups and that they share a conserved backbone featuring an F-like conjugation module that has divergent components responsible for regulation and mating pair stabilization. Further epidemiological studies of IncFIIK plasmids are required due to the sample bias of K. pneumoniae genomes in public databases. This study provides insights into the evolution and structures of IncFIIK plasmids.
INTRODUCTION
Multiple-antibiotic resistance in bacteria has emerged as an urgent threat to human health (1, 2). Plasmids are important factors that promote the adaptive evolution of bacterial hosts. The IncF plasmid family plays a critical role in the dissemination of antibiotic resistance in Enterobacteriaceae (3). In particular, the IncFIIK plasmids are usually found to be associated with the worldwide dissemination of extended-spectrum β-lactamases (ESBLs) and carbapenemases in Klebsiella pneumoniae clinical isolates. For example, the ESBL gene blaCTX-M-15 and the carbapenemase gene blaKPC in K. pneumoniae are frequently found on IncFIIK plasmids (4–11).
IncFII is a major plasmid incompatibility group recognized in Enterobacteriaceae (12). The IncFII replicon (replication region) is regulated by an antisense-RNA, CopA, that inhibits the synthesis of the replication protein, as exemplified by plasmid Rl from Escherichia coli (13). IncFIIK replicons are a group of homogeneous members of the divergent IncFII replicons (3) that were initially defined according to the phylogenetic relatedness of IncFII replicons. All early members (plasmids pKPN3, pKPN4, pKP3-94, pKP91, and pGSH500) are from K. pneumoniae. To date, IncFIIK plasmids are identified mostly in K. pneumoniae and are important due to their role as a reservoir for antibiotic resistance genes in clinical isolates. IncFIIK plasmids commonly possess multiple replicons. IncFIIK replicons are often associated with FIB replicons, with types FIBpKPQIL and FIBpKPN being found more frequently, which also distinguish two prevalent K. pneumoniae plasmid types, pKPQIL and pKPN3, respectively (14). There have been 17 known alleles of the IncFIIK locus (https://pubmlst.org/plasmid/). IncFIIK plasmids are usually large (over 100 kb) but heterogeneous in size and possess mosaic structures. Many antibiotic resistance IncFIIK plasmids are capable of conjugation, which is attributed to the homologous transfer regions that they carry (4, 5, 15–18).
IncFIIK plasmids can be recognized based on the IncF replicon sequence typing (RST) scheme (3). Plasmid typing is a powerful tool to study the molecular epidemiology of transferable plasmids. A PCR-based replicon typing (PBRT) scheme was initially developed to detect the major incompatibility groups in Enterobacteriaceae (19), but it did not detect IncFIIK plasmids. The further-developed RST scheme solves the problems associated with the lack of resolution of the former PBRT scheme for typing IncF plasmids (3). A commercial PBRT kit has been devised based on the upgraded PBRT scheme (20). The recent Web tools PlasmidFinder and pMLST (plasmid multilocus sequence typing) allow plasmid replicon analysis and typing of high-throughput sequencing data (21). In addition, a degenerate primer MOB typing method targeting relaxase genes instead of replicons has also been proposed to classify gammaproteobacterial plasmids (22). Although the typing schemes and methods are evolving, the IncF RST scheme was specifically designed for IncF plasmids. PlasmidFinder, which contains in silico plasmid replicon probes (sequences specific for plasmid replicons), and pMLST, which contains IncF RST locus alleles for subtyping, provide resources for the in silico typing of IncFIIK plasmids.
With the increase in the amount of whole-genome/plasmid sequencing data, there is a need for large-scale plasmid analysis, which currently is rare (23, 24). Comparative genomic studies of mobile genetic elements (MGEs) have provided insights into common structural features, diverse gene pools, biological traits, and their evolution (25, 26). In particular, studies are needed to understand the impact of IncFIIK plasmids on the phylogeny and evolution of K. pneumoniae strains and the expansion of particular multidrug-resistant strains.
In this study, we performed in silico typing and comparative analysis of IncFIIK plasmids using bacterial genome and plasmid sequences available in the NCBI database, based on the IncF RST scheme. We analyzed the distribution of IncFIIK plasmids and compared the IncFIIK alleles, the conserved conjugation machineries (backbones), and the profiles of resistance determinants among IncFIIK plasmids. This study provides important insights into the phylogeny and evolution of IncFIIK plasmids and further addresses their role in the acquisition and spread of resistance genes.
RESULTS
In silico typing of IncFIIK plasmids.
By using the pMLST and PlasmidFinder tools (see Fig. S1 and S2 in the supplemental material), the genome sequences of 1,441 unique strains spanning 19 Klebsiella species (Data Set S1) were analyzed. We identified 1,069 IncFIIK replicons in 918 (63.7%) genomes (Data Set S2 and Table S1). Specifically, out of the 1,258 K. pneumoniae genomes covering 226 known STs (sequence types) (Data Set S3), 868 (69.0%) that fall into 148 STs (65.5%) had IncFIIK replicons detected. This ratio was higher than the average for other Klebsiella species. In the 79 complete Klebsiella genomes, we found that 21.8% (44/202) of the plasmids belonged to the IncFIIK group. Among the 110 fully sequenced Klebsiella plasmids that were not in the NCBI genome database (Data Set S1), 27 (24.5%) were typed as IncFIIK plasmids (Data Set S4), which were all from K. pneumoniae strains. In non-Klebsiella bacteria, only six IncFIIK plasmids were found (Data Set S4).
Divergent evolution and distribution of IncFIIK alleles.
Besides the 17 IncFIIK alleles already known, we uncovered 16 new alleles (Fig. 1). Single nucleotide variations (SNVs) accounted for the majority of the variations of these alleles. Deletion and insertion events were also found. These alleles fell into three groups, according to the phylogenetic analysis (Fig. 1; see also Fig. S3 in the supplemental material). We also counted the occurrence of each allele and found that K1, K2, and K5 were the three most detected ones (Fig. 1). Interestingly, nearly all IncFIIK types with >5 occurrences in the studied K. pneumoniae genomes were widespread in different STs (Data Set S2 and Fig. S4). For example, IncFIIK1, IncFIIK2, and IncFIIK5 replicons were found in 32, 51, and 35 K. pneumoniae STs, respectively, out of the 226 sequenced STs. IncFIIK13 replicons were the only exception, as they were all found in one K. pneumoniae ST, ST101 (Data Set S2), but note that ST101 strains also contained other IncFIIK replicons.
FIG 1.

Variations, phylogenetic tree, and occurrences of IncFIIK alleles. On the left is the phylogenetic tree of all IncFIIK alleles. The tree was constructed by using the neighbor-joining method with 1,000 bootstrap replicates in MEGA5. The topology was also supported by the maximum likelihood method (see Fig. S3 in the supplemental material). Colors indicate different allele groups and their mapping to PlasmidFinder probes. Percentages next to the brackets are nucleotide identities between the alleles and the best-matching probes. In the middle is the variation panel of the corresponding alleles. Allele K1 is the reference allele. Numbers on the top are positions within the reference allele. Alleles with names in red are the ones already known before this study. On the right are the occurrences of the corresponding alleles on a logarithmic scale. Colors indicate different data sources. Alleles with incomplete sequences were unclassified. Kp, K. pneumoniae.
Characteristics of IncFIIK plasmids.
Given that the considerable gaps in draft genomes did not allow recognition of complete plasmids for full-content comparison, we included the 77 IncFIIK plasmids with full sequences for further analyses (see Data Set S4 in the supplemental material). These plasmids had various lengths (21 to 339 kb; median, 142 kb) and GC contents (48.8% to 56.3%; median, 52.7%) (Fig. 2A and B, respectively) and commonly possessed multiple replicons (89.6%; 69/77) (Fig. 2C and Table S2), which was consistent with previous findings (14). The additional recognizable replicon types included IncFIA, IncFIB, IncFIIYp, IncFIIpCRY, IncR, IncL, and IncN. Note that many of the 77 complete plasmids are linked to clonal complex (CC) 258 strains, and the frequencies of the replicons observed may be biased by the samples. Nevertheless, The IncFIIK replicons have often been found to be associated with IncFIB replicons, especially the IncFIBpKPQIL and IncFIBpKPN types (14). In addition, we did not find that the plasmid length was correlated with replicon number (r2 = 0.165) (Fig. 2D).
FIG 2.
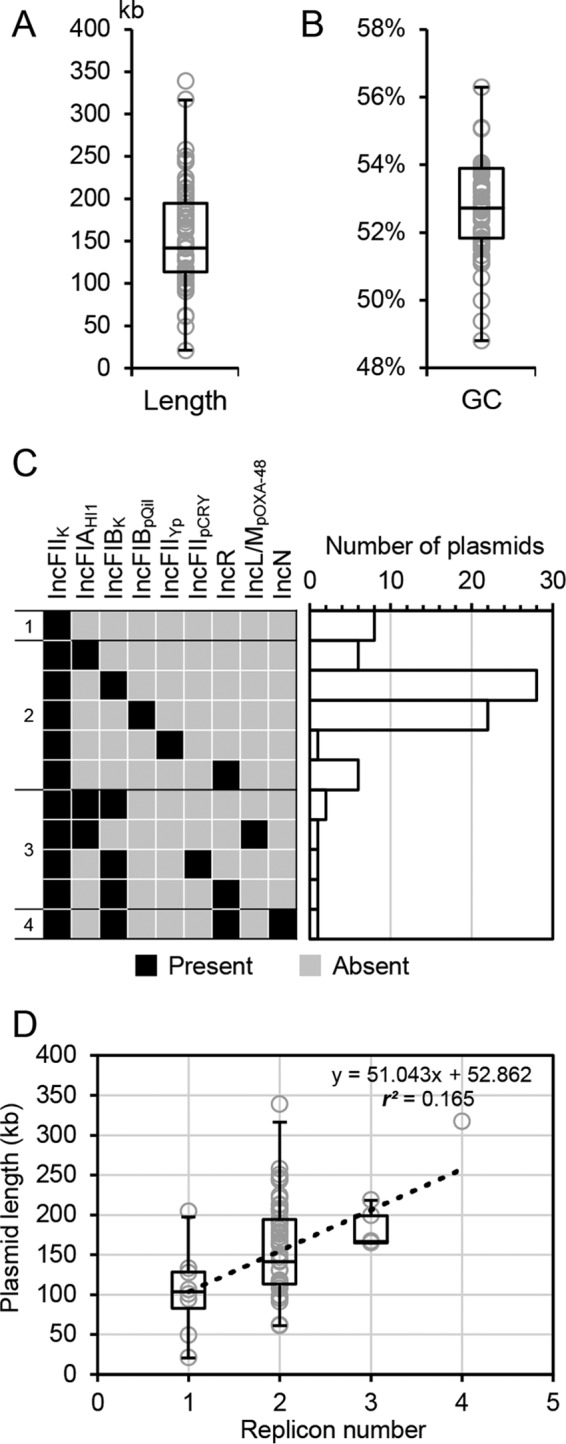
Characteristics of the IncFIIK plasmids. (A) Box plot of the length distribution. (B) Box plot of the GC content distribution. (C) Distribution of different multireplicon plasmids. The matrix on the left shows different multireplicon profiles. Numbers by the left are replicon numbers. The histograms on the right show the numbers of plasmids with the corresponding profiles. (D) Plot of plasmid length by replicon numbers. Box plots of the length distributions by replicon numbers are also given. The dotted line indicates linear fitting. The equation and coefficient of determination (r2) are shown.
IncFIIK plasmids were known carriers of antibiotic resistance genes. Consistently, we identified 327 antibiotic resistance genes or remnants from 75.3% of the studied IncFIIK plasmids (Data Set S5), covering nearly 50 unique genes that encode resistance to a wide spectrum of antibiotics (Fig. 3). Interestingly, among the diverse resistance gene combinations observed, five common clusters could be identified (Fig. 3A), each representing plasmids with specific combinations of different types of genes.
FIG 3.

Antibiotic resistance gene profiles of the IncFIIK plasmids. (A) Profiles of intact, truncated, and/or inactivated resistance genes. On the top are the classes of antibiotics denoted by different colors and the corresponding resistance genes. “Δ” indicates truncated genes. blaOXA-9* and ΔcatB3* in gray blocks and gray lettering are inactivated genes due to point mutations. The dendrogram on the left was generated by the correlation similarity metric and average linkage clustering. NA, not available. (B) Profiles of truncated resistance gene remnants. Plasmid names, GenBank accession numbers (in parentheses), IncFIIK types, K. pneumoniae STs, occurrences of each resistance gene (remnant) among these plasmids, and numbers of resistance genes (remnant) for each plasmid are shown. Plasmids that do not contain annotated resistance genes or remnants are not shown.
IncFIIK plasmids were also known to be plastic. We aligned the reference plasmid pKPHS2 (GenBank accession no. CP003224) (Fig. 4), which has a well-characterized genetic configuration (11), against each of the IncFIIK plasmids and plotted the coverage in function of the position to visualize the degree of conservation. Despite the highly variable nature of IncFIIK plasmids, the 35-kb conjugation module was found to be the most conserved, being shared by at least 87% of IncFIIK plasmids.
FIG 4.

Alignment plot of the IncFIIK plasmids against plasmid pKPHS2. pKPHS2 (GenBank accession no. CP003224) was used as a reference plasmid for alignment. Its gene organization (drawn to scale) and mobile genome are shown on the top. The conjugation module (transfer region) is denoted by green lines. The blue line in the chart below indicates the corresponding regions of pKPHS2 that occurred in how many plasmids (occurrence), while the orange line indicates how many hits of the corresponding region of pKPHS2 were found among the IncFIIK plasmids (coverage, or number of hits). The horizontal axis indicates the coordinates of pKPHS2. The red line highlights the total number of analyzed plasmids (77 plasmids). A zoom-in view of the conjugation module is shown at the bottom.
Evolution of the conserved F-like conjugation modules among IncFIIK plasmids.
Although the transfer regions (see Data Set S6 in the supplemental material) of these plasmids were overall syntenic, there were unconserved gaps and uneven coverage within (Fig. 4). We thus compared the components of the conjugation modules in detail (Fig. 5). The gene organization of the conjugation module was nearly identical to that of plasmid F, and the counterparts shared 60% to 70% amino acid identities (Fig. 5A and Fig. S5). Each plasmid contained one F-like conjugation module, except for pKPC-727, which contained two. Variations ranging from point mutations to a loss of the entire conjugation module were observed among IncFIIK plasmids (Fig. 5A). Most of the proteins involved in conjugation displayed >90% amino acid identities to their counterparts on pKPHS2. Gene truncation and inactivation due to point mutation, insertion sequence (IS) insertion, and disruption were seen. Some single genes were occasionally missing, such as trbF and traT. Truncation of the conjugation module occurred at a low frequency, being seen in only four modules. The ygfA-traMJYA regions were found to be missing in two pKPN-498 plasmids (identical but from different strains), while large-fragment deletions with only a few components left were observed in two other plasmids. The entire conjugation module was absent in six plasmids, two of which were from Enterobacter cloacae. These variations may affect or abolish the conjugative function in different ways. Most of the protein components were conserved, whereas TraM, TraJ, TraY, TraV, TraG, TraS, TrbE, and TrbF exhibited lower and various degrees of conservation (Fig. 5B). TraM, TraJ, and TraY are responsible for the regulation of the F-like type IV secretion system (T4SS) (27); TraV is a lipoprotein that anchors TraK as part of an envelope-spanning structure (28); and TraG and TraS are involved in entry exclusion (29), while TrbE and TrbF are nonessential components (28). The proteins constituting the conjugation module were generally conserved at a high level, with a few exceptions, which were found mainly in non-K. pneumoniae strains (Fig. 5C). The conjugation modules were roughly clustered into two groups based on the protein identity profiles (Fig. 5A). The clustering was consistent with and defined mainly by the homologies of TraM, TraJ, and TraV. No obvious clustering was observed in the phylogenetic tree of the DNA sequences of the transfer regions (Fig. S6). The transfer regions were 28 to 33 kb, with little variation in size (Fig. 5D). The IncFIIK replicons (denoted by the alleles) were located in the downstream vicinity of the tra gene clusters (Fig. 5D).
FIG 5.

Comparison of the conjugation modules of the IncFIIK plasmids. (A) Homology matrix of the conjugation proteins. The color gradient indicates identities at the amino acid level to pKPHS2 proteins. The profile of plasmid F (in red) is shown at the bottom of the matrix. The dendrogram on the left was drawn by the correlation similarity metric and average linkage clustering. Plasmid names, GenBank accession numbers (in parentheses), IncFIIK types, K. pneumoniae STs, occurrences of each gene among these plasmids, and numbers of component genes of each module are shown. Red lettering indicates that the two conjugation modules are from the same plasmid, pKPC-727, which are distinguished by the letters “a” and “b.” Plasmids in the gray blocks are from non-K. pneumoniae strains. NA, not available. (B) Box plot of the identity distribution of each type of protein. The corresponding median and average values are shown on the top, using the same color gradient scheme. (C) Box plot of the identity distribution of proteins constituting the conjugation modules. The corresponding median and average values are shown, using the same color gradient scheme. (D) Positions of the conjugation modules (green or orange, defined by tra cluster orientations) and IncFIIK loci (red, regardless of orientation) on these plasmids (gray).
We then constructed phylogenetic trees of the polymorphic components. The phylogenetic trees of TraM, TraJ, and TraY were similar, and each tree exhibited four major branching patterns, while within each branch, the components were rarely divergent (Fig. 6A). Remarkably, the set of conjugation modules under a major branch of the TraM tree also constituted a major branch of the TraJ or TraY tree. Similar features were found in the phylogenetic trees of TraG and TraS (Fig. 6B). The TraG and TraS trees were different from those of TraM/TraJ/TraY, and each tree exhibited five major branching patterns. TraV, TrbE, and Trb displayed distinct phylogenetic patterns (Fig. S7), suggesting that they might have evolved independently.
FIG 6.

Comparison of selected polymorphic components and oriT of the F-like conjugation modules. (A) Phylogenetic trees of the T4SS regulators TraM, TraJ, and TraY. (B) Phylogenetic trees of TraG and TraS that are involved in entry exclusion. Major branches representing the same sets of conjugation modules in different trees are denoted by identical colors. The trees were constructed by using the neighbor-joining method with 1,000 bootstrap replicates in MEGA5. Plasmids with same names are discriminated by their GenBank accession numbers (in parentheses). The letters “a” and “b” are used to distinguish the two conjugation modules on plasmid pKPC-727, as described in the legend of Fig. 5. (C) Consensus logo of the oriT sequences of IncFIIK plasmids aligned with oriT of plasmid F (with a double-strand sequence). Asterisks denote 100% conserved residues. The height of the letters indicates the degree of conservation of each base. Bits indicate base frequencies on a log2 scale. Vertical lines indicate identical bases between the IncFIIK plasmids and plasmid F. Half-arrows indicate inverted repeats.
The consensus of oriT (origin of transfer) among these conjugation modules was also observed (Fig. 6C). The oriT loci were homologous to oriT of plasmid F. There are two sets of inverted repeats flanking the nic site on plasmid F, whereas on IncFIIK plasmids, one set was degenerated due to SNVs.
DISCUSSION
We have performed large-scale in silico typing and analyses of IncFIIK plasmids to elucidate IncFIIK replicon phylogeny and plasmid structural features. IncFIIK plasmids are prevalent in K. pneumoniae but sparse in other Enterobacteriaceae and almost limited to Klebsiella spp., highlighting that IncFIIK plasmids are strictly narrow-host-range plasmids.
IncFIIK replicons were relatively heterologous, consistent with the diversity observed with the IncFII replicons (3, 13, 30). It has been suggested that the evolution of the IncFII replicons is promoted to broaden compatibility with incoming IncF plasmids (3, 31). Moreover, the IncFIIK alleles can be divided into three lineages with different variation signatures. The PlasmidFinder probes were capable of discriminating these lineages, as one lineage is closely related to probe IncFII(K)_1, while the other two are closely related to probe IncFII_pKP91. Alleles of the IncFII(K)_1 lineage were detected more frequently than those of the IncFII_pKP91 lineages, suggesting that plasmids with the IncFII(K)_1 replicons may have wider dissemination and/or be more adaptive under selective pressure. However, this result may reflect the IncFIIK plasmid distribution only under certain circumstances because there is an inevitable bias in isolation origin in the current genome database (e.g., more clinical isolates than environmental ones or more resistant isolates than susceptible isolates). Indeed, in silico multilocus sequence typing (MLST) showed that the public database contained a large number of CC258 (especially ST258 and blaKPC-associated) genomes (see Data Set S3 in the supplemental material), which might have resulted in overrepresented IncFIIK1 and IncFIIK2 lineages. Further molecular epidemiological investigations of IncFIIK plasmids are required due to the sample bias in the database used, especially by prevalent clinical clones and/or clones from outbreaks. Nevertheless, we have observed that most IncFIIK types are widespread in different K. pneumoniae STs, strongly highlighting the lateral genetic exchange of IncFIIK plasmids in K. pneumoniae.
We further analyzed the structures of IncFIIK plasmids. The IncFIIK plasmids often carry other IncF replicons. However, using current data, we did not find any replicon combination that was correlated with particular ST clones, further suggesting that there might have been genetic exchange between different clones. The multireplicon status has been believed to be a mechanism to broaden the host range by the acquisition of broad-host-range replicons (31) and/or to increase the compatibility of the incoming plasmids with incompatible replicons by using alternative replicons for replication (32). The reason why multireplicon IncFIIK plasmids are still of a narrow host range is likely because most of the additional replicons also belong to the narrow-host-range IncF group. Whether the IncFIIK replicons in the multireplicon plasmids are functional is unclear, but it has been known that IncFII replicons do not participate in the initiation of replication when associated with FIA and/or FIB replicons, consistent with their diversity (3). The diversity of IncFIIK replicons implies that they are usually not responsible for the control of plasmid replication. Indeed, in pKP048, an IncFIIK5 plasmid, the IncR replicon is believed to control replication (4). The number of coexisting replicons does not correlate with the plasmid length. Plasmids are highly mosaic, the contents of which are shaped by various mobile genetic elements (MGEs) with diverse genetic events, resulting in variable plasmid sizes.
A vast pool of diverse antibiotic resistance genes were found among IncFIIK plasmids. Multiresistance regions (MRRs) are usually modular and mosaic (33), but we found that the presence of antibiotic genes may fall into different signature clusters, which could provide hints about the evolutionary trajectories of the MRRs on IncFIIK plasmids. These clusters are not necessarily associated with IncFIIK types. For example, the overrepresented IncFIIK1 and IncFIIK2 replicons spanned different clusters, implying that IncFIIK plasmids are open to diverse antibiotic resistance genes. Given that most of the resistant plasmids are from clinical isolates, our results reiterate the prominent role of IncFIIK plasmids as carriers of diverse antibiotic resistance genes in K. pneumoniae in clinical settings.
Whole-plasmid genome comparison suggests that IncFIIK plasmids are highly variable but commonly share an F-like conjugation module as their backbone. The highly variable regions usually experience extensive gene rearrangements and accommodate diverse resistance-associated ISs, integrons, and/or transposons (33). The conjugation module consisted of oriT, a gene coding for relaxase (TraI), and a tra gene cluster coding for a Tra_F type IV secretion system (T4SS). Many IncFIIK plasmids bearing this transfer region have been reported to be conjugative (4, 5, 15–18). It is very likely that the conjugation modules of IncFIIK plasmids have diverged from the same ancestor of plasmid F. The majority of the genes responsible for conjugation are highly conserved, while variation occurs in the genes responsible for regulation and mating pair stabilization. Although traM, traJ, and traY belong to different transcription units, they were located in tandem and seemed to have coevolved. As the key regulators of the T4SS (27), different types of TraM-TraJ-TraY variants may possess different regulatory capacities, resulting in different conjugation dynamics. The coevolution of the two genes traG and traS, involved in entry exclusion, may have an impact on mating pair stabilization (28). It is likely that genes participating in the shared function have coevolved. Meanwhile, the lack of similarity in the phylogenetic trees of those genes with different functions indicates that these conjugation modules may have gone through modular evolution. Whether plasmid-host interaction and/or coevolution plays a role in this variation awaits investigation. The results may suggest that there were equal selective pressures on these conjugation modules, while individual genes were subject to different evolutionary pressures (25).
The findings of our study will advance our understanding of the evolution and structures of IncFIIK plasmids and expand the application of plasmid typing. Further epidemiological studies of IncFIIK plasmids are required due to the current sample bias of K. pneumoniae genomes, especially overrepresented by clinical isolates of certain STs.
MATERIALS AND METHODS
Genomic data.
Data for complete and draft Klebsiella genomes were retrieved from the NCBI genome database (http://ftp.ncbi.nih.gov/genomes/). Genomes meeting RefSeq exclusion criteria (e.g., genomes containing many frameshifted proteins, a low contig N50, or missing rRNA genes or genomes that were too long) were excluded. For genome assembles of a repeatedly sequenced strain, the one with a higher or the highest assembly level was taken as applicable or otherwise taken randomly. From 1,524 sequenced genomes representing 20 Klebsiella species, 79 complete genomes and 1,362 draft genomes of 1,441 unique strains spanning 19 Klebsiella species were included for analysis (see Data Set S1 in the supplemental material). Among the completely sequenced strains, 62 contained 202 plasmids, while the others contained none (Data Set S1). Meanwhile, 110 fully sequenced Klebsiella plasmids that were not in the NCBI genome database (Data Set S1) were retrieved from the plasmid category of the NCBI RefSeq database (http://ftp.ncbi.nih.gov/refseq/release/plasmid/).
In silico multilocus sequence typing of K. pneumoniae genomes.
Allele sequences of the gapA, infB, mdh, pgi, phoE, rpoB, and tonB loci and multilocus sequence typing (MLST) profile definitions were downloaded from BIGSdb (http://bigsdb.pasteur.fr/) (34). Alleles from the K. pneumoniae genomes were identified with BLASTn, with criteria of 100% identity and 100% coverage. Sequence types (STs) were then defined according to the allele profiles by using an in-house Perl script.
In silico typing of IncFIIK plasmids in Klebsiella genome sequences.
Plasmid replicon probes were downloaded from PlasmidFinder (21), while IncFIIK alleles used for subtyping were obtained from pMLST (http://pubmlst.org/plasmid/). There were 121 replicon probes in PlasmidFinder and 17 IncFIIK alleles (K1 to K17) in pMLST when we conducted this study. Mega BLAST of the 17 alleles against the PlasmidFinder database and phylogenetic analysis of the alleles were performed to determine the best-matching probes and to define the corresponding identity thresholds (see Fig. S1 in the supplemental material).
A two-step in silico IncFIIK replicon typing approach was developed (Fig. S2). The first step was screening of IncFIIK or IncFIIK-like replicons using the PlasmidFinder probes, and the second step was cross-verification and subtyping using the collected IncFIIK alleles. Briefly, DNA sequences (plasmids/scaffolds/contigs) were subjected to Mega BLAST (E value, ≤0.001) against the PlasmidFinder database. Sequences probed by “IncFII(K)_1__CP000648” [IncFII(K)_1 for short] with identities of ≥90% or “IncFII_1_pKP91_CP000966” (IncFII_pKP91 for short) with identities of ≥80% as the best hits were selected. The probed regions with short flanks were then subjected to Mega BLAST (E value, ≤0.001; identity, ≥90%) against the IncFIIK alleles selected based on the probing results (Fig. S2). Plasmids with hits were defined as IncFIIK plasmids. Other replicons on these plasmids were typed by using other PlasmidFinder probes. Manual review was finally conducted. Given that IncFIIK replicons are conserved, the identity thresholds that we used were strict to ensure specificity. All in-house computer scripts in this study were written in Perl unless otherwise specified.
In silico typing of IncFIIK plasmids in non-Klebsiella bacteria.
IncFIIK plasmids in non-Klebsiella bacteria were identified from the NCBI nucleotide collection (nr) online with its own BLASTn program according to the described strategy (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The PlasmidFinder tool was used to identify other replicons in the analyzed plasmid sequences.
Annotation of antibiotic resistance genes.
Antibiotic resistance genes were annotated by using Mega BLAST (E value, ≤0.0001; identity, ≥70%) against the ResFinder database (35, 36), followed by manual review.
Comparative genomics of IncFIIK plasmids.
The IncFIIK2 plasmid pKPHS2 (GenBank accession no. CP003224) of the well-studied K. pneumoniae strain HS11286 (11, 37) was used as a reference plasmid for comparison. The genome of HS11286 is the representative genome of K. pneumoniae in the NCBI database, and pKPHS2 is a well-documented blaKPC-2 carrier (11). All fully sequenced IncFIIK plasmids were aligned with pKPHS2 by using Mega BLAST (E value, ≤0.001). The number of hits of different regions of pKPHS2 were counted to identify conserved regions. The conjugation module of each plasmid was compared with that of pKPHS2, gene by gene. Type IV secretion systems (T4SSs) were annotated by using SecReT4 (38) when needed. Putative oriT was predicted by BLASTn (E value, ≤0.001) against oriT of plasmid F (GenBank accession no. AP001918) (30).
Multiple-sequence alignment and phylogenetic analysis.
Multiple-sequence alignments of short DNA sequences (IncFIIK alleles and oriT) and protein sequences were performed with MUSCLE v3.7 (39), while those of long DNA sequences (regions of the conjugation module) were performed with Kalign v2.04 (40). The MEME suite was used to visualize consensus sequences where applicable (41). Inferred phylogenetic trees were constructed by using the neighbor-joining method and/or the maximum likelihood method with 1,000 bootstrap replicates in MEGA5 (42).
Dendrogram.
A dendrogram was generated by using the “heatplot” function of the R package made4 (43), which draws dendrograms using the correlation similarity metric and average linkage clustering.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China to D.B. (no. 81702037), Q.W. (no. 81472501), J.-J.L. (no. 81703567), and Z.-K.S. (no. 81603166) and from the 973 program, Ministry of Science and Technology, China (no. 2015CB554202), to H.-Y.O.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00764-18.
REFERENCES
- 1.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J Jr, Infectious Diseases Society of America. 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 2.Ventola CL. 2015. The antibiotic resistance crisis. Part 1: causes and threats. P T 40:277–283. [PMC free article] [PubMed] [Google Scholar]
- 3.Villa L, Garcia-Fernandez A, Fortini D, Carattoli A. 2010. Replicon sequence typing of IncF plasmids carrying virulence and resistance determinants. J Antimicrob Chemother 65:2518–2529. doi: 10.1093/jac/dkq347. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Y, Yu D, Wei Z, Shen P, Zhou Z, Yu Y. 2010. Complete nucleotide sequence of Klebsiella pneumoniae multidrug resistance plasmid pKP048, carrying blaKPC-2, blaDHA-1, qnrB4, and armA. Antimicrob Agents Chemother 54:3967–3969. doi: 10.1128/AAC.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leavitt A, Chmelnitsky I, Carmeli Y, Navon-Venezia S. 2010. Complete nucleotide sequence of KPC-3-encoding plasmid pKpQIL in the epidemic Klebsiella pneumoniae sequence type 258. Antimicrob Agents Chemother 54:4493–4496. doi: 10.1128/AAC.00175-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dolejska M, Brhelova E, Dobiasova H, Krivdova J, Jurankova J, Sevcikova A, Dubska L, Literak I, Cizek A, Vavrina M, Kutnikova L, Sterba J. 2012. Dissemination of IncFIIK-type plasmids in multiresistant CTX-M-15-producing Enterobacteriaceae isolates from children in hospital paediatric oncology wards. Int J Antimicrob Agents 40:510–515. doi: 10.1016/j.ijantimicag.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz-Garbajosa P, Curiao T, Tato M, Gijon D, Pintado V, Valverde A, Baquero F, Morosini MI, Coque TM, Canton R. 2013. Multiclonal dispersal of KPC genes following the emergence of non-ST258 KPC-producing Klebsiella pneumoniae clones in Madrid, Spain. J Antimicrob Chemother 68:2487–2492. doi: 10.1093/jac/dkt237. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Chavda KD, Melano RG, Jacobs MR, Levi MH, Bonomo RA, Kreiswirth BN. 2013. Complete sequence of a blaKPC-2-harboring IncFIIK1 plasmid from a Klebsiella pneumoniae sequence type 258 strain. Antimicrob Agents Chemother 57:1542–1545. doi: 10.1128/AAC.02332-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lohr IH, Hulter N, Bernhoff E, Johnsen PJ, Sundsfjord A, Naseer U. 2015. Persistence of a pKPN3-like CTX-M-15-encoding IncFIIK plasmid in a Klebsiella pneumonia ST17 host during two years of intestinal colonization. PLoS One 10:e0116516. doi: 10.1371/journal.pone.0116516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansour W, Grami R, Ben Haj Khalifa A, Dahmen S, Chatre P, Haenni M, Aouni M, Madec JY. 2015. Dissemination of multidrug-resistant blaCTX-M-15/IncFIIk plasmids in Klebsiella pneumoniae isolates from hospital- and community-acquired human infections in Tunisia. Diagn Microbiol Infect Dis 83:298–304. doi: 10.1016/j.diagmicrobio.2015.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Bi D, Jiang X, Sheng ZK, Ngmenterebo D, Tai C, Wang M, Deng Z, Rajakumar K, Ou HY. 2015. Mapping the resistance-associated mobilome of a carbapenem-resistant Klebsiella pneumoniae strain reveals insights into factors shaping these regions and facilitates generation of a ‘resistance-disarmed’ model organism. J Antimicrob Chemother 70:2770–2774. doi: 10.1093/jac/dkv204. [DOI] [PubMed] [Google Scholar]
- 12.Carattoli A. 2009. Resistance plasmid families in Enterobacteriaceae. Antimicrob Agents Chemother 53:2227–2238. doi: 10.1128/AAC.01707-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blomberg P, Nordstrom K, Wagner EG. 1992. Replication control of plasmid R1: RepA synthesis is regulated by CopA RNA through inhibition of leader peptide translation. EMBO J 11:2675–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navon-Venezia S, Kondratyeva K, Carattoli A. 2017. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev 41:252–275. doi: 10.1093/femsre/fux013. [DOI] [PubMed] [Google Scholar]
- 15.Sandegren L, Linkevicius M, Lytsy B, Melhus A, Andersson DI. 2012. Transfer of an Escherichia coli ST131 multiresistance cassette has created a Klebsiella pneumoniae-specific plasmid associated with a major nosocomial outbreak. J Antimicrob Chemother 67:74–83. doi: 10.1093/jac/dkr405. [DOI] [PubMed] [Google Scholar]
- 16.Huang TW, Chen TL, Chen YT, Lauderdale TL, Liao TL, Lee YT, Chen CP, Liu YM, Lin AC, Chang YH, Wu KM, Kirby R, Lai JF, Tan MC, Siu LK, Chang CM, Fung CP, Tsai SF. 2013. Copy number change of the NDM-1 sequence in a multidrug-resistant Klebsiella pneumoniae clinical isolate. PLoS One 8:e62774. doi: 10.1371/journal.pone.0062774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Chavda KD, Melano RG, Jacobs MR, Koll B, Hong T, Rojtman AD, Levi MH, Bonomo RA, Kreiswirth BN. 2014. Comparative genomic analysis of KPC-encoding pKpQIL-like plasmids and their distribution in New Jersey and New York hospitals. Antimicrob Agents Chemother 58:2871–2877. doi: 10.1128/AAC.00120-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berger S, Alauzet C, Aissa N, Henard S, Rabaud C, Bonnet R, Lozniewski A. 2013. Characterization of a new blaOXA-48-carrying plasmid in Enterobacteriaceae. Antimicrob Agents Chemother 57:4064–4067. doi: 10.1128/AAC.02550-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carattoli A, Bertini A, Villa L, Falbo V, Hopkins KL, Threlfall EJ. 2005. Identification of plasmids by PCR-based replicon typing. J Microbiol Methods 63:219–228. doi: 10.1016/j.mimet.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 20.Carloni E, Andreoni F, Omiccioli E, Villa L, Magnani M, Carattoli A. 2017. Comparative analysis of the standard PCR-based replicon typing (PBRT) with the commercial PBRT-KIT. Plasmid 90:10–14. doi: 10.1016/j.plasmid.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Carattoli A, Zankari E, Garcia-Fernandez A, Voldby Larsen M, Lund O, Villa L, Moller Aarestrup F, Hasman H. 2014. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alvarado A, Garcillan-Barcia MP, de la Cruz F. 2012. A degenerate primer MOB typing (DPMT) method to classify gamma-proteobacterial plasmids in clinical and environmental settings. PLoS One 7:e40438. doi: 10.1371/journal.pone.0040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamminen M, Virta M, Fani R, Fondi M. 2012. Large-scale analysis of plasmid relationships through gene-sharing networks. Mol Biol Evol 29:1225–1240. doi: 10.1093/molbev/msr292. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez-Lopez R, de Toro M, Moncalian G, Garcillan-Barcia MP, de la Cruz F. 2016. Comparative genomics of the conjugation region of F-like plasmids: five shades of F. Front Mol Biosci 3:71. doi: 10.3389/fmolb.2016.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wozniak RA, Fouts DE, Spagnoletti M, Colombo MM, Ceccarelli D, Garriss G, Dery C, Burrus V, Waldor MK. 2009. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet 5:e1000786. doi: 10.1371/journal.pgen.1000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanza VF, de Toro M, Garcillan-Barcia MP, Mora A, Blanco J, Coque TM, de la Cruz F. 2014. Plasmid flux in Escherichia coli ST131 sublineages, analyzed by plasmid constellation network (PLACNET), a new method for plasmid reconstruction from whole genome sequences. PLoS Genet 10:e1004766. doi: 10.1371/journal.pgen.1004766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frost LS, Koraimann G. 2010. Regulation of bacterial conjugation: balancing opportunity with adversity. Future Microbiol 5:1057–1071. doi: 10.2217/fmb.10.70. [DOI] [PubMed] [Google Scholar]
- 28.Lawley TD, Klimke WA, Gubbins MJ, Frost LS. 2003. F factor conjugation is a true type IV secretion system. FEMS Microbiol Lett 224:1–15. doi: 10.1016/S0378-1097(03)00430-0. [DOI] [PubMed] [Google Scholar]
- 29.Audette GF, Manchak J, Beatty P, Klimke WA, Frost LS. 2007. Entry exclusion in F-like plasmids requires intact TraG in the donor that recognizes its cognate TraS in the recipient. Microbiology 153:442–451. doi: 10.1099/mic.0.2006/001917-0. [DOI] [PubMed] [Google Scholar]
- 30.Frost LS, Ippen-Ihler K, Skurray RA. 1994. Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol Rev 58:162–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osborn AM, da Silva Tatley FM, Steyn LM, Pickup RW, Saunders JR. 2000. Mosaic plasmids and mosaic replicons: evolutionary lessons from the analysis of genetic diversity in IncFII-related replicons. Microbiology 146(Part 9):2267–2275. doi: 10.1099/00221287-146-9-2267. [DOI] [PubMed] [Google Scholar]
- 32.Bergquist PL, Lane HE, Malcolm L, Downard RA. 1982. Molecular homology and incompatibility in the IncFI plasmid group. J Gen Microbiol 128:223–238. [DOI] [PubMed] [Google Scholar]
- 33.Partridge SR. 2011. Analysis of antibiotic resistance regions in Gram-negative bacteria. FEMS Microbiol Rev 35:820–855. doi: 10.1111/j.1574-6976.2011.00277.x. [DOI] [PubMed] [Google Scholar]
- 34.Jolley KA, Maiden MC. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zankari E, Hasman H, Kaas RS, Seyfarth AM, Agerso Y, Lund O, Larsen MV, Aarestrup FM. 2013. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J Antimicrob Chemother 68:771–777. doi: 10.1093/jac/dks496. [DOI] [PubMed] [Google Scholar]
- 37.Liu P, Li P, Jiang X, Bi D, Xie Y, Tai C, Deng Z, Rajakumar K, Ou HY. 2012. Complete genome sequence of Klebsiella pneumoniae subsp. pneumoniae HS11286, a multidrug-resistant strain isolated from human sputum. J Bacteriol 194:1841–1842. doi: 10.1128/JB.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bi D, Liu L, Tai C, Deng Z, Rajakumar K, Ou HY. 2013. SecReT4: a Web-based bacterial type IV secretion system resource. Nucleic Acids Res 41:D660–D665. doi: 10.1093/nar/gks1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lassmann T, Frings O, Sonnhammer EL. 2009. Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res 37:858–865. doi: 10.1093/nar/gkn1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey TL, Johnson J, Grant CE, Noble WS. 2015. The MEME suite. Nucleic Acids Res 43:W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Culhane AC, Thioulouse J, Perriere G, Higgins DG. 2005. MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics 21:2789–2790. doi: 10.1093/bioinformatics/bti394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.