

Abstract
Cellular susceptibility to viral infections is in part determined by the presence of a host cellular receptor. Here we use murine norovirus as a model to uncover an unappreciated connection between an intracellular lipid biosynthetic enzyme and a receptor conformation permissive for viral infection. The serine palmitoyltransferase (SPT) complex is required for de novo sphingolipid biosynthesis and we find that its absence impairs the ability of murine norovirus to bind and enter cells. While, the SPT complex is dispensable for the surface expression of the norovirus receptor, CD300lf, SPT activity is required for CD300lf to adopt a conformation permissive for viral binding. Addition of extracellular ceramide to SPT deficient cells chemically complements both the conformational changes of CD300lf and the cellular susceptibility to murine norovirus infection. Taken together, these data indicate that intracellular sphingolipid biosynthesis regulates the conformation of the murine norovirus receptor, and therefore the tropism of murine norovirus. This indicates that intracellular biosynthetic pathways can regulate viral tropism even when the receptor for a virus is expressed on the target cell surface.
Introduction
Noroviruses (NoV) are non-enveloped, positive-stranded RNA viruses. Human noroviruses (HNoV) are a leading cause of gastroenteritis worldwide1. While HNoV infections are typically self-limiting, in immunocompromised patients, infections can be prolonged, severe, and even fatal2. Currently, there are no approved vaccines or antivirals for NoV therapy, largely owing to the difficulty in culturing HNoV in vitro and the strict species tropism of noroviruses that leads to a lack of robust replication of HNoV in small animal models3.
One barrier for NoV infection is at the step of viral entry as direct delivery of viral genomes into cells enables viral replication4–6. Previous work has shown that HNoV binds to histo-blood group antigens (HBGAs) and susceptibility to specific HNoV strains is correlated with host HBGA status, although HBGAs have proved unable to account for all aspects of HNoV tropism and entry7,8. Unlike HNoV, murine norovirus (MNoV) grows robustly both in vitro and in laboratory strains of mice3,9,10. We recently leveraged this robust replication to perform a whole-genome CRISPR screen to identify essential host genes required for MNoV replication. We identified the cell surface protein CD300lf as a proteinaceous receptor for MNoV5. CD300lf mediates binding of virus to the cell surface and is necessary for viral entry and replication in vitro and in vivo5. Importantly, expression of CD300lf in human cells is sufficient for MNoV to replicate in human cells5. These findings indicate that the intracellular replication machinery for NoV is conserved across species. However, it remains unclear what additional cellular factors cooperate with proteinaceous receptors to enable NoV entry.
In our initial screen for genes required for MNoV replication we reported that two genes, Sptlc1 and Sptlc2 were important for MNoV replication5. These proteins are essential members of the serine palmitoyltransferase (SPT) complex, which catalyzes the first and rate-limiting step in de novo ceramide and sphingolipid biosynthesis11. Ceramide and sphingolipids are important lipid mediators of membrane fluidity and dynamics12,13. Ceramide also can function as a signaling lipid involved in cellular survival, metabolic homeostasis, and inflammation14–16. However, the exact function of the SPT complex during NoV replication is unknown.
Here, we define a critical role for sphingolipid biosynthesis for MNoV infection and viral entry. Furthermore we determine that the step in viral infection at which de novo sphingolipid biosynthesis is required is the binding of the virus to cells, a step also requiring the MNoV receptor CD300lf. Surprisingly, Sptlc2 deficiency did not lead to abnormal trafficking or localization of CD300lf, but altered the conformation of CD300lf so as to not be recognized by MNoV or a conformation dependent antibody. Chemical complementation of the Sptlc2 deficient cells altered the CD300lf conformation and restored MNoV susceptibility. Taken together, these data demonstrate that the lipid composition of host cells is a critical determinant of MNoV entry and that intracellular enzymes can regulate the conformation of viral receptors, providing an additional mechanism to regulate viral tropism.
Results
de novo sphingolipid biosynthesis is required for MNoV cellular binding
The SPT complex consists of two proteins, Sptlc1 and Sptlc2. The Sptlc2 protein is the catalytic subunit while Sptlc1 is required for the stability of Sptlc2 in cells11. Therefore, we generated Sptlc2 deficient BV2 cells (BV2ΔSptlc2) using CRISPR/Cas9 technology (Supplementary Figure 1). We then tested the ability of MNoV strains that cause acute, systemic infection (MNoVCW3) or persistent, enteric infection (MNoVCR6) to replicate in Sptlc2 deficient cells. Whereas wild type BV2 cells produced high titers of MNoVCW3 and MNoVCR6 strains of virus, BV2ΔSptlc2 cells were deficient in MNoV production both after a single (12 hours) or multiple (24 hours) cycles of replication (Figure 1a). Importantly, the growth of both strains of MNoV was restored upon expression of Sptlc2 cDNA (Figure 1b). A mutation that abolished Sptlc2 catalytic activity, Sptlc2R507A, was unable to rescue MNoV replication in BV2ΔSptlc2 cells, indicating that the SPT complex must be enzymatically active in order to support MNoV replication (Figure 1b)17. Also, treatment of BV2 cells with myriocin, a potent inhibitor of the SPT enzyme, reduced the number of MNoV infected cells (Figure 1c).
Figure 1: de novo ceramide biosynthesis is required for efficient MNoV infection, binding, and entry.
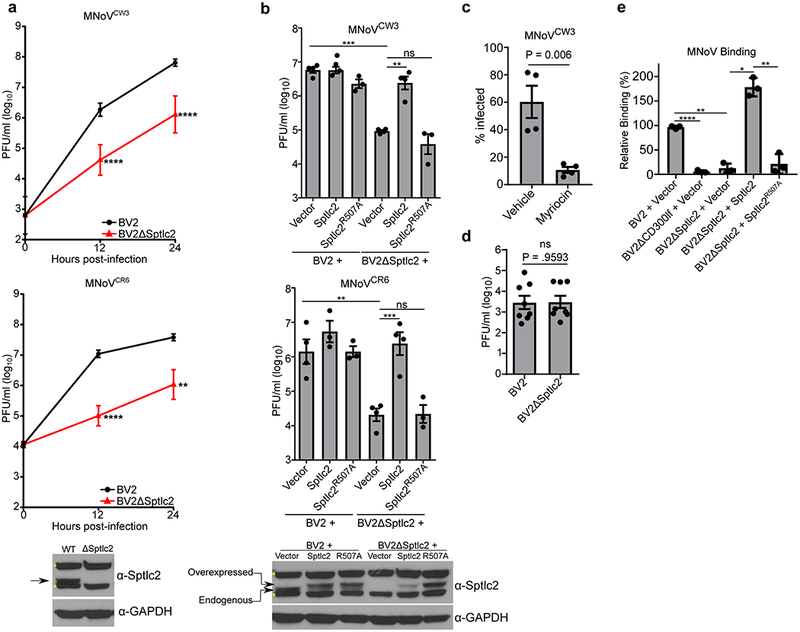
(a) BV2 or BV2ΔSptlc2 cells were challenged with MNoV at a multiplicity of infection (MOI) of 0.05 with strains MNoVCW3 (Top) or MNoVCR6 (Bottom). Viral production was measured using plaque assay (PFU; plaque forming units) at indicated time points. Time point 0 represents input inoculum. Below is a representative western blot for indicated proteins. Arrow indicates expected molecular weight and asterisks denote non-specific bands. Data are shown as means ± SEM from three independent experiments and data were analyzed by unpaired two-sided t-test. **p<0.01, ****p<0.0001.
(b) Wild type BV2 or BV2ΔSptlc2 cells expressing either an empty vector, Sptlc2, or Sptlc2R507A were challenged with MNoV at a multiplicity of infection (MOI) of 0.05 with strains MNoVCW3 (top) or MNoVCR6 (bottom). Viral production was measured using plaque assay at 12 hours post-infection. Results are shown are means ± SEM from three independent experiments and data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Below is a representative western blot from complementation experiment in for indicated proteins. Arrow indicates expected molecular weight and asterisks denote non-specific bands.
(c) MNoVCW3 infection of BV2 cells treated with vehicle (Methanol) or DMSO or Myriocin (25 μm) 24 hours prior to challenge. Infection was measured by FACS for intracellular production of VP1. Data are shown as means ± SEM from four independent experiments and data were analyzed by unpaired two-sided t-test
(d) BV2 or BV2ΔSptlc2 cells were transfected with viral RNA from MNoVCW3 and harvested 12 hours post-transfection. Viral production was measured by plaque assay. Data are shown as means ± SEM from three independent experiments and data were analyzed by unpaired two-sided t-test; ns = not significant
(e) Indicated cell lines were assayed for MNoVCW3 binding using quantitative polymerase chain reaction and normalized to the median of BV2 + Vector for each experiment. Data are shown as means ± SEM from three independent experiments and data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Having established a step in sphingolipid biosynthesis as required for efficient MNoV replication, we next identified the stage of the viral life cycle that is impaired in BV2ΔSptlc2 cells. Equivalent levels of infectious virus were produced after transfection of MNoVCW3 viral RNA into wild type and BV2ΔSptlc2 cells indicating that sphingolipid biosynthesis is required for MNoV entry (Figure 1d). We then tested the ability of MNoVCW3 to bind to cells defective in sphingolipid biosynthesis. Wild type BV2 cells had significantly enhanced binding compared to both BV2ΔCD300lf and BV2ΔSptlc2 cells (Figure 1e). This binding defect in BV2ΔSptlc2 cells was rescued by expression of a wild type Sptlc2 construct but not a catalytically inactive Sptlc2 (Figure 1e). These findings are consistent with a model in which Sptlc2 is required for efficient binding and entry of cells while post-entry replication of MNoV is not effected.
Sptlc2 deficiency alters the conformation of CD300lf but not the surface localization.
As CD300lf expression is essential for MNoV binding, we next tested the hypothesis that Sptlc2 is required for cell surface expression of this viral receptor. The amount of CD300lf on the surface of BV2ΔSptlc2 cells was equivalent to wild type and complemented cells as measured by flow cytometry, and total CD300lf protein levels were equivalent as measured by western blot (Figure 2a-c). Therefore, Sptlc2 deficiency does not alter the amount or the cell surface localization of the MNoV receptor.
Figure 2: Sptlc2 is not required for CD300lf surface localization.
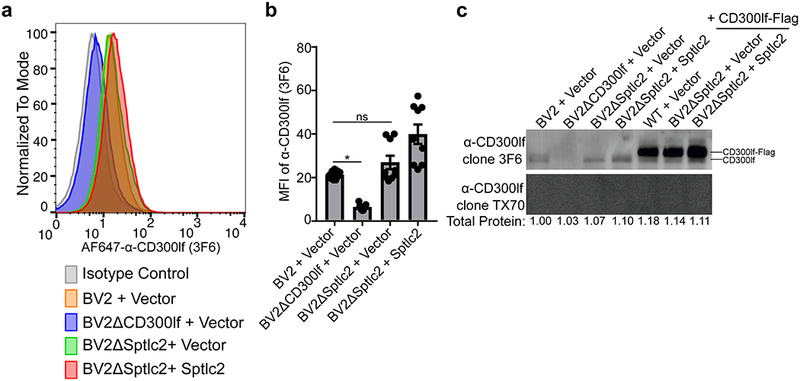
(a) Histogram of a representative FACS experiment from three independent experiments looking at the CD300lf surface levels using AF647-α-CD300lf antibody 3F6 with indicated cell lines.
(b) Quantification of mean fluorescence intensity (MFI) of α-CD300lf antibody 3F6 from three independent experiments. Data are shown as means ± SEM from three independent experiments and data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. *p<0.05, ns = not significant.
(c) A representative western blot of indicated cell lysates with CD300lf antibodies 3F6 (4 μg/ml, top) and TX70 (4 μg/ml, bottom). Below each lane is listed the total protein measured in each lane relative to WT + Vector. The inability of TX70 to recognize CD300lf when overexpressed by western blot but is able to recognize CD300lf by FACS indicates that the molecule is conformation dependent. Data is representative of three independent experiments.
Because CD300lf can bind ceramide18, a lipid whose synthesis requires SPT activity, we wanted to test if the conformation of CD300lf is altered in cells deficient in ceramide production. We found that the TX70 antibody recognizes CD300lf by flow cytometry but not by western blot (Figure 2c and Figure 3a and 3b). Based on these observations, the recognition of CD300lf by TX70 is context and most likely conformation dependent and thus can be used as a tool to probe the configuration of CD300lf in sphingolipid and ceramide deficient cells. To test this hypothesis we stably expressed a CD300lf construct with a C-terminal Flag-tag in both wild type and BV2ΔSptlc2 cells (Figure 3a). We used this approach because we were unable to detect endogenous CD300lf using TX70 in BV2 cells (data not shown). This approach also allowed us to stain for both total CD300lf using intracellular staining for the flag epitope and the amount of cell surface expressed CD300lf using the TX70 conformation sensitive antibody. While both wild type and BV2ΔSptlc2 cells had similar levels of intracellular staining of the flag epitope, the CD300lf-conformational dependent antibody was significantly impaired in its ability to recognize CD300lf in BV2ΔSptlc2 cells (Figure 3b and 3c). Additionally, TX70 staining in BV2ΔSptlc2 + CD300lf-Flag cells could be rescued by expression of Sptlc2 although the total expression of CD300lf-Flag in all cells in this experiment was lower than the previous experiment due to use of a different CD300lf-Flag construct in conjunction with the Sptlc2-complementation constructs (Figure 3d). Additionally, treatment of BV2 CD300lf-Flag cells with myriocin, an SPT inhibitor, decreased the TX70 staining compared to vehicle control (Figure 3e and 3f). The defect in TX70 recognition of the CD300lf-Flag was not due to improper trafficking or localization as both cell surface biotinylation and fluorescence microscopy experiments demonstrated that CD300lf-Flag is at the cell surface (Figure 3g and 3h). Taken together, these data suggest that Sptlc2 is dispensable for CD300lf surface expression, but is required for a specific CD300lf structural configuration that is jointly recognized by a conformational dependent antibody and MNoV.
Figure 3: Ceramide biosynthesis is required for a functional CD300lf conformation.
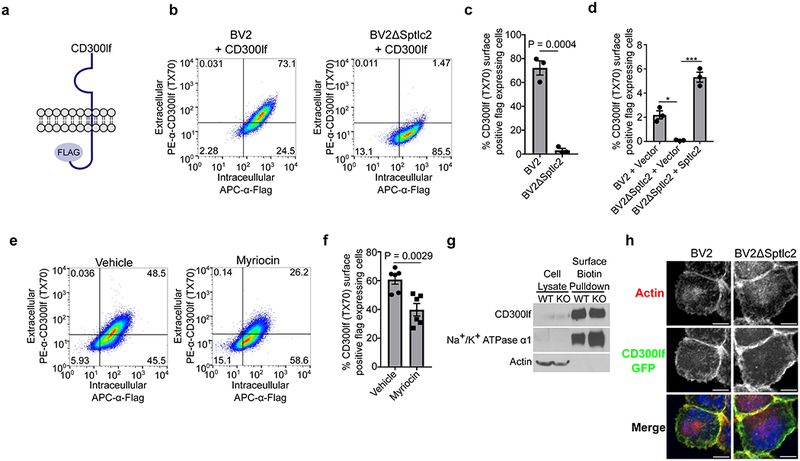
(a) Cartoon diagram of the CD300lf-Flag construct used. Data are representative from three independent experiments.
(b) FACS plots of indicated cells stained first with the conformational dependent antibody PE-α-CD300lf antibody TX70 (y-axis) prior to intracellular staining of APC-α-Flag (x-axis).
(c) Quantification of the percentage of α-CD300lf antibody TX70 positive pCDH-CD300lf-Flag-T2A-Puromycin transduced cells that express Flag for the indicated cells. Data are shown as means ± SEM from three independent experiments and data were analyzed by unpaired two-sided t-test
(d) Quantification of percentage of α-CD300lf antibody TX70 positive pCMV-CD300lf-Flag-P2A-Blasticidin transduced Sptlc2 complemented cells that express Flag. Data are shown as means ± SEM from three independent experiments and data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. *p<0.05, ***p<0.001, ns = not significant
(e) FACS plots of pCDH-CD300lf-T2A-Puromycin cells treated with vehicle or myriocin for 24 hours prior to staining with the conformational dependent antibody PE-α-CD300lf antibody TX70 (y-axis) prior to intracellular staining of APC-α-Flag (x-axis). Data are representative from three independent experiments.
(f) Quantification of the percentage of α-CD300lf antibody TX70 positive pCDH-CD300lf-T2A-Puromycin transduced cells that express Flag for the indicated treatments. Data are shown as means ± SEM from three independent experiments and data were analyzed by unpaired two-sided t-test
(g) Representative western blot of whole cell lysate (left) or purified cell surface biotinylated proteins (right) in BV2 or BV2ΔSptlc2 cells overexpressing CD300lf-FLAG. Na+/K+ ATPase α1 (cell surface) and actin (intracellular) are used as controls. Data are representative from three independent experiments.
(h) Representative microscopy images of BV2 or BV2ΔSptlc2 expressing a CD300lf-Flag-GFP (green) transgene. Cells were stained with AlexaFluor594-Phalloidin (red) to illustrate the outline of cell and DAPI (Blue) to highlight the nucleus. Scale bar represents 10 μm. Data are representative from three independent experiments.
Chemical complementation of Sptlc2 deficient cells changes the CD300lf conformation and restores MNoV infection
Having established that genetic disruption of the SPT complex altered MNoV infection and changed the conformation of CD300lf at the cell surface, we tested the hypothesis that this effect is due to the lack of sphingolipids and thus examined whether we could chemically complement the genetic deficiency in BV2ΔSpltlc2 cells. We chose to use a synthetic, soluble ceramide, C2 ceramide, to deliver to cells due to ceramide’s synthesis requiring SPT activity and the solubility of the C2 ceramide, which can traverse the lipid bilayers of cells. Addition of C2 ceramide to BV2ΔSptlc2 restored MNoVCW3 infectivity as measured by flow cytometry (Figure 4a). Also, C2 ceramide treatment restored the ability of the conformation-specific antibody TX70 to recognize a CD300lf-Flag transgene in BV2ΔSptlc2 cells (Figure 4b and 4c). Taken together, our genetic and chemical data indicate an essential role for sphingolipid biosynthesis, which occurs intracellularly, in regulating the conformation of CD300lf, which likely contributes to MNoV binding and cellular tropism.
Figure 4: Chemical complementation restores MNoV infection and a CD300lf conformation in Sptlc2 deficient cells.
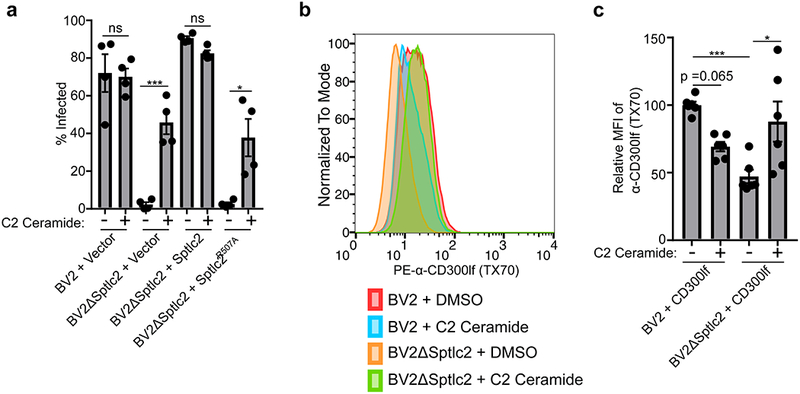
(a) MNoVCW3 infection of indicated cell lines either treated with DMSO or C2 ceramide. Infection was measured by FACS for intracellular production of VP1. Data are shown as means ± SEM from three independent experiments and data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. *p<0.05, ***p<0.001, ns = not significant.
(b) Histogram of a representative histogram from three independent experiments looking at the CD300lf conformation-dependent antibody TX70 ability to stain indicated cells treated with either DMSO or C2 ceramide.
(c) Quantification of mean fluorescence intensity (MFI) of α-CD300lf antibody TX70 from three independent experiments. MFI within an experiment was normalized to BV2 + DMSO. Data are shown as means ± SEM from three independent experiments and data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. *p<0.05, ***p<0.001, ns = not significant.
Discussion:
A key factor in determining the cells, tissues, and species a virus can infect is the ability of viral proteins to engage cellular receptors. Here we demonstrate that the essential and rate limiting enzyme for de novo ceramide and sphingolipid biosynthesis, SPT, is required for efficient MNoV entry and binding at least in part through controlling the conformation of a protein receptor. As norovirus entry is one limiting factor for in vitro replication, these findings may have implications for the development of efficient human norovirus (HNoV) culture systems4. Significant effort in identifying cellular models that enable HNoV replication have focused on potential receptors including HBGAs; however, our data shows that intracellular factors may contribute to the susceptibility of cells to norovirus infection19,20. As HBGAs alone are not sufficient to explain HNoV cellular tropism, it is tempting to speculate that unappreciated intracellular pathways that regulate HBGA accessibility or the conformation of unidentified protein receptors regulate HNoV tropism and underlie the difficulty in establishing robust and tractable HNoV cell culture systems.
Most investigations into the relationship between viral entry and sphingolipids have identified a direct interaction between viruses and host lipids or describe an endosomal escape mechanism leveraging the unique geometry of ceramide in lipid bilayers21–23. We now provide evidence to support a new model in which the intracellular production of sphingolipids is required for a functional conformation of a viral receptor. Our data indicates that CD300lf is expressed equivalently on the surface of wild type and Sptlc2 deficient cells (Figure 2a-c). Importantly, we and others have previously demonstrated that even minimal surface expression of CD300lf renders cells susceptible to MNoV infection5,6. Rather surprisingly, here we uncover an unappreciated connection between the conformation of CD300lf and sphingolipid production that is associated with recognition by both the virus for binding to cells and a conformation specific anti-receptor antibody. The molecular details underlying the Sptlc2-dependent conformation of CD300lf are currently unknown. We previously mapped the critical regions of MNoV receptor activity of CD300lf to the amino acids that compose the CD300lf lipid binding pocket5,24. Taken together these findings suggest the possibility that ceramide or a sphingolipid controls the conformation of CD300lf through its interactions with this ligand binding pocket; however, our data does not exclude the possibility that sphingolipids interact specifically with the transmembrane domain, as observed for other proteins25. It is also possible that the ceramide-dependent conformation of CD300lf is a function of ceramide and/or sphingolipid molecules regulating the formation of membrane microdomains such as lipid rafts which may facilitate CD300lf clustering or controlling interactions with additional proteins at the cell surface26.
Previous work has shown that post-translational modifications of viral receptors by intracellular enzymes can regulate cellular susceptibility27,28. Here however, we report an intracellular biosynthetic enzyme that alters the conformation of a cell surface viral receptor protein. To the best of our knowledge this is this first report of a non-covalent modification of a receptor that alters viral susceptibility. More broadly, these findings suggest that the blockade in binding and establishing infection for certain viruses is not the absence of a receptor but an intracellular deficiency that renders the receptor nonpermissive for viral engagement.
Methods:
Cells
BV2 cells previously karyotyped and confirmed to replicate MNoV5 and 293T (ATCC) cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), and 10 mM HEPES. 2.5 μg/ml of puromycin (Sigma Aldrich) and 5 μg/ml blasticidin (Invitrogen) were added as indicated.
BV2ΔCD300lf cells have been described previously5. BV2ΔSptlc2 cells were generated at the Genome Engineering and iPSC center at Washington University School of Medicine. BV2 cells were nucleofected with Cas9 and a Sptlc2-specific sgRNA (GATATCTTCGAGATTTCTTGAGG). Cells were then single cell sorted and genomic DNA was extracted. DNA was amplified using primers forward (AGCATGGCCTCGGTGTTCCATTGGT) and reverse (AGTCAAAGCAATCCTCCTGCCTCAA). Clones were screened for frameshifts by sequencing the target region with Illumina MiSeq at approximately 500× coverage (Supplemental Figure 1). All cell lines tested and verified to be free of mycoplasma contamination.
MNoV Assays
MNoVCW3 (Gen bank accession no. EF014462.1) and MNoVCR6 (Gen bank accession no. JQ237823) were generated by transfecting MNoV cDNA clones into 293T cells as described previously 29. For MNoV growth curves, 5 × 104 cells infected in suspension with MNoVCW3 or MNoVCR6 at an MOI of 0.05 in a well of a 96-well plate. Plates were frozen at 0, 12, or 24 hours post infection at −80°C. Total cell lysate was used in subsequent plaque assays as previously described 30. All infections were done in triplicate in each of at least three independent experiments.
Viral RNA (vRNA) from MNoVCW3 was extracted from cell-free viral preparations using TRIzol (Invitrogen) according to manufacturer instructions. Purified RNA, which includes both host and viral RNAs, was plaqued to ensure complete inactivation of MNoV. 10 μg of this RNA was transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Transfected cells were frozen 12 hours later. Each condition was assayed by plaque assay in triplicate in three independent experiments.
MNoVCW3 binding assays were done as previously described5. Briefly, MNoVCW3 binding to BV2 cells was performed for 1 hour at 4°C in 0.5 ml complete growth. BV2 cells were used at final concentration of 2 ×106 cells/ml, and MNoVCW3 was used at a final concentration of 2.5 × 106 PFU/ml. Cells were centrifuged at 500g for 5 min at 4°C to remove unbound virus. Cells were washed with four 1.0 ml washes with PBS. RNA was extracted with the ZR Viral RNA kit (Zymo Research) according to manufacturer instructions. qPCR was performed as described previously for MNoV and rps29 31. Binding experiments were done in triplicate in each of at least three independent experiments and binding was normalized within an experiment to the ratio of MNoV to rps29.
Plasmids
cDNA for mouse Sptlc2 was obtained from TransOMIC (pCS6 BC003227) and subsequently subcloned into pCDH-MCS-T2A-Puro vector (System Biosciences). Sptlc2R507A was generated through overlap extension PCR. Codon optimized CD300lf-FLAG, described previously 5, was subcloned into pCDH-MCS-T2A-Puro or synthesized into pCMV-CD300lf-Flag-P2A-Blast (VectorBuilder). For the pCMV-CD300lf-Flag-P2A-Blast, we routinely see lower expression compared to the pCDH-CD300lf-T2A-Puromycin. All plasmid constructs were sequenced verified.
Lentiviral transduction
Lentivirus was generated by transfecting lentiviral vectors with packaging vector (psPAX2) and pseudo-typing vector (pCMV-VSV-G) into 293Ts using TransIT-LT1 (Mirus). 48 hours post-transfection, supernatants were collected, filtered through a 0.45 μm filter (Millipore), and added to the indicated BV2 cells. After 48 hours, cells were selected with the appropriate antibiotic.
Antibodies, Flow Cytometry, and Western Blotting
The following antiboides were used for flow cytometry or western blotting as indicated: rabbit α-Sptlc2 (Proteintech), mouse α-GAPDH-HRP (Sigma), rat α-CD300lf-PE clone TX70 (Biolegend), armenian hamster α-CD300lf clone 3F6 (Genentech), rat α-FLAG-PE (Biolegend), rat α-FLAG-APC (Biolegend), mouse anti-VP1-FITC (A6.2 monoclonal antibody conjugated to FITC; 9).
Cells were placed on ice, washed once in PBS, prior to lysis in cold RIPA Buffer (50 mM Tris pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% IGEPAL, 0.5% Sodium Deoxycholate, and 0.1% SDS) with HALT protease and phosphastase inhibitor cocktail (Sigma). Lysates were clarified by centrifugation prior to resolving on SDS-PAGE Stain Free gels (BioRad) and transfer to PVDF membranes. Where indicated, total protein was quantified and used for normalization using a ChemiDoc MP Imaging System (BioRad).
For FACS analysis, cells were isolated and probed for extracellular CD300lf expression prior to being fixed and permeabilized with Cytofix/Cytoperm (BD biosciences). Cells were stained with indicated antibodies intracellularly. Cells were then washed and analyzed on a FACSCalibur flow cytometer (BD). At least 20,000 events were collected per condition. Each experiment was performed in triplicate in each of three independent experiments.
Cell surface biotinylation
For cell surface biotinylation experiments the Pierce Cell Surface Protein Isolation Kit (Thermo Fisher) was used following manufactures guidelines. Briefly, 12e6 BV2 or BV2ΔSptlc2 expressing CD300lf-flag were seeded overnight, washed, and Sulfo-NHS-SS-Biotin added to cells in PBS on ice for 30 minutes. Labeling reaction was quenched prior to collecting and lysing cells. A sample of the clarified lysate was kept for western blot analysis and the remainder was added to streptavidin columns for isolation of biotinylated proteins.
Microscopy
BV2 or BV2ΔSptlc2 cells expressing CD300lf-flag-eGFP were seeded overnight onto glass coverslips in 6-well dishes. Cells were fixed, permeablized with 0.5% Triton X-10 and stained with AlexaFluor555-Phalloidin (ThermoFisher). Coverslipds were mounted onto slides with ProLong Gold Antifade with DAPI (ThermoFisher) and imaged on a Zeiss LSM 880 Confocal Laser Scanning Microscope.
Chemical Inhibitors and Complementation
Myriocin was purchased from Sigma and dissolved in methanol at 1.25 mM. For inhibition studies, cells were seeded overnight and the following day media was changed to contain methanol or myriocin (25 μM). 24 hours later cells were either processed for FACS (Figure 3e and 4F) or infected with MNoVCW3 (Figure 1c) at an MOI of 5. 16–18 hours after infection cells were collected for FACS and stained intracellularly for MNoV capsid production.
C2 ceramide (d18:1/2:0) was purchased from Avanti Lipids and resuspended in DMSO. 2e6 cells/well were seeded in a six-well plate overnight. Media was removed and cells were subsequently washed with PBS. For infection assays, cells were incubated with gentle rocking at room temperature for one hour with 500 μM C2 ceramide or DMSO with 10e6 PFU in a total volume of 500 μl of complete media. Media was removed, cells were washed with PBS and fresh media was added to the cells. After 16–18 hours, cells were collected for FACS and stained intracellularly for MNoV capsid production. A similar experimental setup was used for for chemical complementation of the conformational dependent antibody recognition except no virus was added and cells were only incubated for 45 minutes at room temperature prior to washing.
Supplementary Material
Acknowledgements
We would like to thank Leon Hsieh for technical assistance for MNoV binding assay. R.C.O. was supported by NIH grant K99 DK116666. C.B.W was supported by NIH grant K08 AI128043. H.W.V. was supported by NIH grants U19 AI10972505 and R01 AI127552.
Footnotes
Data Availability
The data that support the findings of this study are available from the corresponding authors upon request.
Financial Interests
Washington University School of Medicine holds patents on several aspects of murine norovirus. These have been licensed, generating income for the University and the inventors including Dr. Virgin
References:
- 1.Glass RI, Parashar UD & Estes MK Norovirus gastroenteritis. N Engl J Med 361, 1776–1785, doi: 10.1056/NEJMra0804575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bok K & Green KY Norovirus gastroenteritis in immunocompromised patients. The New England journal of medicine 367, 2126–2132, doi: 10.1056/NEJMra1207742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karst SM, Wobus CE, Goodfellow IG, Green KY & Virgin HW Advances in norovirus biology. Cell host & microbe 15, 668–680, doi: 10.1016/j.chom.2014.05.015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guix S et al. Norwalk virus RNA is infectious in mammalian cells. J Virol 81, 12238–12248, doi: 10.1128/JVI.01489-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orchard RC et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 353, 933–936, doi: 10.1126/science.aaf1220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haga K et al. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc Natl Acad Sci U S A 113, E6248–E6255, doi: 10.1073/pnas.1605575113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindesmith L et al. Human susceptibility and resistance to Norwalk virus infection. Nat Med 9, 548–553, doi: 10.1038/nm860 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Kambhampati A, Payne DC, Costantini V & Lopman BA Host Genetic Susceptibility to Enteric Viruses: A Systematic Review and Metaanalysis. Clin Infect Dis 62, 11–18, doi: 10.1093/cid/civ873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wobus CE et al. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS biology 2, e432, doi: 10.1371/journal.pbio.0020432 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karst SM, Wobus CE, Lay M, Davidson J & Virgin HW STAT1-dependent innate immunity to a Norwalk-like virus. Science 299, 1575–1578, doi: 10.1126/science.1077905 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Hanada K Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta 1632, 16–30 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Montes LR, Ruiz-Argüello MB, Goñi FM & Alonso A Membrane restructuring via ceramide results in enhanced solute efflux. J Biol Chem 277, 11788–11794, doi: 10.1074/jbc.M111568200 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Stancevic B & Kolesnick R Ceramide-rich platforms in transmembrane signaling. FEBS Lett 584, 1728–1740, doi: 10.1016/j.febslet.2010.02.026 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fucho R, Casals N, Serra D & Herrero L Ceramides and mitochondrial fatty acid oxidation in obesity. FASEB J 31, 1263–1272, doi: 10.1096/fj.201601156R (2017). [DOI] [PubMed] [Google Scholar]
- 15.Gomez-Muñoz A et al. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res 61, 51–62, doi: 10.1016/j.plipres.2015.09.002 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Galadari S, Rahman A, Pallichankandy S & Thayyullathil F Tumor suppressive functions of ceramide: evidence and mechanisms. Apoptosis 20, 689–711, doi: 10.1007/s10495-015-1109-1 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Lowther J et al. Role of a conserved arginine residue during catalysis in serine palmitoyltransferase. FEBS Lett 585, 1729–1734, doi: 10.1016/j.febslet.2011.04.013 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Izawa K et al. The receptor LMIR3 negatively regulates mast cell activation and allergic responses by binding to extracellular ceramide. Immunity 37, 827–839, doi: 10.1016/j.immuni.2012.08.018 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Jones MK et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346, 755–759, doi: 10.1126/science.1257147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ettayebi K et al. Replication of human noroviruses in stem cell-derived human enteroids. Science, doi: 10.1126/science.aaf5211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider-Schaulies J & Schneider-Schaulies S Sphingolipids in viral infection. Biol Chem 396, 585–595, doi: 10.1515/hsz-2014-0273 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Luisoni S et al. Co-option of Membrane Wounding Enables Virus Penetration into Cells. Cell Host Microbe 18, 75–85, doi: 10.1016/j.chom.2015.06.006 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Otsuki N et al. Both sphingomyelin and cholesterol in the host cell membrane are essential for Rubella virus entry. J Virol, doi: 10.1128/JVI.01130-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson CA et al. Structural basis for murine norovirus engagement of the CD300lf receptor. PNAS (Submitted). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Contreras FX et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 481, 525–529, doi: 10.1038/nature10742 (2012). [DOI] [PubMed] [Google Scholar]
- 26.García-Arribas AB, Alonso A & Goñi FM Cholesterol interactions with ceramide and sphingomyelin. Chem Phys Lipids 199, 26–34, doi: 10.1016/j.chemphyslip.2016.04.002 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Wei W et al. ICAM-5/Telencephalin Is a Functional Entry Receptor for Enterovirus D68. Cell Host Microbe 20, 631–641, doi: 10.1016/j.chom.2016.09.013 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Farzan M et al. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96, 667–676 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Strong DW, Thackray LB, Smith TJ & Virgin HW Protruding domain of capsid protein is necessary and sufficient to determine murine norovirus replication and pathogenesis in vivo. Journal of virology 86, 2950–2958, doi: 10.1128/JVI.07038-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang S et al. Murine norovirus: propagation, quantification, and genetic manipulation. Curr Protoc Microbiol 33, 15K.12.11–61, doi: 10.1002/9780471729259.mc15k02s33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baert L et al. Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Applied and environmental microbiology 74, 543–546, doi:Doi 10.1128/Aem.01039-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.