

Abstract
Background
Fruits of persimmon plants are traditional healthy food in China, Korea and Japan. However, due to the shortage of morphological and DNA markers, the development of persimmon industry has been heavily inhibited.
Results
Chloroplast genomes of Diospyros cathayensis, D. virginiana, D. rhombifolia and D. deyangensis were newly sequenced. Comparative analyses of ten chloroplast genomes including six previously published chloroplast genomes of Diospyros provided new insights into the genome sequence diversity and genomic resources of the genus. Eight hyper-variable regions, trnH-psbA, rps16-trnQ, rpoB-trnC, rps4-trnT-trnL, ndhF, ndhF-rpl32-trnL, ycf1a, and ycf1b, were discovered and can be used as chloroplast DNA markers at/above species levels. The complete chloroplast genome sequences provided the best resolution at inter-specific level in comparison with different chloroplast DNA sequence datasets.
Conclusion
Diospyros oleifera, D. deyangensis, D. virginiana, D. glaucifolia, D. lotus and D. jinzaoshi are important wild species closely related to the cultivated persimmon D. kaki. The hyper-variable regions can be used as DNA markers for global genetic diversity detection of Diospyros. Deeper study on these taxa would be helpful for elucidating the origin of D. kaki.
Electronic supplementary material
The online version of this article (10.1186/s12870-018-1421-3) contains supplementary material, which is available to authorized users.
Keywords: Diospyros, Chloroplast genome, Hyper-variable region, Genetic diversity
Background
Diospyros is the largest genus in the family Ebenaceae, including more than 400 species all over the world, with a wide distribution and cultivation from tropical to temperate zones. Their fruits have been traditional healthy food source in China, Korea and Japan. Ebony trees are valued for their hard, heavy and dark timber. In addition, the bark, leaves, wood, fruits and seeds of several species are the main sources of medicines. Diospyros kaki is an important economic tree crop and widely cultivated species of the genus [1, 2].
In China, cultivation of persimmon plants (Diospyros kaki) for edible fruits and Chinese medicine is commonly one of the best choices for regional development of characteristic fruit tree industry, with different regions having different cultivars. At present, there are about 1000 persimmon cultivars in China. Most of the extant persimmon cultivars are elite plants from natural seedlings or bud mutation. Among these cultivars, a majority is of the pollination- constant and astringent (PCA) type, pollination-constant and non-astringent (PCNA) type of premature persimmon and disease-resistant cultivars are rarer and are highly desirable. In general, closely related wild species possess elite genetic background for qualitative improvement and high-yield breeding of the persimmon crops [1, 2].
Breeding of woody Diospyros plants is time-consuming, commonly around ten years being necessary for evaluating the performance of a hybrid plant. Due to the difficulty of identifying the Diospyros germplasm resource diversities, directive breeding through artificial hybridization is severely inhibited. Therefore, accumulation of genetic information for revealing the genetic diversity of Diospyros plants is in urgent need.
The germplasm resources of Diospyros plants have been documented in aspects of the use and phenotypic characteristics, such as fruit morphology, fruit color, fruit quality and floral characteristics. Diospyros plants have four ploidy levels (2n = 2× = 30; 2n = 4× = 60; 2n = 6× = 90; 2n = 9× = 135) according to their chromosome number [1–7]. However, due to the shortage and limited sensitivity of phenotypic and DNA markers, study on genetic differentiation detection techniques for the huge amount of Diospyros plants worldwide is still a global challenge [8–12].
In Diospyros, molecular resources have been developed in recent years for species identification. Some chloroplast genome markers (such as rbcL, matK, trnH-psbA) and nuclear DNA markers (e.g., internal transcribed spacer of ribosomal DNA, ITS) were used to discriminate plants in Diospyros. However, these markers are low in variability, or have limited resolution and cannot reach the goals [3, 13–18]. Development of more effective DNA barcodes is also important for Diospyros plants.
The angiosperm chloroplast genomes are uniparentally inherited with relatively stable structure [16, 17]. Accordingly, it is considered to be an informative and valuable resource for plant phylogenetic analyses at family/genus/species levels [16, 19–24]. In the past decades, the chloroplast genomes have been proven to be more powerful in revealing phylogeny of plants and resolving previously ambiguous taxonomic and phylogenetic issues [16–28].
In this study, we report newly sequenced complete chloroplast genomes of D. cathayensis, D. deyangensis, D. rhombifolia and D. virginiana. The aims of our study are: (1) to evaluate the variation in Diospyros; and (2) to develop new and efficient cpDNA markers for species identification in Diospyros.
Methods
Sampling and DNA extraction
The fresh leaves of the Diospyros taxa were collected in spring 2016 from the Beijing Botanical Garden of Chinese Academy of Sciences and the National Field Genbank for Persimmons of College of Horticulture, Northwest A&F University, Yangling, Shaanxi, China and were dried immediately using silica gels for DNA extraction. Total genomic DNAs were extracted following the procedure of Plant Genomic DNA Kit (DP305) from Tiangen Biotech (Beijing) Co., Ltd., China.
Chloroplast genome sequencing, assembling and annotation
DNA was sheared to fragments of 400–600 bp with an ultrasonic disruptor. An Illumina paired-end library was constructed with the NEBNext® Ultra™ DNA Library Prep Kit according to the manufacturer’s protocol. Paired-end sequencing (2 × 150 bp) was conducted on an Illumina HiSeq 4000 platform. The high-throughput sequencing data were qualitatively assessed and assembled using SPAdes 3.10.1 [29]. Using the chloroplast genome sequence of Diospyros kaki (GenBank accession No. KT223565) as a reference sequence, we selected chloroplast genome contigs using the BLAST method. The contigs of the chloroplast genomes were assembled using Sequencher (v5.4) with default parameters. Ambiguous nucleotides or gaps and the four junction regions between the IRs and SSC/LSC in the chloroplast genome sequences were further confirmed by PCR amplification and Sanger sequencing with specific primers or the universal primers [20]. After that, all reads were mapped to the spliced chloroplast genome sequence using Geneious 8.1 [30] to avoid assembly errors. Chloroplast genome annotation was performed using the Dual Organellar Genome Annotator (DOGMA) [31]. The chloroplast genome map was drawn using Genome Vx software [32].
Analysis of tandem repeats and single sequence repeats in chloroplast genomes
MISA (MIcroSAtellite; http://pgrc.ipk-gatersleben.de/misa) was used to detect simple sequence repeats (SSR) within the chloroplast genomes. Thresholds for a minimum number of repeat units were established as follows: > 10 for mono-nucleotide, > 5 for di-nucleotide, > 4 for tri-nucleotide, and > 3 for tetra-nucleotide, penta-nucleotide, or hexa-nucleotide SSR. Repeating sequences were scanned over the complete chloroplast DNA sequences, taxon by taxon, using the REPuter program.
Sequence divergence analysis in chloroplast genomes
Four newly sequenced chloroplast genomes and six chloroplast genomes from GenBank were aligned using MAFFT [33] and manually adjusted using Se-Al 2.0 [34]. Variable and parsimony-informative base sites and genetic distance across the complete chloroplast genomes were calculated using MEGA 6.0 software [35]. Sliding window analysis was conducted to generate nucleotide diversity (Pi) of the chloroplast genomes using DnaSP (DNA Sequences Polymorphism version 5.10.01) software [36]. The step size was set to 200 bp, with a 600 bp window length.
Chloroplast DNA barcoding analysis
Distance and tree based methods were used to evaluate discriminatory power of detected hyper variable regions and core DNA barcodes rbcL and matK. The function nearNeighbour of SPIDER was used for Distance method barcoding analyses [37]. Neighbor-Joining (NJ) trees were constructed by each hyper-variable marker and the different marker combinations using MEGA 6.0 based on a K2P distance model [35]. Relative support for the clades of the NJ tree was assessed via 1000 bootstrap replicates.
Phylogenetic analysis based on chloroplast genome sequence data
Phylogenetic trees were constructed by Maximum Parsimony (MP), Maximum Likelihood (ML) and Bayesian Inference (BI) using the entire chloroplast genome.
MP analyses were performed using PAUP v4b10 [38] described in Dong et al. [24]. The ML analyses were conducted using RAxML 8.0 [39]. For ML analyses, the best-fit model, General Time Reversible, with gamma distribution (GTR + G) was used as suggested with 1000 bootstrap replicates. BI method was conducted with MrBayes v3.2 [40]. The Markov Chain Monte Carlo (MCMC) analysis was run for 2 × 5,000,000 generations with trees sampled every 1000 generations, with the first 25% discarded as burn-in. The remaining trees were used to build a 50% majority-rule consensus tree. The stationarity was regarded to be reached when the average standard deviation of split frequencies remained below 0.01.
Results
Chloroplast genome characterization
Previous experiments indicated that chloroplast genome sequences are identical among individual plants of the same species. Therefore, we sequenced four taxa of Diospyros (D. cathayensis, D. virginiana, D. rhombifolia and D. deyangensis) using an Illumina Hiseq 4000 system, 20,675,288 to 33,584,779 paired-end raw reads were obtained (average read length was 150 bp). After mapping the paired-end reads of each Diospyros taxon, 357,721 to 1,338,624 reads were extracted, yielding 340 × to 1271 × coverage (Table 1). The inverted repeat (IR) junction regions in assembled sequences were confirmed by using PCR-based sequencing. High quality chloroplast genome sequences were thus obtained and were used for downstream analyses. The four Diospyros chloroplast genome sequences were deposited in GenBank (accession numbers, MF288575- MF288578).
Table 1.
Summary of the sequencing data for four Diospyros taxa
| No. | Taxon | Raw data no. | Mapped read no. | Mapped to reference genome (%) | Chloroplast genome coverage (×) |
|---|---|---|---|---|---|
| 1 | D. cathayensis | 25,027,276 | 533,606 | 2.13% | 508 |
| 2 | D. deyangensis | 33,584,779 | 1,338,624 | 3.99% | 1271 |
| 3 | D. rhombifolia | 20,675,288 | 915,369 | 4.43% | 873 |
| 4 | D. virginiana | 30,770,623 | 357,721 | 1.16% | 340 |
The ten complete chloroplast genomes of Diospyros investigated in this study ranged from 157,300 base pairs (D. jinzaoshi) to 157,934 base pairs (D. deyangensis) in length. All the chloroplast genomes possessed the typical quadripartite structure of angiosperms, consisting of a pair of the inverted repeat region (IRs: 25,910–26,119 bp) separated by a large single-copy region (LSC: 86,948–87,246 bp) and a small single-copy region (SSC: 18,076–18,485 bp) (Fig. 1; Table 2). Each of the complete chloroplast genomes possesses 113 unique genes arranged in the same order, including 79 protein-coding, 30 tRNA, and 4 rRNA genes. The GC content of each chloroplast genome is identically 37.4% (Table 2). The genomic structure including gene number and gene order was highly conserved within Diospyros (Fig. 1; Table 2).
Fig. 1.

Gene map of Diospyros chloroplast genomes. The genes inside and outside of the circle are transcribed in clockwise and counterclockwise directions, respectively. Genes belonging to different functional groups are shown in different colors. The thick lines indicate the extent of the inverted repeats (IRa and IRb) that separate the genomes into small single-copy (SSC) and large single-copy (LSC) regions
Table 2.
Complete chloroplast genome features of ten Diospyros taxa
| Sample | GenBank accession No. | Genome size (bp) | LSC length (bp) | SSC length (bp) | IR length (bp) | Gene content | Protein coding genes | tRNA genes | rRNA genes | GC content (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| D. discolor | KX426216 | 157,745 | 87,246 | 18,323 | 26,088 | 113 | 79 | 30 | 4 | 37.4 |
| D. cathayensis | MF288576 | 157,689 | 87,176 | 18,349 | 26,082 | 113 | 79 | 30 | 4 | 37.4 |
| D. deyangensis | MF288575 | 157,934 | 87,237 | 18,485 | 26,106 | 113 | 79 | 30 | 4 | 37.4 |
| D. glaucifolia | KM504956 | 157,610 | 86,965 | 18,407 | 26,119 | 113 | 79 | 30 | 4 | 37.4 |
| D. jinzaoshi | KM522848 | 157,300 | 87,010 | 18,076 | 26,107 | 113 | 79 | 30 | 4 | 37.4 |
| D. kaki | KT223565 | 157,784 | 87,059 | 18,505 | 26,110 | 113 | 79 | 30 | 4 | 37.4 |
| D. lotus | KM522849 | 157,597 | 86,948 | 18,411 | 26,119 | 113 | 79 | 30 | 4 | 37.4 |
| D. oleifera | KM522850 | 157,760 | 87,034 | 18,532 | 26,097 | 113 | 79 | 30 | 4 | 37.4 |
| D. rhombifolia | MF288578 | 157,368 | 87,223 | 18,325 | 25,910 | 113 | 79 | 30 | 4 | 37.4 |
| D. virginiana | MF288577 | 157,761 | 87,089 | 18,444 | 26,114 | 113 | 79 | 30 | 4 | 37.4 |
Comparative analysis of chloroplast genomes
The number of simple sequence repeats (SSRs) ranged from 48 (D. cathayensis or D. rhombifolia) to 82 (D. jinzaoshi) among the ten Diospyros taxa. The homopolymer repeat number with the highest variability, ranged from 29 (D. cathayensis) to 70 (D. jinzaoshi), while the number of di-nucleotide, tri-nucleotide or penta-nucleotide repeats had no significant difference among the ten Diospyros taxa (Fig. 2). The homopolymer repeats represented the major source of genetic diversity in Diospyros. In total, 505 SSRs were detected in LSC region, 141 in SSC region and 26 in IR region (Fig. 2).
Fig. 2.

Comparison of simple sequence repeats among ten chloroplast genomes. a. Numbers of SSRs detected in ten Diospyros chloroplast genomes; b. Frequencies of identified SSRs in LSC, IR and SSC regions; c. Numbers of SSR types detected in ten Diospyros chloroplast genomes
Four repeat types were detected in the chloroplast genomes using REPuter software. Forward repeat number ranged from 17 (D. kaki) to 22 (D. deyangensis), which were found to have no significant difference among the ten chloroplast genomes. Tandem repeat number ranged from 20 (D. jinzaoshi) to 29 (D. virginiana). Palindromic repeat number ranged from 18 (D. discolor or D. virginiana) to 33 (D. kaki). The highest value of palindromic repeat number which occurred in D. kaki is significantly different from that of other taxa. The highest tandem repeat number was observed in D. virginiana (Fig. 3).
Fig. 3.
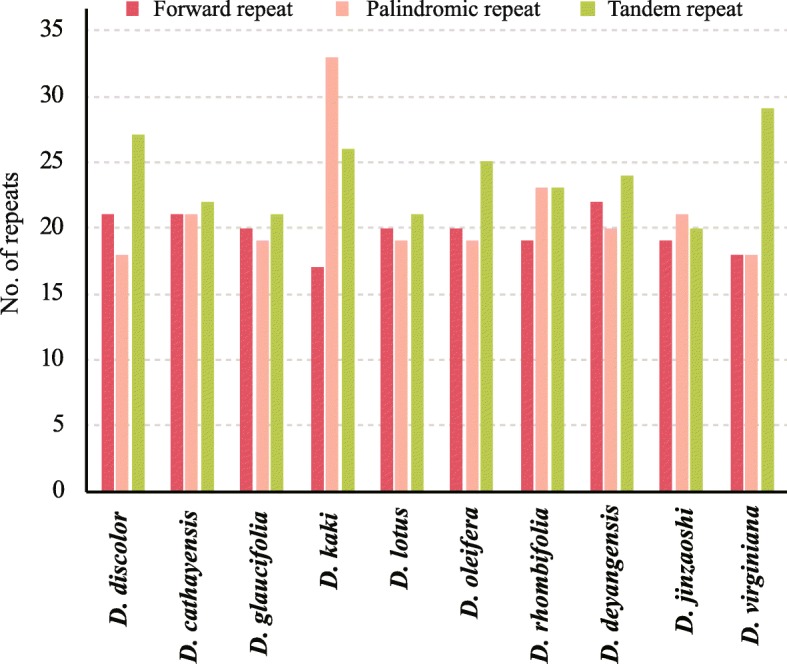
Numbers of repeats in comparison among ten chloroplast genomes
The lowest value (31) of nucleotide substitution number was observed between D. glaucifolia and D. lotus, while the highest value (1493) of nucleotide substitution number occurred between D. cathayensis and D. virginiana, showing a wider range of variability according to the sequence alignment of the ten chloroplast genomes (Table 3).
Table 3.
Numbers of nucleotide substitutions and sequence distance among the complete chloroplast genomes of ten Diospyros taxa
| Taxon | D. kaki | D. oleifera | D. deyangensis | D. jinzaoshi | D. glaucifolia | D. lotus | D. virginiana | D. cathayensis | D. rhombifolia | D. discolor |
|---|---|---|---|---|---|---|---|---|---|---|
| D. kaki | 0.0022 | 0.0022 | 0.0032 | 0.0046 | 0.0046 | 0.0068 | 0.0082 | 0.0081 | 0.0078 | |
| D. oleifera | 346 | 0.0003 | 0.0029 | 0.0043 | 0.0043 | 0.0065 | 0.0079 | 0.0078 | 0.0074 | |
| D. deyangensis | 348 | 42 | 0.0029 | 0.0043 | 0.0043 | 0.0065 | 0.0079 | 0.0077 | 0.0074 | |
| D. jinzaoshi | 500 | 449 | 447 | 0.0043 | 0.0043 | 0.0066 | 0.0080 | 0.0079 | 0.0074 | |
| D. glaucifolia | 723 | 673 | 676 | 673 | 0.0002 | 0.0074 | 0.0089 | 0.0088 | 0.0083 | |
| D. lotus | 725 | 674 | 677 | 676 | 31 | 0.0074 | 0.0088 | 0.0088 | 0.0083 | |
| D. virginiana | 1060 | 1017 | 1018 | 1029 | 1160 | 1161 | 0.0092 | 0.0091 | 0.0088 | |
| D. cathayensis | 1280 | 1229 | 1228 | 1245 | 1381 | 1378 | 1493 | 0.0011 | 0.0070 | |
| D. rhombifolia | 1263 | 1210 | 1207 | 1223 | 1366 | 1363 | 1419 | 168 | 0.0069 | |
| D. discolor | 1211 | 1154 | 1151 | 1158 | 1295 | 1297 | 1366 | 1087 | 1081 |
Notes: The lower triangle shows the number of nucleotide substitutions. The upper triangle indicates the number of sequence distance in complete chloroplast genomes
The sequence distance between Diospyros taxa ranged from 0.0002 to 0.0092. The smallest sequence distance occurred between D. lotus and D. glaucifolia, and the largest sequence distance occurred between D. virginiana and D. cathayensis (Table 3). LSC region was the most rapidly evolving region in the chloroplast genomes, while the IR region was the most slowly evolving region. The evolutionary rate of SSC region is moderate.
Phylogenetic analysis
Phylogenetic analyses indicated that all the ten taxa were clearly discriminated and seven clades were recognized among them (Fig. 4). The cultivars of Diospyros belonged to one clade of D. kaki. The DNA sequence data supported the isolated positions of D. deyangensis and D. jinzaoshi which were regarded previously as cultivars and have been suggested to rank to species level according to morphological and DNA characteristics by other recent researches [2–6]. The clade of D. oleifera and D. deyangensis, and the clade of D. jinzaoshi included the closest wild relatives of the cultivated persimmon plants of D. kaki (Fig. 4). Relatively speaking, the clade of D. lotus and D. glaucifolia, and the clade of D. virginiana have close genetic relationships with D. kaki. D. lotus and D. virginiana produce edible fruits, while D. glaucifolia is mainly used for timber production. D. cathayensis and D. rhombifolia that are genetically close with each other formed one clade. This is identical with their classification based on morphological characteristics. D. discolor is genetically the most distant taxon from the cultivated plants of D. kaki among the taxa investigated in this study.
Fig. 4.

Phylogenetic relationships of ten Diospyros taxa inferred from Maximum Parsimony (MP), Bayesian Inference (BI), and Maximum Likelihood (ML) analyses of the complete chloroplast genomes. ML topology shown with ML bootstrap support value/Bayesian posterior probability/MP bootstrap support value given at each node. Nodes with 100 ML bootstrap support value/1.0 Bayesian posterior probability/100 MP bootstrap support value are not marked
Chloroplast DNA marker development
According to the chloroplast genome sequence alignment of the ten Diospyros taxa, eight hyper-variable regions, trnH-psbA, rps16-trnQ, rpoB-trnC, rps4-trnT-trnL, ndhF, ndhF-rpl32-trnL, ycf1a, and ycf1b were discovered (Fig. 5). These eight sequences could be used as DNA markers for classification and revealing the genetic divergence of the Diospyros taxa, with a high discrimination success ranging from 60 to 100% (Table 4), of which, the sequences of the three most rapidly evolving regions (i.e., rps4-trnT-trnL, ndhF-rpl32-trnL, and ycf1a) were able to discriminate all the taxa investigated in this study.
Fig. 5.

Sliding window analysis of the whole chloroplast genomes of nine Diospyros taxa (window length: 600 bp, step size: 200 bp). X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window
Table 4.
Variability of nine hyper-variable markers and universal chloroplast DNA barcodes (rbcL and matK) in Diospyros
| Marker | Length (bp) | Variable base sites | Informative base sites | Mean distance | Discrimination success (%) based on Distance method | ||
|---|---|---|---|---|---|---|---|
| Number | Percentage (%) | Number | Percentage (%) | ||||
| trnH-psbA | 1011 | 52 | 5.14 | 28 | 2.77 | 0.0178 | 80.0 |
| rps16-trnQ | 2184 | 122 | 5.59 | 65 | 2.98 | 0.0183 | 60.0 |
| rpoB-trnC | 885 | 46 | 5.20 | 22 | 2.49 | 0.0165 | 80.0 |
| rps4-trnT-trnL | 1469 | 72 | 4.90 | 39 | 2.65 | 0.0157 | 100.0 |
| ndhF | 1063 | 59 | 5.55 | 39 | 3.67 | 0.0166 | 80.0 |
| ndhF-rpl32-trnL | 2143 | 122 | 5.69 | 54 | 2.52 | 0.0182 | 100.0 |
| ycf1a | 2141 | 123 | 5.74 | 60 | 2.80 | 0.0181 | 100.0 |
| ycf1b | 1505 | 88 | 5.85 | 41 | 2.72 | 0.0177 | 80.0 |
| Combined | 12,401 | 684 | 5.52 | 348 | 2.81 | 0.0176 | 100.0 |
| rbcL | 1428 | 26 | 1.82 | 15 | 1.05 | 0.0057 | 40.0 |
| matK | 1512 | 46 | 3.04 | 24 | 1.59 | 0.0096 | 80.0 |
In those most rapidly evolving regions, 72, 122 and 123 variable base sites were detected, respectively, of which, 39, 54 and 60 informative base sites, made up 2.52–2.80% in each of the sequences. Comparatively, the commonly recommended DNA fragments (rbcL and matK) achieved only 40% and 80% of discrimination success respectively (Table 4, Additional file 1: Figure S1).
Similar results were obtained when different methods were used for phylogenetic tree reconstruction (Additional file 1: Figure S1). The combined sequence data from the eight hyper-variable cpDNA regions provided a well-resolved phylogenetic topology of the ten Diospyros taxa, similar to the topology obtained using complete chloroplast genome sequences, but with much lower experimental cost (Fig. 6). All the nodes in the phylogenetic tree received high bootstrap value (100%).
Fig. 6.

Phylogenetic relationships of ten Diospyros taxa inferred from Maximum Parsimony (MP), Bayesian Inference (BI), and Maximum Likelihood (ML) analyses using the chloroplast DNA sequences from eight hyper-variable regions
Discussion
Chloroplast genome variation and evolution
In this study, we sequenced four chloroplast genomes of Diospyros using Illumina Hiseq-4000 platforms and compared these genomes with the other six published chloroplast genomes downloaded from GenBank. The chloroplast genomes of Diospyros displayed the typical quadripartite structure of flowering plants, were conservative in gene order and gene content, in comparison with the most lineages of angiosperms. The chloroplast genomes ranged from 157,300 to 157,934 bp in length. IR expansion and contraction and the occurrence of larger indels (insertion/deletion) are considered to be the primary mechanisms affecting length variation of angiosperm chloroplast genomes. Only minor variations were detected at the SC/IR boundaries of the ten Diospyros chloroplast genomes. Occurrence of indels was the main factor effecting the variation of the length in Diospyros chloroplast genomes. Similar to previously published chloroplast genomes of the angiosperms, the Diospyros chloroplast genomes contained more AT content than GC content.
Simple sequence repeats (SSRs) are genetically variable molecular markers and are used widely in population genetics [41, 42], polymorphism investigations, and phylogenetic analyses [43]. Using MISA, the SSRs in the ten Diospyros chloroplast genomes were identified. The number of SSRs ranged from 48 to 82 in Diospyros, similar to those in Lagerstroemia [28]. SSRs with AT richness have been reported in other plants [44, 45]. Homopolymers are the most common SSRs in chloroplast genomes. Since the structure and organization of chloroplast genomes are conservative, SSR primers are transferable across species, genera, and even families. Information involving SSRs in this study will provide useful sources for estimating the genetic diversity and studying phylogenetic relationships among species and genera.
Potential cpDNA barcodes
Diospyros is the largest genus in its family, including more than 400 species all over the world. For effective exploration, conservation, and domestication, accurately identified wild species would provide a clear genetic background of this genus. However, the taxonomic inventory of Diospyros still has a long way to go, because of the vast amount of species with extensive global distribution and the plasticity of the morphological characteristics. DNA barcoding has been largely used as a new biological tool to facilitate accurate species identification [46]. The two chloroplast DNA regions, rbcL and matK, are recommended as core universal DNA barcodes in plants. Therefore, genomic comparative researches of more complete chloroplast genome sequences have become necessary for developing variable DNA barcodes. These mutation “hotspot” regions could provide adequate genetic information for species identification, and can be used to develop novel DNA barcodes [19]. The eight potential mutational hotspots (trnH-psbA, rps16-trnQ, rpoB-trnC, rps4-trnT-trnL, ndhF, ndhF-rpl32-trnL, ycf1a, and ycf1b) identified in this study could be suitable barcodes for plant classification in Diospyros. In previous study, ycf1 gene showed high divergence in chloroplast genome and was recommended as core DNA barcode for plants [22]. Ycf1 gene has been more and more widely applied in plant phylogeny and DNA barcoding studies [47–51]. TrnH-psbA, ndhF and rps16-trnQ were popular candidates for phylogenetic studies [52, 53]. Rps4-trnT-trnL and ndhF-rpl32-trnL were newly identified in this study.
Recently, using the chloroplast genome as a super-barcode for plant species identification was discussed [49]. The analyses on chloroplast genome sequence divergence and phylogeny showed that the chloroplast genome may indeed be useful as a super-barcode for species identification of Diospyros (Table 3 and Fig. 4). Further research is necessary to investigate whether these hyper-variable regions or complete chloroplast genome sequences could be used as reliable and effective DNA barcodes for species of Diospyros. The results obtained in this study have significant value for future studies on global genetic diversity assessment, phylogeny, and population genetics of Diospyros.
Perspectives of persimmon research in future
It is important and fundamental to develop efficient identification methods and elucidate the genetic relationship of Diospyros taxa for planning breeding strategies, intensive management and conservation of Diospyros germplasm resources. The origin of D. kaki and its relationship to other Diospyros species have been also hot issues attracting scientists. Ng [54] suggested that D. oleifera was a parent of D. kaki based on morphological, geographical and cytological evidences. Yonemori et al. [55] suggested that D. virginiana and D. kaki had close relationship, as revealed by restriction fragment length polymorphism of chloroplast DNA. However, Choi et al. [9] considered the relationship between D. virginiana and D. kaki to be possibly phylogentically more remote than others.
The DNA evidences obtained in this study clearly support the previous taxonomic proposals that D. deyangensis and D. jinzaoshi should be raised to species rank based on morphological, molecular characteristics and chromosome numbers [4, 9–12]. In the cpDNA–based tree, the D. kaki lineage and the lineage including the dioecious D. deyangensis and the monoecious D. oleifera shared a common furcation. Interestingly, the chromosome number of D. kaki (2n = 6× = 90) is equal to the sum of the chromosome numbers of D. deyangensis (2n = 4× = 60) and D. oleifera (2n = 2× = 30). This strongly implied that cultivated D. kaki might be resulted from a superposition event of genetic materials between D. deyangensis and D. oleifera through certain genetic mechanism during the evolutionary history of Diospyros plants. D. oleifera is naturally distributed south of the Changjiang River in China, has strong adaptability and resistance to wet condition, and has been used as rootstocks of D. kaki in southern China. Its fruits can also be used for extraction of persimmon paint. D. deyangensis, naturally distributed in Sichuan Province, China, produces red flowers and has fine hairs on the surface of the leaves, shoots, fruits, and some parts of the flowers [7].
Diospyros rhombifolia and D. cathayensis are genetically remote from D. kaki, being identical with the classification based on phenotypic characteristics. D. rhombifolia could not be used as rootstocks or inter-stocks because of its dwarfing effect [2]. D. glaucifolia and D. lotus were clustered together based on cpDNA sequences. They are very similar in phenotypic characteristics [2, 5, 6, 55]. D. lotus has made great contributions as rootstocks in persimmon production areas of northern China because of its highest cold resistance in the genus. The small fruits of D. lotus have been used as food and Chinese medicine with a long history. In southern China, the dioecious species D. glaucifolia is used to produce wood and could also be used as rootstocks. The species boundary between D. lotus and D. glaucifolia should be further studied by sampling more individual plants in future [2, 5, 6, 56].
Elucidation of the questions mentioned above would definitely improve our understanding on phylogeny, relationship, and the origin of the cultivated persimmons in Diospyros, and further accelerating directive breeding of Diospyros plants.
Conclusion
Chloroplast DNA sequences can be used for classification of Diospyros plants at inter-specific level. The results obtained in this study implied that D. oleifera, D. deyangensis, D. virginiana, D. glaucifolia, D. lotus and D. jinzaoshi are important wild species closely related to the cultivated persimmon D. kaki and deeper studies on these taxa would be helpful for understanding the origin of D. kaki. Our results have significant value for global genetic diversity assessment, phylogeny, and population genetics in Diospyros in future.
Additional file
Figure S1. NJ trees of Diospyros taxa based on each of the ten chloroplast barcodes, showing the resolutions of each different locus for revealing the phylogeny. The figures above the lines are the bootstrap support values for the clades. (PDF 506 kb)
Acknowledgements
The authors thank Chao Xu for his kind help in molecular experiments and data analysis, and Jianping Wang and Renzi Wang for their instructions in field investigations.
Funding
The study was financially supported by “Collection, Conservation and Evaluation of Forest Tree Germplasm Resources” (LKZ201496–1-3) of Shandong Provincial Agricultural Elite Varieties Project, grant No. 2017YFD0600604, No. LCNZ2016–36, the National Forest Genetic Resources Platform (2005DKA21003), National Service Sharing Infrastructure of Agricultural Crop Germplasm Resources of Persimmon Resources (NICGR2017–50).
Availability of data and materials
The complete chloroplast genomes of D. cathayensis, D. deyangensis, D. rhombifolia and D. virginiana were submitted to the NCBI database with GenBank accession numbers MF288576 (D. cathayensis), MF288575 (D. deyangensis), MF288578 (D. rhombifolia) and MF288577 (D. virginiana).
Abbreviations
- BI
Bayesian Inference
- cpDNA
Chloroplast DNA
- DnaSP
DNA Sequences Polymorphism
- DOGMA
Dual Organellar Genome Annotator
- GTR
General time reversible
- Indel
Insertion/deletion
- IR
Inverted repeat
- ITS
Internal transcribed spacer of ribosomal DNA
- LSC
Large single copy
- MCMC
Markov Chain Monte Carlo
- ML
Maximum Likelihood
- MP
Maximum Parsimony
- NJ
Neighbor-Joining
- PCA type
Pollination-constant and astringent type
- PCNA type
Pollination-constant and non-astringent type
- Pi
Nucleotide diversity
- rRNA
Ribosomal RNA
- SSC
The small single copy
- SSR
Simple sequence repeats
- tRNA
Transfer RNA
Authors’ contributions
ZS, WD, and WL designed this study; WD and YL designed experiments, sequenced chloroplast genomes; YY, XX, YL, XJ, ZY and ZS conducted field investigations; WD, YL, XJ and ZY analyzed the data; ZS, WD, WL and XJ wrote the manuscript; all authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wenqing Li, Email: wenqingli203@126.com.
Yanlei Liu, Email: liuyanlei@ibcas.ac.cn.
Yong Yang, Email: yang.yong521@163.com.
Xiaoman Xie, Email: xxm529@126.com.
Yizeng Lu, Email: 353081999@qq.com.
Zhirong Yang, Email: zry@ibcas.ac.cn.
Xiaobai Jin, Email: jinxiaobai@ibcas.ac.cn.
Wenpan Dong, Email: wpdong@ibcas.ac.cn.
Zhili Suo, Phone: 13520435137, Email: zlsuo@ibcas.ac.cn.
References
- 1.Lee S, Michael GG, Frank W. In: Ebenaceae. Wu ZY, Raven PH, Hong DY, editors. Beijing/Missouri/St. Louis: Flora of China. Science Press/Botanical Garden Press; 1996. pp. 215–234. [Google Scholar]
- 2.Wang RZ, Yang Y, Li GC. Chinese persimmon germplasm resources. Acta Hort. 1997;(436):43–50.
- 3.Tang DL, Hu Y, Zhang QL, Yang Y, Luo ZR. Discriminant analysis of ‘Jinzaoshi’ from persimmon (Diospyros kaki Thunb.; Ebenaceae): a comparative study conducted based on morphological as well as ITS and matK sequence analyses. Sci Hortic. 2014;168:168–174. doi: 10.1016/j.scienta.2014.01.033. [DOI] [Google Scholar]
- 4.Yang Y, Jing ZB, Ruan XF, Cheng JM. Development of simple sequence repeat markers in persimmon (Diospyros L.) and their potential use in related species. Genet Mol Res. 2015;14:609–618. doi: 10.4238/2015.January.30.2. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Yang TT, Jing ZB. Genetic diversity and taxonomic studies of date plum (Diospyros lotus L.) using morphological traits and SCoT markers. Biochemical Systematics Ecol. 2015;61:253–259. doi: 10.1016/j.bse.2015.06.008. [DOI] [Google Scholar]
- 6.Zhang YF, Hu CQ, Yang Y, Zhu RS, Guo J. Wang RZ. Pollen morphology observation of eight resources in Diospyros. Acta Horticulturae Sinica. 2016; 43 (6): 1167–1174, doi: 10.16420/j.issn.0513-35F3x.2015%2D0741.
- 7.Zhang YF, Yang Y, Guo J, Hu CQ, Zhu RH. Taxonomic status of ‘Deyangshi’ based on chromosome number and SRAP markers. Sci Hortic. 2016;207:57–64. doi: 10.1016/j.scienta.2016.05.006. [DOI] [Google Scholar]
- 8.Mallavadhani UV, Panda AK, Rao YR. Pharmacology and chemotaxonomy of Diospyros. Phytochemistry. 1998;49:901–951. doi: 10.1016/S0031-9422(97)01020-0. [DOI] [PubMed] [Google Scholar]
- 9.Choi YA, Tao R, Yonemori K, Sugiura A. Genomic distribution of three repetitive DNAs in cultivated hexaploid Diospyros spp. (D. kaki and D. virginiana) and their wild relatives. Genes Genet Syst. 2003;78:301–308. doi: 10.1266/ggs.78.301. [DOI] [PubMed] [Google Scholar]
- 10.Geng P, Ruan XF, Yang Y, Zhang HX. Analysis of genetic diversity of Diospyros spp. germplasm resources by using SSR markers. J Northwest A&F University (Natural Science Edition). 2010;38(12):190–6. 10.13207/j.cnki.jnwafu.2010.12.021.
- 11.Jing ZB, Ruan XF, Wang RZ, Yang Y. Genetic diversity and relationships between and within persimmon (Diospyros L.) wild species and cultivated varieties by SRAP markers. Plant Systematics Evol. 2013;299(8):1485–1492. doi: 10.1007/s00606-013-0810-1. [DOI] [Google Scholar]
- 12.Liang JJ, Liang YQ, Fu JM. Genetic relationships of Diospyros kaki and related Diospyros species using chloroplast DNA PCR-RFLP markers. Acta Hortic Sin. 2014;41(12):2474–2480. [Google Scholar]
- 13.Duangjai S, Wallnöffer B, Samuel R, Munzinger J, Chase MW. Generic delimitation and relationships in Ebenaceae sensu lato: evidence from six plastid DNA regions. Am J Bot. 2006;93:1808–1827. doi: 10.3732/ajb.93.12.1808. [DOI] [PubMed] [Google Scholar]
- 14.Duangjai S, Samuel R, Munzinger J, Forest F, Wallnöfer B, Barfuss MH, Fischer G, Chase MW. A multi-locus plastid phylogenetic analysis of the pantropical genus Diospyros (Ebenaceae), with an emphasis on the radiation and biogeographic origins of the new Caledonian endemic species. Mol Phylogenet Evol. 2009;52:602–620. doi: 10.1016/j.ympev.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 15.Du X. Y., Zhang Q. L., Luo Zheng-Rong. Comparison of four molecular markers for genetic analysis in Diospyros L. (Ebenaceae) Plant Systematics and Evolution. 2009;281(1-4):171–181. doi: 10.1007/s00606-009-0199-z. [DOI] [Google Scholar]
- 16.Suo Zhili, Zhang Chuanhong, Zheng Yongqi, He Lixia, Jin Xiaobai, Hou Boxin, Li Jiajue. Revealing genetic diversity of tree peonies at micro-evolution level with hyper-variable chloroplast markers and floral traits. Plant Cell Reports. 2012;31(12):2199–2213. doi: 10.1007/s00299-012-1330-0. [DOI] [PubMed] [Google Scholar]
- 17.Suo ZL, Chen LN, Pei D, Jin XB, Zhang HJ. A new nuclear DNA marker from ubiquitin ligase gene region for genetic diversity detection of walnut germplasm resources. Biotechnol Reports. 2015;5:40–45. doi: 10.1016/j.btre.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suo ZL, Li WY, Jin XB, Zhang HJ. A new nuclear DNA marker revealing both microsatellite variations and single nucleotide polymorphic loci: a case study on classification of cultivars in Lagerstroemia indica L. J Microb Biochem Technol. 2016;8:266–71. 10.4172/1948%2D5948.1000296.
- 19.Dong Wenpan, Liu Jing, Yu Jing, Wang Ling, Zhou Shiliang. Highly Variable Chloroplast Markers for Evaluating Plant Phylogeny at Low Taxonomic Levels and for DNA Barcoding. PLoS ONE. 2012;7(4):e35071. doi: 10.1371/journal.pone.0035071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong WP, Xu C, Cheng T, Lin K, Zhou SL. Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol Evol. 2013;5(5):989–997. doi: 10.1093/gbe/evt063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong WP, Liu H, Xu C, Zuo YJ, Chen ZJ, Zhou SL. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 2014;15:138. 10.1186/s12863-014-0138-z. [DOI] [PMC free article] [PubMed]
- 22.Dong WP, Dong WP, Xu C, Li CH, Sun JH, Zuo YJ, Shi S, Cheng T, Guo JJ, Zhou SL. ycf1, the most promising plastid DNA barcode of land plants. Sci Rep. 2015;5:8348. 10.1038/srep08348. [DOI] [PMC free article] [PubMed]
- 23.Dong Wenpan, Xu Chao, Li Delu, Jin Xiaobai, Li Ruili, Lu Qi, Suo Zhili. Comparative analysis of the complete chloroplast genome sequences in psammophyticHaloxylonspecies (Amaranthaceae) PeerJ. 2016;4:e2699. doi: 10.7717/peerj.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong WP, Xu C, Li WQ, Xie XM, Lu YZ, Liu YL, Jin XB, Suo ZL. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Front Plant Sci. 2017;8:1148. 10.3389/fpls.2017.01148. [DOI] [PMC free article] [PubMed]
- 25.He LX, Suo ZL, Zhang CH, Jin XB, Zhao DX, Zhao XQ, Hou BX, Deng CF. Classification of Chinese medicinal tree peony cultivars based on chloroplast DNA sequences. AASRI Procedia. 2012;1:344–352. doi: 10.1016/j.aasri.2012.06.053. [DOI] [Google Scholar]
- 26.Li WQ, Yang Y, Xie XM, Lu YZ, Chang Q, Jin XB, Suo ZL. Diospyros oleifera and D. deyangensis are revealed as the closest relatives to D. kaki by E3 ubiquitin-protein ligase UPL3 DNA sequences. Hans J. Agricultural Sci. 2018; 8(6): 657–673. 10.12677/hjas.2018.86100. (In Chinese with English abstract).
- 27.Fu Jianmin, Liu Huimin, Hu Jingjing, Liang Yuqin, Liang Jinjun, Wuyun Tana, Tan Xiaofeng. Five Complete Chloroplast Genome Sequences from Diospyros: Genome Organization and Comparative Analysis. PLOS ONE. 2016;11(7):e0159566. doi: 10.1371/journal.pone.0159566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu C, Dong WP, Li WQ, Lu YZ, Xie XM, Jin XB, Shi JP, He KH, Suo ZL. Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front Plant Sci. 2017;8:15. 10.3389/fpls.2017.00015. [DOI] [PMC free article] [PubMed]
- 29.Bankevich Anton, Nurk Sergey, Antipov Dmitry, Gurevich Alexey A., Dvorkin Mikhail, Kulikov Alexander S., Lesin Valery M., Nikolenko Sergey I., Pham Son, Prjibelski Andrey D., Pyshkin Alexey V., Sirotkin Alexander V., Vyahhi Nikolay, Tesler Glenn, Alekseyev Max A., Pevzner Pavel A. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., Drummond A. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyman S. K., Jansen R. K., Boore J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 32.Conant GC, Wolfe KH. GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics. 2008;24(6):861–862. doi: 10.1093/bioinformatics/btm598. [DOI] [PubMed] [Google Scholar]
- 33.Katoh K., Standley D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rambaut A. Se-Al: sequence alignment editor. Version 2.0. Oxford: University of Oxford, Department of Zoology; 1996. [Google Scholar]
- 35.Tamura Koichiro, Stecher Glen, Peterson Daniel, Filipski Alan, Kumar Sudhir. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Librado P., Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 37.Brown SD, Collins RA, Boyer S, Lefort MC, Malumbres-Olarte J, Vink CJ, Cruickshank RH. Spider: an R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Molecular Ecol Resources. 2012;12:562–565. doi: 10.1111/j.1755-0998.2011.03108.x. [DOI] [PubMed] [Google Scholar]
- 38.Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4b10. Sunderland. Massachusetts: Sinauer; 2003. [Google Scholar]
- 39.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 40.Ronquist F, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2002;11:2453–2465. doi: 10.1046/j.1365-294X.2002.01643.x. [DOI] [PubMed] [Google Scholar]
- 42.Provan J, Soranzo N, Wilson NJ, Goldstein DB, Powell W. A low mutation rate for chloroplast microsatellites. Genetics. 1999;153:943–947. doi: 10.1093/genetics/153.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perdereau Aude, Klaas Manfred, Barth Susanne, Hodkinson Trevor R. Plastid genome sequencing reveals biogeographical structure and extensive population genetic variation in wild populations ofPhalaris arundinaceaL. in north-western Europe. GCB Bioenergy. 2016;9(1):46–56. doi: 10.1111/gcbb.12362. [DOI] [Google Scholar]
- 44.Song Y, Wang SJ, Ding YM, Xu J, Li MF, Zhu SF, Chen NZ. Chloroplast genomic resource of Paris for species discrimination. Sci Rep. 2017;7:3427. doi: 10.1038/s41598-017-02083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu XQ, Drew BT, Yang JB, Gao LM, Li DZ. Comparative chloroplast genomes of eleven Schima (Theaceae) species: insights into DNA barcoding and phylogeny. PLoS One. 2017;12:e0178026. doi: 10.1371/journal.pone.0178026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hebert PDN, Cywinska A, Ball SL, DeWaard JR. Biological identifications through DNA barcodes. Proc Royal Soc London Series B-Biological Sciences. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drew BT, Sytsma KJ. Phylogenetics, biogeography, and staminal evolution in the tribe Mentheae (Lamiaceae) Am J Bot. 2012;99:933–953. doi: 10.3732/ajb.1100549. [DOI] [PubMed] [Google Scholar]
- 48.Parks M, Liston A, Cronn R. Newly developed primers for complete ycf1 amplification in Pinus (Pinaceae) chloroplasts with possible family-wide utility. Am J Bot. 2011;98:e185–e188. doi: 10.3732/ajb.1100088. [DOI] [PubMed] [Google Scholar]
- 49.Hernandez-Leon S, Gernandt DS, Perez de la Rosa JA, Jardon-Barbolla L. Phylogenetic relationships and species delimitation in Pinus section Trifoliae inferred from plastid DNA. PLoS One. 2013;8:e70501. doi: 10.1371/journal.pone.0070501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Vazquez L, Chen X, Li H, Zhang H, Liu Z, Zhao G. Development of chloroplast and nuclear DNA markers for Chinese oaks (Quercus subgenus Quercus) and assessment of their utility as DNA barcodes. Front Plant Sci. 2017;8:816. doi: 10.3389/fpls.2017.00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dastpak A, Osaloo SK, Maassoumi AA, Safar KN. Molecular phylogeny of Astragalus sect. Ammodendron (Fabaceae) inferred from chloroplast ycf1 gene. Ann Bot Fenn. 2018;55:75–82. doi: 10.5735/085.055.0108. [DOI] [Google Scholar]
- 52.Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J, Siripun KC, Winder CT, Schilling EE, Small RL. The tortoise and the hare II: relative utility of 21 non-coding chloroplast DNA sequences for phylogenetic analysis. Amer J Bot. 2005;92:142–166. doi: 10.3732/ajb.92.1.142. [DOI] [PubMed] [Google Scholar]
- 53.Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Amer J Bot. 2007;94:275–288. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- 54.Ng FSP. Diospyros roxburghii and the origin of Diospyros kaki. Malays Forest. 1978;41:43–50. [Google Scholar]
- 55.Yonemori K, Kanzaki S, Parfitt DE, Utsunomiya N, Subhadrabandhu S, Sugiura A. Phylogenetic relationship of Diospyros kaki (persimmon) to Diospyros spp. (Ebenaceae) of Thailand and four temperate zone Diospyros spp. from an analysis of RFLP variation in amplified cpDNA. Genome. 1998;41:173–82.
- 56.Zhang HP, Zhang JY, Liu QL. Research progress on the advantages of persimmon germplasm resources and genetic breeding in China. Heilongjiang Agricultural Sci. 2016;9:149–151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NJ trees of Diospyros taxa based on each of the ten chloroplast barcodes, showing the resolutions of each different locus for revealing the phylogeny. The figures above the lines are the bootstrap support values for the clades. (PDF 506 kb)
Data Availability Statement
The complete chloroplast genomes of D. cathayensis, D. deyangensis, D. rhombifolia and D. virginiana were submitted to the NCBI database with GenBank accession numbers MF288576 (D. cathayensis), MF288575 (D. deyangensis), MF288578 (D. rhombifolia) and MF288577 (D. virginiana).