

Abstract
Telomeres are dynamic nucleoprotein-DNA structures that cap and protect linear chromosome ends. Because telomeres shorten progressively with each replication, they impose a functional limit on the number of times a cell can divide. Critically short telomeres trigger cellular senescence in normal cells, or genomic instability in pre-malignant cells, which contribute to numerous degenerative and aging-related diseases including cancer. Therefore, a detailed understanding of the mechanisms of telomere loss and preservation is important for human health. Numerous studies have shown that oxidative stress is associated with accelerated telomere shortening and dysfunction. Oxidative stress caused by inflammation, intrinsic cell factors or environmental exposures, contributes to the pathogenesis of many degenerative diseases and cancer. Here we review the studies demonstrating associations between oxidative stress and accelerated telomere attrition in human tissue, mice and cell culture, and discuss possible mechanisms and cellular pathways that protect telomeres from oxidative damage.
Keywords: telomeres, oxidative stress, oxidative DNA damage, base excision repair
Graphical Abstract
1. Introduction
Telomeres address two critical problems posed by the organization of mammalian genomes into linear chromosomes, thereby placing them at the forefront of aging and cancer research. First, the inability to complete DNA replication to the very chromosome ends leads to gradual loss of about 20–50 base pairs every cell division. Thus, progressive chromosome shortening occurs during cell replication, and is observed with aging (Harley et al., 1990). Germ cells, some stem cells, and most cancer cells prevent shortening by expressing the enzyme telomerase, which lengthens chromosomes by adding consecutive 5′-TTAGGG-3′ repeats. In humans, telomeres consist of approximately 10–15 kilobases of TTAGGG duplex repeats and end in a single stranded 3′ tail, which serves as the substrate for telomerase (Palm and de Lange, 2008). However, most human somatic cells lack telomerase, and when the telomeres reach a critically short length, they can no longer perform the second crucial function – chromosome end protection. Critically short telomeres lack sufficient binding sites for the six-member protein complex, termed shelterin, which remodels telomeres into a “capped” structure. Loss of the telomere “cap” exposes the chromosome end to potential degradation, and false recognition by the DNA damage response proteins as chromosome breaks (d’Adda di Fagagna et al., 2003; de Lange, 2009). In normal cells this triggers senescence and loss of replicative capacity. Senescent cells accumulate with age, secrete inflammatory cytokines, and have well-established roles in promoting degenerative diseases and pathology with aging (reviewed in (Campisi et al., 2011)). However, pre-malignant cells lacking a functional p53 pathway bypass senescence and continue dividing. The uncapped ends are processed by the DNA double strand break repair machinery leading to chromosome fusions and rampant chromosomal instability, which kills most cells, but drives malignant transformation in the survivors (Artandi et al., 2000). Most survivors upregulate telomerase to maintain telomeres for unlimited proliferation, but about 15% use the alternative lengthening of telomeres (ALT) pathway (reviewed in (Opresko and Shay, 2017)). Given the critical roles of telomeres in cancer and aging, the rate at which telomeres shorten can profoundly influence genome stability, as well as the overall health and well being of an organism (i.e. healthspan).
Are telomeres “rusting” as we age? While numerous genetic and environmental factors are associated with accelerated telomere shortening, the most commonly cited underlying mechanism is oxidative stress. Oxidative stress results from an imbalance between the production of reactive oxygen species (ROS) and cellular antioxidant defenses, and contributes to the pathogenesis of numerous human diseases including cancer. These include neurological, pulmonary, and cardiovascular diseases, along with atherosclerosis, diabetes, obesity and arthritis (reviewed in (Hegde et al., 2012; Lonkar and Dedon, 2011; Malinin et al., 2011; Mao et al., 2017; Reuter et al., 2010)) (Figure 1). For most of these diseases the primary source of ROS is inflammation and/or mitochondrial dysfunction, since ROS is generated by immune cells in response to infection or injury and from oxygen metabolism. ROS is elevated at sites of chronic inflammation, and is prevalent in chronic inflammatory diseases such as ulcerative colitis, Barrett’s esophagus and hepatitis (Lonkar and Dedon, 2011). Chronic inflammation is proposed to cause more than 20% of cancers based on epidemiology studies, and is a major risk factor for aging-related diseases (Bouvard et al., 2009; Lonkar and Dedon, 2011). Oxidative stress is also associated with numerous environmental exposures including radon, cigarette smoke, pollution, toxic metals, ultraviolet light and pesticides (Aseervatham et al., 2013; Baltazar et al., 2014; Poljsak and Fink, 2014). Here again, inflammation and mitochondrial dysfunction are often proposed to be responsible for the elevated ROS. Numerous studies have shown that most of the degenerative and inflammatory diseases, and many cancers, characterized by oxidative stress are also associated with accelerated telomere shortening (Sanders et al., 2011; Valdes et al., 2005; Zhang et al., 2016)(Figure 1). These studies are based on measurement of average telomere length in white blood cells (WBC), inflamed tissues and tumors from patients (see below). In addition, many of the environmental exposures that lead to ROS elevation, are also associated with shortened telomeres in epidemiology studies based on WBC analysis (Martens and Nawrot, 2016; Valdes et al., 2005; Zhang et al., 2013). In this review we will discuss the evidence from studies in human tissues, animal models and cell culture that oxidative stress accelerates telomere shortening and dysfunction. We will then elaborate on possible mechanisms, and will focus primarily the processing of oxidative damage to DNA bases in telomeres.
Figure 1.

Diseases associated with oxidative stress, chronic inflammation and shortened telomeres. The schematic summarizes various diseases that are characterized by elevated reactive oxygen species as well as shortened average telomere lengths as measured either in white blood cells or in the affected/inflamed tissues, compared to unaffected individuals or tissues. See text for details and citations.
2. Oxidative stress and accelerated telomere shortening or dysfunction
2.1 Evidence from human studies
A recent review documented that 6 out of 8 human population studies reported markers for oxidative stress correlated with shorter average telomere lengths, as measured in WBCs (Reichert and Stier, 2017). Similar correlations were observed in populations with higher perceived psychological stress and higher inflammatory loads (Epel et al., 2004; O’Donovan et al., 2011). While interesting, it is difficult to derive mechanistic information from these types of correlative studies. More convincing evidence for direct roles of oxidative stress in telomere length homeostasis comes from studies of inflamed tissues. Chronic inflammatory diseases are characterized by elevated ROS and development of pre-neoplastic lesions that can progress to cancer. Ulcerative colitis (UC) and Barrett’s esophagus patients present with shortened telomeres in affected mucosa compared to normal stroma and controls from unaffected individuals, as revealed by telomere quantitative fluorescence in situ hybridization (qFISH) or high resolution Southern blot (Finley et al., 2006; Letsolo et al., 2017; O’Sullivan et al., 2002). Shorter telomeres correlate with greater chromosomal instability and elevated infiltrating leukocytes (i.e. inflammation) (Baird et al., 2003; Risques et al., 2011). Shorter telomeres are observed in inflamed livers from chronic hepatitis and liver cirrhosis, and in atherosclerotic lesions, compared to unaffected tissues (Aikata et al., 2000; Nzietchueng et al., 2011; Rey et al., 2017). Hyper-proliferation of cells can contribute to telomere shortening. However, in a recent review Graham and Meeker argue that at normal telomere shortening rates, prostate cells would require ~100 years of proliferation to achieve the critical telomere lengths observed in prostate cancer (Graham and Meeker, 2017). They propose oxidative stress in prostatic inflammatory atrophy and precancerous prostatic intraepithelial neoplasia (PIN) cooperates with increased proliferation to accelerate telomere shortening. Finally, critically short telomeres have a well-recognized role in both sporadic and familial pulmonary fibrosis. Familial forms are caused by mutations in telomere maintenance genes (Armanios and Blackburn, 2012). Mechanisms of telomere shortening in sporadic disease are not understood, but may relate to oxidative stress (Kliment and Oury, 2010). Shortened telomeres are observed in alveolar epithelium from sporadic IPF lungs compared to controls, and in alveolar type 2 cells from fibrotic areas compared to non-fibrotic areas (Alder et al., 2008; Snetselaar et al., 2017). In addition, pulmonary vascular endothelial cells from patients with chronic obstructive pulmonary disease (COPD) showed shorter telomeres compared to normal controls, and telomere length correlated with inflammatory loads (Amsellem et al., 2011). Numerous examples of telomere length analysis in chronic inflammatory diseases consistently reveal shortened telomeres in inflamed tissues, compared to normal tissue from the same patient or from unaffected individuals. More mechanistic studies are required to determine how inflammation and oxidative stress accelerate telomere shortening.
2.2 Evidence from mouse models
Studying factors that influence telomere length in mice is challenging because laboratory inbred mice have very long telomeres (~20–40 kb). To overcome this barrier, studies have used late generation telomerase deficient mice bred to harbor shorter telomeres, or the wild-derived CAST/Ei strain which possess telomere lengths similar to humans. Chronically exposing CAST/Ei mice to L-buthione sufoximine (BSO), which depletes glutathione antioxidant, accelerated telomere shortening in fat, skin, tail, and testis, and increased the level of oxidized proteins (Cattan et al., 2008). Other tissues, including lung, heart and liver, did not show BSO-induced changes in average telomere length. The authors suggested some tissues may have higher antioxidant capacities. Alternatively, average telomere length measurement by Southern blot may lack the sensitivity required to detect telomere changes. The few critically short telomeres that are sufficient to trigger senescence (Kaul et al., 2012) may not alter average telomere lengths. A recent study provides evidence that chronic inflammation in mice leads to telomere dysfunction and premature aging. Jurk and colleagues examined Nfkb1 knock out mice that lack NF-κB proteins p105 and p50, which regulate inflammatory gene expression (Jurk et al., 2014). These mice showed inflammatory and premature aging phenotypes, as well as increased cell senescence and telomere dysfunction, that were rescued with anti-inflammatory and antioxidant treatments (Jurk et al., 2014). Telomere dysfunction was marked by localization of the DNA damage response protein 53BP1 to telomeres, while changes in average telomere length were not observed. Lack of detectable length changes may be due to the very long telomeres in this inbred mouse strain.
Studies in telomerase deficient mice (Tert−/− or Terc−/−) that are bred for shortened telomeres have revealed interesting links between telomere dysfunction and mitochondrial dysfunction in highly proliferative organs as well as more quiescent organs. Analysis of hematopoietic cells, heart, and liver in these mice showed hallmarks of mitochondria dysfunction including decreased mitochondrial biogenesis, reduced oxidative phosphorylation and ATP generation, and increased ROS (Sahin et al., 2011). The mitochondrial effects were mediated by suppression of transcriptional co-activators PGC-1α/β through telomere dysfunction-induced activation of p53 (Sahin et al., 2011). Importantly, because dysfunctional mitochondria generate more ROS, this sets up a vicious cycle in which telomeres may suffer further insult from oxidative damage. Recent studies confirm that the cross-talk between telomere dysfunction and mitochondrial dysfunction impacts the heart. Duchenne muscular dystrophy (DMD) can be recapitulated in transgenic mice lacking both dystrophin and telomerase that are bred to possess shortened telomeres, resembling human telomere length (Chang et al., 2016). Loss of dystrophin alone is insufficient to induce DMD symptoms in mice with long telomeres, indicating that “humanized” telomere lengths are essential for fully recapitulating DMD. Telomere shortening occurred in the non-proliferating cardiomyocytes, and critically short telomeres activated p53-mediated repression of PGC-1α/β, triggering mitochondrial dysfunction, which was suppressed with a mitochondrial anti-oxidant (Chang et al., 2016). Whether protecting the mitochondria also prevents telomere shortening in this model has yet to be examined. More work is required to determine how oxidative stress promotes telomere shortening in the absence of proliferation, and whether this effect extends to other non-proliferating cells such as neurons.
2.3 Evidence from cell culture
Numerous studies have reported that oxidative stress and exposure to ROS-generating agents accelerate telomere shortening in cultured human cells. A review of 22 studies consistently showed that mild oxidative stress accelerated telomere shortening in cultured normal human fibroblasts and endothelial cells, whereas antioxidants and free radical scavengers decreased shortening rates and increased proliferative lifespan (von Zglinicki, 2002). In these studies oxidative stress was achieved by culturing cells at 20% O2 or by exposing to oxidants such as hydrogen peroxide. Subsequent studies confirmed that culturing cells at 20% O2 accelerates telomere shortening, compared to physiological 3–5% O2 (Coluzzi et al., 2014; Forsyth et al., 2003; Richter and von Zglinicki, 2007; Wang et al., 2010). Other studies in mesenchymal stem cells showed oxidative stress induces stochastic telomere loss, indicated by an increase in critically short telomeres (Harbo et al., 2012). Just five critically short telomeres can trigger senescence in normal cells (Kaul et al., 2012). Finally, oxidative stress induced by mitochondrial dysfunction in cultured cells also accelerates telomere shortening and/or causes telomere dysfunction, even in cells expressing telomerase (Ahmed et al., 2008; Passos et al., 2007; Saretzki et al., 2003). These studies further support the link between mitochondrial and telomere dysfunction.
3. Mechanisms of oxidative stress induced telomere changes
Several mechanistic models have been proposed to explain how oxidative stress accelerates telomere shortening. One possibility is that oxidative stress triggers cell death and/or senescence, and to compensate the survivors undergo more cell divisions, leading to increased telomere shortening. However, this does not explain accelerated telomere shortening observed under non-cytotoxic and mild oxidative stress conditions (von Zglinicki, 2002). One widely cited model suggests that ROS induces single strand breaks (SSB)s at telomeres directly, or as intermediates in lesion repair, leading to replication fork collapse and telomere loss (Figure 2) (von Zglinicki, 2002). Alternatively, lesions that impede telomere replication can cause an accumulation of unreplicated ssDNA, and manifest as multi-telomeric foci at chromatid ends termed fragile telomeres (Figure 2)(Sfeir et al., 2009). Replication interference does not explain how oxidative stress affects telomeres in non-proliferative cells. Other possibilities are that oxidative lesions interfere with shelterin binding or transcription at telomeres into TERRA transcripts. Finally, processing of oxidative lesions may lead to changes in telomere repeat number. These possibilities are explored in more detail below, along with evidence from studies that support these models.
Figure 2.

Consequences of replication fork stalling and blocks at telomeres. The schematic shows a model for how telomere fragility or telomere losses arise from DNA lesions that stall or block replication fork progression, respectively. DNA replication fork encounters with single strand breaks (SSBs) can cause the fork to collapse into a double strand break. Fragile telomeres manifest as multi-telomeric foci at a chromatid end, and are proposed to result from uncondensed regions arising from accumulated unreplicated ssDNA. Telomeres losses manifest as chromatid ends lacking sufficient telomeric DNA for detection with a telomeric probe. Telomerase can suppress telomere losses by extending a pre-maturely truncated telomere.
3.1 ROS-induced damage to telomeric DNA
Is oxidative damage to telomeric DNA responsible for accelerated telomere shortening under oxidative stress? When various ROS react with DNA they can generate upwards of 100 different types of oxidatively damaged bases (Cadet and Wagner, 2013). These lesions include damaged pyrimidines and purines, as well as SSBs and abasic sites. Guanine is the most susceptible of the natural bases to oxidation, commonly generating 8-oxoguanine (8-oxoG), which is even more sensitive to oxidation, ultimately giving rise to hydantoin lesions (Fleming and Burrows, 2017; Luo et al., 2001). Biochemical studies show TTAGGG repeats are preferred sites for iron binding and iron mediated Fenton reactions, which generate hydroxyl radicals that induce cleavage 5′ of GGG (Henle et al., 1999; Oikawa and Kawanishi, 1999; Oikawa et al., 2001). Consistent with this, several studies reported more SSBs or 8-oxoG lesions in telomeres compared to microsatellite repeats and bulk genomic DNA, after cellular exposures to oxidizing agents (Coluzzi et al., 2014; Petersen et al., 1998; Rhee et al., 2011; Wang et al., 2010). Antioxidant peroxiredoxin 1 (PRDX1), which scavenges H2O2, is enriched at telomeres and PRDX1 loss leads to preferential damage at the telomeres (Aeby et al., 2016). Whether the preferential accumulation of oxidative damage at telomeres is due to increased damage susceptibility and/or decreased repair is unresolved. Indeed, 8-oxoG cannot be repaired in the context of folded telomeric G-quadruplex structures (Zhou et al., 2013). Cellular studies suggest shelterin protein TRF2 may interfere with base excision repair (BER) (Richter et al., 2007), whereas biochemical studies indicate shelterin proteins enhance BER in vitro (Miller et al., 2012). More work is required to determine how efficiently oxidative base damage is repaired at telomeres, compared to elsewhere in the genome.
3.2 Base excision repair of oxidative base damage
Most oxidative lesions are repaired by BER, which is essential for genome stability and for preserving telomeres. These lesions can be cytotoxic or mutagenic, and thereby promote carcinogenesis (for review see (Wallace et al., 2012)). In BER, a mono-functional DNA glycosylase recognizes and removes specific DNA lesions, thereby generating an abasic site. APE1 endonuclease cleaves 5′ of the abasic site to generate an SSBwith a 3′hydroxyl and a 5′ sugar phosphate. DNA polymerase (Pol) β removes the 5′ sugar phosphate via its lyase activity, and fills the gap by templated DNA synthesis. DNA ligase I or III seals the nick (Srivastava et al., 1998). While these core steps are conserved, several variations in BER (sub-pathways) exist, assisted by additional proteins. In mammalian cells the five glycosylases that recognize oxidative base damage include OGG1, NTHL1, NEIL1, NEIL2 and NEIL3 (Krokan and Bjoras, 2013; Wallace, 2013). They are bi-functional glycosylases, meaning that they both remove the damaged base and cleave the DNA backbone 3′ tothe abasic site. The resulting 3′ blocking sugar or phosphate group is removed by APE1 or polynucleotide kinase, respectively (Krokan and Bjoras, 2013; Wallace, 2013). BER normally involves the replacement of a single nucleotide, but if the 5′end is refractory to Pol β processing, then Pol δ or ε adds several nucleotides, generating a displaced DNA flap that is cleaved by FEN1 endonuclease. BER intermediates can be cytotoxic and therefore, efficient hand-off to each downstream processing enzyme is essential (Sobol et al., 2000). XRCC1 scaffolding protein and modifying enzyme Poly(ADP)-Ribose Polymerase 1 (PARP1) have important roles in coordinating and recruiting proteins involved in SSB repair and BER.
3.3 Processing of 8-oxoG at telomeres
Given that 8-oxoG is one of the most common oxidative lesions, multiple pathways exist to deal with this form of damage. When 8-oxoG forms opposite C it is recognized by OGG1 glycosylase, which removes the lesion, generating an abasic site (Figure 3ii). Biochemical studies show OGG1 glycosylase activity is stimulated by APE1, XPC, and NEIL1 (Hill et al., 2001; Mokkapati et al., 2004; Vidal et al., 2001). OGG1 can further process this site with its AP-lyase function, however OGG1 has high affinity for abasic sites, and becomes trapped by its own product (Hill et al., 2001; Morland et al., 2005). More likely, APE1 endonuclease removes the deoxyribose moiety, generating an SSB. However, OGG1 cannot remove 8-oxoG when present in ssDNA or a folded G-quadruplex structure (Zhou et al., 2013). This raises the question of how 8-oxoG is repaired in the ssDNA regions of the telomeric overhang or the t-loop/D-loop structure. Remarkably, an unbiased screen in yeast for genes that alter telomere length revealed that ogg1 deletion strains had longer telomeres than wild type (Askree et al., 2004). An independent study confirmed this result and reported that the lengthening was partly telomerase dependent (Lu and Liu, 2010). The longer telomere phenotype is recapitulated in Ogg1−/− mice in vivo, but when the cells from these mice are cultured in pro-oxidant conditions in vitro they exhibit increased telomere shortening, loss and aberrations compared to wild type (Wang et al., 2010). This may represent a hormesis situation, in which low levels of 8-oxoG at telomeres promotes lengthening, whereas high amounts cause telomere losses and aberrations. Mechanistically, we showed that a single 8-oxoG in telomeric ssDNA disrupts the folded G-quadruplex structures that impede telomerase loading, thereby promoting telomere elongation (Fouquerel et al., 2016a). In contrast, how oxidative stress synergizes with unrepaired 8-oxoG lesions to induce telomere loss is Ogg1−/− cells is unclear, since this lesion is not a strong block to DNA replication or transcription. The increased 8-oxoG at telomeres may lead to shelterin disruption (Lu and Liu, 2010; Opresko et al., 2005) or synergize with other ROS-induced lesions (i.e. SSBs, oxidized pyrimidines) to disrupt telomere processing and/or function.
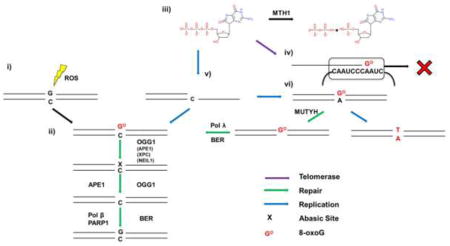
Figure 3.

Processing of 8-oxoG. When 8-oxoG forms opposite C by direct oxidation (i), it is recognized by OGG1 (ii) which removes the modified base. OGG1 or APE1 then processes the abasic site to make it a suitable substrate for the remaining steps of BER. When dGTP is oxidized to 8-oxoGdTP (iii), and escapes degradation by MTH1, incorporation by telomerase results in termination of telomere synthesis (iv). If a DNA polymerase incorporates 8-oxodGTP opposite C and it escapes BER, a subsequent round of replication can result in incorporation of A opposite 8-oxoG (vi). MUTYH is able to excise the A, producing a gap filled in by Pol λ or Pol β, avoiding a G:T/C:A transversion mutation. Proteins in parentheses can stimulate OGG1 activity.
If 8-oxoG in the DNA escapes repair, a round of DNA replication can result in misincorporation of A opposite 8-oxoG (Figure 3vi). This is because 8-oxoG miscodes for A. Changes in the telomeric sequence would disrupt shelterin binding. MUTYH glycosylase excises A opposite 8-oxoG, producing a gap to be filled in by Pol λ or Pol β in BER (Maga et al., 2007; Markkanen, 2017). If this error is not corrected, such mispairing would convert TTA(8-oxoG)GG repeats to TTATGG variants, which are not commonly observed (Lee et al., 2014) (Figure 4B). This suggests that either 8-oxoG is efficiently repaired at telomeres prior to replication, or that MUTYH efficiently removes A opposite 8-oxoG at telomeres to prevent mutations. Sequencing telomeres from oxidative stress conditions in OGG1 and MUTYH singly and doubly deficient cells, is needed to fully elucidate the mutagenic potential of 8-oxoG at telomeres, and the roles for these glycosylases in preserving telomeric repeats.
Figure 4.

Models for 8-oxoG induced mutations at telomeres. A) The schematic shows three possible scenarios of 8-oxodGTP insertion opposite A during telomere replication and the resulting change in telomeric repeat sequence. B) If 8-oxoG forms directly in telomeric repeats, the misincorporation of dATP during telomere replication would alter the telomeric repeat to TTATGG, TTAGTG or TTAGGT depending on the lesion position. C) The variant GTAGGG repeat can arise if telomerase misincorporates 8-oxodGTP opposite rA during telomeric DNA synthesis, and the mispair is extended.
Free dNTPs are even more susceptible to oxidative damage than bases in duplex DNA and chromatin. As such, cells possess sanitases including MTH1, which hydrolyze oxidized dNTPs before they can be incorporated into the genome by DNA polymerases during replication (Figure 3) (Rudd et al., 2016). Oxidized dNTPs may be even more deleterious to telomeres than oxidative base damage within the duplex DNA. First, the preferential insertion of 8-oxodGTP opposite A during replication of the 5–15 kb telomere duplex would convert TTAGGG repeats to GTAGGG and TGAGGG repeats (Figure 4A). Interestingly, these are the most common variant repeats reported from telomere sequencing studies (Lee et al., 2014). Second, telomerase can incorporate 8-oxodGTP during telomere extension, and like most DNA polymerases, it preferentially misincorporates the oxidized dGTP opposite rA (Aeby et al., 2016; Fouquerel et al., 2016a). Lee et al. reported that GTAGGG variants occur more frequently in distal telomere locations in telomerase positive cells, suggesting they are generated by telomerase errors (Lee et al., 2014) (Figure 4C). However, 8-oxodGTP is a telomerase chain terminator in vitro (Aeby et al., 2016; Fouquerel et al., 2016a). Consistent with this, acute MTH1 depletion increases telomere loss and cell death in telomerase positive cancer cells harboring critically short telomeres, but not in cancer cell with longer telomere reserves (Fouquerel et al., 2016a). This suggests that 8-oxodGTP, and potentially oxidized versions of dATP, which are normally removed by MTH1, inhibit telomerase restoration of critically short telomeres. Some cancer cell lines are more sensitive to MTH1 inhibition, compared to normal cells, which may be due differences in the reliance on telomerase activity for short-term survival (Gad et al., 2014). Whether MTH1 inhibition leads to mutagenesis at telomeres remains to be determined. Although, 8-oxodGTP is a telomerase chain terminator, factors such as POT1-TPP1, may assist telomerase extension following misincorporation of damaged dNTPs during telomere extension in cells. Specialized DNA polymerases promote DNA synthesis from damaged bases to enable continued replication (Sale et al., 2012). Whether a similar mechanism exists to assist telomerase extension from a terminal 8-oxoG remains unknown.
3.4 Processing of other oxidized purines
While 8-oxoG is primarily mutagenic, other oxidized purines are more cytotoxic due to their ability to block DNA replication and transcription. Since 8-oxoG has a lower redox potential than the normal unmodified bases, it can be further oxidized to other lesions including spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh) (Luo et al., 2001). These distorting hydantoin lesions impede DNA replication and transcription (Henderson et al., 2003; Kolbanovskiy et al., 2017), but can be removed by any of the three NEIL glycosylases (reviewed in (Wallace, 2013)). Interestingly, NEIL1 and mNeil3 remove Sp and Gh from telomeric G-quadruplex structures and ssDNA, whereas 8-oxoG cannot be removed (Zhou et al., 2013). How BER proceeds in the absence of a templating base is unclear, but the processing of lesions in telomeric ssDNA and G-quadruplexes has important implications for telomere integrity. Cellular studies show NEIL3 localizes to telomeres during the S/G2 cell cycle phase, and NEIL3 depletion increases telomere loss, fusions and aberrations, and consequent anaphase DNA bridges (Zhou et al., 2017). The offending lesion(s) responsible for telomere loss is difficult to define because NEIL glycosylases remove several lesion types including ring opened 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) and oxidized pyrimidines (see below). Interestingly, both Ogg1−/− mice and Neil2−/− mice show increased susceptibility to LPS and oxidative stress induced inflammation (Aguilera-Aguirre et al., 2014; Chakraborty et al., 2015). Culturing cells derived from Neil2−/− mice at 20% O2 increases telomere losses compared to controls, similar to Ogg1−/− cells (see above), as observed by telomere FISH (Chakraborty et al., 2015). Since NEIL2 is implicated in BER during transcription, unrepaired hydantoin lesions may interfere with generation of telomeric TERRA transcripts. Roles for NEIL1 at telomeres have not been examined. However, Neil1, Neil2, and Neil3 triple knock out mice did not exhibit telomere shortening as measure by qPCR (Rolseth et al., 2017). Analyses using more sensitive measurements such as qFISH could be more revealing. Collectively, these studies suggest that NEIL glycoslyases have important roles in protecting telomeres against ROS-induced base damage, particularly to preserve DNA replication and transcription at telomeres.
3.5 Processing of oxidized pyrimidines
Pyrimidine bases are also susceptible to damage by free radicals, giving rise to various lesions including thymine glycol (Tg), 5-hydroxycytosine, and 5-hydroxyuracil. Tg is the most common oxidized thymine lesion and is cytotoxic because it can block DNA replication (McNulty et al., 1998). These modified bases are removed either by NTHL1 glycosylase in duplex DNA, or by NEIL glycosylases in duplex or ssDNA (reviewed in (Krokan and Bjoras, 2013; Wallace, 2013)). However, only Neil3 can excise Tg from a telomeric G-quadruplex and shows a strong preference for excising Tg from telomeric versus non-telomeric duplex DNA (Zhou et al., 2013). Therefore, unrepaired Tg lesions may be partly responsible for the telomere defects observed in Neil3 and/or Neil2 deficient cells (Chakraborty et al., 2015; Zhou et al., 2017). Nth1−/− mice exhibit increased fragile telomeres in vivo, and cells derived from these mice show increased telomere shortening, loss, dysfunction and aberrations when cultured at 20% oxygen, compared to wild type controls (Vallabhaneni et al., 2013), similar to Ogg1−/− cells (see above). Furthermore, telomerase deficiency exacerbates telomere shortening and telomere loss phenotypes in Nth1−/− cells, suggesting telomerase is able to partly restore telomeres rendered critically short by replication failures (Vallabhaneni et al., 2013) (Figure 2). Telomeres from Nth1−/− mice show a higher frequency of oxidized pyrimidines. Biochemical and biophysical studies indicate that unlike 8-oxoG, the presence of a single Tg only slightly disrupts telomeric G-quadruplexes (Lee et al., 2017). However, Tg alters the structural conformations and dynamics of the telomeric G-quadruplex in a manner that favors telomerase binding (Lee et al., 2017). These studies reveal that Tg likely causes telomere defects through interference with telomere replication rather than telomerase inhibition.
4. Roles for other repair proteins in processing oxidative base damage
While the members of the BER pathway are well known, studies show proteins from other repair pathways also promote the repair of oxidative lesions. For example, Nek7 kinase is recruited to telomeres after the localized induction of superoxide anion, and stabilizes shelterin TRF1 (Tan et al., 2017). Xeroderma Pigmentosum (XP) complementation group C protein, which is an integral component of nucleotide excision repair (NER), stimulates the activity of both OGG1 and thymine DNA glycosylase in vitro (D’Errico et al., 2006; Shimizu et al., 2003; Shimizu et al., 2010). Additionally, XPG, a structure-specific endonuclease, and CSB (Cockayne Syndrome Type B), stimulate the activity of NTH1 and NEIL2 glycosylases respectively (Aamann et al., 2014; Klungland et al., 1999). Treatment of cell lines deficient in XPC, XPB, XPD, and CSB with oxidants demonstrated an oxidized purine repair defect, and/or increased cell death and mutagenesis compared with normal cells (D’Errico et al., 2006; Gopalakrishnan et al., 2010; Melis et al., 2013; Tuo et al., 2003). Similarly, several NER proteins have been shown to protect telomeres against oxidative DNA damage. Xpc−/− mouse fibroblasts grown at 20% O2 display a fragile telomere phenotype, that can be rescued at 3% O2 (Stout and Blasco, 2013). Human cells deficient in XPB or XPD show elevated levels of chromosome ends lacking telomeric DNA following H2O2 treatment (Gopalakrishnan et al., 2010). Interestingly, multiple studies reported that XPA does not affect general or telomere specific oxidative repair (D’Errico et al., 2006; Gopalakrishnan et al., 2010; Klungland et al., 1999). Whether these proteins protect telomeres by stimulating BER, or by removing oxidative lesions through NER, remains to be determined.
Mismatch repair also contributes to oxidative damage repair in human cells. Loss of mismatch repair proteins MSH2 or MLH1 elevates both the basal and H2O2 treated levels of 8-oxoG in mouse and human cell lines (Colussi et al., 2002). This effect was reduced when MTH1 was overexpressed, indicating MMR processes 8-oxoG misincorporated during replication or repair events. Interestingly, the MMR complex MutSα recognizes 8-oxoG opposite A or C very poorly, but has strong affinity for 8-oxoG opposite T or G (Larson et al., 2003; Mazurek et al., 2002). While these mispairings are unlikely, 8-oxoG opposite C is the preferred substrate of OGG1, and A opposite 8-oxoG is the preferred substrate of MUTYH. Therefore, it makes sense that cells would have evolved a mechanism for dealing with the remaining two 8-oxoG pairings, via MutSα. These studies also raise the possibility that MMR proteins are also important for protecting telomeres from oxidative base damage.
5. Consequences of defects during repair of oxidative lesions
Processing of oxidative DNA damage can be more detrimental than the initial damage, if toxic intermediates arise in the repair pathway that are not properly resolved. PARP1/2 and poly(ADP-ribosyl)ation (PAR) function in BER following base damage conversion to an SSB (Figure 3). However, roles in preserving telomeres under oxidative stress remains controversial. Two studies reported extensive telomere shortening, as well as telomere losses and chromosomes fusions in cells derived from PARP1−/− mice (d’Adda di Fagagna et al., 1999; Tong et al., 2001). Pharmacologic or genetic inhibition of PARP1 in HeLa and hamster cells, presumably cultured at 20% O2, corroborate these observations (Beneke et al., 2008). Similarly cultured PARP2−/− mouse cells show an increased frequency of telomere losses (Dantzer et al., 2004). Interestingly, telomerase activity was not altered in PARP1−/− or PARP2−/− cells, or in HeLa cells treated with a PARP1 inhibitor, ruling out modulation of telomerase activity by PARP enzymes in these experimental conditions. In contrast, an independent study reported no telomere shortening or increased chromosome fusions in cells derived from PARP1−/− mice generated by disrupting a different gene exon (Samper et al., 2001). However, these cells displayed a significant increase in chromosome end-to-end fusions after 26 population doublings. This suggest culture conditions, such as oxygen levels that can cause oxidative stress, may be responsible for the increased telomere aberrations in the PARP1−/− cells in which toxic BER intermediates such as SSBs can accumulate. Consistent with this, PARP1−/− cells from the above studies show increased telomere losses, end-to-end fusions and telomere shortening, compared to wild type cells, after exposures to ROS-generating agents including H2O2, X-rays or arsenite (Gomez et al., 2006; Poonepalli et al., 2005). Conversely, depletion of poly(ADP-ribose) glycohydrolase (PARG), which is responsible for PAR degradation, protects against irradiation-induced telomere defects (Ame et al., 2009). Finally, PARP1 and PARP2 interact with shelterin TRF2, which may serve to regulate PARP1/2 at damaged telomeres (Dantzer et al., 2004; Gomez et al., 2006). Taken together, these studies reveal that the telomere defects observed in absence of PARP1 or PARP2 most likely arise from aborted BER of base damage or SSBs.
As mentioned above, if the ssDNA intermediates that arise during BER persist or accumulate, they can lead to replication fork collapse and subsequent DSB formation (Ebrahimkhani et al., 2014; Sobol et al., 2000) (Figure 2). Failures in repair of SSBs leads to numerous neurological disorders (reviewed in (Rulten and Caldecott, 2013)). The impact of SSBs or processing of oxidative base damage in telomeres in neural cells remains to be examined. The repair of closely spaced damaged bases (i.e. clustered lesions) on opposing strands can also lead to a DSB (Cannan et al., 2014). Importantly, the efficiency and accuracy of DSB repair at the telomeres may be comprised or altered by the presence of shelterin proteins and the highly repetitive nature of the sequence (reviewed in (Doksani and de Lange, 2014)). Finally, the repair of 8-oxoG though OGG1 mediated BER causes the pathogenic CAG expansion in a mouse model of Huntington’s disease (Kovtun et al., 2007). This raises the possibility that BER processing of 8-oxoG in telomeric repeats may also lead to changes in telomeric repeat number. Similar to CAG repeats, telomeric TTAGGG repeats are also capable of forming alternate structures (i.e. G-quadruplexes) that could impact repair processing (reviewed in (Fouquerel et al., 2016b)).
6. Perspective
Accumulating evidence indicates that oxidative stress correlates with accelerated telomere shortening and dysfunction in studies from human tissues, mouse models, cell culture and biochemical experiments. As discussed in this review, several mechanisms have been proposed to explain how elevated ROS alters telomere length homeostasis; the most prominent being oxidative DNA damage. However, many questions remain. Given that ROS can damage numerous cellular components, it is difficult to determine whether changes in telomere length and integrity under oxidative stress conditions are due to indirect factors, or direct damage to the telomeres. Tools to selectively induce oxidative base damage at telomeres will be useful for elucidating how the formation and processing of oxidative lesions impacts telomere maintenance, cellular function, as well as organism health and aging. Answering the question of whether telomeres are more susceptible to oxidative base damage in cells requires more accurate tools and methods to measure and quantify various types of oxidative lesions at telomeres. This is particularly challenging due to the highly repetitive telomeric sequence, their location at chromosome ends, and their low abundance. Analytical methods such as HPLC-MS require micrograms of DNA. Telomeres are ~0.025% of the genome, meaning 10,000 micrograms of DNA is required to obtain just 2.5 micrograms of telomeres. Possible oxidation of DNA during isolation procedures confounds the ability to accurately measure DNA lesion. The ability to measure different lesion types will be important since cytotoxic lesions, mutagenic lesions, single strand breaks, and repair intermediates can lead to different telomere outcomes. The same features that make damage detection in telomeres difficult also make sequencing telomeres extremely challenging. Advances in next generation sequencing tools and protocols should help overcome these barriers, and will be required to determine whether oxidative base damage leads to mutations in telomeric DNA and accumulation of variant repeats. Future studies are also required to address how oxidative base damage, and the processing of damage by repair enzymes, impact non-proliferating cells including neurons. Activation of DNA repair at the telomeres has the potential to dramatically alter telomere lengths even in the absence of replication. Understanding how the formation and processing of oxidative damage alters telomere length homeostasis and integrity will be valuable for developing intervention strategies that protect telomeres in the face of oxidative stress, and promote healthy aging.
Highlights.
Loss of telomere maintenance contributes ageing-related diseases and carcinogenesis.
Numerous diseases associated with oxidative stress are also associated with shortened telomeres.
Studies in human tissues, mouse models and cell culture provide evidence that oxidative stress is associated with accelerate telomere shortening.
Telomeres are highly sensitive to oxidative DNA damage, which can induce telomere losses and dysfunction.
Base excision repair of oxidative damage is essential for telomere maintenance.
Acknowledgments
We apologize to those investigators whose work was not cited in the interest of preparing a concise review. Research in the Opresko lab is supported by the National Institute of Environmental Health (ES R01ES022944, R21/33ES025606, ES028242, the National Institute of General Medicine (R43GM108187), and the National Cancer Institute CA207342.
Footnotes
Conflict of interest statement
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aamann MD, Hvitby C, Popuri V, Muftuoglu M, Lemminger L, Skeby CK, Keijzers G, Ahn B, Bjørås M, Bohr VA, et al. Cockayne Syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech Ageing Dev. 2014;135:1–14. doi: 10.1016/j.mad.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeby E, Ahmed W, Redon S, Simanis V, Lingner J. Peroxiredoxin 1 Protects Telomeres from Oxidative Damage and Preserves Telomeric DNA for Extension by Telomerase. Cell Rep. 2016;17:3107–3114. doi: 10.1016/j.celrep.2016.11.071. [DOI] [PubMed] [Google Scholar]
- Aguilera-Aguirre L, Bacsi A, Radak Z, Hazra TK, Mitra S, Sur S, Brasier AR, Ba X, Boldogh I. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-kappaB pathway. J Immunol. 2014;193:4643–4653. doi: 10.4049/jimmunol.1401625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121:1046–1053. doi: 10.1242/jcs.019372. [DOI] [PubMed] [Google Scholar]
- Aikata H, Takaishi H, Kawakami Y, Takahashi S, Kitamoto M, Nakanishi T, Nakamura Y, Shimamoto F, Kajiyama G, Ide T. Telomere reduction in human liver tissues with age and chronic inflammation. Exp Cell Res. 2000;256:578–582. doi: 10.1006/excr.2000.4862. [DOI] [PubMed] [Google Scholar]
- Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ame JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci. 2009;122:1990–2002. doi: 10.1242/jcs.039115. [DOI] [PubMed] [Google Scholar]
- Amsellem V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, Validire P, Stern JB, Noureddine H, Sapin E, Rideau D, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184:1358–1366. doi: 10.1164/rccm.201105-0802OC. [DOI] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- Aseervatham GS, Sivasudha T, Jeyadevi R, Arul Ananth D. Environmental factors and unhealthy lifestyle influence oxidative stress in humans--an overview. Environ Sci Pollut Res Int. 2013;20:4356–4369. doi: 10.1007/s11356-013-1748-0. [DOI] [PubMed] [Google Scholar]
- Askree SH, Yehuda T, Smolikov S, Gurevich R, Hawk J, Coker C, Krauskopf A, Kupiec M, McEachern MJ. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc Natl Acad Sci U S A. 2004;101:8658–8663. doi: 10.1073/pnas.0401263101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird DM, Rowson J, Wynford-Thomas D, Kipling D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet. 2003;33:203–207. doi: 10.1038/ng1084. [DOI] [PubMed] [Google Scholar]
- Baltazar MT, Dinis-Oliveira RJ, de Lourdes Bastos M, Tsatsakis AM, Duarte JA, Carvalho F. Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases--a mechanistic approach. Toxicol Lett. 2014;230:85–103. doi: 10.1016/j.toxlet.2014.01.039. [DOI] [PubMed] [Google Scholar]
- Beneke S, Cohausz O, Malanga M, Boukamp P, Althaus F, Burkle A. Rapid regulation of telomere length is mediated by poly(ADP-ribose) polymerase-1. Nucleic Acids Res. 2008;36:6309–6317. doi: 10.1093/nar/gkn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, et al. A review of human carcinogens--Part B: biological agents. Lancet Oncol. 2009;10:321–322. doi: 10.1016/s1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol. 2013:5. doi: 10.1101/cshperspect.a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21:354–359. doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannan WJ, Tsang BP, Wallace SS, Pederson DS. Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excision repair of clustered oxidative damages. J Biol Chem. 2014;289:19881–19893. doi: 10.1074/jbc.M114.571588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattan V, Mercier N, Gardner JP, Regnault V, Labat C, Maki-Jouppila J, Nzietchueng R, Benetos A, Kimura M, Aviv A, et al. Chronic oxidative stress induces a tissue-specific reduction in telomere length in CAST/Ei mice. Free Radic Biol Med. 2008;44:1592–1598. doi: 10.1016/j.freeradbiomed.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Wakamiya M, Venkova-Canova T, Pandita RK, Aguilera-Aguirre L, Sarker AH, Singh DK, Hosoki K, Wood TG, Sharma G, et al. Neil2-null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J Biol Chem. 2015;290:24636–24648. doi: 10.1074/jbc.M115.658146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AC, Ong SG, LaGory EL, Kraft PE, Giaccia AJ, Wu JC, Blau HM. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2016;113:13120–13125. doi: 10.1073/pnas.1615340113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi C, Parlanti E, Degan P, Aquilina G, Barnes D, Macpherson P, Karran P, Crescenzi M, Dogliotti E, Bignami M. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr Biol. 2002;12:912–918. doi: 10.1016/s0960-9822(02)00863-1. [DOI] [PubMed] [Google Scholar]
- Coluzzi E, Colamartino M, Cozzi R, Leone S, Meneghini C, O’Callaghan N, Sgura A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS One. 2014;9:e110963. doi: 10.1371/journal.pone.0110963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Hande MP, Tong WM, Lansdorp PM, Wang ZQ, Jackson SP. Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat Genet. 1999;23:76–80. doi: 10.1038/12680. [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- D’Errico M, Parlanti E, Teson M, de Jesus BM, Degan P, Calcagnile A, Jaruga P, Bjørås M, Crescenzi M, Pedrini AM, et al. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006;25:4305–4315. doi: 10.1038/sj.emboj.7601277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer F, Giraud-Panis MJ, Jaco I, Ame JC, Schultz I, Blasco M, Koering CE, Gilson E, Menissier-de Murcia J, de Murcia G, et al. Functional interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol Cell Biol. 2004;24:1595–1607. doi: 10.1128/MCB.24.4.1595-1607.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, de Lange T. The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb Perspect Biol. 2014;6:a016576. doi: 10.1101/cshperspect.a016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimkhani MR, Daneshmand A, Mazumder A, Allocca M, Calvo JA, Abolhassani N, Jhun I, Muthupalani S, Ayata C, Samson LD. Aag-initiated base excision repair promotes ischemia reperfusion injury in liver, brain, and kidney. Proc Natl Acad Sci U S A. 2014;111:E4878–4886. doi: 10.1073/pnas.1413582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101:17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley JC, Reid BJ, Odze RD, Sanchez CA, Galipeau P, Li X, Self SG, Gollahon KA, Blount PL, Rabinovitch PS. Chromosomal instability in Barrett’s esophagus is related to telomere shortening. Cancer Epidemiol Biomarkers Prev. 2006;15:1451–1457. doi: 10.1158/1055-9965.EPI-05-0837. [DOI] [PubMed] [Google Scholar]
- Fleming AM, Burrows CJ. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic Biol Med. 2017;107:35–52. doi: 10.1016/j.freeradbiomed.2016.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth NR, Evans AP, Shay JW, Wright WE. Developmental differences in the immortalization of lung fibroblasts by telomerase. Aging Cell. 2003;2:235–243. doi: 10.1046/j.1474-9728.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- Fouquerel E, Lormand J, Bose A, Lee HT, Kim GS, Li J, Sobol RW, Freudenthal BD, Myong S, Opresko PL. Oxidative guanine base damage regulates human telomerase activity. Nat Struct Mol Biol. 2016a;23:1092–1100. doi: 10.1038/nsmb.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouquerel E, Parikh D, Opresko P. DNA damage processing at telomeres: The ends justify the means. DNA Repair (Amst) 2016b doi: 10.1016/j.dnarep.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Strom CE, Svensson LM, Schultz N, Lundback T, Einarsdottir BO, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508:215–221. doi: 10.1038/nature13181. [DOI] [PubMed] [Google Scholar]
- Gomez M, Wu J, Schreiber V, Dunlap J, Dantzer F, Wang Y, Liu Y. PARP1 Is a TRF2-associated poly(ADP-ribose)polymerase and protects eroded telomeres. Mol Biol Cell. 2006;17:1686–1696. doi: 10.1091/mbc.E05-07-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan K, Low GKM, Ting APL, Srikanth P, Slijepcevic P, Hande MP. Hydrogen peroxide induced genomic instability in nucleotide excision repair-deficient lymphoblastoid cells. Genome Integr. 2010;1:16. doi: 10.1186/2041-9414-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham MK, Meeker A. Telomeres and telomerase in prostate cancer development and therapy. Nat Rev Urol. 2017;14:607–619. doi: 10.1038/nrurol.2017.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbo M, Koelvraa S, Serakinci N, Bendix L. Telomere dynamics in human mesenchymal stem cells after exposure to acute oxidative stress. DNA Repair (Amst) 2012;11:774–779. doi: 10.1016/j.dnarep.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech Ageing Dev. 2012;133:157–168. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- Henle ES, Han Z, Tang N, Rai P, Luo Y, Linn S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J Biol Chem. 1999;274:962–971. doi: 10.1074/jbc.274.2.962. [DOI] [PubMed] [Google Scholar]
- Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. 2014;2:4172. doi: 10.1038/ncomms5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2012;13:52–59. doi: 10.1038/embor.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliment CR, Oury TD. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic Biol Med. 2010;49:707–717. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungland A, Höss M, Gunz D, Constantinou A, Clarkson SG, Doetsch PW, Bolton PH, Wood RD, Lindahl T. Base excision repair of oxidative DNA damage activated by XPG protein. Mol Cell. 1999;3:33–42. doi: 10.1016/s1097-2765(00)80172-0. [DOI] [PubMed] [Google Scholar]
- Kolbanovskiy M, Chowdhury MA, Nadkarni A, Broyde S, Geacintov NE, Scicchitano DA, Shafirovich V. The Nonbulky DNA Lesions Spiroiminodihydantoin and 5-Guanidinohydantoin Significantly Block Human RNA Polymerase II Elongation in Vitro. Biochemistry. 2017;56:3008–3018. doi: 10.1021/acs.biochem.7b00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson ED, Iams K, Drummond JT. Strand-specific processing of 8-oxoguanine by the human mismatch repair pathway: inefficient removal of 8-oxoguanine paired with adenine or cytosine. DNA Repair (Amst) 2003;2:1199–1210. doi: 10.1016/s1568-7864(03)00140-x. [DOI] [PubMed] [Google Scholar]
- Lee HT, Bose A, Lee CY, Opresko PL, Myong S. Molecular mechanisms by which oxidative DNA damage promotes telomerase activity. Nucleic Acids Res. 2017;45:11752–11765. doi: 10.1093/nar/gkx789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Hills M, Conomos D, Stutz MD, Dagg RA, Lau LM, Reddel RR, Pickett HA. Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 2014;42:1733–1746. doi: 10.1093/nar/gkt1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsolo BT, Jones RE, Rowson J, Grimstead JW, Keith WN, Jenkins GJ, Baird DM. Extensive telomere erosion is consistent with localised clonal expansions in Barrett’s metaplasia. PLoS One. 2017;12:e0174833. doi: 10.1371/journal.pone.0174833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int J Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Liu Y. Deletion of Ogg1 DNA glycosylase results in telomere base damage and length alteration in yeast. EMBO J. 2010;29:398–409. doi: 10.1038/emboj.2009.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- Malinin NL, West XZ, Byzova TV. Oxidation as “the stress of life”. Aging (Albany NY) 2011;3:906–910. doi: 10.18632/aging.100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Gu C, Chen D, Yu B, He J. Oxidative stress-induced diseases and tea polyphenols. Oncotarget. 2017;8:81649–81661. doi: 10.18632/oncotarget.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markkanen E. Not breathing is not an option: How to deal with oxidative DNA damage. DNA Repair (Amst) 2017;59:82–105. doi: 10.1016/j.dnarep.2017.09.007. [DOI] [PubMed] [Google Scholar]
- Martens DS, Nawrot TS. Air Pollution Stress and the Aging Phenotype: The Telomere Connection. Curr Environ Health Rep. 2016;3:258–269. doi: 10.1007/s40572-016-0098-8. [DOI] [PubMed] [Google Scholar]
- Mazurek A, Berardini M, Fishel R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. J Biol Chem. 2002;277:8260–8266. doi: 10.1074/jbc.M111269200. [DOI] [PubMed] [Google Scholar]
- McNulty JM, Jerkovic B, Bolton PH, Basu AK. Replication inhibition and miscoding properties of DNA templates containing a site-specific cis-thymine glycol or urea residue. Chem Res Toxicol. 1998;11:666–673. doi: 10.1021/tx970225w. [DOI] [PubMed] [Google Scholar]
- Melis JP, Kuiper RV, Zwart E, Robinson J, Pennings JL, van Oostrom CT, Luijten M, van Steeg H. Slow accumulation of mutations in Xpc−/− mice upon induction of oxidative stress. DNA Repair (Amst) 2013;12:1081–1086. doi: 10.1016/j.dnarep.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AS, Balakrishnan L, Buncher NA, Opresko PL, Bambara RA. Telomere proteins POT1, TRF1 and TRF2 augment long-patch base excision repair in vitro. Cell Cycle. 2012;11:998–1007. doi: 10.4161/cc.11.5.19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokkapati SK, Wiederhold L, Hazra TK, Mitra S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry. 2004;43:11596–11604. doi: 10.1021/bi049097i. [DOI] [PubMed] [Google Scholar]
- Morland I, Luna L, Gustad E, Seeberg E, Bjørås M. Product inhibition and magnesium modulate the dual reaction mode of hOgg1. DNA Repair (Amst) 2005;4:381–387. doi: 10.1016/j.dnarep.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Nzietchueng R, Elfarra M, Nloga J, Labat C, Carteaux JP, Maureira P, Lacolley P, Villemot JP, Benetos A. Telomere length in vascular tissues from patients with atherosclerotic disease. J Nutr Health Aging. 2011;15:153–156. doi: 10.1007/s12603-011-0029-1. [DOI] [PubMed] [Google Scholar]
- O’Donovan A, Pantell MS, Puterman E, Dhabhar FS, Blackburn EH, Yaffe K, Cawthon RM, Opresko PL, Hsueh WC, Satterfield S, et al. Cumulative inflammatory load is associated with short leukocyte telomere length in the Health, Aging and Body Composition Study. PLoS One. 2011;6:e19687. doi: 10.1371/journal.pone.0019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan JN, Bronner MP, Brentnall TA, Finley JC, Shen WT, Emerson S, Emond MJ, Gollahon KA, Moskovitz AH, Crispin DA, et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat Genet. 2002;32:280–284. doi: 10.1038/ng989. [DOI] [PubMed] [Google Scholar]
- Oikawa S, Kawanishi S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999;453:365–368. doi: 10.1016/s0014-5793(99)00748-6. [DOI] [PubMed] [Google Scholar]
- Oikawa S, Tada-Oikawa S, Kawanishi S. Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry. 2001;40:4763–4768. doi: 10.1021/bi002721g. [DOI] [PubMed] [Google Scholar]
- Opresko PL, Fan J, Danzy S, Wilson DM, 3rd, Bohr VA. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005;33:1230–1239. doi: 10.1093/nar/gki273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opresko PL, Shay JW. Telomere-associated aging disorders. Ageing Res Rev. 2017;33:52–66. doi: 10.1016/j.arr.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen S, Saretzki G, von Zglinicki T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp Cell Res. 1998;239:152–160. doi: 10.1006/excr.1997.3893. [DOI] [PubMed] [Google Scholar]
- Poljsak B, Fink R. The protective role of antioxidants in the defence against ROS/RNS-mediated environmental pollution. Oxid Med Cell Longev. 2014;2014:671539. doi: 10.1155/2014/671539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poonepalli A, Balakrishnan L, Khaw AK, Low GK, Jayapal M, Bhattacharjee RN, Akira S, Balajee AS, Hande MP. Lack of poly(ADP-ribose) polymerase-1 gene product enhances cellular sensitivity to arsenite. Cancer Res. 2005;65:10977–10983. doi: 10.1158/0008-5472.CAN-05-2336. [DOI] [PubMed] [Google Scholar]
- Reichert S, Stier A. Does oxidative stress shorten telomeres in vivo? A review. Biol Lett. 2017:13. doi: 10.1098/rsbl.2017.0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Quintavalle C, Burmeister K, Calabrese D, Schlageter M, Quagliata L, Cathomas G, Diebold J, Molinolo A, Heim MH, et al. Liver damage and senescence increases in patients developing hepatocellular carcinoma. J Gastroenterol Hepatol. 2017;32:1480–1486. doi: 10.1111/jgh.13717. [DOI] [PubMed] [Google Scholar]
- Rhee DB, Ghosh A, Lu J, Bohr VA, Liu Y. Factors that influence telomeric oxidative base damage and repair by DNA glycosylase OGG1. DNA Repair (Amst) 2011;10:34–44. doi: 10.1016/j.dnarep.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter T, Saretzki G, Nelson G, Melcher M, Olijslagers S, von Zglinicki T. TRF2 overexpression diminishes repair of telomeric single-strand breaks and accelerates telomere shortening in human fibroblasts. Mech Ageing Dev. 2007;128:340–345. doi: 10.1016/j.mad.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Richter T, von Zglinicki T. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp Gerontol. 2007;42:1039–1042. doi: 10.1016/j.exger.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Risques RA, Lai LA, Himmetoglu C, Ebaee A, Li L, Feng Z, Bronner MP, Al-Lahham B, Kowdley KV, Lindor KD, et al. Ulcerative colitis-associated colorectal cancer arises in a field of short telomeres, senescence, and inflammation. Cancer Res. 2011;71:1669–1679. doi: 10.1158/0008-5472.CAN-10-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolseth V, Luna L, Olsen AK, Suganthan R, Scheffler K, Neurauter CG, Esbensen Y, Kusnierczyk A, Hildrestrand GA, Graupner A, et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci Rep. 2017;7:4384. doi: 10.1038/s41598-017-04472-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd SG, Valerie NC, Helleday T. Pathways controlling dNTP pools to maintain genome stability. DNA Repair (Amst) 2016;44:193–204. doi: 10.1016/j.dnarep.2016.05.032. [DOI] [PubMed] [Google Scholar]
- Rulten SL, Caldecott KW. DNA strand break repair and neurodegeneration. DNA Repair (Amst) 2013;12:558–567. doi: 10.1016/j.dnarep.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samper E, Goytisolo FA, Menissier-de Murcia J, Gonzalez-Suarez E, Cigudosa JC, de Murcia G, Blasco MA. Normal telomere length and chromosomal end capping in poly(ADP-ribose) polymerase-deficient mice and primary cells despite increased chromosomal instability. J Cell Biol. 2001;154:49–60. doi: 10.1083/jcb.200103049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders JL, Iannaccone A, Boudreau RM, Conley YP, Opresko PL, Hsueh WC, Cummings SR, Cawthon RM, Harris TB, Nalls MA, et al. The association of cataract with leukocyte telomere length in older adults: defining a new marker of aging. J Gerontol A Biol Sci Med Sci. 2011;66:639–645. doi: 10.1093/gerona/glr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saretzki G, Murphy MP, von Zglinicki T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell. 2003;2:141–143. doi: 10.1046/j.1474-9728.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Iwai S, Hanaoka F, Sugasawa K. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 2003;22:164–173. doi: 10.1093/emboj/cdg016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Uchimura Y, Dohmae N, Saitoh H, Hanaoka F, Sugasawa K. Stimulation of DNA Glycosylase Activities by XPC Protein Complex: Roles of Protein-Protein Interactions. J Nucleic Acids. 2010;2010 doi: 10.4061/2010/805698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snetselaar R, van Batenburg AA, van Oosterhout MFM, Kazemier KM, Roothaan SM, Peeters T, der van Vis JJ, Goldschmeding R, Grutters JC, van Moorsel CHM. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS One. 2017;12:e0189467. doi: 10.1371/journal.pone.0189467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- Stout GJ, Blasco MA. Telomere length and telomerase activity impact the UV sensitivity syndrome xeroderma pigmentosum C. Cancer Res. 2013;73:1844–1854. doi: 10.1158/0008-5472.CAN-12-3125. [DOI] [PubMed] [Google Scholar]
- Tan R, Nakajima S, Wang Q, Sun H, Xue J, Wu J, Hellwig S, Zeng X, Yates NA, Smithgall TE, et al. Nek7 Protects Telomeres from Oxidative DNA Damage by Phosphorylation and Stabilization of TRF1. Mol Cell. 2017;65:818–831 e815. doi: 10.1016/j.molcel.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong WM, Hande MP, Lansdorp PM, Wang ZQ. DNA strand break-sensing molecule poly(ADP-Ribose) polymerase cooperates with p53 in telomere function, chromosome stability, and tumor suppression. Mol Cell Biol. 2001;21:4046–4054. doi: 10.1128/MCB.21.12.4046-4054.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Bohr VA, Dizdaroglu M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 2003;17:668–674. doi: 10.1096/fj.02-0851com. [DOI] [PubMed] [Google Scholar]
- Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, Aviv A, Spector TD. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366:662–664. doi: 10.1016/S0140-6736(05)66630-5. [DOI] [PubMed] [Google Scholar]
- Vallabhaneni H, O’Callaghan N, Sidorova J, Liu Y. Defective repair of oxidative base lesions by the DNA glycosylase Nth1 associates with multiple telomere defects. PLoS Genet. 2013;9:e1003639. doi: 10.1371/journal.pgen.1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal AE, Hickson ID, Boiteux S, Radicella JP. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- Wallace SS. DNA glycosylases search for and remove oxidized DNA bases. Environ Mol Mutagen. 2013;54:691–704. doi: 10.1002/em.21820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rhee DB, Lu J, Bohr CT, Zhou F, Vallabhaneni H, de Souza-Pinto NC, Liu Y. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010;6:e1000951. doi: 10.1371/journal.pgen.1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Rane G, Dai X, Shanmugam MK, Arfuso F, Samy RP, Lai MK, Kappei D, Kumar AP, Sethi G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res Rev. 2016;25:55–69. doi: 10.1016/j.arr.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lin S, Funk WE, Hou L. Environmental and occupational exposure to chemicals and telomere length in human studies. Occup Environ Med. 2013;70:743–749. doi: 10.1136/oemed-2012-101350. [DOI] [PubMed] [Google Scholar]
- Zhou J, Chan J, Lambele M, Yusufzai T, Stumpff J, Opresko PL, Thali M, Wallace SS. NEIL3 Repairs Telomere Damage during S Phase to Secure Chromosome Segregation at Mitosis. Cell Rep. 2017;20:2044–2056. doi: 10.1016/j.celrep.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Liu M, Fleming AM, Burrows CJ, Wallace SS. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J Biol Chem. 2013;288:27263–27272. doi: 10.1074/jbc.M113.479055. [DOI] [PMC free article] [PubMed] [Google Scholar]