

Abstract
Host pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) detect viruses and other pathogens, inducing production of cytokines that cause inflammation and mobilize cells to control infection. Vaccinia virus (VACV) encodes proteins that antagonize these host innate immune responses, and elucidating the mechanisms of action of these viral proteins helped shed light on PRR signaling mechanisms. The VACV virulence factor E3 is one of the most intensely studied VACV proteins and has multiple effects on host cells, many of which cannot be explained by the currently known cellular targets of E3. Here, we report that E3 expression in human monocytes alters TLR2- and TLR8-dependent cytokine induction, and particularly inhibits interleukin (IL)-6. Using MS, we identified DExD/H-box helicase 9 (DHX9) as an E3 target. Although DHX9 has previously been implicated as a PRR for sensing nucleic acid in dendritic cells, we found no role for DHX9 as a nucleic acid-sensing PRR in monocytes. Rather, DHX9 suppression in these cells phenocopied the effects of E3 expression on TLR2- and TLR8-dependent cytokine induction, in that DHX9 was required for all TLR8-dependent cytokines measured, and for TLR2-dependent IL-6. Furthermore, DHX9 also had a cell- and stimulus-independent role in IL-6 promoter induction. DHX9 enhanced NF-κB–dependent IL-6 promoter activation, which was directly antagonized by E3. These results indicate new roles for DHX9 in regulating cytokines in innate immunity and reveal that VACV E3 disrupts innate immune responses by targeting of DHX9.
Keywords: toll-like receptor (TLR), signal transduction, pattern recognition receptor (PRR), poxvirus, NF-κB (NF-KB), interleukin 6 (IL-6), innate immunity, host-pathogen interaction, DExD/H-box helicase (DHX9), Immune evasion
Introduction
A cellular innate immune response is triggered by the sensing of pathogen-associated molecular patterns (PAMPs)3 or molecules released from dying cells (damage-associated molecular patterns, DAMPs), which bind to sensors on or within monocytes, macrophages, and dendritic cells (DCs). PAMP and DAMP sensors are mainly pattern recognition receptors (PRRs), which when activated signal via transcription factors such as NF-κB and interferon regulatory factors to induce innate immune effectors genes (1). These genes encode interferons, chemokines (e.g. IP10 and RANTES), and pro-inflammatory cytokines (e.g. TNFα, IL-6, and IL-1β), which mobilize both innate immune cells and T cells to control infection. The prototypical PRR family are the membrane-bound Toll-like receptors (TLRs) that bind conserved molecules from bacteria, viruses, and parasites (2). Other PRRs include the cytosolic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) that bind bacterial products, RIG-I-like receptors (RLRs) that bind viral RNA (1), and cytosolic DNA sensors such as AIM2, IFII6, and cGAS that bind host and pathogen DNA (3–5).
To overcome PRR-stimulated host defense, viruses have evolved proteins that both antagonize and subvert PRR-dependent gene induction (6, 7). Studying how these viral proteins interact with host molecules provides an understanding of viral pathogenesis but can also reveal important aspects of host immune signaling mechanisms, which have been selected for viral antagonism over evolutionary time. For example, we and others have discovered several poxviral proteins that target host proteins subsequently implicated in PRR signaling pathways (6, 7). One such host protein is the DEAD box RNA helicase DDX3, which vaccinia virus (VACV) protein K7 interacts to inhibit RLR responses (8).
VACV protein E3 is a multifunctional protein that is vital for viral replication and pathogenesis (9). E3 is comprised of an N-terminal Z-DNA–binding domain (DNABD) and a C-terminal dsRNA–binding motif (DSRM), both of which are required for virulence (10). E3 inhibits dsRNA-dependent protein kinase (PKR) function by sequestering dsRNA via the DSRM, but also by directly binding to PKR (11, 12). Other functions of the DSRM include inhibition of the RNA Pol III dsDNA sensing pathway (13), and antagonism of the antiviral protein ISG15 (14). A recent study revealed that many of E3's biological functions do not involve dsRNA binding because mutants of E3 that no longer bound dsRNA were still able to affect viral replication in cell culture, modulate apoptosis, and inhibit production of PRR-stimulated cytokines such as IL-6 (15). Therefore, it is likely that more host protein targets remain to be identified for E3, which would shed further light on how viruses modulate cellular signaling pathways.
Here we show that expression of E3 in human monocytes led to altered PRR-dependent cytokine production, and in particular suppression of IL-6 induction by TLRs. Using MS, the RNA helicase DHX9 (also called RNA helicase A) was identified as an E3-interacting protein. DHX9 has previously been implicated as a PRR for sensing nucleic acid in DCs. However, here we found no role for DHX9 in nucleic acid sensing in monocytes. Rather, suppression of DHX9 expression phenocopied the effect of E3 expression on cytokine and chemokine production stimulated by TLR2, TLR8, and TLR4. Further work showed a role for DHX9 in NF-κB–dependent IL-6 promoter induction, which was antagonized by E3. These results suggest a new role for DHX9 in innate immune cytokine regulation and reveal a further mechanism whereby VACV via E3 disrupts innate immunity, via targeting of DHX9.
Results
E3 inhibits cytokine production from human monocytes
E3 has previously been shown to modulate cytokine induction in virally-infected cells (15–17). To determine whether E3 directly regulated PRR-stimulated cytokine production, we wanted to express E3 in isolation from virus, in an immune-relevant cell line model. Therefore, we generated human THP-1 monocytes cells stably expressing E3 using a retroviral expression system (Fig. 1A). PRR-stimulated cytokine production (TNFα, IL-6, IP10, and RANTES) was assessed in these cells by ELISA. To focus on the effects of E3 independent of sequestering of nucleic acids, the nonnucleic acid PRR ligands Pam3Csk4, CL075, and lipopolysaccharide (LPS) were used to stimulate TLR2-, TLR8-, and TLR4-dependent gene induction, respectively. CL075 is a specific agonist for TLR8 in human monocytes (18). Interestingly, expression of E3 in cells affected cytokine production in stimulus- and gene-specific manners. Compared with the other ligands, LPS only stimulated low production of TNFα, IP-10, and RANTES, which was not affected by E3 expression, whereas LPS-stimulated IL-6 was barely detectable (Fig. 1, B–E). Upon TLR2 stimulation of E3-expressing cells, IL-6 production was strongly inhibited, TNFα production was enhanced, and chemokine induction (IP-10, RANTES) was unaffected (Fig. 1, B–E). In contrast, all TLR8-stimulated cytokines measured (TNFα, IL-6, IP10, and RANTES) were potently inhibited in cells expressing E3 compared with cells transduced with empty vector (Fig. 1, B–E). Thus, E3 antagonizes specific PRR-stimulated cytokine induction in monocytes. The varied effects of E3 on some cytokines and lack of effect on others implied that inhibitory effects were not due to toxicity or a general suppression of all genes.
Figure 1.

Effect of E3 expression on TLR-stimulated cytokine induction in human monocytes. A, THP-1 cells were transduced with retroviruses encoding HA-tagged E3 or empty vector (EV). After cell selection, lysates were generated and immunoblotted for HA-E3 and β-actin, alongside a lysate from HEK293T cells transiently expressing E3. B–E, E3- or EV-expressing THP-1 cells were stimulated with 2 μg/ml of Pam3CSK4 (PAM3), 2.5 μg/ml of CLO75, 100 ng/ml of LPS or medium alone. The supernatants were removed 24 h later and TNFα (B), IL-6 (C), IP10 (D), and RANTES (E) secretion were measured by ELISA. The data are mean ± S.D. of triplicate samples and is representative of three experiments. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with EV-containing THP-1 cells. N.D., not detected.
E3 interacts with host helicase DHX9
E3 has been shown to interact with both host nucleic acid and proteins, as described above. The fact that specific Pam3Csk4 and CL075 responses were affected by E3 suggested that E3 was targeting host protein(s) to modulate TLR signaling outcomes. To explore what protein targets of E3 in cells might explain E3 modulation of cytokine production, we infected murine embryonic fibroblasts with vaccinia virus (WR, Western reserve strain), and used an anti-E3 antibody to immunoprecipitate E3-associated proteins. As a control, we compared the anti-E3 antibody immunoprecipitate from WR-infected cells to that derived from cells infected with vaccinia virus WR lacking the gene that encodes E3 (ΔE3L). Immunoprecipitated proteins from WR- and ΔE3L-infected cells were then separated by SDS-PAGE (Fig. 2A), and gel slices were prepared and trypsinized. Peptides eluted from gel slices were analyzed by LC–MS. Several potential E3-interacting proteins were identified, by nature of them being detected in the resolved immunoprecipitate from WR- but not ΔE3L-infected cells. One such protein, DHX9, was of particular interest because it was highly abundant in the WR sample (Fig. 2A), and because it has been implicated in the regulation of gene transcription and translation (19). Thus, here we focused on DHX9 as a potential protein target to explain E3 immunomodulation. We confirmed that E3 could interact with DHX9 in human cells by endogenous IP of E3 from virally-infected HEK293T cells followed by immunoblot for DHX9 (Fig. 2B, top panel). Importantly, in THP-1 cells, where expression of E3 affected cytokine induction (Fig. 1), after vaccinia virus infection a clear interaction was seen between endogenous DHX9 and virally expressed E3 (Fig. 2C). Ectopically expressed E3 in isolation from virus also interacted with endogenous DHX9 (Fig. 2D). Furthermore, both full-length E3 and the isolated DSRM of E3 interacted with DHX9, whereas the isolated DNABD of E3 did not (Fig. 2E). Hence E3 interacts with host DHX9 via the DSRM.
Figure 2.

E3 interacts with DHX9 via its RNA-binding domain. A, murine embryonic fibroblasts were infected with WT VACV WR or VACV lacking the E3L gene (ΔE3). Cell lysates were prepared and subject to immunoprecipitation with anti-E3 antibody. Lysates were resolved by SDS-PAGE and gel slices were analyzed for interacting proteins by MS. The position of a band corresponding to DHX9 is indicated. B, HEK293T cells were infected with VACV WR or ΔE3 and the lysate was subjected to immunoprecipitation with anti-E3 antibody, followed by immunoblotting with antibodies to E3 or DHX9 as indicated. C, THP-1 cells were infected with VACV WR for 16 h and lysates were subjected to immunoprecipitation with anti-DHX9 or IgG antibody, followed by immunoblotting with antibodies to E3 or DHX9 as indicated. D, HEK293T cells were transfected (+) or not (−) with pCMV-E3-HA and lysates subjected to immunoprecipitation by anti-DHX9 antibody, followed by blotting with antibodies to DHX9 or HA as indicated. E, schematic shows E3 domain structure and truncations used. HEK293T cells were transfected with empty vector or plasmid encoding HA-E3(1–190), HA-E3(1–83), or HA-E3(84–190), and incubated for 48 h. Cell lysates were then subjected to IP using DHX9 or IgG antibody and then resolved by SDS-PAGE. E3 and DHX9 interactions were analyzed by immunoblotting for HA and DHX9, respectively. Representative of three experiments.
Suppression of DHX9 expression mimics the effect of E3 on TLR-induced cytokines
To determine whether DHX9 had a role in production of cytokines affected by E3, DHX9 loss-of-function studies were employed. Because DHX9 is an essential gene in both mouse and human (20, 21), we used shRNA knock-down to partially deplete protein expression of DHX9, rather than gene targeting of DHX9. Thus, an inducible shRNA lentiviral expression system was used to reduce DHX9 expression in real time. Cell lines were generated using a pTRIPZ lentiviral system in which doxycycline-inducible shRNA could be introduced into THP-1 cells. Three stable shRNA-expressing cell lines were generated: Fig. 3A shows that 72 h treatment of cells with doxycycline led to strong suppression of DHX9 protein expression in cells transduced with lentiviruses encoding two distinct DHX9-targeting shRNAs (A7 and G2), but not in cells transduced with lentiviruses encoding a nonsilencing control (NSC). Similar to E3-expressing monocytes (Fig. 1), these cells were stimulated with TLR agonists and cytokine production was monitored. A remarkably similar profile of regulation of TLR-stimulated cytokines was observed in cells expressing DHX9 shRNA, compared with cells expressing E3 (Fig. 1) in that suppression of DHX9 expression inhibited production of all four TLR8-stimulated cytokines, and of TLR2-stimulated IL-6 production, whereas TLR2-stimulated TNFα production was enhanced, and IP-10 and RANTES unaffected (Fig. 3, B–E). The low LPS responses were unaffected by suppression of DHX9 expression, except in the case of IL-6, which was inhibited (Fig. 3, B–E). The correlation between the effects of expression of E3 (Fig. 1) and suppression of DHX9 expression (Fig. 3), which exactly phenocopied each other, strongly suggest that E3 modulated TLR-stimulated cytokines via targeting of DHX9.
Figure 3.

Effect of DHX9 shRNA on TLR-stimulated cytokine induction in human monocytes. A, THP-1 cells were transduced with lentiviruses encoding DHX9-targeting shRNA (A7 and G2) or a NSC shRNA. After transduction, the THP-1 cells were incubated in 1 μg/ml of puromycin for 1 week. Each THP-1 cell line was either left untreated (−) or incubated with 1 μg/ml of doxycycline (+) for 72 h to induce the required shRNA. Lysates were generated, resolved by SDS-PAGE, and immunoblotted for DHX9 and tubulin. B–E, doxycycline-treated THP-1 cells expressing shRNAs NSC, A7, or G2 were stimulated with 2 μg/ml of Pam3CSK4 (PAM3), 2.5 μg/ml of CLO75, 100 ng/ml of LPS or medium alone. The supernatants were removed 24 h later and TNFα (B), IL-6 (C), IP10 (D), and RANTES (E) secretions were measured by ELISA. The data are mean ± S.D. of triplicate samples and is representative of three experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with NSC. N.D., not detected.
No role for DHX9 in DNA or RNA sensing in monocytes
Previous work implicated DHX9 as a cytosolic nucleic acid sensor for both DNA and RNA in DCs, operating upstream of either the TLR adaptor protein MyD88 for DNA, or the RLR adaptor MAVS for RNA (22, 23). Therefore, we also tested whether suppression of DHX9 expression impacted DNA and RNA sensing in monocytes. Cytokine production was assessed from THP-1 cells transfected with two types of immune stimulatory DNA to activate cytosolic DNA sensing pathways, and also with cGAMP, which is a direct ligand for STING, the adaptor protein for cGAS. Surprisingly, there was no significant difference in RANTES or IP10 production from cells expressing DHX9 shRNA compared with cells expressing NSC shRNA for any of the agonists tested (Fig. 4, A and B), suggesting no role for DHX9 in DNA sensing or in STING function in monocytes. RNA sensing was examined by stimulating cells with Sendai virus, whose defective interfering particles are sensed by RLRs in human cells. There was no difference in responses between NSC cells and two lines expressing DHX9 shRNA, except in the case of IL-6, where potent inhibition was observed (Fig. 4, C–F). These results suggest that DHX9 does not function as a nucleic acid sensor in human monocytes, but rather has more “downstream” gene-specific roles. Of note, we found that IL-6 production by all ligands tested (TLR2, TLR8, TLR4, and RNA virus) depended on DHX9.
Figure 4.

DHX9 does not act as a PRR for DNA or RNA in monocytes. A and B, THP-1 cells expressing doxycycline-inducible DHX9-specific shRNA (A7 and G2) or NSC shRNA, were incubated with 1 μg/ml of doxycycline for 72 h, and then transfected 24 h later with 1 μg/ml of 70-mer, 5 μg/ml of cGAMP, 1 μg/ml of poly(dA:dT) or Lipofectamine only (Lipo only). The supernatants were removed 24 h later and RANTES (A) and IP10 (B) secretions were measured by ELISA. C–F, cells were mock-infected (Mock) or infected with Sendai virus (SeV). The supernatants were removed 24 h later and TNFα, IL-6, IFNα, IP10. and RANTES secretions were measured by ELISA. The data are mean ± S.D. of triplicate samples and is representative of three experiments. ****, p < 0.0001 compared with NSC. N.D., not detected.
Stimulus-independent role for DHX9 in IL-6 production in human cells
We tested whether the requirement of DHX9 for IL-6 production was true in other human cell types. For this, BEAS-2B bronchial epithelial cells were used, because they produce IL-6 in response to PAMPs and pro-inflammatory cytokines. The lentiviral pTRIPZ system was again employed to generate cell lines stably expressing doxycycline-inducible DHX9 shRNAs (A7 and D11) or a NSC shRNA. Similar to THP-1 cells, doxycycline treatment strongly reduced DHX9 protein expression in the DHX9 shRNA-expressing BEAS-2B cells compared with the NSC cell line (Fig. 5A). Doxycycline-treated cells were stimulated with a range of IL-6-inducing ligands for 24 h (Pam3Csk4 for TLR2 and IL-1, dsRNA for TLR3, transfected dsRNA and transfected dsDNA). In all cases, IL-6 secretion depended on DHX9 because significant inhibition of IL-6 production was seen in A7 and D11 cells compared with NSC cells for all ligands tested (Fig. 5B). Furthermore, an alternative gain-of-function approach to assessing DHX9 also provided evidence of a role for DHX9 in IL-6 production, because transfection of HeLa cells with an expression vector encoding DHX9 led to a plasmid dose-dependent enhancement of MyD88-stimulated IL-6 production (Fig. 5C). Thus, DHX9 is required for IL-6 production by diverse innate immune ligands, and in different human cell types.
Figure 5.
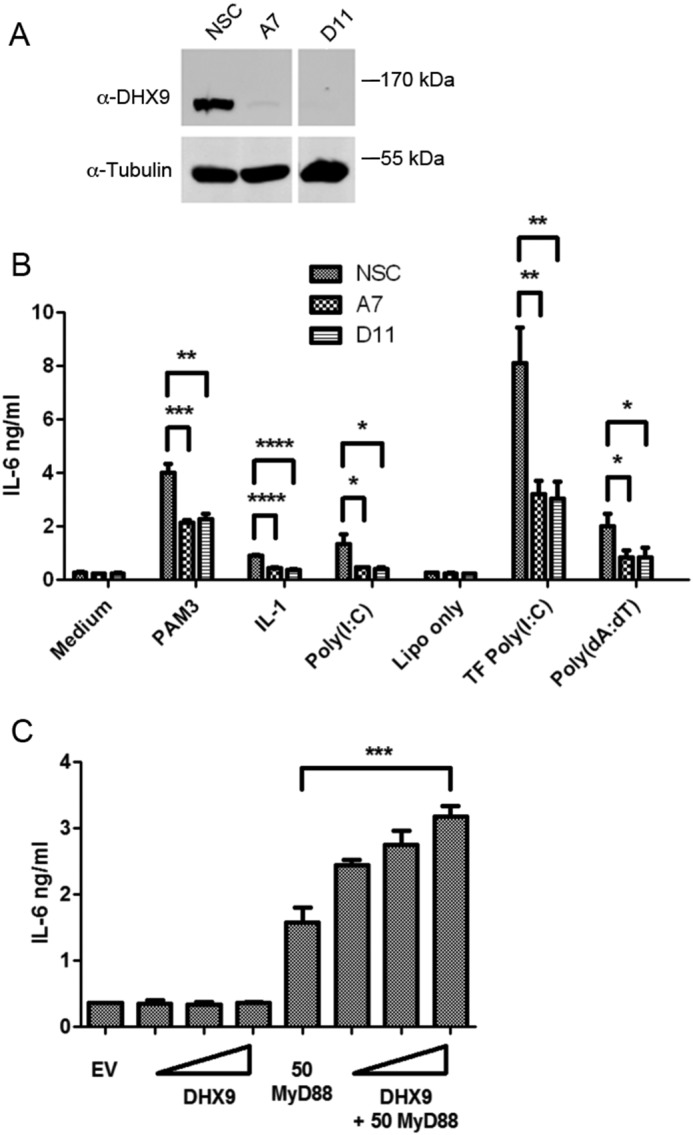
Stimulus-independent role for DHX9 in IL-6 production in human cells. A, BEAS-2B cells were transduced with lentiviruses encoding DHX9-targeting shRNA (A7 or D11) or a NSC shRNA. After puromycin selection, the cells were incubated with 1 μg/ml of doxycycline for 72 h to induce the required shRNA. Lysates were generated and immunoblotted with anti-DHX9 and anti-tubulin antibody. B, BEAS-2B cells expressing doxycycline-inducible shRNAs were incubated with 1 μg/ml of doxycycline for 72 h and then stimulated with medium, 10 μg/ml of Pam3CSK4 (PAM3), 100 ng/ml of IL-1, 12.5 μg/ml of poly(I:C), or transfected (TF) with Lipofectamine only (Lipo only), 25 μg/ml of poly(I:C) or 1 μg/ml of poly(dA:dT) for 24 h. Supernatants were removed and IL-6 secretion was measured by ELISA. C, HeLa cells were transfected with empty vector (EV), 50 ng of MyD88 or 25, 50, or 100 ng of DHX9 expression vector (wedge) as indicated. Supernatants were removed 48 h after transfection and analyzed for IL-6. Data are mean ± S.D. of triplicate samples and is representative of three (B) or two (C) experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
DHX9 regulates the IL-6 promoter in an NF-κB–dependent manner
Because production of IL-6 by every stimulus tested depended on DHX9, we focused on understanding the role of DHX9 in IL-6 regulation. DHX9 has been shown to regulate both transcription and translation (19), so we first determined whether IL-6 mRNA induction in response to TLR ligands was DHX9-dependent. In NSC THP-1 cells, TLR2 or TLR8 stimulation led to time-dependent induction of IL-6 mRNA, and for both TLR ligands this was significantly impaired in the two independent cell lines expressing DHX9 shRNA, A7 and G2 (Fig. 6, A and B). This suggested a requirement of DHX9 in transcriptional induction of the IL-6 gene. The IL-6 promoter is regulated and induced by TLRs upon activation of both p38 MAP kinase and NF-κB (24), so therefore we tested whether activation of these signaling pathways by TLR2 were affected by loss of DHX9 expression. Fig. 7 shows that this was not the case, because the time course of appearance of phospho-p38 in NSC and G2 cells stimulated with TLR2 ligand was very similar. Also, NF-κB activation, measured both by monitoring IκBα degradation, and p65 phosphorylation, was normal in G2 cells (Fig. 7). These data, together with the lack of effect of suppression of DHX9 expression on other TLR-dependent p38- and NF-κB–regulated genes (Fig. 3) strongly suggested a role for DHX9 in IL-6 induction downstream of cytosolic signaling pathways.
Figure 6.

DHX9 is required for TLR2- and TLR8-stimulated IL-6 mRNA induction. A and B, THP-1 cells expressing doxycycline-inducible DHX9-specific shRNA (A7 and G2) or NSC shRNA were incubated with 1 μg/ml of doxycycline for 72 h, and then stimulated with 2 μg/ml of Pam3CSK4 (PAM3) or 2.5 μg/ml of CLO75 for the indicated times. IL-6 mRNA was assayed by quantitative PCR. Data are presented as relative expression of IL-6 normalized to β-actin compared with nonstimulated NSC shRNA-producing cells. The data are mean ± S.D. of triplicate samples and is representative of three experiments. ***, p < 0.001 compared with NSC.
Figure 7.
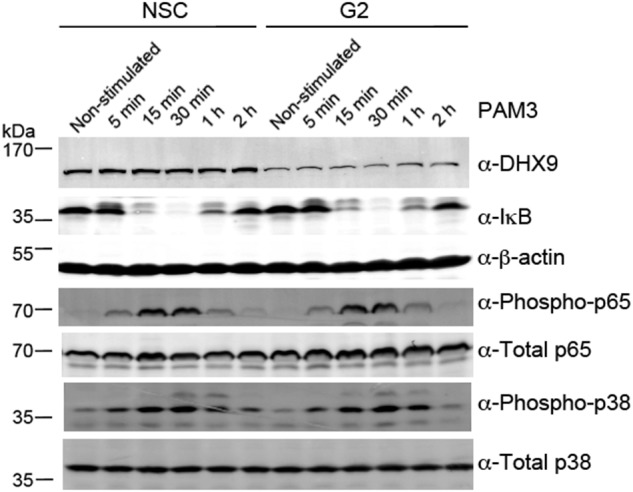
DHX9 is not required for TLR2-stimulated NF-κB or p38 MAPK activation. THP-1 cells expressing doxycycline-inducible DHX9-specific shRNA (G2) or NSC shRNA, were incubated with 1 μg/ml of doxycycline for 72 h, and then stimulated with 2 μg/ml of Pam3CSK4 (PAM3) for the indicated times. Cell lysates were harvested and immunoblotted for DHX9, IκB, β-actin, phospho-p65, total p65, phospho-p38, and total p38 as indicated. These data are representative of three experiments.
We therefore next focused on the IL-6 promoter itself. To assess the direct effects of DHX9 on this promoter, an IL-6 promoter-dependent luciferase reporter was employed (Fig. 8A). Ectopic expression of DHX9 alone led to a small but significant induction of this reporter, as did expression of MyD88 (Fig. 8B). Interestingly, co-expression of DHX9 and MyD88 led to a synergistic induction of the IL-6 promoter (Fig. 8B), and this stimulation was largely reduced when the NF-κB element (Fig. 8C) but not the AP-1 element (Fig. 8D) was deleted from the IL-6 promoter. These data indicate that DHX9 has a promoter-proximal role in induction of the IL-6 promoter, which depends on the presence of the NF-κB element within that promoter.
Figure 8.
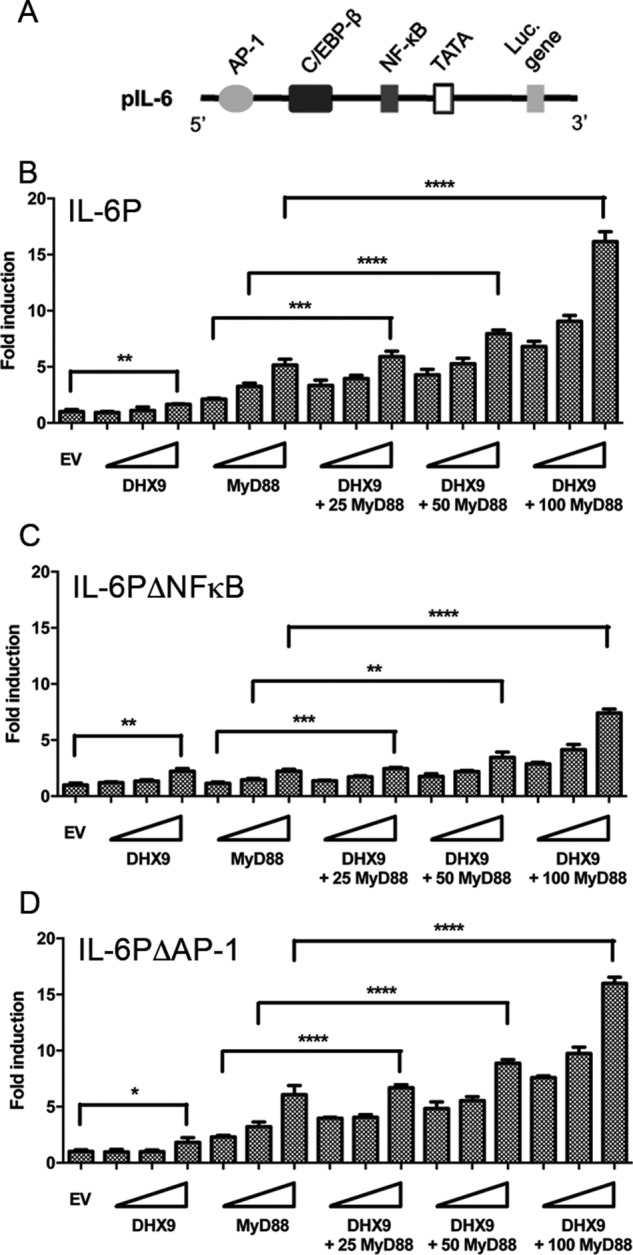
DHX9 regulates the IL-6 promoter in an NF-κB–dependent manner. A, schematic of human IL-6 promoter luciferase reporter. B–D, HeLa cells were transfected with WT IL-6 promoter reporter gene (IL-6P) (B), an IL-6 promoter reporter lacking the κB-binding site (IL-6PΔNF-κB) (C), or an IL-6 promoter reporter lacking the AP1-binding site (IL-6PΔAP1) (D), together with empty vector (EV) or increasing amounts of DHX9 and/or MyD88 expression vector. Wedges indicate increasing amounts of vector encoding DHX9 or MyD88 (25, 50, and 100 ng). 48 h after transfection, the cells were lysed and assessed for reporter gene activity. Data are shown as relative fold-induction normalized to EV only-transfected cells. The data are mean ± S.D. of triplicate samples and is representative of two experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with EV alone-transfected cells or cells transfected with only MyD88 expression vector.
The ability of DHX9 to enhance TLR-stimulated NF-κB–dependent transcription was further explored by use of a reporter gene under the control of an isolated NF-κB element. Fig. 9, A and B, shows that induction of this reporter by TLR8 and TLR2 ligands was synergistically enhanced by ectopic expression of DHX9. This synergistic effect of DHX9 expression was also apparent when the NF-κB–dependent reporter was induced by direct expression of MyD88 (Fig. 9C). To confirm that the positive effects of DHX9 on NF-κB–dependent gene induction were proximal to NF-κB at the promoter, the effect of DHX9 on direct induction of the reporter by NF-κB subunit p65 was examined. Fig. 9D shows that similar to the case for MyD88 expression, direct expression of p65 induced the NF-κB–dependent reporter, and this was synergistically enhanced by DHX9. Furthermore, the ability of p65 to transactivate a Gal4-dependent promoter reporter was also positively affected by DHX9 because MyD88-dependent p65 transactivation was also synergistically enhanced by DHX9 expression (Fig. 9E). Together these data reveal a promoter-proximal role for DHX9 in stimulation of the IL-6 promoter, and of NF-κB–dependent transactivation.
Figure 9.
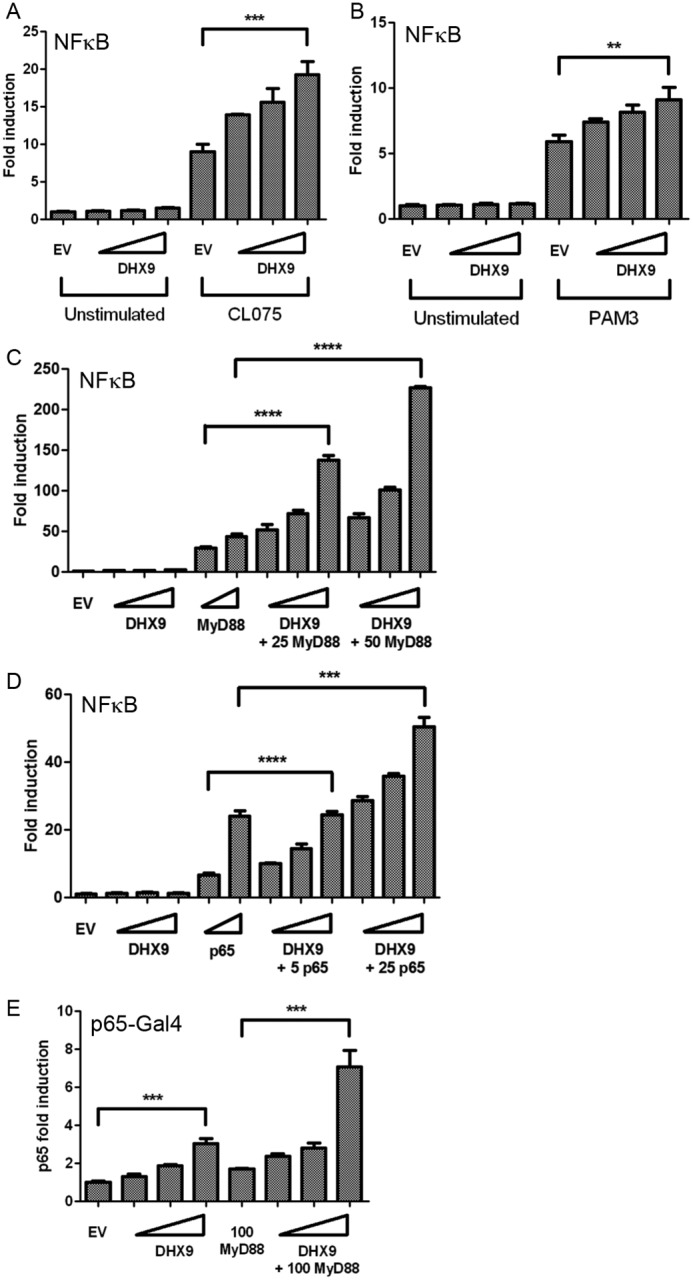
DHX9 enhances NF-κB–dependent promoter activation. A and B, HEK293-TLR8 cells (A) or HEK293-TLR2 cells (B) were transfected with NF-κB–dependent reporter gene and empty vector (EV) or increasing amounts of DHX9 expression vector. Wedges indicate increasing amounts of DHX9 (25, 50, and 100 ng). 24 h after transfection cells were stimulated with 2.5 μg/ml of CL075 (A) or 2 μg/ml of Pam3CSK4 (B). The cells were lysed 24 h after stimulation and assessed for reporter gene activity. C–E, HeLa cells were transfected with NF-κB–dependent reporter gene (C and D) or Gal4-p65 with pFR luciferase (E) and empty vector (EV) or the indicated amounts (ng) of MyD88, p65, or DHX9 expression vectors. Wedges indicate increasing amounts of DHX9 (25, 50, and 100 ng), MyD88 (25 and 50 ng), or p65 (5 and 25 ng). The cells were lysed 48 h after transfection and the lysates were assayed for reporter gene activity. Data are shown as relative fold-induction normalized to unstimulated, EV only-transfected cells. The data are mean ± S.D. of triplicate samples and are representative of three experiments. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with EV (A and B), MyD88 only (C and E), or p65 only (D).
E3 antagonizes DHX9-dependent NF-κB transactivation
Finally, we assessed the effect of expression of E3 on the ability of DHX9 to stimulate NF-κB–dependent gene induction events, to determine whether E3 directly inhibited the positive effect of DHX9 on NF-κB–dependent transactivation. E3 expression was found to significantly suppress the ability of DHX9 to synergize with MyD88 for IL-6 production (Fig. 10A). We also found that E3 could significantly inhibit the positive effect of DHX9 on TLR8-stimulated NF-κB–dependent reporter gene induction (Fig. 10B). Finally, we observed a direct effect of E3 on DHX9-dependent NF-κB transactivation because E3 significantly suppressed the enhancement of p65-stimulated NF-κB–dependent reporter gene induction by DHX9 (Fig. 10C). Together, these data reveal a role for DHX9 in PRR-stimulated gene regulation, and strongly suggest that E3 antagonizes DHX9-dependent innate immune gene induction.
Figure 10.
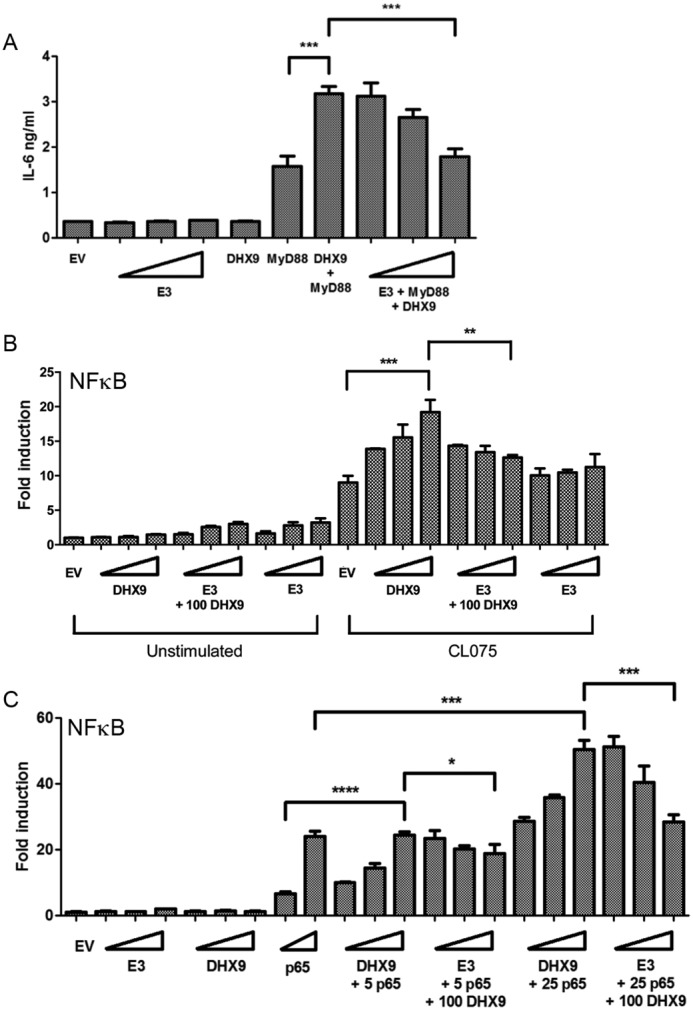
E3 antagonizes DHX9-dependent NF-κB transactivation. A, HeLa cells were transfected with E3, DHX9, or MyD88 expression vectors. Wedges indicate increasing amounts of E3 (25, 50, and 100 ng). E3 was transfected in increasing doses with and without 100 ng of DHX9 and 50 ng of MyD88. The supernatants were removed 48 h after transfection and IL-6 protein secretion was measured by ELISA. The data are mean ± S.D. of triplicate samples and is representative of two experiments. ***, p < 0.001 compared with cells transfected with MyD88 or both DHX9 and MyD88. B and C, HEK293-TLR8 (B) or HeLa (C) cells were transfected with NF-κB–dependent reporter gene and empty vector (EV) or the indicated amounts (ng) of DHX9, p65, or E3 expression vector. Wedges indicate increasing amounts of DHX9 (25, 50, and 100 ng), p65 (5 and 25 ng), or E3 (25, 50, and 100 ng). 24 h later the HEK TLR8 cells were either unstimulated or stimulated with 2.5 μg/ml of CL075 (B). After a further 24 h, the cells were lysed and the lysates were assayed for reporter gene activity. Data are shown as relative fold-induction normalized to unstimulated, EV only-transfected cells. The data are mean ± S.D. of triplicate samples and is representative of three experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with stimulated, EV only transfected cells or cells transfected with the highest dose of DHX9 expression vector (B) or p65-transfected cells or cells transfected with p65 and the highest dose of DHX9 expression vector (C).
Discussion
Because poxviruses target critical nodes in innate immune signaling, determining the host targets of VACV proteins with immunomodulatory properties has proved useful in elucidating in detail innate immune signaling mechanisms. In this regard, E3, which is one of the most intensely studied VACV proteins, has multiple effects on host cells, many of which cannot be explained by the known cellular targets of E3. Here we identify DHX9 as a new target for E3 in human cells, and propose that E3 antagonism of DHX9 function is a viral strategy to modulate innate immune cytokine production, and in particular to suppress IL-6 induction.
DHX9 belongs to the DExD/H-box family of proteins, originally shown to function in RNA metabolism, but now known to have broad roles in viral replication, innate immunity, cell signaling, and gene induction (19). DHX9 is a case in point, having been shown to bind both ssRNA and ssDNA, to unwind double-stranded nucleic acids (25, 26), to sense both dsDNA and dsRNA in DCs (22, 23), to shuttle between the cytoplasm and nucleus (27), and to participate in transcription and translation of both cellular and retroviral mRNAs (28, 29). In fact, many viruses are reported to hijack DHX9 and use it to enhance viral replication, transcription, and translation, including influenza A virus, HIV-1, human T cell leukemia virus type 1, foot-and-mouth disease virus, and hepatitis C virus (30–34).
However, VACV E3 represents the first definitive example of viral antagonism of DHX9 to suppress PRR-stimulated cytokine induction. Interestingly, DHX9 has been identified as a substrate for PKR, the best-defined protein target of E3 (35). Phosphorylation of DHX9 by PKR antagonized the capacity of DHX9 to stimulate transactivation of HIV-1 genes (35, 36). Whether E3 can bind PKR and DHX9 simultaneously, or whether DHX9 has a role in VACV gene transactivation or VACV replication, remains to be determined. There is a real possibility that DHX9 has a role in VACV replication because it has been shown that for the poxvirus myxoma virus, DHX9 is a pro-viral effector that promotes viral replication in THP-1 cells (37). That study also showed that the myxoma virus protein M029, which is a truncated orthologue of E3, targeted DHX9 to conscript the host helicase for proviral effects (37). Targeting of DHX9 by two distinct but related poxviral proteins attests to the importance of this viral-host protein interaction through evolution. Furthermore, M029 lacks the N-terminal DNABD found in E3, but has an intact C-terminal DSRM (38), consistent with the fact that we found that E3 interacts with DHX9 via the DSRM and not the DNABD (Fig. 2E). Although both E3 and DHX9 can bind to RNA, benzonase treatment of cell lysates to degrade nucleic acids did not prevent the E3-DHX9 interaction (data not shown) suggesting that it is not dependent on RNA.
Examining the role of E3 in isolation in human monocytes allowed us to directly assess the effect of E3 on PRR-stimulated cytokine induction, and to subsequently implicate DHX9 in specific E3-antagonized pathways. This showed that E3 expression very effectively inhibited PRR-stimulated IL-6 production, which required DHX9. VACV E3 was previously demonstrated to inhibit IL-6 induction in infected cells by an unidentified largely PKR-independent mechanism (16). E3 targeting of IL-6 in vivo is likely important because IL-6–deficient mice have been reported to support up to a 1000-fold greater viral replication in the lungs, due to a defective cytotoxic T cell response against VACV (39). One mechanism revealed, which could explain the role for DHX9 in IL-6 induction, was enhancement of NF-κB–dependent promoter transactivation, which was shown to be directly antagonized by E3 (Fig. 10). The ability of DHX9 to enhance IL-6 promoter induction was largely dependent on the presence of the NF-κB element within that promoter (Fig. 8), and that DHX9 acts very proximal to NF-κB was shown by the fact that DHX9 enhanced NF-κB–dependent reporter gene expression initiated by direct expression of the p65 NF-κB subunit (Fig. 9D). In fact, the ability of a p65-Gal4 fusion protein to transactivate a Gal4-dependent promoter was also synergistically enhanced by DHX9 expression (Fig. 9E). The precise mechanism by which DHX9 facilitates this transcriptional activity of NF-κB has yet to be fully elucidated. DHX9 has been shown to mediate an association between the coactivator CREB-binding protein (CBP) and RNA polymerase II (40), and NF-κB p65 mediates maximal IL-6 promoter activation via CBP for at least some ligands that induce IL-6 (41).
Furthermore, DHX9 has been identified as an interaction partner for p65, and capable of enhancing NF-κB dependent reporter gene expression induced by p65, TNFα, or NIK (42). Specifically, DHX9 was shown to associate with the transactivation domain of p65 (42). Also, DHX9 can interact with RNA polymerase II to enhance CBP-dependent transcription (43). Therefore, it is likely that in the case of the IL-6 promoter, PRR-stimulated NF-κB–dependent promoter stimulation, via CBP, requires, or is enhanced by, the presence of DHX9 proximal to NF-κB and CBP on the promoter.
A second function for DHX9 revealed by E3 antagonism of PRR-stimulated cytokine production was in the induction of TLR8-dependent cytokines. In contrast to the case for IL-6, this function seemed to be stimulus- and not promoter-specific because although DHX9 was required for maximal CL075-stimulated TNFα, IP10, and RANTES production, and each of these were inhibited by E3, DHX9 was not required for, and E3 did not inhibit, TLR2-dependent production of these cytokines (Fig. 3). The mechanism whereby DHX9 contributes to TLR8-dependent cytokine induction is not clear, and there are a number of possibilities. Because all the genes tested do have NF-κB–binding elements in their promoters, it could be that TLR8-stimulated NF-κB–dependent genes are more dependent on an NF-κB·DHX9·CBP complex than similar genes induced by TLR2, but this seems unlikely. Another possibility is that DHX9 here has a cytosolic role in TLR8 signaling, and cytosolic roles for DHX9 in innate immune signaling have been proposed previously, in terms of nucleic acid sensing (22). In fact, Kim et al. (22) showed that DHX9 could interact with MyD88, which they interpreted as MyD88 being an adaptor protein for DHX9-dependent dsDNA sensing. It is therefore possible that a DHX9-MyD88 interaction may enhance endosomal TLR signaling (for example, by TLR8) but not be required for cell membrane TLR signaling (for example, by TLR2).
Finally, in contrast to previous reports in DCs (22, 23), we found no role for DHX9 in nucleic acid sensing in monocytes, although as expected from our IL-6 promoter studies, RNA virus-stimulated IL-6 production was DHX9-dependent (Fig. 4D). In fact, one of the key genes used as a readout of nucleic acid sensing in the studies in DCs was IL-6. Because we found a stimulus- and cell-independent role for DHX9 in IL-6 induction (e.g. Fig. 5), at least some of the data implicating DHX9 as an innate immune nucleic acid sensor of both dsDNA and dsRNA could be re-interpreted in the context of a role for DHX9 in IL-6 promoter induction. In conclusion, our study demonstrates a broad role for DHX9 in innate immune cytokine regulation and reveals a new mechanism whereby VACV, via E3, disrupts innate immunity by targeting DHX9.
Experimental procedures
Cell culture
THP-1 human monocytic cells were maintained in RPMI containing 10% (v/v) fetal calf serum, 2 mm l-glutamine, and 50 μg/ml of gentamicin. BEAS-2B human bronchial epithelial cells, HeLa cells, and HEK293T cells were maintained in Dulbecco's modified Eagle's medium containing 10% (v/v) fetal calf serum and 10 μg/ml of ciprofloxacin. HEK293 cells stably transfected with TLR2 (HEK293–TLR2) or TLR8 (HEK293–TLR8) were cultured in selection antibiotics to maintain TLR expression (1 mg/ml of G418 or 10 μg/ml of blasticidin, respectively).
PRR agonists and viruses
Cell agonists used were Pam3CSK4 (Invivogen), CL075 (Invivogen), ultrapure LPS (Alexis Biochemicals), a 70-mer dsDNA (70-mer (44)), poly(dA:dT), poly(I:C) (both Sigma), cGAMP (Invivogen), Sendai virus (ECACC), and IL-1α (PBL Interferon Source). 70-mer, poly(dA:dT), and cGAMP were delivered to cells by transfection using Lipofectamine 2000 (Invitrogen). VACV WR strain was a gift from G. Smith (University of Cambridge) and VACV lacking the gene E3L (ΔE3L) was a gift from B. Jacobs (ASU Biodesign Institute, Arizona State University).
Plasmids
E3L was cloned from VACV WR DNA into pCMV-HA (pCMV-HA-E3). E3(1–83) and E3(84–190) were subcloned from pCMV-HA-E3. DHX9 in pcDNA3 (43) was a gift from S. Aratani (St. Marianna University School of Medicine, Kanagawa, Japan). Expression plasmids for MyD88 and p65, and the NF-κB-luc reporter plasmid were described previously (45). Gal4-p65 and pFR-luc were from Stratagene. Plasmids used for retroviral expression were pTRIPZpuro for lentiviral shRNA-inducible expression (Thermo Scientific), psPAX2 lentiviral packaging vector (Addgene), pMD2.G for expression of VSV-G envelope (Addgene), and pMSCVneo for stable expression of E3, VSVg, and Gag-pol (gifts from K Fitzgerald, University of Massachusetts Medical School). The IL-6 promoter reporter gene plasmids were gifts from M. Wewers (Ohio State University).
Generation of THP-1 cells stably expressing E3
E3 was subcloned from pCMV-HA into pMSCVneo (pMSCV-E3). HEK293T cells were transfected with 4 μg of pMSCV-E3, 1 μg of VSVg, and 1 μg of Gag-pol. Cells were also transfected with pMSCV empty vector (pMSCV-EV), VSVg, and Gag-pol. The supernatants were collected and then THP-1 cells were transduced with the retroviral supernatants using 0.5 μg of Polybrene. Selection of the cells was carried out using 1 mg/ml of G418. After selection, lysates of pMSCV-EV- and pMSCV-E3-infected THP-1 cells were generated and the expression of E3 was tested by immunoblotting for HA.
Generation of cells inducibly expressing DHX9 shRNA
To generate THP-1 and BEAS-2B cells stably expressing doxycycline-inducible shRNA, shRNA encoding-lentiviruses were first generated in HEK293T cells. DHX9-targeting shRNA pTRIPZ vectors (Thermo Scientific Open Biosystems), and a NSC shRNA designed with minimal homology to known mammalian genes were used. HEK293T cells were transfected with 4 μg of shRNA-expressing pTRIPZ vector, 3 μg of psPAX2, and 1 μg of pMD2. 48 h later, the supernatants were harvested and replaced with fresh media. The lentivirus-containing supernatants were centrifuged and then filtered through 0.45-μm filters. They were stored at 4 °C until use. The supernatants were harvested again at 72 h, centrifuged, and filtered. THP-1 or BEAS-2B cells were seeded at 2 × 106 cells per 10-cm plate. 24 h later, THP-1 cells were centrifuged at 1000 × g for 5 min and resuspended in 50% (v/v) complete RPMI (5 ml) and 50% (v/v) lentivirus supernatant medium (5 ml) with 4 μg of Polybrene. Adherent BEAS-2B cells had their supernatant pipetted off and then 50% (v/v) complete Dulbecco's modified Eagle's medium and 50% (v/v) lentivirus supernatant (with Polybrene) was gently added to the cells. The cells were incubated at 37 °C for 48 h. The transduction step was then repeated in the absence of Polybrene. After a further 48 h, the cells were selected by the addition of 1 μg/ml of puromycin. The cells were then left for 5–7 days at 37 °C and tested for shRNA expression. shRNA expression was assayed by incubating the cells in 1 μg/ml of doxycycline for 24, 48, and 72 h and looking for expression of green fluorescent protein by fluorescent microscopy. After confirmation of green fluorescent protein expression upon the addition of doxycycline, DHX9 protein and mRNA knockdown were assayed. A total of eight clones were tested and the three clones (A7, G2, and D11) found to give the most effective knockdown of DHX9 mRNA and protein were later employed, alongside the NSC cell line. The targets sequences of the DHX9 shRNAs were: A7, 5′-ATGACATAAACAACATCGT-3′, G2, 5′-TGCACTTCTTGTTCTTCCT-3′, and D11, 5′-TTCGTTCAATTGAGACATG-3′).
Western blotting and co-immunoprecipitation
Antibodies used were anti-HA (mouse, Covance), anti-β-actin (mouse, Sigma), anti-E3 (a gift from B. Jacobs ASU Biodesign Institute, Arizona State University), anti-DHX9 (rabbit, Novus Biologicals), anti-tubulin (mouse, Millipore), anti-IκBα (mouse, gift from R. T. Hay, University of Dundee), anti-phospho-Ser536-p65 (rabbit, Cell Signaling), anti-p65 (mouse, Santa Cruz), anti-phospho-p38 MAPK (rabbit, Cell Signaling), and anti-p38 MAPK (rabbit, Cell Signaling). After cell transfection or stimulation, lysates were prepared and proteins were separated by SDS-PAGE. Resolved proteins were transferred to polyvinylidene difluoride membranes and immunoblotted with the indicated antibodies.
For E3-DHX9 interaction studies in HEK293T cells, E3-expressing vectors were transfected into cells using GeneJuice® (Novagen). 48 h following transfection, the cells were lysed with 1% (v/v) NP-40 lysis buffer. Lysates were pre-cleared using a 50% (v/v) slurry of protein A-Sepharose beads (Sigma) and then immunoprecipitated with the appropriate antibody. To examine E3-DHX9 interaction in virally-infected human monocytes, 100 million undifferentiated THP-1 cells were infected with VACV for 16 h. Cells were then lysed in lysis buffer containing 50 mm Hepes, 1 mm EDTA, 10% glycerol, 1% Nonidet P-40, 300 mm NaCl together with 10 μl/ml of aprotinin, 1 mm phenylmethylsulfonyl fluoride, and 1 mm sodium orthovanadate. Clarified lysates were immunoprecipitated using either 1.6 μg of anti-DHX9 antibody (Novus Biologicals catalog number NB110-40579) or 1.6 μg of rabbit IgG. Resulting immunoprecipitates were subjected to SDS-PAGE and immunoblotted for either E3 or DHX9.
ELISA
Cell culture supernatants were assayed for the presence of TNFα, IL-6, IP10 (CXCL10), and RANTES (CCL5) by ELISA kits (R&D Systems).
Analysis of E3 protein interactome
Murine embryonic fibroblasts were infected with VACV or ΔE3L and 24 h later cell lysates were prepared and immunoprecipitated with anti-E3 Ab. Lysates were then subjected to SDS-PAGE and Coomassie staining. Gel slices were prepared, and proteins in gel slices were reduced by treatment with DTT and then alkylated by treatment with iodoacetamide. Gel slices were then subject to trypsin digest and peptides were eluted. Peptides were identified by LC–MS by FingerPrints Proteomics (University of Dundee, UK).
Quantitative RT-PCR
IL-6 mRNA expression was analyzed by RT-PCR, as previously described (46). The mRNA expression levels were normalized to β-actin mRNA and the respiratory quotient values were calculated. The mean respiratory quotient values calculated from three corresponding replicates was presented as fold-induction relative to the control sample set to 1. Error bars represent the standard deviation calculated using GraphPad Prism 5 software. Primers used were: IL-6 F, 5′-GGTACATCCTCGACGGCATCT-3′; R, 5′-GTGCCTCTTTGCTGCTTTCAC-3′; β-actin F, 5′-CGCGAGAAGATGACCCAGATC-3′; R, 5′-GCCAGAGGCGTACAGGGATA-3′.
Reporter gene assays
Cells were seeded in 96-well plates (HeLa cells at 0.5 × 105 cells/ml, HEK293 cells at 1 × 105 cells/ml) and transfected 24 h later with expression vectors and luciferase reporter genes indicated in the figure legends, using GeneJuice®. In all cases, 20 ng/well of phRL-TK reporter gene was cotransfected to normalize data for transfection efficiency. For IL-6 promoter or NF-κB reporter gene assays, 60 ng of reporter gene was used. For the p65 transactivation assay, 3 ng of a vector encoding p65-Gal4 fusion protein was used in combination with 60 ng of pFR-luciferase reporter. The total amount of DNA per transfection was kept constant at 230 ng by addition of pCMV empty vector. Cells were lysed in Passive Lysis Buffer (Promega) and whole cell lysates were analyzed for luciferase activity. Firefly luciferase activity was normalized to Renilla luciferase activity, and data are expressed as the mean fold-induction, relative to control levels, for a representative experiment performed in triplicate.
Statistical analysis
All data were analyzed with GraphPad Prism version 5.02 software. Statistical analysis was performed using the two-tailed unpaired Student's t test.
Author contributions
A. D., S. E. K., M. C., and A. G. B. investigation; A. D., S. E. K., M. C., and A. G. B. methodology; A. D., S. E. K., M. C., and A. G. B. writing-original draft; A. D., M. C., and A. G. B. writing-review and editing; A. G. B. conceptualization; A. G. B. supervision; A. G. B. funding acquisition.
This work was supported by Irish Health Research Board Grant PHD/2007/09 and Science Foundation Ireland Grants 11/PI/1056 and 16/IA/4376. The authors declare that they have no conflicts of interest with the contents of this article.
- PAMP
- pathogen-associated molecular pattern
- DAMP
- damage-associated molecular pattern
- DC
- dendritic cell
- RANTES
- regulated on activation normal T cell expressed and secreted
- LPS
- lipopolysaccharide
- CBP
- cAMP-response element-binding protein
- DHX9
- DExD/H-box helicase 9
- DNABD
- Z-DNA–binding domain
- DSRM
- dsRNA–binding motif
- NF-κB
- nuclear factor κB
- NSC
- non-silencing control
- PKR
- dsRNA-dependent protein kinase
- PRR
- pattern recognition receptor
- RLR
- RIG-I-like receptor
- TLR
- Toll-like receptor
- VACV
- vaccinia virus
- WR
- Western Reserve strain
- ssRNA
- single-stranded RNA
- NSC
- nonsilencing control
- IP
- immunoprecipitation
- IL
- interleukin
- EV
- empty vector
- TNFα
- tumor necrosis factor α.
References
- 1. Gürtler C., and Bowie A. G. (2013) Innate immune detection of microbial nucleic acids. Trends Microbiol. 21, 413–420 10.1016/j.tim.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Neill L. A., Golenbock D., and Bowie A. G. (2013) The history of Toll-like receptors: redefining innate immunity. Nat. Rev. Immunol. 13, 453–460 10.1038/nri3446 [DOI] [PubMed] [Google Scholar]
- 3. Paludan S. R., and Bowie A. G. (2013) Immune sensing of DNA. Immunity 38, 870–880 10.1016/j.immuni.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Almine J. F., O'Hare C. A., Dunphy G., Haga I. R., Naik R. J., Atrih A., Connolly D. J., Taylor J., Kelsall I. R., Bowie A. G., Beard P. M., and Unterholzner L. (2017) IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 8, 14392 10.1038/ncomms14392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cai X., Chiu Y. H., and Chen Z. J. (2014) The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296 10.1016/j.molcel.2014.03.040 [DOI] [PubMed] [Google Scholar]
- 6. Bowie A. G., and Unterholzner L. (2008) Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922 10.1038/nri2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brady G., and Bowie A. G. (2014) Innate immune activation of NFκB and its antagonism by poxviruses. Cytokine Growth Factor Rev. 25, 611–620 10.1016/j.cytogfr.2014.07.004 [DOI] [PubMed] [Google Scholar]
- 8. Schröder M., Baran M., and Bowie A. G. (2008) Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKvarϵ-mediated IRF activation. EMBO J. 27, 2147–2157 10.1038/emboj.2008.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kwon J. A., and Rich A. (2005) Biological function of the vaccinia virus Z-DNA-binding protein E3L: gene transactivation and antiapoptotic activity in HeLa cells. Proc. Natl. Acad. Sci. U.S.A. 102, 12759–12764 10.1073/pnas.0506011102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brandt T. A., and Jacobs B. L. (2001) Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 75, 850–856 10.1128/JVI.75.2.850-856.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang H. W., Watson J. C., and Jacobs B. L. (1992) The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 89, 4825–4829 10.1073/pnas.89.11.4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharp T. V., Moonan F., Romashko A., Joshi B., Barber G. N., and Jagus R. (1998) The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: implications for PKR autoregulation. Virology 250, 302–315 10.1006/viro.1998.9365 [DOI] [PubMed] [Google Scholar]
- 13. Valentine R., and Smith G. L. (2010) Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J. Gen. Virol. 91, 2221–2229 10.1099/vir.0.021998-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guerra S., Cáceres A., Knobeloch K. P., Horak I., and Esteban M. (2008) Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog. 4, e1000096 10.1371/journal.ppat.1000096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dueck K. J., Hu Y. S., Chen P., Deschambault Y., Lee J., Varga J., and Cao J. (2015) Mutational analysis of vaccinia virus E3 protein: the biological functions do not correlate with its biochemical capacity to bind double-stranded RNA. J. Virol. 89, 5382–5394 10.1128/JVI.03288-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Myskiw C., Arsenio J., van Bruggen R., Deschambault Y., and Cao J. (2009) Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-κB, and IRF3 pathways. J. Virol. 83, 6757–6768 10.1128/JVI.02570-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langland J. O., Kash J. C., Carter V., Thomas M. J., Katze M. G., and Jacobs B. L. (2006) Suppression of proinflammatory signal transduction and gene expression by the dual nucleic acid binding domains of the vaccinia virus E3L proteins. J. Virol. 80, 10083–10095 10.1128/JVI.00607-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorden K. B., Gorski K. S., Gibson S. J., Kedl R. M., Kieper W. C., Qiu X., Tomai M. A., Alkan S. S., and Vasilakos J. P. (2005) Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J. Immunol. 174, 1259–1268 10.4049/jimmunol.174.3.1259 [DOI] [PubMed] [Google Scholar]
- 19. Ranji A., and Boris-Lawrie K. (2010) RNA helicases: emerging roles in viral replication and the host innate response. RNA Biol. 7, 775–787 10.4161/rna.7.6.14249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee C. G., da Costa Soares V., Newberger C., Manova K., Lacy E., and Hurwitz J. (1998) RNA helicase A is essential for normal gastrulation. Proc. Natl. Acad. Sci. U.S.A. 95, 13709–13713 10.1073/pnas.95.23.13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang T., Birsoy K., Hughes N. W., Krupczak K. M., Post Y., Wei J. J., Lander E. S., and Sabatini D. M. (2015) Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 10.1126/science.aac7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim T., Pazhoor S., Bao M., Zhang Z., Hanabuchi S., Facchinetti V., Bover L., Plumas J., Chaperot L., Qin J., and Liu Y. J. (2010) Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 107, 15181–15186 10.1073/pnas.1006539107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Z., Yuan B., Lu N., Facchinetti V., and Liu Y. J. (2011) DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J. Immunol. 187, 4501–4508 10.4049/jimmunol.1101307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiménez-Dalmaroni M. J., Gerswhin M. E., and Adamopoulos I. E. (2016) The critical role of Toll-like receptors: from microbial recognition to autoimmunity: a comprehensive review. Autoimmun. Rev. 15, 1–8 10.1016/j.autrev.2015.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee C. G., and Hurwitz J. (1992) A new RNA helicase isolated from HeLa cells that catalytically translocates in the 3′ to 5′ direction. J. Biol. Chem. 267, 4398–4407 [PubMed] [Google Scholar]
- 26. Huang M., and Mitchell B. S. (2008) Guanine nucleotide depletion mediates translocation of nucleolar proteins, including RNA helicase A (DHX-9). Nucleosides Nucleotides Nucleic Acids 27, 704–711 10.1080/15257770802145132 [DOI] [PubMed] [Google Scholar]
- 27. Tang H., Gaietta G. M., Fischer W. H., Ellisman M. H., and Wong-Staal F. (1997) A cellular cofactor for the constitutive transport element of type D retrovirus. Science 276, 1412–1415 10.1126/science.276.5317.1412 [DOI] [PubMed] [Google Scholar]
- 28. Tettweiler G., and Lasko P. (2006) A new model for translational regulation of specific mRNAs. Trends Biochem. Sci. 31, 607–610 10.1016/j.tibs.2006.09.008 [DOI] [PubMed] [Google Scholar]
- 29. Hartman T. R., Qian S., Bolinger C., Fernandez S., Schoenberg D. R., and Boris-Lawrie K. (2006) RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 13, 509–516 10.1038/nsmb1092 [DOI] [PubMed] [Google Scholar]
- 30. Lin L., Li Y., Pyo H. M., Lu X., Raman S. N., Liu Q., Brown E. G., and Zhou Y. (2012) Identification of RNA helicase A as a cellular factor that interacts with influenza A virus NS1 protein and its role in the virus life cycle. J. Virol. 86, 1942–1954 10.1128/JVI.06362-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bolinger C., Sharma A., Singh D., Yu L., and Boris-Lawrie K. (2010) RNA helicase A modulates translation of HIV-1 and infectivity of progeny virions. Nucleic Acids Res. 38, 1686–1696 10.1093/nar/gkp1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolinger C., Yilmaz A., Hartman T. R., Kovacic M. B., Fernandez S., Ye J., Forget M., Green P. L., and Boris-Lawrie K. (2007) RNA helicase A interacts with divergent lymphotropic retroviruses and promotes translation of human T-cell leukemia virus type 1. Nucleic Acids Res. 35, 2629–2642 10.1093/nar/gkm124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawrence P., and Rieder E. (2009) Identification of RNA helicase A as a new host factor in the replication cycle of foot-and-mouth disease virus. J. Virol. 83, 11356–11366 10.1128/JVI.02677-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Isken O., Baroth M., Grassmann C. W., Weinlich S., Ostareck D. H., Ostareck-Lederer A., and Behrens S. E. (2007) Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13, 1675–1692 10.1261/rna.594207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sadler A. J., Latchoumanin O., Hawkes D., Mak J., and Williams B. R. (2009) An antiviral response directed by PKR phosphorylation of the RNA helicase A. PLoS Pathog. 5, e1000311 10.1371/journal.ppat.1000311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reddy T. R., Tang H., Xu W., and Wong-Staal F. (2000) Sam68, RNA helicase A and Tap cooperate in the post-transcriptional regulation of human immunodeficiency virus and type D retroviral mRNA. Oncogene 19, 3570–3575 10.1038/sj.onc.1203676 [DOI] [PubMed] [Google Scholar]
- 37. Rahman M. M., Liu J., Chan W. M., Rothenburg S., and McFadden G. (2013) Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 9, e1003465 10.1371/journal.ppat.1003465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Myskiw C., Arsenio J., Hammett C., van Bruggen R., Deschambault Y., Beausoleil N., Babiuk S., and Cao J. (2011) Comparative analysis of poxvirus orthologues of the vaccinia virus E3 protein: modulation of protein kinase R activity, cytokine responses, and virus pathogenicity. J. Virol. 85, 12280–12291 10.1128/JVI.05505-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kopf M., Baumann H., Freer G., Freudenberg M., Lamers M., Kishimoto T., Zinkernagel R., Bluethmann H., and Köhler G. (1994) Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368, 339–342 10.1038/368339a0 [DOI] [PubMed] [Google Scholar]
- 40. Nakajima T., Uchida C., Anderson S. F., Lee C. G., Hurwitz J., Parvin J. D., and Montminy M. (1997) RNA helicase A mediates association of CBP with RNA polymerase II. Cell 90, 1107–1112 10.1016/S0092-8674(00)80376-1 [DOI] [PubMed] [Google Scholar]
- 41. Vanden Berghe W., De Bosscher K., Boone E., Plaisance S., and Haegeman G. (1999) The nuclear factor-κB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J. Biol. Chem. 274, 32091–32098 10.1074/jbc.274.45.32091 [DOI] [PubMed] [Google Scholar]
- 42. Tetsuka T., Uranishi H., Sanda T., Asamitsu K., Yang J. P., Wong-Staal F., and Okamoto T. (2004) RNA helicase A interacts with nuclear factor κB p65 and functions as a transcriptional coactivator. Eur. J. Biochem. 271, 3741–3751 10.1111/j.1432-1033.2004.04314.x [DOI] [PubMed] [Google Scholar]
- 43. Aratani S., Fujii R., Oishi T., Fujita H., Amano T., Ohshima T., Hagiwara M., Fukamizu A., and Nakajima T. (2001) Dual roles of RNA helicase A in CREB-dependent transcription. Mol. Cell Biol. 21, 4460–4469 10.1128/MCB.21.14.4460-4469.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Unterholzner L., Keating S. E., Baran M., Horan K. A., Jensen S. B., Sharma S., Sirois C. M., Jin T., Latz E., Xiao T. S., Fitzgerald K. A., Paludan S. R., and Bowie A. G. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 10.1038/ni.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brady G., Haas D. A., Farrell P. J., Pichlmair A., and Bowie A. G. (2017) Molluscum contagiosum virus protein MC005 inhibits NF-κB activation by targeting NEMO-regulated IκB kinase activation. J. Virol. 91, e00545–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Güurtler C., Carty M., Kearney J., Schattgen S. A., Ding A., Fitzgerald K. A., and Bowie A. G. (2014) SARM regulates CCL5 production in macrophages by promoting the recruitment of transcription factors and RNA polymerase II to the Ccl5 promoter. J. Immunol. 192, 4821–4832 10.4049/jimmunol.1302980 [DOI] [PMC free article] [PubMed] [Google Scholar]