

Abstract
A formal total synthesis of the antitumor marine natural product (rac)‐renieramycin T, which possesses a characteristic ecteinascidin‐type A ring in the renieramycin–saframycin core skeleton, was elaborated. The key steps in the synthesis of (rac)‐renieramycin T are a modified Pictet–Spengler cyclization of dialkylated oxomalonate derivatives and decarboxylation via a monocarboxylic acid derivative followed by stereocontrolled protonation of the enol intermediate. A key intermediate in our previous synthesis of renieramycin T was used, and the formal synthesis was accomplished in 21 steps from a known piperazine‐2,5‐dione derivative.
Keywords: cyclization, decarboxylation, fused-ring systems, natural products, total synthesis
1. Introduction
Many 1,2,3,4‐tetrahydroisoquinolines exhibit various bioactivities, such as antitumor, antibacterial, antiviral, anticoagulant, anti‐inflammatory, anti‐Alzheimer, and anticonvulsant activities.1 Among them, members of the saframycin, renieramycin, and ecteinascidin families have captured intense attention due to their interesting biological activities and intriguing structures (Figure 1). Particularly, novel marine‐derived ecteinascidin 7432 (2, Trabectedin, Yondelis) has superior antitumor activity and was approved by the European Commission in 20073 and the US Food and Drug Administration (FDA) in 20154 for the treatment of soft‐tissue sarcomas.
Figure 1.
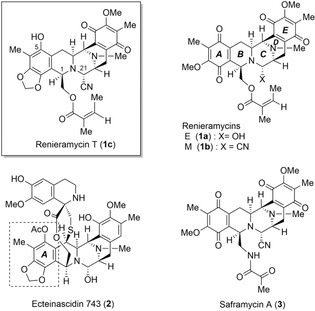
Structures of 1,2,3,4‐tetrahydroisoquinoline natural products.
A large number of renieramycin marine natural products5 have been discovered in sponges of genera Reniera,6 Xestospongia,7 Haliclona,8 Cribrochalina,9 and Neopetrosia 10 in miniscule amounts. In our continuing chemical studies on renieramycins,11 we found that the transformation of an unstable aminal group in a natural product, such as renieramycin E (1 a), into an aminonitrile group by pretreatment with KCN prior to extraction and isolation furnished many kinds of stable renieramycin derivatives, such as renieramycin M (1 b).12 We also elucidated the chemical structure of renieramycin T (1 c), which was isolated from the blue sponge Xestospongia sp. in Thailand11c and the Philippines.11b Renieramycin T (1 c) possesses a highly substituted phenol in the terminal A ring and a condensed 1,3‐dioxole ring, which are exactly the same as those in ecteinascidins. As the chemical structure of 1 c is the first example of a hybrid pentacyclic core, we are very interested in the biosynthetic pathway and the biological activity of 1 c and its derivatives. To date, two total syntheses of (−)‐1 c by us and Chen's group have been reported.13 However, we focused on an alternative route for supplying a large amount of 1 c to promote research of structure–activity relationships. Herein, we report a formal total synthesis of 1 c, which includes the Pictet–Spengler reaction of a primary amine with an oxomalonic acid ester followed by decarboxylation and stereocontrolled protonation at C1 of the enol intermediate from the less‐hindered face.14
2. Results and Discussion
We embarked on an alternative total synthesis of 1 c on the basis of our previous synthetic studies on saframycin antitumor antibiotics (Scheme 1).15 Condensation of highly functionalized benzaldehyde 4 16 with known piperazine‐2,5‐dione derivative 5 15b afforded Z isomer 6 in 79 % yield. Chemoselective reduction of the carbonyl group activated by an isopropyloxycarbonyl group in 6, followed by treatment with trifluoroacetic acid (TFA) gave cyclized product 7 (78 %, two steps). Removal of the isopropoxycarbonyl group in 7 with TFA and H2SO4 gave a secondary amine that was treated with a NaBH3CN–aqueous formaldehyde system to provide tertiary amine 8 in 68 % overall yield. Hydrogenation (2.8 MPa) of the exo olefin in 8 on 20 % Pd(OH)2 in EtOH at 80 °C along with hydrogen attack from the less‐hindered α face gave 9 as a single isomer in 77 % yield. At that point, we thought it would be important to distinguish the two phenolic OH groups in both terminal rings, because regioselective oxidation was required to prepare p‐quinone in the E ring phenol at a later stage. Thus, the phenolic OH group of the E ring of 9 was protected with an allyl group, and the tosyl group in the A ring was removed with KOH/H2O to increase its reactivity for the Pictet–Spengler cyclization to give 10 in 80 % over two steps.
Scheme 1.
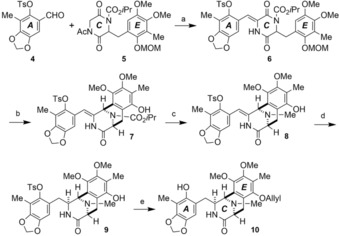
Preparation of tricyclic lactam 10. Reagents and conditions: a) tBuOK, tBuOH, CH2Cl2, 0 °C, 1 h, 79 %; b) 1) Li(tBuO)3AlH, THF, 25 oC, 6 h; 2) TFA, CH2Cl2, 25 oC, 3 h, 78 % (two steps); c) 1) H2SO4, TFA, anisole, 25 °C, 7.5 h; 2) NaBH3CN, 37 % aq HCHO, MeOH, AcOH, 25 °C, 2 h, 68 % (two steps); d) H2 (2.8 MPa), 20 % Pd(OH)2/C, EtOH, 80 °C, 27 h, 77 %; e) 1) allyl bromide, K2CO3, acetone, reflux, 5.5 h; 2) aq KOH, EtOH, reflux, 3 h, 80 % (two steps). Ts=tosyl, Ac=acetyl, MOM=methoxymethyl.
With precursor 10 in hand, we focused on the construction of a pentacyclic core by using the Pictet–Spengler cyclization (Scheme 2). Partial reduction of the lactam carbonyl group in 10, followed by the introduction of a cyanide group gave aminonitrile 11. We had hoped that the aminonitrile would be more reactive than lactam 10 in the Pictet–Spengler cyclization. However, it was revealed that the Pictet–Spengler cyclization of 11 with even commonly used simple aldehydes,15c for example, the reaction of 11 with benzoyloxyacetaldehyde, did not proceed at all.17 Furthermore, substrate decomposition was observed if harsher reaction conditions were used. It was clarified that the aminonitrile moiety of 11 was relatively unstable under acidic or high‐temperature conditions. Therefore, we abandoned this route at this stage.
Scheme 2.
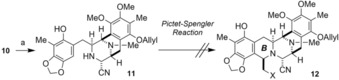
Cyclization attempt by using the Pictet–Spengler reaction of aminonitrile 11. Reagents and conditions. a) Cp2ZrHCl, THF, −20 to 0 °C, 1 h; aq KCN, 25 °C, 4 h, 96 %. Cp=η5‐cyclopentadienyl.
This problem was solved by applying our protocol to the total synthesis of saframycin A (3, Scheme 3).18 Primary amine I did not have a relatively unstable aminonitrile group, and so steric repulsion would be reduced. Thus, it would be easy to construct desired pentacyclic core IV by using a three‐step sequence through compound II, which includes the Pictet–Spengler cyclization with oxomalonic acid ester to install a diester unit, decarboxylation, and stereoselective protonation from the convex face of these “V”‐shaped bis‐1,2,3,4‐tetrahydroisoquinoline natural products.
Scheme 3.
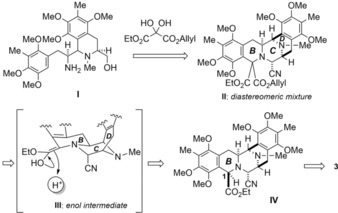
Recently established strategy for construction of the B ring of bis‐tetrahydroisoquinoline natural products, represented by saframycin A (3).
Activation of the lactam carbonyl group in 10 with di‐tert‐butyl dicarbonate (Boc2O) gave 13 in 72 % yield according to a protocol independently outlined by Fukuyama and Stoltz19 (Scheme 4). Reductive cleavage of the lactam ring in 13 with NaBH4 in EtOH, followed by treatment with TFA in CH2Cl2 gave primary amine 14 (73 %, two steps). Pictet–Spengler cyclization of 14 with allyl ethyl oxomalonate hydrate20 furnished diester 15 as an inseparable diastereomeric mixture (1:1) in 80 % yield. Then, the phenolic OH group in 15 was protected by a benzyl (Bn) group (83 %), and Swern oxidation of 16 followed by treatment with KCN afforded pentacyclic core 17 (56 %, two steps). Removal of the allyl group in 17 with Pd(PPh3)4 and dimedone, followed by decarboxylation exclusively gave ester 18 as a single diastereomer in 72 % yield.21, 22 The stereochemistry of 18 was determined by nuclear Overhauser enhancement (NOE) experiments, which indicated that 18 had a syn relationship between the C1 and C3 diaxial protons.
Scheme 4.
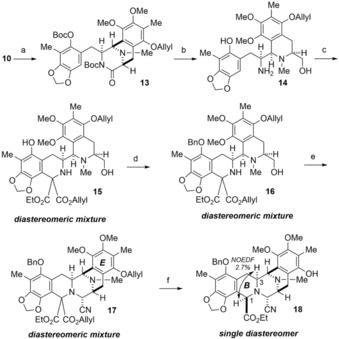
Construction of the B ring. Reagents and conditions. a) Boc2O, 4‐(dimethylamino)pyridine (DMAP), MeCN, reflux, 57.5 h, 72 %; b) 1) NaBH4, EtOH, 25 °C, 3 h; 2) TFA, CH2Cl2, 25 °C, 1.5 h, 73 % (two steps); c) 1‐allyl 3‐ethyl 2,2‐dihydroxymalonate, TFA, AcOH, 25 °C, 6 h, 80 %; d) BnBr, K2CO3, acetone, 25 °C, 8 h, 83 %; e) 1) Swern oxidation; 2) aq KCN, AcOH, THF, 25 °C, 2 h, 56 % (two steps); f) 1) Pd(PPh3)4, dimedone, THF, 25 °C, 1 h; 2) CHCl3, reflux, 2 h, 72 % (two steps).
Finally, reduction of ester 18 (71 %), followed by oxidative demethylation of 19 into quinone ring afforded 20 (51 %) (Scheme 5). This is the key intermediate in our total synthesis;13a its 1H NMR, 13C NMR, and IR spectroscopy data; MS data; and TLC behavior were identical to those of an authentic sample upon comparison Thus, we accomplished a formal synthesis of (rac)‐renieramycin T (1 c).
Scheme 5.
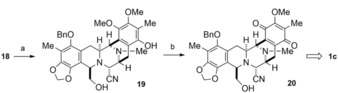
Formal synthesis of (rac)‐renieramycin T (1 c). Reagents and conditions. a) LiBH4, MeOH, THF, 25 °C, 3 h, 71 %; b) aq ceric ammonium nitrate (CAN), THF, 0 °C, 20 min, 51 %.
3. Conclusions
We accomplished a formal synthesis of (rac)‐renieramycin T (1 c). Whereas the aromatic ring having a 1,3‐dioxole ring suppressed reactivity during the Pictet–Spengler cyclization, reductive cleavage of the lactam ring in 13 afforded primary amine 14. Then, treatment of 14 with allyl ethyl oxomalonate in the Pictet–Spengler reaction, followed by decarboxylation and stereoselective protonation of the resulting enol intermediate from the less‐hindered face produced the desired bis‐1,2,3,4‐tetrahydroisoquinoline intermediate.
Ways of utilizing this strategy for the synthesis of ecteinascidins along with fennebricin B23 are under investigation in our laboratory.
Experimental Section
General Methods
All reactions involving air‐ and moisture‐sensitive reagents were performed in oven‐dried glassware and by using standard syringe‐septum cap techniques. All reactions were monitored by thin‐layer chromatography (silica gel GF254) examined under UV light (λ=254 nm). Flash column chromatography was performed on Merck Silica Gel (230–400 mesh) with the solvent indicated. IR spectra were obtained with a Shimadzu Prestige‐21/IR Affinity‐1 Fourier Transform Infrared (FTIR) spectrometer. 1H NMR and 13C NMR spectroscopic data were recorded with a JEOL ECS‐400 spectrometer at 400 MHz for 1H and 100 MHz for 13C and with a JEOL AL‐400 spectrometer at 400 MHz for 1H and 100 MHz for 13C. NMR spectra were measured in CDCl3, and the chemical shifts were recorded in δ H values relative to (CH3)4Si as the internal standard. Low‐ and high‐resolution mass (HRMS) mass spectra were recorded with a JMS‐700 instrument with a direct inlet system operating at 70 eV.
Syntheses
(Z)‐Isopropyl 2‐[4,5‐dimethoxy‐2‐(methoxymethoxy)‐3‐methylbenzyl]‐5‐{[7‐methyl‐6‐(tosyloxy)benzo[d][1,3]dioxol‐5‐yl]methylene}‐3,6‐dioxopiperazine‐1‐carboxylate (6)
A solution of tBuOK in tBuOH (1 m, 30 mL, 30 mmol) was added dropwise to a stirred solution of aldehyde 4 (8.36 g, 25 mmol) and acetate 5 (11.66 g, 25 mmol) in CH2Cl2 (100 mL) at 0 °C over 1 h, and the mixture was stirred at 0 °C for 1 h. The mixture was diluted with saturated aqueous NH4Cl solution (200 mL) and extracted with CH2Cl2 (3×250 mL). The combined extract was washed with brine (200 mL), dried, and concentrated in vacuo to give a residue (18.26 g), which was subjected to flash column chromatography on SiO2 (500 g) with hexane/EtOAc (3:2) to afford 6 (14.62 g, 79 %) as a colorless amorphous powder; R f=0.35 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ=7.62 (d, J=8.5 Hz, 2 H, C6 H 4CH3), 7.24 (d, J=8.5 Hz, 2 H, C6 H 4CH3), 7.10 (br s, 1 H, NH), 6.41 (s, 1 H, 4“‐H), 6.34 (s, 1 H, 6′‐H), 6.15 (s, 1 H, 5a‐H), 6.07 (d, J=1.2 Hz, 1 H, 2”‐H), 6.05 (d, J=1.2 Hz, 1 H, 2“‐H), 5.17 (sept, J=5.7 Hz, 1 H, CH(CH3)2), 5.03 (t, J=4.9 Hz, 1 H, 2‐H), 4.88 (d, J=5.9 Hz, 1 H, OCH 2OCH3), 4.82 (d, J=5.9 Hz, 1 H, OCH 2OCH3), 3.73 (s, 3 H, 5′‐OCH3), 3.58 (s, 3 H, OCH2OCH 3), 3.53 (s, 3 H, 4′‐OCH3), 3.30 (d, J=4.9 Hz, 2 H, 2a‐H2), 2.34 (s, 3 H, C6H4CH 3), 2.27 (s, 3 H, 7”‐CH3), 2.00 (s, 3 H, 3′‐CH3), 1.43 (d, J=5.7 Hz, 3 H, CH(CH 3)2), 1.41 ppm (d, J=5.7 Hz, 3 H, CH(CH 3)2); 13C NMR (100 MHz, CDCl3): δ=164.9 (C‐3), 157.4 (C‐6), 152.2 (CO2), 149.5 (C‐2′ or 5′), 149.3 (C‐2′ or 5′), 148.1 (C‐4′ or 7“a), 148.0 (C‐4′ or 7”a), 145.6 (C‐3“a), 145.4 (C 6H4CH3), 141.8 (C‐6”), 132.3 (C 6H4CH3), 130.1 (C 6H4CH3), 128.7 (C 6H4CH3), 125.9 (C‐3′), 124.7 (C‐5), 122.3 (C‐1′), 119.7 (C‐5“), 117.1 (C‐7”), 113.9 (C‐5a), 112.9 (C‐6′), 104.6 (C‐4“), 102.3 (C‐2”), 100.0 (CH2OCH3), 72.0 (CH(CH3)2), 60.1 (4′‐OCH3), 59.9 (C‐2), 57.6 (CH2OCH3), 55.8 (5′‐OCH3), 33.8 (C‐2a), 21.8 (CH(CH3)2), 21.8 (CH(CH3)2), 21.5 (C6H4 CH3), 10.9 (7“‐CH3), 10.4 ppm (3′‐CH3); FTIR (KBr): =1773, 1694, 1479, 1420, 1375, 1281, 1233, 1196, 1173, 1103, 1080, 1061, 1022, 972, 806 cm−1; MS (EI): m/z (%): 741 (40), 740 (100) [M]+, 569 (33), 568 (31), 499 (31), 467 (55), 319 (21), 287 (59), 230 (27), 225 (43), 220 (23), 219 (46), 191 (23), 190 (22), 181 (73); HRMS (EI): m/z: calcd for C36H40O13N2S: 740.2251 [M]+; found: 740.2250.
(Z)‐Isopropyl (1R*,5S*)‐7‐hydroxy‐9,10‐dimethoxy‐8‐methyl‐2‐ {[7‐methyl‐6‐(tosyloxy)‐benzo[d][1,3]dioxol‐5‐yl]methylene}‐ 4‐oxo‐1,2,3,4,5,6‐hexahydro‐1,5‐iminobenzo[d]azocine‐11‐ carboxylate (7)
Li(tBuO)3AlH (6.36 g, 25 mmol) was added to a stirred solution of 6 (3.70 g, 5 mmol) in THF (170 mL) at 0 °C over 40 min, and the mixture was stirred at 25 °C for 6 h. The mixture was diluted by adding a saturated aqueous Rochelle salt solution (100 mL) and was then extracted with CHCl3 (3×300 mL). The combined extract was washed with brine (300 mL), dried, and concentrated in vacuo to give a residue, which was used in the next step without further purification. TFA (22.5 mL) was added to a stirred solution of the above product (3.71 g) in CH2Cl2 (45 mL), and the mixture was stirred at 25 °C for 3 h. The mixture was concentrated in vacuo, and the residue was diluted with H2O (200 mL). Then, the mixture was brought to pH 9 with concd. NH4OH (25 mL) and was extracted with CH2Cl2 (3×300 mL). The combined extract was washed with brine (300 mL), dried, and concentrated in vacuo to give a residue (3.75 g), which was subjected to column chromatography on SiO2 (100 g) with CHCl3/EtOAc (4:1) to afford tricyclic lactam 7 (2.64 g, 78 %, two steps) as a pale‐yellow amorphous powder. As it was a mixture of rotational isomers, the 1H NMR and 13C NMR spectra are both extremely complex at 25 °C in CDCl3; R f=0.23 (CHCl3/EtOAc 4:1); 1H NMR (400 MHz, [D6]DMSO, 100 °C): δ=8.67 (s, 1 H, OH or NH), 7.99 (s, 1 H, OH or NH), 7.74 (d, J=8.0 Hz, 2 H, C6 H 4CH3), 7.44 (d, J=8.0 Hz, 2 H, C6 H 4CH3), 6.57 (s, 1 H, 4′‐H), 6.02 (s, 2 H, 2′‐H2), 5.72 (s, 1 H, 1‐H), 5.65 (s, 1 H, 2a‐H), 4.89 (sept, J=5.3 Hz, 1 H, CH(CH3)2), 4.81 (dd, J=5.2, 2.3 Hz, 1 H, 5‐H), 3.77 (s, 3 H, 10‐OCH3), 3.72 (s, 3 H, 9‐OCH3), 2.92 (d, J=2.3 Hz, 1 H, 6‐Hα), 2.91 (d, J=5.2 Hz, 1 H, 6‐Hβ), 2.44 (s, 3 H, C6H4CH 3), 2.07 (s, 3 H, 8‐CH3), 1.99 (s, 3 H, 7′‐CH3), 1.25 (d, J=5.3 Hz, 3 H, CH(CH 3)2), 1.24 ppm (d, J=5.3 Hz, 3 H, CH(CH 3)2); 13C NMR (100 MHz, [D6]DMSO, 100 °C): δ=167.6 (C‐1), 152.5 (CO2), 149.1 (C‐7 or 9), 148.3 (C‐7 or 9), 145.4 (C‐7′a), 145.1 (C 6H4CH3), 144.6 (C‐3′a), 142.5 (C‐10a), 139.6 (C‐6′), 133.9 (C‐2 or 5′ or 7′), 132.4 (C 6H4CH3), 129.4 (C 6H4CH3), 127.5 (C 6H4CH3), 124.0 (C‐8 or 6a or 10a), 121.7 (C‐2 or 5′ or 7′), 118.5 (C‐8 or 6a or 10a), 114.8 (C‐8 or 6a or 10a), 114.0 (C‐2 or 5′ or 7′), 106.0 (C‐4′), 101.3 (C‐2′), 101.2 (C‐2a), 68.8 (CH(CH3)2), 59.5 (10‐OCH3), 59.2 (9‐OCH3), 51.6 (C‐5), 48.8 (C‐1), 26.0 (C‐6), 21.2 (CH(CH3)2), 21.2 (CH(CH3)2), 20.6 (C6H4 CH3), 9.8 (7′‐CH3), 8.7 ppm (8‐CH3); FTIR (KBr): =3379, 1684, 1476, 1458, 1420, 1373, 1352, 1298, 1275, 1248, 1217, 1192, 1180, 1167, 1109, 1078, 1024, 1001, 816, 808 cm−1; MS (EI): m/z (%): 680 (13) [M]+, 526 (41), 525 (100), 440 (23), 439 (92), 221 (13), 220 (54), 205 (11); HRMS (EI): m/z: calcd for C34H36O11N2S: 680.2040 [M]+; found: 680.2039.
(Z)‐{(1R*,5S*)‐1,2,3,4,5,6‐Hexahydro‐2‐[7‐hydroxy‐9,10‐di‐ methoxy‐8,11‐dimethyl‐4‐oxo‐3,4,5,6‐tetrahydro‐1,5‐imino‐ benzo‐[d]azocin‐2(1H)‐ylidene]methyl}‐4‐methylbenzo‐[d][1,3]‐ dioxol‐5‐yl 4‐methylbenzenesulfonate (8)
Anisole (1.1 mL, 10 mmol) and concd. H2SO4 (2.4 mL) were successively added to a stirred solution of 7 (1.36 g, 2 mmol) in TFA (47.6 mL) at 0 °C, and the mixture was stirred at 25 °C for 7.5 h. After the mixture was diluted with H2O (800 mL) at 0 °C, it was made alkaline with concd. NH4OH (75 mL) and was then extracted with CHCl3 (2×700 mL) and, finally, CHCl3/MeOH (9:1, 2×700 mL). The combined extract was dried and concentrated in vacuo to give a residue, which was used in the next step without further purification. An analytical sample of the secondary amine was obtained as a yellow amorphous powder by column chromatography with EtOAc/MeOH (19:1); R f=0.39 (EtOAc/MeOH 19:1); 1H NMR (400 MHz, CDCl3): δ=7.77 (d, J=8.3 Hz, 2 H, C6 H 4CH3), 7.33 (d, J=8.3 Hz, 2 H, C6 H 4CH3), 7.22 (br s, 1 H, NH), 6.42 (s, 1 H, 4′‐H), 5.97 (d, J=1.1 Hz, 1 H, 2′‐H), 5.96 (d, J=1.1 Hz, 1 H, 2′‐H), 5.71 (s, 1 H, 2a‐H), 4.77 (s, 1 H, 1‐H), 3.98 (br s, 1 H, 5‐H), 3.85 (s, 3 H, 10‐OCH3), 3.78 (s, 3 H, 9‐OCH3), 3.01–2.92 (overlapped, 2 H, 6‐H2), 2.46 (s, 3 H, C6H4CH 3), 2.14 (s, 3 H, 8‐CH3), 1.94 ppm (s, 3 H, 7′‐CH3); 13C NMR (100 MHz, CDCl3): δ=171.4 (C‐4), 149.7 (C‐9), 148.0 (C‐7), 146.4 (C‐7′a), 145.6 (C‐5′), 145.4 (C‐3′), 143.8 (C‐10), 140.8 (C‐6′), 136.9 (C‐2), 134.1 (C 6H4CH3), 129.7 (C 6H4CH3), 128.2 (C 6H4CH3), 125.6 (C‐6a or 10a), 121.9 (C 6H4CH3), 117.7 (C‐8), 115.5 (C‐7′), 114.6 (C‐6a or 10a), 106.1 (C‐4′), 101.9 (C‐2′), 101.3 (C‐2a), 60.2 (10‐CH3), 60.1 (9‐OCH3), 52.4 (C‐5), 48.8 (C‐1), 27.0 (C‐6), 21.7 (C6H4 CH3), 10.5 (7′‐CH3), 8.8 ppm (8‐CH3); FTIR (KBr): =3305, 1670, 1476, 1460, 1418, 1350, 1219, 1192, 1179, 1076 cm−1; MS (EI): m/z (%): 594 (8) [M]+, 440 (31), 439 (100), 221 (13), 220 (50); HRMS (EI): m/z: calcd for C30H30O9N2S: 594.1672 [M]+; found: 594.1669.
A 37 % aqueous solution of formaldehyde (8 mL), NaBH3CN (1.51 g, 24 mmol), and AcOH (26.3 mL) were successively added to a stirred solution of the above product (1.00 g) in MeOH (100 mL) at 0 °C, and the mixture was then stirred at 25 °C for 2 h. The mixture was diluted with H2O (200 mL), it was made alkaline with concentrated NH4OH (40 mL) and extracted with CHCl3 (3×300 mL). The combined extract was washed with brine (200 mL), dried, and concentrated in vacuo to give a residue (2.92 g), which was subjected to column chromatography on SiO2 (60 g) with hexane/EtOAc (13:7) to afford 8 (833.8 mg, 68 %, two steps) as a colorless amorphous powder; R f=0.23 (hexane/EtOAc 9:1); 1H NMR (400 MHz, CDCl3): δ=7.73 (d, J=8.5 Hz, 2 H, C6 H 4CH3), 7.30 (d, J=8.5 Hz, 2 H, C6 H 4CH3), 6.44 (s, 1 H, 4′‐H), 5.96 (s, 2 H, 2′‐H2), 5.78 (s, 1 H, 2a‐H), 4.83 (br s, 1 H, NH), 4.53 (s, 1 H, 1‐H), 3.85 (s, 3 H, 10‐OCH3), 3.78 (s, 3 H, 9‐OCH3), 3.64 (d, J=7.2 Hz, 1 H, 5‐H), 3.07 (dd, J=17.0, 7.2 Hz, 1 H, 6‐Hα), 2.93 (d, J=17.0 Hz, 1 H, 6‐Hβ), 2.56 (s, 3 H, NCH3), 2.46 (s, 3 H, C6H4CH 3), 2.15 (s, 3 H, 8‐CH3), 1.92 ppm (s, 3 H, 7′‐CH3); 13C NMR (100 MHz, CDCl3): δ=169.9 (C‐4), 149.8 (C‐9), 147.7 (C‐7), 146.4 (C‐7′a), 145.6 (C‐3′a or C 6H4CH3), 145.4 (C‐3′a or C 6H4CH3), 143.9 (C‐10), 140.6 (C‐6′), 134.6 (C‐2 or 6a), 133.7 (C 6H4CH3), 129.8 (C 6H4CH3), 128.3 (C 6H4CH3), 126.0 (C‐10a), 122.3 (C‐5′), 117.1 (C‐8), 115.8 (C‐7′), 113.9 (C‐2 or 6a), 106.2 (C‐4′), 103.9 (C‐2a), 101.9 (C‐2′), 60.3 (10‐OCH3), 60.2 (9‐OCH3), 59.1 (C‐5), 55.5 (C‐1), 41.5 (NCH3), 26.7 (C‐6), 21.7 (C6H4 CH3), 10.4 (7′‐CH3), 8.7 ppm (8‐CH3); FTIR (KBr): =1476, 1460, 1418, 1371, 1352, 1215, 1192, 1179, 1165, 1115, 1078, 1063 cm−1; MS (EI): m/z (%): 608 (7) [M]+, 454 (28), 453 (100), 235 (14), 234 (47); HRMS (EI): m/z: calcd for C31H32O9N2S: 608.1829 [M]+; found: 608.1829.
{(1R*,2S*,5S*)‐[7‐Hydroxy‐9,10‐dimethoxy‐8,11‐dimethyl‐4‐ oxo‐1,2,3,4,5,6‐hexahydro‐1,5‐iminobenzo[d]azocin‐2‐yl]‐ methyl}‐4‐methylbenzo[d][1,3]dioxol‐5‐yl 4‐methylbenzene‐ sulfonate (9)
A suspension of 8 (608.7 mg, 1 mmol) in EtOH (25 mL) was hydrogenated over 20 % Pd(OH)2 on carbon (280.9 mg) at 80 °C for 27 h under a hydrogen atmosphere (2.8 MPa). The catalyst was removed by filtration, and the residue trapped by the filter paper was washed with CHCl3 and MeOH. The combined filtrate was concentrated in vacuo to give a residue, which was subjected to column chromatography on SiO2 (15 g) with EtOAc/MeOH (9:1) to afford 9 (472.3 mg, 77 %) as a colorless amorphous powder; R f=0.23 (EtOAc/MeOH 19:1); 1H NMR (400 MHz, CDCl3): δ=7.66 (d, J=8.0 Hz, 2 H, C6 H 4CH3), 7.33 (d, J=8.0 Hz, 2 H, C6 H 4CH3), 6.49 (s, 1 H, 4′‐H), 5.97 (d, J=1.5 Hz, 1 H, 2′‐H), 5.96 (d, J=1.5 Hz, 1 H, 2′‐H), 4.21–4.14 (overlapped, 2 H, 1‐H & 2‐H), 3.83 (s, 3 H, 10‐OCH3), 3.82 (s, 3 H, 9‐OCH3), 3.59 (d, J=7.2 Hz, 1 H, 5‐H), 3.48 (dd, J=14.6, 2.4 Hz, 1 H, 2a‐Hα), 2.96 (dd, J=17.2, 7.2 Hz, 1 H, 6‐Hα), 2.78 (d, J=17.2 Hz, 1 H, 6‐Hβ), 2.47 (s, 3 H, C6H4CH 3), 2.45 (s, 3 H, NCH3), 2.24–2.15 (overlapped, 1 H, 2a‐Hβ), 2.19 (s, 3 H, 8‐CH3), 1.95 ppm (s, 3 H, 7′‐CH3); 13C NMR (100 MHz, CDCl3): δ=172.2 (C‐4), 149.8 (C‐9), 147.9 (C‐7), 146.1 (C‐7′a), 145.7 (C‐3′a), 145.3 (C 6H4CH3), 144.8 (C‐10), 141.2 (C‐6′), 133.7 (C 6H4CH3), 129.9 (C 6H4CH3), 127.9 (C 6H4CH3), 124.6 (C‐5′), 121.8 (C‐6a or 10a), 117.5 (C‐8), 115.4 (C‐7′), 114.6 (C‐6a or 10a), 107.5 (C‐4′), 101.8 (C‐2′), 60.5 (9‐OCH3), 60.2 (10‐OCH3), 58.1 (C‐5), 55.9 (C‐2), 54.4 (C‐1), 40.1 (NCH3), 33.3 (C‐2a), 23.1 (C‐6), 21.8 (C6H4 CH3), 10.6 (7′‐CH3), 8.9 ppm (8‐CH3); FTIR (KBr): =3364, 2936, 1736, 1661, 1476, 1456, 1418, 1344, 1194, 1180, 1076, 1055 cm−1; MS (EI): m/z (%): 610 (9) [M]+, 456 (10), 455 (19), 235 (28), 234 (100); HRMS (EI): m/z: calcd for C31H34O9N2S: 610.1985 [M]+; found: 610.1987.
(1R*,2S*,5S*)‐7‐(Allyloxy)‐2‐[(6‐hydroxy‐7‐methylbenzo[d]‐ [1,3]dioxol‐5‐yl)methyl]‐9,10‐dimethoxy‐8,11‐dimethyl‐ 2,3,5,6‐tetrahydro‐1,5‐iminobenzo[d]azocin‐4(1H)‐one (10)
A solution of 9 (1.36 g, 2.23 mmol) in acetone (110 mL) was stirred in the presence of K2CO3 (1.55 g, 11.14 mmol) at 0 °C, allyl bromide (385 μL, 4.45 mmol) was added over 10 min, and the mixture was heated at reflux for 5.5 h. The mixture was filtered, and the combined filtrate was concentrated in vacuo to give a residue, which was used in the next step without further purification. An analytical sample was obtained as a colorless amorphous powder by column chromatography with EtOAc to EtOAc/MeOH (19:1); R f=0.21 (CHCl3/MeOH 97:3); 1H NMR (400 MHz, CDCl3): δ=7.65 (d, J=8.5 Hz, 2 H, C6 H 4CH3), 7.34 (d, J=8.5 Hz, 2 H, C6 H 4CH3), 6.50 (s, 1 H, 4′‐H), 6.09 (ddt, J=17.2, 10.7, 5.4 Hz, 1 H, OCH2CH=CH2), 5.96 (d, J=1.2 Hz, 1 H, 2′‐H), 5.95 (d, J=1.2 Hz, 1 H, 2′‐H), 5.45 (dq, J=17.2, 1.5 Hz, 1 H, OCH2CH=CH 2), 5.27 (dq, J=10.7, 1.5 Hz, 1 H, OCH2CH=CH 2), 5.20 (s, 1 H, NH), 4.33 (ddt, J=12.7, 5.4, 1.5 Hz, 1 H, OCH 2CH=CH2), 4.28 (ddt, J=12.7, 5.4, 1.5 Hz, 1 H, OCH 2CH=CH2), 4.20–4.15 (overlapped, 2 H, 1‐H and 2‐H), 3.87 (s, 3 H, 10‐OCH3), 3.82 (s, 3 H, 9‐OCH3), 3.54 (d, J=7.1 Hz, 1 H, 5‐H), 3.45 (dd, J=14.4, 1.7 Hz, 1 H, 2a‐Hα), 3.07 (dd, J=18.0, 7.1 Hz, 1 H, 6‐Hα), 2.95 (d, J=18.0 Hz, 1 H, 6‐Hβ), 2.47 (s, 6 H, C6H4CH 3 & NCH3), 2.22 (s, 3 H, 8‐CH3), 2.25–2.18 (overlapped, 1 H, 2a‐Hβ), 1.94 ppm (s, 3 H, 7′‐CH3); 13C NMR (100 MHz, CDCl3): δ=172.1 (C‐4), 151.2 (C‐7), 149.8 (C‐9), 147.4 (C‐10), 146.1 (C‐3′a or 7′a), 145.6 (C‐3′a or 7′a), 145.4 (C 6H4CH3), 141.1 (C‐6′), 133.8 (OCH2 CH=CH2), 133.6 (C 6H4CH3), 129.9 (C 6H4CH3), 127.8 (C 6H4CH3), 125.0 (C‐8), 124.6 (C‐5′), 122.4 (C‐6a or 10a), 122.3 (C‐6a or 10a), 117.1 (OCH2CH=CH2), 115.3 (C‐7′), 107.5 (C‐4′), 101.8 (C‐2′), 72.9 (OCH2CH=CH2), 60.2 (9‐OCH3), 60.1 (10‐OCH3), 58.2 (C‐5), 55.5 (C‐2), 54.4 (C‐1), 40.3 (NCH3), 33.3 (C‐2a), 24.2 (C‐6), 21.7 (C6H4 CH3), 10.6 (7′‐CH3), 9.7 ppm (8‐CH3); FTIR (KBr): =1674, 1476, 1450, 1414, 1358, 1339, 1192, 1180, 1113, 1076 cm−1; MS (EI): m/z (%): 650 (18) [M]+, 610 (14), 609 (38), 496 (22), 495 (38), 275 (29), 274 (100), 235 (12), 234 (39), 233 (17), 219 (11), 218 (36); HRMS (EI): m/z: calcd for C34H38O9N2S: 650.2298 [M]+; found: 650.2294.
A solution of KOH (7.35 g, 0.111 mmol) in H2O (33 mL) was added dropwise to a stirred solution of the above product (1.54 g) in EtOH (33 mL) at 25 °C, and the mixture was heated at reflux for 3 h. The mixture was diluted with H2O (130 mL), neutralized with 6 m aq HCl solution, and extracted with CHCl3 (3×200 mL). The combined extract was washed with brine (200 mL), dried, and concentrated in vacuo to give a residue (1.10 g), which was subjected to column chromatography on SiO2 (30 g) with CHCl3/MeOH (97:3) to afford 10 (889.1 mg, 80 %, two steps) as a brown amorphous powder; R f=0.24 (CHCl3/MeOH 97:3); 1H NMR (400 MHz, CDCl3): δ=6.39 (s, 1 H, 4′‐H), 6.06 (ddt, J=17.6, 10.6, 5.5 Hz, 1 H, OCH2CH=CH2), 5.91 (br s, 1 H, NH), 5.87 (d, J=1.4 Hz, 1 H, 2′‐H), 5.85 (d, J=1.4 Hz, 1 H, 2′‐H), 5.41 (br dd, J=17.6, 1.4 Hz, 1 H, OCH2CH=CH 2), 5.24 (br dd, J=10.7, 1.4 Hz, 1 H, OCH2CH=CH 2), 4.30 (ddt, J=12.5, 5.5, 1.4 Hz, 1 H, OCH 2CH=CH2), 4.27 (ddt, J=12.5, 5.5, 1.4 Hz, 1 H, OCH 2CH=CH2), 4.21–4.18 (overlapped, 2 H, 1‐H & 2‐H), 3.83 (s, 3 H, 10‐OCH3), 3.80 (s, 3 H, 9‐OCH3), 3.55 (d, J=7.2 Hz, 1 H, 5‐H), 3.20 (d, J=14.5 Hz, 1 H, 2a‐Hα), 3.06 (dd, J=17.5, 7.2 Hz, 1 H, 6‐Hα), 2.95 (d, J=17.5 Hz, 1 H, 6‐Hβ), 2.48 (s, 3 H, NCH3), 2.21 (s, 3 H, 8‐CH3), 2.10 (s, 3 H, 7′‐CH3), 2.02 ppm (dd, J=14.5, 10.4 Hz, 1 H, 2a‐Hβ); 13C NMR (100 MHz, CDCl3): δ=172.3 (C‐4), 151.3 (C‐7), 149.8 (C‐9), 147.3 (C‐10 or 6′), 147.1 (C‐10 or 6′), 145.5 (C‐7′a), 140.8 (C‐3′a), 133.7 (OCH2 CH=CH2), 125.0 (C‐8), 122.5 (C‐6a or 10a), 122.4 (C‐6a or 10a), 117.3 (OCH2CH=CH2), 115.6 (C‐5′), 107.8 (C‐7′), 107.5 (C‐4′), 100.8 (C‐2′), 73.0 (OCH2CH=CH2), 60.2 (10‐OCH3), 60.1 (9‐OCH3), 58.0 (C‐5), 56.0 (C‐2), 54.4 (C‐1), 40.3 (NCH3), 32.4 (C‐2a), 24.2 (C‐6), 9.7 (8‐CH3), 9.0 ppm (7′‐CH3); FTIR (KBr): =3370, 2936, 1655, 1476, 1456, 1412, 1339, 1252, 1238, 1184, 1111, 1092, 1076, 1057 cm−1; MS (EI): m/z (%): 497 (29), 496 (100) [M]+, 456 (18), 455 (48), 275 (28), 274 (89), 260 (14), 234 (39), 233 (24), 219 (13), 218 (47); HRMS (EI): m/z: calcd for C27H32N2O7: 496.2210 [M]+; found: 496.2211.
(1R*,2S*,4R*,5S*)‐7‐(Allyloxy)‐2‐[(6‐hydroxy‐7‐methylbenzo[d]‐ [1,3]dioxol‐5‐yl)methyl]‐9,10‐dimethoxy‐8,11‐dimethyl‐1,2,3,‐ 4,5,6‐hexahydro‐1,5‐iminobenzo[d]azocine‐4‐carbo‐nitrile (11)
A suspension of Cp2ZrHCl (31.9 mg, 0.12 mmol) in THF (0.5 mL) was added to a stirred solution of 10 (19.9 mg, 0.04 mmol) in THF (1 mL) at −20 °C, and the mixture was warmed to 0 °C over 1 h. A solution of KCN (20.8 mg, 0.32 mmol) in H2O (640 μL) was added to the above solution, and the mixture was stirred at 25 °C for 4 h. The mixture was diluted with saturated aqueous NaHCO3 solution (10 mL) and extracted with CHCl3 (3×15 mL). The combined extract was washed with brine (10 mL), dried, and concentrated in vacuo to give a residue (21.5 mg), which was subjected to column chromatography on SiO2 (6 g) with CHCl3 to give 11 (19.4 mg, 96 %) as a brown amorphous powder; R f=0.40 (hexane/CHCl3 1:4); 1H NMR (400 MHz, CDCl3): δ=6.43 (s, 1 H, 4′‐H), 6.09 (ddt, J=17.3, 10.6, 5.4 Hz, 1 H, OCH2CH=CH2), 5.86 (d, J=1.4 Hz, 1 H, 2′‐H), 5.85 (d, J=1.4 Hz, 1 H, 2′‐H), 5.44 (dq, J=17.3, 1.5 Hz, 1 H, OCH2CH=CH 2), 5.28 (dq, J=10.6, 1.5 Hz, 1 H, OCH2CH=CH 2), 4.34 (ddt, J=12.7, 5.4, 1.5 Hz, 1 H, OCH 2CH=CH2), 4.29 (ddt, J=12.7, 5.4, 1.5 Hz, 1 H, OCH 2CH=CH2), 4.08 (d, J=2.7 Hz, 1 H, 1‐H), 3.97 (d, J=1.5 Hz, 1 H, 4‐H), 3.82 (s, 3 H, 10‐OCH3), 3.80 (s, 3 H, 9‐OCH3), 3.64 (dt, J=10.2, 2.7 Hz, 1 H, 2‐H), 3.28 (dd, J=7.8, 1.5 Hz, 1 H, 5‐H), 3.03 (dd, J=18.7, 7.8 Hz, 1 H, 6‐Hα), 2.91 (dd, J=14.9, 2.7 Hz, 1 H, 2a‐Hα), 2.45 (d, J=18.7 Hz, 1 H, 6‐Hβ), 2.32 (s, 3 H, NCH3), 2.21 (s, 3 H, 8‐CH3), 2.08 (s, 3 H, 7′‐CH3), 2.02 ppm (dd, J=14.9, 10.2 Hz, 1 H, 2a‐Hβ); 13C NMR (100 MHz, CDCl3): δ=150.6 (C‐7), 150.0 (C‐9), 148.2 (C‐6′), 147.6 (C‐10), 145.4 (C‐7′a), 140.2 (C‐3′a), 133.7 (OCH2 CH=CH2), 124.5 (C‐8), 123.0 (C‐6a), 122.0 (C‐10a), 119.0 (CN), 117.3 (OCH2CH=CH2), 115.8 (C‐5′), 108.8 (C‐7′), 107.2 (C‐4′), 100.7 (C‐2′), 72.9 (OCH2CH=CH2), 60.4 (10‐OCH3), 60.2 (9‐OCH3), 57.7 (C‐2), 56.6 (C‐1), 53.9 (C‐5), 53.5 (C‐4), 42.0 (NCH3), 34.2 (C‐2a), 21.3 (C‐6), 9.7 (8‐CH3), 9.1 ppm (7′‐CH3); FTIR (KBr): =3447, 2936, 2361, 1458, 1412, 1250, 1111, 1092, 1074, 1055, 1042, 1020 cm−1; MS (FAB): m/z (%): 508 [M+H]+; HRMS (FAB): m/z: calcd for C28H34N3O6: 508.2448 [M]+; found: 508.2452.
(1R*,2S*,5S*)‐tert‐Butyl 7‐(allyloxy)‐2‐{(6′‐[(tert‐butoxy‐ carbonyl)oxy‐7′‐methylbenzo‐[d][1,3]dioxol‐5‐yl]methyl}‐ 9,10‐dimethoxy‐8,11‐dimethyl‐4‐oxo‐1,2,5,6‐tetrahydro‐1,5‐ iminobenzo[d]azocine‐3(4H)‐carboxylate (13)
A mixture of 10 (568.5 mg, 1.14 mmol) and DMAP (279.4 mg, 2.29 mmol, 2 equiv) in MeCN (11.5 mL) was cooled at 0 °C; then, Boc2O (5.3 mL, 22.90 mmol, 20 equiv.) was added to the mixture, which was heated at reflux for 17.5 h. As the starting material still remained, as indicated by TLC monitoring, DMAP (279.4 mg, 2.29 mmol, 2 equiv.) was added to the mixture at 25 °C, and the reaction mixture was heated under reflux for an additional 40 h. The mixture was diluted with H2O (30 mL) and extracted with CHCl3 (3×50 mL). The combined extract was washed with brine (50 mL), dried, and concentrated in vacuo to give a residue, which was subjected to column chromatography on SiO2 (15 g) with hexane/EtOAc (11:9) to afford 13 (570.9 mg, 72 %) as a brown amorphous powder; R f=0.30 (hexane/EtOAc 11:9); 1H NMR (400 MHz, CDCl3): δ=6.55 (s, 1 H, 4′‐H), 6.07 (ddt, J=17.3, 10.6, 5.4 Hz, 1 H, CH2CH=CH2), 5.90 (s, 2 H, 2′‐H2), 5.42 (dq, J=17.3, 1.5 Hz, 1 H, CH2CH=CH 2), 5.26 (dq, J=10.6, 1.5 Hz, 1 H, CH2CH=CH 2), 4.99 (q, J=6.2 Hz, 1 H, 2‐H), 4.39 (dd, J=6.2, 1.5 Hz, 1 H, 1‐H), 4.32 (ddt, J=12.4, 5.4, 1.5 Hz, 1 H, CH 2CH=CH2), 4.26 (ddt, J=12.4, 5.4, 1.5 Hz, 1 H, CH 2CH=CH2), 3.77 (dd, J=7.7, 1.5 Hz, 1 H, 5‐H), 3.72 (s, 3 H, 9‐OCH3), 3.61 (s, 3 H, 10‐OCH3), 3.07 (dd, J=18.5, 7.7 Hz, 1 H, 6H‐α), 2.96 (dd, J=18.5, 1.5 Hz, 1 H, 6‐Hβ), 2.91 (dd, J=15.6, 6.2 Hz, 1 H, 2a‐Hα), 2.46 (s, 3 H, NCH3), 2.18 (s, 3 H, 8‐CH3), 2.05 (s, 3 H, 7′‐CH3), 1.99 (dd, J=15.6, 6.2 Hz, 1 H, 2a‐Hβ), 1.49 (s, 9 H, C(CH 3)3), 1.31 ppm (s, 9 H, C(CH 3)3); 13C NMR (100 MHz, CDCl3): δ=173.2 (C‐4), 151.6 (CO2), 151.5 (CO2), 151.1 (C‐7), 149.8 (C‐9), 147.6 (C‐10), 144.7 (C‐7′a), 144.3 (C‐3′a), 142.6 (C‐6′), 133.7 (CH2 CH=CH2), 125.1 (C‐8), 124.0 (C‐5′), 122.9 (C‐10a), 121.8 (C‐6a), 117.3 (CH2CH=CH2), 112.9 (C‐7′), 105.7 (C‐4′), 101.2 (C‐2′), 83.5 (C(CH3)3), 82.9 (C(CH3)3), 73.1 (CH2CH=CH2), 60.1 (10‐OCH3), 59.8 (9‐OCH3), 59.8 (C‐5), 57.8 (C‐2), 54.0 (C‐1), 40.0 (NCH3), 32.3 (C‐2a), 27.6 (C(CH3)3), 27.5 (C(CH3)3), 22.7 (C‐6), 9.6 (8‐CH3), 9.4 ppm (7′‐CH3); FTIR (KBr): =1757, 1732, 1697, 1477, 1456, 1412, 1395, 1369, 1341, 1275, 1256, 1152, 1099, 1063 cm−1; MS (EI): m/z (%): 696 (4) [M]+, 596 (13), 555 (13), 497 (14), 496 (47), 455 (24), 275 (31), 274 (100), 271 (14), 260 (11), 234 (32), 233 (22), 232 (12), 219 (11), 218 (38), 57 (14); HRMS (EI): m/z: calcd C37H48N2O11: 696.3258 [M]+; found: 696.3256.
1. ‐(2′S*)‐(1“R,3”S)‐[(5“‐Allyloxy‐3”‐hydroxymethyl‐7“, 8”‐di‐ methoxy‐2“,6”‐dimethyl‐1“,2”,3“,4”‐tetrahydroisoquinolin‐ 1“‐yl)‐2′‐aminoethyl]‐4‐methylbenzo[d][1,3]dioxol‐5‐ol (14)
NaBH4 (892.4 g, 23.59 mmol) was added to a stirred solution of 13 (821.8 mg, 1.179 mmol) in EtOH (12 mL) at 0 °C, and the mixture was stirred at 25 °C for 3 h. The mixture was diluted with saturated aqueous NH4Cl (10 mL) and H2O (40 mL) at 0 °C and extracted with CHCl3 (3×50 mL). The combined extract was washed with brine (30 mL), dried, and concentrated in vacuo to give a residue, which was used in the next step without further purification. An analytical sample of the protected compound (857.3 mg) was obtained as a colorless amorphous powder by column chromatography with hexane/EtOAc (13:7); R f=0.21 (hexane/EtOAc 7:3); 1H NMR (400 MHz, CDCl3): δ=6.60 (s, 1 H, 4“‐H), 6.05 (ddt, J=17.1, 10.8, 5.6 Hz, 1 H, CH2CH=CH2), 5.90 (d, J=1.4 Hz, 1 H, 2”‐H), 5.88 (d, J=1.4 Hz, 1 H, 2“‐H), 5.38 (dd, J=17.1, 1.4 Hz, 1 H, CH2CH=CH 2), 5.22 (dd, J=10.8, 1.4 Hz, 1 H, CH2CH=CH 2), 4.49 (br d, J=9.5 Hz, 1 H, 1‐H), 4.27 (dd, J=12.5, 5.6 Hz, 1 H, CH 2CH=CH2), 4.12 (dd, J=12.5, 5.6 Hz, 1 H, CH 2CH=CH2), 3.88–3.76 (overlapped, 1 H, 2‐H), 3.86 (s, 3 H, 8′‐OCH3), 3.83 (s, 3 H, 7′‐OCH3), 3.77 (d, J=9.5 Hz, 1 H, 1′‐H), 3.48 (dd, J=11.3, 3.3 Hz, 1 H, 2‐H), 3.13 (br d, J=13.1 Hz, 1 H, 3′a‐H), 2.88 (dd, J=15.5, 4.8 Hz, 1 H, 4′‐H), 2.75 (dd, J=15.5, 12.6 Hz, 1 H, 4′‐H), 2.50 (s, 3 H, NCH3), 2.38 (m, 1 H, 3′‐ and 3′a‐H), 2.17 (s, 3 H; 6′‐CH3), 2.06 (s, 3 H, 7”‐CH3), 1.57 (s, 9 H, C(CH 3)3), 1.14 ppm (s, 9 H, C(CH 3)3); 13C NMR (100 MHz, CDCl3): δ=155.0 (CO), 151.9 (CO), 150.0 (C‐5′), 149.0 (C‐7′), 146.5 (C‐8′), 145.0 (C‐7“a), 144.6 (C‐3”a), 142.4 (C‐6“), 133.9 (CH2 CH=CH2), 126.7 (C‐4′a or 8′a), 125.9 (C‐4′a or 8′a), 124.0 (C‐6′ or 5”), 123.8 (C‐6′ or 5“), 117.4 (CH2CH=CH2), 112.9 (C‐7”), 106.8 (C‐4“), 101.2 (C‐2”), 83.5 (C(CH3)3), 78.3 (C(CH3)3), 74.1 (CH2CH=CH2), 64.4 (C‐2), 63.7 (C‐1′), 63.1 (C‐3′), 60.5 (8′‐OCH3), 60.1 (7′‐OCH3), 55.6 (C‐1), 46.6 (NCH3), 32.6 (C‐3′a), 28.1 (C(CH3)3), 27.6 (C(CH3)3), 24.4 (C‐4′), 9.50 (6′‐CH3), 9.45 ppm (7“‐CH3); FTIR (KBr): =3422, 2978, 2930, 1751, 1715, 1516, 1477, 1458, 1369, 1281, 1258, 1153, 1101, 1063 cm−1; MS (FAB): m/z (%): 701 [M+H]+; HRMS (FAB): m/z: calcd for C37H53N2O11: 701.3649 [M]+; found: 701.3646.
TFA (18 mL) was added to a stirred solution of the crude product in CH2Cl2 (36 mL) at 0 °C, and the mixture was stirred at 25 °C for 1.5 h. The mixture was diluted with H2O (40 mL) at 0 °C, made alkaline with concentrated NH4OH (30 mL), and extracted with CHCl3 (3×80 mL). The combined extract was washed with brine (60 mL), dried, and concentrated in vacuo to give a residue (568.6 mg), which was subjected to column chromatography on SiO2 (15 g) with CHCl3/MeOH (97:3) to afford 14 (433.2 mg, 73 %, two steps) as a brown amorphous powder; R f=0.19 (CHCl3/MeOH 97:3); 1H NMR (400 MHz, CDCl3): δ=6.44 (s, 1 H, 7‐H), 6.05 (ddt, J=17.0, 10.4, 5.6 Hz, 1 H, CH2CH=CH2), 5.87 (d, J=1.4 Hz, 1 H, 2‐H), 5.82 (d, J=1.4 Hz, 1 H, 2‐H), 5.37 (dq, J=17.0, 1.4 Hz, 1 H, CH2CH=CH 2), 5.25 (dq, J=10.4, 1.4 Hz, 1 H, CH2CH=CH 2), 4.25 (ddt, J=11.1, 5.6, 1.4 Hz, 1 H, CH 2CH=CH2), 4.19 (ddt, J=11.1, 5.6, 1.4 Hz, 1 H, CH 2CH=CH2), 3.84 (s, 3 H, 8“‐OCH3), 3.80 (s, 3 H, 7”‐OCH3), 3.79 (dd, J=10.5, 4.0 Hz, 1 H, 3“a‐H), 3.67 (d, J=9.4 Hz, 1 H, 1”‐H), 3.46 (dd, J=10.5, 2.6 Hz, 1 H, 3′a‐H), 3.14 (d, J=13.6 Hz, 1 H, 1′‐H), 2.95 (d, J=11.0 Hz, 1 H, 4“‐H), 2.75 (dd, J=13.6, 9.4 Hz, 1 H, 1′‐H), 2.69 (t, J=9.4 Hz, 1 H, 2′‐H), 2.51 (s, 3 H, NCH3), 2.42–2.32 (overlapped, 2 H, 3”‐ & 4“‐H), 2.21 (s, 3 H, 6”‐CH3), 2.11 ppm (s, 3 H, 4‐CH3); 13C NMR (100 MHz, CDCl3): δ=150.6 (C‐5 or 5“), 150.4 (C‐5 or 5”), 149.7 (C‐7“), 146.9 (C‐8”), 145.4 (C‐3a), 139.3 (C‐7a), 133.5 (CH2 CH=CH2), 126.7 (C‐8“a), 124.8 (C‐4”a or 6“), 124.6 (C‐4”a or 6“), 117.7 (CH2CH=CH2), 117.6 (C‐6), 109.3 (C‐4), 107.0 (C‐7), 100.5 (C‐2), 74.5 (CH2CH=CH2), 65.0 (C‐1”), 63.9 (C‐3“a), 63.0 (C‐3”), 60.7 (8“‐OCH3), 60.1 (7”‐OCH3), 59.1 (C‐2′), 46.6 (NCH3), 38.2 (C‐1′), 24.3 (C‐4“), 9.7 (6”‐CH3), 9.3 ppm (4‐CH3); FTIR (KBr): =3441, 3370, 2938, 2866, 1470, 1414, 1248, 1115, 1094, 1057, 934 cm−1; MS (EI): m/z (%): 500 (1) [M]+, 307 (18), 306 (100), 234 (15), 218 (10); HRMS (EI): m/z: calcd C27H36N2O7: 500.2523 [M]+; found: 500.2520.
(6aS*,7R*,13S*,14R*,16R*)‐Ethyl 5‐(benzyloxy)‐14‐cyano‐11‐ hydroxy‐8,9‐dimethoxy‐4,10,17‐trimethyl‐6a,7,12,13,14,16‐ hexahydro‐6H‐7,13‐iminobenzo[4,5]azocino‐[1,2‐b]‐[1,3]‐ dioxolo[4,5‐h]isoquinoline‐16‐carboxylate (18)
14→15: A solution of 1‐allyl 3‐ethyl 2,2‐dihydroxymalonate20 (238.2 mg, 1.167 mmol) in TFA (6 mL) was added to a stirred solution of 14 (116.8 mg, 0.233 mmol) in AcOH (1.5 mL), and the mixture was stirred at 25 °C for 6 h. The mixture was diluted with H2O (60 mL) at 0 °C, made alkaline with concentrated NH4OH (11 mL), and extracted with CHCl3 (3×80 mL). The combined extract was washed with brine (40 mL), dried, and concentrated in vacuo to give a residue (358.9 mg), which was subjected to column chromatography on SiO2 (10 g) with hexane/EtOAc (2:3) to afford an inseparable 1:1 diastereomer mixture of 15 (124.9 mg, 80 %) as a yellow amorphous powder.
15→16: BnBr (533 μL, 4.40 mmol) was added to a stirred solution of 15 (147.1 mg, 0.22 mmol) in the presence of K2CO3 (763.9 mg, 5.50 mmol) in acetone (55 mL), and the mixture was stirred at 25 °C for 8 h. The mixture was filtered, and the filtrate was concentrated in vacuo to give a residue, which was subjected to column chromatography on SiO2 (6 g) with hexane/EtOAc (1:1) to afford an inseparable 1:1 diastereomer mixture of 16 (137.2 mg, 83 %) as a yellow amorphous powder.
16→17: (COCl)2 (25 μL, 0.3 mmol) was added to a stirred solution of DMSO (43 μL, 0.6 mmol) in CH2Cl2 (1 mL) at −78 °C, and the mixture was stirred at −78 °C for 15 min. A solution of 16 (45.5 mg, 60 μmol) in CH2Cl2 (1 mL) was added to the above solution at −78 °C over 10 min, and the mixture was stirred at −78 °C for 4 h. Et3N (167 μL, 1.2 mmol) was then added to the mixture at −78 °C over 5 min, and stirring was continued at −78 °C for 30 min. After the mixture was warmed to 25 °C over a period of 3 h and stirred for 2 h, it was diluted with saturated aqueous NaHCO3 solution (5 mL) at 0 °C and extracted with CHCl3 (3×10 mL). The combined extract was washed with brine (10 mL), dried, and concentrated in vacuo to give a residue that was used in the next step without purification. A solution of KCN (31.9 mg, 0.48 mmol) in H2O (960 μL) was added to a stirred solution of the crude product (63.4 mg) in THF (1 mL) in the presence of AcOH (381 μL, 6.6 mmol) at 0 °C, and the mixture was stirred at 25 °C for 2 h. The mixture was diluted with saturated aqueous NaHCO3 (5 mL) at 0 °C and extracted with CHCl3 (3×10 mL). The combined extract was washed with brine (10 mL), dried, and concentrated in vacuo to give a residue (41.5 mg), which was subjected to column chromatography on SiO2 (6 g) with hexane/EtOAc (3:1) to afford an inseparable 1:1 diastereomer mixture of 17 (25.8 mg, 56 %, two steps) as a colorless amorphous powder.
17→18: A solution of Pd(PPh3)4 (16.4 mg, 13.8 μmol) in THF (1.0 mL) was added to a stirred solution of 17 (35.2 mg, 46 μmol) and dimedone (32.6 mg, 0.23 mmol) in THF (2.0 mL) under an argon atmosphere, and the mixture was stirred at 25 °C for 1 h. After the mixture was concentrated in vacuo, the resulting residue was dissolved in CHCl3 (2.5 mL) and heated at reflux for 2 h. The mixture was concentrated in vacuo to give a residue, which was subjected to column chromatography on SiO2 (6 g) with CHCl3/EtOAc (4:1) to afford 18 (21.2 mg, 72 %, two steps) as a pale‐yellow amorphous powder; R f=0.20 (CHCl3/EtOAc 4:1); 1H NMR (400 MHz, CDCl3): δ=7.56 (m, 2 H, C6H5), 7.46 (m, 2 H, C6H5), 7.40 (m, 1 H, C6H5), 5.94 (d, J=1.4 Hz, 1 H, 2‐H), 5.89 (d, J=1.4 Hz, 1 H, 2‐H), 4.70 (d, J=10.4 Hz, 1 H, PhCH2), 4.58 (d, J=10.4 Hz, 1 H, PhCH2), 4.47 (s, 1 H, 16‐H), 4.39 (br s, 1 H, OH), 4.27 (d, J=2.5 Hz, 1 H, 14‐H), 4.07 (dq, J=10.9, 7.1 Hz, 1 H, CH 2CH3), 4.04 (br d, J=2.6 Hz, 1 H, 7‐H), 3.95 (dq, J=10.9, 7.1 Hz, 1 H, CH 2CH3), 3.77 (s, 3 H, 8‐OCH3), 3.72 (s, 3 H, 9‐OCH3), 3.43 (br d, J=8.1 Hz, 1 H, 13‐H), 3.30 (dd, J=15.4, 2.6 Hz, 1 H, 6H‐α), 3.20 (dt, J=11.7, 2.6 Hz, 1 H, 6a‐H), 2.91 (dd, J=17.6, 8.1 Hz, 1 H, 12‐Hα), 2.33 (d, J=17.6 Hz, 1 H, 12‐Hβ), 2.32 (s, 3 H, NCH3), 2.17 (s, 3 H, 4‐CH3), 2.13 (s, 3 H, 10‐CH3), 2.02 (dd, J=15.4, 11.7 Hz, 1 H, 6‐Hβ), 1.04 ppm (t, J=7.1 Hz, 3 H, CH2CH 3); 13C NMR (100 MHz, CDCl3): δ=170.5 (CO), 149.0 (C‐9), 148.2 (C‐5), 146.5 (C‐11), 144.9 (C‐8), 144.4 (C‐3a), 140.0 (C‐16b), 137.1 (C6H5), 128.6 (C6H5), 128.5 (C6H5), 128.3 (C6H5), 123.0 (C‐7a), 121.1 (C‐5a or 16a), 117.6 (CN), 116.1 (C‐11a), 115.6 (C‐10), 113.1 (C‐4), 109.9 (C‐5a or 16a), 101.4 (C‐2), 75.3 (PhCH2), 61.3 (CH2CH3), 61.1 (C‐14), 60.3 (C‐16, 8 & 9‐OCH3), 56.8 (C‐6a & 7), 54.9 (C‐13), 41.8 (NCH3), 26.3 (C‐6), 21.0 (C‐12), 13.7 (CH2 CH3), 9.4 (4‐CH3), 8.6 ppm (10‐CH3); FTIR (KBr): =3437, 2934, 2228, 1728, 1456, 1431, 1416, 1344, 1254, 1109, 1092, 1074, 1028 cm−1; MS (EI): m/z (%): 641 (7) [M]+, 543 (14), 541 (17), 523 (19), 451 (23), 450 (18), 274 (40), 235 (33), 234 (100); HRMS (EI): m/z: calcd for C36H39N3O8: 641.2737 [M]+; found: 641.2735.
(6aS*,7R*,13S*,14R*,16R*)‐5‐(Benzyloxy)‐11‐hydroxy‐16‐ (hydroxymethyl)‐8,9‐dimethoxy‐4,10,17‐trimethyl‐6a,7,12,‐ 13,14,16‐hexahydro‐6H‐7,13‐iminobenzo[4,5]azocino‐[1,2‐b]‐ [1,3]‐dioxolo[4,5‐h]isoquinoline‐14‐carbonitrile (19)
LiBH4 (6.4 mg, 0.281 mmol) was added to a stirred solution of 18 (18.0 mg, 28.1 μmol) in THF (1 mL) and MeOH (11 μL, 0.281 mmol), and the mixture was stirred at 25 °C for 3 h. After the mixture was diluted with brine (5 mL) slowly at 0 °C, it was extracted with CHCl3 (2×10 mL) and then CHCl3/MeOH (19:1, 2×10 mL). The combined extract was dried and concentrated in vacuo to give a residue (19.6 mg), which was subjected to column chromatography on SiO2 (6 g) with CHCl3/EtOAc (3:2) to afford 19 (11.9 mg, 71 %) as a colorless amorphous powder; R f=0.24 (hexane/EtOAc 2:3); 1H NMR (400 MHz, CDCl3): δ=7.56 (m, 2 H, C6H5), 7.46 (m, 2 H, C6H5), 7.40 (m, 1 H, C6H5), 5.95 (d, J=1.4 Hz, 1 H, 2‐H), 5.89 (d, J=1.4 Hz, 1 H, 2‐H), 4.67 (d, J=10.4 Hz, 1 H, PhCH2), 4.58 (br s, 1 H, OH), 4.55 (d, J=10.4 Hz, 1 H, PhCH2), 4.08 (d, J=2.3 Hz, 1 H, 14‐H), 4.07 (d, J=2.5 Hz, 1 H, 7‐H), 4.01 (t, J=3.3 Hz, 1 H, 16‐H), 3.77 (s, 3 H, 8‐OCH3), 3.70 (s, 3 H, 9‐OCH3), 3.68 (br d, J=11.3 Hz, 1 H, CH 2OH), 3.50 (dd, J=11.3, 3.3 Hz, 1 H, CH 2OH), 3.47 (br d, J=9.0 Hz, 1 H, 13‐H), 3.31 (dt, J=12.8, 2.5 Hz, 1 H, 6a‐H), 3.31 (dd, J=16.0, 2.5 Hz, 1 H, 6H‐α), 2.99 (dd, J=18.1, 9.0 Hz, 1 H, 12‐Hα), 2.40 (d, J=18.1 Hz, 1 H, 12‐Hβ), 2.36 (s, 3 H, NCH3), 2.16 (s, 3 H, 4‐CH3), 2.14 (s, 3 H, 10‐CH3), 1.93 ppm (dd, J=16.0, 12.8 Hz, 1 H, 6‐Hβ); 13C NMR (100 MHz, CDCl3): δ=149.3 (C‐9), 148.2 (C‐5), 146.7 (C‐11), 145.0 (C‐8), 144.5 (C‐3a), 139.1 (C‐16b), 137.1 (C6H5), 128.6 (C6H5), 128.4 (C6H5), 128.3 (C6H5), 123.2 (C‐7a), 121.1 (C‐5a), 117.7 (CN), 116.0 (C‐10), 115.6 (C‐11a), 113.4 (C‐16a), 112.5 (C‐4), 101.2 (C‐2), 75.1 (PhCH2), 63.4 (CH2OH), 60.3 (8 & 9‐OCH3), 60.0 (C‐14), 58.1 (C‐16), 56.9 (C‐7), 56.5 (C‐6a), 54.9 (C‐13), 41.8 (NCH3), 26.3 (C‐6), 21.3 (C‐12), 9.3 (4‐CH3), 8.7 ppm (10‐CH3); FTIR (KBr): =3447, 2934, 2228, 1456, 1431, 1418, 1344, 1109, 1092, 1074 cm−1; MS (EI): m/z (%): 599 (1) [M]+, 572 (17), 544 (17), 543 (47), 338 (41), 264 (18), 248 (10), 236 (18), 235 (28), 234 (100), 218 (10), 91 (10); HRMS (EI): m/z: calcd for C34H37N3O7: 599.2632 [M]+; found: 599.2629.
(6aS*,7R*,13S*,14R*,16R*)‐5‐(Benzyloxy)‐16‐(hydroxymethyl)‐ 9‐methoxy‐4,10,17‐trimethyl‐8,11‐dioxo‐6a,7,8,11,12,13,14,16‐ octahydro‐6H‐7,13‐iminobenzo[4,5]azocino[1,2‐b][1,3]‐ dioxolo[4,5‐h]isoquinoline‐14‐carbonitrile (20)
A solution of CAN (21.6 mg, 37.5 μmol) in H2O (700 μL) was added to a stirred solution of 19 (9.00 mg, 15 μmol) in THF (2.1 mL) at 0 °C, and the mixture was stirred at 0 °C for 20 min. The mixture was diluted with H2O (5 mL) and then extracted with EtOAc (3×10 mL). The combined extract was washed with brine (5 mL), dried, and concentrated in vacuo to give a residue (9.8 mg), which was subjected to column chromatography on SiO2 (6 g) with hexane/EtOAc (1:1) to afford 20 (4.43 mg, 51 %) as a yellow gummy solid. Compound 20 was identical with an authentic sample13a on direct comparison of the characterization data (1H NMR, 13C NMR, IR, MS) and TLC behavior; R f=0.20 (hexane/EtOAc 3:2); 1H NMR (400 MHz, CDCl3): δ=7.50–7.36 (m, 5 H, C6H5), 5.98 (d, J=1.4 Hz, 1 H, 2‐H), 5.90 (d, J=1.4 Hz, 1 H, 2‐H), 4.66 (d, J=10.6 Hz, 1 H, PhCH2), 4.60 (d, J=10.6 Hz, 1 H, PhCH2), 4.15 (d, J=2.5 Hz, 1 H, 14‐H), 4.04 (t, J=4.3 Hz, 1 H, 16‐H), 4.01 (br d, J=2.7 Hz, 1 H, 7‐H), 3.94 (s, 3 H, 9‐OCH3), 3.71 (br d, J=10.8 Hz, 1 H, CH 2OH), 3.51(m, 1 H, CH 2OH), 3.39 (br d, J=7.6 Hz, 1 H, 13‐H), 3.18 (dt, J=12.1, 2.7 Hz, 1 H, 6a‐H), 3.04 (dd, J=15.1, 2.7 Hz, 1 H, 6H‐α), 2.82 (dd, J=20.8, 7.6 Hz, 1 H, 12‐Hα), 2.30 (s, 3 H, NCH3), 2.29 (d, J=20.8 Hz, 1 H, 12‐Hβ), 2.16 (s, 3 H, 4‐CH3), 1.95 (s, 3 H, 10‐CH3), 1.66 ppm (dd, J=15.1, 12.1 Hz, 1 H, 6‐Hβ); 13C NMR (100 MHz, CDCl3): δ=186.5 (C‐11), 182.5 (C‐8), 155.3 (C‐9), 148.2 (C‐5), 144.9 (C‐3a), 141.3 (C‐11a), 139.2 (C‐16b), 136.7 (C6H5), 136.2 (C‐7a), 128.6 (C6H5), 128.6 (C‐10), 128.5 (C6H5), 128.3 (C6H5), 120.6 (C‐5a), 117.4 (CN), 112.6 (C‐4 & 16a), 101.3 (C‐2), 75.5 (PhCH2), 65.2 (CH2OH), 60.9 (9‐OCH3), 59.8 (C‐14), 58.5 (C‐16), 56.0 (C‐6a), 54.8 (C‐7 or 13), 54.7 (C‐7 or 13), 41.5 (NCH3), 27.7 (C‐6), 21.5 (C‐12), 9.4 (4‐CH3), 8.7 ppm (10‐CH3); FTIR (KBr): =2936, 1653, 1614, 1456, 1429, 1306, 1105, 1092 cm−1; MS (FAB): m/z (%): 583 [M+H]+; HRMS (FAB): m/z: calcd for C33H33N3O7: 583.2319 [M]+; found: 584.2391.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was partially supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan through Grants‐in‐Aid (Nos. 15K07873 and 18K06561) for Scientific Research.
S. Kimura, N. Saito, ChemistryOpen 2018, 7, 764.
References
- 1. Singh I. P., Shah P., Expert Opin. Ther. Pat. 2017, 27, 17–36. [DOI] [PubMed] [Google Scholar]
- 2. Le V. H., Inai M., Williams R. M., Kan T., Nat. Prod. Rep. 2015, 32, 328–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Medicines Agency, Assessment Report for Yondelis, London, UK, 2009.
- 4.U. S. Food and Drug Administration, FDA Approves New Therapy for Certain Types of Advanced Soft Tissue Sarcoma, Silver Spring, MD, 2015.
- 5. Scott J. D., Williams R. M., Chem. Rev. 2002, 102, 1669–1730. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Frincke J. M., Faulkner D. J., J. Am. Chem. Soc. 1982, 104, 265–269; [Google Scholar]
- 6b. He H. Y., Faulkner D. J., J. Org. Chem. 1989, 54, 5822–5824. [Google Scholar]
- 7. Davidson B. S., Tetrahedron Lett. 1992, 33, 3721–3724. [Google Scholar]
- 8. Parameswaran P. S., Naik C. G., Kamat S. Y., Pramanik B. N., Indian J. Chem. 1998, 37B, 1258–1263. [Google Scholar]
- 9. Pettit G. R., Knight J. C., Collins J. C., Herald D. L., Pettit R. K., Boyd M. R., Young V. G., J. Nat. Prod. 2000, 63, 793–798. [DOI] [PubMed] [Google Scholar]
- 10. Oku N., Matsunaga S., van Soest R. W. M., Fusetani N., J. Nat. Prod. 2003, 66, 1136–1139. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Saito N., Hiramatsu A., Hirade H., Kubota M., Toyoshima R., Fujino A., Sirimangkalakitti N., Suwanborirux K., Concepcion G. P., Heterocycles 2017, 95, 748–752; [Google Scholar]
- 11b. Tatsukawa M., Punzalan L. L. C., Magpantay H. D. S., Villaseñor I. M., Concepcion G. P., Suwanborirux K., Yokoya M., Saito N., Tetrahedron 2012, 68, 7422–7428; [Google Scholar]
- 11c. Daikuhara N., Tada Y., Yamaki S., Charupant K., Amnuoypol S., Suwanborirux K., Saito N., Tetrahedron Lett. 2009, 50, 4276–4278; [Google Scholar]
- 11d. Amnuoypol S., Suwanborirux K., Pummangura S., Kubo A., Tanaka C., Saito N., J. Nat. Prod. 2004, 67, 1023–1028. [DOI] [PubMed] [Google Scholar]
- 12. Suwanborirux K., Amnuoypol S., Plubrukarn A., Pummangura S., Kubo A., Tanaka C., Saito N., J. Nat. Prod. 2003, 66, 1441–1446. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Yokoya M., Toyoshima R., Suzuki T., Le V. H., Williams R. M., Saito N., J. Org. Chem. 2016, 81, 4039–4047; [DOI] [PubMed] [Google Scholar]
- 13b. Jia J., Chen R., Liu H., Li X., Jia Y., Chen X., Org. Biomol. Chem. 2016, 14, 7334–7344. [DOI] [PubMed] [Google Scholar]
- 14.For simplicity, all numbering used in this manuscript followed that shown in Figure 1, whereas IUPAC names and numbering are used in the Supporting Information.
- 15.
- 15a. Saito N., Tashiro K., Maru Y., Yamaguchi K., Kubo A., J. Chem. Soc. Perkin Trans. 1 1997, 53–69; [Google Scholar]
- 15b. Yokoya M., Shinada-Fujino K., Yoshida S., Mimura M., Takada H., Saito N., Tetrahedron 2012, 68, 4166–4181; [Google Scholar]
- 15c. Kimura S., Kawai S., Azuma M., Umehara Y., Koizumi Y., Yokoya M., Saito N., Heterocycles 2015, 90, 327–343. [Google Scholar]
- 16. Saito N., Seki R., Kameyama N., Sugimoto R., Kubo A., Chem. Pharm. Bull. 2003, 51, 821–831. [DOI] [PubMed] [Google Scholar]
- 17.We thought that the methylenedioxy bridge would make the A ring slightly bent, thereby decreasing the electron-donating ability. See also ref. [16].
- 18. Kimura S., Saito N., Tetrahedron, 2018, 74, 4504–4514. [Google Scholar]
- 19.
- 19a. Fukuyama T., Yang L., Ajeck K. L., Sachleben R. A., J. Am. Chem. Soc. 1990, 112, 3712–3713; [Google Scholar]
- 19b. Ashley E. R., Cruz E. G., Stoltz B. M., J. Am. Chem. Soc. 2003, 125, 15000–15001. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Hediger M. F., Bioorg. Med. Chem. 2004, 12, 4995–5010; [DOI] [PubMed] [Google Scholar]
- 20b. Tani S. (Ihara Chemical Industry), Patent WO2010150548A1, 2010.
- 21.As compounds 15–17 were inseparable mixtures of diastereomers, none of the signals in the 1H NMR and 13C NMR spectra were split. Thus, we decided to present the spectral information in compound 18.
- 22.We found that sodium p-toluenesulfinate was the most effective additive, as it exclusively afforded the desired product in the total synthesis of saframycin A (3). However, whereas the transformation of 17 into 18 in the presence of sodium p-toluenesulfinate led to removal of the allyl group in the diester at C1 and to the decarboxylation of 17, the allyl group in the E ring could not be removed. Finally, we found that sodium p-toluenesulfinate and dimedone were used in the removal of allyl groups and the decarboxylation sequence of 17, and desired ethyl ester 18 was obtained in 67 % overall yield in a one-pot process.
- 23.Additional renieramycin-type marine natural products fennebricins A and B were discovered from the blue sponge Xestospongia sp. in the South China Sea;
- 23a. He W. F., Li Y., Feng M. T., Gavagnin M., Mollo E., Mao S. C., Guo Y. W., Fitoterapia 2014, 96, 109–114; [DOI] [PubMed] [Google Scholar]
- 23b. Huang R.-Y., Chen W.-T., Kurtán T., Mándi A., Ding J., Li J., Li X.-W., Guo Y.-W., Future Med. Chem. 2016, 8, 17–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary