

Abstract
Introduction:
Proteins have been historically regarded as “nature’s robots”: Molecular machines that are essential to cellular/extracellular physical mechanical properties and catalyze key reactions for cell/system viability. However, these robots are kept in check by other protein-based machinery to preserve proteome integrity and stability. During aging, protein homeostasis is challenged by oxidation, decreased synthesis, and increasingly inefficient mechanisms responsible for repairing or degrading damaged proteins. In addition, disruptions to protein homeostasis are hallmarks of many neurodegenerative diseases and diseases disproportionately affecting the elderly.
Areas covered:
Here we summarize age- and disease-related changes to the protein machinery responsible for preserving proteostasis and describe how both aging and disease can each exacerbate damage initiated by the other. We focus on alteration of proteostasis as an etiological or phenomenological factor in neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s, along with Down syndrome, ophthalmic pathologies, and cancer.
Expert commentary:
Understanding the mechanisms of proteostasis and their dysregulation in health and disease will represent an essential breakthrough in the treatment of many (senescence-associated) pathologies. Strides in this field are currently underway and largely attributable to the introduction of high-throughput omics technologies and their combination with novel approaches to explore structural and cross-link biochemistry.
Keywords: proteostasis, aging, neurodegeneration, metabolism, advanced omics
1. Introduction
Protein homeostasis networks are regulated by cell stress signaling pathways that respond to events such as unfolded/misfolded proteins in the endoplasmic reticulum or accumulation of toxic protein aggregates. Dysfunction of these quality control mechanisms and resultant intracellular accumulation of abnormal proteins in the forms of protein inclusions and aggregates occur in almost all tissues of an aged organism [1]. Age is indeed the strongest risk factor for many diseases including neurodegenerative disorders, coronary heart disease, and cancer [2–4]. Aging is a complex, multifactorial process characterized by a progressive decline in physiological functions at multiple levels [5]. Maintenance of the aging transcriptome, proteome, and metabolome is essential to preserve cell functionality and the ability to respond and adapt to tissue-specific chronic and acute stressors. In mammals, hallmarks of aging tissues include declining rates of self-renewal capability (e.g., protein homeostasis dysfunction [6, 7], stem cell exhaustion [8]), accumulation of damage to DNA (e.g., epigenetic alterations [9]) and proteins (e.g. carbonylation), genomic instability [10-12], impaired mitochondrial metabolism [13], and increased levels of reactive oxygen species and reactive nitrogen species (ROS and RNS, respectively) [12, 14], among other traits (reviewed in depth here [15]). Exploration of these attributes in mice has identified relationships among senescence, transcriptional regulation, increased mutational burden, and maintenance of proteome integrity – processes normally regulated by a dedicated set of molecular machinery that collectively maintain protein homeostasis, also termed proteostasis. Disrupted proteostasis also underlies many neurodegenerative diseases and cancers, illustrating the complex interplay between aging and pathology. In this review, we discuss the role of proteostasis in aging and age-associated disease states by examining protein synthesis and degradation, chaperone activity, aggregation, age-correlated modifications, and cellular metabolism (Figure 1). The biological mechanisms and function of each aspect of the proteostasis machinery are areas of high importance in cell and tissue physiology, and thus each is broadly studied and rapidly evolving. In light of space limitations, we focus here on introducing each area with details pertinent to understanding disruptions and imbalance in the context of disease, highlighting the most recent year’s results, and pointing the reader to recent reviews focused on each specific topic.
Figure 1.
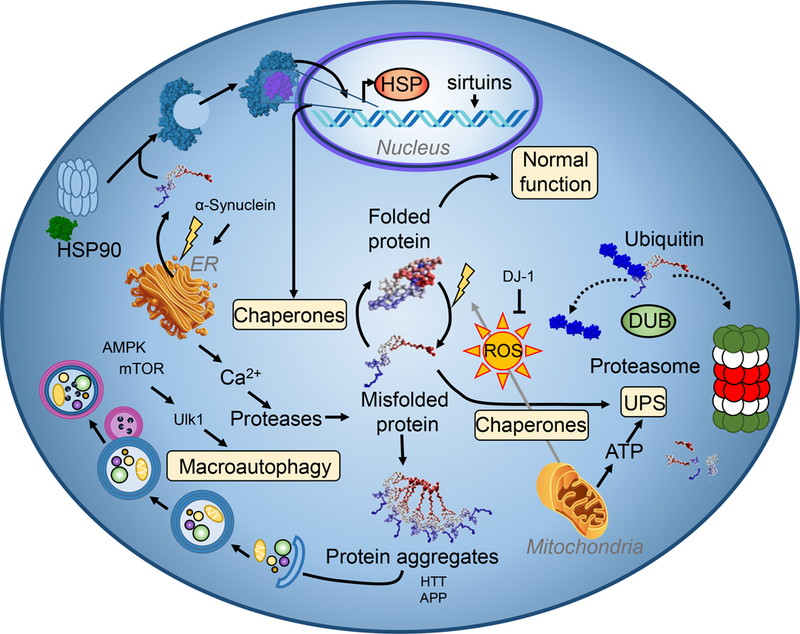
Overview of protein quality control machinery. To preserve proteome integrity, cells contain a diverse set of proteins that work together to ensure proper structure and localization of nascent and existing proteins. Figure adapted from Henning and Brundel [157].
2. Cellular proteome imbalance results through numerous mechanisms in aging
2.1. Mitochondria and oxidative stress
Aging cells are widely known to possess less functional mitochondria and higher basal levels of ROS than their younger counterparts, two closely related attributes [16, 17]. Exposure to ROS and RNS results from exogenous sources such as radiation (e.g., gamma rays in therapy, sunlight), smoking, and other environmental sources, accumulating in cells over time. Importantly, there are numerous endogenous routes of ROS generation, including uncoupling of the electron transport chain (ETC) (i.e., reaction of leaked electrons with O2 to produce superoxide) and by the activity of the NADPH oxidases [18]. Once formed, these species react very rapidly with each other and/or nearby biomolecules, often culminating in irreversible damage to DNA, proteins, and lipids [19]. Cells are armed with efficient small molecule (e.g., glutathione, ascorbate) and enzymatic (e.g., peroxiredoxins, glutathione peroxidase, catalase, superoxide dismutase) tools to counteract ROS/RNS formation, but these systems can become overwhelmed and/or incapacitated with acute or chronic high levels of oxidants. Mitochondrial DNA and proteins are particularly sensitive to the effects of ROS, as two dominant sources of ROS are the ETC complexes I and III. Furthermore, mitochondrial DNA does not have the protection of histones and has less repair machinery than exists for nuclear DNA [20].In the context of aging, inefficiencies in the ETC combined with the accumulation of environmental ROS exposure lead to oxidative damage of mitochondrial DNA and proteins, ultimately manifesting in a decreased capacity for efficient and robust ATP production by oxidative phosphorylation (see [21] for a recent review on mitochondrial function in the context of aging). Mitochondria are equipped with quality control machinery for the recognition of dysfunction and subsequent degradation through mitophagy. These processes are mediated by several protein complexes, including NIX (also known as BNIP3L) and Parkin/PINK1, and share in common the recognition of depolarized or damaged mitochondria. NIX plays key roles in processes such as erythropoiesis, removing mitochondria as reticulocytes develop into mature red blood cells [22], and in attenuating ischemia-reperfusion injury in the brain [23]. BNIP3 expression is regulated by hypoxia-inducible factor-1(HIF-1), and thus BNIP3-mediated mitophagy plays a key role in cancer biology and numerous hypoxia-associated diseases ([24], reviewed in [25]). Parkin/PINK1 is a better characterized pathway involved in both steady state mitophagy and disease states, with particular implications in Parkinson’s disease.
Mitophagy is a highly selective process, and in the Parkin/PINK1 pathway, PINK1 serves as a transmembrane protein anchored in the outer mitochondrial membrane. Under normal basal conditions, PINK1 is cleaved by mitochondrial proteases [26], and the nascent C-terminal fragment localizes to the cytosol (with the help of chaperone Hsp90 [27]) where it is degraded by the ubiquitin-proteasome system. This process is inhibited by membrane depolarization, accumulating intact PINK1 [28], which phosphorylates Mfn2 and facilitates the binding of Parkin, an E3 ubiquitin ligase. Together, PINK1 and Parkin respectively phosphorylate and ubiquitinate numerous targets, ultimately leading to mitochondrial degradation [29]. One such ubiquitination target of Parkin is HIF-1, and Parkin expression was very recently shown to have an inverse correlation with both HIF-1 levels and metastasis in human breast cancer tissue [30]. As mentioned previously, with aging comes increased dysfunction in mitochondrial ETC proteins; imbalanced turnover has also been noted in proteins involved in the mitophagy process, for example in the mouse cerebellum [31] and aging skeletal muscle from several mammals [32].An area of continued focus regarding the Parkin/PINK1 pathway are its roles in regulating basal mitophagy/proteostasis as opposed to activation only in response to a stress signal. Until the last two years, it was widely agreed that this pathway was required for mitophagy in both healthy and pathological conditions, however new evidence has emerged with the help of sophisticated experimental techniques that eliminate the use of strategies that may have confounded mitochondrial physiology such as protein overexpression, mitochondrial uncoupling agents, and addition of ROS to cells. With newer approaches, basal mitophagy in Drosophila was not found to be significantly affected by the loss of either Parkin or PINK1, though cognitive and motor deficits were noted [33]. A study of PINK1 wild type (WT) and knockout mice using fluorescence reporter mito-QC similarly concluded that basal mitophagy in highly metabolically active tissues such as brain, heart, liver, kidney, and retina are all unaffected by the lack of PINK1. PINK1 expression was confirmed in these tissues in the WT animals, and furthermore, microglial mitophagy was also unaltered in the mice lacking PINK1 [34]. The precise roles of Parkin/PINK1 are also a topic of much investigation in the cardiovascular system and will be discussed further in that section.
The Parkin/PINK1 pathway of mitophagy is susceptible to damage by ROS/RNS, for example, by S-nitrosation of PINK1 at Cys568. This modification, observed in a human neural cell line, was observed in response to treatment by S-nitrosocysteine (CysNO) and induction of neuronal nitric oxide synthase (nNOS), and though chemically reversible, impaired mitophagy through several routes including the inhibition of PINK1 kinase activity and decreased translocation of Parkin to the mitochondrial membrane [35]. Together, Parkin and PINK1 play crucial roles in both normal developmental processes, such as in cardiac tissue, and in diseases of proteostasis imbalance like Parkinson’s disease. The role of mitophagy mediated by Parkin and PINK1 are discussed in more detail in those contexts (Sections 4–5).
Another negative consequence of ROS accumulation is the activation of purine deaminases (e.g. AMP deaminase), which deplete the reservoir of high-energy phosphate purines, such as ATP. These actions also pave the way for the generation of pro-oxidants through purine catabolism, exemplified by the generation of hydrogen peroxide via xanthine oxidase [36]. ATP depletion is particularly deleterious in that it serves as the key energy source for maintaining proteostasis balance. A lack of adequate cellular ATP supply necessitates a shift toward the preservation of the most critical processes to maintain cellular homeostasis, thereby hindering ATP-dependent reactions in other non-essential roles such as the ubiquitin-proteasome protein degradation system [37]. Increased steady state levels of ROS coupled with diminished ATP levels exacerbate the aging quality control mechanisms that impact many aspects of proteostasis. While further work is required to understand its role in age-associated dysregulation of proteostasis, modification of ETC efficiency through the maintenance of mitochondrial supercomplexes may provide a mechanism to minimize the damaging effects associated with these processes [38–40].
2.2. Folding – chaperones and endoplasmic reticulum
Proteome integrity is maintained by a network of quality control machinery responsible for the accurate translation of mRNA, polypeptide processing and folding, intracellular trafficking, and recognition and catabolism of damaged protein molecules. Chaperone proteins are specifically involved in vital roles of protein folding and transport. Disease-associated disruptions to the chaperone network have been identified, though a larger challenge lies in determining the specific mechanisms by which invading cells (e.g. cancer, viruses) hijack chaperone proteins or alter their expression. Earlier this year, expression profiles for chaperone genes were prepared for >10,000 patient samples including 22 solid cancers, allowing for stratification of cancer-related alterations and recognition of widespread patterns such as the decreased levels of small heat shock proteins across all cancers investigated [41].
In mammals, the heat shock response (HSR), triggered by acute stresses such as heat, hypoxia, unfolded proteins, and the accumulation of protein aggregates (also referred to as aggresomes), utilizes the transcription factor HSF-1 to upregulate expression of heat shock proteins (Hsp) along with other proteins necessary for eliminating damaged biomolecules and restoring normal cellular function [42]. The Hsp family of proteins are found in every cell with the cytosol, mitochondria, and endoplasmic reticulum (ER) each having a dedicated Hsp system with basal housekeeping roles [43] in addition to stress-induction mediated by HSR. Cell types constitutively exposed to oxidative stress such as the iron-rich oxygen-carrying erythrocytes have a unique Hsp specialization, where the system mediates a “save or sacrifice” response [44]. Hsp proteins bind to unfolded or misfolded proteins, working in conjunction with co-chaperone proteins and foldase enzymes to refold proteins in a process that often requires ATP. Hsp90 chaperones bind HSF-1 in a negative feedback manner; recognition and binding to a damaged protein by Hsp90 releases HSF-1 for translocation to the nucleus and transcriptional activation of HSR genes [45].
The endoplasmic reticulum (ER), home to the synthesis of luminal, membrane, and secretory proteins, as well as the folding of nascent proteins translated in the ribosome, maintains its own response pathway for proteotoxic stress. The ER utilizes a more oxidizing environment than other cellular compartments that facilitates disulfide bond formation by protein disulfide isomerase (PDI) during folding. ER stress occurs in situations of unfolded protein accumulation, disruptions in cellular Ca2+ levels (as the ER maintains the largest cellular Ca2+ pool [46]), and unbalanced oxygen homeostasis that causes hypoxia or oxidative stress [47]. The ER is equipped to remedy these acute challenges using members of the Hsp family. In addition, damaged proteins may be retro-translocated out of the ER with the help of chaperones and into the cytosol for proteasomal degradation, a process termed ER-associated protein degradation (ERAD) [48]. However, stressors that are insurmountable or that cause global disruptions to folding activate the unfolded protein response (UPR) pathway. The UPR involves 3 transmembrane proteins that act as separate arms of the pathway and coordinate to increase the expression of ER homeostasis genes, decrease translation of other proteins to offset protein load, and expand ER size [49, 50]. Increased ER size does not require the activity of UPR chaperone proteins but is selectively induced by UPR signaling [51]. Cells whose UPR machinery cannot efficiently repair ER damage are directed to apoptosis (detailed here [52]).
Like other proteostasis mechanisms, the UPR is adversely affected during aging by a decrease in the abundance of ER chaperone proteins (along with increased oxidative damage), increased expression of pro-apoptotic proteins, and disrupted phosphorylation status of proteins involved in translation [53]. It was recently demonstrated in mouse liver that the 3 UPR proteins can be differentially modulated in aging in a manner that may be beneficially altered by lifelong exercise [54]. Studies of over-nutrition and obesity have frequently noted a chronically active but less effective UPR [55]. Interestingly, the mitochondria mounts its own UPR defense mechanism that is initiated by the activity of proteases on damaged mitochondrial proteins (reviewed in detail [56]).
2.3. Protein degradation systems
In the event of irreparable protein damage, cells employ several tools to sequester and destroy the damaged molecule to protect healthy organelles and preserve lifespan. The two systems, the ubiquitin-proteasome system (UPS) and the lysosomal-autophagy pathway, each utilize highly coordinated networks of proteins dedicated to the recognition of damage, transport to – or synthesis of – the appropriate proteolytic organelle, and lysis of covalent protein bonds. The UPS aids in the homeostasis of short-lived cellular peptides and proteins by tagging damaged proteins with one or more ubiquitin molecules (76 amino acids) at lysine residues, then transporting labeled proteins to the 26S proteasome for degradation. Conjugation of ubiquitin moieties to protein lysines occurs in a step-wise ATP-dependent fashion facilitated by ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2), and ubiquitin-ligating (E3) enzymes. There exist distinct proteasome forms that demonstrate preference for metabolizing mildly oxidized proteins or ubiquitinated proteins [57]. Critically, the ubiquitination process and proteasome function are both ATP-dependent. Moreover, oxidation can inhibit ubiquitin-activating enzymes (E1s) and ubiquitin-conjugating enzymes (E2s) via modification (e.g., glutathionylation, nitrosation) of active site cysteine residues along with noted negative impacts on proteasome function [57]. The UPS is critical for homeostasis maintenance in a variety of cell types and helps to preserve cell vitality by degrading proteins of the apoptotic machinery when they are not needed [58]. UPS activity is increased in stem cells (reviewed in [59]), including human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) as a strategy of preserving plasticity. Recently, several E3 ubiquitin ligases were identified as elevated in hESCs compared to differentiated cells as one of the possible mechanisms underlying enhanced UPS [60]. In the event of severe or prolonged cellular stress, the capacity of the UPS to clear ubiquitinated proteins can be exceeded. In such cases, these proteins are directed to autophagy.
The lysosomal-autophagy pathway targets misfolded proteins, ubiquitinated proteins, and larger molecular structures, such as aggresomes and even damaged organelles. There exist several types: chaperone-mediated authophagy (CMA), microautophagy, and macroautophagy. In CMA, protein chaperones assist in recognition of damaged cytosolic proteins bearing the KFERQ motif and translocation to the lysosome surface, where they bind to protein Lamp2a and transported into the lysosome for degradation. CMA requires the sophisticated coordination and crosstalk of many proteins; these essential molecular interactions have been recently reviewed in detail [61]. Microautophagy involves direct lysosomal absorption of smaller cellular debris, whereas macroautophagy, the dominant form referred to as simply “autophagy”, requires the synthesis of a double membrane around the damaged molecules to form an autophagosome that is later engulfed by the lysosome. The lysosome contains at least 60 unique hydrolase enzymes [62] and maintains an acidic pH of 4.5–5 for proteolysis through the proton pump activity of vacuolar-type H+-ATPases (V-ATPases). As such, chemical integrity of the lysosome is reliant on an adequate ATP supply.
Autophagy occurs at low basal levels in cells to maintain homeostasis, and is activated in response to stresses like starvation, infection, and hypoxia. The autophagy process is indispensable for stem cell metabolism and preservation of their stemness and self-renewal properties (reviewed extensively in [63]). Expeditious activation of autophagy occurs as a result of protein post-translational modifications, of which many are known; sustained activation occurs instead through transcriptional regulation [64]. The small molecule end-products of autophagy, including amino acids, sugars, and other metabolites, are released into the cytosol to serve as substrates in energy metabolism and other anabolic processes in an effort to maintain cellular function despite environmental or microenvironmental stressors (see [65] and [66] for detailed reviews linking metabolism and autophagy). Autophagy activity decreases with age and is hampered in age-related diseases such as cancer [67, 68]. As just one of the many examples, mutation or damage to tumor suppressor protein p53 facilitates its accumulation in the cytosol, preventing its ability to activate the transcription of autophagy genes [64, 68].Chaperones and proteolysis systems are profoundly affected during aging. HSR repression begins at the onset of reproductive maturity in Caenorhabditis elegans (C. elegans) via epigenetic modification of histone H3 [69]. Both the UPS and the lysosomal system have diminished proteolysis capacities in aging organisms and their protein machineries are each susceptible to aging-related damage through oxidation, conjugation to lipid peroxidation products [70], and/or protein cross-linking [71-73]. Though spatially distinct, the lysosome and proteasome are interrelated and have been shown to compensate, at least partially, when one is compromised [74, 75]. One example of this interplay was revealed in a study earlier this year of N-terminal arginylation, in which arginine is added to specific residues of protein N-termini exposed by endopeptidase activity. Though a comprehensive picture of the cellular consequences of arginylation is not yet established, experiments in this study found that proteins containing this modification are detected by N-recognins that direct the protein to the UPS or, when the UPS is inhibited or compromised, by p62 with resulting autophagy [76]. Such observations illustrate compensatory mechanisms within proteostasis and how damaged or dysfunctional proteins may be processed differentially in context-dependent manners. Further illustrating the interconnections between these two proteolytic hubs, the transcription factor homeodomain-interacting protein kinase 1 (HPK-1) was recently identified as preserving proteostasis in C. elegans by both suppressing an inhibitory post-translational modification to HSF-1 and, separately, by regulating the expression of genes involved in the autophagy pathway, particularly autophagosome formation [77].
3. The interplay between cellular bioenergetics and protein quality control
Since the energy derived from metabolic pathways must be partitioned into cellular activities that maintain homeostasis, synthesize new biomolecules, and repair damages ones, the links between metabolism and proteostasis are becoming intuitively clear. For instance, the role autophagy plays in maintaining a functional proteome is influenced by many signaling pathways that can be routed through the activities of mammalian target of rapamycin (mTOR) and adenosine monophosphate-activated protein kinase (AMPK) (extensively reviewed in [78]). Functioning as a nutrient sensor, mTOR is a serine/threonine kinase with a variety of downstream targets, including the autophagy-regulating protein Ulk1 [79]. Through phosphorylation of Ser757, mTOR inhibits Ulk1 activity thereby decreasing cellular autophagy. Conversely, AMPK functions as a cellular energy sensor activated in response to increased AMP:ATP ratios [80] as well as through adenosine signaling pathways [81]. It too acts upon Ulk1, though phosphorylation of Ser317 and Ser777 activates autophagosome formation thereby promoting autophagy. As such, the functional interplay between mTOR and AMPK has become an attractive therapeutic target for anti-aging strategies [82, 83]. In addition to direct modulation of these protein activities with the use of compounds such as rapamycin and metformin (among others), recent studies have highlighted a strategy of immunomodulation with the implementation of cytokine-based therapies, such as the anti-inflammatory cytokine IL-37 [84]. While considerable work remains to provide more direct links among immunomodulation, energy homeostasis, and aging, these studies illustrate a potential area of therapeutic intervention for diseases dependent on age-associated dysregulation of proteostasis.
In addition to mTOR and AMPK, autophagy can also be modulated by polyamines. These small polybasic molecules are produced as an off-shoot of the urea cycle and exert multiple effects on DNA and RNA, as well as ROS scavenging and pH balance [85]. In addition to these roles, their ability to alter properties of autophagy have also been demonstrated [86]. Because the levels of these molecules decline with age [87], a process also associated with the onset of neurodegenerative disease [88], alterations to polyamine levels by way of pharmacological [89] or dietary [90] intervention deems a promising avenue to address complications of aging and associated disease.
Another family of proteins important in the connection between nutrient metabolism and aging are the sirtuins. Consisting of seven members, these proteins function as NAD+-dependent histone deacetylases that have been shown to modulate proteostasis by way of autophagy regulation [91]. The discovery that NAD+ levels decrease with age in the tissues of both model organisms and humans [92] suggests that sirtuin function may also become impaired with aging. Indeed, restoring NAD+ to more youthful levels has been shown to provide beneficial effects in the context of age-dependent diseases [93]. As NAD+ levels depend heavily on the ability of cells to convert tryptophan into nicotinamide, it is interesting that tryptophan metabolism is also altered with age [94] and the ratio of the alternative tryptophan catabolite kynurenine to its precursor is associated with species longevity [95]. In addition, metagenomic profiling of gut microbiota in elderly and centenarians revealed preferential consumption of tryptophan, thus diminishing its bioavailability for important biological processes such as nicotinamide production [96].
The metabolic impairments that contribute to age-associated pathologies similarly affect inflammatory processes. An increasingly appreciated factor in age-associated dysregulation of proteostasis is chronic, low-grade inflammation, the collective effects of which are referred to as inflammaging. First termed in 2000 [97], inflammaging is believed to involve, in large part, the systemic accumulation of cell debris arising from damaged/dead cells and organelles (extensively reviewed in [98]). As processes such as autophagy [67, 99] and the ubiquitin-proteasome system [100] decline with age, an imbalance arises between the synthesis and removal of cellular molecules that results in accumulation of misfolded/damaged proteins. Classified as damage associated molecular patterns (DAMPs), these molecules are recognized by a variety of immune receptors, including pattern recognition receptors (PRRs), dendritic cell receptors, and scavenger receptors that initiate an immune response to clear the damaged components in avoidance of excessive inflammation. However, as proteostasis becomes impaired and this balance tips, a type of autoimmune response is initiated that propagates additional tissue damage. Consequently, inflammaging has been associated with many age-related diseases, including Alzheimer’s disease [101], Parkinson’s disease, amyotrophic lateral sclerosis, multiple sclerosis, atherosclerosis, heart disease, age-related macular degeneration [102], type II diabetes [103], osteoporosis and insulin resistance [104], and cancer [105, 106]. Future studies will be able to expand upon the connections between metabolism and dysregulated proteostasis in these diseases. While many of these processes are heterogeneous, high-throughput methodologies [107] will enable the processing of larger, and thus more highly-powered sample sets that will allow for improved detection of biological phenomena associated with aging.
The slow but certain decline of robustness in proteostasis mechanisms that occurs during aging renders cells less able to combat challenges, which, therefore makes the organism more susceptible to disease progression. We will next highlight the role of proteostasis in diseases with a defined genetic component along with several metabolic diseases. These diseases are grouped by those that are directly associated with aging (i.e., having age as a primary risk factor) and those that are exacerbated by aging, but with onsets earlier in life (Figure 2). Independent of etiology, disrupted and/or dysfunctional proteostasis plays a role in nearly all pathologies and aging serves as a notorious priming event that weakens cellular defenses and, in particular, feeds the proteostasis imbalance.
Figure 2.
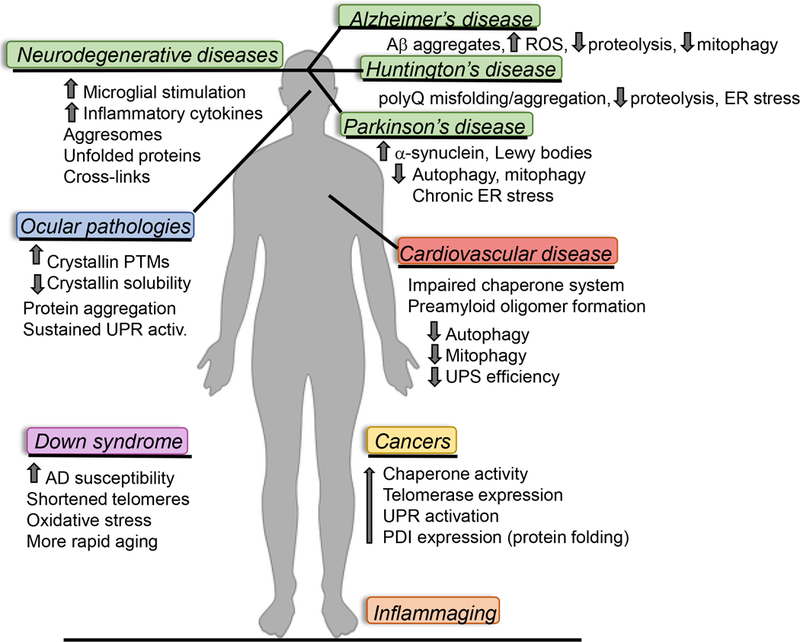
Aging and age-related diseases adversely impact proteostasis mechanisms in a multitude of tissues throughout the human body; only several are shown here. (Ab = amyloid beta, AD = Alzheimer’s disease, ER = endoplasmic reticulum, PDI = protein disulfide isomerase, PTM = post-translational modification, ROS = reactive oxygen species, UPR = unfolded protein response, UPS = ubiquitin-proteosome system)
4. Disrupted proteostasis in neurodegenerative diseases
Altered cellular proteostasis underlies the pathophysiology of many neurodegenerative diseases (NDDs) such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, which disproportionately affect elderly populations. The physiological signature of Down syndrome is also marked by imbalanced proteostasis and accelerated aging processes. These diseases have in common a sustained stimulation of microglia (reviewed in [108]), the innate immune system in the brain, along with increased levels of inflammatory cytokines such as tumor necrosis factor (TNF), interleukin 1 (IL-1), and interleukin 6 (IL-6). Additionally, NDDs are marked by propensities for protein covalent cross-links mediated by transglutaminase [109] and large intracellular concentrations of proteins with unfolded regions; in some cases these outcomes are a result of familial or sporadic genetic mutations. Disordered, unfolded protein regions are typically handled by cellular chaperones, but have a tendency in these disease states to overwhelm the capacity of the chaperone system, leading to a ‘gain of function’ where nascent exposed regions bind indiscriminately to proteins, organelles, and biomolecules with similar polarities, ultimately leading to the formation of aggregates or amyloids. Protein aggregates, sometimes termed aggresomes, obstruct the normal function of the organelle in which they reside and routinely lead to cell death. The formation of amyloids and aggresomes in neurons disrupt synaptic transmission and cell metabolism; subsequent neuronal death underlies brain degeneration and dementia.
4.1. Alzheimer’s disease
Alzheimer’s disease (AD) and frontal lobe dementia are diseases characterized by the accumulation of misfolded aggregates of amyloid β (A) and hyperphosphorylated tau in the brain, which form plaques and tangles, respectively [110, 111]. A peptides are produced by processing of the amyloid precursor protein (APP) by and secretase, which occurs for 10% of translated APP [112]. In contrast, APP processing by secretase generates soluble APP. In the context of AD, APP has received much less attention than A but is noted to confer protection from chronic and acute hypoxia and traumatic brain injury, improving neuronal survival by a multitude of pathways that include preservation of calcium homeostasis [113-115]. To further illustrate the beneficial role of APP, mutants of this protein in the mouse hippocampus impaired mitophagy via reduced levels of PINK1 and Mfn2 [116]. Unlike its protein of origin, APP, A has a strong propensity to aggregate; in AD brains, A accumulates both intra- and extracellularly leading to aggregate structures and amyloid plaques. Extracellular A may be imported into the cell, and is known as a late indicator of AD. Intracellular A is observed earlier in AD onset and has garnered attention only more recently [117]. A aggregates accumulate in lysosomes, mitochondria, and other organelles [118, 119], bind metals and generate ROS [120], and cause metabolic dysfunction [121], triggering apoptosis and resulting in tissue degeneration. Even soluble A peptides can induce organelle damage and alter proteostasis, as demonstrated for the ER [122]. Accumulated intracellular amyloid can also induce other proteins to form amyloid, a process termed cross-interaction, which serves as a chain reaction further disrupting cellular function [123]. In addition, the increased levels of insoluble aggregated proteins experienced during aging are sufficient to induce A aggregation [124]. APP expression is increased in stress situations and throughout aging to confer neuroprotection but conversely can also lead to accumulation of A peptides by and/or secretase activity [112].
In an effort to combat the etiology of AD, there have been numerous investigations to elucidate the precise molecular factors responsible for A aggregation and amyloid formation along with the lack of effective proteolysis mechanisms to eliminate or suppress these structures. In terms of aggregate structure, features such as overall size, peptide conformation, and quaternary structure are challenging to assess but critical for understanding the factors driving their formation and accumulation. The lysosomal-autophagy system is hampered in AD as indicated by both the presence of mutations in autophagy-related genes [125] and the observation of many autophagosomes in AD neurons [126]. These autophagosomes were rich in A, suggesting robustness in sequestering A but ineffective lysosome-mediated proteolysis. A subsequent study revealed increased lysosome pH in both fibroblasts of AD patients and neurons of an AD mouse model as an underlying cause of diminished proteolysis [127]. In a separate study using a C. elegans model of AD, induced expression of A led to a disrupted status of the UPR, though neurotoxicity was delayed perhaps by blocking ER synthesis of toxic proteins [128]. ER stress increased secretase activity and therefore A levels, illustrating another feed-forward mechanism of neuronal destruction by proteostasis imbalance [129] (see [130] for a detailed new review on ER stress and the UPR in AD). Expression of mitophagy-related genes is decreased in AD, and mitochondria in affected patients suffer from slower turnover and protein dynamics [131]. Interestingly, the effects of A spread beyond proteostasis disruption, interfering with cellular metabolism as well. For instance, accumulation of A in red blood cells occurs in an age-dependent manner [132], adversely impacting the activities of glycolytic enzymes responsible for the production of important hemoglobin allosteric modulators such as ATP and 2,3-diphosphoglycerate [133-135], and leading to impaired oxygen delivery and resultant tissue hypoxia [136].
Individuals with Down syndrome (DS) are uniquely predisposed to the development of AD as the gene encoding APP resides on chromosome 21, the trisomy of which is found in DS. The additional copy of chromosome 21 results in an overexpression of APP in brains of individuals with DS as well as higher steady state neuronal levels of A in comparison to disomy 21 individuals [137]. The concentration of toxic A increases with age, and though APP expression does not appear to change in an age-dependent fashion in DS, secretase activity decreases with age thus shifting APP toward toxic A formation [138, 139]. Sadly, even very young children with DS experience high intracellular A, and most DS patients are diagnosed with AD by age 40, an onset at least 10 years earlier than the general population [140]. Shortened telomeres have also been implicated in the early onset of AD associated with DS [141]. Head et al. recently reviewed the biochemistry and pathology induced by A in the context of DS [140]. Proteostasis imbalance has been documented in lymphoblastoid cell lines from DS patients and mouse models of DS [142] along with chronic oxidative stress [143] and protein oxidation in the brain [137, 144].
Interestingly, AD pathology in neural tissue also progresses in a predictable manner as a result of mutations in one or more genes (APP and/or others) ultimately leading to toxic levels of A [112]. A recent study of healthy post-mortem brains investigated the role of proteostasis factors in disease progression and found that regions of the brain with protein expression profiles that suggested a likelihood for aggregate formation, as well as those involved in proteostasis, were particularly susceptible to neurodegeneration in AD [145]. Efforts to combat or prevent AD onset have focused on acetylcholinesterase inhibitors [146] and targets of the cholinergic system [147], N-methyl-D-aspartate (NMDA) receptor antagonism to reduces neural A levels [148], exercise to prevent hippocampal mitochondria dysfunction [149], and erythrocyte membrane-encapsulated celecoxib to promote neurogenesis via prostaglandins [150]. In contrast, attempts to inhibit secretase have not proven clinically effective [151].
4.2. Parkinson’s disease
Parkinson’s disease (PD) is a rarer but well-known NDD associated with involuntary shaking, cognitive decline, depression, impaired sense of smell, and disturbances in normal sleep patterns. The prevalence of PD dramatically increases with age [152] and is marked by mutations in the proteins synuclein and/or leucine-rich repeat kinase 2 (LRRK2) that are inherited or the result of genome damage by traumatic brain injury, exposure to pesticides, previous diagnosis of melanoma and other causes [153]. Though LRRK2 mutations are more common [154], the precise molecular mechanisms that subsequently result in PD are not fully clear, though damage in NF-B signaling has been implicated [155]. Regardless of protein mutational status, the common hallmark of PD is neuronal synuclein accumulation and the formation of insoluble fibrils termed Lewy bodies. Such aggregates indicate disrupted proteostasis and in turn further hamper numerous proteostasis mechanisms culminating in the death of dopaminergic neurons and degeneration in the substantia nigra. Mutations in other proteins have also been noted, for example in the mitophagy enzymes Parkin and PINK1 and the antioxidant protein DJ-1, leading to forms of early onset PD [156, 157]. Oxidative and nitrosative stresses play key roles in the pathogenesis of PD. As one example, cysteine thiol oxidation disrupts transferrin binding of iron, releasing Fe2+, a source for further ROS formation [158]. Residues of Parkin and PINK1 have also been demonstrated as targets for oxidation by NO (derived from CysNO or nNOS [35]) or superoxide [159], affecting both their localizations and enzymatic activities [160, 161] in manners that impair mitophagy. In the context of NDDs, uneffective mitophagy function allows damaged and dysfunctional neurons to accumulate, further obstructing turnover and cell survival processes.
Accumulation and aggregation of synuclein affects the cellular proteostasis network at multiple levels in PD-affected neurons. The lysosome-autophagy pathway is thought to be the dominant clearing mechanism for synuclein (rather than the UPS) [162], however, animal models and studies of PD patients have indicated decreased levels of autophagy enzymes and accumulation of autophagosomes in the substantia nigra of PD-afflicted brains [163]. Mutations have also been noted in the GBA gene, which encodes glucocerebrosidase (GCase), a lysosomal enzyme [164]. Increased levels of mutant synuclein occur in the extracellular space and in the ER, causing chronic ER stress and sustained UPR activation [165-167]. Mitochondrial metabolism is adversely affected as evidenced by decreased activity of ETC complex I in PD [168, 169], mutations in mitochondrial DNA and proteins, alterations in size and shape, and other attributes (reviewed in [169]). Furthermore, a C. elegans model of PD revealed chronic activation of the mitochondrial UPR leading to the loss of dopaminergic neurons in a non-apoptotic manner [170].
Therapies for PD involve the management of symptoms as there is not currently a means of halting disease progression. Treatment with dopamine precursor l-DOPA has been successful at partially alleviating symptoms and is now formulated with inhibitors of dopamine-metabolizing enzymes to extend its half-life [171]. An alternative strategy to increase DJ-1 expression using phenylbutyrate has shown to be neuroprotective in both cell culture and animal models [172]. Very recently, type II diabetes drug exenatide has shown promise in a clinical trial for the treatment of PD though further investigations are needed to examine its role in mechanistically altering disease status versus amelioration of symptoms [173]. Another recent success involved treatment with nitrite, which lead to the reversible and protective S-nitrosation of complex I and activation of transcription factor Nrf2 [174]. Expression of Nrf2 in PD neurons leads to more IBs to sequester mutant LRRK2 and enhanced degradation of synuclein [175]. Lastly, treatment with chaperones has been shown to decrease ER stress and slow the progression of motor dysfunction in vitro. Potential targets of proteostasis modulation in the context of PD are the subjects of recent reviews [171, 176].
4.3. Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant inherited neurological disorder involving an expansion of CAG repeats in the huntingtin (HTT) gene, resulting in a polyQ region near the amino terminus of the HTT protein. The polyQ region confers proteotoxicity with 36 or more glutamine repeats as the propensities for misfolding and non-selective binding to other biomolecules are increased. Clinical symptoms include chorea, cognitive decline, and behavioral changes; alterations in glucose metabolism (reviewed in [177]) and in the retina [178] have also been noted. Disease onset and severity are directly correlated to the number of Q residues present, where onset is earlier, and severity is greater with increasing length of the polyQ region. Similar to AD, HD pathology and neurodegeneration proceed in a predictable manner of affected brain regions [179]. Mutant HTT (mHTT), with toxic polyQ levels, exists in 3 distinct states in the brain: soluble monomers, soluble oligomers, and insoluble aggregates which form inclusion bodies (IBs) in neurons. The factors governing the distribution of mHTT into these cellular fractions are not fully clear but likely involve mHTT concentrations, the extent of polyQ sequence, post-translational modifications of HTT and other proteins, and the activity of proteostasis machinery.
The formation of mHTT aggregates is believed to be an initial neuronal survival pathway intended to sequester and destroy misfolded protein by the UPS and autophagy. In fact, oligomeric mHTT has been noted as more cytotoxic than IBs which is also supported by observations that IB formation is not necessary for proteotoxicity in HD [180]. Soluble mHTT induces ER stress, upregulating all branches of the UPR [181]. Efforts to understand the modulation of mHTT solubility have identified ubiquitin-conjugating enzyme (E2 family) Ube2W as at least one of the key players. Ube2W targets the amino terminus of protein substrates, where the polyQ expansion occurs. Ube2W−/− neurons with mHTT exposure experienced an increased death rate but had fewer IBs and more soluble HTT [182]. Hsp70 was decreased under these conditions but other Hsps (40, 60, and 90) and autophagy protein p62 were unchanged. It is important to note that Ube2W contains an active site Cys required for ubiquitin conjugation; increased levels of oxidants experienced during aging could impair this enzyme and exacerbate neurodegeneration associated with polyQ expansion.
Studies of IB components have identified mHTT, oftentimes ubiquitinated, along with ubiquitin and members of the proteasomal and autophagic systems as localizing in these structures [183, 184]. Once formed, IBs are detrimental to the proteostasis system; not only are proteins sequestered from their functional location, but IBs disrupt the chaperone system, bind and repress transcription factors, impair the UPS, and stunt proteolysis, leading to cell death [185]. Mouse models of HD have revealed diminished proteasome activity in several brain regions along with a dampened HSR [186, 187]. Additionally, studies in HD patients and mouse models demonstrate decreased neuronal proteasome activity in comparison to tissues not expressing mHTT (reviewed in [188]). Unfortunately, treatment options for HD are mainly limited to symptom management, such as alleviating chorea and improving mood. Tissue transglutaminase inhibition has been shown to decrease polyQ aggregation [189]; these, along with immune-based strategies [190] are being pursued as potential treatments for HD patients.
5. Proteostasis disruptions in other age-related diseases
5.1. Cardiovascular diseases
Cardiovascular disease is among the leading causes of mortality worldwide. Alterations of the mechanisms of proteostasis in cardiovascular diseases have been extensively and recently reviewed [191]. Common phenotypes observed in cardiovascular disease involve loss of protein patency in the heart resulting from genetic dysregulation and/or environmental factors triggering cardiac aging. Disease progression also involves impairment of chaperones, the UPS, autophagy, mitophagy, and loss of sarcomeric and cytoskeletal proteins, all leading to cardiomyocyte senescence [191].
Mitophagy in the heart is an essential process of both normal development and a gatekeeper against cardiovascular disease. Maturity of cardiac cells and tissue from the neonate to adulthood is marked by a shift from glycolysis to mitochondrial oxidative metabolism [192] facilitated by robust mitophagy and not metabolic reprogramming [193]. The high energy demands of the adult heart manifest in an increased mitochondrial capacity and thus an increased reliance for highly functional quality control systems [194]. The Parkin/PINK1 pathway has been implicated as essential for cardiac function. As just one example, PINK1 −/− mice experienced ventricular dysfunction and cardiac hypertrophy by 2 months of age.
Characteristics of heart tissue in these mice also included increased fibrosis and oxidative stress, impaired mitochondrial function, and enhanced levels of apoptosis in the cardiomyocytes [195]. However, more recent studies have questioned such observations, revealing that in basal mitophagy processes, the Parkin/PINK1 pathway is dispensable [196], but is crucial in stress responses in the heart. For example, increased expression of Parkin leads to enhanced mitophagy, presumably for cardioprotection, in mouse cardiomyocytes exposed to stress via myocardial infarction [194]. Additionally, decreased levels of PINK1 have been noted in the left ventricle of end-stage heart failure patients [195]. A more expansive look at the role of mitophagy in cardiovascular disease is found in a recent review [197]. Additional work to come in this area will further elucidate the roles of Parkin/PINK1 and mitophagy as a whole in the heart, along with identifying the particular details of how this pathway is induced in response to age and cardiac-specific stressors.
Protein aggregation is a key feature of some cardiac diseases, such as heart failure, in a matter similar to NDDs where protein misfolding or lack of folding facilitates the binding and aggregation of hydrophobic regions, impairing normal clearance mechanisms and interfering with proper organ function. In the context of heart failure, these aggregates form structures called cardiac preamyloid oligomers (PAOs). A very recent study identified phosphorylation of the protein desmin at Ser31 as a molecular seed for the formation of PAO structures observed in heart failure in both canines and humans when desmin’s primary chaperone is absent or non-functional [198]. Monophosphorylation prevents phosphorylation at two other desmin residues by kinase GSK3 to provide the soluble, physiological proteoform, thus singly phosphorylated desmin is more susceptible to aggregation [199]. Additionally, positron emission tomography was established for the first time as a suitable imaging technique for the visualization of PAOs in mice [198].
Similarly, impairment of proteostasis has been associated with multiple (age-related) respiratory pathologies, including pulmonary hypertension, chronic obstructive pulmonary disease, cystic fibrosis and pulmonary fibrosis, as extensively reviewed [200-203]. Of note, individuals with DS are more susceptible to develop comorbidities associated with cognitive impairment and altered proteostasis, such as Alzheimer’s disease, but also respiratory complications and pulmonary hypertension [204].
5.2. Ocular pathologies linked to aging and imbalanced proteostasis
Age-related diseases with clear associations to the proteostasis network also affect the eye. The formation of cataracts, or opaque lenses, leads to impaired vision affecting, disproportionately, the aging population and requires surgical replacement. The lens is comprised predominantly of crystallin, an 80 kDa protein that packs into complexes of approximately 800 kDa which assemble in an ordered manner to form the lens. The concentration of crystallin in the lens is on the order of 400 mg/mL, the highest protein concentration of any tissue [205]. A stringent molecular order is required to ensure lens transparency and the passage of light through the lens and cornea to reach photoreceptors in the retina. Lens proteostasis is therefore paramount to function but as post-mitotic cells must maintain transparency, much of the proteostasis machinery and cellular organelles are absent (reviewed in [206]). In addition to its structural role in the lens, crystallin is a chaperone member of the small Hsp family and its holdase activity helps to maintain the integrity of the lens over years of light exposure and normal aging, however, this protein is not immune to the accumulation of PTMs over time [205]. Such PTMs include oxidative damage, glycation, deamidation, phosphorylation, and others, ultimately decreasing crystallin solubility and leading to its aggregation [207-209]. Crystallin aggregation scatters light and causes sustained ER UPR activation, and leading to apoptosis and visual impairment [210]. Proteostasis is a crucial factor in other eye pathologies as well. Mutations in photoreceptor protein rhodopsin cause the misfolding and aggregation that underlie retinitis pigmentosa; well over 100 mutations are known. Additionally, A accumulation plays key roles in the pathologies of glaucoma and age-related macular degeneration. Eye diseases mediated by disrupted proteostasis have been recently reviewed in detail [208].
5.3. Proteostasis in cancer
Our final area of focus will examine the role of proteostasis in cancer development and progression. Carcinogenesis occurs following mutations in tumor suppressor genes and/or activation of oncogenes that are propagated and expanded as the affected cells experience a rewiring of gene expression, metabolism, and cell growth. In contrast to aging, where proteostasis mechanisms are downregulated and lose efficiency, cancer cells and tumors demonstrate markedly increased and more capable proteostasis networks utilized to support rapid growth and evade normal aging and cell death mechanisms. Enhanced proteostasis in cancer is a culmination of adaptations throughout the protein machinery (which have been recently reviewed in detail); cancerous tumors have increased chaperone activity [211], increased levels of telomerase to delay chromosome aging [212], upregulated UPR (ER) [213], overexpression of folding protein PDI [214], and a strong reliance on the lysosome-autophagy system [215]. Proteolysis is also manipulated to allow for continual cell division and evasion of checkpoint mechanisms.
A critical interaction of proteostasis and cancer progression lies in the homeostasis of transcription factors that respond to stresses, alter metabolism, and affect cell lifespan. Modulation of transcription factors such as tumor suppressor p53, HSF-1, forkhead box (Fox) proteins, and Nrf2 through altered expression or mutations can disrupt normal proteostasis and facilitate carcinogenesis [216, 217]. For example, levels of Nrf2, which induces the expression of chaperones, proteasome components, and antioxidant enzymes, are increased in many cancers [217, 218]. Decreased p53 levels allow for the propagation of malignant cells, and inactivation of p53 by a corrupted chaperone system has been shown to facilitate angiogenesis via vascular endothelial growth factor (VEGF) and nitric oxide synthase (NOS) [211] (see [219] for a recent review of the interplay between proteostasis and angiogenic factors). Mutations in p53 are many and are well-documented to underlie numerous cancers. In a similar manner as for many NDDs, mutations in this protein can lead to aggregation and the formation of nuclear IBs, as has been shown for at least 6 cancer types [220]. Nuclear IBs can activate HSF-1 and also decrease function of the proteasome [220]. Proteasome dynamics is crucial for regulating cellular concentrations of transcription factors, as these proteins are short lived and degraded by the UPS following activation of DNA promoter regions [221]. Cells have the capacity to turn off or decrease proteolysis under stress conditions to extend the life of transcription factors involved in stress responses; this is manipulated in cancer cells and underlies many of their well-known growth characteristics.
By affecting the interplay between redox poise and anabolic reactions, activation of the pentose phosphate pathway by p53 family members (e.g., p63 [222]) and upstream kinases such as ATM [223] influences the cellular antioxidant potential and impacts ATP generation and drug resistance capacity in acute myeloid leukemia [224], a disease of older people uncommon before age 45 (average patient age is 67) according to the American Cancer Society. Mutations to ATM, such as in ataxia telangiectasia result in premature aging syndrome [225] and altered proteostasis [226].
Proteasome inhibition, once a limited strategy for cancer treatment due to cross-reactivity with healthy cells, is again gaining attention. Bortezomib, the first FDA-approved anti-cancer therapy targeting proteostasis, is used to treat multiple myeloma and mantle cell lymphoma by inhibiting the 26S proteasome [227, 228]. Unfortunately, bortezomib alone is not effective against solid tumors and even hematological cancers can develop resistance. Bortezomib when used in conjunction with nutlin-3 is efficacious against cell lines of solid tumors by disrupting proteostasis in the ER and mitochondria [228]. Inhibition of E3 ubiquitin ligases is also utilized for the treatment of hematological cancers [221]. Recently, Hsp70 inhibitors showed success in treating cancer by impairing mitochondrial proteostasis, exploiting the increased expression of stress-induced Hsp70 in cancer cells compared to normal cells [229]. Lastly, quinolone-8-thiol (8TQ) and capzimin have been described as disrupting ubiquitination leading to cell death in several cancer cell lines [230].
6. Omics techniques for assessing proteostasis during aging and in disease
Preservation or enhancement of proteostasis maintenance mechanisms throughout life improves resistance to stress and is sufficient to slow down aging [231]. Recent research, however, has only begun to elucidate the age-dependent changes that modulate lifespan, susceptibility to disease, and response to treatment. These studies are challenging due to heterogeneous aging processes, difficulties associated with implementing longitudinal studies that can compensate for biological variability (i.e., sampling from the same organism over time versus concurrent sampling from young and old populations), and the lack of methods sensitive enough to quantify subtle proteomic changes. Integrated omics approaches that combine data from genomics, transcriptomics, proteomics, and metabolomics studies to understand proteostasis in aging and age-associated disease hold enormous promise, but remain technically challenging and require large, focused teams of diversely skilled researchers.To date, few proteomics studies have broadly profiled aging in mammalian tissues. Previous transcriptomic and proteomic studies have revealed that aging in rats differentially affects tissues and organs [232]. Perhaps the most in-depth proteomic profile of systemic aging was derived from work that utilized stable isotope labeled amino acids in cell culture (SILAC) quantitation and high resolution mass spectrometry (MS) to compare global proteomes of mice aged 5 or 26 months [233]. More than 4000 proteins were quantified, representing the majority of tissue protein mass, though the expression levels many proteins were unchanged. Other studies focused on tissue-specific aging have used alternative proteomic approaches such as two dimensional gel electrophoresis or iTRAQ labeling and matrix-assisted laser desorption/ionization (MALDI)-based quantitative MS to study the effects of senescence on the left rat heart ventricle, finding differential expression of metabolic enzymes, structural and antioxidant proteins [234]. Additionally, a single proteomic time-course study of the aging mouse brain found that aging is indeed associated with a reduction in the abundance of proteasomal subunits and an accumulation of non-functional proteins and fragments [235]. While the findings of these studies have been informative, they have been somewhat limited by data-dependent LC-MS/MS approaches which are biased toward the detection of the most abundant biomolecules in a complex mixture.
A recent surge of advances in quantitative proteomics has resulted in both greatly improved experimental capabilities and, more importantly, the appreciation of this scientific approach to provide fundamentally new knowledge. Thus, while relative quantitation approaches are appropriate for discovery studies aimed at identifying markers of the aging proteome, absolute quantification of proteomic changes using isotope-labeled internal standard peptides (e.g., AQUA or quantitative concatamer - QconCAT) have begun to show early success. For example, we have utilized the QconCAT approach to characterize extracellular matrix (ECM) proteins that have been implicated in diseases that disproportionately affect aging populations, such as cancer, idiopathic pulmonary fibrosis (IPF), and cardiac disease [200-202]. Additional studies that have applied compartment-resolved ECM proteomics to identify novel modulators of pulmonary fibrosis in the aging murine lung have revealed that compartment-specific ECM proteins are dynamically regulated upon lung injury and repair [236, 237]. In another area of specialized proteomics, Perluigi et al. utilized redox proteomics in the analysis of human brains to reveal a disturbance in both proteostasis and autophagy mechanisms in DS. Indeed, others have shown that oxidative stress occurs early in life and can even be detected in the amniotic fluid of a DS pregnancy [238].
Covalent cross-linking of proteins provides the structural meshwork needed to support resident cells and tissues and is altered with age and disease. Cross-links are formed enzymatically [239] or via chemical reactions driven by advanced glycation end products (AGEs), and may contribute to tissue aging [240-242]. While proteomic approaches aimed at characterizing ECM composition have shown success in identifying new proteomic signatures [243-246], methods to characterize protein-protein cross-links in the ECM have yet to be widely adopted for analyzing tissue samples. Identification of these interactions is important given the role that fibrillar collagen, the most abundant component of the ECM and a common target for cross-linking, plays in providing the structural support for tissues. Importantly, past analyses of AGEs and collagen cross-links in human bone and articular cartilage have identified age-related changes in the content of these important biomolecules, implying a possible mechanism by which age serves as a risk factor for osteoarthritis [247, 248]. For instance, in both bone and cartilage, mature crosslinks such as lysyl pyridinoline reach a maximum by 10–15 years of age [249]. In contrast, immature cross-links in bone collagen decrease sharply in abundance between birth and 25 years, but still persist through adult life. While mature crosslinks increase, total crosslinks (i.e., immature and mature) decrease overall with age [249]. This effect may help to explain, in part, why the bones in older individuals are stiffer but actually more brittle, and thus more likely to fracture under stress. In order to apply crosslinking analysis to softer, less collagenous tissues (i.e., breast, brain and solid tumors) the development of more sensitive MS-based approaches will likely be necessary. Despite documented ECM composition changes occurring during the development and progression of NDDs [250, 251] (i.e., Alzheimer’s, Parkinson’s, Huntington’s, Down syndrome), the role of crosslinking during disease progression is currently unknown and thus, development of a sensitive MS-based approach would dramatically expand upon knowledge in this area.
7. Expert commentary
In the book Nature’s Robots: A History of Proteins, Tanford and Reynolds outline a fascinating parallel between proteins and automatons that perform programmed functions [252]. Indeed, protein “robots” are key structural and functional components involved in all biological activities from signal transmission to immune function, from vision to mobility. As such, it is fascinating to note how Asimov’s famous “Three Laws of Robotics” [253] may be applied to proteins, whereas a close check of protein half-life and homeostasis is relevant to the health of the whole individual. This is even more relevant when considering the emerging correlation between human aging and dysregulated protein homeostasis in erythrocytes, the most abundant host cell in the human body that is devoid of nuclei and organelles (i.e., lacking de novo protein synthesis capacity) and thus relying on calcium-dependent or ubiquitin-proteasome-dependent proteolytic activity to regulate its lifespan and function [254]. Of all cell types, erythrocytes are most exposed to chronic oxidative stress due to their oxygen carrier function and abundance of iron loaded-hemoglobin, which catalyzes Fenton and Haber-Weiss reactions during aging. Increased red blood cell clearance [255] and impaired hematopoiesis towards the myeloid lineage [256] contribute to anemia, which serves as a hallmark of aging and inflammation. Of note, red blood cell lifespan decreases with age, and many age-associated diseases are characterized by a reduced lifespan of erythrocytes (e.g., Down syndrome [254]) and increased oxidative stress (e.g., senile dementia [257]). In addition, alterations of iron homeostasis and onset of iron-dependent mechanisms of non-apoptotic cell death (i.e., ferroptosis) have been increasingly appreciated in aging/altered proteostasis-associated diseases such as Alzheimer’s disease [258]. As red blood cells play a key function in oxygen transport and delivery, it is fascinating to hypothesize an etiological contribution of pathological proteostasis (in red blood cell biology) and increased ferroptosis as a hallmark of respiratory complications, which are common comorbidities in the elderly population. Indeed, a role has been recently highlighted for inflammatory complications and altered proteostasis in many lung diseases, such as cystic fibrosis, idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease (as extensively reviewed [203]).
8. Five-year view
The introduction of (high-throughput) quantitative omics approaches has provided new toolsets to investigate protein homeostasis and dysfunction in health and disease. Advancements in proteomics protocols targeting the insoluble proteome fraction have expanded our capacity to explore protein and peptide cross links with unprecedented specificity and sensitivity [243]. As novel bioinformatics tools are continuously being developed to identify protein native or chemically-induced cross-links to probe protein structures [259], advances in our understanding of aging and proteostasis-related diseases are expected in the near future. Such tools have already been implemented with success to investigate, for example, dysregulated extracellular matrix deposition and crosslinking which underlies tissue fibrosis at different stages of pancreatic cancer [246]. It is thus easy to anticipate that similar applications will be crucial in the next five years of studying tissue fibrosis in cancer, pulmonology, (skin and organ) aging, inflammaging and red blood cell senescence (especially in patients with neurocognitive impairment).
Key issues.
Despite knowledge that the biological process of aging is believed to be the result of an accumulation of cellular damage to biomolecules, quantitative information from high-throughput proteomics on physiological concentrations and ratios of polypeptides in various compartments of the human cell during the aging remains understudied.
While impaired proteostasis has been clearly associated with numerous age-related diseases, it is unclear whether and to what extent molecular salvage mechanisms may ensue to prevent the onset of such comorbidities. For example, while accumulation of amyloid protein aggregates has been identified as an etiological factor of Alzheimer’s disease, impaired protein homeostasis (i.e., extra copies) of amyloid precursor protein (coded by a gene on chromosome 21) do not necessarily result in Alzheimer’s disease in the Trisomy 21 (Down syndrome) population.
Mechanisms of ferroptosis and their potential mechanistic involvement in dysregulated protein homeostasis have not been yet extensively investigated.
Omics technologies and bioinformatics tools to enrich (e.g., high-pH reversed-phase fractionation [260]) and characterize protein/peptide/amino acid cross-links have been introduced only in recent years. Though promising, the application of these tools to the study of altered proteostasis in aging and disease is only in its infancy.
Acknowledgments
Funding
This article was supported by funds from the Linda Crnic Institute for Down Syndrome, the 2017 Webb-Waring Biomedical Research Award sponsored by the Boettcher Foundation (both to A. D’Alessandro) and by U.S National Institutes of Health grant: NIH T32 HL007171 (to A.S. Barrett).
Footnotes
Declaration of interest
Though unrelated to the contents of the manuscript, the authors disclose that A. D’Alessandro, T. Nemkov, K.C. Hansen are part of Omix Technologies, Inc. A. D’Alessandro is also an advisory board member for New Health Sciences, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Squier TC. Oxidative stress and protein aggregation during biological aging. Exp Gerontol. 2001;36(9):1539–1550. [DOI] [PubMed] [Google Scholar]
- 2.Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22(17):R741–R752. [DOI] [PubMed] [Google Scholar]
- 3.Wilson PW, D’Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97(18):1837–1847. [DOI] [PubMed] [Google Scholar]
- 4.Mariani E, Polidori M, Cherubini A, et al. Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chrom B 2005;827(1):65–75. [DOI] [PubMed] [Google Scholar]
- 5.Rodríguez-Rodero S, Fernández-Morera JL, Menéndez-Torre E, et al. Aging genetics and aging. Aging Dis. 2011;2(3):186. [PMC free article] [PubMed] [Google Scholar]
- 6.Powers ET, Morimoto RI, Dillin A, et al. Biological and chemical approaches to diseases of proteostasis deficiency. Ann Rev Biochem. 2009;78:959–991. [DOI] [PubMed] [Google Scholar]
- 7.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. [DOI] [PubMed] [Google Scholar]
- 8.Shaw AC, Joshi S, Greenwood H, et al. Aging of the innate immune system. Curr Opin Immunol. 2010;22(4):507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talens RP, Christensen K, Putter H, et al. Epigenetic variation during the adult lifespan: cross‐sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11(4):694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moskalev AA, Shaposhnikov MV, Plyusnina EN, et al. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev. 2013;12(2):661–684. [DOI] [PubMed] [Google Scholar]
- 11.Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010;11(8):567–578. [DOI] [PubMed] [Google Scholar]
- 12.Hoeijmakers JH. DNA damage, aging, and cancer. New Eng J Med 2009;361(15):1475–1485. [DOI] [PubMed] [Google Scholar]
- 13.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science. 2011;333(6046):1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harman D The free radical theory of aging: Effect of age on serum copper levels. J Gerontol. 1965;20(2):151–153. [DOI] [PubMed] [Google Scholar]
- 15.López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153(6):1194–1217.**This seminal paper is the most comprehensive publication to date that dissects the connections between the candidate hallmarks and their relative contributions to aging with the final goal of identifying pharmaceutical targets to improve human health during aging.
- 16.Sastre J, Pallardo FV, Garcia de la Asuncion J, et al. Mitochondria, oxidative stress and aging. Free Radic Res. 2000;32(3):189–198. [DOI] [PubMed] [Google Scholar]
- 17.Dai D-F, Chiao YA, Marcinek DJ, et al. Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan. 2014;3:6–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Alessandro A, El Kasmi KC, Plecita-Hlavata L, et al. Hallmarks of pulmonary hypertension: Mesenchymal and inflammatory cell metabolic reprogramming. Antioxid Redox Signal. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reisz JA, Bansal N, Qian J, et al. Effects of ionizing radiation on biological molecules: Mechanisms of damage and emerging methods of detection. Antioxid Redox Signal. 2014;21(2):260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ott C, Grune T. Protein oxidation and proteolytic signalling in aging. Curr Pharm Des. 2014;20(18):3040–3051. [DOI] [PubMed] [Google Scholar]
- 21.Srivastava S The mitochondrial basis of aging and age-related disorders. Genes (Basel). 2017;8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan Y, Zheng Y, Zhang X, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13(10):1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97(16):9082–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Paik SG. Regulation of BNIP3 in normal and cancer cells. Mol Cells. 2006;21(1):1–6. [PubMed] [Google Scholar]
- 26.Greene AW, Grenier K, Aguileta MA, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13(4):378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem. 2008;106(1):464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin SM, Lazarou M, Wang C, et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191(5):933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441(7097):1157–1161. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Zhang C, Zhao Y, et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun. 2017;8(1):1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Popa-Wagner A, Sandu RE, Cristin C, et al. Increased degradation rates in the components of the mitochondrial oxidative phosphorylation chain in the cerebellum of old mice. Front Aging Neurosci. 2018;10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drake JC, Yan Z. Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J Physiol. 2017;595(20):6391–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JJ, Sanchez-Martinez A, Zarate AM, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27(2):439–449.e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh CK, Sultan A, Platzer J, et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017;21(8):2171–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nemkov T, Sun K, Reisz JA, et al. Hypoxia modulates the purine salvage pathway and decreases red blood cell and supernatant levels of hypoxanthine during refrigerated storage. Haematologica. 2017, ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17(7):1807–1819. [DOI] [PubMed] [Google Scholar]
- 38.Hatle KM, Gummadidala P, Navasa N, et al. MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol. 2013;33(11):2302–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Champagne DP, Hatle KM, Fortner KA, et al. Fine-tuning of CD8(+) T cell mitochondrial metabolism by the respiratory chain repressor MCJ dictates protection to influenza virus. Immunity. 2016;44(6):1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milenkovic D, Blaza JN, Larsson N-G, et al. The enigma of the respiratory chain supercomplex. Cell Metab. 2017;25(4):765–776. [DOI] [PubMed] [Google Scholar]
- 41.Hadizadeh Esfahani A, Sverchkova A, Saez-Rodriguez J, et al. A systematic atlas of chaperome deregulation topologies across the human cancer landscape. PLoS Comput Biol. 2018;14(1):e1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dayalan Naidu S, Dinkova-Kostova AT. Regulation of the mammalian heat shock factor 1. FEBS J 2017;284(11):1606–1627.*A review of HSF-1 and its key role in proteostasis maintenance
- 43.Solis EJ, Pandey JP, Zheng X, et al. Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol Cell. 2016;63(1):60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Alessandro A, Righetti PG, Zolla L. The red blood cell proteome and interactome: an update. J Proteome Res. 2010;9(1):144–163. [DOI] [PubMed] [Google Scholar]
- 45.Kalmar B, Greensmith L, Section I: Heat shock proteins and neurodegenerative diseases, in Heat shock proteins and the brain: Implications for neurodegenerative diseases and neuroprotection, IR B, Editor. 2008, Springer: Dordrecht. [Google Scholar]
- 46.Carreras-Sureda A, Pihan P, Hetz C. Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium. 2017, ahead of print. [DOI] [PubMed] [Google Scholar]
- 47.Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015;5(6):586–597. [DOI] [PubMed] [Google Scholar]
- 48.Kalies KU, Allan S, Sergeyenko T, et al. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 2005;24(13):2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–96. [DOI] [PubMed] [Google Scholar]
- 50.Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355(6355):33–45. [DOI] [PubMed] [Google Scholar]
- 51.Schuck S, Prinz WA, Thorn KS, et al. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187(4):525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–335. [DOI] [PubMed] [Google Scholar]
- 53.Frakes AE, Dillin A. The UPRER: Sensor and coordinator of organismal homeostasis. Mol Cell. 2017;66(6):761–771. [DOI] [PubMed] [Google Scholar]
- 54.Kristensen CM, Brandt CT, Ringholm S, et al. PGC-1alpha in aging and lifelong exercise training-mediated regulation of UPR in mouse liver. Exp Gerontol. 2017;98:124–133. [DOI] [PubMed] [Google Scholar]
- 55.Han J, Kaufman RJ. Measurement of the unfolded protein response to investigate its role in adipogenesis and obesity. Methods Enzymol. 2014;538:135–150. [DOI] [PubMed] [Google Scholar]
- 56.van der Bliek AM, Sedensky MM, Morgan PG. Cell biology of the mitochondrion. Genetics. 2017;207(3):843–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83(3–4):301–310. [DOI] [PubMed] [Google Scholar]
- 58.Gupta I, Singh K, Varshney NK, et al. Delineating crosstalk mechanisms of the ubiquitin proteasome system that regulate apoptosis. Front Cell Dev Biol. 2018;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi J, Baek KH. Cellular functions of stem cell factors mediated by the ubiquitin-proteasome system. Cell Mol Life Sci. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saez I, Koyuncu S, Gutierrez-Garcia R, et al. Insights into the ubiquitin-proteasome system of human embryonic stem cells. Sci Rep. 2018;8(1):4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Catarino S, Pereira P, Girao H. Molecular control of chaperone-mediated autophagy. Essays Biochem. 2017;61(6):663–674. [DOI] [PubMed] [Google Scholar]
- 62.Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77:57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boya P, Codogno P, Rodriguez-Muela N. Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development. 2018;145(4). [DOI] [PubMed] [Google Scholar]
- 64.Pietrocola F, Izzo V, Niso-Santano M, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23(5):310–322. [DOI] [PubMed] [Google Scholar]
- 65.Galluzzi L, Pietrocola F, Levine B, et al. Metabolic control of autophagy. Cell. 2014;159(6):1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16(8):461–472. [DOI] [PubMed] [Google Scholar]
- 67.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. [DOI] [PubMed] [Google Scholar]
- 68.Rybstein MD, Bravo-San Pedro JM, Kroemer G, et al. The autophagic network and cancer. Nat Cell Biol. 2018;20(3):243–251. [DOI] [PubMed] [Google Scholar]
- 69.Labbadia J, Morimoto RI. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol Cell. 2015;59(4):639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang S, Eitan E, Mattson MP. Early involvement of lysosome dysfunction in the degeneration of cerebral cortical neurons caused by the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 2017;140(6):941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hohn A, Jung T, Grimm S, et al. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radic Biol Med. 2011;50(5):585–591. [DOI] [PubMed] [Google Scholar]
- 72.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275(40):31505–31513. [DOI] [PubMed] [Google Scholar]
- 73.Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol. 2005;40(8–9):622–633. [DOI] [PubMed] [Google Scholar]
- 74.Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447(7146):859–863. [DOI] [PubMed] [Google Scholar]
- 75.Ji CH, Kwon YT. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells. 2017;40(7):441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoo YD, Mun SR, Ji CH, et al. N-terminal arginylation generates a bimodal degron that modulates autophagic proteolysis. Proc Natl Acad Sci U S A. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Das R, Melo JA, Thondamal M, et al. The homeodomain-interacting protein kinase HPK-1 preserves protein homeostasis and longevity through master regulatory control of the HSF-1 chaperone network and TORC1-restricted autophagy in Caenorhabditis elegans. PLoS Genetics. 2017;13(10):e1007038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Finkel T The metabolic regulation of aging. Nat Med. 2015;21(12):1416–1423.**In-depth review of age-related changes in metabolism
- 79.Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herzig S, Shaw RJ. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu H, Zhang Y, Wu H, et al. Beneficial role of erythrocyte adenosine A2B receptor-mediated AMP-activated protein kinase activation in high-altitude hypoxia. Circulation. 2016;134(5):405–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burkewitz K, Zhang Y, Mair WB. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20(1):10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kennedy BK, Lamming DW. The mechanistic target of rapamycin: The grand conducTOR of metabolism and aging. Cell Metab. 2016;23(6):990–1003.**Recent review of rapamycin’s role in increasing lifespan.
- 84.Cavalli G, Justice JN, Boyle KE, et al. Interleukin 37 reverses the metabolic cost of inflammation, increases oxidative respiration, and improves exercise tolerance. Proc Natl Acad Sci U S A. 2017;114(9):2313–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kusano T, Yamaguchi K, Berberich T, et al. Advances in polyamine research in 2007. J Plant Res. 2007;120(3):345–350. [DOI] [PubMed] [Google Scholar]
- 86.Eisenberg T, Knauer H, Schauer A, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11(11):1305–1314. [DOI] [PubMed] [Google Scholar]
- 87.Minois N, Carmona-Gutierrez D, Madeo F. Polyamines in aging and disease. Aging (Albany NY). 2011;3(8):716–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gupta VK, Scheunemann L, Eisenberg T, et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat Neurosci. 2013;16(10):1453–1460. [DOI] [PubMed] [Google Scholar]
- 89.Casero RA, Jr., Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6(5):373–390. [DOI] [PubMed] [Google Scholar]
- 90.Atiya Ali M, Poortvliet E, Stromberg R, et al. Polyamines in foods: development of a food database. Food Nutr Res. 2011;55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ng F, Tang BL. Sirtuins’ modulation of autophagy. J Cell Physiol. 2013;228(12):2262–2270. [DOI] [PubMed] [Google Scholar]
- 92.Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metab. 2015;22(1):31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24(8):464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van der Goot AT, Nollen EA. Tryptophan metabolism: entering the field of aging and age-related pathologies. Trends Mol Med. 2013;19(6):336–344. [DOI] [PubMed] [Google Scholar]
- 95.Ma S, Yim SH, Lee SG, et al. Organization of the mammalian metabolome according to organ function, lineage specialization, and longevity. Cell Metab. 2015;22(2):332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rampelli S, Candela M, Turroni S, et al. Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging. 2013;5(12):902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Franceschi C, Bonafe M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. [DOI] [PubMed] [Google Scholar]
- 98.Franceschi C, Garagnani P, Vitale G, et al. Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab. 2017;28(3):199–212. [DOI] [PubMed] [Google Scholar]
- 99.Cuervo A, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24(1):92–104.**A detailed review of chaperone-mediated autophagy with special focus on cancer and neurodegenerative diseases.
- 100.Chondrogianni N, Gonos ES. Proteasome function determines cellular homeostasis and the rate of aging. Adv Exp Med Biol. 2010;694:38–46. [DOI] [PubMed] [Google Scholar]
- 101.Giunta B, Fernandez F, Nikolic WV, et al. Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation. 2008;5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boren E, Gershwin ME. Inflamm-aging: autoimmunity, and the immune-risk phenotype. Autoimmun Rev. 2004;3(5):401–406.*The initial paper coining inflammaging, describing the aging immune system and development of autoimmune diseases.
- 103.Prattichizzo F, De Nigris V, La Sala L, et al. “Inflammaging” as a druggable target: A senescence-associated secretory phenotype-centered view of type 2 diabetes. Oxid Med Cell Longev. 2016;2016:1810327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lencel P, Magne D. Inflammaging: the driving force in osteoporosis? Med Hypotheses. 2011;76(3):317–321. [DOI] [PubMed] [Google Scholar]
- 105.Henry CJ, Marusyk A, DeGregori J. Aging-associated changes in hematopoiesis and leukemogenesis: What’s the connection? Aging. 2011;3(6):643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Henry CJ, Casás-Selves M, Kim J, et al. Aging-associated inflammation promotes selection for adaptive oncogenic events in B cell progenitors. J Clin Invest. 2015;125(12):4666–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nemkov T, Hansen KC, D’Alessandro A. A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun Mass Spectrom. 2017;31(8):663–673.*Description of a high-throughput metabolics approach useful for metabolic flux experiments and profiling metabolism in many biological contexts.
- 108.Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. 2017. [DOI] [PubMed] [Google Scholar]
- 109.Iannaccone M, Titta F, Serretiello E, et al. Possible physiopathological effects of the transglutaminase activity on the molecular mechanisms responsible for human neurodegenerative diseases. Recent Pat CNS Drug Discov. 2014;9(2):76–84. [DOI] [PubMed] [Google Scholar]
- 110.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14(2):45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Poon HF, Farr SA, Thongboonkerd V, et al. Proteomic analysis of specific brain proteins in aged SAMP8 mice treated with alpha-lipoic acid: implications for aging and age-related neurodegenerative disorders. Neurochemistry international. 2005;46(2):159–168. [DOI] [PubMed] [Google Scholar]
- 112.Penke B, Bogar F, Fulop L. beta-Amyloid and the pathomechanisms of Alzheimer’s disease: A comprehensive view. Molecules. 2017;22(10).*Nicely details the effects - positive and negative - of increased APP expression induced by stress.
- 113.Muller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. 2017;18(5):281–298. [DOI] [PubMed] [Google Scholar]
- 114.Mockett BG, Richter M, Abraham WC, et al. Therapeutic potential of secreted amyloid precursor protein APPsalpha. Front Mol Neurosci. 2017;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hefter D, Draguhn A. APP as a protective factor in acute neuronal insults. Front Mol Neurosci. 2017;10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Manczak M, Kandimalla R, Yin X, et al. Hippocampal mutant APP and amyloid beta induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 117.Friedrich RP, Tepper K, Ronicke R, et al. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A. 2010;107(5):1942–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Manzoni C, Colombo L, Bigini P, et al. The molecular assembly of amyloid abeta controls its neurotoxicity and binding to cellular proteins. PLoS One. 2011;6(9):e24909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Penke B, Toth AM, Foldi I, et al. Intraneuronal beta-amyloid and its interactions with proteins and subcellular organelles. Electrophoresis. 2012;33(24):3608–3616. [DOI] [PubMed] [Google Scholar]
- 120.Cheignon C, Tomas M, Bonnefont-Rousselot D, et al. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2017;14:450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Volgyi K, Haden K, Kis V, et al. Mitochondrial proteome changes correlating with beta-amyloid accumulation. Mol Neurobiol. 2017;54(3):2060–2078. [DOI] [PubMed] [Google Scholar]
- 122.Lai CS, Preisler J, Baum L, et al. Low molecular weight Abeta induces collapse of endoplasmic reticulum. Mol Cell Neurosci. 2009;41(1):32–43. [DOI] [PubMed] [Google Scholar]
- 123.Luo J, Warmlander SK, Graslund A, et al. Cross-interactions between the Alzheimer Disease amyloid-beta peptide and other amyloid proteins: A further aspect of the amyloid cascade hypothesis. J Biol Chem. 2016;291(32):16485–16493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Groh N, Buhler A, Huang C, et al. Age-dependent protein aggregation initiates amyloid-beta aggregation. Front Aging Neurosci. 2017;9:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. [DOI] [PubMed] [Google Scholar]
- 126.Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wolfe DM, Lee JH, Kumar A, et al. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37(12):1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Safra M, Ben-Hamo S, Kenyon C, et al. The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans. J Cell Sci. 2013;126(Pt 18):4136–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohta K, Mizuno A, Li S, et al. Endoplasmic reticulum stress enhances gamma-secretase activity. Biochem Biophys Res Commun. 2011;416(3–4):362–366. [DOI] [PubMed] [Google Scholar]
- 130.Rahman S, Archana A, Jan AT, et al. Dissecting endoplasmic reticulum unfolded protein response (UPR(ER)) in managing clandestine modus operandi of Alzheimer’s disease. Front Aging Neurosci. 2018;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martin-Maestro P, Gargini R, Garcia E, et al. Slower dynamics and aged mitochondria in sporadic Alzheimer’s disease. Oxid Med Cell Longev. 2017;2017:9302761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kiko T, Nakagawa K, Satoh A, et al. Amyloid β levels in human red blood cells. PLoS One. 2012;7(11):e49620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kaminsky Y, Poghosyan A, Tikhonova L, et al. Glycolytic and proteolytic metabolism in erythrocytes from elderly and demented patients. Am J Neuroprot Neuroregen. 2012;4(1):73–77. [Google Scholar]
- 134.Kosenko EA, Aliev G, Tikhonova LA, et al. Antioxidant status and energy state of erythrocytes in Alzheimer dementia: probing for markers. CNS Neurol Disord Drug Targets. 2012;11(7):926–932. [DOI] [PubMed] [Google Scholar]
- 135.Kaminsky YG, Reddy VP, Ashraf GM, et al. Age-related defects in erythrocyte 2,3-diphosphoglycerate metabolism in dementia. Aging Dis. 2013;4(5):244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aliev G, Palacios HH, Walrafen B, et al. Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int J Biochem Cell Biol. 2009;41(10):1989–2004. [DOI] [PubMed] [Google Scholar]
- 137.Cenini G, Dowling AL, Beckett TL, et al. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta. 2012;1822(2):130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nistor M, Don M, Parekh M, et al. Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain. Neurobiol Aging. 2007;28(10):1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Webb RL, Murphy MP. beta-Secretases, Alzheimer’s disease, and Down syndrome. Curr Gerontol Geriatr Res. 2012;2012:362839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Head E, Helman A, Powell D, et al. Down syndrome, beta-amyloid and neuroimaging. Free Radic Biol Med. 2017.*Review of amyloid beta in Down syndrome, including its presence, modifications, and approaches for detection.
- 141.Jenkins EC, Marchi EJ, Velinov MT, et al. Longitudinal telomere shortening and early Alzheimer’s disease progression in adults with down syndrome. Am J Med Genet B Neuropsychiatr Genet. 2017. [DOI] [PubMed] [Google Scholar]
- 142.Aivazidis S, Coughlan C, Rauniyar A, et al. The burden of trisomy 21 disrupts the proteostasis network in Down syndrome. PLoS One. 2017;12(4):e0176307.*Investigation of proteostasis machinery, and sensitivity to disruption, in humans with Down syndrome.
- 143.Perluigi M, Di Domenico F, Buttterfield DA. Unraveling the complexity of neurodegeneration in brains of subjects with Down syndrome: insights from proteomics. Proteomics Clin Appl. 2014;8(1–2):73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Di Domenico F, Coccia R, Cocciolo A, et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim Biophys Acta - Molecular Basis of Disease. 2013;1832(8):1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Freer R, Sormanni P, Vecchi G, et al. A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease. Sci Adv. 2016;2(8):e1600947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mao F, Wang H, Ni W, et al. Design, synthesis, and biological evaluation of orally available first-generation dual-target selective inhibitors of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5) for the treatment of Alzheimer’s Disease. ACS Chem Neurosci. 2017, ahead of print. [DOI] [PubMed] [Google Scholar]
- 147.Hampel H, Mesulam MM, Cuello AC, et al. Revisiting the cholinergic hypothesis in Alzheimer’s disease: Emerging evidence from translational and clinical research. Alzheimers Dement. 2017. [DOI] [PubMed] [Google Scholar]
- 148.Takahashi-Ito K, Makino M, Okado K, et al. Memantine inhibits beta-amyloid aggregation and disassembles preformed beta-amyloid aggregates. Biochem Biophys Res Commun. 2017;493(1):158–163. [DOI] [PubMed] [Google Scholar]
- 149.Bo H, Kang W, Jiang N, et al. Exercise-induced neuroprotection of hippocampus in APP/PS1 transgenic mice via upregulation of mitochondrial 8-oxoguanine DNA glycosylase. Oxid Med Cell Longev. 2014;2014:834502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guo JW, Guan PP, Ding WY, et al. Erythrocyte membrane-encapsulated celecoxib improves the cognitive decline of Alzheimer’s disease by concurrently inducing neurogenesis and reducing apoptosis in APP/PS1 transgenic mice. Biomaterials. 2017;145:106–127. [DOI] [PubMed] [Google Scholar]
- 151.Agholme L, Clarin M, Gkanatsiou E, et al. Low-dose gamma-secretase inhibition increases secretion of Abeta peptides and intracellular oligomeric Abeta. Mol Cell Neurosci. 2017. [DOI] [PubMed] [Google Scholar]
- 152.Pringsheim T, Jette N, Frolkis A, et al. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2014;29(13):1583–1590. [DOI] [PubMed] [Google Scholar]
- 153.Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016;15(12):1257–1272. [DOI] [PubMed] [Google Scholar]
- 154.Clark LN, Wang Y, Karlins E, et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. 2006;67(10):1786–1791. [DOI] [PubMed] [Google Scholar]
- 155.Lopez de Maturana R, Lang V, Zubiarrain A, et al. Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J Neuroinflammation. 2016;13(1):295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256–259. [DOI] [PubMed] [Google Scholar]
- 157.Nash Y, Schmukler E, Trudler D, et al. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J Neurochem. 2017. [DOI] [PubMed] [Google Scholar]
- 158.Mastroberardino PG, Hoffman EK, Horowitz MP, et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol Dis. 2009;34(3):417–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xiao B, Deng X, Lim GGY, et al. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 2017;8(10):e3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xiao B, Goh JY, Xiao L, et al. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem. 2017;292(40):16697–16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Barodia SK, Creed RB, Goldberg MS. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res Bull. 2017;133:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xilouri M, Brekk OR, Stefanis L. alpha-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47(2):537–551. [DOI] [PubMed] [Google Scholar]
- 163.Chu Y, Dodiya H, Aebischer P, et al. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35(3):385–398. [DOI] [PubMed] [Google Scholar]
- 164.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fernandes HJ, Hartfield EM, Christian HC, et al. ER stress and autophagic perturbations lead to elevated extracellular alpha-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 2016;6(3):342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Colla E, Coune P, Liu Y, et al. Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J Neurosci. 2012;32(10):3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Colla E, Jensen PH, Pletnikova O, et al. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci. 2012;32(10):3301–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schapira AH. Mitochondrial dysfunction in Parkinson’s disease. Cell Death Differ. 2007;14(7):1261–1266. [DOI] [PubMed] [Google Scholar]
- 169.Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139 Suppl 1: 216–231. [DOI] [PubMed] [Google Scholar]
- 170.Martinez BA, Petersen DA, Gaeta AL, et al. Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of Parkinson’s disease. J Neurosci. 2017, ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- 172.Zhou W, Bercury K, Cummiskey J, et al. Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J Biol Chem. 2011;286(17):14941–14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Milanese C, Tapias V, Gabriels S, et al. Mitochondrial complex I reversible S-nitrosation improves bioenergetics and is protective in Parkinson’s disease. Antioxid Redox Signal. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Skibinski G, Hwang V, Ando DM, et al. Nrf2 mitigates LRRK2- and alpha-synuclein-induced neurodegeneration by modulating proteostasis. Proc Natl Acad Sci U S A. 2017;114(5):1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Friesen EL, De Snoo ML, Rajendran L, et al. Chaperone-based therapies for disease modification in Parkinson’s disease. Parkinsons Dis. 2017;2017:5015307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Montojo MT, Aganzo M, Gonzalez N. Huntington’s disease and diabetes: Chronological sequence of its association. J Huntingtons Dis. 2017;6(3):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pearl JR, Heath LM, Bergey DE, et al. Enhanced retinal responses in Huntington’s disease patients. J Huntingtons Dis. 2017;6(3):237–247. [DOI] [PubMed] [Google Scholar]
- 179.Rub U, Vonsattel JP, Heinsen H, et al. The neuropathology of Huntington’s disease: Classical findings, recent developments and correlation to functional neuroanatomy. Adv Anat Embryol Cell Biol. 2015;217:1–146. [PubMed] [Google Scholar]
- 180.Takahashi T, Kikuchi S, Katada S, et al. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17(3):345–356. [DOI] [PubMed] [Google Scholar]
- 181.Leitman J, Ulrich Hartl F, Lederkremer GZ. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;4:2753. [DOI] [PubMed] [Google Scholar]
- 182.Wang B, Zeng L, Merillat SA, et al. The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington’s disease protein, huntingtin. Neurobiol Dis. 2017;109(Pt A):127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–548. [DOI] [PubMed] [Google Scholar]
- 184.He WT, Xue W, Gao YG, et al. HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci Rep. 2017;7(1):14797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Koyuncu S, Fatima A, Gutierrez-Garcia R, et al. Proteostasis of huntingtin in health and disease. Int J Mol Sci. 2017;18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhou H, Cao F, Wang Z, et al. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003;163(1):109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Neueder A, Gipson TA, Batterton S, et al. HSF1-dependent and -independent regulation of the mammalian in vivo heat shock response and its impairment in Huntington’s disease mouse models. Sci Rep. 2017;7(1):12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Finkbeiner S, Mitra S. The ubiquitin-proteasome pathway in Huntington’s disease. ScientificWorldJournal. 2008;8:421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Prime ME, Andersen OA, Barker JJ, et al. Discovery and structure-activity relationship of potent and selective covalent inhibitors of transglutaminase 2 for Huntington’s disease. J Med Chem. 2012;55(3):1021–1046. [DOI] [PubMed] [Google Scholar]
- 190.Colpo GD, Stimming EF, Rocha NP, et al. Promises and pitfalls of immune-based strategies for Huntington’s disease. Neural Regen Res. 2017;12(9):1422–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. 2017;14(11):637–653.**Very recent review detailing disrupted proteostasis in cardiac pathologies
- 192.Lopaschuk GD, Spafford MA, Marsh DR. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol. 1991;261(6 Pt 2):H1698–1705. [DOI] [PubMed] [Google Scholar]
- 193.Gong G, Song M, Csordas G, et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350(6265):aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shi R, Guberman M, Kirshenbaum LA. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc Med. 2017. [DOI] [PubMed] [Google Scholar]
- 195.Billia F, Hauck L, Konecny F, et al. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108(23):9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dorn GW, 2nd. Parkin-dependent mitophagy in the heart. J Mol Cell Cardiol. 2016;95:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120(11):1812–1824. [DOI] [PubMed] [Google Scholar]
- 198.Rainer PP, Dong P, Sorge M, et al. Desmin phosphorylation triggers preamyloid oligomers formation and myocyte dysfunction in acquired heart failure. Circ Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Agnetti G, Halperin VL, Kirk JA, et al. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc Res. 2014;102(1):24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Johnson TD, Hill RC, Dzieciatkowska M, et al. Quantification of decellularized human myocardial matrix: a comparison of six patients. Proteomics-Clinical Applications. 2016;10(1):75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature medicine. 2016(22.5):497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Calle EA, Hill RC, Leiby KL, et al. Targeted proteomics effectively quantifies differences between native lung and detergent-decellularized lung extracellular matrices. Acta Biomaterialia. 2016;46:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Balch WE, Sznajder JI, Budinger S, et al. Malfolded protein structure and proteostasis in lung diseases. Am J Respir Crit Care Med. 2014;189(1):96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.King P, Tulloh R. Management of pulmonary hypertension and Down syndrome. Int J Clin Pract Suppl. 2011(174):8–13. [DOI] [PubMed] [Google Scholar]
- 205.Clark AR, Lubsen NH, Slingsby C. sHSP in the eye lens: crystallin mutations, cataract and proteostasis. Int J Biochem Cell Biol. 2012;44(10):1687–1697. [DOI] [PubMed] [Google Scholar]
- 206.Bassnett S On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res. 2009;88(2):133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Peschek J, Braun N, Rohrberg J, et al. Regulated structural transitions unleash the chaperone activity of alphaB-crystallin. Proc Natl Acad Sci U S A. 2013;110(40):E3780–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Surguchev A, Surguchov A. Conformational diseases: looking into the eyes. Brain Res Bull. 2010;81(1):12–24. [DOI] [PubMed] [Google Scholar]
- 209.Thornell E, Aquilina A. Regulation of alphaA- and alphaB-crystallins via phosphorylation in cellular homeostasis. Cell Mol Life Sci. 2015;72(21):4127–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tang HZ, Yang LM. Activation of the unfolded protein response in aged human lenses. Mol Med Rep. 2015;12(1):389–393. [DOI] [PubMed] [Google Scholar]
- 211.Rappa F, Farina F, Zummo G, et al. HSP-molecular chaperones in cancer biogenesis and tumor therapy: an overview. Anticancer Res. 2012;32(12):5139–5150. [PubMed] [Google Scholar]
- 212.Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350(6265):1193–1198. [DOI] [PubMed] [Google Scholar]
- 213.Alasiri G, Fan LY, Zona S, et al. ER stress and cancer: The FOXO forkhead transcription factor link. Mol Cell Endocrinol. 2017. [DOI] [PubMed] [Google Scholar]
- 214.Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. 2014;19(3):222–240. [DOI] [PubMed] [Google Scholar]
- 215.White E The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46.*This recent review discusses the mechanisms by which dynamic regulation of cell autophagy can promote or inhibit tumorigenesis and progression.
- 216.Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7(11):847–859. [DOI] [PubMed] [Google Scholar]
- 217.Sklirou A, Papanagnou ED, Fokialakis N, et al. Cancer chemoprevention via activation of proteostatic modules. Cancer Lett. 2017. [DOI] [PubMed] [Google Scholar]
- 218.Kwak MK, Wakabayashi N, Greenlaw JL, et al. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol. 2003;23(23):8786–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Devisscher L, Vieri M, Logue SE, et al. Targeting the angio-proteostasis network: Combining the forces against cancer. Pharmacol Ther. 2016;167:1–12.*A review of angiogenic factors and their roles in cancer development, particularly in the context of proteostasis.
- 220.De Smet F, Saiz Rubio M, Hompes D, et al. Nuclear inclusion bodies of mutant and wild-type p53 in cancer: a hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J Pathol. 2017;242(1):24–38. [DOI] [PubMed] [Google Scholar]
- 221.Maneix L, Catic A. Touch and go: Nuclear proteolysis in the regulation of metabolic genes and cancer. FEBS Lett. 2016;590(7):908–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.D’Alessandro A, Amelio I, Berkers CR, et al. Metabolic effect of TAp63α: enhanced glycolysis and pentose phosphate pathway, resulting in increased antioxidant defense. Oncotarget. 2014;5(17):7722–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Cheng Q, Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle. 2010;9(3):472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Gregory MA, D’Alessandro A, Alvarez-Calderon F, et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc Natl Acad Sci U S A. 2016;113(43):E6669–E6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Polubotko EA, Smirnova NV, Pleskach NM, et al. Premature aging syndrome in ataxia telangiectasia patients. Cell Tissue Biol. 2009;3(5):491. [Google Scholar]
- 226.Poletto M, Yang D, Fletcher SC, et al. Modulation of proteostasis counteracts oxidative stress and affects DNA base excision repair capacity in ATM-deficient cells. Nucleic Acids Res. 2017;45(17):10042–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bonvini P, Zorzi E, Basso G, et al. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007;21(4):838–842. [DOI] [PubMed] [Google Scholar]
- 228.Lee DM, Kim IY, Seo MJ, et al. Nutlin-3 enhances the bortezomib sensitivity of p53-defective cancer cells by inducing paraptosis. Exp Mol Med. 2017;49(8):e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Leu JI, Barnoud T, Zhang G, et al. Inhibition of stress-inducible HSP70 impairs mitochondrial proteostasis and function. Oncotarget. 2017;8(28):45656–45669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Li J, Yakushi T, Parlati F, et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat Chem Biol. 2017;13(5):486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Rodriguez KA, Valentine JM, Kramer DA, et al. Determinants of rodent longevity in the chaperone-protein degradation network. Cell Stress Chaperones. 2016;21(3):453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Herndon LA, Schmeissner PJ, Dudaronek JM, et al. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419(6909):808. [DOI] [PubMed] [Google Scholar]
- 233.Walther DM, Mann M. Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Molecular & Cellular Proteomics. 2011;10(2):M110. 004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Grant JE, Bradshaw AD, Schwacke JH, et al. Quantification of protein expression changes in the aging left ventricle of Rattus norvegicus. Journal of proteome research. 2009;8(9):4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Tsugita A, Kawakami T, Uchida T, et al. Proteome analysis of mouse brain: Two‐dimensional electrophoresis profiles of tissue proteins during the course of aging. Electrophoresis. 2000;21(9):1853–1871. [DOI] [PubMed] [Google Scholar]
- 236.Schiller HB, Fernandez IE, Burgstaller G, et al. Time‐and compartment‐resolved proteome profiling of the extracellular niche in lung injury and repair. Mol Syst Biol. 2015;11(7):819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Schiller HB, Fernandez IE, Burgstaller G, et al. , Quantitative Proteomic Profiling Of Extracellular Matrix Composition And Protein Secretion Reveals Novel Modulators Of Pulmonary Fibrosis, in B97. A SCAR IS BORN: NEW INSIGHTS IN LUNG FIBROGENESIS. 2014, Am Thoracic Soc p. A3652–A3652. [Google Scholar]
- 238.Perluigi M, di Domenico F, Fiorini A, et al. Oxidative stress occurs early in Down syndrome pregnancy: a redox proteomics analysis of amniotic fluid. Proteomics Clin Appl. 2011;5(3‐4):167–178. [DOI] [PubMed] [Google Scholar]
- 239.Eyre DR, Paz MA, Gallop PM. Cross-linking in collagen and elastin. Ann Rev Biochem. 1984;53(1):717–748.*Defines and summarizes the molecular mechanisms by which collagen and elastin crosslinks stabilize tissue architectures and are altered during aging.
- 240.Haus JM, Carrithers JA, Trappe SW, et al. Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol (1985). 2007;103(6):2068–2076. [DOI] [PubMed] [Google Scholar]
- 241.Gkogkolou P, Bohm M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinol 2012;4(3):259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Byun K, Yoo Y, Son M, et al. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol Ther. 2017;177:44–55. [DOI] [PubMed] [Google Scholar]
- 243.Barrett AS, Wither MJ, Hill RC, et al. Hydroxylamine chemical digestion for insoluble extracellular matrix characterization. J Proteome Res. 2017;16:4177–4184.*A detailed approach for digestion and characterization of the insoluble extracellular matrix proteome using mass spectrometry.
- 244.Goddard ET, Hill RC, Barrett A, et al. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. Int J Biochem Cell Biol. 2016;81:223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hill RC, Wither MJ, Nemkov T, et al. Preserved proteins from extinct Bison latifrons identified by tandem mass spectrometry; hydroxylysine glycosides are a common feature of ancient collagen. Mol Cell Proteomics. 2015;14(7):1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Buckwalter JA, Woo S, Goldberg V, et al. Soft-tissue aging and musculoskeletal function. J Bone Joint Surg Am. 1993;75(10):1533–1548. [DOI] [PubMed] [Google Scholar]
- 248.Verzijl N, DeGroot J, Ben ZC, et al. Crosslinking by advanced glycation end products increases the stiffness of the collagen network in human articular cartilage: a possible mechanism through which age is a risk factor for osteoarthritis. Arthritis Rheum. 2002;46(1):114–123. [DOI] [PubMed] [Google Scholar]
- 249.Eyre D, Dickson I, Van Ness K. Collagen cross-linking in human bone and articular cartilage. Age-related changes in the content of mature hydroxypyridinium residues. Biochem J. 1988;252:495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Koo EH, Park L, Selkoe DJ. Amyloid beta-protein as a substrate interacts with extracellular matrix to promote neurite outgrowth. Proc Natl Acad Sci U S A. 1993;90(10):4748–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Klier FG, Cole G, Stallcup W, et al. Amyloid β-protein precursor is associated with extracellular matrix. Brain Research. 1990;515(1):336–342. [DOI] [PubMed] [Google Scholar]
- 252.Tanford C, Reynolds J, Nature’s Robots: A History of Proteins. 2001, New York: Oxford University Press. [Google Scholar]
- 253.Asimov I, Runaround, in I, Robot. 1950, Doubleday: New York City. [Google Scholar]
- 254.Culp-Hill R, Zheng C, Reisz JA, et al. Red blood cell metabolism in Down syndrome: Hints on metabolic derangements in aging. Blood Adv. 2017;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Straat M, van Bruggen R, de Korte D, et al. Red blood cell clearance in inflammation. Transfus Med Hemother. 2012;39(5):353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 2017;130(15):1693–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.de Lustig ES, Serra JA, Kohan S, et al. Copper-zinc superoxide dismutase activity in red blood cells and serum in demented patients and in aging. J Neurol Sci. 1993;115(1):18–25. [DOI] [PubMed] [Google Scholar]
- 258.Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Tran BQ, Goodlett DR, Goo YA. Advances in protein complex analysis by chemical cross-linking coupled with mass spectrometry (CXMS) and bioinformatics. Biochim Biophys Acta - Proteins and Proteomics. 2016;1864(1):123–129. [DOI] [PubMed] [Google Scholar]
- 260.Batth TS, Francavilla C, Olsen JV. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J Proteome Res. 2014;13(12):6176–6186. [DOI] [PubMed] [Google Scholar]