

Abstract
Small regulatory RNAs (sRNAs) are an important class of bacterial post-transcriptional regulators that control numerous physiological processes, including stress responses. In Gram-negative bacteria including Escherichia coli, the RNA chaperone Hfq binds many sRNAs and facilitates pairing to target transcripts, resulting in changes in mRNA transcription, translation, or stability. Here, we report that poly(A) polymerase (PAP I), which promotes RNA degradation by exoribonucleases through the addition of poly(A) tails, has a crucial role in the regulation of gene expression by Hfq-dependent sRNAs. Specifically, we show that deletion of pcnB, encoding PAP I, paradoxically resulted in an increased turnover of certain Hfq-dependent sRNAs, including RyhB. RyhB instability in the pcnB deletion strain was suppressed by mutations in hfq or ryhB that disrupt pairing of RyhB with target RNAs, by mutations in the 3′ external transcribed spacer of the glyW-cysT-leuZ transcript (3′ETSLeuZ) involved in pairing with RyhB, or an internal deletion in rne, which encodes the endoribonuclease RNase E. Finally, the reduced stability of RyhB in the pcnB deletion strain resulted in impaired regulation of some of its target mRNAs, specifically sodB and sdhCDAB. Altogether our data support a model where PAP I plays a critical role in ensuring the efficient decay of the 3′ETSLeuZ. In the absence of PAP I, the 3′ETSLeuZ transcripts accumulate, bind Hfq, and pair with RyhB, resulting in its depletion via RNase E-mediated decay. This ultimately leads to a defect in RyhB function in a PAP I deficient strain.
Keywords: poly(A) polymerase, pcnB, Hfq, small RNAs, RNase E
INTRODUCTION
Small regulatory RNAs (sRNAs) are transcripts ranging in size from 50 to 300 nucleotides (nt) that have been shown to regulate nearly every aspect of bacterial behavior and physiology including virulence (Toledo-Arana et al. 2009; Gripenland et al. 2010; Felden et al. 2011; Koo et al. 2011; Bardill and Hammer 2012; Hébrard et al. 2012; Mann et al. 2012; Rutherford and Bassler 2012; Caldelari et al. 2013; Holmqvist and Wagner 2017), biofilm development (Thomason et al. 2012; Jørgensen et al. 2013; Zhao et al. 2013; Parker et al. 2017), antibiotic resistance (Parker and Gottesman 2016; Zhang et al. 2017; Felden and Cattoir 2018), and metabolism (Durand and Storz 2010; Gimpel et al. 2010; Richards and Vanderpool 2011; Salvail and Massé 2012; Bobrovskyy and Vanderpool 2013; McClure et al. 2013; Mandin et al. 2016; Pannuri et al. 2016; Gonzalez et al. 2017). Many sRNAs regulate these processes by recognizing and binding specific target mRNAs through base-pairing resulting in changes in mRNA transcription, translation, or stability depending on the nature of this interaction. For example, sRNAs can inhibit translation by base-pairing with the mRNA translation initiation region directly blocking ribosome access to the Shine–Delgarno sequence (Møller et al. 2002a). Alternatively, negative regulation of gene expression can also be achieved by sRNA-induced RNase E mediated decay of mRNAs (Pfeiffer et al. 2009; Bandyra et al. 2012). Furthermore, sRNAs can positively regulate gene expression by base-pairing with a mRNA as it is being transcribed, preventing intramolecular base-pairing in the 5′ untranslated region (5′-UTR) that would otherwise lead to transcription attenuation (Sedlyarova et al. 2016).
In Escherichia coli and other Gram-negative bacteria, many sRNAs encoded in trans require the RNA-binding protein chaperone Hfq to function (Waters and Storz 2009; Storz et al. 2011). Hfq stabilizes sRNAs by binding and occluding an RNase E cleavage site (Moll et al. 2003), but also serves as a matchmaker by facilitating annealing between sRNAs and their target mRNAs (Møller et al. 2002b; Zhang et al. 2002; Lease and Woodson 2004; Soper and Woodson 2008). Although Hfq has been studied extensively, recent research has identified PNPase, encoding the 3′ → 5′ exoribonuclease polynucleotide phosphorylase, as another mediator of sRNA stability and function (De Lay and Gottesman 2011). PNPase degrades at least some sRNAs not associated with Hfq (Viegas et al. 2007; Andrade et al. 2012). However, PNPase binds and stabilizes many Hfq-dependent sRNAs (Bandyra et al. 2016) and has been further shown to impact sRNA–mRNA pairing (Cameron and De Lay 2016).
The crucial role of PNPase in modulating sRNA stability and function was discovered in a combined genetic selection and screen designed to isolate mutants defective for sRNA function in E. coli. Loss-of-function point mutations identified in pnp interfered with target gene regulation by Hfq-dependent sRNAs including RyhB (De Lay and Gottesman 2011). RyhB is one of the best-characterized sRNAs in E. coli. Under iron replete conditions, RyhB expression is repressed by the iron bound form of the iron sensing transcriptional regulator protein Fur (ferric uptake regulator). Under iron limiting conditions Fur can no longer associate with free Fe2+ efficiently and consequently with DNA, which leads to RyhB expression. RyhB, in turn, acts to modulate iron homeostasis by down-regulating the expression of a large number of mRNAs encoding nonessential iron-containing proteins including sdhCDAB and sodB, which encode succinate dehydrogenase and superoxide dismutase, respectively (Massé and Gottesman 2002; Massé et al. 2003, 2005, 2007; Richards and Vanderpool 2011).
In the same genetic selection and screen that isolated hfq and pnp mutants, independent point mutants were obtained in pcnB, the gene encoding poly(A) polymerase (PAP I), which were not investigated in detail (De Lay and Gottesman 2011). PAP I catalyzes polyadenylation of the 3′ extremities of RNA substrates and has a preference for Rho-independent transcription terminators containing 2–6 nt single-stranded extensions (Mohanty and Kushner 2008, 2011, 2016; Régnier and Hajnsdorf 2013). Poly(A) tails promote transcript decay by providing toe-holds for 3′ → 5′ exoribonucleases like RNase II, RNase R, and PNPase. PAP I interacts with Hfq and PNPase to form a polyadenylation complex. Hfq has been shown to stimulate polyadenylation of mRNAs by PAP I, most likely by binding to and recruiting PAP I to the 3′ ends of RNA substrates (Hajnsdorf and Régnier 2000; Le Derout et al. 2003; Mohanty et al. 2004).
PAP I adds poly(A) tails to many different classes of cellular RNAs (mRNAs, rRNAs, tRNAs, sRNAs, viral RNAs) (Régnier and Hajnsdorf 2013; Mohanty and Kushner 2016), and while the majority of E. coli ORFs undergo polyadenylation under exponential growth conditions, only a small fraction of them are polyadenylated at a specific time (Mohanty and Kushner 2006). Many sRNAs that do not require Hfq for stability and function have been shown to be polyadenylated in vivo, e.g., RNA I, Sok, Oop, SraL, SraG, and GlmY, and are subsequently degraded by exoribonucleases (Régnier and Hajnsdorf 2013; Ruiz-Larrabeiti et al. 2016). Interestingly, previous data have shown that sRNAs that require Hfq for their stability, e.g., MicA and RybB, can also be targeted for degradation by PNPase and PAP I, but only when these sRNAs are not bound by Hfq (Andrade and Arraiano 2008; Andrade et al. 2012; Cameron and De Lay 2016).
In this study, we have further investigated the possible mechanisms by which the PAP I mediated polyadenylation led to a defect in sRNA function. Here, we report that deletion of pcnB encoding PAP I resulted in a significant reduction in RyhB stability and consequently a defect in RyhB-mediated repression of sdhCDAB and sodB transcripts. We provide evidence that the increased turnover of RyhB in a pcnB deletion strain is due to increased accumulation of the 3′ETSLeuZ, which promotes more rapid RyhB degradation by RNase E as a consequence of base-pairing interactions with this sRNA. Finally, we show that PAP I can stabilize another Hfq-dependent sRNA, MicA, but not others (GcvB, CyaR, ChiX, and MgrR), suggesting a specialized role of PAP I in conferring stability to a specific subset of Hfq-dependent sRNAs. This work provides further insight into how yet another protein previously known to be involved in initiating RNA decay contributes to sRNA-dependent gene regulation.
RESULTS
Poly(A) polymerase stabilizes RyhB
In a previous study (De Lay and Gottesman 2011), strains harboring null mutations in hfq, pnp, or pcnB encoding the RNA chaperone Hfq, the exoribonuclease PNPase, or the poly(A) polymerase PAP I, respectively, were recovered in a genetic selection and screen designed to isolate mutants defective for sRNA-mediated gene regulation. The selection was for mutations that allowed an E. coli Δfur strain to grow on minimal succinate medium. In a fur deletion strain, RyhB is constitutively expressed and accumulates under the protection of Hfq. In turn, Hfq promotes base-pairing with the sdhCDAB mRNA, blocking expression of succinate dehydrogenase complex and consequently leading to an inability of a fur mutant to grow on succinate as the sole carbon source (Massé and Gottesman 2002). Inactivation of pnp or hfq caused rapid turnover of RyhB leading to up-regulation of the sdhCDAB transcript, and consequently allowed a Δfur stain to grow on succinate as the sole carbon source (De Lay and Gottesman 2011). Since the point mutations in pcnB allowed growth on succinate minimal medium, we tested the effect of a pcnB deletion on RyhB stability. To test this, we first examined RyhB steady-state levels in exponentially growing cultures of Δfur, Δfur ΔpcnB, and Δfur Δhfq strains. Introduction of a pcnB deletion into the Δfur strain resulted in a decrease in RyhB steady-state levels by 40% to 50% and were comparable to that in a Δfur Δhfq strain (Fig. 1A,B). As expected, RyhB was not detectable in a wild-type fur+ strain (WT). Next, we determined RyhB stability in exponentially growing cultures of the aforementioned strains by blocking transcription initiation by adding rifampicin. RyhB levels were monitored by northern blot analysis of samples taken after transcription inhibition. RyhB was very stable in the Δfur parent background under our experimental conditions, while introduction of an hfq deletion drastically reduced the stability of this sRNA (Table 1; Fig. 1C,D). Interestingly, introduction of a pcnB single deletion reduced RyhB stability significantly, but to a lesser extent than what was observed for an hfq mutant (Table 1; Fig. 1C,D).
FIGURE 1.

RyhB is rapidly degraded in the absence of poly(A) polymerase in an RNase E-dependent mechanism. (A,B) Northern blot analysis to assess RyhB steady-state levels. Overnight cultures of strain NRD1138 (WT), an isogenic Δfur strain (DS024), or derivatives of this Δfur strain harboring deletions in hfq (DS027), pcnB (DS025), rne (DS069), rne and pcnB (DS082), and rne and hfq (DS153) were diluted 200-fold in fresh MOPS EZ rich defined media supplemented with 0.4% glycerol. All cultures were subsequently grown to late exponential phase (OD600 of 1.0), and samples were collected for RNA extraction. (C,D) RNA stability time course experiment to determine the intrinsic stability of RyhB. Briefly, overnight cultures of the Δfur parent (DS024) and its derived mutant strains (Δfur ΔpcnB, DS025; Δfur Δhfq, DS027; Δfur rne-131, DS069; Δfur rne-131 ΔpcnB, DS082; Δfur rne-131 Δhfq, DS153) were grown to OD600 of 1.0 as described above and a culture sample was taken. Rifampicin was added to each culture to stop total transcription, and additional culture samples were taken 2, 5, 10, 15, and 30 min after rifampicin addition. All samples were subjected to RNA extraction and were prepared for northern blot analysis as described in Materials and Methods. Representative northern blots are shown in A and C. (B,D) RyhB signal intensities in the northern blots were quantified and normalized to their corresponding loading controls (ssrA). sRNA decay curves were generated by fitting the normalized signal intensities for each time point. Points and error bars in the curves represent the means and the standard errors (SEM) of at least three independent experiments. RyhB half-life measurements corresponding to RNA stability curves (D) are listed in Table 1.
TABLE 1.
RyhB half-life measurements in various E. coli mutant strains

Since deletion of pcnB led to increased turnover of RyhB, we decided to test whether PAP I was also required for the stability of other Hfq-dependent sRNAs including MicA, GcvB, CyaR, ChiX, and MgrR. We determined the stability of these sRNAs in exponentially growing cultures of a ΔpcnB and a wild-type strain as described above. Deletion of pcnB led to ∼50% reduction in MicA stability compared to the wild-type strain, while there were no significant differences in stability of GcvB, CyaR, ChiX, or MgrR between a wild-type and a ΔpcnB strain (Fig. 2; Supplemental Fig. S1). Overall, our data demonstrated that deletion of pcnB led to increased turnover of at least two Hfq-dependent sRNAs, RyhB and MicA.
FIGURE 2.

A subset of Hfq-dependent sRNAs are unstable in the absence of poly(A) polymerase. RNA stability time-course experiments to determine the intrinsic stabilities of GcvB (A), MicA (B), CyaR (C), ChiX (D), and MgrR (E) sRNAs. (A,B) Strain TC279 (WT; fur+), which has MicA under the control of the ryhB promoter, and an isogenic ΔpcnB strain (ΔpcnB, DS120) were grown to exponential phase. Dipyridyl was added to each culture for 15 min to induce MicA expression, a sample was taken, rifampicin was added to block transcription, and additional samples were taken 1, 2, 4, and 6 min after rifampicin addition. RNA was extracted and northern blot analysis was performed probing for MicA or GcvB as described in Materials and Methods. To determine intrinsic stabilities of CyaR (C), ChiX (D), and MgrR (E), the wild-type strain (NRD1138) and its derived pcnB mutant (NRD1198) were grown to exponential phase, CyaR was induced from its native promoter by cAMP addition, a sample was taken after 15 min of induction, rifampicin was then added, and additional samples were taken 1, 2, 4, and 6 min after transcription inhibition. Northern blot analysis was performed using RNA extracted from these samples probing for CyaR, ChiX, and MgrR. Representative northern blots for each sRNA are shown in Supplemental Figure S1 (Supplemental Information). For the decay curves, sRNA signal intensities from the northern blots were quantified and normalized to their corresponding loading controls (ssrA or 5S). Points and error bars in the curves represent the means and the standard errors (SEM) of at least three independent experiments.
Poly(A) polymerase is important for RyhB-mediated target gene regulation
Based on the results reported above, we investigated whether this defect in RyhB stability in a pcnB deletion mutant led to a subsequent defect in RyhB-dependent target gene regulation. To test this idea, we focused on the two well-characterized RyhB target mRNAs, sodB and sdhCDAB. We determined the relative levels of sodB using northern blotting and sdhCDAB by qRT-PCR using specific primers to probe for transcripts containing sdhC and sdhD in exponential phase cultures of a Δfur strain or derived strain harboring a deletion in pcnB (ΔpcnB) in the presence (+ryhB) or absence (ΔryhB) of ryhB. We observed a greater than fivefold down-regulation of sodB transcript levels by RyhB in the Δfur strain (Fig. 3A,B; compare Δfur versus Δfur ΔryhB). However, in the corresponding isogenic ΔpcnB strains, RyhB-dependent regulation of sodB was only 2.2-fold (Fig. 3A,B; compare Δfur ΔpcnB versus Δfur ΔpcnB ΔryhB). Furthermore, we found that sodB was turned over less rapidly in a ΔpcnB mutant (t1/2 = 4.7 min) relative to a wild-type (t1/2 = 3.4 min) strain upon RyhB induction (Supplemental Fig. S2). The decreased rate of sodB target degradation in a pcnB mutant indicated a defect in RyhB-mediated sodB repression. Similarly, qRT-PCR analysis showed a nearly threefold down-regulation of sdhC transcript levels by RyhB in a Δfur strain background (Fig. 3C; compare Δfur versus Δfur ΔryhB), whereas in the Δfur ΔpcnB strain RyhB expression caused only a 1.7-fold reduction in sdhC levels (Fig. 3C; compare Δfur ΔpcnB versus Δfur ΔpcnB ΔryhB). These results suggested a defect in RyhB-mediated regulation of sdhCDAB in the absence of PAP I. Interestingly, sdhC transcript levels were higher in a Δfur ΔpcnB ΔryhB strain than in the Δfur ΔryhB strain (Fig. 3C), suggesting that PAP I-mediated sdhCDAB regulation was both RyhB-dependent and RyhB-independent. Similar results were also obtained when we probed the sdhCDAB transcript by qRT-PCR using primers specific for sdhD instead of sdhC (Supplemental Fig. S3); i.e., introduction of the pcnB deletion into a Δfur parent background up-regulated sdhD steady-state levels by threefold (Supplemental Fig. S3). Altogether, our results indicated that deletion of pcnB resulted in increased sodB transcript levels as a consequence of reduced RyhB levels, whereas the higher levels of sdhCDAB mRNA in the absence of PAP I were likely due to reduced RyhB levels and loss of PAP I-mediated regulation of the sdhCDAB transcript that was RyhB-independent.
FIGURE 3.
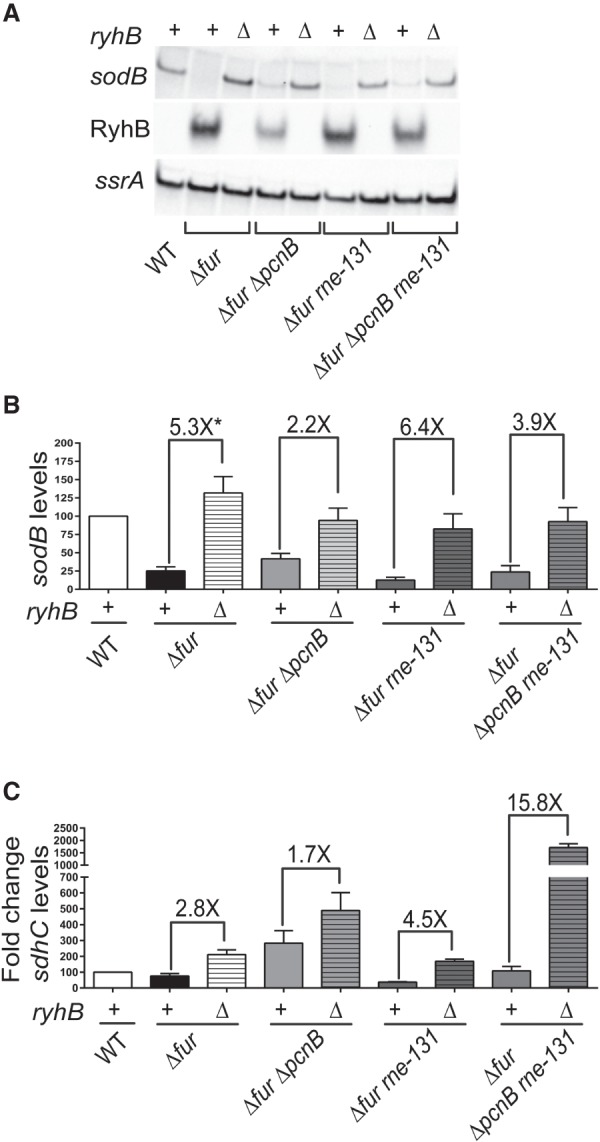
In the absence of poly(A) polymerase, RyhB does not efficiently regulate sodB and sdhCDAB target mRNAs. (A,B) Northern blot analysis was used to determine RyhB and sodB steady state in the wild-type and derived isogenic mutant strains, and (C) qRT-PCR analysis was used to determine sdhC levels. Wild-type parent (WT [fur+]; NRD1138) and its derived isogenic mutants (Δfur, DS024; Δfur ΔpcnB, DS025; Δfur rne-131, DS069; Δfur ΔpcnB rne-131, DS082; Δfur ΔryhB, NRD1546; Δfur ΔpcnB ΔryhB, NRD1547; Δfur rne-131 ΔryhB, NRD1550; Δfur ΔpcnB rne-131 ΔryhB, NRD1551) were diluted 200-fold in fresh MOPS EZ rich defined media supplemented with 0.4% glycerol and grown to late exponential phase (OD600 of 1.0), and samples for RNA extraction were collected. Representative northern blots are shown (A). (B,C) Graphs are presented that display the relative expression levels of sodB and sdhC mRNA in a wild-type strain and derived mutant strains. Briefly, the signal intensities for sodB and sdhC transcripts were first quantified from northern blots or qRT-PCRs. The signal intensity was then normalized to the ssrA transcript level, which served as the loading control, and subsequently the expression level relative to the fur+ (WT) strain NRD1138, which was set at 100%, was calculated. sdhC transcript fold changes relative to NRD1138 were calculated via the ΔΔCt method. Asterisk (*) in B indicates that the calculated fold change is >5.3× since sodB steady-state determination in DS024 (Δfur) strain was not accurate due to very low signal intensity. Data shown in B and C represent the mean (±SEM) of at least three independent experiments. Probes and primers used are listed in Supplemental Table S2.
Finally, we investigated whether PAP I can similarly impact MicA-mediated target gene regulation. To test this, we determined steady-state levels of two MicA-targets ompA and ompX, which encode outer membrane proteins, in a pcnB deletion and wild-type strain under conditions where MicA expression was induced or uninduced. We observed a 4.2-fold and 2.6-fold down-regulation of ompA and ompX transcript levels, respectively, by MicA in the wild-type strain while in the corresponding isogenic ΔpcnB strain, there was only a modest loss in MicA-dependent regulation of ompA (3.3-fold) and ompX (2.2-fold) (Supplemental Fig. S4). Taken together, our data indicated that PAP I plays a crucial role in promoting stability and function of RyhB and MicA.
Hfq mediates accelerated decay of RyhB in the absence of poly(A) polymerase
Based on our observations, we next investigated the mechanism by which PAP I promoted sRNA stability by focusing on RyhB. Since Hfq was essential for RyhB stability, we first determined whether PAP I impacted Hfq protein levels. Hfq protein levels were comparable between a wild-type and pcnB deletion strain under exponential growth conditions, indicating that the decreased stability of RyhB in a pcnB mutant strain was not due to lower levels of Hfq (Fig. 4A,B). Results from previous studies indicated that PAP I together with Hfq mediates efficient polyadenylation at the 3′ ends of Rho-independent terminators which subsequently facilitates Hfq binding to those mRNAs (Hajnsdorf and Régnier 2000; Mohanty and Kushner 2008, 2016; Mohanty et al. 2004; Régnier and Hajnsdorf 2013). We extended this idea to sRNAs and thus tested whether PAP I stabilized RyhB by increasing its binding to Hfq. We compared the total amount of RyhB to the amount immunoprecipitated with Hfq from cell lysates of a wild-type or an isogenic ΔpcnB strain; no significant difference was observed in the amount of RyhB that coimmunoprecipitated with Hfq between the wild-type and ΔpcnB strain (Fig. 4C,D). Altogether, our results indicated that PAP I did not facilitate RyhB binding to Hfq in vivo.
FIGURE 4.

The accelerated decay of RyhB in the absence of poly(A) polymerase is mediated by Hfq. (A,B) Western blot analysis to determine Hfq protein levels. Samples were prepared for western blotting from exponential phase cultures of a fur+ strain (DS090), and its derived strain ΔpcnB (DS092) with anti-Hfq antibody. Protein band intensities were normalized to DnaK detected with an anti-DnaK antibody (Materials and Methods). Representative western blots are shown in A. Quantification of Hfq levels from those western blots normalized to DnaK levels are shown in B. (C,D) Coimmunoprecipitation of RyhB with Hfq. RyhB expression was induced in exponential cultures of a wild-type (NRD1138) and an isogenic ΔpcnB mutant (NRD1198). Hfq was immunoprecipitated with anti-Hfq antibody bound to protein-A-sepharose. RNA extracted from the input and elution fractions were loaded in 1:8 ratio, and RyhB and ssrA levels were determined via northern blot analysis (C). Fold enrichment of RyhB (D) was determined after quantification of the RyhB and ssrA signal intensities in those northern blots as described in Materials and Methods. (E) Schematic showing interactions between RyhB, mRNA targets, and Hfq based on work by Schu and coworkers (Zhang et al. 2013; Schu et al. 2015); a Q8A substitution in the proximal face of Hfq was shown to disrupt RyhB binding, whereas a Y25D substitution in the distal face of Hfq reduced binding of RyhB target mRNAs such as sodB. (F) RNA half-life experiments to determine RyhB stability in the wild-type strain and its derived isogenic hfq and pcnB mutants. Overnight cultures of the wild-type strain (WT [fur+]; NRD1138) and its derived mutants (ΔpcnB, NRD1198; hfqQ8A, DS060; hfqY25D, NRD1410; hfqQ8A ΔpcnB, DS072; hfqY25D ΔpcnB, DS185) were diluted 200-fold in fresh LB media. Cultures were subsequently grown to exponential phase, dipyridyl was added to induce RyhB expression, and a culture sample was taken after 15 min of induction. Rifampicin was added to each culture to stop total transcription, and additional culture samples were taken 1, 2, 4, and 6 min after rifampicin addition. RNA extraction and northern blot analysis were performed as described above. Representative northern blots are shown in Supplemental Figure S5 of Supplemental Information. For decay curves, RyhB signal intensities from the northern blots were quantified and normalized to their corresponding loading controls (SsrA). Data shown in B, D, and F represent the means and the standard errors (SEM) of at least three independent experiments. RyhB half-life measurements corresponding to RNA stability curves are listed in Table 1.
Recent studies directed toward understanding Hfq-RNA interactions have led to the identification of four distinct surfaces on Hfq: the proximal face, distal face, rim, and the C-terminal tail, each possessing unique structural characteristics which promote binding of different RNAs in particular configurations (Vogel and Luisi 2011; Zhang et al. 2013; Schu et al. 2015; Updegrove et al. 2016). sRNAs are classified into two distinct classes, Class I and Class II, based on their mode of Hfq binding. RyhB is a Class I Hfq-binding sRNA shown to bind to sites on the Hfq proximal face via the poly(U) tail of its Rho-independent terminator and the rim via its UA-rich sequence, while RyhB target mRNAs typically interact with the Hfq distal face via repeats of an ARN sequence motif (Fig. 4E). Binding studies with several Hfq mutants have demonstrated that specific conserved residues on the proximal face (Q8, F42, K56) and rim (R16, R17, R19) were important for binding of Class I sRNAs to Hfq, and substitutions in these residues negatively impacted sRNA steady-state levels and stability. On the other hand, specific residues on the Hfq distal face (Y25, I30) were important for cognate mRNA target binding, and substitutions in these residues stabilized Class I sRNAs by protecting them from the subsequent degradation following sRNA–mRNA pairing mediated by Hfq (Zhang et al. 2013; Schu et al. 2015; Updegrove et al. 2016).
Our results so far indicated that PAP I did not impact RyhB binding to Hfq, but at the same time was required to stabilize RyhB in vivo. This observation prompted us to determine whether PAP I was contributing to RyhB stability by impacting the interaction of RyhB target RNAs with Hfq. To test this possibility, we introduced different hfq point mutations (hfqQ8A [proximal face], hfqR17A [rim], and hfqY25D [distal face]) into a wild-type or a ΔpcnB mutant strain and monitored RyhB stability as described above (Fig. 4E,F; Supplemental Fig. S5). Introduction of the hfqY25D mutation not only suppressed the defect in RyhB stability observed for a ΔpcnB mutant (t1/2 = 2.8 min for a ΔpcnB strain versus t1/2 = 13.6 min for the ΔpcnB hfqY25D strain) but further led to a significant increase in the stability of this sRNA compared to the wild-type strain (t1/2 = 7.1 min). RyhB stabilities were comparable between an hfqY25D single and a ΔpcnB hfqY25D double mutant strain (Fig. 4F). In contrast, introduction of the hfqQ8A mutation into the ΔpcnB strain did not suppress the defect in RyhB stability, and RyhB was rapidly degraded in either an hfqQ8A mutant (t1/2 = 1.97 min) or a ΔpcnB hfqQ8A double mutant (t1/2 = 2.1 min) as compared to a wild-type strain (t1/2 = 7.1 min) (Table 1; Fig. 4F). This result was consistent with previous studies showing that an alanine substitution in the Hfq proximal face residue Q8 disrupted Hfq binding to RyhB leading to the rapid degradation of this sRNA (Zhang et al. 2013; Schu et al. 2015). Finally, introduction of the hfqR17A mutation into a wild-type or a ΔpcnB strain background did not significantly decrease RyhB stability relative to that observed in the wild-type strain (Table 1; Supplemental Fig. S5). These data were in agreement with previous studies showing that mutations in the rim-binding residues were not sufficient to lead to a defect in RyhB function (Schu et al. 2015). Overall our data indicated that residue Y25 located on the distal face of Hfq and known to be important for binding targets of RyhB was essential for the decay of RyhB in the absence of PAP I.
Poly(A) polymerase protects RyhB from target pairing mediated decay
RyhB was previously shown to be degraded along with its target RNAs upon sRNA–mRNA pairing (Massé et al. 2003; Lalaouna et al. 2015b). Furthermore, the Y25D substitution in Hfq has been shown to reduce the ability of RyhB to regulate target mRNAs by disrupting Hfq-target RNA interactions (Schu et al. 2015). Thus, based on our results demonstrating the ability of an HfqY25D variant to suppress the defect in RyhB stability observed in a ΔpcnB strain, we hypothesized that RyhB instability in a ΔpcnB strain was a consequence of pairing with target RNAs. To explore this possibility, we constructed a RyhB pairing mutant (ryhBmut) in which bases G and C at positions 44 and 45 were inverted (Fig. 5A). These mutations were previously shown to abolish the ability of RyhB to repress the translation of the open reading frame upstream of fur (uof) and fur (Vecerek et al. 2007). Moreover, these nucleotides in RyhB are located in its seed sequence involved in pairing with other targets including sdhCDAB and sodB mRNAs (Fig. 5A; Massé and Gottesman 2002; Desnoyers and Massé 2012; Peterman et al. 2014; Waters et al. 2017). We assessed whether introducing these mutations into ryhB could suppress the defect in RyhB stability observed in a ΔpcnB strain by monitoring RyhB or RyhBmut steady-state levels in exponential phase cultures of ΔpcnB and pcnB+ strains by northern blot analysis. Interestingly, introduction of the G44C C45G mutations into ryhB in a ΔpcnB mutant background suppressed the defect in RyhB levels. Furthermore, RyhB levels increased by approximately 1.5- and twofold in a ΔpcnB ryhBmut double mutant and a ryhBmut single mutant, respectively, compared to the wild-type strain (Fig. 5B,C). Next, we tested whether the increased levels of RyhB in the ΔpcnB ryhBmut strain compared to the ΔpcnB ryhB+ strain were due to increased stability of the sRNA by monitoring RyhB turnover in exponential phase cultures of these strains after transcription inhibition. As shown in Figure 5D,E and Table 1, the RyhB stability defect observed in a ΔpcnB mutant was completely suppressed by introduction of these mutations in the pairing region of RyhB.
FIGURE 5.

The instability of RyhB in the absence of poly(A) polymerase is due to pairing with target RNAs. (A) Schematic showing complementarity (highlighted in gray) between RyhB and its target mRNA sodB, and the specific mutations (in cyan) introduced in RyhB to create a RyhB variant unable to pair with target mRNAs (RyhBmut). The start codon of sodB is highlighted in red. (B,C) Northern blot analysis to determine the transcript steady-state levels of RyhB targets sodB and uof in a wild-type strain (WT [fur+]; NRD1138) and its derived isogenic mutants (ΔpcnB, NRD1198; ryhBmut, LM11; ryhBmut ΔpcnB) under RyhB inducing and noninducing conditions. Overnight cultures of these strains grown in LB were diluted 200-fold in fresh LB media and grown to log phase and treated with dipyridyl (+) to chelate iron or mock-treated (−). After 15 min of treatment, RNA was extracted from each culture and prepared for northern blot analysis to determine corresponding levels of sodB, uof, RyhB, and ssrA (loading control). uof transcription is driven from two upstream promoters Puof and Pfur to generate the uof-fur and fur mRNAs, respectively, but RyhB specifically interacts with the fur mRNA (C). (D–F) Determination of RyhB intrinsic stability in a wild-type (WT [fur+]; NRD1138) and its derived isogenic mutants (ΔpcnB, NRD1198; Δhfq, DS021; ryhBmut, LM11; ryhBmut ΔpcnB, LM13; rne-131, DS102; rne-131 ΔpcnB, DS106; rne-131 Δhfq, DS130). Strains were subjected to RNA stability time-course experiments as described in the legend of Figure 4. Representative northern blots are shown in D. (E,F) RyhB decay curves were generated as described in Figure 4 and corresponding half-life measurements are listed in Table 1. Points and error bars in the curves represent the means and the standard errors (SEM) of at least three independent experiments.
In parallel, we tested whether RyhBmut was defective in regulating the sodB and fur mRNAs. Northern blot analysis showed that in the wild-type strain sodB mRNA levels were much lower under RyhB inducing conditions (+dipyridyl) than under noninducing conditions (−dipyridyl) in line with its role in negatively regulating the sodB mRNA (Fig. 5B). Consistent with our previous observations, introduction of a pcnB deletion in the wild-type background led to a subsequent defect in RyhB-dependent sodB regulation as indicated by an increased accumulation of sodB transcript under RyhB inducing conditions in a ΔpcnB mutant relative to the wild-type strain (Fig. 5B; Supplemental Fig. S6). In contrast, sodB mRNA levels were comparable in a ryhBmut mutant and a ryhBmut ΔpcnB double mutant strains under both inducing and noninducing conditions consistent with G44 and C45 of RyhB being critical for sRNA–mRNA pairing. Additionally, we found that the fur mRNA, transcribed from the Pfur promoter located within the uof ORF was down-regulated upon RyhB induction in both the wild-type and ΔpcnB strains whereas the level of this transcript was unaffected by RyhBmut expression in ryhBmut and ryhBmut ΔpcnB mutant strains (Fig. 5C). Interestingly, RyhB was not able to facilitate the decay of the uof-fur mRNA transcribed from the Puof promoter located upstream of uof ORF, even though RyhB negatively regulated fur mRNA levels by interacting with the same site. This finding further suggested that the context of RyhB pairing was important for its interaction with certain target transcripts. Taken together our results confirmed that the G44C C45G mutations in RyhBmut block pairing with at least some target mRNAs, indicating that the rapid turnover of RyhB in the absence of PAP I was dependent upon pairing with target RNAs.
The C-terminal domain of RNase E is required for RyhB degradation in the absence of poly(A) polymerase
The C-terminal domain (CTD) of RNase E plays an important role in coupled degradation of RyhB and its target mRNAs following Hfq-mediated pairing, and previous studies have demonstrated that this pairing mediated decay can be suppressed by introduction of an internal deletion in rne (rne-131) (Massé et al. 2003; Prévost et al. 2011; Desnoyers and Massé 2012). This mutation (rne-131) leads to production of an RNase E lacking the CTD. The CTD contains two RNA binding regions, ARRBD (or AR1) and AR2, but also interacts with other proteins including enolase, the RNA helicase RhlB, and PNPase to form the RNA degradosome, the central RNA degradation machine (Vanzo et al. 1998; Callaghan et al. 2004; Morita et al. 2005). To further test this hypothesis that absence of PAP I resulted in accelerated decay of RyhB due to increased target pairing, we assessed the ability of an rne-131 mutant to suppress the defect in RyhB stability in the ΔpcnB strain. Introduction of the rne-131 mutation suppressed the defect in RyhB stability caused by the pcnB deletion in both the fur+ (Table 1; Fig. 5D,F) and Δfur background (Table 1; Fig. 1C,D) leading to a significant increase in RyhB stability in both strains. Additionally, RyhB steady-state levels were up-regulated by 1.9- and 1.5-fold in Δfur rne-131 and Δfur ΔpcnB rne-131 mutants, respectively relative to the Δfur strain (Fig. 1A,B). Interestingly, the rne-131 mutation failed to suppress the RyhB stability defect in a Δhfq (Table 1; Fig. 5D,F) or a Δfur Δhfq strain background (Table 1; Fig. 1C,D), indicating that the CTD of RNase E was not critical for the degradation of sRNAs not associated with Hfq.
Since introduction of the rne-131 mutation into the Δfur ΔpcnB strain suppressed the defect in RyhB stability, we examined whether this mutation also suppressed the defect in regulation of sodB and sdhCDAB mRNAs by RyhB. As shown in Figure 3B, we found that introduction of the rne-131 allele had a modest ability to suppress the defect in RyhB-mediated regulation of sodB in the ΔpcnB strain (compare 2.2-fold repression in the Δfur ΔpcnB strain to 3.9-fold in the Δfur ΔpcnB rne-131 strain). The ability of the rne-131 mutation to suppress the defect in RyhB-mediated regulation of the sdhCDAB transcript was demonstrated by a 15.8-fold decrease in sdhC in the Δfur ΔpcnB rne-131 strain relative to the Δfur ΔpcnB rne-131 ΔryhB strain. In contrast, sdhC levels were 1.7-fold lower in the Δfur ΔpcnB strain relative to the Δfur ΔpcnB ΔryhB strain (Fig. 3C).
Finally, we examined whether the defect in the ability of RyhB to regulate sdhCDAB mRNA in the Δfur ΔpcnB strain relative to the Δfur parental strain had any impact on its ability to grow on succinate as a sole carbon source. As mentioned above, it has been demonstrated that a Δfur mutant is unable to grow on succinate minimal medium due to constitutive RyhB repression of the sdhCDAB transcript. As expected, deletion of hfq in a Δfur background allowed growth on succinate minimal medium with a yield similar to that observed for a wild-type (fur+) strain at the end of 24 h and 48 h of growth (Supplemental Fig. S7). Although the growth yields between the Δfur ΔpcnB and the Δfur Δhfq strains differed significantly at the end of 24 h, the pcnB deletion strain nonetheless reached growth yields comparable to that of an hfq deletion strain after 48 h (Supplemental Fig. S7). Consistently, the rne-131 mutation suppressed the succinate growth phenotype observed for a Δfur ΔpcnB strain as indicated by a failure of the Δfur ΔpcnB rne-131 triple mutant to grow on succinate minimal medium (Supplemental Fig. S7).
Increased levels of 3′ETSLeuZ drive RyhB decay in the absence of poly(A) polymerase
Based on our findings, we hypothesized that PAP I facilitates the decay of a certain pool of RyhB target RNAs, which otherwise accumulate in the absence of PAP I, pair with RyhB, and drive its degradation via an RNase E-dependent decay pathway. Recent RNA-seq studies (Maes et al. 2017) identified a potential list of RNAs that serve as substrates for polyadenylation in E. coli. One of RyhB targets, the LeuZ precursor tRNA encoded by leuZ, was shown to be up-regulated in a ΔpcnB mutant (Maes et al. 2017) and to be polyadenylated downstream from the Rho-independent transcription terminator present at the end of its 3′ external transcribed spacer (3′ETSLeuZ) (Li and Deutscher 2002; Ow and Kushner 2002). Furthermore, recent studies also demonstrated that overexpression of 3′ETSLeuZ results in reduced levels of RyhB via pairing mediated decay (Lalaouna et al. 2015a,b). Based on these data, we first assessed the steady-state levels of 3′ETSLeuZ in a ΔpcnB mutant strain under RyhB inducing (+dipyridyl) and noninducing (−dipyridyl) conditions. Northern blot analysis showed that in the pcnB deletion strain, steady-state levels of 3′ETSLeuZ were consistently higher relative to a wild-type strain in both the presence and absence of RyhB induction (Fig. 6A). Next, we investigated whether the increased levels of 3′ETSLeuZ in a pcnB mutant strain were driving RyhB decay. To test this, we introduced into pcnB+ and ΔpcnB strains a mutant 3′ETSLeuZ (3′ETSleuZ mut) in which 4 nt shown to be critical for pairing with RyhB (Lalaouna et al. 2015b) were replaced with the complementary base (Fig. 6B). These nucleotide changes in the 3′ETSLeuZ unexpectedly resulted in a decrease in expression to levels undetectable by northern blots (Fig. 6C; Supplemental Fig. S8); however, this result did not preclude us from assessing the impact of the 3′ETSLeuZ on RyhB expression in the pcnB+ and ΔpcnB strains. To test this, we then assayed for RyhB stability after transcription inhibition in the pcnB+ and ΔpcnB strains that express the wild-type or mutated 3′ETSLeuZ. As shown in Figure 6D,E and Table 1, the RyhB stability defect observed in a ΔpcnB mutant was completely suppressed by introduction of these mutations in the pairing region of 3′ETSLeuZ. Consistently, the introduction of the 3′ETSleuZ mut also restored the defect in RyhB steady-state levels of a ΔpcnB mutant to wild-type levels (Fig. 6C; Supplemental Fig. S8). Taken together our data demonstrated that the increased decay of RyhB in the absence of PAP I was due to pairing with 3′ETSLeuZ.
FIGURE 6.

3′ETSLeuZ drives RyhB decay in the absence of poly(A) polymerase. (A) Northern blot analysis to determine the steady-state levels of 3′ETSLeuZ and RyhB in a wild-type (WT [fur+]; NRD1138) and the derived ΔpcnB strain (NRD1198) under RyhB inducing and noninducing conditions. Experiments were performed as described in the legend of Figure 5B,C. The primary glyW-cystT-leuZ transcript and the derived processing products that were detected in the northern blot are shown to the right of blot image. (B) Schematic showing complementarity (highlighted in gray) between RyhB and its target 3′ETSLeuZ, and the specific mutations (in orange) introduced in 3′ETSLeuZ to create a LeuZ variant (3′ETSLeuZ mut) unable to pair with RyhB sRNA. (C) Northern blot analysis to determine steady-state levels of 3′ETSLeuZ in wild-type and derived isogenic pcnB and 3′ETSleuZ mutant strains under RyhB inducing and noninducing conditions, as described above. Northern blots showing transcript steady-state levels of 3′ETSLeuZ and RyhB in a WT (fur+; NRD1138) and derived isogenic mutants 3′ETSleuZ WT (fur+ leuZ-kan; NRD1579), 3′ETSleuZ mut (fur+ 3′ETSleuZ mut-kan; NRD1581), ΔpcnB (fur+ ΔpcnB; NRD1198), ΔpcnB 3′ETSleuZ WT (fur+ ΔpcnB leuZ-kan; NRD1585), ΔpcnB 3′ETSleuZ mut (fur+ ΔpcnB 3′ETSleuZ mut-kan; NRD1587). ssrA was used as the loading control. Experiment was performed in triplicate and representative blots are shown. LeuZ term probe (Supplemental Table S2) was used to determine LeuZ and 3′ETSLeuZ levels. (D,E) Determination of RyhB intrinsic stability in a strain encoding the wild-type 3′ETSLeuZ (3′ETSleuZ WT; NRD1579) or mutant 3′ETSLeuZ (3′ETSleuZ mut; NRD1585) and derived ΔpcnB strains, NRD1581 and NRD1587, respectively. RNA stability time-course experiments were performed as described in the legend of Figure 4. Representative northern blots are shown in C. (D) RyhB decay curves were generated as described in Figure 4 and corresponding half-life measurements are listed in Table 1. RyhB decay curves of WT (NRD1138) and its derived isogenic ΔpcnB strain (NRD1198) are included in D as a reference. Points and error bars in the curves represent the means and the standard errors (SEM) of at least three independent experiments. Probes used are listed in Supplemental Table S2.
DISCUSSION
More than two decades ago, a role for PAP I in mediating the decay of anti-sense sRNAs was discovered (He et al. 1993; Xu et al. 1993). A contemporaneous study demonstrated that PAP I also drives mRNA decay by providing a signal for other ribonucleases (O'Hara et al. 1995). Subsequent work demonstrated a role for PAP I in mediating the decay of anti-sense sRNAs and sRNAs not associated with Hfq (Dam Mikkelsen and Gerdes 1997; Söderbom et al. 1997; Söderbom and Wagner 1998; Szalewska-Palasz et al. 1998; Viegas et al. 2007; Maes et al. 2017). Here, we provide for the first time evidence that PAP I increases the stability of two Hfq-binding sRNAs, RyhB and MicA. Absence of PAP I led to a decrease in RyhB stability (Figs. 1D, 4F) resulting in a defect in RyhB-mediated gene regulation (Fig. 3). These findings indicated a previously unknown role of PAP I in regulating sRNA function and thus raised interesting questions about the mechanism by which this protein can contribute to sRNA stability.
Mechanism of poly(A) polymerase mediated stabilization of sRNAs
How does PAP I stabilize Hfq-dependent sRNAs such as RyhB? One possibility was that PAP I-dependent stabilization of RyhB resulted from direct polyadenylation of the sRNA leading to an increase in sRNA binding to Hfq, thereby protecting it against RNase E cleavage. This mechanism would be analogous to the mechanism that has been described for PAP I in assisting mRNAs in binding Hfq, i.e., Rho-independent terminators at the 3′ ends of mRNAs are preferentially polyadenylated by the Hfq-PAP I complex, which subsequently results in formation of a stable complex between Hfq and mRNAs (Mohanty and Kushner 2008; Mohanty et al. 2004; Régnier and Hajnsdorf 2013). However, our data does not support this hypothesis. Firstly, we failed to detect poly(A) tails at the ends of full-length RyhB sRNA using 3′ RACE (Supplemental Fig. S9). This result is consistent with the findings of a recent study examining the global landscape of polyadenylated RNAs in E. coli, in which no Hfq-dependent sRNAs were among the polyadenylated transcripts identified (Maes et al. 2017). In fact, the sRNAs that were found to be polyadenylated in this study were either anti-sense sRNAs (i.e., sRNAs that regulate mRNAs transcribed from the opposing DNA strand) or sRNAs that bind the RNA chaperone ProQ. Secondly, the relative amounts of RyhB that coimmunoprecipitated with Hfq between a ΔpcnB mutant and a wild-type strain under iron starvation were not significantly different (Fig. 4C,D). Altogether, these findings indicated that PAP I mediated stabilization of RyhB was indirect.
Our finding that the RyhB stability defect in a ΔpcnB mutant was suppressed by introduction of a Y25D substitution in the distal face of Hfq (Fig. 4E,F) which is known to be important for facilitating sRNA–mRNA target interactions (Zhang et al. 2013; Schu et al. 2015) instead pointed toward a model where PAP I is stabilizing RyhB by protecting it from pairing-mediated decay. Further support for this model came from our results showing that RyhB stability in the ΔpcnB mutant increased to wild-type levels (Table 1; Fig. 5D,E) by introduction of a mutation in ryhB (ryhBmut) that blocked pairing with its target RNAs (Fig. 5A–C).
What was the RNA target that was accumulating in the absence of PAP I and driving the decay of RyhB? Over a hundred distinct RNA targets have been shown to be regulated by RyhB (Massé et al. 2005; Lalaouna et al. 2015b; Wang et al. 2015; Melamed et al. 2016); therefore, there were many possible candidates that could have been driving the decay and depletion of RyhB in the ΔpcnB strain background. Interestingly, recent work by Maes et al. (2017) identified 14 distinct RNA targets of RyhB that were subjected to polyadenylation in E. coli. Furthermore, a subset of these RyhB targets such as leuZ, cspB, sdhC, sdhD, sdhA, sdhB, fur, and uof were also reported to be up-regulated in the absence of PAP I (Mohanty and Kushner 2006; Maes et al. 2017). Of note here is the LeuZ precursor tRNA encoded by leuZ, which was one of the RyhB targets that was highly up-regulated in a ΔpcnB mutant (Maes et al. 2017). Past studies also pointed toward a critical role of PAP I in promoting the degradation of the 3′ETSLeuZ by exoribonucleases following its excision from the LeuZ precursor tRNA by RNase E (Li and Deutscher 2002; Ow and Kushner 2002). More importantly, it was recently shown that 3′ETSLeuZ can pair with RyhB sRNA and down-regulate RyhB transcript levels, which in turn leads to decreased regulation of RyhB targets including the sodB and sdhCDAB mRNAs (Lalaouna et al. 2015a,b). Based on these findings, we hypothesized that PAP I promotes the exoribonucleolytic decay of the 3′ETSLeuZ through polyadenylation of this transcript (Fig. 7A). Furthermore, we postulated that in the absence of PAP I, 3′ETSLeuZ accumulates and drives RNase E-mediated decay of RyhB as a result of base-pairing with this sRNA leading to loss of regulation of its other target mRNAs including sodB and sdhCDAB (Fig. 7B). In support of this hypothesis, we found that introduction of a mutation in 3′ETSleuZ (3′ETSleuZ mut) that was previously shown to block pairing of 3′ETSLeuZ with RyhB sRNA (Lalaouna et al. 2015b) suppressed the RyhB stability defect of a ΔpcnB mutant (Table 1; Fig. 6D,E) and led to a significant increase in RyhB steady-state levels (Fig. 6C; Supplemental Fig. S8). Decreased RyhB stability in the ΔpcnB mutant subsequently resulted in an impaired regulation of the sodB and sdhCDAB mRNAs (Figs. 3, 5B; Supplemental Fig. S2, S3, S6, S7). This reduced regulation of sdhCDAB by RyhB in the absence of PAP I has a measurable impact on E. coli physiology resulting in the ability of a ΔpcnB Δfur strain to grow on succinate as the sole carbon source (Supplemental Fig. S7).
FIGURE 7.

Model for poly(A) polymerase-mediated regulation of RyhB stability. (A) Poly(A) polymerase (PAP I) stabilizes RyhB by facilitating the degradation of 3′ETSLeuZ (green-colored RNA) through the addition of poly(A) tails, which provide a toe-hold for exoribonucleases. As a result, RyhB is abundant and able to bind and efficiently regulate other mRNA targets (collectively represented by the red-colored RNA), which are subsequently degraded by RNase E along with RyhB. (B) In a strain lacking poly(A) polymerase, 3′ETSLeuZ (green-colored RNA) accumulates and binds to RyhB leading to RNase E-mediated decay of this sRNA. RyhB levels are thereby reduced, and certain mRNA targets (red-colored RNA) accumulate that would otherwise be negatively regulated by this sRNA.
Interestingly, several examples have now been described of target RNAs that drive the decay of sRNAs resulting in reduced regulation of other mRNAs. The first reported example in this category in E. coli and Salmonella enterica was described by Plumbridge et al. (2014) who demonstrated that pairing of an Hfq-dependent sRNA (ChiX) with one target mRNA (chbBC) drove the decay of that sRNA. This depletion of ChiX resulted in impaired regulation of another one of its targets, the chiP mRNA (Figueroa-Bossi et al. 2009). Subsequently, Miyakoshi et al. (2015) demonstrated that the SroC sRNA generated in the decay of the gltIJKL transcript in E. coli and Salmonella enterica functioned to titrate GcvB levels (Miyakoshi et al. 2015).
Mechanisms by which poly(A) polymerase and Hfq block RNase E-mediated decay of sRNAs are distinct
The endoribonuclease RNase E is a key protein involved in degrading both Hfq-dependent sRNAs and cleaving target mRNAs upon sRNA–mRNA pairing (De Lay et al. 2013). The N-terminal domain of RNase E contains the catalytic site, but can also bind RNA through its S1 domain and the 5′ sensor domain as well as its active site residues (Garrey et al. 2009). The C-terminal domain (CTD) of RNase E contains two RNA binding sites, the ARRBD and AR2 regions, in addition to binding sites for other proteins including enolase, the RNA helicase RhlB, and the 3′ → 5′ exoribonuclease PNPase (Mackie 2013). The results of two previous studies suggested that Hfq interacts directly with the CTD of RNase E (Morita et al. 2005; Ikeda et al. 2011), but detailed biochemical studies indicate that Hfq interacts with the CTD via RNA (Worrall et al. 2008). The CTD was also previously shown to be important for coupled degradation of RyhB with its target mRNAs (Massé et al. 2003; Desnoyers and Massé 2012). It is notable that the defect in RyhB stability that was observed in a ΔpcnB mutant was suppressed by introduction of an rne-131 allele encoding an RNase E lacking the CTD (Figs. 1C,D, 5D,F), whereas introduction of an rne-131 mutation failed to suppress the RyhB stability defect of an Δhfq mutant (Figs. 1C,D, 5D,F). Since RyhB decay in the ΔpcnB strain was due to pairing with the 3′ETSLeuZ, these results demonstrated a requirement for the CTD of RNase E for degradation of RyhB upon pairing with the 3′ETSLeuZ, which may indicate that one or both of the RNA binding regions, ARRBD and AR2, mediate the recruitment of this sRNA to RNase E after pairing with target RNAs. However, an alternative interpretation of our results is that the RNA degradosome was required for the degradation of RyhB after sRNA–mRNA pairing as the CTD is important for the assembly of the RNA degradosome, which includes enolase, the RNA helicase RhlB, and PNPase. In contrast, the inconsequential role of the CTD in decay of RyhB in the Δhfq strain may signify that the N-terminal catalytic domain of RNase E is sufficient to cleave RyhB when it is not associated with Hfq or RNA targets.
Impact of poly(A) polymerase on a subset of Hfq-dependent sRNAs
As mentioned above, sRNAs can be classified into two distinct classes based on their interactions with Hfq (Zhang et al. 2013; Schu et al. 2015). Class I sRNAs interact with proximal face and rim of Hfq, and their mRNA targets bind the distal face of Hfq (Fig. 4E). In contrast, Class II sRNAs interact with the proximal and distal faces of Hfq, and their target mRNAs bind along the rim. The fact that deletion of pcnB led to a defect in the stability of MicA in addition to RyhB (two Class I sRNAs) but did not significantly impact the turnover of GcvB or Class II sRNAs such as ChiX, CyaR, and MgrR (Fig. 2A–E; Supplemental Fig. S1) is interesting. Whether this difference in the ability of PAP I to impact the stability of MicA and RyhB, but not ChiX, CyaR, and MgrR was due to the fact that these sRNAs interact with Hfq differently or was a coincidence will need to be resolved in future studies. Regardless, the ability of PAP I to impact a specific subset of sRNAs by a mechanism that is distinct from Hfq suggests an additional layer of complexity in sRNA-mediated gene regulation.
MATERIALS AND METHODS
Bacterial strains and growth conditions
All strains and plasmids used in this study are derivatives of E. coli K-12 strain MG1655 (rph-1) and are listed in Supplemental Table S1. Primers used for strain construction are listed in Supplemental Table S2. Strain construction is described in Supplemental Materials and Methods.
All strains were grown in liquid medium or agar plates containing either Lennox broth (LB), M9 minimal medium supplemented with 0.001% vitamin B1 and 0.2% glucose or succinate, or MOPS EZ rich defined medium (Teknova) supplemented with 0.4% glycerol instead of glucose. Antibiotics were used in the following final concentrations: ampicillin, 100 µg/mL; kanamycin, 25 µg/mL; chloramphenicol, 25 µg/mL or 10 µg/mL (for any mutant(s) containing hfq deletion), tetracycline, 12.5 µg/mL, zeocin, 25 µg/mL, and rifampicin, 250 µg/mL. 2, 2′ dipyridyl was added to liquid medium at a final concentration of 250 µM. All liquid cultures and bacteria on solid medium were grown aerobically at 37°C. Overnight cultures were diluted 1:200 fold in appropriate medium and grown until desired densities were reached. Growth was determined by measuring the optical densities of liquid cultures at 600 nm (OD600). Cultures were considered to be in exponential phase when they reached OD600 between 0.3 and 0.4 in LB or OD600 of ∼1.0 in MOPS EZ rich defined medium.
RNA extraction
Total RNA was extracted from exponential phase cultures growing either in LB or in MOPS EZ rich defined medium using hot phenol lysis method described previously (Massé et al. 2003). Briefly 700 µL of samples were removed from growing cultures and added to a mixture containing 800 µL of acid phenol–chloroform-isoamyl alcohol (pH of 4.3; Fisher Scientific) and 100 µL of lysis buffer (320 mM sodium acetate [pH 4.6], 8% SDS, 16 mM EDTA) equilibrated to 65°C. Samples were mixed at 65°C for 5 min and centrifuged for 30 min at 4°C to separate phases. The upper aqueous phase was extracted a second time with equal volume of neutral phenol–chloroform-isoamyl alcohol (pH of 6.7; Fisher Scientific). RNA was alcohol-precipitated and resuspended in DEPC-treated water. RNA concentration was measured using Nano Drop 2000 (Thermo Fisher Scientific).
RNA stability assay
To determine RNA stabilities for sRNAs in different mutants constructed in the fur+ background (Supplemental Table S1) all cultures were grown to exponential phase. RyhB or MicA expression were induced from a ryhB promoter by the addition of dipyridyl (iron chelator) while CyaR expression was induced from its native promoter by the addition of cyclic AMP (Adenosine 3′, 5′- cyclic monophosphate sodium salt monohydrate, Sigma) at a 5 mM final concentration. Under exponential growth conditions expression of MgrR, GcvB, and ChiX is constitutive. After 15 min of sRNA expression a culture sample (T0) was collected. Following that, rifampicin was added to inhibit all further transcription and additional samples were collected 1, 2, 4, and 6 min after rifampicin addition. To determine RyhB stability in mutants constructed in a fur deletion background strains were grown in MOPS EZ rich defined medium to exponential phase and a culture sample was collected (T0). Rifampicin was added to all cultures to inhibit transcription and additional samples were collected at 2, 5, 10, 15, and 30 min after rifampicin addition. All samples were subjected to RNA extraction as described above.
Northern blot analysis
Two micrograms of each RNA sample was loaded on 5% or 10% Criterion TBE-urea precast gels (Bio-Rad) and electrophoresed at 70 V. Next, the RNA samples were transferred to a Zeta-Probe GT membrane (Bio-Rad) using a Trans-Blot SD semidry transfer apparatus (Bio-rad) following manufacturer's guidelines. Transferred RNA was UV crosslinked and hybridized overnight with 100 ng/mL of 5′ biotinylated DNA probe (Supplemental Table S2) in ULTRAhyb (Ambion) hybridization buffer at 42°C. Blots were developed using a BrightStar BioDetect kit protocol (Ambion), imaged with a ChemiDoc MP imager (Bio-Rad) and quantified using Image Lab software version 5.2.1 (Bio-Rad). Signal intensity corresponding to each sRNA or mRNA was normalized to that of either ssrA or 5S rRNA, which served as internal loading controls. Decay curves corresponding to RNA stability time course experiments were generated by using GraphPad Prism version 5.0.
Quantitative RT-PCR (qRT-PCR) analysis
RNA extracted from exponential growth phase cultures as described above was subjected to DNase treatment (DNase Turbo; Ambion) following manufacturer's protocol. Sample mixtures (total reaction volume of 100 µL) were incubated for 1 h at 37°C and reaction was stopped by addition of 100 µL of DEPC-treated water and 200 µL of neutral phenol–chloroform-isoamyl alcohol (Fisher). DNase treated RNA samples were phenol extracted, alcohol precipitated and RNA concentration was measured as described above. Samples were then tested for the presence of any contaminating DNA by PCR using gene specific qRT-PCR primers (Supplemental Table S2) before proceeding to downstream applications. One microgram of DNA-free RNA was reverse transcribed using random hexamers and Superscript III Reverse Transcriptase (RT) (Invitrogen) following manufacturer's protocol. For each sample a no RT (NRT) control reaction was performed. cDNA samples were diluted 1:100-fold and 2 µL of diluted samples (resulting from both RT- and NRT- PCR) were used in a qRT-PCR reaction mixture containing 10 µL of iTaq Universal SYBR Green Supermix (Bio-Rad) and 2 µL each of 4 µM qPCR primers (Supplemental Table S2). A single no template control (NTC) for each qPCR primer pair used in this study was also included. Data were collected using an CFX connect Real Time thermocycler (Bio-Rad) running the SYBR Green with melt curve program modified as per the manufacturer's recommendations. Reactions were performed with two technical duplicates using cDNA samples from at least three independent biological replicates per strain and ssrA was used as the internal reference for normalization. The ΔΔCt method was used to calculate fold changes of transcripts corresponding to target genes in different sets of mutants relative to the wild-type parent. Statistical analysis was performed using one-way ANOVA (and nonparametric) in GraphPad Prism version 5.0.
Protein extraction and western blot analysis
Overnight cultures of wild-type and mutant strains were subcultured into fresh 10 mL LB and grown to exponential phase. Protein extraction was performed as described previously (De Lay and Gottesman 2011). Briefly, 1.0 mL of culture from each strain was subjected to TCA precipitation, washed once with 80% cold acetone solution, air dried, suspended in 2× Laemmli sample buffer (Bio-Rad) containing freshly added 5% (vol/vol) β-mercaptoethanol and heated at 95°C for 10 min. Total protein amount was normalized to corresponding OD600 of each specific culture. Approximately 0.08 OD600 units of total protein from each strain was separated on a 4% stacking 10% resolving SDS-PAGE gel in 1× Tris-glycine SDS buffer at 120 V. Fractionated protein was then transferred to a 0.45 μm PVDF membrane (Thermo Scientific) at 15 V for 30 min using Trans-Blot SD semidry transfer apparatus (Bio-rad) following manufacturer's guidelines.
Hfq was detected using 1:5000 dilution of preabsorbed anti-Hfq antiserum obtained from Dr. Susan Gottesman (NCI) and goat anti-rabbit IgG secondary antibody. For detection of DnaK (loading control) 1:10,000 dilution of mouse anti-DnaK monoclonal antibody (Abcam) and anti-mouse goat secondary antibody (Santa Cruz Biotechnologies, Inc.) were used following manufacturer's guidelines. All secondary antibodies were conjugated to alkaline phosphatase and were visualized by using Immun-Star AP substrate (Bio-Rad) and ChemiDoc MP imager (Bio-Rad). Signal intensity was quantitated using Image Lab software (Bio-Rad).
Hfq coimmunoprecipitation
Immunoprecipitation with Hfq was performed as described previously (Bandyra et al. 2016) with few modifications. Briefly, overnight cultures were diluted 200-fold into 30 mL of fresh LB liquid medium and grown to an OD600 of ∼0.3. Dipyridyl was added to induce RyhB expression for 15 min and 25 mL aliquots of cells were pelleted, washed, and frozen as previously described (Zhang et al. 2003). Immunoprecipitations were performed as previously described (Zhang et al. 2002) using anti-Hfq antiserum obtained from Dr. Susan Gottesman (NCI). RNA was isolated by phenol extraction and northern blots were performed as described in Materials and Methods.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gisela Storz for her comments on the manuscript. This work, including the efforts of D.S., L.M.M., T.A.C., and N.R.D., was funded by the McGovern Medical School (University of Texas Health Science Center at Houston: Startup Funds) and HHS|NIH|National Institute of General Medical Sciences (NIGMS; R01GM121368). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.067181.118.
Freely available online through the RNA Open Access option.
REFERENCES
- Andrade JM, Arraiano CM. 2008. PNPase is a key player in the regulation of small RNAs that control the expression of outer membrane proteins. RNA 14: 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade JM, Pobre V, Matos AM, Arraiano CM. 2012. The crucial role of PNPase in the degradation of small RNAs that are not associated with Hfq. RNA 18: 844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyra KJ, Said N, Pfeiffer V, Górna MW, Vogel J, Luisi BF. 2012. The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol Cell 47: 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyra KJ, Sinha D, Syrjanen J, Luisi BF, De Lay NR. 2016. The ribonuclease polynucleotide phosphorylase can interact with small regulatory RNAs in both protective and degradative modes. RNA 22: 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardill JP, Hammer BK. 2012. Non-coding sRNAs regulate virulence in the bacterial pathogen Vibrio cholerae. RNA Biol 9: 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovskyy M, Vanderpool CK. 2013. Regulation of bacterial metabolism by small RNAs using diverse mechanisms. Annu Rev Genet 47: 209–232. [DOI] [PubMed] [Google Scholar]
- Caldelari I, Chao Y, Romby P, Vogel J. 2013. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med 3: a010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan AJ, Aurikko JP, Ilag LL, Günter Grossmann J, Chandran V, Kühnel K, Poljak L, Carpousis AJ, Robinson CV, Symmons MF, et al. 2004. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J Mol Biol 340: 965–979. [DOI] [PubMed] [Google Scholar]
- Cameron TA, De Lay NR. 2016. The phosphorolytic exoribonucleases polynucleotide phosphorylase and RNase PH stabilize sRNAs and facilitate regulation of their mRNA targets. J Bacteriol 198: 3309–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam Mikkelsen N, Gerdes K. 1997. Sok antisense RNA from plasmid R1 is functionally inactivated by RNase E and polyadenylated by poly(A) polymerase I. Mol Microbiol 26: 311–320. [DOI] [PubMed] [Google Scholar]
- De Lay N, Gottesman S. 2011. Role of polynucleotide phosphorylase in sRNA function in Escherichia coli. RNA 17: 1172–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay N, Schu DJ, Gottesman S. 2013. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J Biol Chem 288: 7996–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnoyers G, Massé E. 2012. Noncanonical repression of translation initiation through small RNA recruitment of the RNA chaperone Hfq. Genes Dev 26: 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Storz G. 2010. Reprogramming of anaerobic metabolism by the FnrS small RNA. Mol Microbiol 75: 1215–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felden B, Cattoir V. 2018. Bacterial adaptation to antibiotics through regulatory RNAs. Antimicrobial Agents Chemother 62: e02503-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felden B, Vandenesch F, Bouloc P, Romby P. 2011. The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog 7: e1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Bossi N, Valentini M, Malleret L, Fiorini F, Bossi L. 2009. Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev 23: 2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrey SM, Blech M, Riffell JL, Hankins JS, Stickney LM, Diver M, Hsu YH, Kunanithy V, Mackie GA. 2009. Substrate binding and active site residues in RNases E and G: role of the 5′-sensor. J Biol Chem 284: 31843–31850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpel M, Heidrich N, Mäder U, Krügel H, Brantl S. 2010. A dual-function sRNA from B. subtilis: SR1 acts as a peptide encoding mRNA on the gapA operon. Mol Microbiol 76: 990–1009. [DOI] [PubMed] [Google Scholar]
- Gonzalez GM, Durica-Mitic S, Hardwick SW, Moncrieffe MC, Resch M, Neumann P, Ficner R, Görke B, Luisi BF. 2017. Structural insights into RapZ-mediated regulation of bacterial amino-sugar metabolism. Nucleic Acids Res 45: 10845–10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripenland J, Netterling S, Loh E, Tiensuu T, Toledo-Arana A, Johansson J. 2010. RNAs: regulators of bacterial virulence. Nat Rev Microbiol 8: 857–866. [DOI] [PubMed] [Google Scholar]
- Hajnsdorf E, Régnier P. 2000. Host factor Hfq of Escherichia coli stimulates elongation of poly(A) tails by poly(A) polymerase I. Proc Natl Acad Sci 97: 1501–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Söderbom F, Wagner EG, Binnie U, Binns N, Masters M. 1993. PcnB is required for the rapid degradation of RNAI, the antisense RNA that controls the copy number of ColE1-related plasmids. Mol Microbiol 9: 1131–1142. [DOI] [PubMed] [Google Scholar]
- Hébrard M, Kröger C, Srikumar S, Colgan A, Händler K, Hinton JC. 2012. sRNAs and the virulence of Salmonella enterica serovar Typhimurium. RNA Biol 9: 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist E, Wagner EGH. 2017. Impact of bacterial sRNAs in stress responses. Biochem Soc Trans 45: 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Yagi M, Morita T, Aiba H. 2011. Hfq binding at RhlB-recognition region of RNase E is crucial for the rapid degradation of target mRNAs mediated by sRNAs in Escherichia coli. Mol Microbiol 79: 419–432. [DOI] [PubMed] [Google Scholar]
- Jørgensen MG, Thomason MK, Havelund J, Valentin-Hansen P, Storz G. 2013. Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev 27: 1132–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JT, Alleyne TM, Schiano CA, Jafari N, Lathem WW. 2011. Global discovery of small RNAs in Yersinia pseudotuberculosis identifies Yersinia-specific small, noncoding RNAs required for virulence. Proc Natl Acad Sci 108: E709–E717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaouna D, Carrier MC, Massé E. 2015a. The shock absorber: preventing sRNA transcriptional noise. Cell Cycle 14: 2539–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaouna D, Carrier MC, Semsey S, Brouard JS, Wang J, Wade JT, Massé E. 2015b. A 3′ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol Cell 58: 393–405. [DOI] [PubMed] [Google Scholar]
- Le Derout J, Folichon M, Briani F, Deho G, Régnier P, Hajnsdorf E. 2003. Hfq affects the length and the frequency of short oligo(A) tails at the 3′ end of Escherichia coli rpsO mRNAs. Nucleic Acids Res 31: 4017–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lease RA, Woodson SA. 2004. Cycling of the Sm-like protein Hfq on the DsrA small regulatory RNA. J Mol Biol 344: 1211–1223. [DOI] [PubMed] [Google Scholar]
- Li Z, Deutscher MP. 2002. RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA 8: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie GA. 2013. RNase E: at the interface of bacterial RNA processing and decay. Nat Rev Microbiol 11: 45–57. [DOI] [PubMed] [Google Scholar]
- Maes A, Gracia C, Innocenti N, Zhang K, Aurell E, Hajnsdorf E. 2017. Landscape of RNA polyadenylation in E. coli. Nucleic Acids Res 45: 2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandin P, Chareyre S, Barras F. 2016. A regulatory circuit composed of a transcription factor, IscR, and a regulatory RNA, RyhB, controls Fe-S cluster delivery. mBio 7: e00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann B, van Opijnen T, Wang J, Obert C, Wang YD, Carter R, McGoldrick DJ, Ridout G, Camilli A, Tuomanen EI, et al. 2012. Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog 8: e1002788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Gottesman S. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci 99: 4620–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Escorcia FE, Gottesman S. 2003. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17: 2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Vanderpool CK, Gottesman S. 2005. Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol 187: 6962–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Salvail H, Desnoyers G, Arguin M. 2007. Small RNAs controlling iron metabolism. Curr Opin Microbiol 10: 140–145. [DOI] [PubMed] [Google Scholar]
- McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41: e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed S, Peer A, Faigenbaum-Romm R, Gatt YE, Reiss N, Bar A, Altuvia Y, Argaman L, Margalit H. 2016. Global mapping of small RNA-target interactions in bacteria. Mol Cell 63: 884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakoshi M, Chao Y, Vogel J. 2015. Regulatory small RNAs from the 3′ regions of bacterial mRNAs. Curr Opin Microbiol 24: 132–139. [DOI] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR. 2006. The majority of Escherichia coli mRNAs undergo post-transcriptional modification in exponentially growing cells. Nucleic Acids Res 34: 5695–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR. 2008. Rho-independent transcription terminators inhibit RNase P processing of the secG leuU and metT tRNA polycistronic transcripts in Escherichia coli. Nucleic Acids Res 36: 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR. 2011. Bacterial/archaeal/organellar polyadenylation. Wiley Interdiscip Rev RNA 2: 256–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR. 2016. Regulation of mRNA decay in bacteria. Annu Rev Microbiol 70: 25–44. [DOI] [PubMed] [Google Scholar]
- Mohanty BK, Maples VF, Kushner SR. 2004. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli. Mol Microbiol 54: 905–920. [DOI] [PubMed] [Google Scholar]
- Moll I, Afonyushkin T, Vytvytska O, Kaberdin VR, Bläsi U. 2003. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 9: 1308–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller T, Franch T, Udesen C, Gerdes K, Valentin-Hansen P. 2002a. Spot 42 RNA mediates discoordinate expression of the E. coli galactose operon. Genes Dev 16: 1696–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller T, Franch T, Hojrup P, Keene DR, Bachinger HP, Brennan RG, Valentin-Hansen P. 2002b. Hfq: a bacterial Sm-like protein that mediates RNA–RNA interaction. Mol Cell 9: 23–30. [DOI] [PubMed] [Google Scholar]
- Morita T, Maki K, Aiba H. 2005. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev 19: 2176–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara EB, Chekanova JA, Ingle CA, Kushner ZR, Peters E, Kushner SR. 1995. Polyadenylylation helps regulate mRNA decay in Escherichia coli. Proc Natl Acad Sci 92: 1807–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow MC, Kushner SR. 2002. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev 16: 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannuri A, Vakulskas CA, Zere T, McGibbon LC, Edwards AN, Georgellis D, Babitzke P, Romeo T. 2016. Circuitry linking the catabolite repression and Csr global regulatory systems of Escherichia coli. J Bacteriol 198: 3000–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker A, Gottesman S. 2016. Small RNA regulation of TolC, the outer membrane component of bacterial multidrug transporters. J Bacteriol 198: 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker A, Cureoglu S, De Lay N, Majdalani N, Gottesman S. 2017. Alternative pathways for Escherichia coli biofilm formation revealed by sRNA overproduction. Mol Microbiol 105: 309–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterman N, Lavi-Itzkovitz A, Levine E. 2014. Large-scale mapping of sequence-function relations in small regulatory RNAs reveals plasticity and modularity. Nucleic Acids Res 42: 12177–12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer V, Papenfort K, Lucchini S, Hinton JC, Vogel J. 2009. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat Struct Mol Biol 16: 840–846. [DOI] [PubMed] [Google Scholar]
- Plumbridge J, Bossi L, Oberto J, Wade JT, Figueroa-Bossi N. 2014. Interplay of transcriptional and small RNA-dependent control mechanisms regulates chitosugar uptake in Escherichia coli and Salmonella. Mol Microbiol 92: 648–658. [DOI] [PubMed] [Google Scholar]
- Prévost K, Desnoyers G, Jacques JF, Lavoie F, Massé E. 2011. Small RNA-induced mRNA degradation achieved through both translation block and activated cleavage. Genes Dev 25: 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Régnier P, Hajnsdorf E. 2013. The interplay of Hfq, poly(A) polymerase I and exoribonucleases at the 3′ ends of RNAs resulting from Rho-independent termination: a tentative model. RNA Biol 10: 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards GR, Vanderpool CK. 2011. Molecular call and response: the physiology of bacterial small RNAs. Biochim Biophys Acta 1809: 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Larrabeiti O, Plágaro AH, Gracia C, Sevillano E, Gallego L, Hajnsdorf E, Kaberdin VR. 2016. A new custom microarray for sRNA profiling in Escherichia coli. FEMS Microbiol Lett 363: fnw131. [DOI] [PubMed] [Google Scholar]
- Rutherford ST, Bassler BL. 2012. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med 2: a012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvail H, Massé E. 2012. Regulating iron storage and metabolism with RNA: an overview of posttranscriptional controls of intracellular iron homeostasis. Wiley Interdiscip Rev RNA 3: 26–36. [DOI] [PubMed] [Google Scholar]
- Schu DJ, Zhang A, Gottesman S, Storz G. 2015. Alternative Hfq-sRNA interaction modes dictate alternative mRNA recognition. EMBO J 34: 2557–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlyarova N, Shamovsky I, Bharati BK, Epshtein V, Chen J, Gottesman S, Schroeder R, Nudler E. 2016. sRNA-mediated control of transcription termination in E. coli. Cell 167: 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderbom F, Wagner EG. 1998. Degradation pathway of CopA, the antisense RNA that controls replication of plasmid R1. Microbiol 144(Pt 7): 1907–1917. [DOI] [PubMed] [Google Scholar]
- Söderbom F, Binnie U, Masters M, Wagner EG. 1997. Regulation of plasmid R1 replication: PcnB and RNase E expedite the decay of the antisense RNA, CopA. Mol Microbiol 26: 493–504. [DOI] [PubMed] [Google Scholar]
- Soper TJ, Woodson SA. 2008. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA 14: 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43: 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalewska-Palasz A, Wrobel B, Wegrzyn G. 1998. Rapid degradation of polyadenylated oop RNA. FEBS Lett 432: 70–72. [DOI] [PubMed] [Google Scholar]
- Thomason MK, Fontaine F, De Lay N, Storz G. 2012. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol. Microbiol 84: 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, et al. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459: 950–956. [DOI] [PubMed] [Google Scholar]
- Updegrove TB, Zhang A, Storz G. 2016. Hfq: the flexible RNA matchmaker. Curr Opin Microbiol 30: 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch HM, Carpousis AJ. 1998. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev 12: 2770–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecerek B, Moll I, Bläsi U. 2007. Control of Fur synthesis by the non-coding RNA RyhB and iron-responsive decoding. EMBO J 26: 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas SC, Pfeiffer V, Sittka A, Silva IJ, Vogel J, Arraiano CM. 2007. Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 35: 7651–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9: 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Rennie W, Liu C, Carmack CS, Prévost K, Caron MP, Massé E, Ding Y, Wade JT. 2015. Identification of bacterial sRNA regulatory targets using ribosome profiling. Nucleic Acids Res 43: 10308–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136: 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters SA, McAteer SP, Kudla G, Pang I, Deshpande NP, Amos TG, Leong KW, Wilkins MR, Strugnell R, Gally DL, et al. 2017. Small RNA interactome of pathogenic E. coli revealed through crosslinking of RNase E. EMBO J 36: 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrall JA, Howe FS, McKay AR, Robinson CV, Luisi BF. 2008. Allosteric activation of the ATPase activity of the Escherichia coli RhlB RNA helicase. J Biol Chem 283: 5567–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Lin-Chao S, Cohen SN. 1993. The Escherichia coli pcnB gene promotes adenylylation of antisense RNAI of ColE1-type plasmids in vivo and degradation of RNAI decay intermediates. Proc Natl Acad Sci 90: 6756–6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Wassarman KM, Ortega J, Steven AC, Storz G. 2002. The Sm-like Hfq protein increases OxyS RNA interaction with target mRNAs. Mol Cell 9: 11–22. [DOI] [PubMed] [Google Scholar]
- Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. 2003. Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol 50: 1111–1124. [DOI] [PubMed] [Google Scholar]
- Zhang A, Schu DJ, Tjaden BC, Storz G, Gottesman S. 2013. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J Mol Biol 425: 3678–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YF, Han K, Chandler CE, Tjaden B, Ernst RK, Lory S. 2017. Probing the sRNA regulatory landscape of P. aeruginosa: post-transcriptional control of determinants of pathogenicity and antibiotic susceptibility. Mol Microbiol 106: 919–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Koestler BJ, Waters CM, Hammer BK. 2013. Post-transcriptional activation of a diguanylate cyclase by quorum sensing small RNAs promotes biofilm formation in Vibrio cholerae. Mol Microbiol 89: 989–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.