

The long-term use of antibiotics has led to the emergence of multidrug-resistant bacteria. A promising strategy to combat bacterial infections aims at hampering their adaptability to the host environment without affecting growth.
KEYWORDS: Pseudomonas aeruginosa, antivirulence, biofilm, clofoctol, clotrimazole, cystic fibrosis, drug repurposing, miconazole, pqs, quorum sensing
ABSTRACT
The long-term use of antibiotics has led to the emergence of multidrug-resistant bacteria. A promising strategy to combat bacterial infections aims at hampering their adaptability to the host environment without affecting growth. In this context, the intercellular communication system quorum sensing (QS), which controls virulence factor production and biofilm formation in diverse human pathogens, is considered an ideal target. Here, we describe the identification of new inhibitors of the pqs QS system of the human pathogen Pseudomonas aeruginosa by screening a library of 1,600 U.S. Food and Drug Administration-approved drugs. Phenotypic characterization of ad hoc engineered strains and in silico molecular docking demonstrated that the antifungal drugs clotrimazole and miconazole, as well as an antibacterial compound active against Gram-positive pathogens, clofoctol, inhibit the pqs system, probably by targeting the transcriptional regulator PqsR. The most active inhibitor, clofoctol, specifically inhibited the expression of pqs-controlled virulence traits in P. aeruginosa, such as pyocyanin production, swarming motility, biofilm formation, and expression of genes involved in siderophore production. Moreover, clofoctol protected Galleria mellonella larvae from P. aeruginosa infection and inhibited the pqs QS system in P. aeruginosa isolates from cystic fibrosis patients. Notably, clofoctol is already approved for clinical treatment of pulmonary infections caused by Gram-positive bacterial pathogens; hence, this drug has considerable clinical potential as an antivirulence agent for the treatment of P. aeruginosa lung infections.
INTRODUCTION
The discovery and development of new drugs for use in humans is a challenging task that usually requires decade-long laboratory experimentation followed by extensive clinical trials. This process is time-consuming and necessitates substantial economic investments with a high-risk of failure mostly due to the poor pharmacological and pharmaceutical properties of newly identified bioactive molecules. This is particularly discouraging for antibiotic discovery since the investment required cannot be adequately recovered because of the high rate at which resistance emerges (1). As a consequence, while the spread of multiresistant pathogens is accelerating at an unprecedented rate, the antibiotic discovery pipeline is running dry, with 15 big pharmaceutical companies of 18 abandoning antibacterial discovery programs in the last decade (2, 3).
The search for off-target activities in drugs already approved for human use is a promising strategy that could reduce the time and costs generally associated with conventional drug discovery processes, with a high probability of yielding bioavailable and safe compounds which can more easily and swiftly move into clinical trials (4, 5).
A number of studies have shown the promise of drug repurposing strategies for the identification of new antibacterial drugs (6, 7). Examples are gallium nitrate and 5-fluorouracil, conventionally used for the treatment of hypercalcemia and cancer, respectively, which display growth-inhibitory activities against certain Gram-negative and Gram-positive pathogens (8, 9). An alternative approach to the development of new antimicrobials is the inhibition of bacterial virulence, rather than growth (10). Recently, antivirulence activities have been identified in drugs already approved for use in humans (11). As an example, the antifungal compound 5-fluorocytosine inhibits virulence factor production in the Gram-negative human pathogen Pseudomonas aeruginosa both in vitro and in a mouse model of lung infection (12). Since antivirulence drugs attenuate rather than kill pathogens, they should in principle combat bacterial infections without exerting the strong selective pressure for resistance imposed by bactericidal antibiotics (10). The emergence of resistance is less likely to occur for drugs targeting bacterial social behaviors, such as the production of secreted virulence factors. Indeed, resistant mutants expressing extracellular factors that are shared by the members of the entire bacterial population are unlikely to experience a fitness advantage relative to susceptible clones (13). In this context, quorum sensing (QS) is considered to be a promising target for the identification and development of antivirulence drugs, since this intercellular communication system positively controls the expression of virulence factors in a number of different human pathogens, including P. aeruginosa (14, 15).
P. aeruginosa is one of the most problematic human pathogens in industrialized countries, since it causes a variety of severe infections, especially among hospitalized and immunocompromised patients (16, 17). These infections are difficult to treat due to the intrinsic and acquired antibiotic resistance of P. aeruginosa (18) that is further compounded by its ability to form antibiotic tolerant biofilms (19). P. aeruginosa is the predominant cause of morbidity and mortality in individuals with cystic fibrosis (CF), since it forms biofilms, thereby establishing chronic lung infections that are impossible to eradicate with antibiotic treatment (20). The necessity of new therapeutic options for the treatment of P. aeruginosa infections was highlighted in a recent World Health Organization report in which this pathogen is top ranked among pathogens for which new antibiotics are urgently needed (Priority 1: Critical [http://www.who.int/en/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed]).
As a consequence of its importance as a human pathogen, P. aeruginosa has been adopted as a model organism for QS inhibition studies. This bacterium is endowed with a complex QS network consisting of four interconnected systems (i.e., las, rhl, pqs, and iqs), which collectively control social behaviors and the expression of virulence determinants, such as secreted virulence factors, swarming motility, and biofilm formation (21, 22). Over the last decade, numerous compounds interfering with the P. aeruginosa QS circuitry have been identified, and their effectiveness as antivirulence drugs both in vitro and in vivo has boosted the research in the field (23). Unfortunately, most of the drugs identified thus far are cytotoxic or display unfavorable pharmacological properties, thus limiting their transfer to clinical practice (15).
To combine the advantages of drug-repurposing with the antivirulence approach, we previously showed that the anthelmintic drug niclosamide has potent antivirulence activity against P. aeruginosa (24). Niclosamide targets the las QS system, thereby decreasing the expression of las-controlled virulence factors and protecting Galleria mellonella larvae from P. aeruginosa infection (24).
In the present study we searched for inhibitors of the pqs QS system of P. aeruginosa among drugs already approved for human use.
The pqs QS system of P. aeruginosa is based on 2-alkyl-4-quinolones (AQs) as signal molecules, namely, 2-heptyl-3-hydroxy-4-quinolone (PQS), and its immediate precursor 2-heptyl-4-hydroxyquinoline (HHQ). Both HHQ and PQS can bind to and activate the transcriptional regulator PqsR (also known as MvfR). The PqsR/HHQ and PqsR/PQS complexes bind the PpqsA promoter region and trigger the transcription of the pqsABCDE-phnAB operon, coding for the enzymes required for the synthesis of HHQ. HHQ is in turn oxidized to PQS by the monooxygenase PqsH. Therefore, in common with other QS systems, HHQ and PQS act as autoinducers by generating an autoinductive feedback loop that accelerates their synthesis (25–28).
While HHQ only activates the expression of the pqsABCDE-phnAB operon, PQS has additional functionalities; it is an iron chelator, it participates in the formation of outer membrane vesicles, and it controls the expression of virulence genes via a PqsR-independent pathway (28–31).
The mechanism of action of the protein coded by the fifth gene of the pqsABCDE-phnAB operon, PqsE, is still poorly understood. PqsE is a pathway-specific thioesterase, which contributes to the synthesis of HHQ, although loss of its function can be compensated for by other thioesterases in a pqsE mutant (27). Notably, PqsE also positively controls the expression of multiple virulence factors in a P. aeruginosa genetic background in which it cannot participate in AQ biosynthesis, indicating that this protein has additional functions (29, 32, 33).
Overall, P. aeruginosa mutants defective in AQ synthesis/reception or in PqsE are severely attenuated in different plant and animal experimental models of infection (33–38). Moreover, AQs are detectable in the sputum, blood, and urine of individuals with CF, and their presence correlates with clinical status (39).
In this study, a convenient screening system has been developed and used to select for U.S. Food and Drug Administration (FDA)-approved drugs targeting the pqs QS system at multiple levels. This screening campaign led to the identification of the antifungal drugs clotrimazole and miconazole, as well as clofoctol, an antimicrobial compound commonly used to treat lung infections caused by Gram-positive bacteria, as inhibitors of pqs signaling, probably targeting the PqsR receptor protein. Phenotypic analyses performed in the laboratory strain PAO1 and in P. aeruginosa isolates from CF patients support the antivirulence potential of clofoctol, the most active inhibitor.
RESULTS
Development of a coculture-based system for monitoring pqs signaling activity.
A reporter system for monitoring the activity of the pqs QS system has been developed. This is based on the coculture between wild-type P. aeruginosa PAO1 (herein referred to as PAO1) and the AQ biosensor strain P. aeruginosa ΔpqsA PpqsA::luxCDABE (here referred to as AQ-Rep; see Table S1 in the supplemental material). AQ-Rep cannot synthesize AQs due to deletion of the pqsA biosynthetic gene and emits light only in response to exogenously provided AQs due to PqsR-dependent activation of the PpqsA::luxCDABE transcriptional fusion integrated in a neutral chromosomal site (31). Therefore, in the PAO1/AQ-Rep coculture system the AQ signal molecules produced by PAO1 induce bioluminescence, and hence pqs inhibitors interfering with each step of the pqs signaling circuit, including AQ biosynthesis or response, should reduce bioluminescence (Fig. 1A).
FIG 1.

Validation of the screening system. (A) Schematic representation of the coculture-based reporter system. The P. aeruginosa PAO1 strain (PAO1) produces AQ signal molecules which activate PpqsA::luxCDABE transcription, that results in light emission in the biosensor strain AQ-Rep. Drugs interfering with AQ biosynthesis or response are expected to reduce bioluminescence in the PAO1/AQ-Rep coculture, relative to the untreated samples. (B) Activity of the PAO1/AQ-Rep coculture system treated with the indicated concentrations of the pqs inhibitors methyl anthranilate (white bars) or farnesol (gray bars). The bioluminescence of the untreated PAO1/AQ-Rep coculture normalized to the cell density is considered 100%.
Preliminary experiments directed toward setting up the screening system revealed that maximal response of AQ-Rep to exogenous PQS was obtained after 5 h of incubation in microtiter plates (Fig. S1A), when this biosensor strain was inoculated at an optical density at 600 nm (OD600) wavelength of 0.1 (Fig. S1B). Cocultivation of AQ-Rep and PAO1 at different ratios and in different culture conditions showed that the highest bioluminescence signal was registered when AQ-Rep and PAO1 were inoculated in an ∼3:1 ratio (AQ-Rep and PAO1 OD600s of 0.1 and 0.03, respectively) (Fig. S1C), and the resulting coculture was incubated at 37°C with shaking (Fig. S1D). Therefore, the screening campaign has been set up under the above conditions to maximize the biosensor responsiveness to AQs and possibly to drugs interfering with AQ signaling.
The functionality of the PAO1/AQ-Rep coculture system for the identification of anti-pqs drugs was assessed using the commercially available compounds methyl anthranilate and farnesol. Methyl anthranilate inhibits AQs biosynthesis by competing with the HHQ precursor anthranilate for binding to PqsA (40), while farnesol decreases the expression of HHQ biosynthetic genes via an unknown mechanism (41). As expected, both methyl anthranilate and farnesol reduced bioluminescence from the PAO1/AQ-Rep coculture in a dose-dependent manner, with a 50% inhibitory concentration (IC50) of ca. 1 mM (Fig. 1B), in accordance with literature data (40, 41).
Identification of new anti-pqs drugs.
The PAO1/AQ-Rep coculture system was used to screen a library of 1,600 FDA-approved compounds with known biological activities selected for their high chemical and pharmacological diversity and safety in humans (PHARMAKON). In the primary screening, each drug was tested at two different concentrations, 20 and 200 μM, for the ability to reduce bioluminescence in the PAO1/AQ-Rep coculture. Since compounds from the library are dissolved in dimethyl sulfoxide (DMSO), untreated samples containing the same amount of DMSO as the treated samples were used as controls. Cell density and bioluminescence of the untreated samples were considered 100%, and the criteria for the selection of anti-pqs drugs were (i) inhibition of bioluminescence of ≥20% at 20 μM, (ii) inhibition of bioluminescence of ≥60% at 200 μM, and (iii) reduction in the cell density of ≤10% at both 20 and 200 μM. This primary screening led to the selection of 17 hits meeting these criteria (Fig. S2A) and possibly endowed with pqs inhibitory activity.
However, reduced bioluminescence in the samples treated with the selected drugs could be due to their effects on the enzymes involved in light generation or on ATP levels (42, 43). Since inhibition of PpqsA promoter activity in P. aeruginosa should decrease the production of the AQs HHQ and PQS, a secondary screening to test the ability of the 17 hits to reduce AQ production in PAO1 was performed. In this case, AQ levels were measured by means of the AQ-Rep biosensor strain in the spent medium from PAO1 cultures grown for 16 h in Luria-Bertani broth (LB) supplemented with the selected hits at concentrations of 20 or 200 μM or with corresponding amounts of DMSO. This analysis revealed that only three drugs specifically reduced the production of AQs in PAO1: clotrimazole, clofoctol, and miconazole (I-3, I-9, and I-14 in Fig. S2B, respectively). Two of the drugs identified, clotrimazole and miconazole, are antifungal compounds (44–47), while clofoctol is an antibacterial drug with efficacy in Gram-positive human lung infections (48–50) (Table 1).
TABLE 1.
Anti-pqs compounds identified by screening the PHARMAKON library of FDA-approved drugs

To confirm the results of the primary and secondary screening, clotrimazole, clofoctol, and miconazole were purchased from an alternative supplier (Sigma-Aldrich). These drugs did not inhibit PAO1 growth in Muller-Hinton broth or LB even at the highest concentration achievable in solution (i.e., MIC for clotrimazole, >1.6 mM; MICs for miconazole and clofoctol, >6.4 mM). Moreover, these drugs did not alter the growth profile of wild-type PAO1 and of the AQ-Rep biosensor strain up to the maximum concentration used in the primary and secondary screenings (i.e., 200 μM; Fig. S3).
The pqs inhibitory activity of the drug hits was retested in the PAO1/AQ-Rep coculture assay. Dose-response inhibition of PpqsA promoter activity was observed for the three drugs (Fig. 2A). These data generated IC50s of 39, 20, and 27 μM for clotrimazole, clofoctol, and miconazole, respectively (Table 1). The three hits had no effect on bioluminescence in a P. aeruginosa strain in which the expression of the luxCDABE operon for light emission is independent on the activity of the pqs signaling system (Fig. S4), ruling out the possibility that the inhibitory activity on the PAO1/AQ-Rep coculture was due to nonspecific inhibition of bioluminescence. Moreover, the three drugs confirmed their ability to reduce AQ production in PAO1 in a dose-dependent manner (Fig. 2B) in accordance with the repressive effect exerted on the PpqsA promoter.
FIG 2.
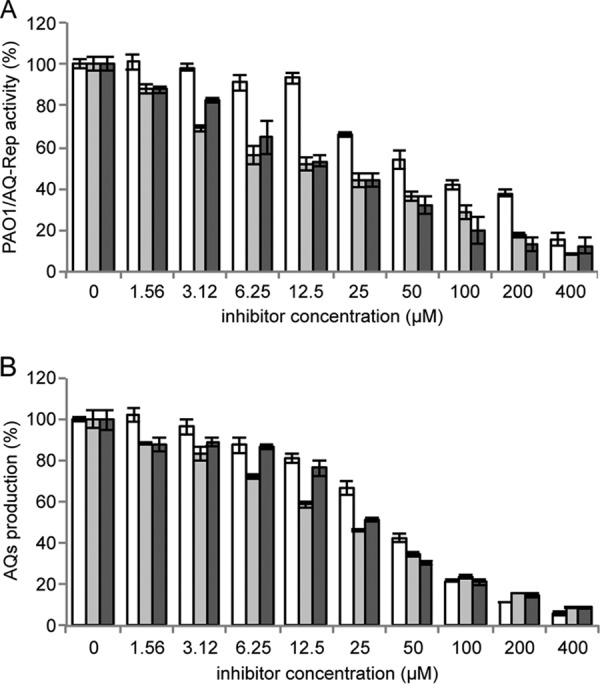
Clotrimazole, clofoctol, and miconazole inhibit PpqsA activity and AQ production. Effect of clotrimazole (white bars), clofoctol (light gray bars), and miconazole (dark gray bars) on the PAO1/AQ-Rep coculture system. Bioluminescence of the untreated PAO1/AQ-Rep coculture normalized to cell density is considered 100%. (B) Effect of clotrimazole (white bars), clofoctol (light-gray bars), and miconazole (dark gray bars) on AQ production in PAO1. The level of AQs produced by untreated PAO1 is considered 100%. For both panels A and B, the average from at least three independent experiments is reported, along with the SD.
The QS cascade in P. aeruginosa is a complex network of interwoven and hierarchical QS circuits (21, 22), and hence the effect of some compounds on the pqs QS system may be due to altered activity of the las and/or rhl QS systems. In particular, the las QS system is required for full activation of the pqs QS systems (36, 51–53), while RhlR has a negative impact on the pqs system by repressing PQS signal production through interference with the expression of pqsR and pqsABCDE (36, 54–56). Hence, reduced activity of the pqs QS system could be due to a negative or a positive effect of the hits on the las or the rhl QS systems, respectively. Therefore, possible effect of the three hits on these QS systems was investigated by using las- and rhl-specific biosensor strains. Clotrimazole, clofoctol, and miconazole did not decrease light emission in a reporter system in which the PAO1 wild type and the las-specific biosensor strain PA14 ΔlasI PrsaL::luxCDABE were cocultured (Fig. S5A) (57). Conversely, the three compounds slightly decreased (from 15 to 30% at 200 μM) light emission from a coculture system based on the PAO1 wild type and on the rhl-specific biosensor strain PAO1 ΔrhlI PrhlA::luxCDABE (Fig. S5B) (24). These data demonstrate that clotrimazole, clofoctol, and miconazole do not affect the las QS system, while these drugs have a slight negative effect on the rhl QS system. Considering that (i) the repressive effect exerted by the hits on the pqs QS system (Fig. 2A) occurs at lower concentration and is more pronounced than the repressive effect exerted by the same molecules on the rhl QS system (Fig. S5B) and that (ii) the pqs system exerts a positive effect on the rhl system (54, 58), these data support a primary activity of the hits on the pqs QS system that consequently reduces rhl activity. Overall, these data confirm that clotrimazole, clofoctol, and miconazole exert an anti-pqs activity without altering P. aeruginosa growth.
Characterization of the mechanism of action of the newly identified pqs inhibitors.
The inhibition of PpqsA activity in the PAO1/AQ-Rep coculture system (Fig. 2A) may be due to inactivation of AQ biosynthesis in the PAO1 strain or of AQ reception in both PAO1 and AQ-Rep strains (Fig. 1A). Similarly, the reduced AQ levels in PAO1 (Fig. 2B) could also be due to inhibition of either AQ biosynthesis or response, due to the PqsR-dependent regulatory loop governing transcription of the HHQ biosynthetic enzymes (36, 55).
To discriminate between these two possibilities, the effect of the three drugs on AQ production was tested in a PAO1 ΔpqsA ΔpqsH double-mutant strain (ΔpqsAH; Table S1) carrying the pFD-pqsABCD plasmid for constitutive expression of the HHQ biosynthetic enzymes. In this genetic background, in which AQ production does not depend on the ability of AQs to activate PpqsA via PqsR, the inhibitors did not reduce AQ levels, demonstrating that they do not affect the functionality of the enzymes required for HHQ biosynthesis (Fig. 3A). Moreover, the inhibitors were effective in reducing bioluminescence emission by the AQ-Rep biosensor strain grown in the presence of synthetic PQS (Fig. 3B), suggesting that the inhibitors target the PqsR-dependent AQ response rather than biosynthesis.
FIG 3.
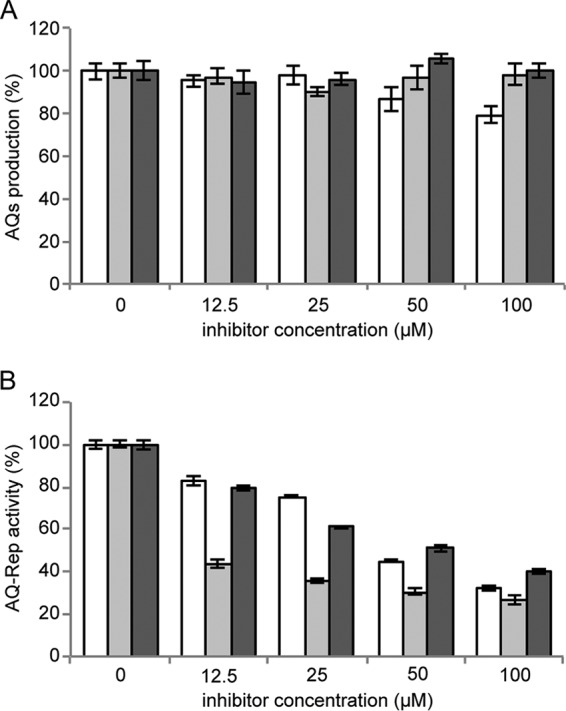
Clotrimazole, clofoctol, and miconazole inhibit AQ reception. (A) Production of AQs in P. aeruginosa PAO1 ΔpqsAH(pFD-pqsABCD) grown for 16 h in LB in the absence or in the presence of clotrimazole (white bars), clofoctol (light gray bars), and miconazole (dark gray bars). The AQ level measured in the untreated sample is considered 100%. (B) Activity of the AQ-Rep biosensor strain grown in LB supplemented with 10 μM synthetic PQS and clotrimazole (white bars), clofoctol (light gray bars), or miconazole (dark gray bars). The bioluminescence of the untreated AQ-Rep biosensor normalized to its cell density is considered 100%. For both panels A and B, the averages from at least three independent experiments are reported, along with the SD.
To validate this hypothesis, we investigated the effect of the hits on the levels of pqsR mRNA and PqsR protein. As shown in Fig. 4A, real-time reverse transcription-PCR (RT-PCR) analysis revealed that the hits do not affect pqsR mRNA levels. Moreover, Western immunoblotting showed that the inhibitors do not reduce PqsR protein levels in a PAO1 ΔpqsA ΔpqsH ΔpqsR triple mutant strain (ΔpqsAHR; Table S1) carrying the pPqsR-6H plasmid for IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible expression of a 6×His-tagged variant of PqsR (Fig. 4B) (59). Actually, clotrimazole increased PqsR levels, indicating that this drug has a positive effect on the translation of the pqsR mRNA or on PqsR stability. However, clotrimazole decreased PpqsA activity (Fig. 2A) and AQ production (Fig. 2B) and reduced the mRNA level of pqs-controlled genes, as demonstrated by real-time RT-PCR analysis performed on total mRNA extracted from PAO1 wild type grown in the absence or in the presence of 100 μM clotrimazole (Fig. S6). Overall, despite increasing PqsR level, clotrimazole seems to hamper the ability of this transcriptional regulator to activate gene expression.
FIG 4.
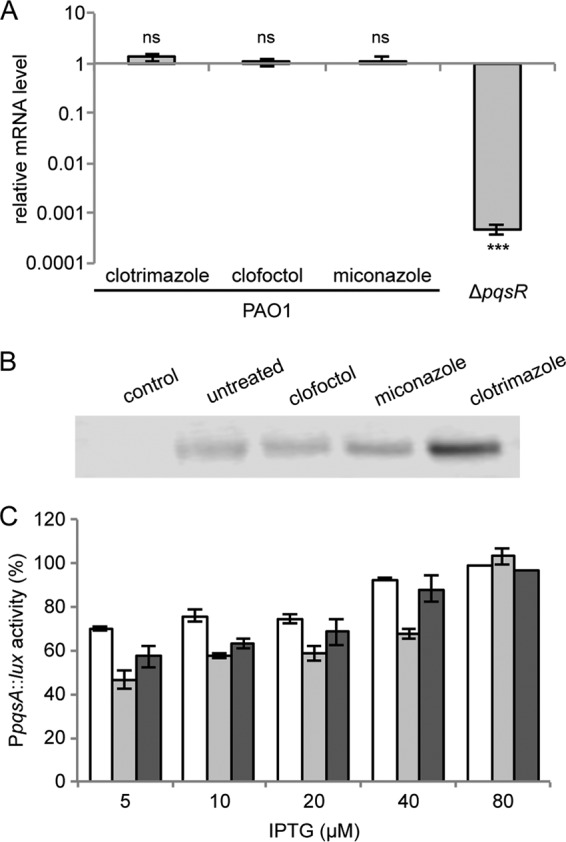
Clotrimazole, clofoctol, and miconazole inhibit PqsR functionality. (A) Real-time RT-PCR analysis showing the mRNA level of pqsR in PAO1 cultures treated with 100 μM concentrations of the indicated drugs relative to untreated PAO1 cultures. The PAO1 ΔpqsR strain was used as a negative control. The average from three independent experiments is reported, along with the SD. ns, nonsignificant difference; ***, P < 0.001 (ANOVA). (B) Western immunoblotting performed with anti-6×His antibody on crude protein extracts of PAO1 ΔpqsAHR(pPqsR-6H) grown in LB supplemented with 10 μM PQS and 20 μM IPTG, in the absence (untreated) or in the presence of the indicated drugs (100 μM). The PAO1 ΔpqsAHR strain carrying the empty vector pME6032 was used as a control. The data are representative of three independent experiments. (C) Effect of 100 μM clotrimazole (white bars), clofoctol (light gray bars), and miconazole (dark gray bars) on PpqsA::lux activity in the PAO1 ΔpqsA ΔpqsH ΔpqsR mutant carrying the pPqsR-6H plasmid, grown in LB supplemented with 10 μM PQS and different concentrations of IPTG as indicated in the graph. The averages from three independent experiments are reported, along with the SD.
To support PqsR as a target of the hits, we investigated their ability to reduce light emission from the PpqsA::luxCDABE transcriptional fusion in a PAO1 triple-mutant strain unable to synthesize AQs and to produce PqsR (i.e., PAO1 ΔpqsAHR), carrying the pPqsR-6H plasmid for IPTG-inducible expression of PqsR. Cultures of this strain were grown in LB supplemented with 10 μM PQS to induce PpqsA activity, with a fixed concentration of the hits (100 μM), and with increasing concentrations of IPTG. The rationale of this experiment is that increased expression of PqsR, due to increased concentration of IPTG, should decrease the repressive effect exerted by the hits on PpqsA, if PqsR is the target of the hits. As shown in Fig. 4C, the inhibitory effect exerted by the hits on PpqsA activity decreased in parallel to increasing IPTG concentration in the growth medium, thus supporting PqsR as their molecular target. Overall, these data indicate that each of the hits acts downstream of pqsR expression, likely hampering PqsR functionality.
To support the hypothesis that the inhibitors directly interact with PqsR, molecular docking simulations were performed based on the crystal structure of the PqsR coinducer binding domain (CBD) in the apo form (PDB 4JVC) (59). To increase the reliability of the simulations, the docking search space encompassed the entire CBD of PqsR, i.e., a “blind” docking procedure was carried out. Amino acid residues previously reported to be involved in the binding of the natural ligand 2-nonyl-4-hydroxy-quinoline (NHQ) to the PqsR CBD (59) were considered flexible (see Materials and Methods for details). This analysis indicated that the three hits bind PqsR with high affinity at the same site as the natural ligand NHQ (Fig. 5) with predicted ΔG values for binding of clotrimazole, clofoctol, and miconazole being −8.4, −9.8, and −8.5 kcal/mol, respectively. Interestingly, these values are lower than the predicted ΔG value for binding of NHQ (−7.9 kcal/mol; Table 1). Similar results were obtained when using the PqsR CBD structure bound to NHQ (PDB 4JVD) (59), from which the ligand was removed. In the latter case, the ΔG values for the binding of clotrimazole, clofoctol, miconazole, and NHQ were −9.4, −9.9, −8.1, and −8.1 kcal/mol, respectively. Finally, maintaining all the CBD residues in a fixed position yielded very similar results (data not shown). Interestingly, in each case the predicted affinity of the hits for PqsR parallels their efficacy as pqs inhibitors (Table 1).
FIG 5.
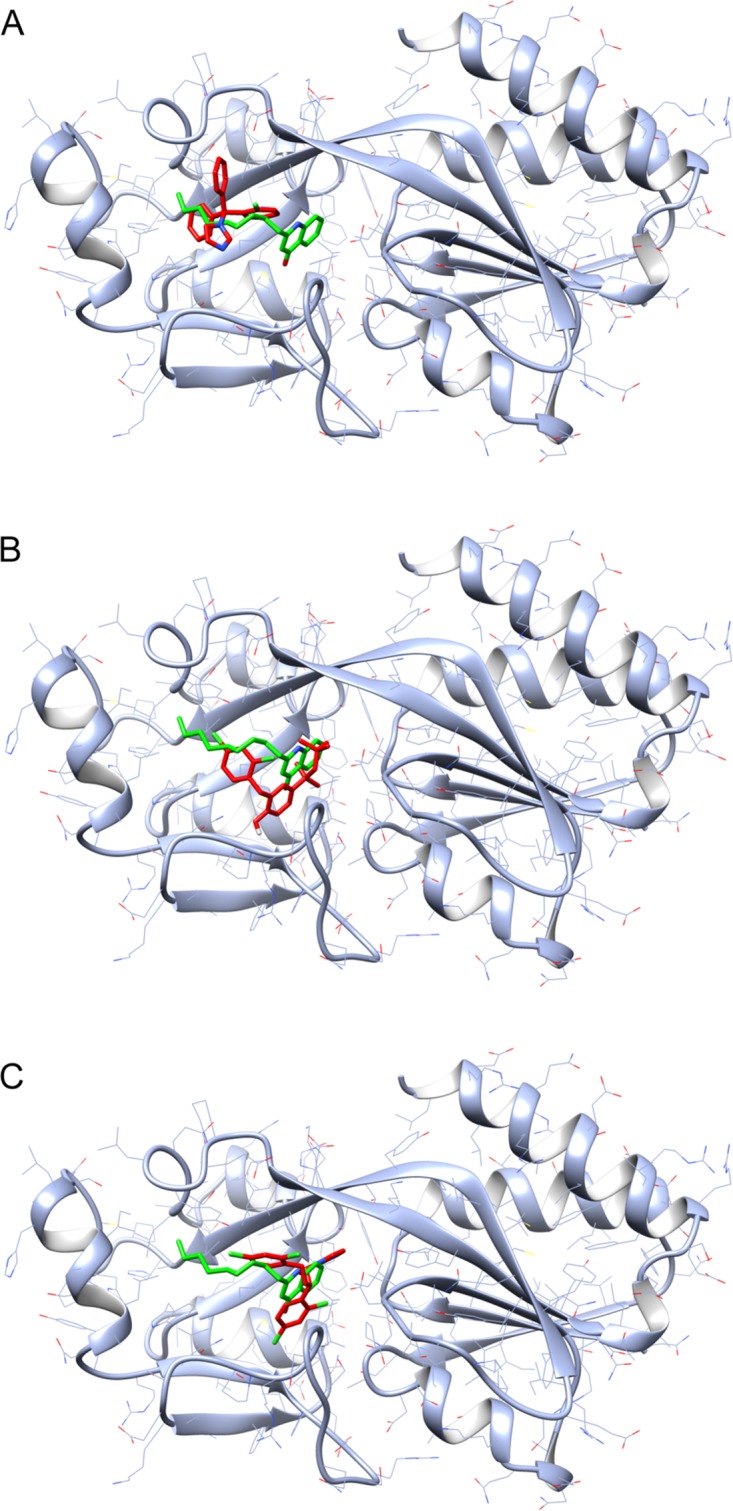
Putative complexes formed by clotrimazole, clofoctol, and miconazole with the PqsR CBD. Schematic representations of the complexes formed by clotrimazole (A), clofoctol (B), and miconazole (C) with the PqsR CBD, obtained by molecular docking simulations (see Materials and Methods for details), are shown. The three drugs are represented in red, while the natural ligand NHQ is represented in green.
Overall, these data suggest that the newly identified inhibitors could be endowed with a similar mechanism of action, that is to hamper PqsR functionality by competing with AQ agonists for PqsR binding. Also, the evidence that clotrimazole increases PqsR level (Fig. 4B) while hampering its ability to drive AQ production (Fig. 2B) and to activate pqs-controlled genes (Fig. 2A and S6) supports direct interaction of this hit to PqsR.
Notably, both activity assays and in silico predictions indicate that clofoctol has greater inhibitory activity relative to miconazole and clotrimazole (Table 1). To support competitive binding of PQS and clofoctol to PqsR, the ability of this drug to repress PpqsA activity was evaluated in the AQ-Rep biosensor grown in the presence of a range of concentrations of the native PqsR agonist PQS. This competition assay revealed the reduced ability of clofoctol to inhibit PpqsA activity in the presence of increasing concentrations of PQS (Fig. S7), in accordance with the activity of clofoctol as a competitive antagonist of the PQS receptor protein PqsR.
Clofoctol inhibits the expression of pqs-controlled virulence phenotypes.
By hampering the ability of PqsR to activate the transcription of the pqsABCDE-phnAB operon, clofoctol is expected to reduce the expression of virulence traits controlled by both PQS and PqsE in P. aeruginosa. First of all, since the assays previously performed to assess the effect of clofoctol on AQ production did not discriminate between HHQ and PQS, these QS signal molecules were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of spent media from PAO1 cultures treated with a range of concentrations of clofoctol. As shown in Fig. 6A, this analysis confirmed that clofoctol inhibits AQ production in P. aeruginosa, with both HHQ and PQS concentrations being significantly reduced by the drug.
FIG 6.

Clofoctol inhibits the expression of pqs-controlled virulence traits. (A) Concentrations of HHQ (white bars) and PQS (gray bars) measured by LC-MS/MS on supernatants of PAO1 cultures grown for 16 h in LB in the absence or in the presence of clofoctol at the indicated concentrations. The averages from three independent experiments are reported, along with the SD. **, P = 0.0062; ***, P < 0.001 (ANOVA). (B to D) Effect of 100 μM clofoctol on pyocyanin production (B), swarming motility (C), and biofilm formation (D) in PAO1. The same phenotypes were evaluated in the ΔpqsR mutant as a control. For pyocyanin production in panel B, the averages from three independent experiments are reported with the SD, and representative supernatants are shown in the inset picture. ***, P < 0.001 (ANOVA). For the swarming motility (panel C) and biofilm formation (panel D), representative pictures of three independent experiments are shown. (E) Real-time RT-PCR analysis showing the mRNA levels of the indicated genes in PAO1 treated with 100 μM clofoctol (white bars) and in the ΔpqsR mutant (gray bars) relative to untreated PAO1. The averages from three independent experiments are reported with the SD. **, P = 0.0012; ***, P < 0.001 (ANOVA).
With respect to the effect of clofoctol on PQS- and PqsE-controlled virulence determinants, phenotypic analyses revealed that 100 μM clofoctol leads to >80% reduction in pyocyanin (Fig. 6B) and considerably reduced swarming motility (Fig. 6C). Moreover, 100 μM clofoctol significantly reduced biofilm formation in a PAO1 strain constitutively expressing green fluorescent protein (GFP) via the pMRP9-1 plasmid (60) (Fig. 6D). Notably, the effect of clofoctol on the tested phenotypes in PAO1 mimicked deletion of the pqsR gene (ΔpqsR; Fig. 6B to D), in accordance with the hypothesis that PqsR is the clofoctol target.
Subsequently, real-time RT-PCR analyses were performed to examine the effect of clofoctol on the expression of pqs-controlled virulence genes (28). The PQS-dependent pvdS and pchR genes code for the PvdS and PchR regulatory proteins required for the synthesis of the siderophores pyoverdine and pyochelin, respectively (28, 61); the PqsE-dependent lecA gene codes for the LecA lectin involved in the formation of antibiotic-resistant biofilms (28, 62). As a control, the mRNA level of pqsA was also measured. Real-time RT-PCR analyses showed that clofoctol significantly decreased the mRNA level of each of the genes tested, in agreement with the downregulation observed in a PAO1 ΔpqsR mutant strain (Fig. 6E). The negative effect exerted by clofoctol on lecA transcription was also confirmed by promoter activity assay showing reduced activity of the PlecA::luxCDABE transcriptional fusion in PAO1 cultures treated with clofoctol (Fig. S8). Overall, these data support clofoctol as an antivirulence agent active against the P. aeruginosa pqs QS system.
Clofoctol protects Galleria mellonella larvae from P. aeruginosa infection and inhibits the pqs QS system in CF clinical isolates.
The antivirulence activity of clofoctol was tested in G. mellonella larvae, an insect infection model which correlates well with P. aeruginosa mouse infection models (63). First, G. mellonella was infected with ca. 10 cells of P. aeruginosa PAO1 or of the isogenic ΔpqsR mutant and incubated at 37°C for 120 h. As shown in Fig. 7A, mutation of pqsR significantly reduced the ability of P. aeruginosa to kill the larvae, demonstrating the suitability of this insect model to investigate the antivirulence potential of drugs targeting PqsR.
FIG 7.
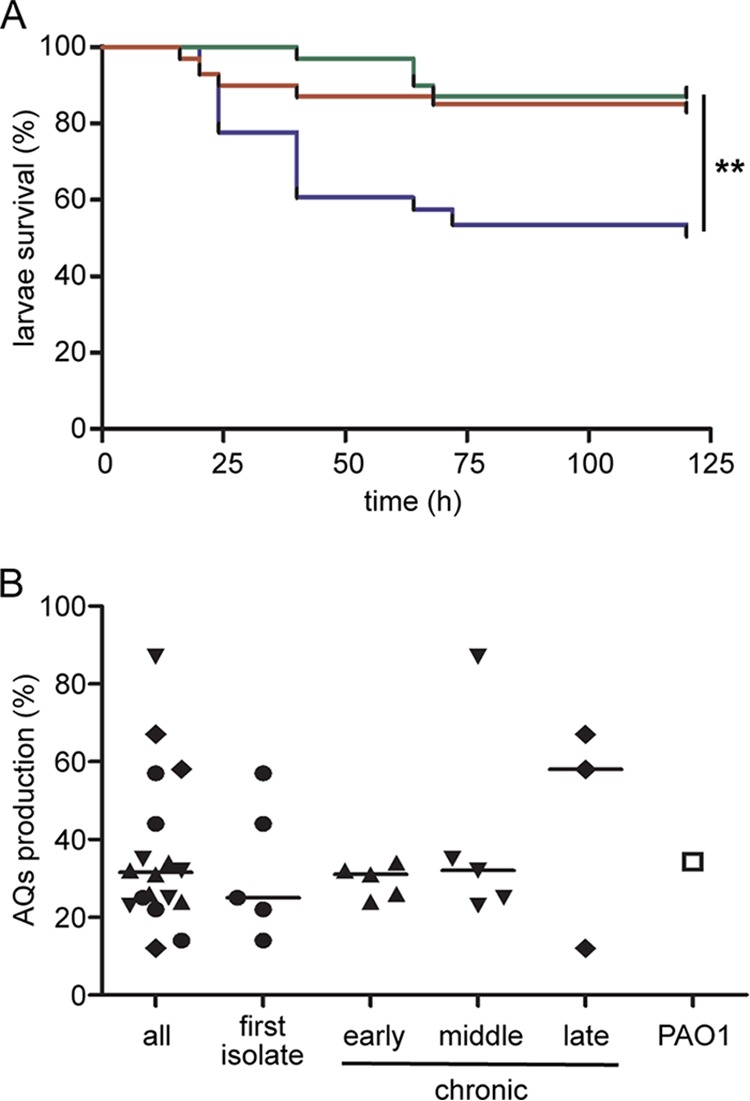
Clofoctol displays an antivirulence effect in vivo and inhibits the pqs QS system in P. aeruginosa CF clinical isolates. (A) Kaplan-Meier plot shows the percentage survival of G. mellonella larvae inoculated with P. aeruginosa PAO1 (blue line), with PAO1 and clofoctol at a final concentration of 100 μM (red line), or with ΔpqsR mutant (green line). The mean survival rate calculated from four independent experiments performed on at least 30 larvae per condition is reported. **, P = 0.0033 for PAO1 versus PAO1 plus clofoctol and P = 0.0016 for PAO1 versus ΔpqsR mutant (ANOVA). (B) Dot plot showing the inhibition of AQ production in P. aeruginosa CF isolates (filled symbols) and P. aeruginosa PAO1 (open square) treated with 100 μM clofoctol, relative to the untreated samples, which were considered 100%. Black lines represent median values: all, 31.4%; first isolate, 25.2%; early chronic, 31.1%; middle chronic, 32.1%; and late chronic, 57.8%. The AQ production in treated PAO1 cultures was 34.3% relative to untreated PAO1. Differences between the median values are not statistically significant. Mean results from three independent experiments are reported.
Since the average weight of G. mellonella larvae was ca. 500 mg, and arbitrarily assuming uniform dispersal of injected bacteria and clofoctol in 500 μl of larval volume (64, 65), 10 μl of saline containing 5 mM clofoctol was injected to yield 100 μM clofoctol in each larva. Preliminarily, we verified that the injection of 10 μl of saline containing 5 mM clofoctol did not affect the survival of uninfected larvae and that 2 h of incubation of P. aeruginosa with 5 mM clofoctol did not affect P. aeruginosa growth and viability (data not shown). Then, G. mellonella larvae were inoculated with P. aeruginosa PAO1 in the absence or in the presence of clofoctol. The treatment with clofoctol led to a survival percentage of 87%, similar to that observed with the ΔpqsR mutant (83%), while only 50% of untreated G. mellonella larvae survived PAO1 infection (Fig. 7A). Overall, these data demonstrate that clofoctol attenuates P. aeruginosa PAO1 lethality in G. mellonella.
To verify that clofoctol is active also against clinical P. aeruginosa strains, its ability to reduce AQ production was evaluated in a collection of 20 P. aeruginosa isolates from the lungs of CF patients, grouped into four categories with respect to the stage of infection (Table S2). A preliminary analysis revealed that only two strains isolated from patients with more than 15 years of chronic infection (chronic late group) did not produce detectable levels of AQs (Table S2); hence, these strains should be considered resistant to the antivirulence effect of clofoctol. The remaining 18 clinical isolates were grown in LB for 24 h in the absence or presence of 100 μM clofoctol, and the AQ concentration was determined in the corresponding spent media by using the AQ-Rep biosensor. Residual AQ production was estimated for each treated isolate relative to the amount of AQ detected in the corresponding untreated sample, considered 100%. Notably, clofoctol decreased AQ production in each of the clinical isolates tested, with a reduction ranging from 12.7 to 88.4% (Fig. 7B). The median reduction in AQ production in the tested isolates was 68.6% and hence comparable to the reduction in AQ levels measured in PAO1 treated with 100 μM clofoctol under the same conditions (65.7%; Fig. 7B). Differences in the median reduction values among the analyzed groups were not statistically significant. Moreover, differences in the median reduction of AQ production were not significant also when grouping the isolates according to their antibiotic resistance profiles (Table S2). Indeed, the median reductions of AQ levels were 71.6 and 67.4% in 4 antibiotic-susceptible and in 12 antibiotic-resistant strains, respectively (Fig. S9). Also the two multidrug-resistant or extensively drug-resistant strains analyzed in this study were susceptible to clofoctol, with reductions in AQ levels of 56.5 and 88.4%, respectively (Fig. S9). Although performed on a limited number of clinical isolates, this analysis indicates that clofoctol is effective in blocking the pqs QS system in CF strains, irrespective of their adaptation to the host environment during long-lasting chronic lung infection and of their antibiotic resistance profiles.
DISCUSSION
As a consequence of widespread antibiotic resistance, inhibition of virulence rather than growth has become a viable approach for combatting bacterial infections with lower selective pressure for emergence of resistance (10). In particular, in vitro evolution experiments suggest that resistant mutants will not emerge for drugs targeting public goods, such as virulence factors that are secreted and shared between individuals (66). Moreover, since antivirulence drugs target specific bacterial functions required for infection, these molecules are not expected to impact the beneficial resident microbiota relative to that of antibiotics (11, 13).
In many bacterial pathogens, QS positively controls the expression of multiple secreted virulence factors; hence, this communication system is considered a promising target for the development of antivirulence agents (23, 67). Since P. aeruginosa has four interconnected QS systems that positively control the production of virulence factors and biofilm formation, most of the research on QS inhibition has focused on this bacterium as a model system. Indeed, several molecules inhibiting the las QS system of P. aeruginosa have been identified (23, 67). Recently, a number of studies have described inhibitors of the pqs QS system. The pqs system positively controls the expression of multiple virulence determinants, including secreted virulence factors and biofilm formation, and pqs mutant strains display attenuated virulence in plant and animal models of infection (32–35, 38). Moreover, the pqs system is active during P. aeruginosa infections in humans (39, 68, 69).
Inhibitors of the pqs system were previously identified among analogs of anthranilate, the substrate of PqsA in the first step of the biosynthetic route leading to AQ production (37, 40). Subsequently, compounds binding to the AQ-biosynthetic enzyme PqsD were shown to act as potent pqs inhibitors, with IC50s in the low micromolar range (from 1 to 14 μM) (70, 71). The possibility of interfering with the pqs system via enzymatic degradation of the AQ signals, rather than via small molecules targeting their biosynthesis, was also explored, and PQS degrading activity has been described in Arthrobacter nitroguajacolicus and Achromobacter xylosoxidans (72, 73). However, the majority of anti-pqs molecules identified so far are competitive inhibitors of the transcriptional regulator PqsR. Potent PqsR antagonists with IC50s ranging from 0.4 to 38.5 μM have been found among analogs of the natural agonists HHQ and PQS (59, 74–76). Whole-cell high-throughput screening and structure-activity relationship analyses led to the identification of benzamide-benzimidazole PqsR inhibitors with low IC50s (<1 μM), some of which also inhibited the PqsBC complex (77–79). Also, 2-sulfonylpyrimidines were identified as hampering both AQ reception and biosynthesis (80). Overall, a number of reports validated the antivirulence potential of anti-pqs molecules, showing their ability to reduce the expression of pqs-controlled virulence traits both in vitro and in animal models of infection. Despite the promise of anti-pqs agents for the treatment of P. aeruginosa infections, to the best of our knowledge none of these molecules has thus far entered clinical trials. This is probably due to the poor pharmacological properties of the inhibitors, including possible cytotoxicity, and to the lack of ADME-TOX studies required for their evaluation in humans. In this context, searching for off-target activities in drugs already approved for use in humans represents a potential shortcut for developing new anti-pqs molecules that could move straight into clinical trials.
In this study, a drug-repurposing approach led to the identification of three promising anti-pqs drugs already used in humans by screening a library of 1,600 FDA-approved compounds (Table 1; Fig. S2). Data on the acute and chronic toxicity are already available for these drugs, as well as information on their pharmacokinetics. Clotrimazole and miconazole are antifungal drugs used in humans to treat ringworm, pityriasis versicolor, vaginal and oral candidiasis, and skin yeast infections (44, 45, 81, 82). They both alter the permeability of the fungal cell wall by binding to phospholipids and inhibiting the biosynthesis of ergosterol and other sterols required for fungal cell membrane integrity (83, 84). Miconazole displays its activity by inhibiting fungal peroxidases, which results in peroxide-mediated cell death (83). Both of these drugs are mainly administered as creams or ointments; thus, their current formulations could be particularly suitable for the topical treatment of chronic wound infections caused by P. aeruginosa (85, 86). However, this opportunistic pathogen is a main cause of lung infections especially in individuals with CF, where it establishes chronic infections that can last for decades (87). The use of clotrimazole and miconazole to treat P. aeruginosa lung infections would require their reformulation as inhalable nanosuspensions, an approach that has recently demonstrated its value for repurposing the anthelmintic drug niclosamide as an anti-QS agent against P. aeruginosa (24, 88).
Of the 1,600 compounds tested in this screening campaign, the most promising anti-pqs drug was clofoctol, an antimicrobial used for the treatment of acute and chronic upper respiratory tract infections and for tracheobronchial infections caused by Gram-positive pathogens, especially staphylococci, pneumococci, and streptococci (48, 50). Clofoctol is also used in preventive and curative treatment of otolaryngology and stomatology (89). The mechanism of action of this drug as an antimicrobial is still poorly understood, but a detrimental effect of clofoctol on membrane and cell wall biosynthesis in Gram-positive bacteria has been reported (49, 90). Clofoctol is usually administered as suppositories since it is well absorbed through the rectal mucosa and rapidly spreads through the tissues, reaching the highest concentrations in the respiratory system (91). Since clofoctol mainly acts in the airways, it is potentially valuable as a future treatment of P. aeruginosa lung infections. Notably, clofoctol is used to treat infections in infants, and this is another advantageous feature considering that in CF a P. aeruginosa lung infection is established in early life (92).
Overall, despite their lower potency compared to other pqs inhibitors described thus far, the anti-pqs drugs identified in this study have considerable potential for human use and could be directly tested in clinical trials or serve as chemical scaffolds for future drug optimization programs.
With respect to the mechanism of action of the three FDA-approved drugs, they all affect PqsR functionality, probably by competing with the natural ligands HHQ and PQS for the PqsR ligand-binding site (Fig. 3, 4, and S7). This hypothesis is supported by docking simulations, which predict that all three compounds bind to the PqsR coinducer binding domain in the same binding site as the natural ligand NHQ (Fig. 5). This result was somehow unexpected, since the PAO1/AQ-Rep coculture used in the screening campaign should primarily identify molecules affecting both AQ biosynthesis and AQ reception (Fig. 1A). Indeed, this coculture-based reporter system was functional in identifying the PqsA-inhibitor methyl anthranilate (Fig. 1B). Intriguingly, the anti-QS activity of the anthelmintic drug niclosamide was discovered using a coculture-based reporter system similar to the one deployed in this work. In common with clofoctol, niclosamide inhibited the QS signal molecule response rather than biosynthesis (24). Therefore, the selection of drugs targeting QS receptors could be a bias intrinsic to the screening system used. In fact, in coculture-based screening systems, drugs interfering with QS signal molecule receptor would have a dual outcome since they would block both QS signal receptor and consequently signal biosynthesis in the wild type, as well as inhibiting the QS receptor in the reporter strain. Conversely, an inhibitor of QS signal molecule biosynthesis would only affect the functionality of the P. aeruginosa wild-type strain. Hence, the PAO1/AQ-Rep coculture system may offer a more sensitive screen for PqsR inhibitors than for those that inhibit AQ biosynthesis, so that only drugs targeting PqsR will meet the selection criteria for the primary screen.
Since each of the hits identified in this study are likely to target PqsR, we focused our attention on the most potent inhibitor, clofoctol (Table 1).
Different elements of the pqs QS system have recently been shown to control distinct virulence traits. In particular, the PQS signal molecule drives the expression of genes required for the biosynthesis of siderophores and of genes coding for PrpL and AprX proteases, and exotoxin S, while PqsE is required for the production of pyocyanin, LecA and LecB lectins, hydrogen cyanide, rhamnolipids, and ChiC chitinase (28). With regard to pleiotropic virulence phenotypes such as swarming motility and biofilm formation, these appear to be regulated by both PQS and PqsE (33). Consistent with the activity of clofoctol as a PqsR inhibitor, the expression of both PQS-controlled virulence traits, such as the expression of genes required for siderophore biosynthesis (Fig. 6E), and of PqsE-dependent phenotypes, including pyocyanin production (Fig. 6B) and expression of the lecA gene (Fig. 6E and S8), were inhibited. Moreover, clofoctol reduced both swarming motility and biofilm formation (Fig. 6C and D). Notably, clofoctol exerted an antivirulence effect in vivo, since this drug-attenuated P. aeruginosa infection in G. mellonella larvae (Fig. 7A).
A major concern with respect to the use of anti-QS drugs for the treatment of CF pulmonary infection originates from evolutionary selection driving P. aeruginosa adaptation to the CF lung. Indeed, during chronic infections, CF isolates accumulate mutations that reduce the production of virulence factors, lead to the formation of mucoid biofilms, increase antibiotic resistance mainly as a consequence of efflux pump overexpression, and in some cases inactivate QS systems (93–96). Since P. aeruginosa QS-defective mutants should be considered resistant to anti-QS drugs, the suitability of QS inhibition for CF therapy is under debate. However, most studies have focused on the inactivation of the las QS system in chronic CF isolates, while little attention has so far been given to the pqs QS system (97–100). The evidence that AQs have been identified in the sputum of CF patients with both intermittent and chronic P. aeruginosa infections demonstrates unequivocally that the pqs QS system is active in the CF lung (68, 69, 99, 101). In addition, AQs can be detected in the sputum, plasma, and urine of ca. 80% of CF patients suffering with P. aeruginosa chronic lung infections. The levels of the AQ molecule NHQ increased at the start of a pulmonary exacerbation and positively correlated with quantitative measures of P. aeruginosa cells in the lung (39). This evidence is consistent with the results obtained in this study, since only 2 of the 20 clinical isolates tested did not produce detectable levels of AQs (Table S2). Notably, clofoctol reduced functionality of the pqs QS system in all the pqs-proficient CF isolates, irrespective of their antibiotic resistance profiles (Fig. 7B and S9).
Future analyses performed on a larger panel of P. aeruginosa clinical isolates from both CF and chronic wound patients and in vivo assays in murine models of infection are required to better assess the suitability of clofoctol, clotrimazole, and miconazole for the treatment of P. aeruginosa chronic infections. However, the results of this work should encourage further preclinical studies to aid transfer of the newly identified pqs inhibitors from the laboratory into clinical practice.
MATERIALS AND METHODS
Bacterial strains, media, and chemicals.
The bacterial strains, clinical isolates, plasmids, and oligonucleotides used in this study are listed in Tables S1, S2, S3, and S4, respectively. Bacterial strains were routinely grown at 37°C in Luria-Bertani broth (LB) with aeration and, when necessary, antibiotics were added at the following concentrations: tetracycline, 200 μg/ml; carbenicillin, 150 μg/ml; gentamicin, 100 μg/ml; and kanamycin, 200 μg/ml. When necessary, IPTG (isopropyl-β-d-thiogalactopyranoside) was added at the concentrations indicated in the text. Muller-Hinton broth and M9 minimal medium supplemented with 20 mM glucose as a carbon source were used in the MIC assay (Clinical and Laboratory Standards Institute) and in the biofilm assay, respectively. Synthetic HHQ and PQS stock solutions were prepared in methanol. Clotrimazole, clofoctol, and miconazole were purchased from Sigma-Aldrich and dissolved in dimethyl sulfoxide (DMSO).
Primary screening for the identification of pqs inhibitors.
P. aeruginosa PAO1 and the AQ-Rep biosensor strain (PAO1 ΔpqsA PpqsA::luxCDABE) were grown overnight at 37°C on LB agar plates. Bacteria were scraped from plate surfaces and diluted in LB to optical density at 600 nm (OD600) wavelengths of 0.1 and 0.03 for the biosensor and PAO1 strains, respectively (procedure modified from [57]). Aliquots (200 μl) of the coculture were grown at 37°C in 96-well microtiter plates in LB supplemented with each compound of the PHARMAKON library (20 μM and 200 μM). The OD600 values and relative light units (RLU) were measured after 5 h of incubation by using a Wallac 1420 Victor3V multilabel plate reader (Perkin-Elmer). Eight samples grown in the presence of DMSO (0.2 or 2%) were used as controls in each microtiter plate. The reporter activity was determined as the RLU/OD600 for each sample. The residual reported activity was determined in treated samples relative to control samples grown in the presence of DMSO, for which the value was considered 100%.
A similar approach was used to investigate the effect of the hits on the las and rhl QS systems. In this case, cocultures of the P. aeruginosa PAO1 wild-type strain and of the PA14-R3 (PA14 ΔlasI PrsaL::luxCDABE [57]) or the C4-Rep (PAO1 ΔrhlI PrhlA::luxCDABE; 24) biosensor strains were used, respectively.
Quantification of AQs.
Levels of AQ signal molecules in treated-P. aeruginosa PAO1 culture supernatants were determined by using the reporter strain AQ-Rep, as previously described (102). Bacterial cultures were grown in 96-well microtiter plates at 37°C with shaking. Supernatants were collected after 16 h for the experiments shown in Fig. 2B, 3A, and S2, or after 24 h for experiments shown in Fig. 7B and S9, to allow optimal AQ production in slow-growing clinical isolates. Briefly, 10 μl of culture supernatant was added to 190 μl of LB inoculated with AQ-Rep biosensor (final OD600 = 0.1) in 96-well microtiter plates. Microtiter plates were incubated at 37°C with gentle shaking, and the OD600 and RLU values were measured after 5 h of incubation. A calibration curve was generated by growing the AQ-Rep biosensor in the presence of increasing concentrations of synthetic HHQ or PQS; the resulting dose-response curve was used to calculate the concentration of the AQ signals in each culture supernatant.
AQs produced by P. aeruginosa PAO1 were also quantified in by LC-MS/MS analysis, as previously described (103). Briefly, PAO1 was inoculated into 5 ml of LB in the absence or in the presence of 100 μM clofoctol. After 16 h of incubation at 37°C with shaking, the cell density of the culture was recorded, and the supernatants were filter sterilized. Supernatants were solvent extracted with ethyl acetate, dried under vacuum, and redissolved in methanol prior to quantitative analysis by LC-MS/MS. For each sample, a supernatant concentration of HHQ and PQS was calculated by comparing analytic peak areas with a matched calibration line.
Pyocyanin production, swarming motility, and biofilm formation assays.
Pyocyanin was extracted and quantified from P. aeruginosa PAO1 and ΔpqsR grown in LB supplemented with 100 μM clofoctol or with DMSO as a control, as previously described (104). Swarming motility assays were performed on swarming plates (0.8% [wt/vol] nutrient broth N.2, 0.5% [wt/vol] glucose, 0.5% [wt/vol] bacteriological agar). Plates were supplemented with or without clofoctol (100 μM). After 16 h of growth at 37°C, swarming motility was directly observed at the air-agar interface.
For microscopic visualization of biofilms, P. aeruginosa PAO1 or ΔpqsR strains constitutively expressing GFP via the pMRP9-1 plasmid (60) were grown in an 8-well chamber slide, as previously described (105), with minor modifications. Briefly, bacterial cells were inoculated at an OD600 of 0.02 in 700 μl of M9 minimal medium supplemented with 20 mM glucose as carbon source, in the absence or in the presence of 100 μM clofoctol. Cultures were incubated at 30°C for 24 h to allow the adhesion of the bacterial cells to the glass surface. To maintain bacterial viability, the medium was changed every 24 h. Biofilm formation was examined after 3 days incubation by using the Leica TCS SP5 confocal microscope.
Western immunoblotting.
Crude protein extracts were collected from the P. aeruginosa PAO1 ΔpqsA ΔpqsH ΔpqsR triple-mutant strain carrying the pPqsR-6H plasmid grown in LB supplemented with 10 μM PQS and 20 μM IPTG, in the absence or in the presence of 100 μM clotrimazole, clofoctol, or miconazole. The P. aeruginosa PAO1 ΔpqsA ΔpqsH ΔpqsR strain carrying the pME6032 empty vector was used as a control. A Bradford assay (106) was used to quantify and normalize total protein content in the samples. Western immunoblotting was performed using a standard technique (107) with mouse anti-6His antibody (1:5,000; Sigma-Aldrich) and goat anti-mouse IgG horseradish peroxidase-conjugate as secondary antibody (1:6,000; Bio-Rad Laboratories). Final development was performed with Amersham ECL chemiluminescent reagents (Amersham Biosciences). A C-DiGit blot scanner (LI-COR Biosciences) was used for data acquisition.
RNA extraction and real-time RT-PCR analysis.
P. aeruginosa PAO1 and ΔpqsR were inoculated at an OD600 of 0.02 in 5 ml of LB in the absence or in the presence of 100 μM clotrimazole, clofoctol, or miconazole. Cultures were grown at 37°C with vigorous shaking until they reached an OD600 of 2.0, and then 1 ml of cells was harvested by centrifugation and resuspended in 2 ml of RNAprotect bacterial reagent (Qiagen). Total RNA extraction was performed with an RNeasy Mini Columns kit (Qiagen) according to the manufacturer's instructions, including the on-column DNase I digestion step. In addition, eluted RNA was treated for 1 h at 37°C with DNase Turbo (0.2 U per μg of RNA; Ambion) and with SUPERase-In (0.4 U per μg of RNA; Ambion). DNase I was removed using the RNeasy column purification kit (Qiagen). Purified RNA was quantified using the NanoDrop 2000 spectrophotometer (Thermo-Fisher Scientific). The absence of genomic DNA in the RNA samples was verified by PCR performed with the primers FWPpqsL and RVPpqsL (Table S4). cDNA synthesis was performed with the iScript reverse transcription supermix for RT-qPCR kit (Bio-Rad Laboratories) according to the manufacturer's instructions and quantified with NanoDrop 2000. Real-time RT-PCRs were performed using an iTaq Universal SYBR Green Supermix kit (Bio-Rad Laboratories) according of the manufacturer's instructions, and the Rotor Gene 6000 thermocycler (Corbett Research). Primers employed in real-time RT-PCR analysis were designed using the Primer-BLAST software (www.ncbi.nlm.nih.gov/tools/primer-blast) and are listed in Table S4. The reaction procedure involved incubation at 95°C for 1 min and 40 cycles of amplification at 95°C for 10 s and 60°C for 45 s. Fluorescence was registered in the last 15 s of the 60°C step. 16S rRNA was chosen as an internal control (housekeeping gene) to normalize the real-time RT-PCR data in each single run and to calculate the relative fold change in gene expression by using the 2−ΔΔCT method. The average data and standard deviations (SD) were calculated from three independent experiments.
Galleria mellonella killing assay.
The G. mellonella killing assay was performed as previously described (63, 65), with minor modifications. Briefly, G. mellonella caterpillars in the final instar larval stage (average weight, 486 ± 67 mg) were infected with 10 μl of saline containing about 10 bacterial cells in the absence or in the presence of 5 mM clofoctol. Although PAO1 cells were incubated in the presence of clofoctol for less than 5 min before injection, preliminary assays showed that 5 mM clofoctol treatment (for up to 24 h) does not significantly affect PAO1 cell or larval viability (data not shown). G. mellonella larvae were incubated at 37°C in petri dishes (ten larvae per dish) and monitored for 120 h. Larvae were considered dead when they did not respond to gentle prodding. At least 30 larvae per condition were used in four independent experiments. Survival curves for the G. mellonella killing assay were generated using the Kaplan-Meier method.
Molecular docking simulations.
Molecular docking simulations were carried out using DockingApp (108), a user friendly interface for the docking program AutoDock Vina (109). In all simulations, the search space (docking grid) included the whole PqsR coinducer binding domain (CBD) structure in order to carry out “blind” predictions of the “hit” compound binding sites.
Simulations were carried out on the apo (PDB 4JVC) and holo (PDB 4JVD) forms of the protein (59), both by keeping all protein residues rigid and by allowing flexibility only of the residues previously reported to be involved in PqsR binding to the natural ligand NHQ (i.e., ILE 149, ALA 168, VAL 170, ILE 186, LEU 189, LEU 207, LEU 208, PHE 221, ILE 236, TYR 258, ASP 264, and THR 265) (52).
Statistical analysis.
Statistical analysis was performed with the software GraphPad Prism 5, using one-way analysis of variance (ANOVA), followed by Tukey-Kramer multiple comparison tests. Differences with a P value of <0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Siri Ram Chhabra and Alex Truman (Centre for Biomolecular Sciences, University of Nottingham, Nottingham, United Kingdom) for HHQ and PQS synthesis and Susanne Fetzner (Institute of Molecular Microbiology and Biotechnology, University of Münster, Münster, Germany) for kindly providing the pBBR-pqsABCD plasmid.
This study was supported by the Italian Ministry for Education, University and Research (RBFR10LHD1_002 to G.R.), the Italian Cystic Fibrosis Research Foundation (FFC 21/2015 and FFC 18/2017 to P.V.; FFC 17/2018 to L.L.), Regione Lazio (LR 13/2008–FILAS-RU-2014-1009 to P.V.), and the Biotechnology and Biological Sciences Research Council, United Kingdom (BB/F014392/1 to P.W.). The Grant of Excellence Department, MIUR-Italy (ARTICOLO 1, COMMI 314-337 LEGGE 232/2016), is also gratefully acknowledged.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01296-18.
REFERENCES
- 1.Fernandes P, Martens E. 2017. Antibiotics in late clinical development. Biochem Pharmacol 133:152–163. doi: 10.1016/j.bcp.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 2.Ventola CL. 2015. The antibiotic resistance crisis: part 1: causes and threats. P T 40:277–283. [PMC free article] [PubMed] [Google Scholar]
- 3.Mohr KI. 2016. History of antibiotics research. Curr Top Microbiol Immunol 398:237–272. doi: 10.1007/82_2016_499. [DOI] [PubMed] [Google Scholar]
- 4.Ashburn TT, Thor KB. 2004. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 5.Mullard A. 2012. Drug repurposing programmes get lift off. Nat Rev Drug Discov 11:505–506. doi: 10.1038/nrd3776. [DOI] [PubMed] [Google Scholar]
- 6.Rangel-Vega A, Bernstein LR, Mandujano-Tinoco EA, García-Contreras SJ, García-Contreras R. 2015. Drug repurposing as an alternative for the treatment of recalcitrant bacterial infections. Front Microbiol 6:282. doi: 10.3389/fmicb.2015.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savoia D. 2016. New antimicrobial approaches: reuse of old drugs. Curr Drug Targets 17:731–738. doi: 10.2174/1389450116666150806124110. [DOI] [PubMed] [Google Scholar]
- 8.Gieringer JH, Wenz AF, Just HM, Daschner FD. 1986. Effect of 5-fluorouracil, mitoxantrone, methotrexate, and vincristine on the antibacterial activity of ceftriaxone, ceftazidime, cefotiam, piperacillin, and netilmicin. Chemotherapy 32:418–424. doi: 10.1159/000238445. [DOI] [PubMed] [Google Scholar]
- 9.Minandri F, Bonchi C, Frangipani E, Imperi F, Visca P. 2014. Promises and failures of gallium as an antibacterial agent. Future Microbiol 9:379–397. doi: 10.2217/fmb.14.3. [DOI] [PubMed] [Google Scholar]
- 10.Rasko DA, Sperandio V. 2010. Antivirulence strategies to combat bacteria-mediated disease. Drug Discov 9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 11.Rampioni G, Visca P, Leoni L, Imperi F. 2017. Drug repurposing for antivirulence therapy against opportunistic bacterial pathogens. Emerg Top Life Sci 1:13–22. doi: 10.1042/ETLS20160018. [DOI] [PubMed] [Google Scholar]
- 12.Imperi F, Massai F, Facchini M, Frangipani E, Visaggio D, Leoni L, Bragonzi A, Visca P. 2013. Repurposing the antimycotic drug flucytosine for suppression of Pseudomonas aeruginosa pathogenicity. Proc Natl Acad Sci U S A 110:7458–7463. doi: 10.1073/pnas.1222706110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen RC, Popat R, Diggle SP, Brown SP. 2014. Targeting virulence: can we make evolution-proof drugs? Nat Rev Microbiol 12:300–308. doi: 10.1038/nrmicro3232. [DOI] [PubMed] [Google Scholar]
- 14.Brannon JR, Hadjifrangiskou M. 2016. The arsenal of pathogens and antivirulence therapeutic strategies for disarming them. Drug Des Devel Ther 10:1795–1806. doi: 10.2147/DDDT.S98939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maura D, Ballok AE, Rahme LG. 2016. Considerations and caveats in antivirulence drug development. Curr Opin Microbiol 33:41–46. doi: 10.1016/j.mib.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 17.Pendleton JN, Gorman SP, Gilmore BF. 2013. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther 11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- 18.Aloush V, Navon-Venezia S, Seigman-Igra Y, Cabili S, Carmeli Y. 2006. Multidrug-resistant Pseudomonas aeruginosa: risk factors and clinical impact. Antimicrob Agents Chemother 50:43–48. doi: 10.1128/AAC.50.1.43-48.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciofu O, Tolker-Nielsen T, Jensen P∅, Wang H, Høiby N. 2015. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Deliv Rev 85:7–23. doi: 10.1016/j.addr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 20.Lund-Palau H, Turnbull AR, Bush A, Bardin E, Cameron L, Soren O, Wierre-Gore N, Alton EW, Bundy JG, Connett G, Faust SN, Filloux A, Freemont P, Jones A, Khoo V, Morales S, Murphy R, Pabary R, Simbo A, Schelenz S, Takats Z, Webb J, Williams HD, Davies JC. 2016. Pseudomonas aeruginosa infection in cystic fibrosis: pathophysiological mechanisms and therapeutic approaches. Expert Rev Respir Med 10:685–697. doi: 10.1080/17476348.2016.1177460. [DOI] [PubMed] [Google Scholar]
- 21.Williams P, Cámara M. 2009. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr Opin Microbiol 12:182–191. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Lee J, Zhang L. 2015. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 6:26–41. doi: 10.1007/s13238-014-0100-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rampioni G, Leoni L, Williams P. 2014. The art of antibacterial warfare: deception through interference with quorum sensing-mediated communication. Bioorg Chem 55:60–68. doi: 10.1016/j.bioorg.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Imperi F, Massai F, Ramachandran Pillai C, Longo F, Zennaro E, Rampioni G, Visca P, Leoni L. 2013. New life for an old drug: the anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob Agents Chemother 57:996–1005. doi: 10.1128/AAC.01952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heeb S, Fletcher MP, Chhabra SR, Diggle SR, Williams P, Cámara M. 2011. Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev 35:247–274. doi: 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dulcey CE, Dekimpe V, Fauvelle DA, Milot S, Groleau MC, Doucet N, Rahme LG, Lépine F, Déziel E. 2013. The end of an old hypothesis: the Pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem Biol 20:1481–1491. doi: 10.1016/j.chembiol.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drees SL, Fetzner S. 2015. PqsE of Pseudomonas aeruginosa acts as pathway-specific thioesterase in the biosynthesis of alkylquinolone signaling molecules. Chem Biol 22:611–618. doi: 10.1016/j.chembiol.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Rampioni G, Falcone M, Heeb S, Frangipani E, Fletcher MP, Dubern JF, Visca P, Leoni L, Cámara M, Williams P. 2016. Unravelling the genome-wide contributions of specific 2-alkyl-4-quinolones and PqsE to quorum sensing in Pseudomonas aeruginosa. PLoS Pathog 12:e1006029. doi: 10.1371/journal.ppat.1006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bredenbruch F, Nimtz M, Wray V, Morr M, Müller R, Häussler S. 2005. Biosynthetic pathway of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines. J Bacteriol 187:3630–3635. doi: 10.1128/JB.187.11.3630-3635.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mashburn LM, Whiteley M. 2005. Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature 437:422–425. doi: 10.1038/nature03925. [DOI] [PubMed] [Google Scholar]
- 31.Diggle SP, Matthijs S, Wright VJ, Fletcher MP, Chhabra SR, Lamont IL, Kong X, Hider RC, Cornelis P, Cámara M, Williams P. 2007. The Pseudomonas aeruginosa 4-quinolone signal molecules HHQ and PQS play multifunctional roles in quorum sensing and iron entrapment. Chem Biol 14:87–96. doi: 10.1016/j.chembiol.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 32.Hazan R, He J, Xiao G, Dekimpe V, Apidianakis Y, Lesic B, Astrakas C, Déziel E, Lépine F, Rahme LG. 2010. Homeostatic interplay between bacterial cell-cell signaling and iron in virulence. PLoS Pathog 6:e1000810. doi: 10.1371/journal.ppat.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rampioni G, Pustelny C, Fletcher MP, Wright VJ, Bruce M, Rumbaugh KP, Heeb S, Cámara M, Williams P. 2010. Transcriptomic analysis reveals a global alkyl-quinolone-independent regulatory role for PqsE in facilitating the environmental adaptation of Pseudomonas aeruginosa to plant and animal hosts. Environ Microbiol 12:1659–1673. doi: 10.1111/j.1462-2920.2010.02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao H, Krishnan G, Goumnerov B, Tsongalis J, Tompkins R, Rahme LG. 2001. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc Natl Acad Sci U S A 98:14613–14618. doi: 10.1073/pnas.251465298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Déziel E, Gopalan S, Tampakaki AP, Lépine F, Padfield KE, Saucier M, Xiao G, Rahme LG. 2005. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI, or the production of N-acyl-l-homoserine lactones. Mol Microbiol 55:998–1014. [DOI] [PubMed] [Google Scholar]
- 36.Xiao G, He J, Rahme LG. 2006. Mutation analysis of the Pseudomonas aeruginosa mvfR and pqsABCDE gene promoters demonstrates complex quorum-sensing circuitry. Microbiology 152:1679–1686. doi: 10.1099/mic.0.28605-0. [DOI] [PubMed] [Google Scholar]
- 37.Lesic B, Lépine F, Déziel E, Zhang J, Zhang Q, Padfield K, Castonguay MH, Milot S, Stachel S, Tzika AA, Tompkins RG, Rahme LG. 2007. Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog 3:1229–1239. doi: 10.1371/journal.ppat.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubern JF, Cigana C, De Simone M, Lazenby J, Juhas M, Schwager S, Bianconi I, Döring G, Eberl L, Williams P, Bragonzi A, Cámara M. 2015. Integrated whole-genome screening for Pseudomonas aeruginosa virulence genes using multiple disease models reveals that pathogenicity is host specific. Environ Microbiol 17:4379–4393. doi: 10.1111/1462-2920.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barr HL, Halliday N, Cámara M, Barrett DA, Williams P, Forrester DL, Simms R, Smyth AR, Honeybourne D, Whitehouse JL, Nash EF, Dewar J, Clayton A, Knox AJ, Fogarty AW. 2015. Pseudomonas aeruginosa quorum sensing molecules correlate with clinical status in cystic fibrosis. Eur Respir J 46:1046–1054. doi: 10.1183/09031936.00225214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calfee MW, Coleman JP, Pesci EC. 2001. Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 98:11633–11637. doi: 10.1073/pnas.201328498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cugini C, Calfee MW, Farrow JM 3rd, Morales DK, Pesci EC, Hogan DA. 2007. Farnesol, a common sesquiterpene, inhibits PQS production in Pseudomonas aeruginosa. Mol Microbiol 65:896–906. doi: 10.1111/j.1365-2958.2007.05840.x. [DOI] [PubMed] [Google Scholar]
- 42.Hakkila K, Maksimow M, Karp M, Virta M. 2002. Reporter genes lucFF, luxCDABE, gfp, and DsRed have different characteristics in whole-cell bacterial sensors. Anal Biochem 301:235–242. doi: 10.1006/abio.2001.5517. [DOI] [PubMed] [Google Scholar]
- 43.Jansson JK. 2003. Marker and reporter genes: illuminating tools for environmental microbiologists. Curr Opin Microbiol 6:528–529. doi: 10.1016/j.mib.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 44.Clayton YM, Connor BL. 1973. Comparison of clotrimazole cream, Whitfield's ointment and Nystatin ointment for the topical treatment of ringworm infections, pityriasis versicolor, erythrasma and candidiasis. Br J Dermatol 89:297–303. doi: 10.1111/j.1365-2133.1973.tb02978.x. [DOI] [PubMed] [Google Scholar]
- 45.Sawyer PR, Brogden RN, Pinder RM, Speight TM, Avery GS. 1975. Clotrimazole: a review of its antifungal activity and therapeutic efficacy. Drugs 9:424–447. doi: 10.2165/00003495-197509060-00003. [DOI] [PubMed] [Google Scholar]
- 46.Shellow WV. 1982. 2% Miconazole nitrate powder in aerosol spray form: its efficacy in treating tinea pedis. J Int Med Res 10:28–31. doi: 10.1177/030006058201000105. [DOI] [PubMed] [Google Scholar]
- 47.Ahmed TA, El-Say KM, Mahmoud MF, Samy AM, Badawi AA. 2012. Miconazole nitrate oral disintegrating tablets: in vivo performance and stability study. AAPS PharmSciTech 13:760–771. doi: 10.1208/s12249-012-9798-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buogo A. 1981. Trials of the in vitro antibacterial activity of clofoctol and pharmacokinetic features. G Ital Chemioter 28:65–71. [PubMed] [Google Scholar]
- 49.Yablonsky F, Simonnet G. 1982. Action of clofoctol on bacterial cell wall synthesis. J Pharmacol 13:515–524. [PubMed] [Google Scholar]
- 50.Danesi R, Gasperini M, Senesi S, Freer G, Angeletti CA, Del Tacca M. 1988. A pharmacokinetic study of clofoctol in human plasma and lung tissue by using a microbiological assay. Drugs Exp Clin Res 14:39–43. [PubMed] [Google Scholar]
- 51.Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. 2002. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J Bacteriol 184:6472–6480. doi: 10.1128/JB.184.23.6472-6480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Déziel E, Lépine F, Milot S, He J, Mindrinos MN, Tompkins RG, Rahme LG. 2004. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc Natl Acad Sci U S A 101:1339–1344. doi: 10.1073/pnas.0307694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gilbert KB, Kim TH, Gupta R, Greenberg EP, Schuster M. 2009. Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol Microbiol 73:1072–1085. doi: 10.1111/j.1365-2958.2009.06832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKnight SL, Iglewski BH, Pesci EC. 2000. The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol 182:2702–2708. doi: 10.1128/JB.182.10.2702-2708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wade DS, Calfee MW, Rocha ER, Ling EA, Engstrom Coleman JP, Pesci EC. 2005. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J Bacteriol 187:4372–4380. doi: 10.1128/JB.187.13.4372-4380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brouwer S, Pustelny C, Ritter C, Klinkert B, Narberhaus F, Häussler S. 2014. The PqsR and RhlR transcriptional regulators determine the level of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa by producing two different pqsABCDE mRNA isoforms. J Bacteriol 196:4163–4171. doi: 10.1128/JB.02000-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massai F, Imperi F, Quattrucci S, Zennaro E, Visca P, Leoni L. 2011. A multitask biosensor for micro-volumetric detection of N-3-oxo-dodecanoyl-homoserine lactone quorum sensing signal. Biosens Bioelectron 26:3444–3449. doi: 10.1016/j.bios.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 58.Diggle SP, Winzer K, Chhabra SR, Worrall KE, Cámara M, Williams P. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol Microbiol 50:29–43. doi: 10.1046/j.1365-2958.2003.03672.x. [DOI] [PubMed] [Google Scholar]
- 59.Ilangovan A, Fletcher M, Rampioni G, Pustelny C, Rumbaugh K, Heeb S, Cámara M, Truman A, Chhabra SR, Emsley J, Williams P. 2013. Structural basis for native agonist and synthetic inhibitor recognition by the Pseudomonas aeruginosa quorum sensing regulator PqsR (MvfR). PLoS Pathog 9:e1003508. doi: 10.1371/journal.ppat.1003508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- 61.Visca P, Leoni L, Wilson MJ, Lamont IL. 2002. Iron transport and regulation, cell signaling and genomics: lessons from Escherichia coli and Pseudomonas. Mol Microbiol 45:1177–1190. doi: 10.1046/j.1365-2958.2002.03088.x. [DOI] [PubMed] [Google Scholar]
- 62.Diggle SP, Stacey RE, Dodd C, Cámara M, Williams P, Winzer K. 2006. The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ Microbiol 8:1095–1104. doi: 10.1111/j.1462-2920.2006.001001.x. [DOI] [PubMed] [Google Scholar]
- 63.Jander G, Rahme LG, Ausubel FM. 2000. Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol 182:3843–3845. doi: 10.1128/JB.182.13.3843-3845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rampioni G, Schuster M, Greenberg EP, Zennaro E, Leoni L. 2009. Contribution of the RsaL global regulator to Pseudomonas aeruginosa virulence and biofilm formation. FEMS Microbiol Lett. 301:210–217. doi: 10.1111/j.1574-6968.2009.01817.x. [DOI] [PubMed] [Google Scholar]
- 65.Rampioni G, Pillai CR, Longo F, Bondì R, Baldelli V, Messina M, Imperi F, Visca P, Leoni L. 2017. Effect of efflux pump inhibition on Pseudomonas aeruginosa transcriptome and virulence. Sci Rep 7:11392. doi: 10.1038/s41598-017-11892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mellbye B, Schuster M. 2011. The sociomicrobiology of antivirulence drug resistance: a proof of concept. mBio 2:5. doi: 10.1128/mBio.00131-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.LaSarre B, Federle MJ. 2013. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev 77:73–111. doi: 10.1128/MMBR.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Machan ZA, Taylor GW, Pitt TL, Cole PJ, Wilson R. 1992. 2-Heptyl-4-hydroxyquinoline N-oxide, an antistaphylococcal agent produced by Pseudomonas aeruginosa. J Antimicrob Chemother 30:615–623. doi: 10.1093/jac/30.5.615. [DOI] [PubMed] [Google Scholar]
- 69.Collier DN, Anderson L, McKnight SL, Noah TL, Knowles M, Boucher R, Schwab U, Gilligan P, Pesci EC. 2002. A bacterial cell to cell signal in the lungs of cystic fibrosis patients. FEMS Microbiol Lett. 215:41–46. doi: 10.1111/j.1574-6968.2002.tb11367.x. [DOI] [PubMed] [Google Scholar]
- 70.Storz MP, Maurer CK, Zimmer C, Wagner N, Brengel C, de Jong JC, Lucas S, Müsken M, Häussler S, Steinbach A, Hartmann RW. 2012. Validation of PqsD as an anti-biofilm target in Pseudomonas aeruginosa by development of small-molecule inhibitors. J Am Chem Soc 134:16143–16146. doi: 10.1021/ja3072397. [DOI] [PubMed] [Google Scholar]
- 71.Weidel E, de Jong JC, Brengel C, Storz MP, Braunshausen A, Negri M, Plaza A, Steinbach A, Müller R, Hartmann RW. 2013. Structure optimization of 2-benzamidobenzoic acids as PqsD inhibitors for Pseudomonas aeruginosa infections and elucidation of binding mode by SPR, STD NMR, and molecular docking. J Med Chem 56:6146–6155. doi: 10.1021/jm4006302. [DOI] [PubMed] [Google Scholar]
- 72.Pustelny C, Albers A, Büldt-Karentzopoulos K, Parschat K, Chhabra SR, Cámara M, Williams P, Fetzner S. 2009. Dioxygenase-mediated quenching of quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem Biol 16:1259–1267. doi: 10.1016/j.chembiol.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 73.Soh EY, Chhabra SR, Halliday N, Heeb S, Müller C, Birmes FS, Fetzner S, Cámara M, Chan KG, Williams P. 2015. Biotic inactivation of the Pseudomonas aeruginosa quinolone signal molecule. Environ Microbiol 17:4352–4365. doi: 10.1111/1462-2920.12857. [DOI] [PubMed] [Google Scholar]
- 74.Klein T, Henn C, de Jong JC, Zimmer C, Kirsch B, Maurer CK, Pistorius D, Müller R, Steinbach A, Hartmann RW. 2012. Identification of small-molecule antagonists of the Pseudomonas aeruginosa transcriptional regulator PqsR: biophysically guided hit discovery and optimization. ACS Chem Biol 7:1496–1501. doi: 10.1021/cb300208g. [DOI] [PubMed] [Google Scholar]
- 75.Zender M, Klein T, Henn C, Kirsch B, Maurer CK, Kail D, Ritter C, Dolezal O, Steinbach A, Hartmann RW. 2013. Discovery and biophysical characterization of 2-amino-oxadiazoles as novel antagonists of PqsR, an important regulator of Pseudomonas aeruginosa virulence. J Med Chem 56:6761–6774. doi: 10.1021/jm400830r. [DOI] [PubMed] [Google Scholar]
- 76.Lu C, Kirsch B, Maurer CK, de Jong JC, Braunshausen A, Steinbach A, Hartmann RW. 2014. Optimization of anti-virulence PqsR antagonists regarding aqueous solubility and biological properties resulting in new insights in structure-activity relationships. Eur J Med Chem 79:173–183. doi: 10.1016/j.ejmech.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 77.Starkey M, Lepine F, Maura D, Bandyopadhaya A, Lesic B, He J, Kitao T, Righi V, Milot S, Tzika A, Rahme L. 2014. Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog 10:e1004321. doi: 10.1371/journal.ppat.1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maura D, Drees SL, Bandyopadhaya A, Kitao T, Negri M, Starkey M, Lesic B, Milot S, Déziel E, Zahler R, Pucci M, Felici A, Fetzner S, Lépine F, Rahme LG. 2017. Polypharmacology approaches against the Pseudomonas aeruginosa MvfR regulon and their application in blocking virulence and antibiotic tolerance. ACS Chem Biol 12:1435–1443. doi: 10.1021/acschembio.6b01139. [DOI] [PubMed] [Google Scholar]
- 79.Maura D, Rahme LG. 2017. Pharmacological inhibition of the Pseudomonas aeruginosa MvfR quorum sensing system interferes with biofilm formation and potentiates antibiotic-mediated biofilm disruption. Antimicrob Agents Chemother 61:e01362-17. doi: 10.1128/AAC.01362-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomann A, de Mello Martins AG, Brengel C, Empting M, Hartmann RW. 2016. Application of dual inhibition concept within looped autoregulatory systems toward antivirulence agents against Pseudomonas aeruginosa infections. ACS Chem Biol 11:1279–1286. doi: 10.1021/acschembio.6b00117. [DOI] [PubMed] [Google Scholar]
- 81.De Cremer K, Lanckacker E, Cools TL, Bax M, De Brucker K, Cos P, Cammue BP, Thevissen K. 2015. Artemisinins, new miconazole potentiators resulting in increased activity against Candida albicans biofilms. Antimicrob Agents Chemother 59:421–426. doi: 10.1128/AAC.04229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang LW, Fu JY, Hua H, Yan ZM. 2016. Efficacy and safety of miconazole for oral candidiasis: a systematic review and meta-analysis. Oral Dis 22:185–195. doi: 10.1111/odi.12380. [DOI] [PubMed] [Google Scholar]
- 83.Fothergill AW. 2006. Miconazole: a historical perspective. Expert Rev Anti Infect Ther 4:171–175. doi: 10.1586/14787210.4.2.171. [DOI] [PubMed] [Google Scholar]
- 84.Crowley PD, Gallagher HC. 2014. Clotrimazole as a pharmaceutical: past, present and future. J Appl Microbiol 117:611–617. doi: 10.1111/jam.12554. [DOI] [PubMed] [Google Scholar]
- 85.Osmon S, Ward S, Fraser VJ, Kollef MH. 2004. Hospital mortality for patients with bacteremia due to Staphylococcus aureus or Pseudomonas aeruginosa. Chest 125:607–616. doi: 10.1378/chest.125.2.607. [DOI] [PubMed] [Google Scholar]
- 86.Driscoll JA, Brody SL, Kollef MH. 2007. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- 87.Lyczak JB, Cannon CL, Pier GB. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect 2:1051–1060. doi: 10.1016/S1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 88.Costabile G, d'Angelo I, Rampioni G, Bondì R, Pompili B, Ascenzioni F, Mitidieri E, d'Emmanuele di Villa Bianca R, Sorrentino R, Miro A, Quaglia F, Imperi F, Leoni L, Ungaro F. 2015. Toward repositioning niclosamide for antivirulence therapy of Pseudomonas aeruginosa lung infections: development of inhalable formulations through nanosuspension technology. Mol Pharm 12:2604–2617. doi: 10.1021/acs.molpharmaceut.5b00098. [DOI] [PubMed] [Google Scholar]
- 89.Danesi R, Del Tacca M. 1985. Clinical study on the efficacy of clofoctol in the treatment of infectious respiratory diseases. Int J Clin Pharmacol Res 5:175–179. [PubMed] [Google Scholar]
- 90.Yablonsky F. 1983. Alteration of membrane permeability in Bacillus subtilis by clofoctol. J Gen Microbiol 129:1089–1095. [DOI] [PubMed] [Google Scholar]
- 91.Del Tacca M, Danesi R, Senesi S, Gasperini M, Mussi A, Angeletti CA. 1987. Penetration of clofoctol into human lung. J Antimicrob Chemother 19:679–683. doi: 10.1093/jac/19.5.679. [DOI] [PubMed] [Google Scholar]
- 92.Lyczak JB, Cannon CL, Pier GB. 2002. Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Winstanley C, Fothergill JL. 2009. The role of quorum sensing in chronic cystic fibrosis Pseudomonas aeruginosa infections. FEMS Microbiol 290:1–9. doi: 10.1111/j.1574-6968.2008.01394.x. [DOI] [PubMed] [Google Scholar]
- 94.Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Høiby N, Molin S. 2012. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 95.Kamath KS, Pascovici D, Penesyan A, Goel A, Venkatakrishnan V, Paulsen IT, Packer NH, Molloy MP. 2016. Pseudomonas aeruginosa cell membrane protein expression from phenotypically diverse cystic fibrosis isolates demonstrates host-specific adaptations. J Proteome Res 15:2152–2163. doi: 10.1021/acs.jproteome.6b00058. [DOI] [PubMed] [Google Scholar]
- 96.Winstanley C, O'Brien S, Brockhurst MA. 2016. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol 24:327–337. doi: 10.1016/j.tim.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoffman LR, Kulasekara HD, Emerson J, Houston LS, Burns JL, Ramsey BW, Miller SI. 2009. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J Cyst Fibros 8:66–70. doi: 10.1016/j.jcf.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bjarnsholt T, Jensen P∅, Jakobsen TH, Phipps R, Nielsen AK, Rybtke MT, Tolker-Nielsen T, Givskov M, Høiby N, Ciofu O, Scandinavian Cystic Fibrosis Study Consortium . 2010. Quorum sensing and virulence of Pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PLoS One 5:e10115. doi: 10.1371/journal.pone.0010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiricny N, Molin S, Foster K, Diggle SP, Scanlan PD, Ghoul M, Johansen HK, Santorelli LA, Popat R, West SA, Griffin AS. 2014. Loss of social behaviours in populations of Pseudomonas aeruginosa infecting lungs of patients with cystic fibrosis. PLoS One 9:e83124. doi: 10.1371/journal.pone.0083124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Feltner JB, Wolter DJ, Pope CE, Groleau MC, Smalley NE, Greenberg EP, Mayer-Hamblett N, Burns J, Déziel E, Hoffman LR, Dandekar AA. 2016. LasR variant cystic fibrosis isolates reveal an adaptable quorum-sensing hierarchy in Pseudomonas aeruginosa. mBio 7:e01513-16. doi: 10.1128/mBio.01513-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guina T, Purvine SO, Yi EC, Eng J, Goodlett DR, Aebersold R, Miller SI. 2003. Quantitative proteomic analysis indicates increased synthesis of a quinolone by Pseudomonas aeruginosa isolates from cystic fibrosis airways. Proc Natl Acad Sci U S A 100:2771–2776. doi: 10.1073/pnas.0435846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fletcher MP, Diggle SP, Crusz SA, Chhabra SR, Cámara M, Williams P. 2007. A dual biosensor for 2-alkyl-4-quinolone quorum-sensing signal molecules. Environ Microbiol. 9:2683–2693. doi: 10.1111/j.1462-2920.2007.01380.x. [DOI] [PubMed] [Google Scholar]
- 103.Ortori CA, Dubern JF, Chhabra SR, Cámara M, Hardie K, Williams P, Barrett DA. 2011. Simultaneous quantitative profiling of N-acyl-l-homoserine lactone and 2-alkyl-4(1H)-quinolone families of quorum-sensing signaling molecules using LC-MS/MS. Anal Bioanal Chem 399:839–850. doi: 10.1007/s00216-010-4341-0. [DOI] [PubMed] [Google Scholar]
- 104.Essar DW, Eberly L, Hadero A, Crawford IP. 1990. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J Bacteriol 172:884–900. doi: 10.1128/jb.172.2.884-900.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jurcisek JA, Dickson AC, Bruggeman ME, Bakaletz LO. 2011. In vitro biofilm formation in an 8-well chamber slide. J Vis Exp 47:e2481. doi: 10.3791/2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 107.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 108.Di Muzio E, Toti D, Polticelli F. 2017. DockingApp: a user friendly interface for AutoDock Vina. J Comput Aided Mol Des 31:213–218. doi: 10.1007/s10822-016-0006-1. [DOI] [PubMed] [Google Scholar]
- 109.Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem 31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.