

Abstract
Background:
Cardiac dysfunction is a major component of sepsis-induced multi-organ failure in critical care units. Changes in cardiac autophagy and its role during sepsis pathogenesis have not been clearly defined. Targeted autophagy-based therapeutic approaches for sepsis are not yet developed.
Methods:
Beclin-1-dependent autophagy in the heart during sepsis and the potential therapeutic benefit of targeting this pathway were investigated in a mouse model of lipopolysaccharide (LPS)-induced sepsis.
Results:
LPS induced a dose-dependent increase in autophagy at low doses, followed by a decline that was in conjunction with mTOR activation at high doses. Cardiac-specific overexpression of Beclin-1 promoted autophagy, suppressed mTOR signaling, improved cardiac function, and alleviated inflammation and fibrosis after LPS challenge. Haplosufficiency for beclin 1 resulted in opposite effects. Beclin-1 also protected mitochondria, reduced the release of mitochondrial DAMPs, and promoted mitophagy via PINK1-Parkin but not adaptor proteins in response to LPS. Injection of a cell-permeable Tat-Beclin-1 peptide to activate autophagy improved cardiac function, attenuated inflammation, and rescued the phenotypes caused by beclin 1 deficiency in LPS-challenged mice.
Conclusions:
These results suggest that Beclin-1 protects the heart during sepsis and that the targeted induction of Beclin-1 signaling may have important therapeutic potential.
Keywords: Beclin-1, autophagy, mitophagy, cardiac dysfunction, sepsis, LPS
Introduction
Sepsis is a leading cause of death in intensive care units1, and cardiac dysfunction is an identified serious component of the multi-organ failure associated with this critical condition2. Mitochondria comprise about 30% of myocardial volume3. Using both in vitro and in vivo models, we have focused on understanding how sepsis deregulates mitochondrial signaling and thereby leads to cardiac dysfunction. We have shown that impairments in mitochondrial structure and function, in association with overproduction of mitochondrial reactive oxygen species (mtROS) and generation of mitochondria-derived danger-associated molecular patterns (DAMPs), play a pivotal role in inducing cardiac inflammation and functional deficiencies during sepsis4–7.
The quality and quantity of mitochondria are tightly regulated. Dysfunctional mitochondria can be segregated and eliminated through autophagy8, a lysosome-dependent process of removing damaged proteins and organelles9. Autophagy impacts a diverse range of cellular responses, such as metabolic balance10, cell fate11 and inflammation12. Under normal physiological responses or mild stress, autophagy provides a level of control to promote survival and is therefore adaptive. However, under severe or chronic stress, either excessive autophagic activity or inadequate autophagy can lead to massive self-degradation or the accumulation of toxic materials respectively; either of these outcomes is maladaptive and can ultimately cause cell death13.
Clinical and preclinical studies indicate that sepsis triggers autophagy in multiple organs including the heart14–16. However, our current understanding of the role of autophagy in the pathogenesis of sepsis remains limited and inconclusive. Evidence obtained in vivo using a mouse cecal ligation and puncture (CLP) sepsis model17 and in vitro using cultured, lipopolysaccharide (LPS)-challenged cardiomyocytes18 suggests that stimulating autophagy via pharmacological approaches protects the myocardium, thus providing evidence that autophagy is an adaptive response. Conversely, a recent publication showed that reducing autophagy using an autophagy inhibitor or antioxidants improved cardiac contractility in a mouse model of LPS-induced sepsis, supporting the conclusion that autophagy is maladaptive19. The discrepancy of these observations may be due to the differences in experimental settings, in which the severity of sepsis, drug specificity, and timing of delivery differed.
Beclin-1, one of the first mammalian autophagy effectors identified20, 21, is ubiquitously expressed. Homozygous deletion of the beclin-1 gene results in early embryonic lethality22. Beclin-1 functions during the initiation of autophagy through interaction with PtdIns(3)-kinase (Vps34)23. Together, this protein complex initiates the nucleation step of autophagy to begin autophagic flux and also participates in later steps involving the fusion of autophagosomes to lysosomes24–26. Our studies, summarized in this report, were aimed at addressing how autophagic activity changes in response to the severity of sepsis, and on determining the role of Beclin-1 in the autophagic response of the septic heart. The investigation was performed using a mouse model of sepsis induced by LPS, a known toxic component of gram-negative bacteria. Transgenic mice with cardiac-specific overexpression of Beclin-1 and heterozygous deficiency were used to increase and decrease Beclin-1. The role of Beclin-1 in quality control of cardiac mitochondria through mitophagy, a process of selective degradation of mitochondria by autophagy, was also examined. Lastly, the therapeutic potential of pharmacologically inducing autophagy using a novel peptide derived from Beclin-1 was evaluated27, 28.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure.
A detailed Methods section is available in the online-only Data Supplement. Animal experiments were reviewed and approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee and conformed to all guidelines for animal care as outlined by the American Physiology Society and the National Institutes of Health.
Statistical analysis.
Results were expressed as mean ± SEM using the indicated number of experiments or mice. Non-parametric Mann–Whitney U tests were applied for comparisons between groups. A log-rank test was applied in the survival study to compare survival curves. All computations were conducted in the R environment, version 3.4.3, and the R packages “survival” (version 2.41–3) and “coin” (version 1.2–2) were used. Differences were considered statistically significant when p ≤ 0.05.
Results
There are dose-dependent changes in cardiac autophagic flux during LPS-induced sepsis.
Mice were given intraperitoneal (i.p.) injections of either LPS (0.25–15 mg/kg) or vehicle PBS in shams. Heart tissue was harvested 18 hours post challenge. LC3II, a marker of autophagosome formation, was examined by Western blot. Compared to shams, the level of LC3II was higher in LPS-challenged animals (Figure 1A). Interestingly, LC3II was increased dose dependently in response to low doses of LPS, 0.25–2.5 mg/kg, but this was followed by a gradual decline in response to higher doses of 5–15 mg/kg. Consistent with an increase in autophagy, p62/SQSTM1, a polyubiquitin-binding protein that is degraded during autophagy, showed the reverse trend. LPS had modest effect on Beclin-1 levels. These data suggest that LPS stimulates dose-dependent changes in cardiac autophagy, initially activating, then declining at high doses.
Figure 1.
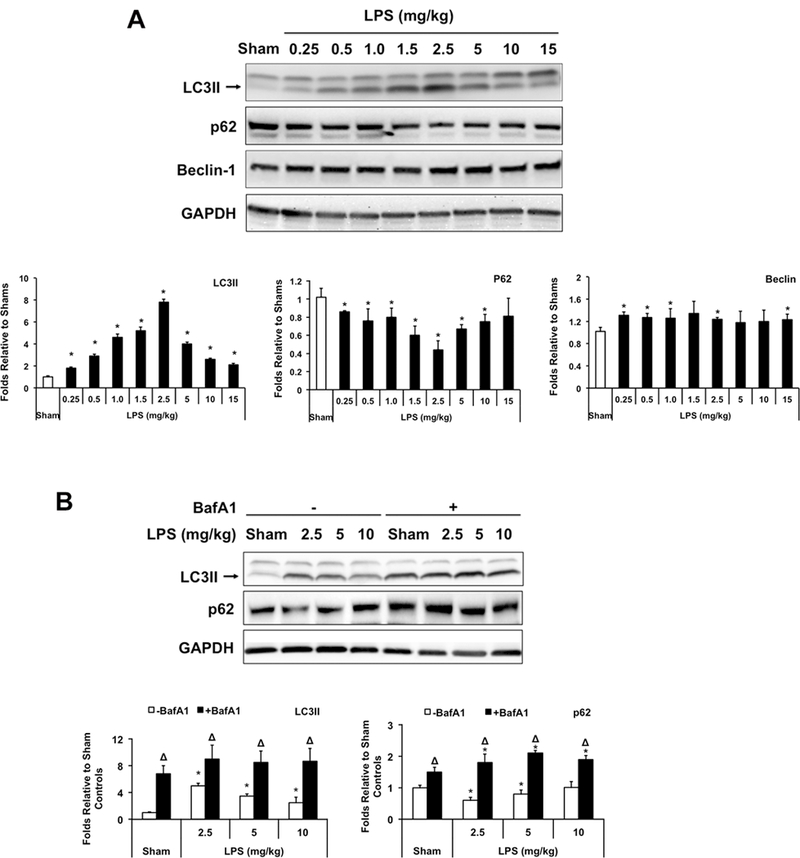
Changes of cardiac autophagy in response to different doses of LPS. Wild-type C57BL/6 mice were given LPS via i.p. at indicated doses, heart tissues were harvested 18 hours later and total tissue lysates were prepared. A. Levels of LC3II, p62, Beclin and GAPDH were analyzed by Western blots using GAPDH as a loading control. B. LPS-induced autophagy flux was further confirmed by comparison of LC3II and p62 between animals with and without treatment of bafilomycin A1 (BafA1, 1.5 mg/kg). Results were quantified by densitometry and expressed as fold changes relative to shams. All values are means ± SEM. Significant differences are shown as * for sham vs. LPS-treated and Δ for vehicle vs. BafA1-treated groups (p < 0.05, n = 5, Mann-Whitney U test).
To confirm the above trend of LC3II representing the activation of autophagy rather than a blockage in autophagosomal maturation, we treated mice with Bafilomycin A1 (BafA1), an inhibitor that blocks the fusion of autophagosomes with lysosomes29. As expected, BafA1 caused significant accumulations of both LC3II and p62 in sham and LPS-challenged animals, indicating that a LPS-induced increase in LC3II was not due to a downstream inhibition in autophagic flux (Figure 1B).
These data demonstrate that LPS induces a dose-dependent autophagic flux in the heart. This reaction is comprised of two phases: a steady state increase when LPS doses are low, modeling less severe or mild sepsis, and a gradual decline when LPS doses are high, resembling events during severe sepsis.
Changing Beclin-1 levels alters the autophagic response and mTOR signaling in the heart in response to LPS.
To examine the role of autophagy in cardiac dysfunction after sepsis, we analyzed mouse lines that had either cardiac-specific overexpression of Beclin-1 under the α-myosin heavy chain promoter (Becn1-Tg) or were haploinsufficient for beclin 1 (Becn1+/−). At baseline, wild type (WT), Becn1-Tg, and Becn1+/− mice were indistinguishable and displayed similar levels of physical activity. As anticipated, Beclin-1 levels were lower than WT in the hearts of Becn1+/− mice but higher in the Becn1-Tg (Figure 2A). Mice were treated with LPS in a dosage range sufficient to cause a detectable decline in cardiac function, 2.5–10 mg/kg (see data in Figure 3). The level of LC3II was higher in the Becn1-Tg compared to WT both at baseline and in response to LPS. A stronger degradation of p62 was observed in parallel, consistent with an increase in autophagic flux in Becn1-Tg mice (Figure 2B). Conversely, these responses in the hearts of the Becn1+/− animals were significantly blunted. These results confirmed previously reported differences in autophagy phenotypes between these mouse lines 30.
Figure 2.
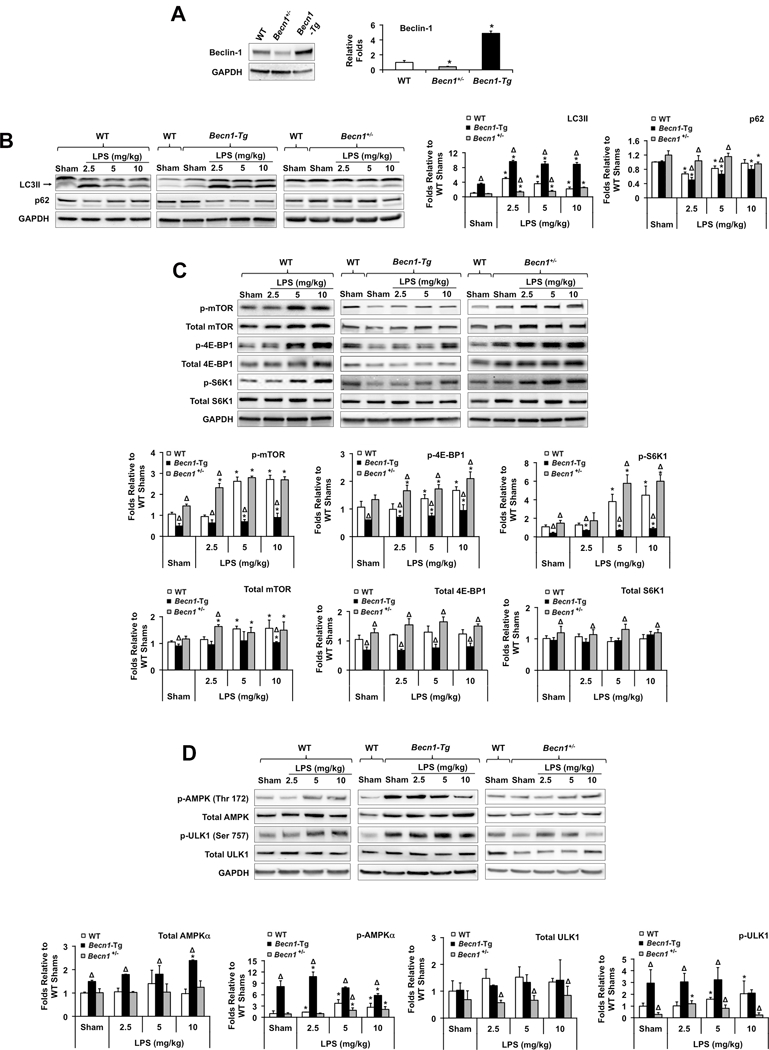
LPS-induced changes of cardiac autophagy and mTOR signaling in WT, Becn1-Tg, and Becn1+/− mice. Mice were given LPS via i.p. at indicated doses, heart tissues were harvested 18 hours later and total tissue lysates were prepared. Expression levels of Beclin-1 were confirmed (A), levels of LC3II and p62 (B), total and phosphorylated mTOR pathway molecules (C), and total and phosphorylated AMPKα and ULK1 (D) were examined by Western blots using GAPDH as a loading control. Results obtained by densitometry were expressed as fold changes relative to WT shams. All values are means ± SEM. Significant differences are shown as * for sham vs. LPS-treated and Δ for WT vs. Becn1-Tg or Becn1+/− groups (p < 0.05, n = 5, Mann-Whitney U test).
Figure 3.
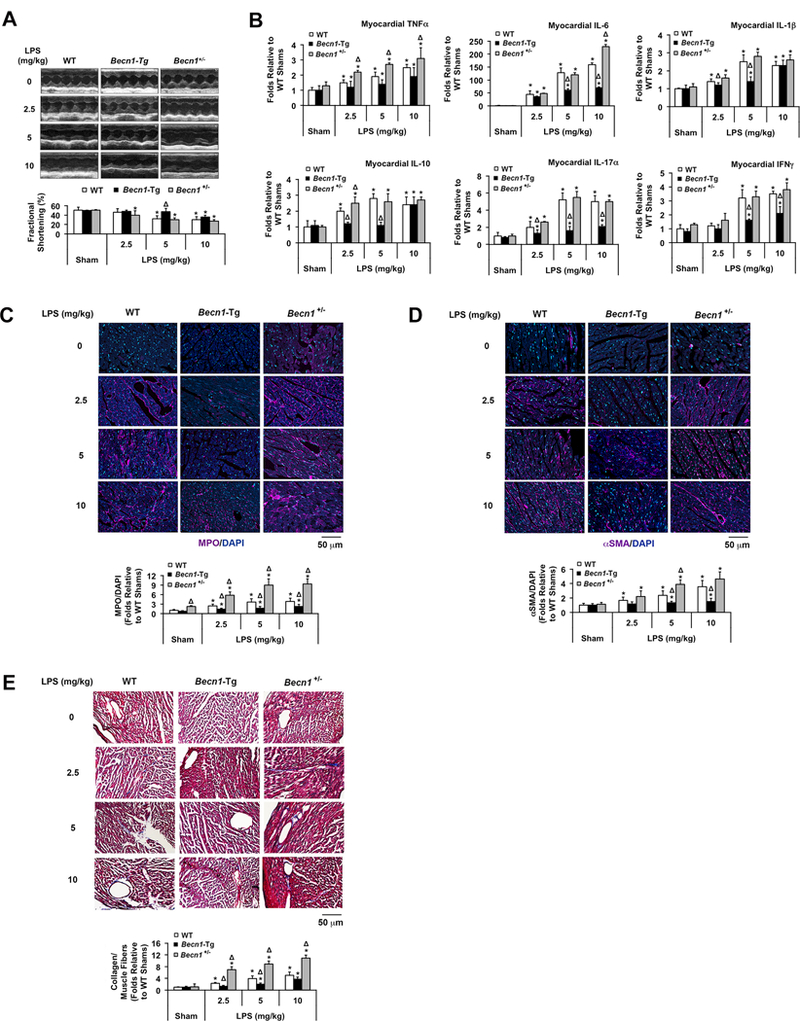
Evaluation of outcomes in LPS-challenged WT, Becn1-Tg, and Becn1+/− mice. Mice were given LPS via i.p. at indicated doses and analyzed 18 hours post challenge. A. Cardiac function was examined by echocardiography (n=5). B. Cytokines were measured in the heart tissue lysates by multiplex ELISA assays (n=5). C. Heart tissue sections were immune-stained with myeloperoxidase (MPO; purple) and co-stained with DAPI (blue) to show nucleus. D. Heart tissue sections were immune-stained with αSMA (purple) and co-stained with DAPI (blue) to show nucleus. E. Histological trichrome stain was applied to the heart tissue sections to visualize collagenous connective tissue fibers (blue). In C-E, images are representative of n ≥ 3 animals per group, and results were quantified using NIS Elements microscope imaging software. All values are means ± SEM. Significant differences are shown as * for sham vs. LPS-treated and Δ for WT vs. Becn1-Tg or Becn1+/− groups (p < 0.05, Mann-Whitney U test).
The kinase mammalian target of rapamycin (mTOR) is a key regulatory point suppressing Beclin-1-dependent autophagic activity31. We examined the activation of mTOR and its downstream factors, including eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and the p70 ribosomal S6 kinase 1 (S6K1)32, in the hearts of sham and LPS-challenged mice. Higher doses of LPS, including 5 mg/kg and 10 mg/kg, significantly increased phosphorylation of all three effectors (Figure 2C), indicating activations of mTOR signaling in WT hearts. However, at the lower dose of LPS, 2.5 mg/kg, where we previously showed that autophagy was most robust, LPS had little, or no effect on the phosphorylation of mTOR and its downstream factors. These results show the correlation between the activation of mTOR signaling and the decline in inducing cardiac autophagy in response to higher LPS dosages. In addition, since a significant difference in mTOR activation between shams and low-dose LPS-treated groups, such as 2.5 mg/kg, was not detectable, the data suggest a possibility that the activation of autophagy by LPS at low doses might be mediated through an mTOR-independent mechanism.
The activation of mTOR was significantly blunted in the hearts of Becn1-Tg, but was much stronger in Becn1+/− mice as compared to WT (Figure 2C). Thus, the activation of mTOR was inversely correlated with autophagic activities, suggesting that enhancing Beclin-1 signaling can suppress mTOR activation, thereby sustaining autophagy even under conditions of severe sepsis. Additionally, the signals of AMP-activated protein kinase (AMPK) and Unc-51 like-autophagy-activating kinase 1 (ULK1) in the hearts of these mice were compared. The levels of phosphorylation at the active sites of these two kinases were significantly higher in Becn1-Tg but lower in Becn1+/− mice (Figure 2D), suggesting that Beclin-1 involves a positive feedback regulation of autophagy by enhancing AMPK and ULK1. This result also suggests a mechanism of Beclin-1’s suppression of mTOR activation.
Increasing Beclin-1 levels improves cardiac outcomes in LPS-induced sepsis.
We examined heart performance by echocardiography in sham and LPS-challenged WT, Becn1-Tg, and Becn1+/− mice. LPS challenge of ≥5 mg/kg substantially reduced fractional shortening (FS%) 18 hours post LPS challenge in WT mice (Figure 3A). However, a lower dose of 2.5 mg/kg LPS was sufficient to impact cardiac function in Becn1+/− mice. Conversely, only the highest dose tested, 10 mg/kg LPS reduced FS% in Becn1-Tg mice. The decline in cardiac function in WT mice treated with 5 mg/kg LPS occurred in concert with the failure in induction of autophagic flux (Figure 1) and the activation of mTOR signaling (Figure 2C). With the observed preservation of cardiac function in the Becn1-Tg mice, these results suggest that Beclin-1-dependent autophagy is an adaptive, protective response to sepsis in the heart.
As an additional index of damage, we examined cytokine levels in the heart tissue of WT, Becn1-Tg, and Becn1+/− mice (Figure 3B). Consistent with the literature, LPS dose-dependently increased inflammatory cytokines in the heart. The overexpression of Beclin-1 significantly decreased cytokine levels, with the greatest reduction found in IL-6, IL-17α and IFN-λ. The haploinsufficiency of Becn1 led to elevated cytokines following LPS challenges.
Myocardial content of myeloperoxidase, a marker of neutrophil accumulation33, was examined in WT, Becn1-Tg, and Becn1+/− mice by immunohistochemistry. LPS stimulated significant neutrophil infiltration in the heart, which was worsened in Becn1+/− but much less dramatic in Becn1-Tg mice (Figure 3C).
Fibrosis is often a consequence of inflammation. We examined cardiac fibrosis in WT, Becn1-Tg, and Becn1+/− mice using the presence of known markers, α-smooth muscle actin (αSMA) and the deposition of collagen34. LPS induced a progressive increase in cardiac content of αSMA in WT hearts. This response was significantly reduced in Becn1-Tg mice but elevated in Becn1+/− mice (Figure 3D). Masson’s trichrome staining to detect collagen in the heart tissues showed a similar pattern in the fibrotic response (Figure 3E). These data are consistent with Beclin-1 activity protecting the myocardium from fibrotic injury during sepsis.
Increasing Beclin-1 levels preserves cardiac mitochondrial quality during sepsis.
We have previously observed that sepsis damages mitochondrial structure and results in a deficiency in cardiac mitochondrial function4, 5, 7. Electron microscopy (EM) was used to compare mitochondrial morphology in the left ventricular wall of hearts from WT, Becn1-Tg, and Becn1+/− mice at baseline and following LPS. In WT hearts, an increase in LPS dosage led to a dose-dependent increase in the frequency of isolated, small, rounded mitochondria, and the loss of clearly defined cristae structures of the inner mitochondrial membrane (Figure 4A). These changes were less evident and required higher doses of LPS to manifest in Becn1-Tg mice. Conversely, these disruptions were more severe in the Becn1+/− mice.
Figure 4.
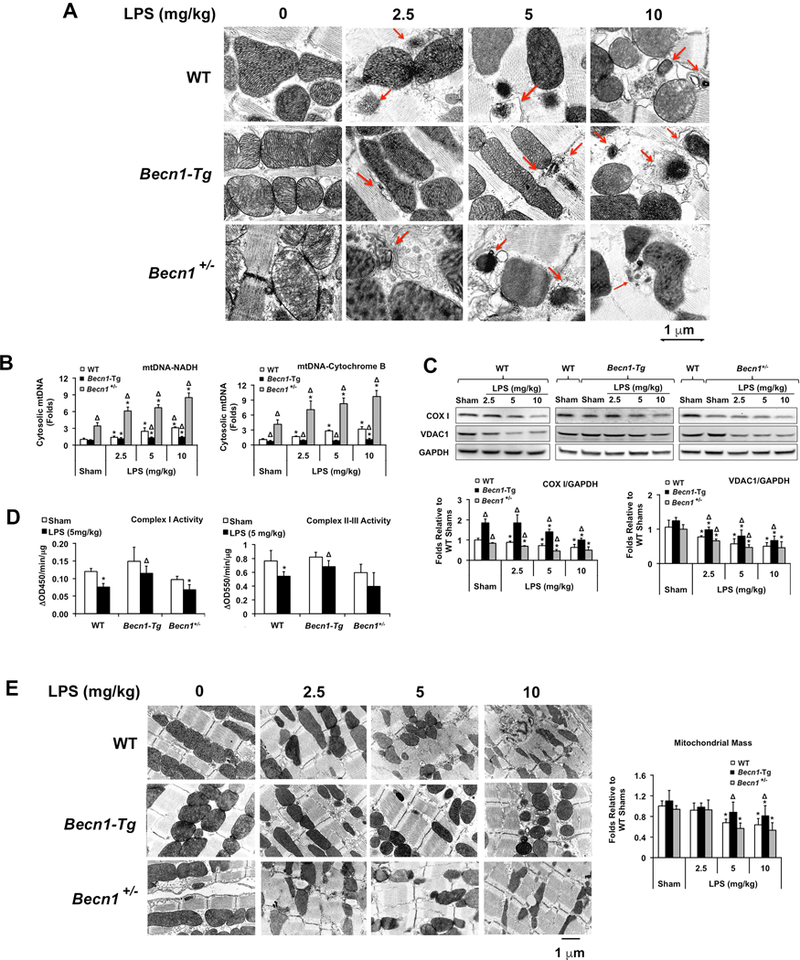
LPS-induced changes in mitochondrial structure and function in the heart of WT, Becn1-Tg, and Becn1+/− mice. Mice were given LPS via i.p. at indicated doses; heart tissues were harvested 18 hours later. A. Ultrastructure of myocardial mitochondria was observed by transmission electron microscope (EM). Red arrows indicated various stages of mitochondrial degradation. These images are representative of n≥3 animals per group. B. Total DNA was isolated from equal amount of cytosolic fractions and an exogenous internal positive control (IPC) DNA was spiked into all samples prior to DNA isolation as a positive control. Real-time PCR assays were performed using primers against mouse mtDNA NADH, cytochrome B or IPC. Results were expressed as a ratio of a target mtDNA to IPC. C. Levels of mtDNA encoded COX 1 and nuclear DNA encoded VDAC1 were examined in heart lysates by Western blots using GAPDH as a loading control. Results obtained by densitometry were expressed as fold changes relative to WT shams. D. Mitochondrial fractions were subjected to the measurements of complex I-V activities. E. Representative EM images of myocardial mitochondria at indicated magnitude. Mitochondrial mass was compared between groups. All values are means ± SEM. Significant differences are shown as * for sham vs. LPS-treated and Δ for WT vs. Becn1-Tg or Becn1+/− groups (p <0.05, n = 5, Mann-Whitney U test).
In response to sepsis or other trauma conditions, mitochondrial DNA (mtDNA) fragments can be released from damaged mitochondria into the cytoplasm and/or the extracellular space, and they function as a type of DAMP to stimulate inflammation35–37. We quantified the level of free mtDNA in the cytoplasm of the hearts of WT, Becn1-Tg, and Becn1+/− mice following an LPS challenge. There was a dose-dependent release of mtDNA in WT hearts, which was suppressed in the Becn1-Tg but exaggerated in the Becn1+/− mice (Figure 4B).
To evaluate changes in mitochondrial content, protein levels of mtDNA-encoded cytochrome c oxidase I (COX I) and nuclear DNA-encoded voltage-dependent anion-selective channel 1 (VDAC-1) were assessed (Figure 4C). LPS triggered a dose-dependent decrease in the abundance of these two mitochondrial proteins, suggesting a LPS-induced reduction in mitochondria content in the heart, consistent with our previous report of mitochondrial loss obtained in a pneumonia-related sepsis model35. Mitochondrial loss was alleviated in Becn1-Tg mice but accentuated in Becn1+/− mice. Similarly, mitochondrial function, measured as complex I and complex II-III activity, was better preserved in the Becn1-Tg mice as compared to WT following an LPS challenge of 5 mg/kg, a dose which clearly caused cardiac dysfunction in WT but not Becn1-Tg mice (Figure 4D). Comparison of mitochondrial mass between groups based on EM images additionally confirmed the above results (Figure 4E).
Beclin-1 increases LC3II associated with mitochondria and factors of PINK1-Parkin pathway but suppresses an increase in mitophagy receptors following LPS.
Damaged mitochondria with lost membrane potential or accumulated misfolded proteins can be cleared by PINK1(PTEN-induced putative kinase 1)-Parkin mitophagy38–40. Alternatively, mitophagy receptors in the outer mitochondrial membrane, such as BNIP3L/NIX, BNIP3 and FUNDC1, can bind directly to LC3 and direct mitochondria to autophagosomes for degradation41. We examined whether these mitophagy pathways are differentially stimulated in WT, Becn1-Tg, and Becn1+/− mice in response to LPS.
LPS triggered a gradual increase in LC3II that was associated with mitochondrial fractions in WT hearts, a response that was greater in Becn1-Tg mice but lower in the Becn1+/− mice (Figure 5A). This detected trend of mitophagy was confirmed by co-immunostaining the heart tissue with antibodies against mitochondrial protein mitofusin 2 (Mfn2) and lysosomal protein Lamp1 (Figure 5B). Co-localization of mitochondria and lysosomes, shown in white and pale green, was increased with LPS challenge, and this trend was evidently stronger in Becn1-Tg mice but weaker in Becn1+/− mice. These results are also consistent with the observation obtained under electron microscopy that there were more mitophagosomes and mitolysosomes, in which mitochondria were engulfed by and closely associated with the membrane structures of autosomes, in Becn1-Tg mice, but more lysed, deformed mitochondria in Becn1+/− mice (red arrows; Figure 4, and quantifications in Figure 5C).
Figure 5.
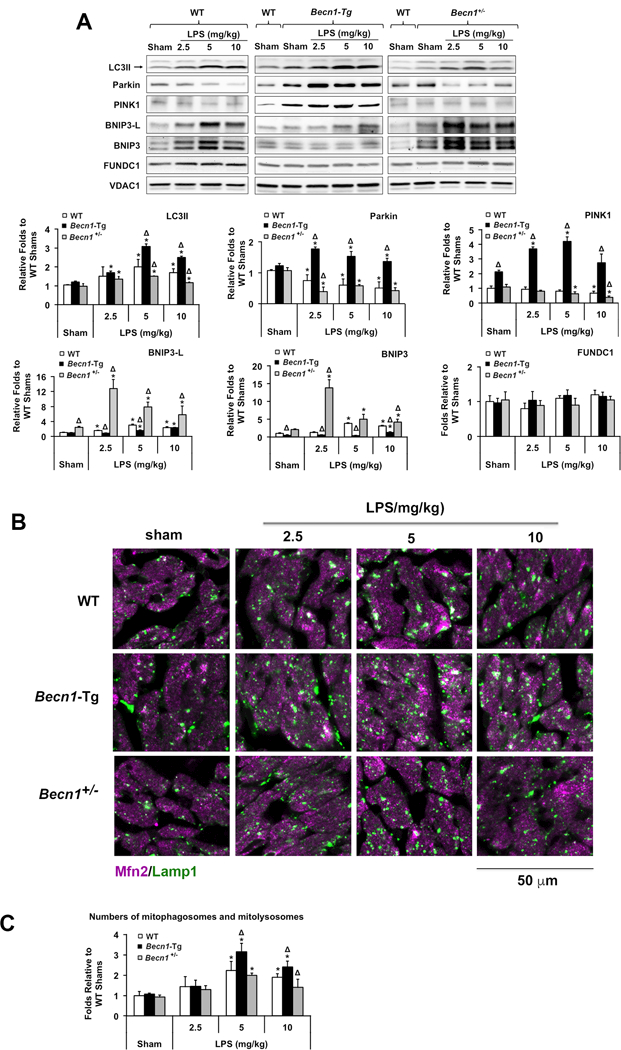
LPS-induced changes in mitophagy signaling molecules in the heart of WT, Becn1-Tg, and Becn1+/− mice. Mice were given LPS via i.p. at indicated doses, heart tissues were harvested 18 hours later. A. Levels of mitochondria-associated LC3II, Parkin, PINK1, BNIP3L, BNIP3, and FUNDC1 were examined by Western blots using VDAC1 as a loading control in mitochondrial fractions. Results obtained by densitometry were expressed as fold changes relative to WT shams. B. Heart tissue sections were co-stained with antibodies against Lamp1 (green) and Mfn2 (purple). Colors in white and pale green indicate the overlay of these two signals. Images are representative of n ≥ 3 animals per group. C. Number counts of mitophagosomes and mitolysosomes based on EM images. In A and C, all values are means ± SEM. Significant differences are shown as * for sham vs. LPS-treated and Δ for WT vs. Becn1-Tg or Becn1+/− groups (p < 0.05, n = 5, Mann-Whitney U test).
Interestingly, mitochondria-associated PINK1 and Parkin were highest in the Becn1-Tg mice and their levels were elevated or maintained following LPS treatment, whereas these signals decreased following LPS in both WT and Becn1+/− (Figure 5A). In contrast, LPS induced increases in mitochondrial-localized BNIP3L and BNIP3 in both WT and Becn1+/− hearts. The increases were most pronounced in the Beclin-1 deficient Becn1+/− hearts, but barely detectable in the Becn1-Tg mice. No significant changes were found in the levels of FUNDC1. Consistent with a previous report that Parkin is protective in the heart during LPS-induced sepsis42, our data suggest that Beclin-1 may help to suppress the activation of receptor-mediated mitophagy while facilitating a more adaptive mitophagic response via PINK1/Parkin.
Therapeutic potential of a Beclin-1-activating peptide.
A cell-permeable, autophagy-inducing peptide, Tat-beclin-1 (TB-peptide), has been shown to have antiviral activities27 and to attenuate cardiac dysfunction after pressure overload43. In our investigation, a recently developed, optimized version of the TB-peptide28 was given to mice via i.p.. Based on publications27, 28, 43, we chose a dose that was able to induce sufficient autophagy without causing detectable toxicity. As expected, TB-peptide dramatically enhanced cardiac autophagy, as shown by an increase in LC3II and a decrease in p62, in both sham animals and those given a sublethal LPS challenge of 5 mg/kg (Figure 6A). The TB-peptide significantly improved cardiac function (Figure 6B) and reduced increases in circulating cytokines, especially TNF-α, IFN-γ, IL-17α and IL-6, following LPS (Figure 6C). TB-peptide rescued the phenotypes of Becn1+/− mice, confirming that the peptide’s mechanism of action is mediated though a direct-targeted stimulation of Beclin-1-dependent autophagy.
Figure 6.
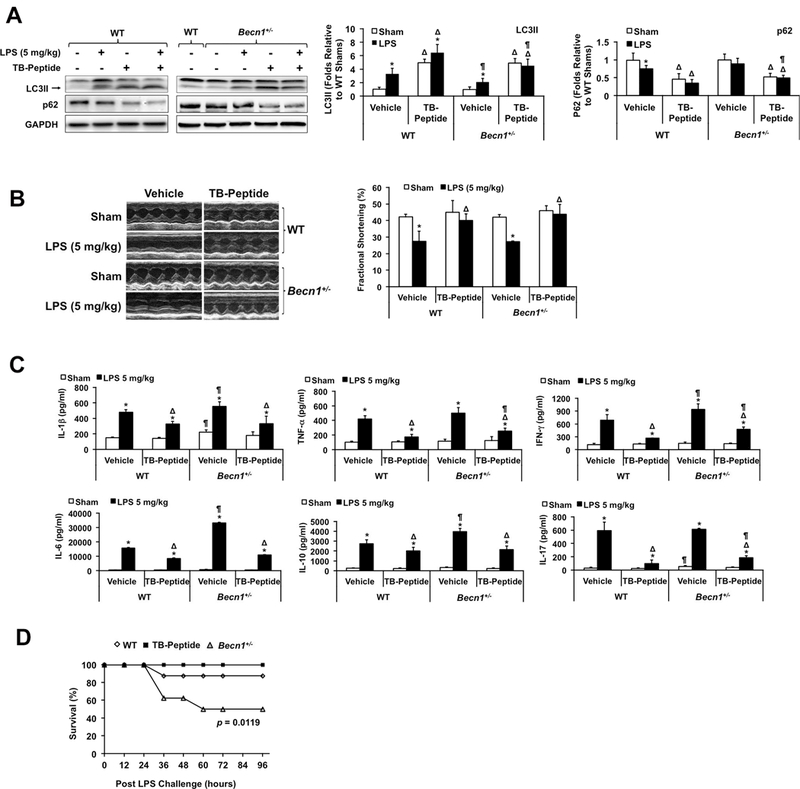
Effects of Tat-beclin-1 peptide (TB-peptide) in LPS-challenged mice. WT and Becn1+/− mice were given 5 mg/kg (A-C) or 10 mg/kg (D) LPS i.p. TB-peptide, 16 mg/kg, was administered i.p. 30 minutes post LPS challenge. Cardiac function was measured and tissues collected 18 hours post LPS challenge. A. Levels of LC3II, p62 and GAPDH were analyzed by Western blots using GAPDH as a loading control in heart tissue lysates. B. Cardiac function was examined by echocardiography. C. Cytokines in serum were measured by ELISA assays. In A-C, all values are means ± SEM. Significant differences are shown as * for with sham vs. LPS-treated, Δ for vehicle vs. TB-peptide-treated, and ¶ for WT vs. Becn1+/− groups (p < 0.05, n = 5, Mann-Whitney U test). D. Survival was monitored in LPS (10 mg/kg)-challenged WT ± TB-peptide and Becn1+/− mice, and * indicates a statistical significance when compared with WT (p < 0.05, n = 12, log-rank test).
When mice were challenged with 10 mg/kg LPS, there was a significant decrease in the survival of Becn1+/− mice compared to WT (Figure 6D), indicating the importance of Beclin-1-dependent processes in sepsis. However, TB-peptide did not significantly improve the survival of WT mice when LPS was increased to a lethal dose of 15 mg/kg (data not shown). Taken together, these data suggest that enhancing Beclin-1-dependent autophagy by TB-peptide holds a therapeutic potential for alleviating secondary organ failure during sepsis.
Discussion
This investigation was aimed at determining the role of Beclin-1-dependent autophagy in the heart during sepsis and to test the therapeutic potential of targeting this pathway using a mouse model of LPS-induced sepsis. We found that LPS triggered a dose-dependent autophagic response (Figure 1). When LPS was given at low doses, modeling mild sepsis, autophagy increased proportionally to the magnitude of the insult. However, under conditions of severe sepsis, this capability declined proportionally to the severity of the insult. The failure of autophagy induction at high-dose LPS challenges correlated with the activation of mTOR signaling (Figure 2) and the appearance of clinical signs of pathogenesis, manifested as a decline in cardiac function, elevation in inflammation, and evidence of fibrotic injury (Figure 3). Our observations in the heart are consistent with previous studies in the liver showing increased autophagy in the early phases of sepsis followed by a decline near late-stage organ failure in a mouse CLP sepsis model15, 44. Thus, the changes we have documented in the heart may mimic the progression of systemic organ failure.
Previous studies have used pharmacological interventions to examine the role of autophagy in sepsis15, 44–48. Our studies, however, are the first use of genetically modified mouse strains with cardiac-specific overexpression of Beclin-1 (Becn1-Tg) and global depletion of Beclin-1 (Becn1+/−) to manipulate levels of autophagy in vivo. The presence of the Becn1-Tg increased the signs of autophagy, suppressed mTOR signaling, enhanced AMPK and ULK1 activation, preserved cardiac function, and reduced evidence of cardiac inflammation and fibrosis in response to LPS (Figures 2 and 3). Becn1+/− mice showed opposite phenotypes in the above measurements, and additionally, they displayed a reduced survival rate at higher doses of LPS (Figure 6D). These results suggest that Beclin-1-dependent autophagy is an adaptive, protective response in the heart during sepsis. Lastly, an intervention by TB-peptide alleviated LPS-induced outcomes in both WT and Becn1+/− mice, suggesting that this novel peptide possesses a therapeutic potential for sepsis (Figure 6).
The mTOR pathway is a well-known upstream nodal point that acts to inhibit autophagy49. In our experimental settings, we were unable to detect significant changes in mTOR activation in the heart of animals challenged by low-dose LPS, such as 2.5 mg/kg, compared with shams. However, the attenuation of autophagy in response to high-dose LPS, such as 5 mg/kg and 10 mg/kg, is clearly associated with mTOR activation (Figure 2C). We propose that sepsis-induced autophagy is an initial adaptive defense mechanism in early stages, whereas during severe stages of sepsis, mTOR activation inhibits beneficial autophagic activities, which then become maladaptive. The data may also suggest that the activation of autophagy in early sepsis is mTOR-independent. Recent studies have identified a number of autophagy pathways whose actions are mTOR-independent, such as the intracellular inositol-IP3 pathway50, Ca2+-calpain pathway51, cAMP-exchange protein activated by cAMP (Epac)-PLCε-IP3 pathway51, leucine-rich repeat kinase 252 , and trehalose53, etc. Whether sepsis utilizes any of these signals or other unknown mechanisms to operate autophagy machinery in the heart as well as in other organs remains to be investigated. Additionally, while mTOR suppresses autophagy by inhibiting ULK1, the activating kinase that lies upstream of Beclin-154, our demonstration here shows that mTOR activation is suppressed in the Becn1-Tg hearts (Figure 2C). The underlying mechanism of this Beclin-1 action on mTOR may involve the activation of AMPK and ULK1 (Figure 2D). Together, these results suggest a positive feedback regulation of autophagy by Beclin-1.
Sepsis-induced systemic inflammatory syndrome is a result of combined actions of PAMPs (pathogen-associated molecular patterns) and DAMPs. Recognized by pattern recognition receptors (PPRs), such as toll-like receptors, RIG-I-like receptors, and NOD-like receptors, PAMPs and DAMPs initiate an overwhelming production of cytokines through NF-κB, MAPKs, inflammasomes and caspases55, 56. LPS triggers cytokine production via binding to the TLR4/CD14/MD2 receptor complex on cellular membranes and/or to caspase inside of cells, resulting in the activation of NF-κB and inflammasomes57, 58. This action sequentially causes the release of DAMPs from activated, injured, or necrotic cells56, 59. LPS also stimulates mtROS overproduction, leading to mitochondrial damage60 and a release of mitochondrial DAMPs including mtDNA fragments36, N-formyl peptides61, mtROS35, cardiolipin62, ATP63, mitochondrial transcription factor A64, and cytochrome c65.
Autophagy presents a control over inflammation by limiting the availability of inflammasome activators and/or components66, and by reducing mitochondrial DAMPs via mitophagy37. However, an increase in autophagy under certain severe or prolonged stress may exacerbate unwanted outcomes due to deregulated degradation of cellular components, resulting in aggressive inflammation12, 30. Our previous investigations have shown that cardiac inflammation during sepsis and burn trauma is mediated at least partially by the activation of TLR4/CD14 signaling6 and the increase in mitochondrial DAMPs5, 35. In this present investigation, using an LPS-induced sepsis model, we observed that increasing Beclin-1-dependent autophagy alleviates sepsis outcomes, shown by improved contractility, attenuated cytokines, and reduced fibrotic injury in the heart (Figure 3). However, this benefit comes with certain limitations, since in response to high-dose LPS (10 mg/kg), overwhelming inflammation still occurs in Becn1-Tg mice even when significant autophagic activity was provided. Thus, inflammation remains, at least in part, a major cause of cardiac dysfunction in Becn1-Tg mice. It is uncertain whether the increase in autophagy itself contributes to heart failure in this group of animals. Signaling crosstalk between autophagy and inflammation happens dynamically throughout the development of various disease conditions12. How autophagy alters individual inflammation pathways via inflammasomes, caspases, NFκB and/or MAPKs, etc. at different stages of sepsis remains to be defined.
As evidenced in this study, damage in cardiac mitochondria progresses with the severity of sepsis, shown by the disruption of mitochondrial structure, a reduction in metabolic function, and the release of mtDNA fragments to the cytosol, which can subsequently act as a type of mitochondrial DAMP to promote inflammation35–37. These manifestations of mitochondrial damage following LPS challenge were all reduced in the Becn1-Tg but increased in Becn1+/− mice (Figure 4). Additionally, we observed more mitophagosomes/mitolysosomes in Becn1-Tg mice when compared with WT and Becn1+/− mice, suggesting a functional significance of Beclin-1 in the clearance of dysfunctional mitochondria via mitophagy in the myocardium during sepsis. Since combined signals from PAMPs and DAMPs stimulate inflammation in the heart after sepsis, it is not a surprise that levels of cytosolic mtDNA measured are not proportional to the production of total cytokines in the heart tissue measured in Figure 3. In addition to mtDNA, other types of mitochondrial damage, such as a deficiency in energy supply and alterations in mitochondrial metabolisms, are considered significant impact factors in cardiac dysfunction after sepsis. Therefore, in Beclin1+/− mice, LPS triggers an aggressive accelerated overall mitochondrial deterioration that contributes to the animals’ rapid decline in cardiac performance via multiple pathways.
A striking difference in the spectrum of proteins involved in mitophagy was found when comparing mice with increased Beclin-1 availability versus those deficient in Beclin-1 (Figure 5). In the mitochondrial fractions isolated from Becn1+/− hearts, there was a robust increase in mitophagy adaptor proteins BNIP3L and BNIP3, occurring with reduced evidence of activity along the PINK1-Parkin axis. The converse was observed in Becn1-Tg hearts that showed little increase in the association of BNIPL3 or BNIP3 with mitochondria but a drastic elevation in PINK1 and Parkin. PINK1 recruits Parkin to damaged mitochondria via phosphorylated Mfn238–40. In our experimental setting, we were unable to detect the phosphorylated form of either Parkin or Mfn2 in the heart tissue of Becn1-Tg mice using the published approach of Phos-tag Western blotting38, 39. However, we realized that the detection of phosphorylated Parkin or Mfn2 was accomplished in cultured cells with an overexpression of protein targets38, 39. We suspect that the current method may have a limitation in detecting low contents of phosphorylation in in vivo models when PINK1, Parkin and Mfn2 are expressed at endogenous physiological levels. Based on ongoing investigation in our lab (data not shown), we hypothesize that the stimulation of PINK1-Parkin mitophagy by Beclin-1 may involve mitochondria-associated membranes (MAMs). MAMs are specialized subcellular domains that physically link mitochondria with the endoplasmic reticulum (ER)67, and they are essential to mitochondrial health68, 69. Beclin-1 and PINK1 are found to re-localize to MAMs during mitophagy initiation, and via direct interaction, they promote ER-mitochondria tethering and the formation of autophagosome precursors to facilitate mitophagy70, 71. Beclin-1 was also found to interact with Parkin, and this association is required for the progress of PINK1-Parkin mitophagy72. Thus, the PINK1-Parkin mitophagy pathway is more accessible for activation when Beclin-1 signaling is upregulated. Intriguingly, recent studies suggest that PINK1/Parkin exert an additional control over mitochondrial quality in addition to mitophagy. Parkin positively regulates mitochondrial biogenesis through proteasomal degradation of its substrate PARIS, a zinc-finger protein73, 74. Following PINK1-Parkin mitophagy, increases in mitochondrial and lysosomal biogenesis via transcription factors, Nrf2 and TFEB, were detected in neuroblastoma cells75. In vivo, a lack of Parkin expression in the ventral midbrain resulted in decreases in mitochondrial size, number, and mitochondrial biogenesis makers, together with declines in mitochondrial respiration73. Whether similar mechanisms are utilized to regulate mitochondria in septic hearts remains for further investigation. In this report, we observed that the quality control of mitochondria was improved, as well as cardiac outcomes, in Becn1-Tg mice under LPS challenge (Figures 3 and 4). The result suggests a signaling pathway from Beclin-1 to PINK1/Parkin that protects cardiac mitochondria during sepsis.
The stimulation of BNIP3 is a pathological response under conditions of hypoxia76 and ischemia/reperfusion77. It is regulated transcriptionally via hypoxia-inducible factor 178 and FOXO3a79 and/or by posttranslational oxidation via ROS77. However, the mechanism for how Beclin-1 suppresses BNIP3 signaling remains to be determined. According to literature, BNIP3 functions as a dual regulator in mitochondria. Upon activation, BNIP3 prompts the opening of the mitochondrial permeability transition pore (mPTP), recruits LC3 for autophagosome formation, and initiates mitophagy80–82. This protein also causes mitochondrial dysfunction and subsequently induces cell death via the activation of downstream effectors Bax/Bak under stress conditions76. Both BINP3-induced mitophagy and mitochondrial damage were observed in the myocardium80, 83. It was further revealed that transcriptional upregulation of BNIP3 by FOXO3a caused a decrease in mitochondrial membrane potential and an increase in mitochondrial Ca2+, leading to mitochondrial fragmentation and apoptosis of cardiomyocytes79. Hence, the effect of BNIP3 accumulation on mitochondria is a result of the balance between its double actions. In the hearts of LPS-induced septic mice, the increase in mitochondrial BNIP3 may aggravate detrimental mitochondrial deficiency while the induced mitophagy is not strong enough to clear damaged mitochondria. Our data suggest that Beclin-1 may have a control to mitigate BNIP3 signaling.
Taken together, we consider that the improved status of the mitochondria in the hearts of Becn1-Tg mice is likely the result of several folds of signaling regulations. While PINK1-Parkin mitophagy facilitates the clearance of damaged mitochondria, both the AMPK/ULK1 pathway and PINK1/Parkin pathway have the capability to promote mitochondrial biogenesis73–75, 84–86. These effects act together, leading to preservation of the mass of functional mitochondria during sepsis in Becn1-Tg mice. In contrast, an enhanced mitochondrial BNIP3 signal and low levels of AMPK and ULK1 activation, together with the stronger inflammatory responses, aggravate the deterioration of cardiac mitochondria in WT and Becn1+/− mice under challenge by LPS.
Nonetheless, our observations suggest that Beclin-1-dependent processes may help suppress the processing of cardiac mitochondria via mitophagy adaptor receptor pathways while supporting PINK1/Parkin-mediated quality control and preventing the release of mitochondrial DAMPs to promote inflammation. This conclusion is consistent with previous reports that Parkin is at least partially protective in the heart during LPS-induced sepsis42.
The induction of autophagy by TB-peptide preserved cardiac function and suppressed the increase in circulating cytokines following LPS challenge (Figure 6). This peptide was able to rescue the phenotypes of Becn1+/− mice, supporting its mechanism of action by directly targeting the Beclin-1 pathway. Previous preclinical studies evaluating autophagy as a therapeutic approach for sepsis were mainly focused on the mTOR inhibitor rapamycin17, 45–48. Though certain beneficial effects were observed in a mouse CLP-sepsis model17, 45, 48, adverse effects of rapamycin on lung injury were reported in a LPS-sepsis model46, 47. Since mTOR is involved in the regulation of a variety of pathways87, rapamycin impacts a broad spectrum of cellular functions in addition to autophagy and may cause unwanted toxicity46. In this regard, altering Beclin-1 levels provides a more targeted approach for modulating autophagy. Thus, TB-peptide, used by itself or in combination with other therapies, may prove to be a potent beneficial therapeutic approach in the treatment of sepsis. However, it should be noted that Beclin-1 is involved in other cellular functions, such as endocytosis88. We will investigate these aspects in future studies to fully understand the role of Beclin-1 in sepsis.
Supplementary Material
Clinical Perspective
What Is New?
We examined the status of cardiac autophagy and its role during sepsis pathogenesis using a rodent lipopolysaccharide (LPS)-induced sepsis model.
Forced overexpression of Beclin-1 in the heart promotes autophagy and mitophagy, protects mitochondria, improves cardiac function, and alleviates inflammation and fibrosis after LPS challenge, whereas haplosufficiency for beclin 1 results in the opposite effects.
Injection of a cell-permeable Tat-Beclin-1 peptide improves outcomes in LPS-challenged animals.
What Are the Clinical Implications?
Promoting Beclin-1-dependent signaling may be a novel and effective intervention to alleviate organ dysfunction caused by insufficient and maladaptive autophagy during severe sepsis.
Acknowledgements
We thank Drs. Juquan Song, Ryan Huebinger, Joshua Gatson (Surgery, UTSW), Thomas Gillette, and Sean Xiang Luo (Cardiology, UTSW) for discussions about experiments, Dr. Hong Zhu (Clinical Sciences, UTSW) for consultation about statistical analysis, and Dave Primm for editing this manuscript.
Sources of Funding
This work was supported by National Institute of Health grant 1R01GM111295–01 (to Q.S.Z.), RO1HL109471 (to Z.P.L.), GM115473 (to Y.X), HL097768 and HL102478 (to B.A.R.), U19AI199725 (to B.L.), and a Foundation Leducq Transatlantic Network of Excellence in Cardiovascular Research Award (to B.L.).
Footnotes
Data Availability
The data supporting the findings of this study are available from the corresponding author upon request.
Disclosures
None.
References
- 1. Angus DC, Pereira CA and Silva E. Epidemiology of severe sepsis around the world. Endocr Metab Immune Disord Drug Targets. 2006;6:207–212. [DOI] [PubMed] [Google Scholar]
- 2. Zanotti-Cavazzoni SL and Hollenberg SM. Cardiac dysfunction in severe sepsis and septic shock. Curr Opin Crit Care. 2009;15:392–397. [DOI] [PubMed] [Google Scholar]
- 3. Kayar SR and Banchero N. Volume density and distribution of mitochondria in myocardial growth and hypertrophy. Respir Physiol. 1987;70:275–286. [DOI] [PubMed] [Google Scholar]
- 4. Zang Q, Maass DL, Tsai SJ and Horton JW. Cardiac mitochondrial damage and inflammation responses in sepsis. Surg Infect (Larchmt). 2007;8:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zang QS, Sadek H, Maass DL, Martinez B, Ma L, Kilgore JA, Williams NS, Frantz DE, Wigginton JG, Nwariaku FE, Wolf SE and Minei JP. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am J Physiol Heart Circ Physiol. 2012;302:H1847–H1859. [DOI] [PubMed] [Google Scholar]
- 6. Zang QS, Maass DL, Wigginton JG, Barber RC, Martinez B, Idris AH, Horton JW and Nwariaku FE. Burn serum causes a CD14-dependent mitochondrial damage in primary cardiomyocytes. Am J Physiol Heart Circ Physiol. 2010;298:H1951–H1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zang QS, Martinez B, Yao X, Maass DL, Ma L, Wolf SE and Minei JP. Sepsis-induced cardiac mitochondrial dysfunction involves altered mitochondrial-localization of tyrosine kinase Src and tyrosine phosphatase SHP2. PloS one. 2012;7:e43424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE and Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mizushima N and Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kubli DA and Gustafsson AB. Cardiomyocyte health: adapting to metabolic changes through autophagy. Trends endocrinol metab. 2014;25:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishida K, Yamaguchi O and Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res. 2008;103:343–351. [DOI] [PubMed] [Google Scholar]
- 12. Levine B, Mizushima N and Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie M, Morales CR, Lavandero S and Hill JA. Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol. 2011;26:216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM and Hsu C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37:289–296. [DOI] [PubMed] [Google Scholar]
- 15. Chien WS, Chen YH, Chiang PC, Hsiao HW, Chuang SM, Lue SI and Hsu C. Suppression of autophagy in rat liver at late stage of polymicrobial sepsis. Shock. 2011;35:506–511. [DOI] [PubMed] [Google Scholar]
- 16. Watanabe E, Muenzer JT, Hawkins WG, Davis CG, Dixon DJ, McDunn JE, Brackett DJ, Lerner MR, Swanson PE and Hotchkiss RS. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab Invest. 2009;89:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsieh CH, Pai PY, Hsueh HW, Yuan SS and Hsieh YC. Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg. 2011;253:1190–1200. [DOI] [PubMed] [Google Scholar]
- 18. Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC and Gottlieb RA. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470–H479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turdi S, Han X, Huff AF, Roe ND, Hu N, Gao F and Ren J. Cardiac-specific overexpression of catalase attenuates lipopolysaccharide-induced myocardial contractile dysfunction: role of autophagy. Free Radic Biol Med. 2012;53:1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B and Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H and Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. [DOI] [PubMed] [Google Scholar]
- 22. Yue Z, Jin S, Yang C, Levine AJ and Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Nat Acad Sci USA. 2003;100:15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kihara A, Kabeya Y, Ohsumi Y and Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO J. 2001;2:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T and Yoshimori T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. [DOI] [PubMed] [Google Scholar]
- 25. Itakura E, Kishi C, Inoue K and Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N and Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV and Levine B. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi F, Jacquelot N, Yamazaki T, Senovilla L, Marino G, Aranda F, Durand S, Sica V, Chery A, Lachkar S, Sigl V, Bloy N, Buque A, Falzoni S, Ryffel B, Apetoh L, Di Virgilio F, Madeo F, Maiuri MC, Zitvogel L, Levine B, Penninger JM and Kroemer G. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell. 2016;30:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R and Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. [DOI] [PubMed] [Google Scholar]
- 30. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clinical Invest. 2007;117:1782–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jung CH, Ro SH, Cao J, Otto NM and Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laplante M and Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bedard PM, Zweiman B and Atkins PC. Quantitation by myeloperoxidase assay of neutrophil accumulation at the site of in vivo allergic reactions. J Clin Immunol. 1983;3:84–89. [DOI] [PubMed] [Google Scholar]
- 34. Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T and Yoshida T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP and Zang QS. Mitochondrial ROS Induces Cardiac Inflammation via a Pathway through mtDNA Damage in a Pneumonia-Related Sepsis Model. PloS one. 2015;10:e0139416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K and Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I and Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen Y and Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S and Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin SM and Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moyzis AG, Sadoshima J and Gustafsson AB. Mending a broken heart: the role of mitophagy in cardioprotection. Am J Physiol Heart Circ Physiol. 2015;308:H183–H192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Piquereau J, Godin R, Deschenes S, Bessi VL, Mofarrahi M, Hussain SN and Burelle Y. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy. 2013;9:1837–1851. [DOI] [PubMed] [Google Scholar]
- 43. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B and Sadoshima J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation. 2016;133:1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi W, Watanabe E, Fujimura L, Watanabe-Takano H, Yoshidome H, Swanson PE, Tokuhisa T, Oda S and Hatano M. Kinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsis. Crit Care. 2013;17:R160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu W, Guo J, Mu J, Tian L and Zhou D. Rapamycin Protects Sepsis-Induced Cognitive Impairment in Mouse Hippocampus by Enhancing Autophagy. Cell Mol Neurobiol. 2017;37:1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fielhaber JA, Carroll SF, Dydensborg AB, Shourian M, Triantafillopoulos A, Harel S, Hussain SN, Bouchard M, Qureshi ST and Kristof AS. Inhibition of mammalian target of rapamycin augments lipopolysaccharide-induced lung injury and apoptosis. J Immunol. 2012;188:4535–4542. [DOI] [PubMed] [Google Scholar]
- 47. Yan Z, Xiaoyu Z, Zhixin S, Di Q, Xinyu D, Jing X, Jing H, Wang D, Xi Z, Chunrong Z and Daoxin W. Rapamycin attenuates acute lung injury induced by LPS through inhibition of Th17 cell proliferation in mice. Sci Rep. 2016;6:20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin CW, Lo S, Perng DS, Wu DB, Lee PH, Chang YF, Kuo PL, Yu ML, Yuan SS and Hsieh YC. Complete activation of autophagic process attenuates liver injury and improves survival in septic mice. Shock. 2014;41:241–249. [DOI] [PubMed] [Google Scholar]
- 49. Kim YC and Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ and Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O’Kane CJ, Floto RA and Rubinsztein DC. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manzoni C, Mamais A, Roosen DA, Dihanich S, Soutar MP, Plun-Favreau H, Bandopadhyay R, Hardy J, Tooze SA, Cookson MR and Lewis PA. mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1. Sci Rep. 2016;6:35106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sarkar S, Davies JE, Huang Z, Tunnacliffe A and Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. [DOI] [PubMed] [Google Scholar]
- 54. Cao Y and Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839–849. [DOI] [PubMed] [Google Scholar]
- 55. Kawai T and Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Internat Immunol. 2009;21:317–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen GY and Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ and Lee JO. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. [DOI] [PubMed] [Google Scholar]
- 58. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L and Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. [DOI] [PubMed] [Google Scholar]
- 59. Seong SY and Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. [DOI] [PubMed] [Google Scholar]
- 60. Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE and Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. [DOI] [PubMed] [Google Scholar]
- 61. Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S, Huang Q and Wewers MD. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit Care Med. 2009;37:2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chakraborty K, Raundhal M, Chen BB, Morse C, Tyurina YY, Khare A, Oriss TB, Huff R, Lee JS, St Croix CM, Watkins S, Mallampalli RK, Kagan VE, Ray A and Ray P. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat Commun. 2017;8:13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schwiebert EM and Zsembery A. Extracellular ATP as a signaling molecule for epithelial cells. Biochimica et biophysica acta. 2003;1615:7–32. [DOI] [PubMed] [Google Scholar]
- 64. Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC and Crouser ED. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol. 2012;189:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Eleftheriadis T, Pissas G, Liakopoulos V and Stefanidis I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front Immunol. 2016;7:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A and Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nature immunol. 2012;13:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mannella CA, Buttle K, Rath BK and Marko M. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors. 1998;8:225–228. [DOI] [PubMed] [Google Scholar]
- 68. Raturi A and Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochimica et biophysica acta. 2013;1833:213–224. [DOI] [PubMed] [Google Scholar]
- 69. Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR and Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal. 2015;22:995–1019. [DOI] [PubMed] [Google Scholar]
- 70. Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM and Valente EM. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy. 2017;13:654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, Torosantucci L, Cassina L, Russo MA, Dallapiccola B, Valente EM and Casari G. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. [DOI] [PubMed] [Google Scholar]
- 72. Choubey V, Cagalinec M, Liiv J, Safiulina D, Hickey MA, Kuum M, Liiv M, Anwar T, Eskelinen EL and Kaasik A. BECN1 is involved in the initiation of mitophagy: it facilitates PARK2 translocation to mitochondria. Autophagy. 2014;10:1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, Shin JH and Dawson TM. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Nat Acad Sci USA. 2015;112:11696–11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL and Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ivankovic D, Chau KY, Schapira AH and Gegg ME. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136:388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kubli DA, Ycaza JE and Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007;405:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kubli DA, Quinsay MN, Huang C, Lee Y and Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2025–H2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Nat Acad Sci USA. 2000;97:9082–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chaanine AH, Kohlbrenner E, Gamb SI, Guenzel AJ, Klaus K, Fayyaz AU, Nair KS, Hajjar RJ and Redfield MM. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am J Physiol Heart Circ Physiol. 2016;311:H1540–H1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA and Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. [DOI] [PubMed] [Google Scholar]
- 81. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S and Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 83. Regula KM, Ens K and Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. [DOI] [PubMed] [Google Scholar]
- 84. Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M and Yan Z. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8:548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H, Wang Y, Subramaniam S, Chien S and Shyy JY. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci Signal. 2017;10:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shimizu Y, Polavarapu R, Eskla KL, Nicholson CK, Koczor CA, Wang R, Lewis W, Shiva S, Lefer DJ and Calvert JW. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. Journal of molecular and cellular cardiology. 2018;116:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Laplante M and Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M and Vandenabeele P. Beclin1: a role in membrane dynamics and beyond. Autophagy. 2012;8:6–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.