

Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is an important transcription factor that modulates lipid metabolism and inflammation. However, it remains unclear whether PPARγ is involved in modulation of estrogen (E2)-induced inflammation, thus affecting apoptosis of E2-deprived breast cancer cells, MCF-7:5C and MCF-7:2A. Here, we demonstrated that E2 treatment suppressed the function of PPARγ in both cell lines, although the suppressive effect in MCF-7:2A cells was delayed owing to high PPARγ expression. Activation of PPARγ by a specific agonist, pioglitazone selectively blocked the induction of tumor necrosis factor alpha (TNFα) expression by E2, but did not affect other adipose inflammatory genes, such as fatty acid desaturase 1(FADS1) and interleukin-6 (IL-6). This suppression of TNFα expression by pioglitazone was mainly mediated by trans-repression of nuclear factor-κB (NF-κB) DNA-binding activity. A novel finding was that NF-κB functions as an oxidative stress inducer in MCF-7:5C cells but an antioxidant in MCF-7:2A cells. Therefore, the NF-κB inhibitor JSH-23 displayed effects equivalent to those of pioglitazone, with complete inhibition of apoptosis in MCF-7:5C cells, but it increased E2-induced apoptosis in MCF-7:2A cells. Depletion of PPARγ by small interfering RNA or the PPARγ antagonist T0070907 accelerated E2-induced apoptosis, with activation of NF-κB-dependent TNFα and oxidative stress. For the first time, we demonstrated that PPARγ is a growth signal and has potential to modulate NF-κB activity and oxidative stress in E2-deprived breast cancer cell lines. All of these findings suggest that anti-PPARγ therapy is a novel strategy to improve the therapeutic effects of E2-induced apoptosis in E2-deprived breast cancer.
Keywords: peroxisome proliferator-activated receptor γ (PPARγ), nuclear factor-κB (NF-κB), apoptosis, estrogen receptor, breast cancer
Introduction
Anti-hormone therapy is a standard treatment of estrogen receptor (ER)-positive breast cancer (1). However, resistance to this therapy is inevitable. Paradoxically, E2 has been found to be able to induce apoptosis in anti-hormone-resistant models in vivo (2, 3) and in vitro (4, 5). Indeed, E2-induced apoptosis has clinical relevance (6) to treatment of aromatase inhibitor-resistant breast cancer patients (7) and the reduction of breast cancer incidence in postmenopausal women receiving hormone replacement therapy (HRT) with only conjugated equine estrogen (CEE) (8). However, only 30% of these patients benefit from this treatment (7). This clinical fact mandates the investigation of factors that may modify the therapeutic efficacy of E2-induced apoptosis.
Our recent findings demonstrated that a major mechanism by which E2 induces apoptosis is accumulation of stress responses, including endoplasmic reticulum, oxidative, and inflammatory stress (9, 10). Two major cellular organelles: mitochondria and the endoplasmic reticulum have been observed to mediate stress responses (9, 10). Oxidative stress triggered by E2 elevates the production of reactive oxygen species (ROS) and the expression of oxidative stress indicator hemeoxygenase-1(HMOX1) in long-term estrogen deprived (LTED) breast cancer cell lines: MCF-7:5C and MCF-7:2A (10, 11). The endoplasmic reticulum is a critical regulatory site for conveying signals between the nucleus and cytoplasm to induce apoptosis (10, 12). Two sensors of endoplasmic reticulum stress are activated but perform different functions after E2 treatment. One of these sensors, protein kinase RNA-like endoplasmic reticulum kinase (PERK) is responsible for homeostasis of unfolded proteins and is a key driver of E2-induced apoptosis (10, 12). The other sensor, inositol-requiring protein 1 alpha (IRE1α), mainly mediates endoplasmic reticulum-associated degradation of phospholipids (12). Activation of these endoplasmic reticulum stress sensors suggests that abnormal protein folding and lipid metabolism occur after exposure to E2, although the mechanism is unknown.
Aberrant lipid metabolism and uncontrolled endoplasmic reticulum stress are well known to be causative factors that induce inflammatory responses in many diseases (13–15). In line with this, E2 widely activates lipid metabolism-associated genes, including adipogenetic transcription factor CCAAT/enhancer binding protein β (CEBPβ), members of the fatty acid desaturase (FADS) family for arachidonic acid biosynthesis, and the adipose inflammatory factors interleukin (IL)-4 and −6, in LTED breast cancer cell lines (9, 16). Our global gene and microRNA arrays both demonstrated that abnormal lipid metabolism occurs in MCF-7:5C and MCF-7:2A cells, particularly the latter (9, 17). Despite the fact that MCF-7:5C and MCF-7:2A cells are derived from the same parental MCF-7 cells under LTED conditions, NF-κB is constitutively activated in MCF-7:5C cells but not in MCF-7:2A cells (16); while MCF-7:2A cells have a stronger antioxidant system than that MCF-7:5C cells (11). Moreover, cellular redox status has been observed to be closely related to adipogenesis regulated by many transcription factors, such as PPARγ, NF-κB, and nuclear factor erythroid 2-related factor 2 (Nrf2) (18, 19). Nevertheless, whether lipid metabolism-associated transcription factors are involved in the modulation of oxidative stress to affect E2-induced apoptosis remains unclear.
PPARγ plays an important role in the regulation of adipogenesis in mammary glands, as well as in breast cancer cells (20). Substantial evidence demonstrates that PPARγ carries out functional cross-talk with ERα to affect normal mammary development and breast cancer progression (21–23). Additionally, PPARγ is a well-known nuclear factor that regulates the progress of inflammation in a variety of cells, including vascular endothelial cells, intestinal epithelial cells, and macrophages (24–28). Thus, the PPARγ-specific agonist rosiglitazone and pioglitazone (both thiazolidinediones) provide therapeutic benefits on type 2 diabetes mellitus, cardiovascular diseases, colitis, and rheumatoid arthritis (24–29). Trans-suppression of NF-κB by the PPARγ agonist is a major mechanism of inhibition of inflammation (26). Our very recent findings demonstrated that the NF-κB-dependent TNFα axis is activated by PERK kinase in MCF-7:5C cells to mediate E2-induced apoptosis (16). However, it remains unclear the functional relationship between PPARγ and NF-κB in regulation of E2-induced apoptosis in the LTED breast cancer cells.
We sought to further understand how PPARγ modulates inflammatory responses that affect E2-induced apoptosis in the LTED breast cancer cell lines: MCF-7:5C and MCF-7:2A. Our results demonstrated that E2 deprivation alters the expression of PPARγ in breast cancer cells. Activation of PPARγ by its specific agonist pioglitazone suppressed NF-κB DNA-binding activity and NF-κB-dependent TNFα expression. Furthermore, a mechanistic finding was that NF-κB functions as an oxidative stress inducer in MCF-7:5C cells but as an antioxidant in MCF-7:2A cells. Therefore, the NF-κB inhibitor JSH-23 displayed effects equivalent to those of pioglitazone on the two cell lines, by completely blocking apoptosis in MCF-7:5C cells whereas increasing E2-induced apoptosis in MCF-7:2A cells. Further depletion of PPARγ or treatment with the PPARγ antagonist T0070907 activated NF-κB and oxidative stress (30), thereby accelerating E2-induced apoptosis in the two LTED breast cancer cell lines. Collectively, PPARγ is the first identified molecule to counteract E2-induced apoptosis via transcriptional suppression of NF-κB activity and oxidative stress in LTED breast cancer cell lines. Disruption of this suppression by anti-PPARγ therapy has the potential to improve the therapeutic effects of E2-induced apoptosis in endocrine resistant breast cancer.
Materials and Methods
Materials
Estradiol and GW9662 were purchased from Sigma-Aldrich (St. Louis, MO). Pioglitazone and T0070907 were obtained from Tocris. JSH-23 was purchased from CalBiochem. For Western blotting, antibodies against PPARγ, cleaved poly (ADP-ribose) polymerase (PARP), Caspase 7, phosphor-Akt, total-Akt, and IRE1α were obtained from Cell Signaling Technology (Beverly, MA). ERα (sc-544) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture conditions
A panel of breast cancer cell lines were cultured as described previously (31). They included ER-positive (MCF-7, T47D, ZR-75–1, BT-474, MCF-7:5C, and MCF-7:2A) and ER-negative (Sk-Br-3, MDA-MB-231, MCF-7:ICI-R, and T47D:C42) cell lines. MCF-7:5C and MCF-7:2A cells were cloned from long-term E2-deprived (LTED) MCF-7 cells, and T47D:C42 cells were cloned from LTED T47D cells. These three cell lines were maintained in phenol red-free RPMI 1640 supplemented with 10% dextran-coated charcoal-stripped fetal bovine serum. All cell lines were validated according to their short tandem repeat (STR) profiles at The University of Texas MD Anderson Cancer Center Characterized Cell Line Core (CCLC). The STR patterns of all cell lines were consistent with those from the CCLC standard cells (Supplementary Table S1).
Annexin V binding assay to detect apoptosis
A FITC annexin V Detection Kit I (BD Pharmingen) was used to quantify apoptosis of MCF-7:5C and MCF-7:2A cells through flow cytometry according to the manufacturer’s instructions. In brief, MCF-7:5C and MCF-7:2A cells were seeded in 10-cm dishes. The next day, the cells were treated with different compounds for different periods. Cells were suspended in 1× binding buffer, and 1 × 105 cells were stained simultaneously with FITC-labeled annexin V and propidium iodide (PI) for 15 minutes at room temperature. The cells were analyzed using a BD Accuri C6 plus flow cytometer (Becton Dickinson).
NF-κB (p65) Transcription Factor DNA-binding Assay
MCF-7:5C and MCF-7:2A cells were treated with a vehicle control (0.1% DMSO) or pioglitazone (10 µM) at different time points. Nuclear protein was extracted from cells according to the manufacturer’s instruction (Cayman Chemical). NF-κB (p65) DNA-binding activity was detected using an NF-κB (p65) Transcription Factor Assay Kit (Cayman Chemical).
Immunoblotting
Cells were harvested in cell lysis buffer (Cell Signaling Technology, Beverly, MA) supplemented with Protease Inhibitor Cocktail Set I and Phosphatase Inhibitor Cocktail Set II (Calbiochem, San Diego, CA). Immunoblotting was performed as previously described (10).
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA isolated from cells using an RNeasy Micro kit (Qiagen) was converted to first-strand cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR assays were performed with SYBR Green PCR Master Mix (Applied Biosystems) and a QuantStudio 6 Flex real-time PCR System (Applied Biosystems). All primers were synthesized in Integrated DNA Technologies. All data were normalized by 36B4.
PPARγ siRNA transfection
Briefly, cells were seeded in 6-well plates. Next day, cells were transfected with human PPARγ SMARTpool siRNA (Dharmacon, L-003436–00-0005) at 50nM according to manufacturer’s instructions. Targeting sequences were summarized in the following: J-003436–06 CAAAUCACCAUUCGUUAUC, J-003436–07 GACAUGAAUUCCUUAAUGA, J-003436–08 GAUAUCAAGCCCUUCACUA, J-003436–09 GACAGCGACUUGGCAAUAU.
Statistical analysis
All reported values are means ± SD (standard deviation). Statistical comparisons were assessed using two-tailed Student’s t tests. Results were considered statistically significant if the P value was <0.05.
Results
PPARγ functions as a growth signal in breast cancer cell lines.
PPARγ has two isoforms: PPARγ1 is widely expressed in tissues of epithelial origin, whereas PPARγ2 is mainly expressed in adipocytes (32). MCF-7 and T47D are two representative ER-positive breast cancer cell lines. We first measured the expression of PPARγ in endocrine-resistant cell lines derived from parental MCF-7 and T47D cells. MCF-7:5C, MCF-7:2A, and MCF-7:ICI-R cell lines were derived from MCF-7 cells (Fig. 1A and B). ERα expression increased in MCF-7:5C and MCF-7:2A cells; MCF-7:ICI-R cells were ERα-negative (Fig. 1B). These cell lines expressed PPARγ at quite different levels from those in parental MCF-7 cells. PPARγ protein and mRNA expression levels in MCF-7:5C cells were extremely low, whereas MCF-7:2A cells had higher levels of PPARγ1 expression than did MCF-7 cells (Fig. 1A, Supplementary Fig. S1A). MCF-7:ICI-R had the highest levels of PPARγ1 expression among cell lines derived from MCF-7 (Fig. 1B). In contrast with that in MCF-7 cells, ERα expression in T47D cells decreased after 3 days of E2 deprivation and it decreased further to undetectable levels in T47D:C42 cells after LTED (Fig. 1C). Of note, PPARγ1 expression increased remarkably in T47D:C42 cells. To further examine PPARγ expression in breast cancer cells, we compared ERα-positive and -negative cell lines. The results demonstrated that PPARγ1 expression levels were higher in the ERα-negative cell lines (MCF-7:ICI-R, T47D:C42, Sk-Br-3, and MDA-MB-231) than in the ERα-positive cell lines (MCF-7, T47D, ZR-75–1, and BT-474) (Fig. 1B-D), indicating an inverse relationship between ERα and PPARγ expression in breast cancer cells.
Figure 1. Expression of PPARγ and its function in different breast cancer cell lines.
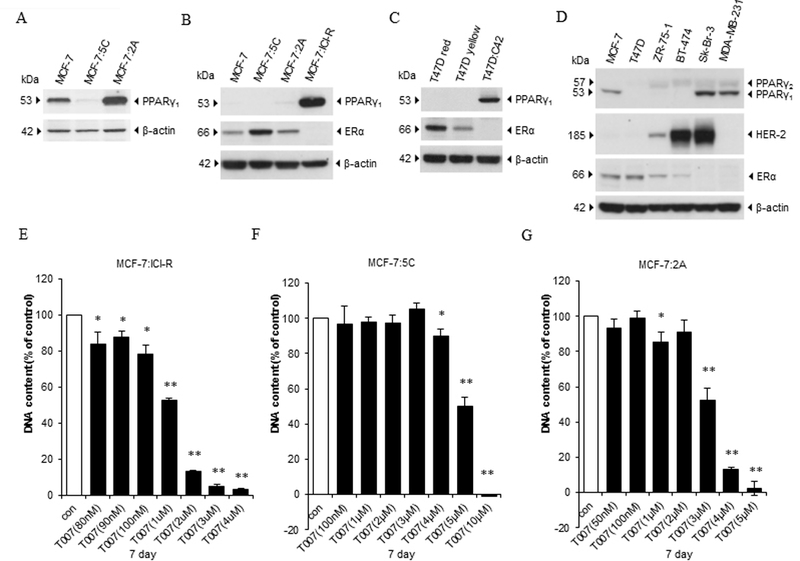
(A) PPARγ protein expression in MCF-7, MCF-7:5C, and MCF-7:2A cells. Cell lysates of three cell lines were harvested for Western blotting. (B) PPARγ and ERα protein expression in MCF-7-derived cell lines. Cell lysates of MCF-7, MCF-7:5C, MCF-7:2A, and MCF-7:ICI-R were harvested for Western blotting. (C) PPARγ and ERα protein expression in T47D-derived cell lines.T47D cells were transferred to E2-free medium for 3 days. Cell lysates of T47D cells cultured in E2-containig medium and E2-free medium were then harvested together with T47D:C42 cells for Western blotting. (D) Expression of PPARγ, HER-2, and ERα in a panel of breast cancer cell lines. Lysates of six cell lines (MCF-7, T47D, ZR-75–1, BT-474, Sk-Br-3, and MDA-MB-231) were harvested for Western blotting. (E) Growth response to T0070907 in MCF-7:ICI-R cells. MCF-7:ICI-R cells were seeded in 24-well plates. Then, cells were treated with a vehicle control (0.1% DMSO) or different concentrations (from 80 nM to 4µM) of T0070907 for 7 days. Cells were harvested for DNA proliferation assay. *P<0.05, **P<0.001. (F) Growth response to T0070907 in MCF-7:5C cells. Cells were treated with a vehicle control (0.1% DMSO) or different concentrations of T0070907 (from 100 nM to 10µM) in 24-well plates for 7 days. Cells were harvested for DNA proliferation assay. *P<0.05, **P<0.001. (G) Growth response to T0070907 in MCF-7:2A cells. Cells were treated with a vehicle control (0.1% DMSO) or different concentrations of T0070907 (from 50 nM to 5µM) in 24-well plates for 7 days. Cells were harvested for DNA proliferation assay. *P<0.05, **P<0.001.
To further examine the function of PPARγ in different breast cancer cell lines, we treated them with a specific PPARγ antagonist, T0070907 at different concentrations for 7 days. Among the MCF-7-derived cell lines, MCF-7:ICI-R cells were the most sensitive to T0070907 with IC50 around 1µM (Fig. 1E-G and Supplementary Fig. S1B). In the two MCF-7-derived LTED breast cancer cell lines, treatment with T0070907 inhibited the growth of MCF-7:2A cells more than that of MCF-7:5C cells (Fig. 1F and 1G and Supplementary Fig. S1C). Also, T0070907 inhibited the growth of E2-deprived cell line T47D:C42 more than that of its parental control T47D cells (Supplementary Fig. S1D). As for the other two ERα-positive cell lines, ZR-75–1 and BT-474, T0070907 markedly inhibited their growth, particularly BT-474 (Supplementary Fig. S1E-F). Two ERα-negative cell lines had distinct responses to the PPARγ antagonist. T0070907 remarkably inhibited Sk-Br-3 cell growth but had no inhibitory effects on the triple-negative MDA-MB-231 cells (Supplementary Fig. S1G-H). These results suggested that PPARγ is an important growth signal in breast cancer cells. We focused below on how PPARγ affected E2-induced apoptosis in the LTED breast cancer cell lines MCF-7:5C and MCF-7:2A.
E2 suppresses the function of PPARγ in the two LTED breast cancer cell lines.
Because T47D:C42 cells are ERα-negative after E2 deprivation and lose response to E2 treatment, we selected MCF-7:5C and MCF-7:2A cells as clinically relevant cell models to investigate how PPARγ modulates E2-induced apoptosis. Both MCF-7 and MCF-7:2A cells mainly expressed PPARγ1, but MCF-7:5C cells had higher expression of PPARγ2 than of PPARγ1 (Fig. 2A-C). After 24 hours of exposure to E2, PPARγ1 expression was quickly downregulated in both MCF-7 and MCF-7:5C cells, whereas it was weakly downregulated in MCF-7:2A cells. With extension of the treatment time to 6 days, E2 started to clearly downregulate PPARγ1 expression in MCF-7:2A cells (Fig. 2D). In line with PPARγ protein expression, E2 continuously downregulated PPARγ mRNA expression in both MCF-7 and MCF-7:5C cells (Fig. 2E and F). Notably, PPARγ mRNA expression was gradually upregulated by E2 in MCF-7:2A cells in the first 3 days of treatment (Fig. 2G). Thus, the two LTED breast cancer cell lines had different responses to E2 regarding of PPARγ expression (Fig. 2H). In addition, PPARγ target gene Acyl-CoA Oxidase 3 (ACOX3), which is involved in degradation of the long branched fatty acids in peroxisomes, was downregulated by E2 at the same rate in three cell lines (Supplementary Fig. S2A-C). These results demonstrated that E2 has the potential to suppress the function of PPARγ in breast cancer cell lines.
Figure 2. Suppression of the function of PPARγ by treatment with E2.
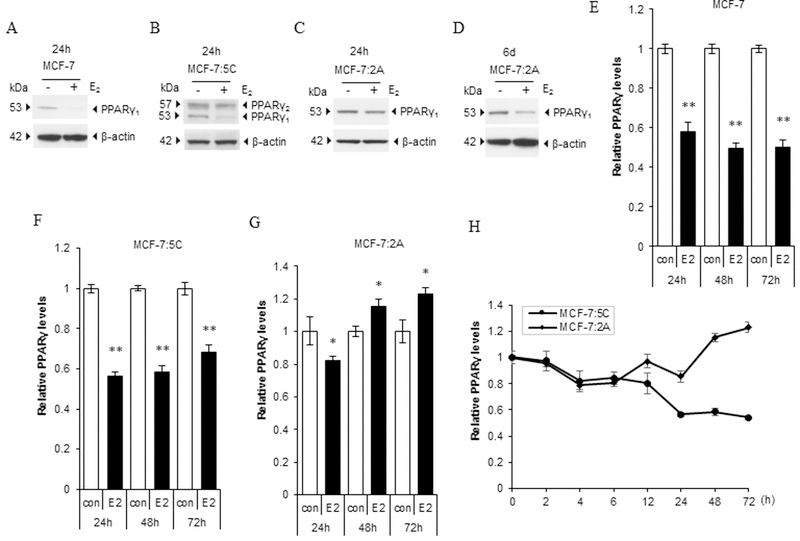
(A-C) PPARγ protein expression after E2 treatment. MCF-7 cells were transferred to E2-free medium for 3 days, and (A) MCF-7, (B) MCF-7:5C, and (C) MCF-7:2A cells were treated with E2 for 24 hours. PPARγ expression was measured using Western blotting. (D) PPARγ protein expression after extension of E2 treatment in MCF-7:2A cells. Cells were treated with E2 for 6 days. PPARγ expression was measured using Western blotting. (E-G) PPARγ mRNA expression after E2 treatment. MCF-7 cells were transferred to E2-free medium for 3 days, and (E) MCF-7, (F) MCF-7:5C, and (G) MCF-7:2A cells were treated with E2 for 24, 48, and 72 hours, respectively. PPARγ expression was quantitated by RT-PCR. *P<0.05, **P<0.001. (H) Time response of PPARγ mRNA expression in MCF-7:5C and MCF-7:2A cells. Two cell lines were treated with E2 for the indicated times. PPARγ expression was quantitated by RT-PCR.
Activation of PPARγ selectively inhibits TNFα inflammatory pathway in the two LTED breast cancer cell lines.
E2 induces expression of a range of inflammatory factors in LTED breast cancer cell lines with different dynamics via ERα (9). TNFα is induced by E2 in MCF-7:5C and MCF-7:2A cells with different peak times (3 days and 9 days, respectively) (10, 11). An opposite response was found in wild-type MCF-7 cells in that E2 decreased TNFα mRNA expression (Supplementary Fig. S3A). To investigate how the PPARγ regulates the inflammatory responses after E2 treatment, we treated MCF-7:5C and MCF-7:2A cells with a specific PPARγ agonist, pioglitazone for different times. As expected, E2 increased TNFα expression in both cell lines (Fig. 3A and F). Pioglitazone reduced the basal levels of TNFα in MCF-7:5C cells but not in MCF-7:2A cells. Combination treatment with E2 and pioglitazone effectively blocked the induction of TNFα by E2 after 3 and 9 days treatment in the two cell lines, respectively. With respect to another TNF family member, LTB, exposure to E2 and pioglitazone had regulatory pattern similar to that for TNFα in MCF-7:5C cells (Supplementary Fig. S3B). However, E2 did not significantly increase LTB expression in MCF-7:2A cells, and pioglitazone did not inhibit LTB expression in MCF-7:2A cells (Supplementary Fig. S3C), indicating different mechanisms of regulating the TNF family members in the two cell lines. As for the adipose inflammatory factors IL-6/IL-6R and FADS1, E2 increased the mRNA expression levels for these factors, but pioglitazone did not affect them. Also, combination treatment did not alter the upregulation of IL-6R/IL-6 or FADS1 expression by E2 in MCF-7:5C and MCF-7:2A cells (Fig. 3B-C, 3G-H and Supplementary Fig. S3D-E). Protein expression of ERα and PPARγ was further measured after E2 or pioglitazone treatment in these two cell lines. E2 reduced both ERα and PPARγ1 protein expression in both cell lines (Fig. 3D and I), but E2 increased PPARγ2 protein expression with shift of band in MCF-7:5C cells after 72 hours of treatment (Fig. 3D). Pioglitazone mildly increased the ERα protein expression in MCF-7:5C but not MCF-7:2A cells. PPARγ1 protein expression increased after pioglitazone treatment, particularly in MCF-7:2A cells (Fig. 3D and I). The combination treatment could not prevent the reduction of ERα or PPARγ1 protein expression by E2. It is known that PPARγ can dimerize with retinoid X receptor (RXR) and bind to estrogen responsive element (ERE) that affects the function of ERα (33). Our results demonstrated that E2 remarkably increased ERE-target gene pS2 expression. Pioglitazone decreased pS2 expression in LTED cell lines. However, pioglitazone increased further pS2 expression after combination treatment with E2 in the two cell lines, particularly in MCF-7:5C cells (Fig. 3E and J). These results suggested that pioglitazone selectively represses TNFα expression induced by E2 not through classic ERE transcriptional pathway.
Figure 3. The PPARγ agonist selectively suppressed induction of TNFα by E2 in LTED breast cancer cell lines.
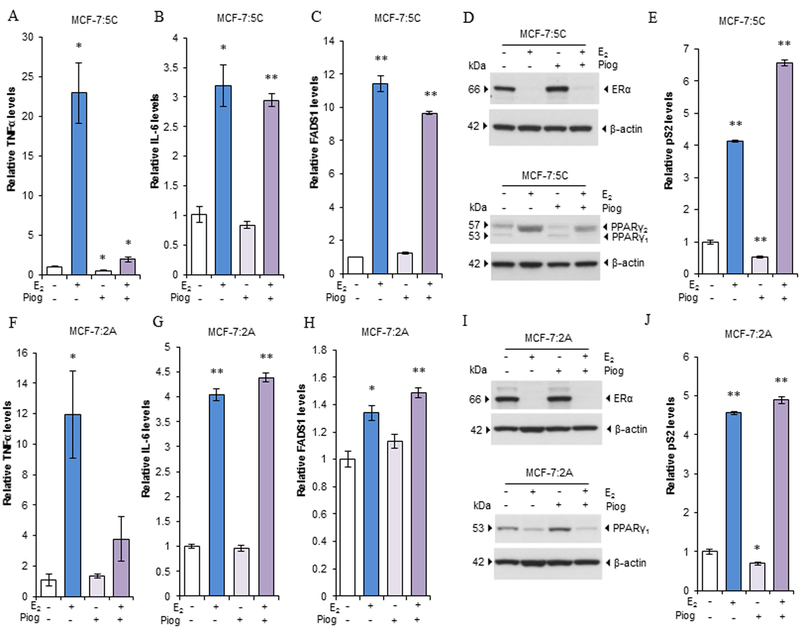
(A-C) Effects on inflammatory factors of pioglitazone in MCF-7:5C cells. Cells were treated with E2 (1 nM), pioglitazone (10 µM), or a combination of them for 72 hours. (A)TNFα, (B) IL-6, and (C) FADS1 expression was quantitated by RT-PCR. * P<0.05, **P<0.001. (D) PPARγ and ERα expression after pioglitazone treatment in MCF-7:5C cells. Cells were treated as described in A-C. Cell lysates were harvested for Western blotting. (E) Expression of pS2 after pioglitazone treatment in MCF-7:5C cells. Cells were treated as described in A-C. pS2 expression was quantitated by RT-PCR. **P<0.001. (F-H) Effects on inflammatory factors of pioglitazone in MCF-7:2A cells. Cells were treated with E2 (1 nM), pioglitazone (10 µM), or a combination of them for 9 days. (F) TNFα, (G) IL-6, and (H) FADS1 expression was quantitated by RT-PCR. *P<0.05, **P<0.001. (I) PPARγ and ERα expression after pioglitazone treatment in MCF-7:2A cells. Cells were treated as described in F-H. Cell lysates were then harvested for Western blotting. (J) Expression of pS2 after pioglitazone treatment in MCF-7:2A cells. Cells were treated as described in F-H for 72 hours. pS2 expression was quantitated by RT-PCR. *P<0.05, **P<0.001.
Activation of PPARγ suppresses NF-κB DNA binding but has different effects on E2-induced apoptosis in the two LTED breast cancer cell lines.
E2 activates NF-κB via increasing DNA binding in LTED breast cancer cells (16). To determine whether pioglitazone selectively blocks TNFα expression via suppression of NF-κB, we used an NF-κB (p65) transcription factor assay kit to assess the NF-κB DNA-binding activity in MCF-7:5C and MCF-7:2A cells. The result demonstrated that MCF-7:5C cells had higher basal NF-κB DNA-binding activity than did MCF-7:2A cells (Supplementary Fig. S3F). After different time treatment, E2 clearly increased the NF-κB DNA-binding activity in MCF-7:5C cells after 72 hours of treatment. Furthermore, pioglitazone effectively blocked nuclear activation of NF-κB in MCF-7:5C cells (Fig. 4A). Apoptosis was detected by annexin V binding assay through flow cytometry. E2 increased annexin V binding in MCF-7:5C cells after 72 hours of treatment. Pioglitazone did not change the annexin V binding compared with control. However, the combination pioglitazone and E2 completely blocked the E2-induced apoptosis in MCF-7:5C cells (Fig. 4B and Supplementary Fig. S4A). Consistent with the annexin V binding results, pioglitazone effectively blocked the cleavage of PARP and caspase 7 activated by E2 in MCF-7:5C cells (Fig. 4C). With respect to MCF-7:2A cells, E2 began to moderately increase NF-κB DNA-binding activity after 6 days of treatment. Also, pioglitazone effectively inhibited the nuclear activation of NF-κB in these cells (Fig. 4D). As for regulation of apoptosis, E2 started to significantly induce apoptosis in MCF-7:2A cells after 6 days of treatment. Of note, pioglitazone also elevated the percentage of annexin V binding in these cells. Combination treatment with E2 and pioglitazone further increased the rate of apoptosis after 6 days treatment (Fig. 4E and Supplementary Fig. S4B). It is known that E2-induced cell death is delayed to 2 weeks in MCF-7:2A cells (11). When the treatment time was prolonged to 9 days, E2 increased the apoptosis rate over that at 6 days of treatment. Pioglitazone increased annexin V binding after 9 days of treatment similarly to 6 days of treatment. Apoptosis was increased after 9 days of combination treatment compared with E2 alone treated group in MCF-7:2A cells (Supplementary Fig. S5A). In line with these results, pioglitazone increased cleaved PARP and caspase-7 caused by E2 in MCF-7:2A cells (Fig. 4F). These results suggested different mechanisms of E2-induced apoptosis in the two LTED cell lines.
Figure 4. Different effects of the PPARγ agonist on E2-induced apoptosis in LTED breast cancer cell lines.
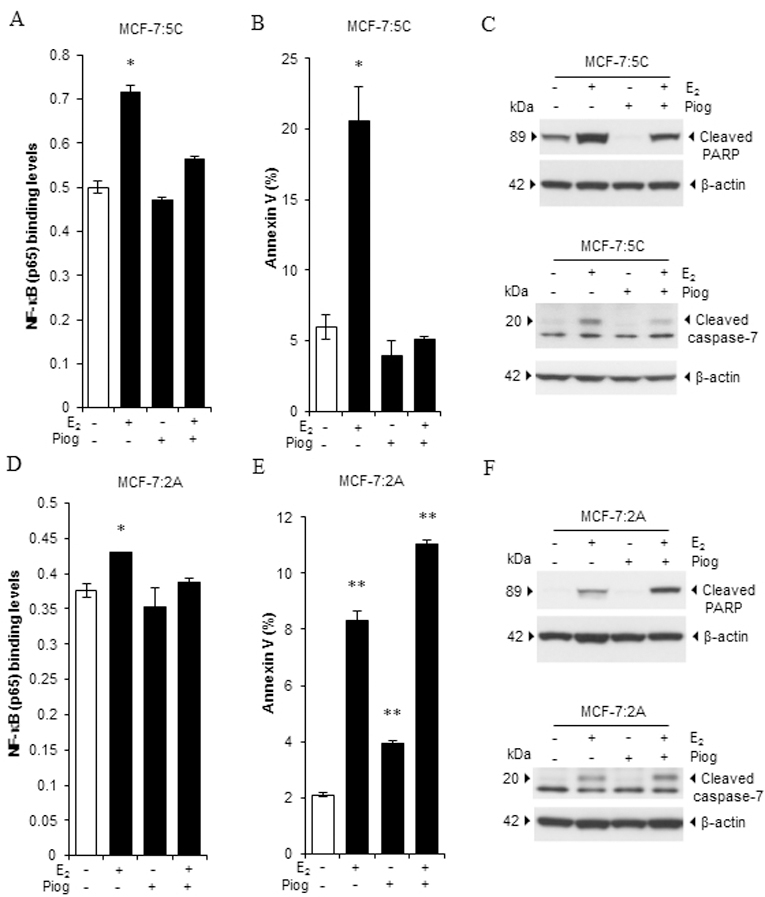
(A) NF-κB DNA-binding activity in MCF-7:5C cells. Cells were treated with E2 (1 nM), pioglitazone (10 µM), or a combination of them for 72 hours. Cells were then harvested for extraction of nuclear protein. The NF-κB DNA-binding activity was measured using an NF-κB (p65) transcription factor assay kit. *P<0.05. (B) Effects of pioglitazone on E2-induced apoptosis in MCF-7:5C cells. Cells were treated as described in A. Next, cells were harvested for annexin V binding assay via flow cytometry. *P<0.05. (C) Expression of apoptotic markers in MCF-7:5C cells. Cells were treated as described in A. Expression of cleaved PARP and caspase 7 was detected using Western blotting. (D) NF-κB DNA-binding activity in MCF-7:2A cells. Cells were treated with E2 (1 nM), pioglitazone (10 µM), or a combination of them for 6 days. Then, cells were harvested for extraction of nuclear protein. The NF-κB DNA-binding activity was measured using an NF-κB (p65) transcription factor assay kit. *P<0.05. (E) Effects of pioglitazone on E2-induced apoptosis in MCF-7:2A cells. Cells were treated as described in D. Then, cells were harvested for annexin V binding assay via flow cytometry. **P<0.001. (F) Expression of apoptotic markers in MCF-7:2A cells. Cells were treated with E2 (1 nM), pioglitazone (10 µM), or a combination of them for 9 days. Expression of cleaved PARP and caspase 7 was detected using Western blotting.
A distinct function of NF-κB modulates oxidative stress in the two LTED breast cancer cell lines.
Oxidative stress pathway is activated by E2 to promote apoptosis (10, 11). To investigate whether PPARγ modulates oxidative stress in breast cancer cell lines, we treated them with E2, pioglitazone, or a combination of the two for different times. Our results demonstrated that E2 decreased expression of the oxidative stress indicator HMOX1, whereas pioglitazone increased it. E2 completely blocked upregulation of HMOX1 by pioglitazone after combination treatment in wild-type MCF-7 (Fig. 5A). MCF-7:5C cells had an opposite response in that E2 increased HMOX1 expression. Pioglitazone did not change the expression of HMOX1, but it effectively blocked oxidative stress in these cells (Fig. 5B). Compared with MCF-7:5C cells, MCF-7:2A cells have a stronger antioxidant system (11). E2 markedly increased HMOX1 expression after 6 days of treatment. Pioglitazone also increased HMOX1 expression in MCF-7:2A cells. Nevertheless, combination treatment with E2 and pioglitazone increased more HMOX1 expression than single compound did in MCF-7:2A cells (Fig. 5C). When we prolonged the treatment to 9 days in MCF-7:2A cells, HMOX1 expression levels were similar to those at 6 days (Supplementary Fig. S5B). Even with different effects on oxidative stress, pioglitazone almost did not inhibit cell growth, nor dramatically affected E2 responsive cell growth in three cell lines (Supplementary Fig. S5C-E). In addition to that in the mitochondria, lipid metabolism in the endoplasmic reticulum affects redox homeostasis (34). The sensor IRE1α is associated with lipid metabolism in LTED breast cancer cells (12). Our results demonstrated that E2 upregulated expression of IRE1α in MCF-7:5C and MCF-7:2A cells. Pioglitazone almost had no effect on IRE1α expression, it did not inhibit upregulation of IRE1α expression after combination with E2 (Supplementary Fig. S5F-G), indicating that pioglitazone does not directly regulate lipid metabolism in the endoplasmic reticulum to alter redox homeostasis. Further experiments demonstrated that NF-κB was a regulatory target for PPARγ to determine the final consequence of oxidative stress in the two LTED breast cancer cells. Our recent publication demonstrated that a specific NF-κB inhibitor JSH-23 effectively blocks NF-κB DNA binding activity (16, 35) and it completely inhibited the oxidative stress induced by E2 in MCF-7:5C cells (Fig. 5D). In contrast, inhibition of NF-κB markedly increased oxidative stress and was additive with E2 in upregulating HMOX1 expression in MCF-7:2A cells (Fig. 5E). Thus, JSH-23 completely blocked E2-induced apoptosis in MCF-7:5C cells (16) but increased E2-induced apoptosis in MCF-7:2A cells (Fig. 5F). For the first time, we identified that NF-κB has distinct roles in the regulation of oxidative stress in two LTED breast cancer cell lines, serving as an oxidative stress inducer in MCF-7:5C and an antioxidant in MCF-7:2A cells (Fig. 5G). This differential modulation of oxidative stress resulted in pioglitazone completely blocking E2-induced apoptosis in MCF-7:5C but not MCF-7:2A cells.
Figure 5. A distinct function of NF-κB modulated oxidative stress in LTED breast cancer cell lines.
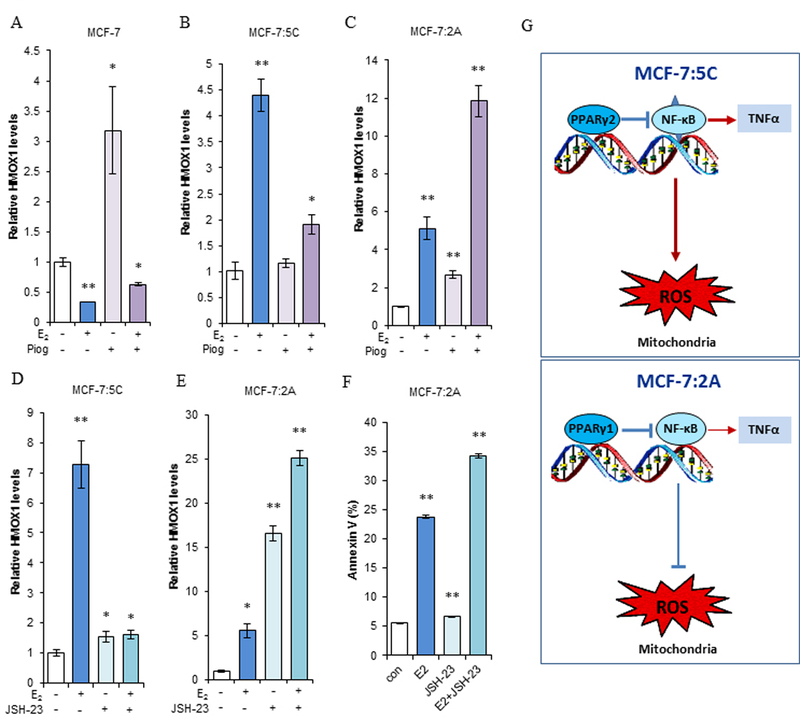
(A-B) Expression of oxidative stress indicator HMOX1. MCF-7 cells were transferred to E2-free medium for 3 days. Then, (A) MCF-7 and (B) MCF-7:5C cells were treated with E2 (1 nM), pioglitazone (10 µM), or a combination of them for 72 hours. Expression of HMOX1 was quantitated by RT-PCR. *P<0.05, **P<0.001. (C) Expression of HMOX1 in MCF-7:2A cells. Cells were treated with the same compounds as in A and B for 6 days. HMOX1 expression was quantitated by RT-PCR. **P<0.001. (D-E) Regulation of oxidative stress by NF-κB. MCF-7:5C and MCF-7:2A cells were treated with E2 (1 nM), JSH-23 (20 µM), or a combination of them for 3 and 6 days, respectively. HMOX1 expression levels were quantitated by RT-PCR. *P<0.05, **P<0.001. (F) E2-induced apoptosis regulated by NF-κB in MCF-7:2A cells. Cells were treated with E2 (1 nM), JSH-23 (20 µM), or a combination of them for 6 days. Then, cells were harvested for an annexin V binding assay via flow cytometry. **P<0.001. (G) Differential regulation of oxidative stress by NF-κB in two LTED breast cancer cells. MCF-7:5C cells have constitutive activation of NF-κB (with triangles) compared with MCF-7:2A cells, which results in high levels of TNFα induction (thick arrow). MCF-7:5C cells express extremely low levels of PPARγ and mainly is PPARγ2 isoform. MCF-7:2A cells mainly express PPARγ1 isoform. Activation of PPARγ suppresses the NF-κB/TNFα axis in two LTED breast cancer cells. However, NF-κB functions as an oxidative stress inducer in MCF-7:5C cells but an antioxidant in MCF-7:2A cells.
Knockdown of PPARγ upregulates apoptosis-related pathways in the two LTED breast cancer cell lines.
As described above, PPARγ has the potential to modulate the function of NF-κB associated inflammation and oxidative stress. MCF-7:5C and MCF-7:2A cells were transfected with specific PPARγ siRNA, which effectively downregulated PPARγ mRNA (Supplementary Fig. S6A-B) and protein expression (Fig. 6A and F). Knockdown of PPARγ resulted in increasing apoptotic marker cleavage of PARP in all cells, whereas decreasing ERα in MCF-7:5C and MCF-7:2A (Fig. 6A and F), but moderately increasing ERα in T47D:C42 cells (Supplementary Fig. S6C). Then, transfected cells were treated with E2 for 48 hours in MCF-7:5C and 72 hours in MCF-7:2A. Compared with scrambled siRNA transfected cells, depletion of PPARγ increased the basal levels of NF-κB in both cell lines (Fig. 6B and G). In addition, E2 treatment increased NF-κB expression in cells with knockdown of PPARγ but not in the scrambled siRNA-transfected cells (Fig. 6B and G). Furthermore, knockdown of PPARγ was synergistic with E2 in remarkably upregulating expression of NF-κB-target gene TNFα, even though E2 weakly increased TNFα expression in MCF-7:5C cells but without any induction in MCF-7:2A cells at this time points (Fig. 6C and H). This demonstrated that PPARγ is a potent repressive factor for E2 to induce TNFα expression. Unexpectedly, knockdown of PPARγ significantly increased expression of oxidative stress indicator HMOX1 (Fig. 6D and I), supporting our conclusion that PPARγ also regulates redox homeostasis and functions as an antioxidant in MCF-7:5C and MCF-7:2A cells. E2 treatment moderately increased HMOX1 expression in PPARγ depleted cells (Fig. 6D and I). Consistent with cleaved PARP, PPARγ siRNA increased the percentage of annexin V and PI staining. Combination with E2 mainly increased PI staining, particularly for MCF-7:5C cells (Supplementary Fig. S6D and E). Importantly, PPARγ siRNA combination with E2 further inhibited more cell growth in two LTED cells after 5 and 7 days treatment, respectively (Fig. 6E and J). These findings indicated that depletion of PPARγ can functionally modulate the extrinsic and intrinsic apoptosis pathways to increase E2-induced apoptosis.
Figure 6. Depletion of PPARγ upregulated apoptosis-related pathways in LTED breast cancer cell lines.
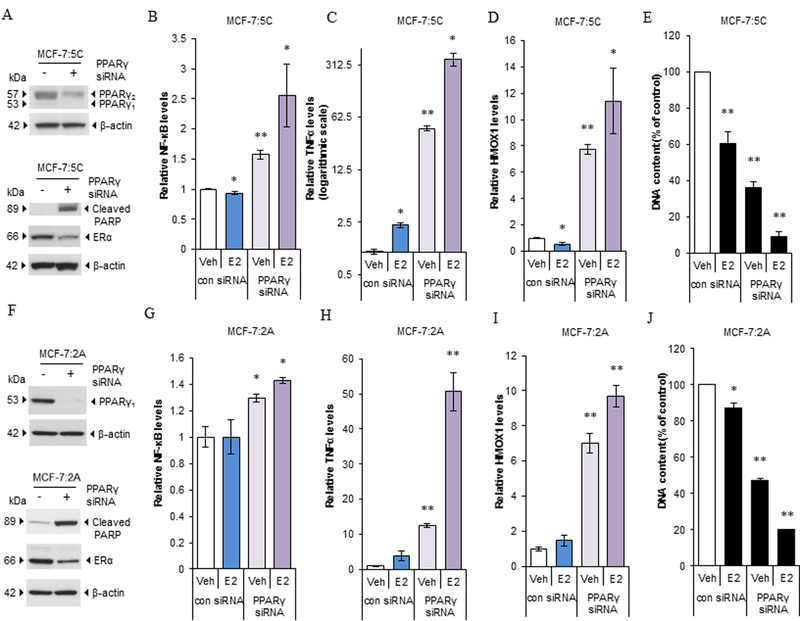
(A) Expression of PPARγ, cleaved PARP, and ERα in MCF-7:5C cells. Cells were transfected with scrambled siRNA or specific PPARγ siRNA for 72 hours. Expression of PPARγ, cleaved PARP, and ERα were measured using Western blotting. (B-D) Alteration of apoptosis-associated pathways. MCF-7:5C cells were transfected with scrambled siRNA or specific PPARγ siRNA for 72 hours. Next, cells were treated with a vehicle control (0.1% EtOH) or E2 (1nM) for 48 hours. Cells were harvested in TRIzol. (B) NF-κB, (C) TNFα, and (D) HMOX1 expression was quantitated by RT-PCR. *P<0.05, **P<0.001 compared with the scrambled siRNA transfected vehicle control. (E) PPARγ siRNA synergized with E2 to inhibit cell growth in MCF-7:5C cells. MCF-7:5C cells were transfected with scrambled siRNA or specific PPARγ siRNA for 3 days. Next, cells were treated with a vehicle control (0.1% EtOH) or E2 (1nM) for 5 days. Cells were harvested for DNA growth assay. **P<0.001 compared with the scrambled siRNA transfected vehicle control. (F) Expression of PPARγ, cleaved PARP, and ERα in MCF-7:2A cells. Cells were double transfected with scrambled siRNA or specific PPARγ siRNA for 5 days. Expression levels of PPARγ, cleaved PARP, and ERα were measured using Western blotting. (G-I) Alteration of apoptosis-associated pathways. MCF-7:2A cells were double transfected with scrambled siRNA or specific PPARγ siRNA for 5 days. Next, cells were treated with a vehicle control (0.1% EtOH) or E2 (1nM) for 72 hours. Cells were harvested in TRIzol. (G) NF-κB, (H) TNFα, and (I) HMOX1 expression levels were quantitated by RT-PCR. *P<0.05, **P<0.001 compared with the scrambled siRNA transfected vehicle control. (J) PPARγ siRNA synergized with E2 to inhibit cell growth in MCF-7:2A cells. MCF-7:2A cells were double transfected with scrambled siRNA or specific PPARγ siRNA for 5 days. Next, cells were treated with a vehicle control (0.1% EtOH) or E2 (1nM) for 7 days. Cells were harvested for DNA growth assay. *P<0.05, **P<0.001 compared with the scrambled siRNA transfected vehicle control.
A PPARγ antagonist promotes E2-induced cell death in the two LTED breast cancer cell lines.
We further treated MCF-7:5C and MCF-7:2A cells with E2, the specific PPARγ antagonist T0070907, or a combination of them for 7 days. E2 significantly decreased the number of MCF-7:5C cells but not MCF-7:2A cells within 1 week (Fig. 7A and G). After 7 days of E2 treatment, many of the MCF-7:5C cells were floating in the culture medium, whereas MCF-7:2A cells remained attached to the bottom of 24-well plate. Different doses of T0070907 remarkably inhibited the growth of both cell lines; the combination treatment clearly inhibited cell growth to a greater extent (Fig. 7A and G). Another PPARγ antagonist GW9662 had the similar effects on increasing E2-induced growth inhibition in two LTED breast cancer cells (30) (Supplementary Fig. S7A-B). The annexin V binding assay demonstrated that T0070907 did not increase apoptosis in the two cell lines (Fig. 7B and H). E2 clearly increased apoptosis after 3 and 6 days of treatment in MCF-7:5C and MCF-7:2A cells, respectively. The combination treatment increased apoptosis in MCF-7:2A cells but not in MCF-7:5C cells (Fig. 7B and H). Further examination of apoptosis signaling pathways showed that T0070907 alone increased expression of TNFα and HMOX1 in MCF-7:2A cells but only increased HMOX1 expression in MCF-7:5C cells. Also, T0070907 was additive with E2 in increasing expression of TNFα and HMOX1 in MCF-7:2A cells but partially blocked TNFα induction in MCF-7:5C cells (Fig. 7C, D, I, and J). With extension of the treatment time, T0070907 (5µM) could not block TNFα expression induced by E2 and remarkably decreased phosphorylation of Akt after 3 days of treatment in MCF-7:5C cells (Supplementary Fig. S7C-D), an important growth pathway in LTED cells (10, 12). Further examination of cleaved PAPR, E2 increased cleaved PARP after 3 and 6 days of treatment in MCF-7:5C and MCF-7:2A cells, respectively. T0070907 remarkably increased cleaved PARP in MCF-7:2A, but not in MCF-7:5C cells. E2 synergized with T0070907 to increase cleaved PARP in MCF-7:2A, but not in MCF-7:5C cells (Fig. 7E and K). T0070907 did not change the expression of PPARγ2 in MCF-7:5C cells (Fig. 7F). Of note, T0070907 had different effects on PPARγ1 expression in the two cell lines. Specifically, it increased PPARγ1 protein expression in MCF-7:5C cells at a dose of IC50 (5µM), whereas a high concentration (10 µM) had almost no effect on PPARγ1 expression (Fig. 7F). By contrast, it clearly decreased PPARγ1 protein expression at a low concentration (2.5µM) in MCF-7:2A cells and was in a dose-dependent manner (Fig. 7L). T0070907 also decreased ERα protein levels in MCF-7:2A cells but it was not altered in MCF-7:5C cells (Fig.7F, and L). As for the regulation of pS2, T0070907 upregulated pS2 expression in the two LTED breast cancer cells (Supplementary Fig. S7E and F). It did not affect the upregulation of pS2 by E2 in MCF-7:5C cells but weakly inhibited pS2 expression induced by E2 in MCF-7:2A cells (Supplementary Fig. S7E- F). These results suggested that different isoforms of PPARγ may affect the therapeutic effect of the PPARγ antagonist. Unlike PPARγ siRNA, T0070907 had differential effects on PPARγ1 protein levels in the two LTED cell lines that leads to distinct response to TNFα induction after combination with E2 treatment. We concluded that downregulation of PPARγ is an effective way to increase E2-induced apoptosis.
Figure 7. The PPARγ antagonist promoted E2-induced cell death in LTED breast cancer cell lines.
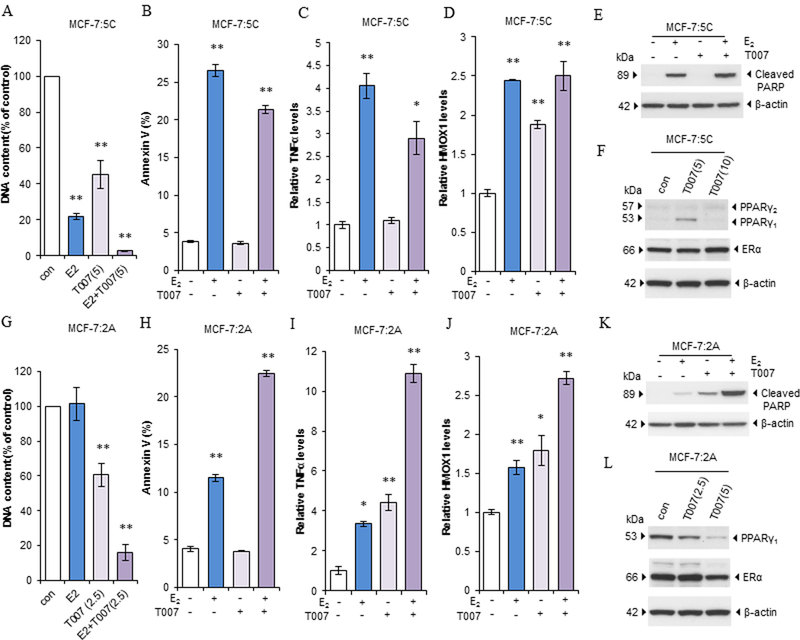
(A) DNA Growth assay in MCF-7:5C cells. Cells were treated with E2 (1 nM), T0070907 (5 µM), or a combination of them for 7 days. Cells were harvested for DNA proliferation assay. **P<0.001. (B) Apoptosis after T0070907 treatment in MCF-7:5C cells. Cells were treated with E2 (1 nM), T0070907 (5 µM), or a combination of them for 72 hours. Cells were then harvested for annexin V binding assay. **P<0.001. (C-D) Alteration of apoptosis-associated pathways in MCF-7:5C cells. Cells were treated as described in B and harvested in TRIzol. (C) TNFα and (D) HMOX1 expression levels were quantitated by RT-PCR. *P<0.05, **P<0.001. (E) T0070907 regulated cleaved PARP in MCF-7:5C cells. Cells were treated with E2 (1 nM), T0070907 (5 µM), or a combination of them for 3 days. Cleaved-PARP was detected using Western blotting. (F) Expression of PPARγ and ERα after T0070907 treatment in MCF-7:5C cells. Cells were treated with a vehicle control (0.1% DMSO) or T0070907 (5, 10 µM) for 72 hours. Expression of PPARγ and ERα was detected using Western blotting. (G) DNA Growth assay in MCF-7:2A cells. Cells were treated with E2 (1 nM), T0070907 (2.5 µM), or a combination of them for 7 days. Cells were harvested for DNA proliferation assay. **P<0.001. (H) Apoptosis after T0070907 treatment in MCF-7:2A cells. Cells were treated with E2 (1 nM), T0070907 (2.5 µM), or a combination of them for 6 days. Cells were harvested for annexin V binding assay. **P<0.001. (I-J) Alteration of apoptosis-associated pathways in MCF-7:2A cells. Cells were treated as described in H and harvested in TRIzol. (I) TNFα and (J) HMOX1 expression levels were quantitated by RT-PCR. *P<0.05, **P<0.001. (K) T0070907 regulated cleaved PARP in MCF-7:2A cells. Cells were treated with E2 (1 nM), T0070907 (2.5 µM), or a combination of them for 3 days. Cleaved-PARP was detected using Western blotting. (L) Expression of PPARγ and ERα after T0070907 treatment in MCF-7:2A cells. Cells were treated with a vehicle control (0.1% DMSO) or T0070907 (2.5, 5 µM) for 72 hours. Expression of PPARγ and ERα was detected using Western blotting.
Discussion
The scientific investigation of E2-induced apoptosis has clinical relevance to treat aromatase inhibitor-resistant breast cancer (7) and decrease the breast cancer incidence in postmenopausal women when conjugated estrogen is given alone as hormone replacement therapy (8). However, the clinical application is limited because of a 30% benefit rate in aromatase inhibitor-resistant breast cancer patients (7). Thus, there is a need to find new targets to improve E2-induced apoptosis. Herein, PPARγ is the first identified transcription factor that suppresses NF-κB and oxidative stress in LTED breast cancer cell lines to counteract E2-induced apoptosis. Inhibition of PPARγ through either depletion of PPARγ or the specific antagonist significantly accelerates E2-induced apoptosis. Thus, PPARγ is a potential target molecule for increasing E2-induced apoptosis in endocrine-resistant breast cancer.
It is known that NF-κB/TNFα and oxidative stress are two key pathways activated by E2 to induce apoptosis in LTED breast cancer cells (10, 11, 16). E2 initially has the potential to suppress the activation of NF-κB whereas the NF-κB DNA-binding activity is increased by E2 via PERK kinase, a sensor of the endoplasmic reticulum stress (16). Nuclear trans-suppression of NF-κB is a fundamental mechanism for the PPARγ agonist to selectively inhibit inflammatory factor TNFα induced by E2 in the two LTED breast cancer cells, as well as in other diseases (23–28). In addition to strictly regulating TNFα, NF-κB is an oxidative stress-responsive transcription factor (19). A novel finding in the present study is that NF-κB displays distinct functions in modulating oxidative stress in the two LTED breast cancer cell lines. Specifically, activated NF-κB causes oxidative stress in MCF-7:5C cells. Thus, inhibition of NF-κB can effectively block extrinsic and intrinsic apoptosis pathways in these cells, thereby completely blocking E2-induced apoptosis. By contrast, the function of NF-κB is more complex in MCF-7:2A cells. NF-κB acts as an antioxidant to protect cells from oxidative stress, which results in increasing oxidative stress after inhibition of NF-kB in MCF-7:2A cells. The ultimate effect of an NF-κB inhibitor on MCF-7:2A cells is accelerating E2-induced apoptosis, even though the NF-κB inhibitor effectively blocks induction of TNFα (16). This result clearly demonstrates that mitochondrial dysfunction dominates E2-induced apoptosis, rather than extrinsic apoptosis pathways in MCF-7:2A cells. How NF-κB differentially modulates oxidative stress in MCF-7:5C and MCF-7:2A cells remains unclear. It is very likely that NF-κB differentially coordinates with other oxidative stress-responsive molecules, such as Nrf2, to modulate redox homeostasis depending on the cellular context (19, 36, 37).
How PPARγ modulates oxidative stress is another crucial mechanism of counteracting E2-induced apoptosis. Depletion of PPARγ or the PPARγ antagonist markedly increases oxidative stress, which indicates that PPARγ functions as a strong antioxidant to defend against oxidative stress in LTED breast cancer cells, particularly MCF-7:2A. A paradoxical result is how both the agonist and antagonist of PPARγ increase oxidative stress in MCF-7:2A cells (Fig. 5C and7J). An unanticipated mechanism in MCF-7:2A cells is that NF-κB acts as an antioxidant, which results in increased oxidative stress after trans-suppression of NF-κB by pioglitazone. Additionally, pioglitazone can activate PPARγ co-activator 1 (PGC-1) to modulate a broad spectrum of genes related to β-oxidation and mitochondrial biogenesis (38, 39). This is a recognized mechanism for PPARγ to transcriptionally modulate the homeostasis of mitochondria (38–41). Estrogen-related receptor (ERR) is closely linked with PGC-1 in modulation of mitochondrial function (39, 40). Thus, pioglitazone increases oxidative stress in cells with relatively high levels of PPARγ, such as MCF-7:2A and wild-type MCF-7 cells (Fig. 5A and C). However, the ultimate cell fate is determined by the function of E2/ERα after co-treatment with the pioglitazone and E2. In wild-type MCF-7 cells, E2 suppresses oxidative stress and completely blocks oxidative stress induced by pioglitazone. By contrast, E2 alone damages mitochondrial function via accumulation of stress in MCF-7:2A cells (11). Under this condition, activation of β-oxidation by pioglitazone further increases the mitochondrial burden in MCF-7:2A cells. Here, it needs to make a note that regulation of oxidative stress is more complex than that of NF-κB/TNFα axis in LTED breast cancer cells (11, 16). In addition to the transcription factors such as Nrf2, NF-κB, and PPARγ (36, 37), there are many metabolic enzymes involved in the maintenance of redox homeostasis (11). Furthermore, the crosstalk between endoplasmic reticulum and mitochondria leads to the dysfunction of mitochondria (16, 33).
We also focused on how ERα cross-talks with PPARγ to ultimately determine the process of E2-induced apoptosis in LTED breast cancer cells. Different from in wild-type breast cancer cell, ERα is over-activated by E2 that leads to the accumulation of apoptosis-associated stress in LTED breast cancer cells (9–12). Although PPARγ can dimerize with RXR and binds to ERE that suppresses the function of ERα (33), our results demonstrated that pioglitazone selectively suppresses of TNFα induced by E2 but further increases ERE-regulated gene pS2 expression, suggesting that transcriptional ERE pathway is not used by PPARγ to regulate TNFα expression. It is consistent with our previous finding that the c-Src inhibitor blocks TNFα-induction and E2-induced apoptosis, but increases ERE transcriptional activity (10, 42). These findings also suggest that E2 separately activates ERE activity and NF-κB in LTED breast cancer cell lines. Importantly, more evidence has indicated that the transcriptional function of PPARγ is integrally regulated through dynamic chromatin remodeling with the alteration of PPARγ expression and the function of target genes (43, 44). Notably, PPARγ has short half-life and its expression is regulated by ubiquitin-proteasome system (44). In support with this view, we observed the shift of PPARγ2 after 72 hours of treatment with E2 in MCF-7:5C cells. This is also a time point occurring endoplasmic reticulum stress-associated degradation (ERAD) of phospholipids induced by E2 (12). All of these results suggested that endoplasmic reticulum and ubiquitin-proteasome system are activated to remove misfolded or short-lived proteins after E2 treatment in LTED breast cancer cells.
Additionally, repression of PPARγ and its target gene by E2 is a direct evidence to support the conclusion that ERα is a crucial transcription factor that modulates lipid metabolism (45, 46). Thus, anti-hormone therapy is a lipid metabolism reprogramming process for breast cancer patients, as well as for postmenopausal women. In particular, PPARγ expression levels are altered after E2 deprivation in MCF-7 and T47D cells. Due to loss of ERα in T47D:C42 cells after E2 deprivation, they do not respond to E2 treatment. However, high expression of PPARγ1 makes T47D:C42 cells more sensitive to the PPARγ antagonist T0070907 than parental cells T47D. Depletion of PPARγ also increased apoptosis with high levels of cleaved PARP in T47D:C42 cells (Supplementary Fig. S6C). Despite of the fact that MCF-7:5C and MCF-7:2A cells are derived from the same parental MCF-7 cells, LTED differently regulates MCF-7:2A cells with higher levels of PPARγ than MCF-7:5C, which results in a stronger antioxidant system in MCF-7:2A cells. The suppressive relationship between PPARγ and NF-κB is also demonstrated in the MCF-7:5C cells which express extremely low PPARγ but have very active NF-κB (16). It remains unclear why MCF-7:5C cells express extremely low PPARγ. Selective promoter use may affect the expression levels of PPARγ (47). Additionally, PPARγ expression is regulated by ubiquitin-proteasome system (44) which clues that ubiquitin-proteasome system might be more active in MCF-7:5C cells. PPARγ1 is widely expressed in tissues of epithelial origin, whereas PPARγ2 is mainly expressed in adipocytes (32). Both E2 and T0070907 mainly reduces PPARγ1 but not PPARγ2 expression. Unlike PPARγ siRNA to effective depletion of PPARγ protein in all breast cancer cells, T0070907 increases the PPARγ1 protein expression in MCF-7:5C cells (Fig. 7F), which leads to the partially blocking the TNFα induction by E2 (Fig. 7C). However, the PPARγ antagonist has anti-proliferative effects (30) to increase E2-induced cell death in MCF-7:5C cells (Fig. 7A and G). Moreover, higher PPARγ2 expression in MCF-7:5C than MCF-7:2A cells renders these cells like adipocytes, and PPARγ2 is potentiated by lipids and lipid-like compounds, such as unsaturated fatty acids (48, 49). Of note is that genes related with fatty acid metabolism are significantly activated by E2 in LTED cells (9). This special function of PPARγ2 may be a mechanism of MCF-7:5C cells to be susceptible to inflammation after exposure to E2.
Collectively, we have demonstrated the enhancement of E2-induced cell death in LTED breast cancer cell lines via anti-PPARγ therapy. This has significance for clinical translation as a component of preemptive salvage therapy (50) to reduce micrometastasis tumor burden in high-risk cancer patients following five years of adjuvant anti-hormone therapy (51). Clinical studies (7) demonstrate the efficacy of low-dose E2 therapy to treat metastatic breast cancer. However, concerns about E2 use in breast cancer patients remain. Importantly, medicinal chemists have already created a new group of medicines, Selective human Estrogen Receptor Partial Agonists (ShERPAs) for clinical evaluation (52). Far more than regulation of apoptosis, PPARγ displays many faces in the process of breast cancer progression (19, 53, 54). We are undertaking investigation on how a PPARγ antagonist significantly inhibits aggressive fulvestrant-resistant breast cancer cells. These ongoing studies will provide an important rationale for using PPARγ antagonists to treat endocrine-resistant breast cancer.
Supplementary Material
Acknowledgements
V.C.J thanks George and Barbara Bush Foundation for Innovative Cancer Research and the benefactors of the Dallas/Ft. Worth Living Legend Chair of Cancer Research for their generous support. The authors thank Donald R. Norwood in the Department of Scientific Publications at MD Anderson Cancer center for editing the manuscript. They also thank the MD Anderson’s Characterized Cell Line Core for validating cell lines, which is funded by NCI #CA16672.
Financial Support:
This work was supported by the NIH/NCI under award number P30- CA016672 (P.W. Pisters), Susan G. Komen for the Cure Foundation under award number SAC100009 (V.C. Jordan), and Cancer Prevention Research Institute of Texas (CPRIT) for the STARs and STARs plus Awards (V.C. Jordan).
Abbreviations list:
- (E2)
Estrogen
- (ER)
Estrogen receptor
- (LTED)
Long-term estrogen deprivation
- (PPARγ)
Peroxisome proliferator-activated receptor γ
- (NF-κB)
Nuclear factor-κB
- (TNFα)
Tumor necrosis factor alpha
- (FADS1)
Fatty acid desaturase 1
- (IL-6)
Interleukin-6
- (ROS)
Reactive oxygen species
- (HMOX1)
Hemeoxygenase-1
- (PERK)
Protein kinase RNA-like endoplasmic reticulum kinase
- (IRE1α)
Inositol-requiring protein 1 alpha
- (CEBPβ) CCAAT/
enhancer binding protein β
- (HRT)
Hormone replacement therapy
- (CEE)
Conjugated equine estrogen
Footnotes
Competing Interest: No potential conflicts of interest were disclosed by the authors.
References:
- 1.Jordan VC, Brodie AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids 2007;72:7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yao K, Lee ES, Bentrem DJ, England G, Schafer JI, O’Regan RM, et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res 2000;6:2028–36. [PubMed] [Google Scholar]
- 3.Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst 2003;95:1586–97. [DOI] [PubMed] [Google Scholar]
- 4.Song RX, Mor G, Naftolin F, McPherson RA, Song J, Zhang Z, et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17 beta-estradiol. J Natl Cancer Inst 2001;93:1714–23. [DOI] [PubMed] [Google Scholar]
- 5.Lewis JS, Meeke K, Osipo C, Ross EA, Kidawi N, Li T, et al. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst 2005; 97:1746–59. [DOI] [PubMed] [Google Scholar]
- 6.Jordan VC. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocr Relat Cancer 2015;22:R1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs. high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA 2009;302:774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson GL, Chlebowski RT, Aragaki AK, Kuller LH, Manson JE, Gass M, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal hysterectomy: extended follow-up of the Women’s Health Initiative Randomised Trial. Lancet Oncol 2012;13:476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ariazi EA, Cunliffe HE, Lewis-Wambi JS, Slifker MJ, Willis AL, Ramos P, et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci USA 2011;108:18879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan P, Griffith OL, Agboke FA, Anur P, Zou X, McDaniel RE, et al. c-Src modulates estrogen-induced stress and apoptosis in estrogen-deprived breast cancer cells. Cancer Res 2013;73:4510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweeney EE, Fan P, Jordan VC. Mechanisms underlying differential response to estrogen-induced apoptosis in long-term estrogen-deprived breast cancer cells. Int J Oncol 2014;44:1529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan P, Cunliffe HE, Maximov PY, Agboke FA, McDaniel RE, Sweeney EE, et al. Integration of downstream signals of insulin-like growth factor-1 receptor by endoplasmic reticulum stress for estrogen-induced growth or apoptosis in breast cancer cells. Mol Cancer Res 2015;13:1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehrke M, Lazar MA. Inflamed about obesity. Nat Med 2004;10:126–7. [DOI] [PubMed] [Google Scholar]
- 14.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010;140:900–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, et al. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015;349:500–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan P, Tyagi AK, Agboke FA, Mathur R, Pokharel N, Jordan VC. Modulation of nuclear factor-kappa B activation by the endoplasmic reticulum stress sensor PERK to mediate estrogen-induced apoptosis in breast cancer cells. Cell Death Discov 2018; 4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoppe R, Fan P, Büttner F, Winter S, Tyagi AK, Cunliffe H, et al. Profiles of miRNAs matched to biology in aromatase inhibitor resistant breast cancer. Oncotarget 2016;7:71235–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim BR, Lee GY, Yu H, Maeng HJ, Oh TJ, Kim KM, et al. Suppression of Nrf2 attenuates adipogenesis and decreases FGF21 expression through PPAR gamma in 3T3-L1 cells. Biochem Biophys Res Commun 2017; pii: S0006–291X:30166–3. [DOI] [PubMed] [Google Scholar]
- 19.Varady J, Eder K, Ringseis R. Dietary oxidized fat activates the oxidative stress-responsive transcription factors NF-κB and Nrf2 in intestinal mucosa of mice. Eur J Nutr 2011;50:601–9. [DOI] [PubMed] [Google Scholar]
- 20.Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, et al. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1998;1:465–70. [DOI] [PubMed] [Google Scholar]
- 21.Yin Y, Yuan H, Zeng X, Kopelovich L, Glazer RZ. Inhibition of peroxisome proliferator-activated receptor gamma increases estrogen receptor-dependent tumor specification. Cancer Res 2009;69:687–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonofiglio D, Gabriele S, Aquila S, Catalano S, Gentile M, Middea E, et al. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator activated receptor gamma signaling. Clin Cancer Res 2005;11:6139–47. [DOI] [PubMed] [Google Scholar]
- 23.Zaytseva YY, Wang X, Southard RC, Wallis NK, Kilgore MW. Down-regulation of PPARgamma1 suppresses cell growth and induces apoptosis in MCF-7 breast cancer cells. Mol Cancer 2008;7:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol 2007;28:551–8. [DOI] [PubMed] [Google Scholar]
- 25.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998;391:82–6. [DOI] [PubMed] [Google Scholar]
- 26.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998;391:79–82. [DOI] [PubMed] [Google Scholar]
- 27.Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, et al. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest 1999; 104:383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han J, Wang D, Ye L, Li P, Hao W, Chen X, et al. Rosmarinic Acid Protects against inflammation and cardiomyocyte apoptosis during myocardial ischemia/reperfusion injury by activating peroxisome proliferator-activated receptor gamma. Front Pharmacol 2017;8:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 2014;20:573–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burton JD, Goldenberg DM, Blumenthal RD. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Res 2008;2008:494161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan P, McDaniel RE, Kim HR, Clagett D, Haddad B, Jordan VC. Modulating therapeutic effects of the c-Src inhibitor via oestrogen receptor and human epidermal growth factor receptor 2 in breast cancer cell lines. Eur J Cancer 2012;48:3488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee R, Jow L, Croston GE, Paterniti JR Jr.. Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid X receptor agonists and antagonists. J Biol Chem 1997;272: 8071–6. [DOI] [PubMed] [Google Scholar]
- 33.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signaling pathways through heterodimer formation of their receptors. Nature 1992;358:771–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 2014;20:1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiel G, Ulrich M, Mukaida N, Rössler OG. Resveratrol stimulation induces interleukin-8 gene transcription via NF-κB. Pharmacol Res 2018;134:238–45. [DOI] [PubMed] [Google Scholar]
- 36.Cuadrado A, Martín-Moldes Z, Ye J, Lastres-Becker I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J Biol Chem 2014;289:15244–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun CC, Li SJ, Yang CL, Xue RL, Xi YY, Wang L, et al. Sulforaphane attenuates muscle inflammation in dystrophin-deficient mdx mice via NF-E2-related factor 2 (Nrf2)-mediated inhibition of NF-κB signaling pathway. J Biol Chem 2015;290:17784–95. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, et al. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res 2014;114:626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L, et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest 2015;125:3461–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, et al. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci USA 2004;101:6472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polvani S, Tarocchi M, Galli A. PPARγ and Oxidative Stress: Con(β) Catenating NRF2 and FOXO. PPAR Res 2012;2012:641087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan P, Agboke FA, McDaniel RE, Sweeney EE, Zou X, Creswell K, et al. Inhibition of c-Src blocks oestrogen-induced apoptosis and restores oestrogen-stimulated growth in long-term oestrogen-deprived breast cancer cells. Eur J Cancer 2014;50:457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eeckhoute J, Oger F, Staels B, Lefebvre P. Coordinated regulation of ppargamma expression and activity through control of chromatin structure in adipogenesis and obesity. PPAR Res 2012;2012 :164140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lefebvre B, Benomar Y, Guédin A, Langlois A, Hennuyer N, Dumont J, et al. Proteasomal degradation of retinoid X receptor alpha reprograms transcriptional activity of PPARgamma in obese mice and humans. J Clin Invest 2010;120:1454–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delitala AP, Steri M, Pilia MG, Dei M, Lai S, Delitala G, et al. Menopause modulates the association between thyrotropin levels and lipid parameters: The SardiNIA study. Maturitas 2016;92:30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nabulsi AA, Folsom AR, White A, Patsch W, Heiss G, Wu KK, et al. Association of hormone-replacement therapy with various cardiovascular risk factors in postmenopausal women. The atherosclerosis risk in communities study investigators. N Engl J Med 1993;328:1069–75. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Southard RC, Kilgore MW. The increased expression of peroxisome proliferator-activated receptor-gamma1 in human breast cancer is mediated by selective promoter usage. Cancer Res 2004;64:5592–6. [DOI] [PubMed] [Google Scholar]
- 48.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994;79:1147–56. [DOI] [PubMed] [Google Scholar]
- 49.Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci USA 1997;94:4318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abderrahman B, Jordan VC. Rethinking Extended Adjuvant Antiestrogen Therapy to Increase Survivorship in Breast Cancer. JAMA Oncol 2018;4:15–6. [DOI] [PubMed] [Google Scholar]
- 51.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med 2017;377:1836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiong R, Patel HK, Gutgesell LM, Zhao J, Delgado-Rivera L, Pham TND, et al. Selective Human Estrogen Receptor Partial Agonists (ShERPAs) for Tamoxifen-Resistant Breast Cancer. J Med Chem 2016;59:219–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell 2005;123:993–9. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Sun Y, Wong J, Conklin DS. PPARγ maintains ERBB2-positive breast cancer stem cells. Oncogene 2013;32:5512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.