

Abstract
Most human pathogenic mutations in 5′ splice sites affect the canonical GT in positions +1 and +2, leading to noncanonical dinucleotides. On the other hand, noncanonical dinucleotides are observed under physiological conditions in ∼1% of all human 5′ss. It is therefore a challenging task to understand the pathogenic mutation mechanisms underlying the conditions under which noncanonical 5′ss are used. In this work, we systematically examined noncanonical 5′ splice site selection, both experimentally using splicing competition reporters and by analyzing a large RNA-seq data set of 54 fibroblast samples from 27 subjects containing a total of 2.4 billion gapped reads covering 269,375 exon junctions. From both approaches, we consistently derived a noncanonical 5′ss usage ranking GC > TT > AT > GA > GG > CT. In our competition splicing reporter assay, noncanonical splicing was strictly dependent on the presence of upstream or downstream splicing regulatory elements (SREs), and changes in SREs could be compensated by variation of U1 snRNA complementarity in the competing 5′ss. In particular, we could confirm splicing at different positions (i.e., −1, +1, +5) of a splice site for all noncanonical dinucleotides “weaker” than GC. In our comprehensive RNA-seq data set analysis, noncanonical 5′ss were preferentially detected in weakly used exon junctions of highly expressed genes. Among high-confidence splice sites, they were 10-fold overrepresented in clusters with a neighboring, more frequently used 5′ss. Conversely, these more frequently used neighbors contained only the dinucleotides GT, GC, and TT, in accordance with the above ranking.
Eukaryotic pre-mRNA contains introns that are removed prior to nuclear export by the splicing process, where splice sites, conserved sequence elements at the exon/intron boundaries, are recognized by the spliceosome.
The vast majority (>99%) of all introns are excised by the major or U2-type spliceosome, associated with a multitude of accessory proteins (Wahl et al. 2009), and most of these introns are delimited by GT-AG dinucleotides. The U2-type spliceosome rarely also recognizes introns flanked by AT-AC dinucleotides (“AT-AC II introns”) (Wu and Krainer 1997). Another rare class of introns (∼0.4%) are removed by the minor U12-type spliceosome (Hall and Padgett 1994, 1996; Turunen et al. 2013) that also recognizes GT-AG or AT-AC (“AT-AC I introns”) (Wu and Krainer 1997) dinucleotides (Patel and Steitz 2003) together with their distinct branch point sequence (Hall and Padgett 1994; Alioto 2007).
Hybrid introns, displaying AT-AG dinucleotides, cannot a priori be assigned to either U2- or U12-type. However, hybrid introns are more likely recognized by the U2-type spliceosome, since U1 and U2 snRNP can bind independently to a 5′ or 3′ half-substrate (Barabino et al. 1990; Michaud and Reed 1993; Tarn and Steitz 1995). In contrast, U12-dependent splicing requires concurrent recognition of the 5′ splice site (5′ss) and the branch point sequence (BPS) by the U11/U12 di-snRNP complex (Frilander and Steitz 1999).
Finally, at least ∼0.9% of all human introns contain 5′ss with GC or, to a much lesser extent, other noncanonical dinucleotides in positions +1/+2 (Sheth et al. 2006). Efficient excision of introns containing 5′ss with noncanonical dinucleotides have been reported (e.g., Twigg et al. 1998; Brackenridge et al. 2003). Although most human GT > TT 5′ splice site mutations disrupt splicing (Krawczak et al. 2007), low activation of a noncanonical TT site was associated with milder clinical disease manifestation in Fanconi Anemia C patients (Hartmann et al. 2010), underlining the clinical importance of noncanonical splice site recognition.
Extensive sequence complementarity between splice junctions in hnRNA molecules and the nucleotide sequence at the 5′ end of U1 snRNA was already observed decades ago (Lerner et al. 1980; Rogers and Wall 1980). These seminal observations were encouraging for numerous biochemical and biological experiments demonstrating that indeed U2-type spliceosome assembly begins with RNA duplex formation between the free 5′ end of U1 snRNA and the 5′ss sequence (Aebi et al. 1986, 1987; Zhuang and Weiner 1986; Siliciano and Guthrie 1988). Beyond the 5′ss consensus sequence, nucleotide complementarity in positions +7 and +8 can also contribute to splicing efficiency (Kammler et al. 2001; Freund et al. 2003, 2005) and compensate for lack of complementarity in the three exonic positions (Hartmann et al. 2008). U1 snRNA complementarity was also found to be a major determinant for 5′ss selection in competition experiments comparing a reference site with various test sites at a fixed position (Eperon et al. 1986; Nelson and Green 1990). Beyond sequence complementarity to U1 snRNA, Zhuang and Weiner (1986) envisioned that RNA duplex formation could be mediated by protein factors in a compensatory way. In fact, nuclear serine- and arginine-rich phosphoproteins (SR proteins) were found to support in vitro pre-mRNA splicing (Fu et al. 1992; Mayeda and Krainer 1992). Exonic mutations were shown to cause alterations in vertebrate splicing patterns, leading to the identification of short purine-rich sequences stimulating splicing even in a heterologous context (Watakabe et al. 1993; Xu et al. 1993). Lavigueur et al. (1993) identified SR proteins as trans-acting factors binding such short purine-rich stretches. Taken together, U1 snRNA duplex stability determines splicing efficiency in concert with adjacent splicing regulatory elements (SREs), acting in a position-dependent manner (Erkelenz et al. 2013a).
Recent availability of transcriptome-wide (“gapped”) exon junction reads provides increasing data volume on noncanonical introns. There are several caveats, however. First, introducing quality scores is necessary to reduce the large proportion of false positive exon junction reads contained in raw transcriptome data (Kaisers et al. 2017b). Second, in order to detect noncanonical sites, it is important to use aligners like STAR, which do not rely upon specific intron flanking dinucleotides and do not require previous splice site annotation (Dobin et al. 2013). In a recent human transcriptome-wide study using the second-generation splice detection algorithm MapSplice (Wang et al. 2010; Parada et al. 2014), a comprehensive overview of high-confidence noncanonical introns was presented; 51% of these splice sites were not previously annotated, and noncanonical 5′ss frequencies were consistent with current Ensembl data.
In the present study, we analyzed noncanonical 5′ss usage both in a splice site competition assay and in a large data set of 54 human fibroblast samples with 2.4 billion exon junction reads. In the splice site competition assay, we systematically examined the impact of competing 5′ss U1 snRNA complementarity and presence of upstream and downstream SREs on noncanonical 5′ss recognition.
Results
Competition assay for determining noncanonical 5′ss efficiency
To experimentally examine noncanonical 5′ss usage, we designed a competition splicing assay comparing noncanonical 5′ss with several canonical 5′ss of different strengths. The GT dinucleotide in intron positions +1/+2 is highly conserved in 5′ss across virtually all eukaryotes (Collins and Penny 2005) and plays a critical part in correct splice site recognition. Examining noncanonical splicing, we therefore chose the minimal change from the canonical consensus sequence and varied only a single nucleotide in one of these GT positions.
More specifically, we used a splicing reporter containing one noncanonical 5′ss separated by a 16-nt-long neutral spacer sequence from the competing canonical site that could be additionally activated by downstream TIA1 binding sites (Fig. 1A). HEK293T cells were transiently cotransfected with each of one of the splicing reporters and the expression plasmid pXGH5 (growth hormone 1, GH1) to allow monitoring transfection efficiencies. Thirty hours post transfection, total RNA was isolated and analyzed by semiquantitative RT-PCR to estimate splicing activity (Fig. 1B).
Figure 1.
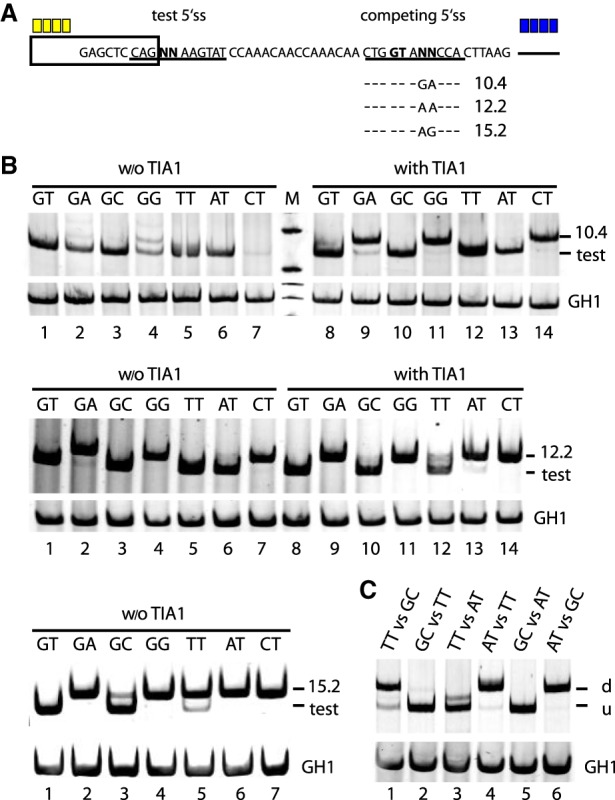
Competition assay for determining noncanonical 5′ss efficiency. (A) Schematic of the HIV-1-based splicing reporter containing the different competitive splice site pairs. Sequence variations (denoted by “NN”) and HBond scores (https://www2.hhu.de/rna/html/hbond_score.php) of the competing canonical splice sites are indicated below. Enhancer repeats are highlighted in yellow (SRSF7) or blue (TIA1). (B,C) RT-PCR analyses of spliced reporter mRNAs. All splicing reporters in B contained SRSF7 splicing enhancer repeats, and the presence or absence of TIA1 repeats is indicated above each panel. Splicing reporters in C contained both SRSF7 and TIA1. (C) The comparison of two noncanonical 5′ss, inserted at the positions of “test” and “competing” 5′ss in A. (u) Upstream site; (d) downstream site. To monitor transfection efficiency, 2.5 × 105 HEK293T cells were transiently transfected with 1 µg of each: the respective HIV-1-based splicing reporter and pXGH5 (expressing human growth hormone 1 [GH1]). RNA was extracted 30 h post transfection and subjected to RT-PCR analysis as described in Methods. All experiments were performed in triplicates (Supplemental Fig. S6).
When the noncanonical 5′ss were tested against a very weak canonical site with an HBond score (HBS) (Supplemental Methods) of 10.4 in the absence of a downstream splicing enhancer, noncanonical 5′ss activation could be observed with varying efficiencies for all noncanonical 5′ss (Fig. 1B, upper panel, lanes 1–7). From the band intensities we could tentatively rank the noncanonical 5′ss usage in the following order: GC > TT ≈ AT > GA > GG > CT. In order to examine this ranking more precisely, we systematically varied the competing 5′ss strength using different splice site sequences and downstream splicing enhancers.
In the presence of downstream TIA1 binding sites, we observed a complete splice site switch from GG, and CT to the competing canonical 5′ss, and an almost complete switch from GA, while the noncanonical GC, TT, and AT splice sites were still exclusively used (Fig. 1B, upper panel, lanes 8–14).
Substituting the weak (HBS 10.4) canonical 5′ss by a slightly stronger one (HBS 12.2), in the absence of downstream TIA1 binding sites, we observed the same splice site switch pattern as in the presence of TIA1 and the weaker 5′ss (Fig. 1B, cf. upper panel, lanes 8–14, with middle panel, lanes 1–7), consistent with the above tentative ranking of noncanonical 5′ss.
In particular, the similarity of splice site switch patterns suggested comparable effects on splice site usage of either the downstream TIA1 binding sites or an increase in U1 snRNA complementarity of the competing 5′ss (HBS difference 1.8 = 12.2 − 10.4).
To further differentiate between the efficiencies of noncanonical 5′ss GC, TT, and AT, which could not be resolved in the previous experiments, we supported splicing at the competing canonical 5′ss (HBS of 12.2) by adding downstream TIA1 binding sites. For the noncanonical AT 5′ss, splice site usage mostly switched to its canonical competitor, whereas the TT splice site was used more than the competing 5′ss. Finally, almost no switch from the GC splice site was observed, indicating the ranking GC > TT > AT (Fig. 1B, lower panel, cf. lanes 10,12,13).
For a crisper differentiation between the noncanonical 5′ss GC and TT, we substituted a competing canonical 5′ss of HBS 15.2 in the absence of any downstream TIA1 binding sites. Indeed, we observed a prominent splice site switch from TT but not GC to its competing canonical 5′ss, while the full complementarity reference GT splice site, as expected, did not switch at all (cf. Fig. 1B, lower left panel, lanes 3,5).
Furthermore, the effects on splice site usage were stronger for the HBS 15.2 splice site than for the HBS 12.2 (HBS difference 3) splice site supported by additional downstream TIA1 binding sites, consistent with the previous observation that the TIA1 effect is comparable to an increase in U1 snRNA complementarity corresponding to an HBS difference of 1.8. Note that the 5′ss substitution from HBS 12.2 to HBS 15.2 was obtained by a single nucleotide change, inducing a stronger effect on splice site usage than adding four downstream TIA1 binding sites.
Finally, in order to directly compare GC, TT, and AT sites with each other, we tested all six pairwise combinations of these three noncanonical 5′ss CAG NNAAGTAT in the same assay (Fig. 1A), where both competing 5′ss were potentially activated by upstream SRSF7 and downstream TIA1 binding sites. In agreement with the previously determined ranking, we found GC > TT, TT > AT, and GC > AT in both available positions (Fig. 1C). Taken together, noncanonical 5′ss efficiency was ranked GC > TT > AT > GA > GG > CT.
Greater enhancer dependency for noncanonical 5′ss than for canonical 5′ss
In the preceding section, we examined noncanonical 5′ss usage in a two-splice-site competition assay. The noncanonical 5′ss we chose had full complementarity to U1 snRNA except in one of the positions +1/+2, and therefore possibly contained additional GT dinucleotides in positions −1/+1 (for TT splice sites only) and +5/+6. In the next step, we applied the concept of splice site competition at these different splicing positions in the presence and absence of the splicing enhancers SRSF7 and TIA1, using the same splicing reporter as before, but without the second splice site and its separating neutral spacer sequence (Fig. 2A). In order to identify the accurate splicing position with single nucleotide precision, we extracted the PCR products from the gel and sequenced them (Fig. 2C; Supplemental Fig. S1).
Figure 2.

Noncanonical 5′ss exhibit a greater enhancer dependency than canonical 5′ss. (A) Schematic of the HIV-1 minigene containing a single noncanonical splice site. Experimentally found splicing registers (−1, +1, +5) are indicated by arrows. Yellow and blue boxes represent upstream (SRSF7) or downstream (TIA1) enhancer repeats. (B) Activation of splice sites in presence or absence of splicing enhancers. RT-PCR analysis was carried out as described before. Used splicing registers (R) are given below the gel image. All experiments were performed in triplicate (Supplemental Fig. S6). (C) Splicing positions mapped by sequencing of the extracted RT-PCR products (for a complete overview, see Supplemental Fig. S1).
The fully complementary canonical 5′ss with GT both at positions +1/+2 and +5/+6 was used even in the absence of any enhancer, and its usage still increased when it was supported by either SRSF7 or TIA1, or both (Fig. 2B, cf. lanes 1 with 2–4). In all events, splicing occurred exclusively at position +1 (Supplemental Fig. S1).
Contrary to the canonical splice site, all noncanonical 5′ss seemed to be strictly enhancer-dependent: Almost no splicing was detected in the absence of SRSF7 and TIA1 if one nucleotide in position +1 or +2 was changed, confirming the special impact of the canonical GT dinucleotide (Fig. 2B, cf. lanes 5,9,13, 17,21,25). This is in agreement with the finding that by far most known disease-causing single base-pair substitutions in 5′ss of human genes occur in one of these two positions (Krawczak et al. 2007).
The noncanonical GC splice site was very weakly used in the absence of SREs and could be strongly activated by either SRE: TIA1 or SRSF7, while splicing occurred exclusively at position +1 (Fig. 2B, cf. lanes 9–12).
The noncanonical 5′ss TT, AT, and GA were activated by the stronger enhancer SRSF7 but not by the weaker TIA1 alone (Fig. 2B, cf. lanes 6,7,18 with 19,22,23) (Erkelenz et al. 2013a). Beyond splicing at the noncanonical dinucleotides in positions +1/+2, different positions were additionally activated in the presence of both SREs: −1 for the TT site; +5 for the AT and GA sites. Note, however, that for the TT site position, −1 was also activated by SRSF7 alone.
The noncanonical 5′ss GG and CT were only considerably activated in the presence of both SRSF7 and TIA1. SRSF7 alone only led to weak splicing activity: at position +1 for GG, but at the competing GT in positions +5/+6 for CT.
Taken together, ranking noncanonical 5′ss according to their degree of dependence on the splicing enhancers SRSF7 and TIA1, we obtained the order GT > GC > (TT, AT, GA) > (GG, CT), consistent with the ranking obtained before. All splice sites except TT spliced exclusively at a single position (“accurate splicing”) in the presence of SRSF7, whereas additional insertion of TIA1 binding sites could lead to inaccurate splicing at an additional position. Independent of SREs, the noncanonical TT site, however, was always spliced inaccurately at both positions −1 and +1, but not at +5 (Fig. 2B, cf. lanes 18–20). This may be due to a property of the splice site sequence CAG TTAAGTAT in our assay: It contained a G in −1 and thus GT dinucleotides at both −1/+1 and +5/+6, and may also form a U1 snRNA duplex by bulging out a single nucleotide (Roca et al. 2012; Tan et al. 2016). We hypothesize that this splicing outcome is the result of a competition between splice site recognition by U1 snRNA duplex formation with the nonbulged (10 complementary nucleotides) or bulged sequence (11 complementary nucleotides) in the presence of this favorable SRE constellation.
Collectively, these results demonstrate that (1) an environmental enrichment of splicing enhancers positively correlates with the overall splicing activation of both canonical and in particular noncanonical splice sites, and that (2) the used splicing position is the result of 5′ss recognition by U1 snRNA base-pairing and the supporting splicing regulatory elements.
Accuracy of noncanonical TT splicing
In order to examine the competition between splicing positions −1 and +1 in noncanonical TT splicing, we selected the human pathogenic FANCC (FA complementation group C) exon 2 splice site mutation c.165 + 1G > T (Hartmann et al. 2010; Roca et al. 2012). We tested a set of eight noncanonical TT splice site variations, downstream from the previously used SRSF7 binding sites in a splicing reporter containing two weak intronic cryptic 5′ss (c1: HBS 7.6 and c2: 12.3) (Fig. 3A).
Figure 3.
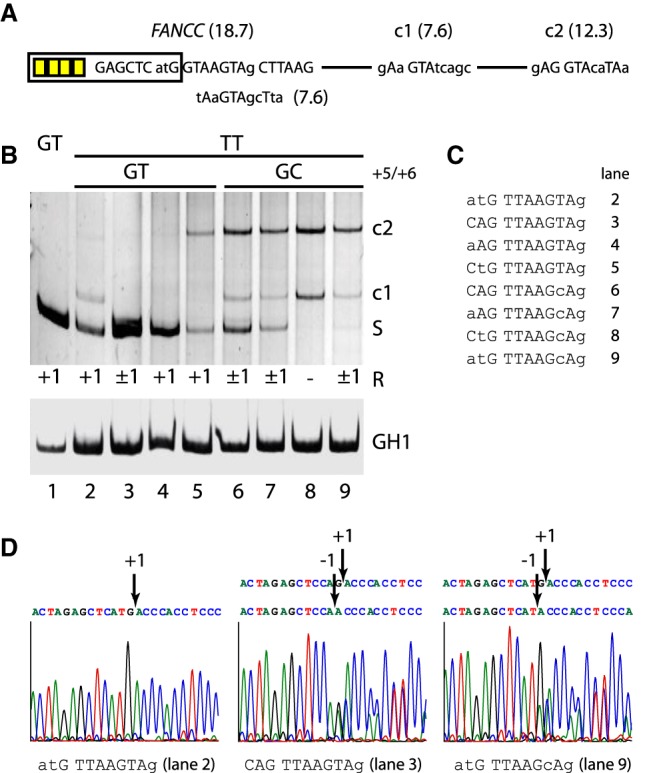
Accuracy of noncanonical TT splicing. (A) Schematic of the splicing reporter containing the human FANCC exon 2 splice site and two weak cryptic splice sites (c1/c2) in the downstream intron. Exonic SRSF7 enhancer sites are indicated by yellow boxes. (B) Different splicing positions (R) obtained by variations of the TT splice site sequence. RT-PCR analysis was performed as described before. All experiments were performed in triplicate (Supplemental Fig. S6). (C) TT splice site sequences from B, lanes 2–9. The human pathogenic FANCC exon 2 c.165 + 1G > T splice site is shown in lane 2. (D) Exemplary splicing positions mapped by sequencing of the extracted RT-PCR products.
All eight variations contained a TT dinucleotide in +1 and +2 and corresponded to the sequence pattern “..G TTAAG.Ag” (Fig. 3B,C). Four splice site variations contained a second GT in +5/+6, and splicing efficiency was further varied through nucleotides −3 and −2.
The TT site CAG TTAAGTAg with the largest set of U1 snRNA complementary nucleotides was “inaccurately” spliced at both positions −1 and +1 (Fig. 3B, lane 3, and 3D, middle), similar to CAG TTAAGTAT in the splicing reporter used in Figure 2B (cf. lane 18). Reducing U1 snRNA complementarity further by variation of positions −3 and −2 but keeping the second GT in +5/+6 activated accurate splicing only in position +1 (Fig. 3B, lanes 2,4,5 and, e.g., 3D, left), together with weak coactivation of a cryptic 5′ss (Fig. 3B, lane 5). No splicing in position +5 could be observed, however.
The remaining four variations, which did not contain a second GT in +5 and +6, exhibited either inaccurate TT splicing (Fig. 3B, lanes 6,7,9 and, e.g., 3D, right) or completely switched to the cryptic splice sites (Fig. 3B, lane 8). Therefore, accurate noncanonical TT splicing at +1 seemed to be dependent on a second GT in +5 and +6 and suboptimal U1 snRNA complementarity.
This dependence could be confirmed for two naturally occurring noncanonical TT splice sites in the human genes FBXO9 (F-box protein 9) and ETV1 (ETS variant 1) (Parada et al. 2014), transferred to the same splicing reporter. The TT site aAG TTAAGTAT (FBXO9) activated TT splicing at both positions −1 and +1, whereas the site aAG TTAAGTtg (ETV1) with fewer complementary nucleotides exclusively activated +1 (Supplemental Fig. S2).
In previous careful and thoroughly carried out analyses, Roca et al. (2012) found the FANCC TT splice site atG TTAAGTAg to be exclusively spliced at GT in positions −1 and +1, however in the context of a different splicing reporter. With the splicing reporters used here, we never observed exclusive GT splicing at −1 in any of the TT sites.
However, using a different splicing reporter with a designer exon (Fig. 4A–C; Brillen et al. 2017) containing two SRSF7 binding sites upstream of the noncanonical TT 5′ss CAG TTAAGTGT, we could also observe nearly exclusive splicing at the GT in position −1 (Fig. 4C, lane 2 and 4D, 2), when a second, weak, U1 binding site AAC GTACGCAG (BsiWI −8/−7 GT) was located 8 nt upstream without being used. This could be due to repeats of the splicing neutral CCAAACAA octamer sequence (Zhang et al. 2009), which were inserted as spacers into the designer exon. Concatenating this octamer with itself created a “CANC” motif at the border, which could serve as an SRSF3 binding site (Hargous et al. 2006). To investigate the impact of SRSF3 binding at the CAAC site overlapping with the BsiWI −8/−7 GT site, we substituted the first cytosine (Fig. 4B, and 4C, lane 3) by adenosine, resulting in first usage of the weak BsiWI −8/−7 GT site (HBS 9.3) and enhanced usage of the upstream splice donor c1 (cf. Fig. 4C, lanes 2,3). This was indeed confirmed using a QIAxcel DNA screening cartridge clearly separating two bands for both BsiWI −8/−7 GT and noncanonical TT splicing (Fig. 4C, top).
Figure 4.

Upstream SREs and a second GT at positions −8 and −7 affect the accuracy of TT splice site usage. (A) Overview of the used designer exon splicing reporter containing either five repeats of the splicing neutral sequence CCAAACAA (white boxes) or two SRSF7 binding sites (yellow box). The underlined bases represent CANC motifs, which arise from concatenating CCAAACAA and can serve as SRSF3 binding sites. (B) Sequences of the exon–intron border in the different designer exon variants. Lane numbers correspond to C and D. Potential SRSF3 binding sites are underlined. Sequence and HBond score of the U1 binding site at position −8 (bold GT) are shown below. Splicing registers at the noncanonical TT are indicated by −1 and +1. (C) HeLa cells were transfected with 1 µg of each of the depicted constructs and 1 µg of GH1, which was used as transfection control. Twenty-four hours post transfection, total RNA was isolated, reverse transcribed, and amplified with specific primer pairs: (DE) #2648/#2649; (S) splice site usage; (c1) c1 usage; (ES) exon skipping; (GH1) #1224/#1225. PCR products were separated either on a 10% PAA gel (bottom), or for higher resolution, on a QIAxcel DNA screening gel cartridge (top). (D) Sequencing results of the splice site usage shown in C. PAA bands were isolated, reamplified with the primer pair #2648/#2649, and sent to sequencing analysis using primer #2648. Blue shades in the sequencing chromatogram represent sequencing uniqueness, and black lines roughly indicate the level of alternative splice site usage. (E) The noncanonical CT splice site sequence CAG CTAAGTAT (cf. Figs. 1, 2) recruits SRSF3 in competition with U1 snRNA binding. In an RNA affinity chromatography assay, substrate RNAs containing a bacteriophage MS2 sequence and either canonical GT or noncanonical CT splice site sequences with otherwise full U1 snRNA complementarity were covalently linked to adipic acid dihydrazide-agarose beads (AB) and incubated with HeLa cell nuclear protein extract. Recombinant bacteriophage MS2 coat protein was added to monitor RNA input. Precipitated proteins were resolved by SDS-PAGE (15%) and detected by immunoblot analysis using anti-SRSF3, anti-SNRPC, or anti-MS2 coat protein antibodies.
The shift to c1 was further enhanced by mutating the next nearest upstream SRSF3 binding site, consistent with reduced silencing of upstream 5′ss c1 (Erkelenz et al. 2013a), and it seemingly occurred at the expense of both BsiWI −8/−7 GT and noncanonical TT site usage (cf. Fig. 4C, lanes 2–4). Sequencing confirmed indeed that the splicing pattern switched from nearly exclusive −1 in lane 2 to a mixture of −1 and BsiWI −8/−7 GT usage in lane 3 (Fig. 4D, 2 and 3).
To further investigate splicing at the noncanonical TT site, the BsiWI site was altered by a single nucleotide substitution, resulting in BsiWI −8/−7 GT > CT. As expected, in this competition situation between a very weak noncanonical CT and a TT site with high U1 snRNA complementarity, we observed splicing exclusively at the TT site (QIAxcel) (Supplemental Fig. S3). Similar to the original BsiWI −8/−7 GT site, we observed an increase in c1 usage upon inactivation of one or both SRSF3 binding sites (Fig. 4C, lanes 5–7).
Sequencing analysis showed mixed usage of both splicing registers at −1 and +1 in the absence of competing BsiWI 5′ss usage (either −8/−7 GT masked by SRSF3 binding or −8/−7 GT > CT). We could only find nearly exclusive use of the −1 register in the TT site, if BsiWI −8/−7 GT was actually used (Fig 4D, 3 and 4).
Finally, we confirmed our hypothesis that SRSF3 binding can compete with U1 snRNP binding using an RNA affinity chromatography assay. In order to optimize U1 snRNP binding, we used the high U1 snRNA complementarity CT site CAG CTAAGTAT containing the CAGC binding motif for SRSF3 as in our experiments shown in Figures 1 and 2. Indeed, Figure 4E demonstrates U1 snRNP binding at the GT site, but not at the CT site, while SRSF3 was found strongly bound to the CT site. This confirms actual binding of SRSF3 to the CAGC motif overlapping the CT site, and thus competition with U1 snRNP binding to this same site, which may in fact explain why highly complementary CT sites were the weakest of all tested noncanonical 5′ splice sites.
These findings are in line with our results (Fig. 2) that not only overall splicing activity, but also the precise splicing position can be affected by flanking splicing enhancers and sequence environment.
RNA-seq data
Complementing our experimental approach, we examined actual noncanonical 5′ splice site usage reflected in our large human RNA-seq transcriptome data set of 54 human fibroblast samples taken from 27 subjects (14 male; 18 samples in each of the three age groups 18–25, 35–49, 60–67) (see Kaisers et al. 2017a). To obtain a measure for 5′ss usage, we extracted gapped reads spanning exon junctions, which we simply denote as “reads” in the following.
In order to focus on 5′ss and reduce additional variability by noncanonical 3′ss, we only analyzed exon junctions with canonical Ensembl annotated AG-type acceptor sites. To retain confirmed 5′ss and suppress false positive events (biological noise), we furthermore excluded exon junctions with fewer than 11 reads, leaving us with 2,369,936,633 reads covering 269,375 exon junctions in 18,633 unique genes expressed in these fibroblast samples. Of these, 125,498 exon junctions (46.5%) from 12,204 genes (65.5%) were detected in all 54 samples; 54% of all exon junctions and 99.4% of associated reads occurred in 50 or more samples.
As an alternative to U1 and U2 snRNP, U11/U12 splicing machinery is used in noncanonical splicing. This minor spliceosome mainly excises AT-AC and AT-AG introns, and its 5′ splice sites frequently share an ATATC motif at position +1. For an adequate comparison with our splicing reporter experiments, which only support U1 snRNA-dependent splicing, we excluded 15 presumed minor spliceosome 5′ss containing an ATATC motif, which on average occurred in 41 samples, from our transcriptome data analysis. Next, we present data on the remaining 269,360 exon junctions. Table 1 presents an exon junction and 5′ss overview of our RNA-seq data.
Table 1.
Overview of exon junction and 5′ss data, presented separately for high-confidence exon junctions with ≥2% of gene expression, and noise candidates

Gene expression and detection level: profiles of the top 10 exon junction reads in a gene
In any given cell type (here, fibroblasts), individual genes have different expression levels. In order to compare splice site usage across genes, detected exon junction reads need to be separately normalized within each gene by the expression level of this particular gene in the studied cell type.
To obtain estimates for gene expression, we determined the maximum number of reads on a single exon junction for each of the 18,633 genes (maximum reads in gene [MRIG]). The MRIGs obeyed a long-tailed distribution with a maximum of 5.4 × 106 and a median of 2859 reads (Fig. 5A). There were very few highly expressed genes: 499 genes (2.7%) had a maximal exon junction with more than 100,000 reads and 5051 genes (27.1%) with more than 10,000 reads on the most detected exon junction. At low expression levels, 6974 genes (37.4%) had a maximal exon junction of less than 1000 reads.
Figure 5.

Distributions of 269,360 exon junctions in 18,633 genes. (A) Distribution of the maximum number of reads on a single exon junction (MRIG) for all 18,633 genes. (B) Profile for top 10 numbers of reads on a single exon junction (mean ± SD). For each gene, exon junctions were ordered by their reads, then normalized by their respective maximum reads in each gene (MRIG) to obtain gene-normalized reads (GNR). Axis labels show the number of genes with at least 1, 2, …, 10 exon junctions. There were, for example, 16,739 genes with two or more exon junctions, and the second highest exon junctions had average GNR of 80%.
In the next step, we examined if all or most exon junctions in a specific gene were detected at similar levels, i.e., with similar read numbers. For each gene, we therefore determined its “exon junction read profile” from its top 10 exon junction reads, normalized by its MRIG. The average gene profile (mean ± standard deviation) shown in Figure 5B gradually declined from 80% of MRIG for the second highest reads in 16,739 genes to 36% for the tenth highest reads in 10,277 genes with at least 10 exon junctions. This decline is in part due to occurrence of “noisy” exon junctions with very few reads and probably reflects variance introduced through both alternative splicing events and nonuniform exon junction detection by deep sequencing. Moreover, it implies that there is no simple cutoff for absolute or relative exon junction reads separating constitutive and alternative splicing events from false positive exon junctions (biological noise).
Noncanonical 5′ exon junctions occur preferentially in highly expressed genes
In order to examine the dependence of noncanonical 5′ splice site usage on gene expression, we determined histograms of maximum reads in gene (MRIG) by counting exon junctions separately for all 16 dinucleotides. Of all 269,360 exon junctions, 6030 (2.2%) had noncanonical dinucleotides, and they occurred in 3901 distinct genes. Typically, these genes contained just one (2666 genes) or two (780 genes) noncanonical exon junctions.
For every individual dinucleotide—except TG—the MRIG distribution exhibited a single maximum (Supplemental Fig. S4A), so that average and standard error of MRIG were meaningful measures. The average gene expression level (MRIG) of all exon junctions with a given dinucleotide (Fig. 6) was lowest for GT (average MRIG 29,505), reflecting the high abundance of GT splice sites in many genes expressed at low levels (cf. Fig. 5A). GC (average MRIG 57,730) and TT (average MRIG 81,621) 5′ splice sites also occurred in genes with low expression levels. On the other hand, average gene expression levels were 23- to 25-fold higher for GG and CT sites than for GT sites. Ranking noncanonical dinucleotides by their average gene expression level, we obtained the same order GT > GC > TT > AT > GA > GG > CT we found in the splicing reporter experiments. This indicates that GC and TT sites were the only noncanonical splice sites detected at lower gene expression levels, while all other noncanonical exon junctions were preferentially detected in highly expressed genes.
Figure 6.
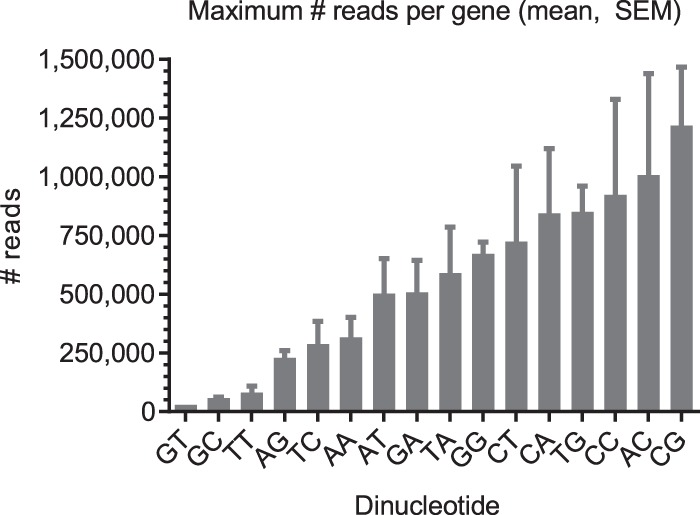
Average gene expression level (maximum number of reads on a single exon junction [MRIG]) for all exon junctions with a given dinucleotide (mean and SEM).
Gene-normalized 5′ splice site usage identifies noise candidates
In our fibroblast data set, exon junction reads widely differed across several orders of magnitude due to differential gene expression and RNA-seq coverage. In order to adequately compare detected splice site usage across genes, we calculated the gene-normalized read percentage (GNR) for each exon junction by dividing numbers of reads by the maximum number of reads (MRIG) in the respective gene: . Obviously, by normalization 0 ≤ GNR ≤ 100. The overall GNR mean and median were 42% and 45%, respectively.
However, the distribution of gene-normalized reads in all 269,360 exon junctions shown in Figure 7 appeared as a superposition of three parts: a power law distribution with exponent −1.7 at low GNR values, a Gaussian-type distribution centered around GNR = 71% with SD 22%, and a single peak at GNR = 100% representing one maximal exon junction per gene. This observation could be confirmed by maximum likelihood estimates for the power law (goodness of fit, R2 = 0.998) and Gaussian distribution (R2 = 0.970) parameters. There was no clear separation between the exponential and Gaussian regions of the distribution.
Figure 7.
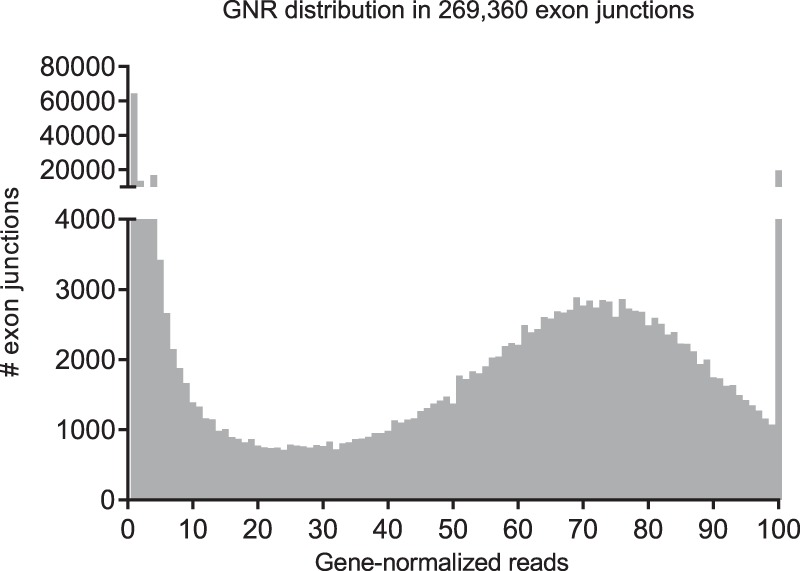
Distribution of gene-normalized reads (GNR) for all 269,360 exon junctions. At less than 2% of MRIG, 77,912 exon junctions (28.9%) were only very weakly used and contributed 0.28% of all reads. Average GNR was 42, and the median was 45. For individual dinucleotides, see Table 2.
However, the first two bins 0%–2% contained significantly more exon junctions (77,912) than any single GNR bin in the rest of the distribution (maximum 6900). We therefore considered these 77,912 exon junctions with usage below 2% of the respective gene expression as “noise candidates.” Occurring in an average of 24 samples, they represented 28.9% of all exon junctions, but accounted for only 0.28% of all reads and 0.35% of all gene-normalized reads. Excluding these noise candidates left 191,448 remaining “high-confidence” exon junctions detected in an average of 47 samples, which depended only weakly on absolute or gene-normalized reads (Supplemental Fig. S4C,D). No matter how much a “high-confidence” exon junction was used in absolute read numbers or relative to its gene expression, it was on average detected in between 40 and 50 samples. After noise candidate removal, only 1892 high-confidence noncanonical exon junctions of the initial 6030 remained (Table 1). Although GT junctions with GNR ≥ 2% were still detected in 47 samples, GC junctions in 44 and TT junctions in 41 samples, AT junctions only occurred in 30 samples (Supplemental Fig. S4B).
Noise candidate exclusion generally reduced the number of noncanonical exon junctions more than threefold compared to the 1.4-fold overall reduction (P < 0.0001, χ2 test). Noise candidates contained 5.3% noncanonical 5′ss—five times the proportion in the 191,448 high-confidence exon junctions with GNR ≥ 2%. Excluding noise candidates also had specific impact on individual dinucleotides. Although the number of GC exon junctions was 2.5-fold lower, the reduction was at least eightfold for all other dinucleotides (Table 2), and eight dinucleotides were no longer detected at all—in particular those with two noncanonical bases (except AG).
Table 2.
Number of noncanonical exon junctions by dinucleotide

High-confidence noncanonical 5′ exon junctions occur at ∼1% independent of gene expression level
We next investigated the hypothesis that noncanonical splicing might in part occur due to biological errors in the process of splice site recognition. This would be reflected in a correlation between gene expression level and noncanonical splicing. We therefore grouped exon junctions by their gene expression level (in logarithmically equidistant MRIG intervals) and counted canonical and noncanonical 5′ exon junctions separately in each MRIG interval. Both canonical and noncanonical exon junction distributions peaked around 6500 MRIG.
The proportion of noncanonical splice sites among all 269,360 exon junctions strongly increased 22-fold from 1% in low expressed genes (147/14,180; MRIG <100) to 22% in highly expressed genes (209/934; MRIG >1,000,000) (Fig. 8, gray bars), and showed a significant linear correlation of r = 0.80. However, in the set of 191,488 high-confidence exon junctions detected at least at 2% of gene expression level, the proportion of noncanonical exon junctions remained constant between 0.4% and 1%, independent of gene expression level (Fig. 8, black bars). Table 3 indeed confirms a fivefold to 75-fold reduction in the number of noncanonical exon junctions for 5051 highly expressed genes with MRIG >10,000 upon noise candidate removal. Noncanonical 5′ splice sites for MRIG <300 were unchanged due to previous exclusion of exon junctions with fewer than 10 reads.
Figure 8.
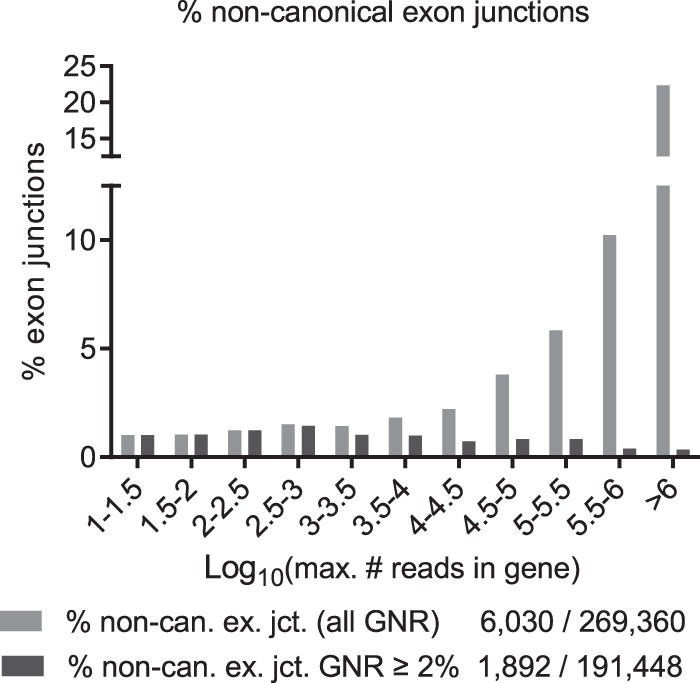
Percentage of noncanonical exon junctions grouped by gene expression level (MRIG, log10 scale, separately normalized for each MRIG range) for all exon junctions (gray, 6030 noncanonical/269,360) and after exclusion of noise candidates (black, 1892 noncanonical/191,448 high-confidence). All exon junction reads were first grouped by MRIG range before normalization. After exclusion of noise, the level of noncanonical exon junctions was ∼0.4%–1% independent of gene expression level.
Table 3.
Number of noncanonical exon junctions by gene expression level (MRIG, log10 scale)

Noncanonical 5′ss are 10-fold overrepresented in secondary 5′ss
In the next step, we examined whether noisy splice site recognition occurred as a general phenomenon or was particularly associated with noncanonical splicing. To this end, we first aggregated the reads on all exon junctions in our RNA-seq data set with the same splice donor, obtaining 222,163 different 5′ splice sites overall and 174,551 5′ss from high-confidence exon junctions detected at least at 2% of gene expression level—after removal of the noise candidates identified above. For every index 5′ splice site, we then collected all neighboring 5′ss within ±11 nt into a cluster. The neighborhood width was chosen equal to twice the maximum number of U1 snRNA complementary nucleotides (11), led by the assumption that this choice left no room for splicing regulatory elements between competing 5′ss. By far, most (209,640) 5′ss were single splice sites, that is, had no neighboring 5′ss within ±11 nt.
Those 5′ splice sites with only less used neighboring 5′ss (or none at all: single 5′ss) were denoted as “primary” 5′ss: These were the most used within such an ±11 nt cluster, and possible candidates for genuine 5′ss recognition mechanisms. Conversely, 5′ss with fewer reads than a neighboring 5′ss were denoted as “secondary” 5′ss, and were candidates hypothetically detected due to splice site competition or inaccurate recognition of a highly used nearby primary 5′ss.
Clustering 5′ splice sites in ±11 nt neighborhoods permitted identifying those 5′ss actually competing with other nearby 5′ss. Overall, only 5.6% (12,523) of all 5′ss occurred in such clusters, and 91% of all such clusters contained just two 5′ss (Table 4).
Table 4.
Percentage of canonical and noncanonical 5′ splice sites in complementing groups of single versus in-cluster 5′ss and primary versus secondary 5′ss, both for all 222,163 5′ss and for 174,551 high-confidence 5′ss with GNR ≥2%

Noncanonical splicing, however, was highly overrepresented (12-fold) in secondary, less used 5′ss: Secondary 5′ss contained 29% noncanonical sites compared to an overall proportion of 2.5% (P < 0.0001, χ2-test). Even more important, a 10-fold overrepresentation of noncanonical splicing among secondary splice sites persisted after removal of noise candidates below 2% of gene expression level: 10% of secondary high-confidence 5′ss were noncanonical sites compared to an overall rate of 1% (P < 0.0001, χ2-test) (Table 4). These 1051 secondary 5′ss were much less detected at GNR of 9.8% of gene expression level compared to an overall average GNR of 64%. Thus, secondary sites were relatively weakly used.
Clustering closely spaced 5′ splice sites and separating the less used from the most used 5′ss in each cluster, we have established an association with noncanonical splicing: The less used (“weaker”) of nearby competing 5′ splice sites were 10 times more likely to have a noncanonical dinucleotide.
Higher 5′ss usage variability across individuals in secondary splice sites
In the complete set of 2090 high-confidence 5′ss in clusters, we examined whether canonical and noncanonical 5′ss were differently regulated across individuals. For 2072 of these 2090 5′ss, we obtained individual reads separately for all 27 subjects. We measured 5′ss usage variability across subjects by the coefficient of variation (CV; standard deviation of all 27 read numbers divided by their mean). Primary sites (CV 0.64 ± 0.38) showed a significantly smaller CV than secondary sites (0.84 ± 0.46; P < 0.0001, Mann–Whitney U test).
Furthermore, for primary 5′ss, CV was lower for canonical (0.63 ± 0.37, N = 977) than for noncanonical (0.79 ± 0.43, N = 57) 5′ splice sites, whereas for secondary sites there was no significant difference in CV. In a two-way ANOVA, we did not detect any significant interaction between these two factors (P > 0.05). Thus, secondary 5′ss had a higher variability across individuals than primary 5′ss, and the variability of noncanonical 5′ss was larger than in canonical sites.
Protein coding relies more on primary than secondary 5′ss
In order to examine potentially different association of primary and secondary 5′ss with protein coding, we tracked all Ensembl transcripts of the 2090 high-confidence 5′ss detected in any of the 1039 clusters. For 1456 high-confidence 5′ss in clusters, we could identify coding and noncoding transcripts. Among the 634 5′ss without Ensembl transcripts, there were 554 secondary (87%) and only 80 primary 5′ss. We denoted a 5′ss as “protein coding,” if we found at least one coding transcript. Whenever we found only nonprotein coding Ensembl transcripts of a 5′ss, we denoted the 5′ss as “noncoding.”
Indeed, 775/1039 primary 5′ss (75%) were protein coding, whereas only 317/1051 secondary 5′ss (30%) were protein coding. Thus, at least part of the secondary sites could contribute to cellular protein synthesis. Conversely, protein coding relied more heavily on primary 5′ss, since 71% of coding 5′ss were primary sites.
Eventually, we examined protein coding properties of associated primary and secondary 5′ss in the same cluster. From 983 pairs with protein coding primary 5′ss, 401 had coding and 124 noncoding secondary 5′ss, and 458 secondary 5′ss were not annotated. The histogram of distance between primary and secondary 5′ss peaked at ±4, ±6, and ±9 nucleotides, in agreement with Dou et al. (2006).
Usage of secondary 5′ss is not random
Usage of secondary splice sites might reflect an intrinsic inaccuracy of the splicing process itself. Assuming that all reads on secondary 5′ splice sites had been caused by erroneous splicing on the associated primary 5′ splice site in the same cluster, the overall splicing error rate could be estimated as the proportion of all 1,253,067 reads on 6434 secondary sites in all 2,369,936,633 reads: Every 1890th read would have occurred in error at a neighboring secondary 5′ site, a “splicing gone wrong.”
However, this interpretation is not compatible with the observation that 94.4% of all 5′ss had no neighbors at all. In fact, 112 highly used 5′ss with more than 1 million reads had no secondary neighbors, although with an average of 1,837,460 reads, we would expect about 970 secondary reads for each of these 112 5′ splice sites. Thus, secondary 5′ss do not occur randomly in the splicing process, but are rather dependent on specific sequence environment, for example, permitting recognition by U1 snRNA and/or splicing regulatory proteins.
Competing high-confidence 5′ splice site usage in clusters
To compare RNA-seq findings to our experimental data obtained from the competition assay, we selected a corresponding RNA-seq data set of all neighboring high-confidence 5′ss pairs. In order to exemplarily confirm that such 5′ss pairs were indeed used, we reamplified RNA from four different individuals and analyzed PCR products by sequencing. In all these cases, we detected a mixture of splice site usage, confirming noncanonical 5′ss usage (Supplemental Fig. S5).
From all 12,523 5′ss occurring in clusters, we analyzed only the 2090 high-confidence 5′ss above 2% of the respective gene expression (GNR ≥ 2%). For each specific dinucleotide, we then averaged gene-normalized reads for all high-confidence 5′ss “competing” in such a pair as measure of their average usage across genes (Fig. 9).
Figure 9.
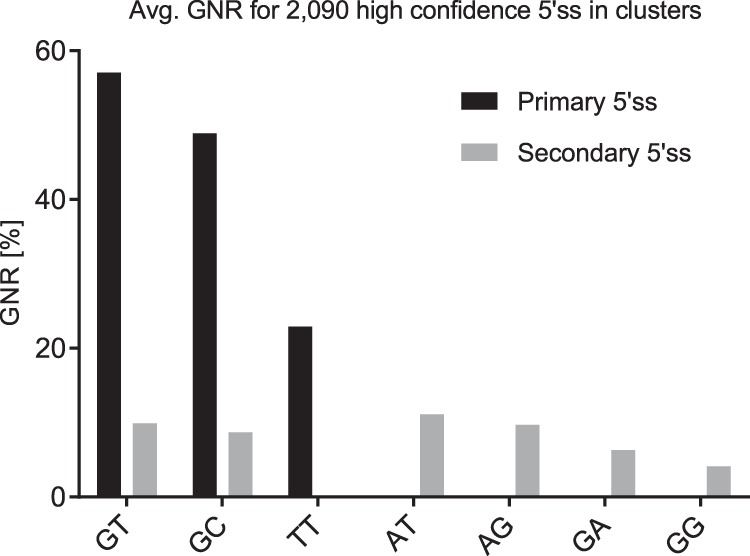
Average gene-normalized exon junction reads (GNR) for all 2090 high-confidence 5′ss with GNR ≥2% that were detected in clusters. Average GNR for primary 5′ss are shown in black bars and secondary 5′ss in gray bars.
Only GT-, GC-, and TT sites occurred as primary sites with average GNR of 57% (GT, N = 982), 49% (GC, N = 54), and 23% (TT, N = 3). Conversely, AT, GA, and GG sites occurred only as secondary, less used part of a pair. Secondary 5′ss were much less used at average GNR levels of 4%–11%. GC and TT sites were the only noncanonical 5′ss that did not require a highly used nearby GT site.
Secondary 5′ss were frequently detected at specific positions relative to primary GT-sites: 1132 secondary GT 5′ss were preferentially found at positions −4/−3 or +5/+6 relative to the respective primary GT in +1/+2. Similarly, 110 secondary GC sites were detected almost exclusively at positions −4/−3. Five secondary AT 5′ss occurred solely at positions −3/−2 or +4/+5, and three GA 5′ss occurred −4 and +6 nucleotides from a primary GT site. Ten secondary GG sites preferentially occurred at −4 and −1. All nine TT sites were primary sites: No neighbors were found next to six TT sites, and three TT sites had secondary GT sites at relative positions −3 and −1. It may be instructive to look at these examples in detail. The TT site in DDX46 (DEAD-box helicase 46) has 50% GNR and its neighboring GT site at position −3 has 16% GNR. In the two other cases in NPIPB3 (nuclear pore complex interacting protein family member B3) and NPIPB4 (nuclear pore complex interacting protein family member B4), the TT site carries 10% GNR, and the neighboring GT sites at position −1 have only 2% GNR. This difference in GNR may be due to nonsense-mediated decay (NMD) consistent with out-of-frame transcript removal, since the difference of splice site positions is no multiple of three. In all these cases, noncanonical secondary sites were found in positions favored by high U1 snRNA complementarity.
Notably, the average GNR rank order of noncanonical 5′ splice sites in 2090 5′ss competing in clusters (GT > GC > TT > AT > GA > GG > CT) was identical to the rank order of our competition assay.
Discussion
In this work, we systematically examined noncanonical 5′ splice site selection, both experimentally using splicing competition reporters, and analyzing a large RNA-seq data set of 54 fibroblast samples from 27 subjects. Both approaches consistently yielded a noncanonical 5′ss usage ranking GC > TT > AT > GA > GG > CT. In our splicing reporter assay, noncanonical splicing required upstream or downstream splicing regulatory elements, and covariation of the competing 5′ss U1 snRNA complementarity could compensate changes in SREs. In particular, we could confirm splicing at different positions (i.e., −1, +1, +5) of a splice site for all noncanonical dinucleotides “weaker” than GC.
We found that SREs and additional U1 snRNA binding sites could influence the selection of the splicing position. For example, for weak noncanonical 5′ss except GC and TT, we saw a shift toward position +5 in the presence of intronic TIA1 enhancers. We speculate that higher enhancer dependencies and weaker direct interactions between the U1 snRNA and noncanonical 5′ss provide the basis for more flexible base-pairing registers (Kondo et al. 2015). Hypothetically, in the presence of multiple enhancers, the actual splicing register might be determined by an equilibrium of exonic and intronic enhancer pulling forces. Furthermore, previous studies indicated that the concurrent binding of more than one U1 snRNP could also influence splice site selection through reciprocal stabilization of U1 snRNPs and SR proteins (Fernandez Alanis et al. 2012; Hodson et al. 2012; Martinez-Pizarro et al. 2018).
In our comprehensive RNA-seq data set analysis, noncanonical 5′ss were preferentially detected in weakly used exon junctions of highly expressed genes. In particular, they were 10-fold overrepresented among all 5′ss with a neighboring, more frequently used 5′ss. Conversely, these more frequently used neighbors contained only the dinucleotides GT, GC, and TT, in accordance with the above ranking.
In genetic disease, human pathogenic 5′ splice site mutations often lead to cryptic 5′ss activation, if the wild type sites are sufficiently weakened (e.g., Spena et al. 2006; Krawczak et al. 2007). 5′ss selection in such a competition situation is determined both by individual 5′ss U1 snRNA complementarity and upstream and downstream splicing regulatory elements differentially acting in both directions (Erkelenz et al. 2013a). In clinically important examples, such cryptic 5′ss even possess noncanonical dinucleotides. Therefore, we designed a competition assay comparing noncanonical 5′ss usage with competing canonical 5′ splice sites of different U1 snRNA complementarities, bracketed between upstream and downstream SREs and spaced by a short 16-nt splicing neutral sequence (Zhang et al. 2009; Arias et al. 2015). Stepwise reduction of the competing canonical 5′ss U1 snRNA complementarity together with variation of the downstream TIA1 elements led to activation of several noncanonical 5′ss and permitted ranking these sites depending on the competition conditions under which their usage first became detectable.
However, this ranking could not simply be explained by, for example, 5′ss MaxEnt score (ME): In competition with the 5′ss CAG GTAAGT (ME 10.86), the 5′ss CTG GTAAGC with ME 8.69 was not used at all, whereas, for example, the noncanonical CAG GCAAGT (ME 3.10) and CAG TTAAGT (ME 2.35, or with a “bulged T”: CCA GTAAGT ME 9.09) were clearly detected. In the same way, the actually used splicing registers (i.e., −1, +1, +5) could not be explained, for example, by nucleotide bulging (Roca et al. 2012; Tan et al. 2016).
The noncanonical GC 5′ss was recognized even in the absence of any SRE, albeit weakly (Fig. 2B, lane 9). It was, however, overlapped by a shifted GT site at position +5/+6, which had the highest U1 snRNA complementarity (HBS 9.3) among all tested noncanonical 5′ss. In conjunction with the hypothesized impact of such a GT dinucleotide observed in Figure 3C, this second GT may first recruit U1 snRNP, which then shifts to the nearby noncanonical 5′ss. A similar constellation was observed in the human fibroblast growth factor receptor gene, where a noncanonical GA site is supported by an upstream GT site (Brackenridge et al. 2003). In a more systematic analysis of 5′ splice site selection by shifted base-pairing to U1 snRNA, an even phylogenetically conserved mechanism for a small subset of very weak 5′ss was revealed (Roca and Krainer 2009).
Recently, using filtered pre-mRNA binding assays, mismatches at the central 5′ss positions +1 and +2 were shown to strongly impair stable RNA duplex formation with U1 snRNA (Kondo et al. 2015). The only exception to this seems to be GC sites, which might be stabilized by an “on-a-par” C·A wobble base pair formed at position +2 (Kondo et al. 2015), possibly explaining the lower SRE dependency observed in this study.
Beyond U1 snRNA base-pairing, stabilization of U1 snRNP is also affected by both external, peripheral splicing enhancing proteins like SRSF7 or TIA1 and U1 snRNP-specific proteins as SNRPC and LUC7-like that are supposed to particularly aid in the selection of weak splice sites by stabilizing the RNA duplex (Plaschka et al. 2018).
TT sites compare favorably to all other tested noncanonical dinucleotides (except GC). Frequently, TT sites possess an extra GT at position −1 potentially providing stabilizing effects exerted by SNRPC (Rosel et al. 2011; Preußer et al. 2014) and LUC7-like. Indeed, in our RNA-seq data set, 37/41 high-confidence noncanonical non-GC 5′ss—in particular all TT sites—contained at least one GT dinucleotide.
Beyond external splicing regulatory proteins, multiple weak U1 snRNA binding sites could synergistically increase the local affinity for U1 snRNP binding and further assembly of the spliceosome. However, the U1 snRNP binding location is not necessarily identical to the cleavage position, which is determined by later interacting spliceosome components such as U6 and U5 snRNA.
Regarding 3′ss selection, it was recently shown by cryoelectron microscopy that non-Watson-Crick base-pairing interactions between the G at position +1 and the G at position −1 of the 3′ss are critical for 3′ splice site selection upon 5′ and 3′ exon ligation (Wilkinson et al. 2017). It is unclear, however, why a +1 T nucleotide exchange is better tolerated than noncanonical GA and GG sites, which are used at lower efficiencies.
In human pathogenic 5′ss mutations, the most frequently affected positions are +1/+2 containing the canonical GT dinucleotide, and TT and AT were found to disrupt splicing more frequently than GG (Krawczak et al. 2007). In contrast, we found higher 5′ss usage with TT and AT than with GG dinucleotides.
Covariation of competing proper 5′ss complementarity to U1 snRNA (“5′ss strength”) and its SRE environment also permitted us to quantify their relative impacts on 5′ss selection in some instances, finding four downstream TIA1 binding sites comparable to an increase in 5′ss complementarity from 10.4 to 12.2. However, expression levels of splicing regulatory proteins typically vary between different cell types and thus contribute to alternative splicing, rendering such a quantification cell type dependent.
Our experimentally determined noncanonical 5′ss ranking reflects splice site usage in a competition situation. To obtain comparative data from the human genome, one cannot rely on splice site annotation in, for example, Ensembl only. In fact, among 341,386 Ensembl annotated introns with a canonical (AG) 3′, TT (121), and CT (131), 5′ss are found at nearly half the abundance of AT (226), GG (222), and GA (215), very different than our experimental ranking. However, Ensembl annotation itself does not properly reflect splice site usage, which may account for this difference. In order to obtain human genome data on 5′ss usage, we therefore used our own comprehensive RNA-seq data from 54 human fibroblast samples (Kaisers et al. 2017a).
To extract reliable noncanonical 5′ss from these data, we used a two-tier method to exclude noisy false positive results: We restricted our analysis exclusively to gapped reads detected at least 10 times and also selected only introns with canonical AG 3′ss. This restriction excludes identification of minor spliceosome AT—AC exon junctions, and it may lead to an underestimation of the AT 5′ss abundance.
In order to adequately compare exon junction reads between genes of different expression levels, we normalized all gapped reads by the largest gapped read in the respective gene, thus obtaining gene-normalized reads (GNR). Based on the GNR histogram, we then excluded exon junctions with GNR <2% from further analysis as noise candidates. Although these least-used exon junctions amounted to 29% of all exon junctions, they were weakly used by definition and represented only 0.28% of all reads. Along similar lines, Parada et al. (2014) denoted splice sites as high-confidence sites that had at least 5% usage compared to the most abundant splice variant. They also retained only introns that were found in at least two independent sources. We did not ab initio require exon junctions to occur in a minimum number of independent samples, but exon junctions with noncanonical dinucleotides except CT were detected in an average number of more than 30 of 54 samples. Notwithstanding that extracting high-confidence sites from large RNA-seq data sets requires some noise removal procedures, general results should be only weakly dependent on specific imposed restrictions. Here, noncanonical exon junctions were five times overrepresented among noise candidates compared to all exon junctions with GNR ≥2%. Similar to Parada et al. (2014), exclusion of noise candidates reduced the number of noncanonical sites to about one-third.
Although canonical GT 5′ss were detected across all genes independent of their expression level, rare noncanonical 5′ss were only detected in highly expressed genes. In fact, average gene expression level revealed the same ranking for noncanonical exon junctions as the splicing competition assay.
In an attempt to mimic the situation of our splicing competition assay, we finally selected all clusters of 5′ss less than 12 nt apart. These 5′ss were thought to be competing with each other without additional splicing regulatory proteins binding in between. Such clusters typically consisted of two 5′ss of different usage: a primary and a secondary 5′ss. Splitting these 5′ss into disjoint groups with specific dinucleotides revealed that only TT, GC, and GT sites occurred as more frequently used 5′ss in a cluster. In contrast, AT, GA, and GG sites occurred only as less frequently used 5′ss. This result suggests a picture where these noncanonical sites may have been used by failure to splice the nearby highly used GT site, a kind of erroneous splicing. On the other hand, TT sites occurred only as the primary, more used 5′ss in a cluster, which cannot be explained by a shifted splicing register.
Methods
Oligonucleotides
Oligonucleotides were obtained from Metabion GmbH.
Primers used for cloning are listed in Supplemental Table S1, and primers used for semiquantitative RT-PCR and RNA affinity chromatography assay are listed in Supplemental Table S2.
Plasmids
All SV-env/eGFP plasmids were cloned by substitution of the SacI/NdeI fragment with PCR products using appropriate forward primers (Supplemental Table S1) and primer #640 as a reverse primer. For insertion of TIA1 high-affinity binding sites into the downstream intron, the AflII/NdeI fragment of the SV-env/eGFP plasmids was replaced by a respective fragment from SV SRSF7(4x) L3 TIA1(4x) env/eGFP (Erkelenz et al. 2013a).
LTR ex2 ex3 FANCC plasmids were generated by cloning Bsu36I/SphI-digested PCR products amplified with a respective forward primer (Supplemental Table S1) and #4641 as a reverse primer using LTR ex2 ex3 FANCC as a template (Hartmann et al. 2010). PCR products were cloned into a LTR ex2 ex3 FANCC (StuI, BsrGI) preclone that was generated by substitution of the StuI/BsrGI fragment with the respective StuI/BsrGI fragment from pNL4-3 (GenBank Accession No. M19921). U1 expression plasmids were cloned as described previously (Erkelenz et al. 2013b).
Designer exon splicing reporters were generated on the basis of an FGB minigene containing only neutral sequences as described before (Brillen et al. 2017). Two SRSF7 binding sites were introduced by substituting the Bsu36I/NotI fragment with the PCR product using the primer pair #4705/#2620. The noncanonical SD site (AA CGTACG CAG ttaagtgt) was cloned by digestion with BsiWI/Bpu10I using primer pairs #5123/#2620. The upstream GT of the BsiWI site was changed to CT (AA CCTACG CAG ttaagtgt) by XhoI/AflIII digestion and substitution of the fragments with PCR products using primer pair #5247/#2620, respectively. These BsiWI −8/−7 GT and BsiWI −8/−7 GT > CT plasmids were used as templates for mutating upstream CANC motifs. The first motif was removed by digestion with XhoI/AflIII using the primer pairs #6025/#2649 (GT −8/−7) or #6027/#2649 (CT −8/−7). The plasmids with mutations of the first and the second CANC motif were cloned by digestion with XhoI/AflIII using the primer pairs #6026/#2649 (GT −8/−7) or #6028/#2649 (CT −8/−7), respectively.
Cell culture and nucleic acid transfections
HEK293T and HeLa cells were maintained in DMEM (Invitrogen) with 10% fetal calf serum (FCS) and 1% penicillin and streptomycin (P/S) (Invitrogen). Plasmid transfections were performed in six-well plates with 2.5 × 105 HEK293T or HeLa cells using TransIT-LT1 reagent (Mirus Bio LLC) in accordance with the manufacturer's instructions.
RNA extraction and semiquantitative RT-PCR
Total RNA samples were collected 30 h post transfection. For RT-PCR analyses, RNA was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen) and Oligo(dT) primer (Invitrogen). For semiquantitative analyses of spliced eGFP mRNAs, cDNA was used in a PCR reaction with primer #3210 and #3211. Splicing of LTR ex2 ex3 FANCC derived reporter mRNAs was analyzed using primer pair #1544/#2851, and designer exon reporters used primer pair #2648/#2649. To control for equal transfection efficiencies, a separate PCR reaction was carried out with primer pair #1224/#1225 detecting coexpressed GH1 mRNA. All primer sequences used for semiquantitative RT-PCR analyses are listed in Supplemental Table S2A. PCR products were separated on 8%–10% nondenaturing polyacrylamide gels and stained with ethidium bromide for visualization.
Reamplification of PAA-separated PCR products
The PAA gel fragments were excised with a clean, sharp scalpel and incubated for 30 min at 50°C in 100 µL diffusion buffer (0.5M ammonium acetate, 10 mM magnesium acetate, 1 mM EDTA, 0.1% SDS). The supernatant was transferred to a new tube and DNA was isolated using the QIAGEN Gel extraction kit according to the manufacturer's protocol and eluted in 30 µL dH2O. The eluted PCR product was reamplified by PCR using the same primer pairs as in the initial PCR, analyzed on a 1% agarose gel, again excised with a clean, sharp scalpel and purified using the QIAGEN Gel extraction kit, eluted in 30 µL dH2O and sent to sequencing.
QIAxcel
For higher resolution 10 µL of the semiquantitative PCR products were separated on a QIAxcel DNA screening gel cartridge, using the QX Size Marker 250 bp – 4 kb (Qiagen).
Covalent coupling of in vitro transcribed RNAs to agarose beads, and RNA affinity chromatography (RAC) assay
To synthesize templates for in vitro transcription, a sense T7 primer (#4825) was annealed to antisense DNA oligonucleotides either containing the canonical GT (#5986) or the noncanonical CT (#5987) splice site sequence. Both oligos also included a T7 polymerase binding site as well as an RNA binding site for the recombinant bacteriophage MS2 coat protein. RNA was synthesized using the RiboMax large-scale RNA production system (Promega) according to the manufacturer's instructions.
Subsequently, synthesized RNA oligonucleotides (3000 pmol) were covalently coupled to adipic acid dihydrazide-agarose beads (Sigma) and were then incubated with 60% HeLa nuclear extract (Cilbiotech) containing recombinant bacteriophage MS2 coat protein to monitor equal precipitation efficiencies. After five stringent washing steps with buffer D containing different concentrations of KCl (0.1, 0.25, 0.5, 0.25, and 0.1 M KCl, together with 20 mM HEPES-KOH [pH 7.9], 5% [vol/vol] glycerol, 0.2 M EDTA, 0.5 mM dithiothreitol, 0.4 M MgCl2), precipitated proteins were eluted in protein sample buffer, heated up to 95°C for 10 min, and loaded onto a 15% SDS-PAGE. Next, proteins were transferred to a nitrocellulose membrane, and the membrane was probed with primary antibodies detecting SRSF3 (abcam, ab198291), SNRPC (anti-U1-C, Sigma, SAB4200188), or bacteriophage MS2 coat protein (Tetracore, TC-7004-002). After incubating the membrane with respective secondary antibodies—anti-Rabbit IgG (H + L) Superclonal, HRP (#A27036, Thermo Fisher), anti-Rat IgG (whole molecule) Peroxidase antibody produced in goat (A9037, Sigma-Aldrich)—the membrane was developed using ECL chemiluminescence reagent (GE Healthcare).
HBond score
The HBond score (HBS) is a dimensionless measure of the overall hydrogen bond pattern binding strength in the 11-nt-long duplex between the 5′ss and the 5′ end of U1 snRNA. For more details see Supplemental Methods. A lookup table with an alphabetically sorted two-column list of all 11-nt sequences and corresponding HBond scores is provided as Supplemental Table S3.
RNA-seq data set
We examined noncanonical 5′ splice site usage in our large human RNA-seq transcriptome data set of 54 human fibroblast samples taken from 27 subjects (14 male; 18 samples in each of the three age groups 18–25, 35–49, 60–67). The raw sequencing data have been deposited during the course of a previous study (Kaisers et al. 2017a) to ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-4652.
For alignment and all subsequent analysis, Human genomic sequence (GRCh38) and annotation data (release 82) were downloaded from Ensembl (Cunningham et al. 2015) and BioMart (Durinck et al. 2005; Guberman et al. 2011). cDNA libraries were synthesized using TruSeq RNA SamplePrep kit (Illumina) according to the manufacturer's protocol. One microgram of total RNA was used for poly(A) RNA enrichment. The samples were amplified on nine Illumina flow cells (v1.5) and sequenced on an Illumina HiSeq 2000 sequencer using TruSeq SBS kits v1. From each lane, the resulting 101-nt sequence reads were converted to FASTQ by CASAVA (1.8.2). Subsequent alignments were calculated on unprocessed FASTQ files. Alignments were calculated using STAR (2.4.1d modified) (Dobin et al. 2013).
Subsequent calculation of splice site localization from gapped (exon junction) reads was done with CRAN package rbamtools (Kaisers et al. 2015). Identified splice sites then were processed using Bioconductor package spliceSites (R package version 1.8.3, https://bioconductor.org/packages/3.2/bioc/html/spliceSites.html) in order to compare the number of exon junction reads for every exon junction and for every biological sample.
To obtain more reliable U2-type 5′ss, we only used introns with canonical AG 3′ss that were annotated in the human reference genome GRCh38/hg38 (Ensembl version 82), delimited by exon junctions covered by at least 10 gapped reads, and which could unambiguously be assigned to either the plus or minus strand.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) under Grant No. SCHA 909/4-1, the German Ministry of Research and Education (BMBF) within the Network Gerontosys consortium on Stromal Aging WP3, Part C (to H.S.), and Jürgen Manchot Stiftung (to L.W., L.M., A.-L.B., and H.S.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.235861.118.
References
- Aebi M, Hornig H, Padgett RA, Reiser J, Weissmann C. 1986. Sequence requirements for splicing of higher eukaryotic nuclear pre-mRNA. Cell 47: 555–565. 10.1016/0092-8674(86)90620-3 [DOI] [PubMed] [Google Scholar]
- Aebi M, Hornig H, Weissmann C. 1987. 5′ cleavage site in eukaryotic pre-mRNA splicing is determined by the overall 5′ splice region, not by the conserved 5′ GU. Cell 50: 237–246. 10.1016/0092-8674(87)90219-4 [DOI] [PubMed] [Google Scholar]
- Alioto TS. 2007. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res 35: D110–D115. 10.1093/nar/gkl796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias MA, Lubkin A, Chasin LA. 2015. Splicing of designer exons informs a biophysical model for exon definition. RNA 21: 213–229. 10.1261/rna.048009.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabino SM, Blencowe BJ, Ryder U, Sproat BS, Lamond AI. 1990. Targeted snRNP depletion reveals an additional role for mammalian U1 snRNP in spliceosome assembly. Cell 63: 293–302. 10.1016/0092-8674(90)90162-8 [DOI] [PubMed] [Google Scholar]
- Brackenridge S, Wilkie AO, Screaton GR. 2003. Efficient use of a ‘dead-end’ GA 5′ splice site in the human fibroblast growth factor receptor genes. EMBO J 22: 1620–1631. 10.1093/emboj/cdg163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillen AL, Walotka L, Hillebrand F, Müller L, Widera M, Theiss S, Schaal H. 2017. Analysis of competing HIV-1 splice donor sites uncovers a tight cluster of splicing regulatory elements within exon 2/2b. J Virol 91: e00389-17 10.1128/JVI.00389-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins L, Penny D. 2005. Complex spliceosomal organization ancestral to extant eukaryotes. Mol Biol Evol 22: 1053–1066. 10.1093/molbev/msi091 [DOI] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S et al. 2015. Ensembl 2015. Nucleic Acids Res 43: D662–D669. 10.1093/nar/gku1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Fox-Walsh KL, Baldi PF, Hertel KJ. 2006. Genomic splice-site analysis reveals frequent alternative splicing close to the dominant splice site. RNA 12: 2047–2056. 10.1261/rna.151106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. 2005. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21: 3439–3440. 10.1093/bioinformatics/bti525 [DOI] [PubMed] [Google Scholar]
- Eperon LP, Estibeiro JP, Eperon IC. 1986. The role of nucleotide sequences in splice site selection in eukaryotic pre-messenger RNA. Nature 324: 280–282. 10.1038/324280a0 [DOI] [PubMed] [Google Scholar]
- Erkelenz S, Mueller WF, Evans MS, Busch A, Schoneweis K, Hertel KJ, Schaal H. 2013a. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 19: 96–102. 10.1261/rna.037044.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkelenz S, Poschmann G, Theiss S, Stefanski A, Hillebrand F, Otte M, Stühler K, Schaal H. 2013b. Tra2-mediated recognition of HIV-1 5′ splice site D3 as a key factor in the processing of vpr mRNA. J Virol 87: 2721–2734. 10.1128/JVI.02756-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez Alanis E, Pinotti M, Dal Mas A, Balestra D, Cavallari N, Rogalska ME, Bernardi F, Pagani F. 2012. An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet 21: 2389–2398. 10.1093/hmg/dds045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund M, Asang C, Kammler S, Konermann C, Krummheuer J, Hipp M, Meyer I, Gierling W, Theiss S, Preuss T et al. 2003. A novel approach to describe a U1 snRNA binding site. Nucleic Acids Res 31: 6963–6975. 10.1093/nar/gkg901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund M, Hicks MJ, Konermann C, Otte M, Hertel KJ, Schaal H. 2005. Extended base pair complementarity between U1 snRNA and the 5′ splice site does not inhibit splicing in higher eukaryotes, but rather increases 5′ splice site recognition. Nucleic Acids Res 33: 5112–5119. 10.1093/nar/gki824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frilander MJ, Steitz JA. 1999. Initial recognition of U12-dependent introns requires both U11/5′ splice-site and U12/branchpoint interactions. Genes Dev 13: 851–863. 10.1101/gad.13.7.851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Mayeda A, Maniatis T, Krainer AR. 1992. General splicing factors SF2 and SC35 have equivalent activities in vitro, and both affect alternative 5′ and 3′ splice site selection. Proc Natl Acad Sci 89: 11224–11228. 10.1073/pnas.89.23.11224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guberman JM, Ai J, Arnaiz O, Baran J, Blake A, Baldock R, Chelala C, Croft D, Cros A, Cutts RJ et al. 2011. BioMart Central Portal: an open database network for the biological community. Database 2011: bar041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SL, Padgett RA. 1994. Conserved sequences in a class of rare eukaryotic nuclear introns with non-consensus splice sites. J Mol Biol 239: 357–365. 10.1006/jmbi.1994.1377 [DOI] [PubMed] [Google Scholar]
- Hall SL, Padgett RA. 1996. Requirement of U12 snRNA for in vivo splicing of a minor class of eukaryotic nuclear pre-mRNA introns. Science 271: 1716–1718. 10.1126/science.271.5256.1716 [DOI] [PubMed] [Google Scholar]
- Hargous Y, Hautbergue GM, Tintaru AM, Skrisovska L, Golovanov AP, Stevenin J, Lian LY, Wilson SA, Allain FH. 2006. Molecular basis of RNA recognition and TAP binding by the SR proteins SRp20 and 9G8. EMBO J 25: 5126–5137. 10.1038/sj.emboj.7601385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann L, Theiss S, Niederacher D, Schaal H. 2008. Diagnostics of pathogenic splicing mutations: Does bioinformatics cover all bases? Front Biosci 13: 3252–3272. 10.2741/2924 [DOI] [PubMed] [Google Scholar]
- Hartmann L, Neveling K, Borkens S, Schneider H, Freund M, Grassman E, Theiss S, Wawer A, Burdach S, Auerbach AD et al. 2010. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am J Hum Genet 87: 480–493. 10.1016/j.ajhg.2010.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson MJ, Hudson AJ, Cherny D, Eperon IC. 2012. The transition in spliceosome assembly from complex E to complex A purges surplus U1 snRNPs from alternative splice sites. Nucleic Acids Res 40: 6850–6862. 10.1093/nar/gks322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisers W, Schaal H, Schwender H. 2015. rbamtools: an R interface to samtools enabling fast accumulative tabulation of splicing events over multiple RNA-seq samples. Bioinformatics 31: 1663–1664. 10.1093/bioinformatics/btu846 [DOI] [PubMed] [Google Scholar]
- Kaisers W, Boukamp P, Stark HJ, Schwender H, Tigges J, Krutmann J, Schaal H. 2017a. Age, gender and UV-exposition related effects on gene expression in in vivo aged short term cultivated human dermal fibroblasts. PLoS One 12: e0175657 10.1371/journal.pone.0175657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisers W, Ptok J, Schwender H, Schaal H. 2017b. Validation of splicing events in transcriptome sequencing data. Int J Mol Sci 18: E1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammler S, Leurs C, Freund M, Krummheuer J, Seidel K, Tange TO, Lund MK, Kjems J, Scheid A, Schaal H. 2001. The sequence complementarity between HIV-1 5′ splice site SD4 and U1 snRNA determines the steady-state level of an unstable env pre-mRNA. RNA 7: 421–434. 10.1017/S1355838201001212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Oubridge C, van Roon AM, Nagai K. 2015. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. eLife 4 10.7554/eLife.04986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. 2007. Single base-pair substitutions in exon–intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat 28: 150–158. 10.1002/humu.20400 [DOI] [PubMed] [Google Scholar]
- Lavigueur A, La Branche H, Kornblihtt AR, Chabot B. 1993. A splicing enhancer in the human fibronectin alternate ED1 exon interacts with SR proteins and stimulates U2 snRNP binding. Genes Dev 7: 2405–2417. 10.1101/gad.7.12a.2405 [DOI] [PubMed] [Google Scholar]
- Lerner MR, Boyle JA, Mount SM, Wolin SL, Steitz JA. 1980. Are snRNPs involved in splicing? Nature 283: 220–224. 10.1038/283220a0 [DOI] [PubMed] [Google Scholar]
- Martinez-Pizarro A, Dembic M, Pérez B, Andresen BS, Desviat LR. 2018. Intronic PAH gene mutations cause a splicing defect by a novel mechanism involving U1snRNP binding downstream of the 5′ splice site. PLoS Genet 14: e1007360 10.1371/journal.pgen.1007360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A, Krainer AR. 1992. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell 68: 365–375. 10.1016/0092-8674(92)90477-T [DOI] [PubMed] [Google Scholar]
- Michaud S, Reed R. 1993. A functional association between the 5′ and 3′ splice site is established in the earliest prespliceosome complex (E) in mammals. Genes Dev 7: 1008–1020. 10.1101/gad.7.6.1008 [DOI] [PubMed] [Google Scholar]
- Nelson KK, Green MR. 1990. Mechanism for cryptic splice site activation during pre-mRNA splicing. Proc Natl Acad Sci 87: 6253–6257. 10.1073/pnas.87.16.6253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada GE, Munita R, Cerda CA, Gysling K. 2014. A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res 42: 10564–10578. 10.1093/nar/gku744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AA, Steitz JA. 2003. Splicing double: insights from the second spliceosome. Nat Rev Mol Cell Biol 4: 960–970. 10.1038/nrm1259 [DOI] [PubMed] [Google Scholar]
- Plaschka C, Lin PC, Charenton C, Nagai K. 2018. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature 559: 419–422. 10.1038/s41586-018-0323-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preußer C, Rossbach O, Hung LH, Li D, Bindereif A. 2014. Genome-wide RNA-binding analysis of the trypanosome U1 snRNP proteins U1C and U1-70K reveals cis/trans-spliceosomal network. Nucleic Acids Res 42: 6603–6615. 10.1093/nar/gku286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca X, Krainer AR. 2009. Recognition of atypical 5′ splice sites by shifted base-pairing to U1 snRNA. Nat Struct Mol Biol 16: 176–182. 10.1038/nsmb.1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca X, Akerman M, Gaus H, Berdeja A, Bennett CF, Krainer AR. 2012. Widespread recognition of 5′ splice sites by noncanonical base-pairing to U1 snRNA involving bulged nucleotides. Genes Dev 26: 1098–1109. 10.1101/gad.190173.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Wall R. 1980. A mechanism for RNA splicing. Proc Natl Acad Sci 77: 1877–1879. 10.1073/pnas.77.4.1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosel TD, Hung LH, Medenbach J, Donde K, Starke S, Benes V, Ratsch G, Bindereif A. 2011. RNA-Seq analysis in mutant zebrafish reveals role of U1C protein in alternative splicing regulation. EMBO J 30: 1965–1976. 10.1038/emboj.2011.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. 2006. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res 34: 3955–3967. 10.1093/nar/gkl556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano PG, Guthrie C. 1988. 5′ splice site selection in yeast: genetic alterations in base-pairing with U1 reveal additional requirements. Genes Dev 2: 1258–1267. 10.1101/gad.2.10.1258 [DOI] [PubMed] [Google Scholar]
- Spena S, Tenchini ML, Buratti E. 2006. Cryptic splice site usage in exon 7 of the human fibrinogen B β-chain gene is regulated by a naturally silent SF2/ASF binding site within this exon. RNA 12: 948–958. 10.1261/rna.2269306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Ho JX, Zhong Z, Luo S, Chen G, Roca X. 2016. Noncanonical registers and base pairs in human 5′ splice-site selection. Nucleic Acids Res 44: 3908–3921. 10.1093/nar/gkw163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. 1995. Modulation of 5′ splice site choice in pre-messenger RNA by two distinct steps. Proc Natl Acad Sci 92: 2504–2508. 10.1073/pnas.92.7.2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen JJ, Niemelä EH, Verma B, Frilander MJ. 2013. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA 4: 61–76. 10.1002/wrna.1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Burns HD, Oldridge M, Heath JK, Wilkie AO. 1998. Conserved use of a non-canonical 5′ splice site (/GA) in alternative splicing by fibroblast growth factor receptors 1, 2 and 3. Hum Mol Genet 7: 685–691. 10.1093/hmg/7.4.685 [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He XP, Mieczkowski P, Grimm SA, Perou CM et al. 2010. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38: e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watakabe A, Tanaka K, Shimura Y. 1993. The role of exon sequences in splice site selection. Genes Dev 7: 407–418. 10.1101/gad.7.3.407 [DOI] [PubMed] [Google Scholar]
- Wilkinson ME, Fica SM, Galej WP, Norman CM, Newman AJ, Nagai K. 2017. Postcatalytic spliceosome structure reveals mechanism of 3′-splice site selection. Science 358: 1283–1288. 10.1126/science.aar3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Krainer AR. 1997. Splicing of a divergent subclass of AT-AC introns requires the major spliceosomal snRNAs. RNA 3: 586–601. [PMC free article] [PubMed] [Google Scholar]
- Xu R, Teng J, Cooper TA. 1993. The cardiac troponin T alternative exon contains a novel purine-rich positive splicing element. Mol Cell Biol 13: 3660–3674. 10.1128/MCB.13.6.3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Arias MA, Ke S, Chasin LA. 2009. Splicing of designer exons reveals unexpected complexity in pre-mRNA splicing. RNA 15: 367–376. 10.1261/rna.1498509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Weiner AM. 1986. A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell 46: 827–835. 10.1016/0092-8674(86)90064-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.