

Abstract
Targeted mutation assessment of 81 genes in 1,021 adults with de novo acute myeloid leukemia (AML) identified recurrent mutations in the neurofibromin 1 (NF1) gene in 52 (5.1%) patients, including 36 (5.2%) younger and 16 (4.8%) older patients, which suggests NF1 belongs to the 20 most frequently mutated genes in adult AML. NF1 mutations were found throughout the gene, and comprised missense, frame-shift and nonsense mutations. One mutation hotspot, at amino acid threonine 676 (Thr676), was found in 27% of AML patients with NF1 mutations. NF1-mutated patients belonged more often to the adverse European LeukemiaNet (ELN) risk category than NF1 wild-type patients. Among patients aged <60 years, the presence of NF1 Thr676 mutations was associated with lower complete remission (CR) rates (P=0.04) and shorter overall survival (OS; P=0.01), as was the presence of any NF1 mutation in patients in the adverse ELN risk category (CR, P=0.05; OS, P<0.001). CR rates were also lower in NF1-mutated patients aged ≥60 years compared with NF1 wild-type patients (P=0.001). In summary, our findings provide novel insights into the frequency of NF1 mutations in AML, and are suggestive of an adverse prognostic impact in patients treated with standard chemotherapy.
INTRODUCTION
The neurofibromin 1 gene (NF1), located at 17q11.2, encodes the RAS GTPase activating protein neurofibromin.1,2 Mutations in NF1 affect the RAS-MAPK-signaling pathway,1,2 and the neurofibromin/RAS-GTPase connection has a crucial role in controlling cell growth and proliferation.3 Germline loss-of-function mutations in NF1 are the molecular cause for neurofibromatosis type 1 (also known as von Recklinghausen disease),4 an autosomal-dominant inherited disorder belonging to the so called “cancer predisposition syndromes.”4,5 Neurofibromatosis type 1 is phenotypically characterized by the presence of café au lait spots, Lisch nodules in the eye, and a highly increased incidence of benign and malignant tumors, including fibromatous skin tumors. Over 1,000 pathogenic variants of NF1 are listed in the Human Gene Mutation Database, and include nonsense mutations, amino acid substitutions (missense mutations) and most commonly sizeable truncations of NF1 due to frameshift mutations. The observation that children carrying germline NF1 mutations have a high likelihood to develop juvenile myelomonocytic leukemia (JMML),5,6 and that the JMML might in some cases actually be the presenting phenotype (without the presence of the aforementioned skin changes)7 highlighted the importance of NF1 and hyperactive RAS signaling in normal myeloid cell growth and also leukemogenesis in murine models.3,8
In addition to the aforementioned germline mutations, somatic mutations of NF1 are commonly found in human cancers.9 Furthermore, transcriptional inactivation of NF1 and small or large rearrangements have been identified as an important mechanism to disturb NF1’s function.6–8,10 While the latter mechanisms have found considerable interest in the acute myeloid leukemia (AML) research community, the effect of somatic gene mutations has largely been neglected. This might be in part due to several early studies,10–12 which suggested that NF1 mutations were very rare in AML. Although NF1 mutations were detected by the recent large sequencing efforts,13,14 the clinical correlates and possible prognostic and/or therapeutic implications of somatic NF1 mutations have, to our knowledge, not yet been assessed.
Using targeted next-generation sequencing (NGS) in a relatively large cohort of 1,021 adults with de novo AML, we detected recurrent mutations in NF1 in 5% of the patients. Among NF1 mutations, a mutation hotspot within the NF1 gene, located at amino acid threonine 676 (Thr676), was found in 27% of AML patients with NF1 mutations. Our data suggest that the presence of NF1 mutations is associated with poor outcome in younger AML patients with adverse genetic features and that NF1 Thr676 mutations may confer adverse prognosis.
METHODS
Patients, treatment, and cytogenetic studies
Pretreatment bone marrow (BM) or blood samples suitable for next-generation sequencing were obtained from 1,021 adults diagnosed with AML who were treated similarly on Cancer and Leukemia Group B (CALGB)/Alliance for Clinical Trials in Oncology (Alliance) trials,15–26 the details of which are provided in the Supplementary Information. Cytogenetic analyses of pretreatment BM and/or blood samples were performed by institutional, CALGB/Alliance-approved laboratories, and the results confirmed by central karyotype review.27 Patients provided written informed consent to participate in protocols CALGB 8461 (cytogenetic studies), CALGB 9665 (leukemia tissue bank) and CALGB 20202 (molecular studies), which involved collection of pretreatment BM and blood samples. Treatment protocols were in accordance with the Declaration of Helsinki and approved by the institutional review boards at each center, and all patients provided written informed consent.
Statistical analyses
Baseline characteristics were compared between patients with NF1 mutations and those with wild-type NF1 using Fisher’s exact test for categorical variables and the Wilcoxon rank-sum test for continuous variables. We used the log-rank test to test survival variables, with Kaplan Meier curves for illustration. A P-value of <0.05 was considered statistically significant. Definitions of the clinical endpoints─complete remission (CR), disease-free survival (DFS) and overall survival (OS)─are provided in the Supplementary Information. The dataset was frozen on February 13, 2017. Data collection and statistical analyses were performed by the Alliance Statistics and Data Center using SAS 9.4.
Molecular analyses
Mononuclear cells were enriched through Ficoll-Hypaque gradient centrifugation and cryopreserved until use. The mutational status of 80 protein-coding genes was determined centrally at The Ohio State University by targeted amplicon sequencing using two different gene panels on the MiSeq platform (Illumina, San Diego, CA; see Supplementary Information for details).28 The MuCor program29 was used for initial data analysis. Details about the variant calling are outlined in the Supplementary Information. In addition to the 80 genes assessed by next-generation sequencing, testing for CEBPA mutations, was performed as previously described,30 thus adding up to 81 total genes assessed in our study. Only patients with biallelic CEBPA mutations were considered as mutated.31 Gene mutations were assigned to functional groups similar to those previously described by the Cancer Genome Atlas Research Network11 as follows: chromatin remodeling (ASXL1, BCOR, BCORL1, EZH2 and SMARCA2), cohesin complex (RAD21, SMC1A, SMC3 and STAG2), kinases (AXL, FLT3-ITD, FLT3-TKD, KIT and TYK2), methylation-related (DNMT3A, IDH1/2, and TET2), NPM1 (NPM1), RAS pathway (CBL, KRAS, NRAS and PTPN11), spliceosome (SF3B1, SRSF2, U2AF1 and ZRSR2), transcription factors (CEBPA, ETV6, GATA2, IKZF1, NOTCH1 and RUNX1) and tumor suppressors (PHF6, TP53 and WT1).
Validation of the Thr676 mutation by Sanger sequencing, both of the primary samples and after Topo-TA cloning, was done using the following primers: NF1_676F, 5′ TTCCACCCTTGACTCTCAGG 3′; NF1_676R, 5′ TTGCTGACAGAGGCAAACTC 3′. To further exclude technical artifacts, blinded analysis of five normal samples were included in the sequencing as negative controls.
The presence of copy number variations (CNVs) was determined by genotyping patient samples with sufficient available material using Illumina Omni-Express SNP arrays, followed by analysis with Illumina GenomeStudio plugin cnvpartition v3.2.0. Copy-neutral loss of heterozygosity (CN-LOH) was set to 1Mb.
RESULTS
Frequency of NF1 mutations in AML patients
We detected 59 NF1 mutations in 52 patients among 1,021 AML patients examined, for an overall frequency of 5.1% (5.2% in younger and 4.8% in older patients), which is higher than that found in previous reports.10–12 Two, three and five different NF1 mutations were found in one patient each. The NF1 mutations were found throughout the gene, and comprised eight nonsense, 21 frame-shift and 29 missense mutations, and one in-frame insertion/deletion (which we combined with the missense mutations for analysis; Supplementary Table S1 and Figure 1a). Three of the mutations were found at locations that were previously described for germline neurofibromatosis patients (Supplementary Figure S1). We observed a relatively high incidence of the mutation hotspot at amino acid threonine 676 (p.Thr676fs*24 [c.2026dupC], Thr676), in which the insertion of an additional cytosine within a mononucleotide run of 7 cytosines by polymerase strand-slippage causes a frame-shift mutation (Figure 1b).
Figure 1.
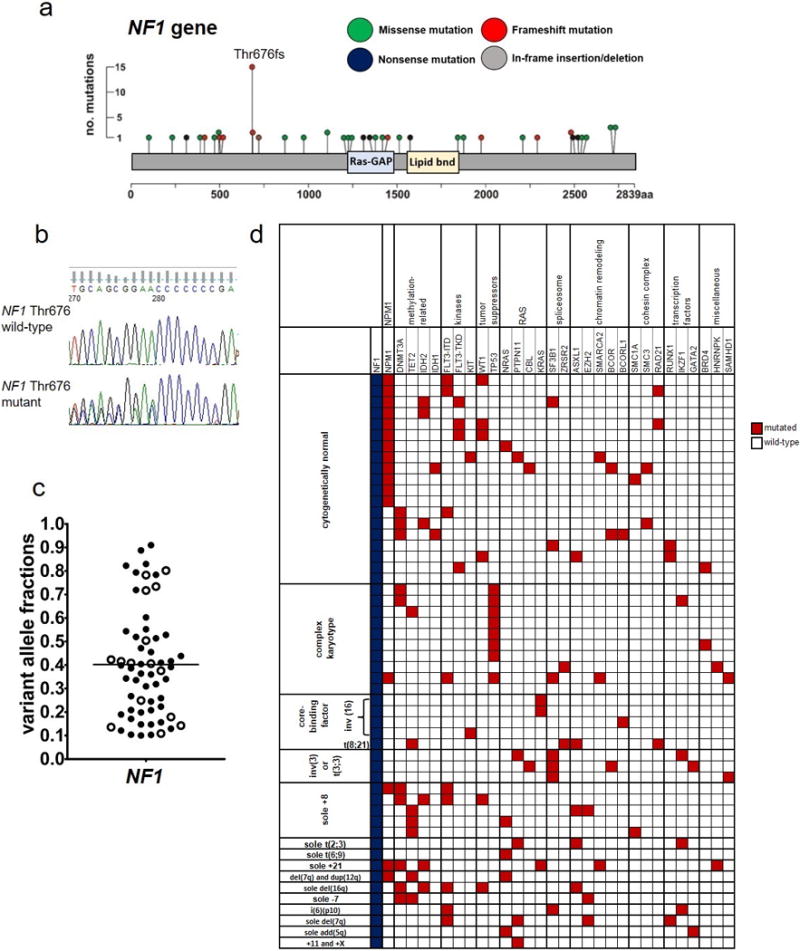
Mutations in the NF1 gene found in 1,021 patients with de novo acute myeloid leukemia. (a) Lollipop plots depicting 59 NF1 mutations detected in 52 patients. No. denotes number; aa, amino acid. (b) Sanger sequencing traces depicting examples of NF1 amino acid Threonine (Thr) 676 wild-type (top) and mutated (bottom) AML cases. (c) Beehive plot depicting variant allele fractions of the detected NF1 mutations. (d) Oncoprint of co-occurring mutations found in at least two AML patients with NF1 mutations (blue color), and of genes classified into the previously described functional groups.11 Each column represents an individual patient. Red color indicates that a gene was found to be mutated in the patient, white indicates wild-type status of the gene. The patients are grouped according to their pretreatment cytogenetic findings. The following genes also found to be mutated are not depicted in the oncoprint because they were detected in single patients (i.e., with a frequency below 2%): AXL and TYK2 (kinases); PHF6 (tumor suppressors); SRSF2 (splicesome); SF1 and SF3A3 (chromatin remodeling); STAG2 (cohesin complex); and CEBPA, ETV6 and NOTCH1 (transcription factors).
The mutations of all NF1 Thr676-mutated samples of which remaining DNA was available (9/14) were validated by Sanger sequencing, with excellent correspondence of observed variant allele fractions (VAFs) in the NGS analyses and peak height in Sanger sequencing (Supplementary Figure S2). Since the premature termination codon resulting from the frameshift mutation rests at least 50bp upstream from an exon-exon junction, the mutant transcript should be subjected to nonsense mediated decay.
The VAFs of NF1 mutations were observed at a median VAF of 0.39, suggesting that these mutations act as driver mutations at least in some leukemic clones (Figure 1c), which is consistent with the previous finding that mutations in NF1 may be present in hematopoietic stem cells.12 Eleven mutations were found at VAFs ≥0.70, possibly indicating loss of heterozygosity.
To determine if this was the case, we tested patients that harbored NF1 mutations with a VAF ≥0.70, as well as those whose VAF was between 0.50 and 0.69, for the presence of copy number variations (CNVs). Seven of 11 patients with NF1 mutations with a VAF ≥0.70 and 5 of 6 patients with NF1 mutations with a VAF 0.50-0.69 had material available for analysis. Indeed, we found evidence of copy number variations in all patients with NF1 mutations with a VAF ≥0.70: 6 patients showed clear deletions across the NF1 gene, and one had a copy-neutral loss of heterozygosity of nearly all of the long arm of chromosome 17 (17q), including NF1 locus. This confirms the notion that VAF ≥0.70 is a good indicator of the presence of copy number variations. In contrast, among patients with NF1 mutations with a VAF 0.50-0.69, only one patient harboring NF1 mutation with VAF of 0.50 had a deletion encompassing the NF1 locus (Supplementary Figure S3).
Finally, we attempted to assess the relative importance of NF1 mutations by comparing their VAFs with the VAFs of other co-occurring mutations in the additional 79 genes that were sequenced on our panel for each patient. Since copy number variants (CNVs) can confound using VAFs for clonality estimates, we limited this analysis to NF1-mutated cases with VAFs <0.60, and excluded NF1-mutated cases that we determined have CNVs in NF1 (Supplementary Figure S3). To account for CNVs in other mutated genes, we excluded mutations with VAFs >0.60 from consideration. Moreover, we also excluded from this comparison those patients who harbored recurrent balanced chromosome abnormalities leading to gene fusions, which are clearly primary, disease defining, used in WHO-classification, genetic rearrangements in these patients [i.e., 4 patients with inv(16)(p13.1q22) (no. 3, 23, 24 and 52 in Supplementary Table S1); 1 patient with t(8;21)(q22;q22) (no. 34); 3 with inv(3)(q21q26.2) (no. 20, 41 and 44) and 1 patient with t(6;9)(p23;q34) (no. 50)]. Among the 33 NF1-mutated patients subject to this analysis, the NF1 mutation was found in the major clone in 45% of patients (n=15; as defined by NF1 being the mutation with either the largest VAF, or being within 0.05 of the largest VAF gene mutation). In the remaining 55% of NF1-mutated patients (n=18), acquisition of the NF1 mutation was likely a later mutational event, indicated by a lower VAF (Supplementary Table 2).
Clinical, cytogenetic and molecular genetic characteristics of NF1-mutated AML patients
NF1-mutated patients had a lower percentage of bone marrow (BM) blasts compared with NF1 wild-type patients (P<0.001), and belonged more often to the adverse risk category (P=0.02) in the 2017 European LeukemiaNet (ELN) risk stratification classification.31 With respect to pretreatment cytogenetic findings in NF1-mutated patients, normal karyotypes were most common [found in 19 (37%) patients], followed by complex karyotypes [10 (19%) patients], sole +8 in at least one clone [5 (10%) patients], inv(16)(p13.1q22) [4 (8%) patients] and inv(3)(q21q26)/t(3;3)(q21;q26) [3 (6%) patients]. The detection of NF1 mutations in patients harboring inv(16)(p13.1q22) and those with inv(3)(q21q26.2)/t(3;3)(q21;q26.2) is consistent with the previous report showing mutations in genes belonging to the RAS pathway in patients with the aforementioned chromosome abnormalities.14 Notably, NF1-mutated patients more often had complex karyotypes (19% vs 9%, P<0.001) and less frequently normal karyotypes (37% vs 54%, P=0.02) than patients with wild-type NF1 (Supplementary Tables S1 and S3).
We next analyzed mutations co-occurring with the NF1 mutations. NF1-mutated patients harbored mutations in tumor suppressor genes more often than patients with wild-type NF1 (29% vs 16%, P=0.02), and also tended to carry mutations in the chromatin remodeling genes (27% vs 16%, P=0.06). The most frequently mutated functional groups in NF1-mutated patients were the methylation-related genes (38% of patients), kinases (33%) and NPM1 (35%), but their frequency did not differ significantly from those in patients with wild-type NF1 (Supplementary Table S4). With respect to single gene mutations, more common in NF1-mutated patients than in patients with wild-type NF1 were mutations in SF3B1 (13% vs 3%, P=0.002), IKZF1 (8% vs 1%, P=0.005), TP53 (17% vs 6%, P=0.007) and HNRNPK (4% vs 1%, P=0.05) genes (Supplementary Table S5).
For better visualization of the mutational spectrum of NF1-mutated patients, we created an oncoprint depicting both co-occurring gene mutations and cytogenetic findings of the patients (Figure 1d). The paucity of co-occurring biallelic CEBPA mutations in cytogenetically normal AML, found only in one NF1-mutated patient, and the absence of KIT mutations in core-binding factor AML seems noteworthy (Figure 1d).
We also assessed whether different NF1 mutation types [Thr676 hotspot mutations, other frameshift and/or nonsense mutations and missense mutations (including one patient with in-frame insertion/deletion) were associated with particular clinical features and/or co-occurring mutations. Patients with Thr676 mutations had lower presenting white blood cell (WBC) counts than patients with NF1 frameshift or nonsense mutations whose WBC counts were lower than those of patients with NF1 missense mutations (NF1 Thr676 mutations vs other frameshift/nonsense mutations vs missense mutations, median, 7.9 × 109/l vs 14.8 × 109/l vs 27.2 × 109/l, P=0.02). Patients with Thr676 mutations also tended to have lower hemoglobin levels at time of diagnosis (8.1 g/dl vs 9.1 g/dl and 9.2 g/dl, P=0.05; Supplementary Table S6). Whereas no patient with NF1 Thr676 mutation and only one patient (9%) with other frameshift/nonsense mutations had extramedullary involvement, 10 patients (40%) with NF1 missense mutations had extramedullary disease (P=0.007), including four patients with splenomegaly and five with lymphadenopathy. With regard to co-occurring mutations, there were no significant differences in the frequencies of gene mutations assigned to the functional groups among patients with the three aforementioned NF1 mutation types (NF1 Thr676 mutations vs other frameshift/nonsense mutations vs missense mutations; Supplementary Table S7).
Treatment outcome of NF1-mutated AML patients
Since patients aged <60 years and those aged ≥60 years were treated differently, the outcome analyses were performed separately for each age group. In younger patients, the presence of NF1 mutations had no significant impact on the patients’ outcome (Table 1, Supplementary Figures S4a and S4b).
Table 1.
Outcomes of AML patients with and without NF1 mutations separately listed for patients aged <60 years and those aged ≥60 years
|
AML patients <60 years
| |||
|---|---|---|---|
| Endpoint |
NF1 mutated (n=34)a |
NF1 wild-type (n=586) |
P-valueb |
|
| |||
| Complete remission, n (%) | 23 (68) | 463 (79) | 0.13 |
|
| |||
| Relapse rate, n (%) | 11 (50)c | 276 (60) | 0.38 |
|
| |||
| Disease-free survival | |||
| Mutated patients (number of eventsd) | 22 (15) | 457 (309) | 0.78 |
| Median, years | 1.5 | 1.4 | |
| % Disease-free at 1 year (95% CI) | 59 (36-76) | 57 (52-61) | |
| % Disease-free at 3 years (95% CI) | 36 (17-56) | 38 (34-42) | |
|
| |||
| Overall survival | |||
| Mutated patients (number of eventse) | 34 (25) | 586 (375) | 0.12 |
| Median, years | 1.6 | 2.2 | |
| % Alive at 1 year (95% CI) | 62 (43-76) | 71 (67-75) | |
| % Alive at 3 years (95% CI) | 38 (22-54) | 45 (41-49) | |
|
| |||
|
AML patients ≥60 years
| |||
| Endpoint |
NF1 mutated (n=14)a |
NF1 wild-type (n=283) |
P-valueb |
|
| |||
| Complete remission, n (%) | 3 (21) | 168 (59) | 0.001 |
|
| |||
| Relapse rate, n (%) | 3 (100) | 139 (84) | 1.00 |
|
| |||
| Disease-free survival | |||
| Mutated patients (number of eventsd) | 3 (3) | 166 (154) | – |
| Median, years | 0.6 | 0.6 | |
| % Disease-free at 1 year (95% CI) | 0 | 34 (27-41) | |
| % Disease-free at 3 years (95% CI) | 0 | 14 (9-20) | |
|
| |||
| Overall survival | |||
| Mutated patients (number of eventse) | 14 (13) | 283 (266) | 0.11 |
| Median, years | 0.4 | 0.8 | |
| % Alive at 1 year (95% CI) | 14 (2-37) | 41 (35-47) | |
| % Alive at 3 years (95% CI) | 14 (2-37) | 15 (12-20) | |
Abbreviations: CI, confidence interval; n, number.
Patients who received allogeneic hematopoietic stem cell transplantation while in first CR (n=3) or did not receive post-remission chemotherapy according to protocol (n=1) were excluded from the outcome analyses.
P-values for categorical variables are from Fisher’s exact test, P-values for time to event variables are from the log-rank test.
One patient who achieved a CR was lost to follow-up.
An event for DFS is relapse or death, patients alive and relapse-free at last follow-up are censored.
An event for OS is death and patients alive at last follow-up are censored.
We also compared the clinical outcome of younger patients who had NF1 mutations with VAF ≥0.50 (n=13) with that of younger patients with NF1 mutation VAF <0.50 (n=21), and found that the former had worse outcome. All 5 patients with VAF ≥0.50 who achieved a CR and had adequate follow-up relapsed, compared with only 35% of patients harboring NF1 mutations with a VAF <0.50 (P=0.04). Patients with VAF ≥0.50 also had shorter overall survival (3-year rates, 23% vs 48%, P=0.03), and tended to have shorter disease-free survival (3-year rates, 0% versus 47%, P=0.09; Supplementary Table S8).
In older patients, the presence of NF1 mutations was associated with a lower probability to achieve a complete remission (CR; P=0.001, Table 1), with only three of 14 (21%) NF1-mutated patients achieving a CR compared with 168 of 283 (59%) patients with wild-type NF1. There was no significant difference in overall survival (OS) between older patients with and those without NF1 mutations (Table 1, Supplementary Figure S4d).
As NF1 mutations were most common in AML patients classified in the favorable and adverse risk categories according to the ELN 2017 classification,31 we investigated whether the presence of NF1 mutations could refine the outcomes within these risk groups. The outcomes of younger patients in the ELN favorable group were not affected by the presence of NF1 mutations (Table 2). However, younger patients in the ELN adverse risk category harboring NF1 mutations had a lower CR rate (20% vs 53%, P=0.05), and shorter OS (median, 0.4 vs 1.0 years, P<0.001) than patients with wild-type NF1. No younger NF1-mutated patient belonging to the ELN adverse risk category who did not receive an allogeneic transplant was alive 1.5 years after diagnosis.
Table 2.
Outcomes of younger (aged <60 years) and older (aged ≥60 years) AML patients with and without NF1 mutations classified into the ELN favorable or adverse risk groups
|
AML patients <60 years classified in the ELN favorable risk group
| |||
|---|---|---|---|
| Endpoint |
NF1 mutated (n=18)a |
NF1 wild-type (n=297)a |
P-valueb |
|
| |||
| Complete remission, n (%) | 18 (100) | 272 (92) | 0.38 |
|
| |||
| Disease-free survival‡ | 0.84 | ||
| Median, years | 3.1 | 4.9 | |
| % Disease-free at 1 year (95% CI) | 72 (46-87) | 68 (62-73) | |
| % Disease-free at 3 years (95% CI) | 50 (26-70) | 52 (46-58) | |
|
| |||
| Overall survival‡ | 0.37 | ||
| Median, years | 4.3 | 13.0 | |
| % Alive at 1 year (95% CI) | 83 (57-94) | 85 (80-88) | |
| % Alive at 3 years (95% CI) | 56 (31-75) | 65 (59-70) | |
|
| |||
|
AML patients <60 years classified in the ELN adverse risk group
| |||
| Endpoint |
NF1 mutated (n=10)a |
NF1 wild-type (n=124)a |
P-valueb |
|
| |||
| Complete remission, n (%) | 2 (20) | 66 (53) | 0.05 |
|
| |||
| Disease-free survivalc | – | ||
| Median, years | 0.4 | 0.7 | |
| % Disease-free at 1 year (95% CI) | 0 | 36 (25-48) | |
| % Disease-free at 3 years (95% CI) | 0 | 12 (6-21) | |
|
| |||
| Overall survivalc | <0.001 | ||
| Median, years | 0.4 | 1.0 | |
| % Alive at 1 year (95% CI) | 20 (3-47) | 51 (42-59) | |
| % Alive at 3 years (95% CI) | 0 | 22 (15-29) | |
|
| |||
|
AML patients ≥60 years classified in the ELN adverse risk group
| |||
| Endpoint |
NF1 mutated (n=10)a |
NF1 wild-type (n=93)a |
P-valueb |
|
| |||
| Complete remission, n (%) | 2 (20) | 39 (42) | 0.31 |
|
| |||
| Disease-free survivalc | – | ||
| Median, years | 0.4 | 0.4 | |
| % Disease-free at 1 year (95% CI) | 0 | 21 (10-35) | |
| % Disease-free at 3 years (95% CI) | 0 | 0 | |
|
| |||
| Overall survivalc | 0.48 | ||
| Median, years | 0.4 | 0.6 | |
| % Alive at 1 year (95% CI) | 10 (1-36) | 27 (18-36) | |
| % Alive at 3 years (95% CI) | 10 (1-36) | 4 (1-10) | |
Abbreviations: CI, confidence interval; ELN, European LeukemiaNet; n, number.
Among patients who achieved a CR, only those who received at least one cycle of postremission chemotherapy according to protocol were included in the outcome analysis.
P-values for complete remission are from Fisher’s exact test, P-values for disease-free and overall survival are from the log-rank test.
Patients who received allogeneic hematopoietic stem cell transplantation in first CR were excluded from disease-free survival and overall survival analyses.
Among older patients in the ELN adverse group, there were no outcome differences between patients with and without NF1 mutations (Table 2). Too few older patients in the ELN favorable group had NF1 mutations to assess the influence of these mutations on the patients’ prognosis.
Lastly, we analyzed associations among the different mutation types and the patients’ outcome. As only 14 patients age ≥60 years were NF1 mutated, we restricted the analysis to patients <60 years of age. Interestingly, while the CR rates and survival of patients with missense mutations (n=16), or other frameshift or nonsense mutations (n=8) did not differ from those of patients with wild-type NF1, patients harboring the Thr676 hotspot mutation (n=10) had a reduced CR rate (50% vs 79%, P=0.04), and shorter OS than patients with wild-type NF1 (median, 0.8 vs 2.2 years, P=0.01, Figure 2b and Supplementary Table S9).
Figure 2.
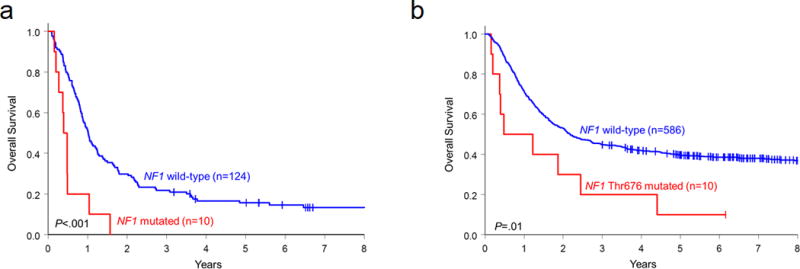
Kaplan-Meyer curves depicting overall survival of patients with de novo AML according to the presence or absence of NF1 mutations. (a) Patients <60 years of age belonging to the ELN 2017 adverse risk group with any NF1 mutation (red) compared with patients without NF1 mutations (blue). (b) Patients <60 years of age with NF1 Thr676 mutations (red) compared with patients without these mutations (blue).
DISCUSSION
Our finding of the relatively frequent occurrence of NF1 mutations in AML patients might suggest a more prominent role of NF1 than previously anticipated. The higher frequency of NF1 mutations in our patient cohort compared with earlier studies10–12 might in part be due to rapidly improving sensitivity of the targeted NGS techniques. Moreover, two recent large NGS studies detected NF1 mutations in only 1%11 and 1.8%14 of their patients, respectively. Notably, neither of these two large studies11,14 captured recurrent mutations in the newly defined mutation hotspot Thr676, which accounts for a large proportion of mutations (24%) in our patient set. The most likely reason for the oversight of the mutation hotspot in previous studies is its location in a mononucleotide repeat, which, for example, is similar to the strand slippage mutation in CTCF in endometrial cancers that was also initially unidentified.32 In contrast, the recently published AACR GENIE dataset33 detected Thr676 mutations in AML, thereby highlighting that the continuous advances in NGS techniques will continue to uncover mutations in difficult and repetitive regions. Interestingly, the Thr676 mutation was also previously detected in patients with juvenile myelomonocytic leukemia,34 thus further supporting the importance of this hotspot in leukemia. Two other studies, which analyzed AML patients with heterozygous NF1 deletions for the presence of NF1 mutations in the remaining coding allele of the gene, only sporadically found these mutations, in one10 and two12 patients, respectively. A third study by Haferlach et al.13 showed that 30% (7/23) of their NF1 deleted de novo AML cases harbored NF1 mutations. However, the Thr676 hotspot was not listed among these mutations.13 However, despite advances in sequencing and variant-calling techniques, a continuous careful evaluation of sequencing data to exclude filtering of such variants will likely still be necessary in the future.
To our knowledge, this is the first report of an adverse prognostic impact of NF1 mutations in younger de novo AML patients classified in the ELN adverse risk group and in patients who harbored NF1 Thr676 mutations. A previous study assessing the prognostic impact of NF1 deletions in de novo AML found no statistically significant differences in CR rates, 3-year relapse-free and overall survival.10 However, Boudry-Labis et al.,10 who performed mutation screening only in NF1-deleted patients, detected only one NF1 Thr676 mutation in their patients, and did not analyze the data in the context of such a prognostic classification as the ELN classification used in our study.
The number of patients analyzed in our study was relatively small, and they were heterogeneous with regard to cytogenetic findings. Thus our findings require corroboration. If confirmed, testing for NF1 mutations might provide additional prognostic information both in AML patients aged <60 years, especially if NF1 mutation is found at a VAF of ≥0.50, and in patients ≥60 years of age. The particularly poor outcome of NF1-mutated patients belonging to the ELN adverse risk category suggests that NF1 mutation status could be considered as part of the mutational risk assessment. Given the importance of VAF in younger patients, and the difficulty assessing the Thr676 mutation hotspot, it might be necessary to use next generation sequencing techniques with high sensitivities in repetitive regions, like the Miseq used in our analysis.
Our findings both with respect to the frequency, the mutation hotspot and the observed adverse prognostic impact of the NF1 mutations may be of potential importance in view of targeted treatment approaches. While Nf1-deficient AML cell lines were shown to confer cytarabine resistance,35 loss or inactivation of NF1 have been previously demonstrated to increase sensitivity to rapamycin-induced apoptosis, suggesting mTOR-directed therapeutics as possible targeted therapeutic options in presence of the NF1 null state.12
Supplementary Material
Acknowledgments
The authors are grateful to the patients who consented to participate in these clinical trials and the families who supported them; to Donna Bucci and the CALGB/Alliance Leukemia Tissue Bank at The Ohio State University Comprehensive Cancer Center, Columbus, OH, for sample processing and storage services and Lisa J. Sterling and Christine Finks for data management. This work was supported in part by grants from the National Cancer Institute (Bethesda, Maryland, USA) [U10CA180821, U10CA180882 (to the Alliance), P30CA016058, P50CA140158, U10CA003927, U10CA032291, U10CA035279, U10CA047545, U10CA059518, U10CA101140, U10CA180850, U10CA180861, U10CA180866, U10CA180867, and U24CA196171]; the Leukemia Clinical Research Foundation; the Warren D. Brown Foundation; the Pelotonia Fellowship Program (to A-KE); and by an allocation of computing resources from The Ohio Supercomputer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no conflicts of interest.
Presented in part at the 59th Annual Meeting of the American Society of Hematology, Atlanta, GA, December 9-12, 2017, and published in abstract form: Eisfeld A-K, Kohlschmidt J, Mrózek K, Mims AS, Walker CJ, Blachly JS et al. Neurofibromin 1 gene mutations are recurrent events in adult patients with acute myeloid leukemia (AML), associate with poor outcome and refine the adverse European Leukemianet (ELN) risk category. Blood 2017; 130(suppl 1): abstract 103.
AUTHOR CONTRIBUTIONS
A-KE, KM, JCB and CDB designed the study. A-KE, KM, AM, CJW, JSB, SEM, AdlC, JCB and CDB contributed to the data interpretation. A-KE, KM, JK and CDB wrote the manuscript. JK and DN performed statistical analysis. A-KE and SO performed laboratory-based research. KM, AJC, BLP, JEK, ESW, RMS, JCB and CDB were involved directly or indirectly in the care of patients and/or sample procurement. All authors reviewed the manuscript and approved its final version.
Supplementary Information is available at Leukemia’s website.
References
- 1.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 2.Buchberg AM, Cleveland LS, Jenkins NA, Copeland NG. Sequence homology shared by neurofibromatosis type-1 gene and IRA-1 and IRA-2 negative regulators of the RAS cyclic AMP pathway. Nature. 1990;347:291–294. doi: 10.1038/347291a0. [DOI] [PubMed] [Google Scholar]
- 3.Largaespada DA, Brannan CI, Jenkins NA, Copeland NG. Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat Genet. 1996;12:137–143. doi: 10.1038/ng0296-137. [DOI] [PubMed] [Google Scholar]
- 4.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 5.Hope DG, Mulvihill JJ. Malignancy in neurofibromatosis. Adv Neurol. 1981;29:33–55. [PubMed] [Google Scholar]
- 6.Side L, Taylor B, Cayouette M, Conner E, Thompson P, Luce M, et al. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med. 1997;336:1713–1720. doi: 10.1056/NEJM199706123362404. [DOI] [PubMed] [Google Scholar]
- 7.Side LE, Emanuel PD, Taylor B, Franklin J, Thompson P, Castleberry RP, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood. 1998;92:267–272. [PubMed] [Google Scholar]
- 8.Largaespada DA, Shaughnessy JD, Jr, Jenkins NA, Copeland NG. Retroviral integration at the Evi-2 locus in BXH-2 myeloid leukemia cell lines disrupts Nf1 expression without changes in steady-state Ras-GTP levels. J Virol. 1995;69:5095–5102. doi: 10.1128/jvi.69.8.5095-5102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Bollag G, Clark R, Stevens J, Conroy L, Fults D, et al. Somatic mutations in the neurofibromatosis 1 gene in human tumours. Cell. 1992;69:275–281. doi: 10.1016/0092-8674(92)90408-5. [DOI] [PubMed] [Google Scholar]
- 10.Boudry-Labis E, Roche-Lestienne C, Nibourel O, Boissel N, Terre N, Perot C, et al. Neurofibromatosis-1 gene deletions and mutations in de novo acute myeloid leukemia. Am J Hematol. 2013;4:306–311. doi: 10.1002/ajh.23403. [DOI] [PubMed] [Google Scholar]
- 11.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parkin B, Ouillette P, Wang Y, Liu Y, Wright W, Roulston D, et al. NF1 inactivation in adult acute myelogenous leukemia. Clin Cancer Res. 2010;16:4135–4147. doi: 10.1158/1078-0432.CCR-09-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haferlach C, Grossmann V, Kohlmann A, Schindela S, Kern W, Schnittger S, et al. Deletion of tumor suppressor gene NF1 occurs in 5% of myeloid malignancies and is accompanied by a mutation in the remaining allele in half of the cases. Leukemia. 2012;26:834–839. doi: 10.1038/leu.2011.296. [DOI] [PubMed] [Google Scholar]
- 14.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolitz JE, George SL, Marcucci G, Vij R, Powell BL, Allen SL, et al. P-glycoprotein inhibition using valspodar (PSC-833) does not improve outcomes for patients under age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808. Blood. 2010;116:1413–1421. doi: 10.1182/blood-2009-07-229492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blum W, Sanford BL, Klisovic R, DeAngelo DJ, Uy G, Powell BL, et al. Maintenance therapy with decitabine in younger adults with acute myeloid leukemia in first remission: a phase 2 Cancer and Leukemia Group B study (CALGB 10503) Leukemia. 2017;31:34–39. doi: 10.1038/leu.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baer MR, George SL, Caligiuri MA, Sanford BL, Bothun SM, Mrózek K, et al. Low-dose interleukin-2 immunotherapy does not improve outcome of patients age 60 years and older with acute myeloid leukemia in first complete remission: Cancer and Leukemia Group B study 9720. J Clin Oncol. 2008;26:4934–4939. doi: 10.1200/JCO.2008.17.0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolitz JE, George SL, Dodge RK, Hurd DD, Powell BL, Allen SL, et al. Dose escalation studies of cytarabine, daunorubicin, and etoposide with and without multidrug resistance modulation with PSC-833 in untreated adults with acute myeloid leukemia younger than 60 years: final induction results of Cancer and Leukemia Group B study 9621. J Clin Oncol. 2004;22:4290–4301. doi: 10.1200/JCO.2004.11.106. [DOI] [PubMed] [Google Scholar]
- 19.Marcucci G, Moser B, Blum W, Stock W, Wetzler M, Koltiz JE, et al. A phase III randomized trial of intensive induction and consolidation chemotherapy ± oblimersen, a pro-apoptotic Bcl-2 antisense oligonucleotide in untreated acute myeloid leukemia patients >60 years old. J Clin Oncol. 2007;25:360s. abstract 7012. [Google Scholar]
- 20.Mayer RJ, Davis RB, Schiffer CA, Berg DT, Powell BL, Schulman P, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. N Engl J Med. 1994;331:896–903. doi: 10.1056/NEJM199410063311402. [DOI] [PubMed] [Google Scholar]
- 21.Moore JO, George SL, Dodge RK, Amrein PC, Powell BL, Kolitz JE, et al. Sequential multiagent chemotherapy is not superior to high-dose cytarabine alone as postremission intensification therapy for acute myeloid leukemia in adults under 60 years of age: Cancer and Leukemia Group B study 9222. Blood. 2005;105:3420–3427. doi: 10.1182/blood-2004-08-2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26:2061–2068. doi: 10.1038/leu.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. 2013;31:923–929. doi: 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stone RM, Berg DT, George SL, Dodge RK, Paciucci PA, Schulman P, et al. Granulocyte-macrophage colony-stimulating factor after initial chemotherapy for elderly patients with primary acute myelogenous leukemia. N Engl J Med. 1995;332:1671–1677. doi: 10.1056/NEJM199506223322503. [DOI] [PubMed] [Google Scholar]
- 25.Moore JO, Dodge RK, Amrein PC, Kolitz J, Lee EJ, Powell B, et al. Granulocyte-colony stimulating factor (filgrastim) accelerates granulocyte recovery after intensive postremission chemotherapy for acute myeloid leukemia with aziridinyl benzoquinone and mitoxantrone: Cancer and Leukemia Group B study 9022. Blood. 1997;89:780–788. [PubMed] [Google Scholar]
- 26.Schiffer CA, Davis RB, Schulman P, Cooper B, Coyle T, Lee E, et al. Intensive post remission therapy of acute myeloid leukemia (AML) with cytoxan/etoposide (CY/VP16) and diazaquone/mitoxantrone (AZQ/MITO) Blood. 1991;78(suppl):460. abstract 1829. [Google Scholar]
- 27.Mrózek K, Carroll AJ, Maharry K, Rao KW, Patil SR, Pettenati MJ, et al. Central review of cytogenetics is necessary for cooperative group correlative and clinical studies of adult acute leukemia: the Cancer and Leukemia Group B experience. Int J Oncol. 2008;33:239–244. [PMC free article] [PubMed] [Google Scholar]
- 28.Eisfeld AK, Mrózek K, Kohlschmidt J, Nicolet D, Orwick S, Walker CJ, et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia. 2017;31:2211–2218. doi: 10.1038/leu.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kroll KW, Eisfeld A-K, Lozanski A, Bloomfield CD, Byrd JC, Blachly JS. MuCor: mutation aggregation and correlation. Bioinformatics. 2016;32:1557–1558. doi: 10.1093/bioinformatics/btw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcucci G, Maharry K, Radmacher MD, Mrózek K, Vukosavljevic T, Paschka P, et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B study. J Clin Oncol. 2008;26:5078–5087. doi: 10.1200/JCO.2008.17.5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Döhner H, Estey E, Grimwade D, Amadori S, Applebaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zighelboim I, Mutch DG, Knapp A, Ding L, Xie M, Cohn DE, et al. High frequency strand slippage mutations in CTCF in MSI-positive endometrial cancers. Hum Mutat. 2014;35:63–65. doi: 10.1002/humu.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang Y-D, Mazor T, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet. 2015;47:1326–1333. doi: 10.1038/ng.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin B, Morgan K, Hasz DE, Mao Z, Largaespada DA. Nf1 gene inactivation in acute myeloid leukemia cells confers cytarabine resistance through MAPK and mTOR pathways. Leukemia. 2006;20:151–154. doi: 10.1038/sj.leu.2404033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.