

ABSTRACT
Renal clear cell carcinoma (RCC) is the most common type of kidney cancer and has a high propensity for metastasis. While treatment with immune checkpoint inhibitors, such as anti-PD-1, have shown modest improvements in survival for RCC, it is difficult to identify responders from non-responders. Attempts to elucidate the mechanisms associated with differential response to checkpoint inhibitors have been limited by small sample size making it difficult to detect meaningful associations. We utilized existing large datasets from The Cancer Genome Atlas (TCGA) to first find predictors of disease aggressiveness in the tumor microenvironment (TME) and hypothesized that these same predictors may influence response to immunotherapy. We found primary metastatic (M1-stage IV) tumors exhibit high immune infiltration, and high TP53-inactivation induced senescence activity compared to non-metastatic (M0-Stage I/II) tumors. Moreover, some TME features inferred from deconvolution algorithms, which differ between M0 and M1 tumors, also influence overall survival. A focused analysis identified interactions between tumor TP53-inactivation induced senescence activity and expression of inflammatory molecules in pre-treatment RCC tumors, which predict both change in tumor size and response to checkpoint blockade therapy. We also noted frequency of inactivating mutations in the protein polybromo-1 (PBRM1) gene was found to be negatively associated with TP53-inactivation induced senescence enrichment. Our findings suggest a mechanism by which tumor TP53-inactivation induced senescence can modulate the TME and thereby influence outcome from checkpoint blockade therapy.
Keywords: deconvolution, renal clear cell carcinoma, cellular senescence, metastasis, molecular modeling, tumor immunobiology
Introduction
Renal clear cell carcinoma (RCC) is the most common type of kidney cancer. It originates from the proximal tubules of the nephron1 and often presents at late stages of illness, such that over one-fourth of individuals diagnosed with RCC present with metastatic disease, which is difficult to treat.2 Limited success in the treatment of aggressive, late-stage RCC tumors has been seen with immune checkpoint blockade therapy, such as Nivolumab, which targets the programmed cell death-1 receptor (PD-1).3 Few studies have examined transcriptomic changes in RCC treated with anti-PD-1 therapy.4,5 The small size of prior studies and limited application of transcriptome analytical tools has made it difficult to find meaningful associations predictive of response to checkpoint blockade therapy.
RCC tumors are known to have increased lymphocytic infiltrates compared to other tumor types with a similar mutation burden, and these high immune infiltrates in RCC tumors are associated with an unfavorable disease prognosis.6,7 Previous studies in RCC have shown CD8 + T cell infiltrates characterized by high expression of immune checkpoint molecules in the absence of active dendritic cells are associated with poor prognosis, while tumors characterized by presence of active dendritic cell infiltration are associated with a good prognosis.8 In RCC as well as other tumor types, tumor-immune interactions can lead to alterations in the cellular state of immune cells in the TME. For example, senescent tumor cells are known to secrete cytokines, which results in the recruitment of innate immune cells as well as the promotion of premalignant tumor growth9 and activation of hypoxic pathways,10 which promote PD-L1 expression and subsequent T cell dysfunction.11 Recent works on the role of cellular senescence activity in tumors point to the induction of a senescence-associated secretory phenotype (SASP) as playing a major role in inducing tumor inflammation.12,13 Specifically, senescence activity is modulated by the innate immunity cystolic DNA-sensors cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING), which induce SASP.14,15 Moreover, SASP, can lead to immune mediated clearance of senescent tumor cells. This clearance is dependent on CD4 + T cell activity as well as activation of dendritic cells (DC) by cGAS, as DC activation increases antigen presentation to CD8 + T cells.
Understanding tumor immune interactions, such as those between senescent tumor cells and immune cells in the TME, is essential for determining immunotherapy outcome. However, immunotherapy datasets often suffer from small sample size (e.g. Miao et al., 2018 with n = 33) making it difficult to discern mechanisms associated with differential response to treatment despite the availability of multi-omics data. To address this limitation, we first aimed to determine predictors of disease aggressiveness from the much larger dataset of RCC available through The Cancer Genome Atlas (TCGA).16 We hypothesized that the same predictors of disease aggressiveness may also predict response to checkpoint blockade therapy. Specifically, we examined immune infiltration and pathway enrichment differences in primary metastatic (M1/Stage IV) and non-metastatic (M0/Stage I-II) RCC tumors in the TCGA dataset to determine predictors of disease aggressiveness. We then used the identified predictors to perform a focused analysis of the Miao et al. dataset, which contains RNA-seq and whole exome seq data on pre-treatment tumors from patients in two cohorts treated with checkpoint blockade therapy, in order to explore mechanisms underlying immunotherapy response in RCC.
Materials and methods
Code and TCGA data availability
Level three RNA-seq and whole exome sequencing datasets from pre-treatment primary renal clear cell carcinoma (denoted KIRC by TCGA)16 and cutaneous skin melanoma (SKCM),17 as well as their corresponding clinical variables were used in this article and can be obtained from The Cancer Genome Atlas (TCGA) Network (last update: 05/27/2016) via FireBrowse. All statistical calculations were done using R 3.1.018 and figures were generated using R packages ggplot2,19 corrplot, and pheatmap, and Adobe Illustrator version 22.0.1. Code for statistical calculations and figures are available upon request.
TCGA RCC clinical data
Tumors were categorized as either M0, M1, or were not included in the study. The following criteria were met for all samples categorized as M0: tumors were pathologically diagnosed via TNM staging as M0 and with a pathological grade of stage 1 or stage 2. The following criteria were met by all samples categorized as M1: tumors were pathologically diagnosed via TNM staging as M1, M1a or M1b, and had a pathological grade of stage 4. We excluded all stage III tumors from the initial analysis as there is often uncertainty associated with whether these tumors are metastatic or not, whereas stage I/II tumors are clearly non-metastatic. From TCGA, 499 KIRC (M1: 79, M0: 297, stage III: 123) were examined.
RCC immunotherapy cohorts
RNA-seq and whole exome sequencing data on pre-treatment stage 4 metastatic RCC tumors from n = 33 patients were published by Miao et al., 20185 and were used in this study to evaluate the relevance of our findings from the TCGA M0 vs. M1 RCC analysis. The Miao et al., 2018 study comprised 16 patients in the discovery cohort and 17 patients in the validation cohort. The discovery cohort was part of the NCT01358721 clinical trial,4 while the validation cohort consists of patients and samples from multiple institutions. Although all tumor tissue samples were analyzed prior to immunotherapy treatment, we note significant clinical (Table 1) and immunological differences (Fig. S2) between the validation and discovery cohorts. Therefore, we adjust for cohort membership, (discovery vs. validation cohort) in all of our regression models and note if enrichment of specific pathways or expression of key molecules differs between the two cohorts (Fig. S2).
Table 1.
Discovery and validation cohort characteristics. Clinical characteristics for all individuals in the discovery and validation immunotherapy cohorts are shown. All patients (n = 33) were diagnosed with stage IV metastatic disease and were treated with immunotherapy agents. RNA-seq and whole exome sequencing (WES) was performed on n = 33 tumor samples prior to immunotherapy treatment. Patients from the discovery cohort belonged to the clinical trial published by Choueiri et al., 2016, while patients from the validation cohort belonged to multiple institutions.
| Cohort | Sample Size | Site of Biopsy | Age | Sex |
First-line treatment |
Immunotherapy Treatment | Therapy Dose |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M | F | Y | N | 0.3 mg/kg | 2 mg/kg | 10 mg/kg | |||||
| Discovery | n = 16 | Soft tissue metastases | 62.88 | n = 13 | n = 3 | n = 3 | n = 13 | Nivolumab n = 16 | n = 4 | n = 5 | n = 10 |
| Validation | n = 17 | Multiple Locations | 63.06 | n = 11 | n = 6 | n = 7 | n = 10 | Nivolumab n = 11 Atezolizumab n = 2 Nivolumab + ipilimumab n = 4 |
Unknown | ||
In the Miao et al., 2018 study, treatment response was categorized using the best RECIST categorization20 and the standard benefit response categorization (clinical benefit, intermediate benefit, or no benefit). In our study, we also categorize response as a binary metric, which groups the best RECIST categories into either Response or Disease. We binned the “clinical response” and “partial response” RECIST categories as “Response”, and the “partial disease” and “stable disease” RECIST categories as “Disease”.
Immune infiltration scores
To determine the degree of immune infiltration in M0 and M1 samples in RCC, we applied the BASE deconvolution algorithm,21 which infers immune infiltration level from bulk tumor gene expression data using immune cell gene expression profiles from the Immunological Genome Project (ImmGen) as a reference.22 Specificity of some immune infiltration scores was confirmed previously by correlating immune infiltration scores with flow cytometry fractions from immune cell mixtures, as well as with tumor purity, as described previously.21,23 We eliminated all immune infiltration scores that positively correlated with tumor purity. To increase our resolution further, we examined how immune cell subtype-specific gene sets were weighted by BASE when calculating the infiltration score for each ImmGen cell type. Of the 226 ImmGen cell phenotypes, we eliminated all immune infiltration scores that positively correlated with tumor purity, and selected cell phenotypes based on their marginal association with metastatic-status (FDR correction at Q = 0.05). To increase our resolution further, we examined how immune cell subtype-specific gene sets were weighted by BASE when calculating the infiltration score for each ImmGen cell type. We correlated ImmGen based weights with the average gene expression of gene sets defining a specific cell type from an external dataset.24 Of the 226 ImmGen cell phenotypes, this resulted in 48 immune cell types in RCC which were inputs for least absolute shrinkage and selection operator (LASSO) penalized regression25 which identifies a parsimonious set of independent immune cell type predictors of metastatic status.
A LASSO penalized logistic regression model was estimated for RCC with metastatic status (M0/M1) as the dependent variable and 48 immune cell infiltration scores as penalized predictors with age and sex as unpenalized predictors. We selected the immune cell types with non-zero coefficient estimates in the LASSO model at the penalization threshold with minimal prediction error via 10-fold cross validation. The tuning parameter selected gives the minimum mean cross-validated error. The LASSO predictors and the clinical covariates age and sex were subsequently utilized in unpenalized multivariate Cox-proportional hazards regression modeling. We then determined if each of the LASSO-selected immune cell types interacted with metastatic status to influence survival. Based on the Cox regression modeling, hazard ratios were determined for each immune cell type with an overall measure of significance for the regression model computed via a likelihood ratio test, where the null model adjusted for age and sex. Additionally, immune infiltration scores of the LASSO predictors were segregated into high and low scores based on the median distribution to determine if enrichment or depletion of specific immune cell types in the microenvironment was predictive of survival. Difference in high-low scores was determined using a log-rank test.
In order to develop a predictive model of survival based on immune infiltration, we implemented backward selection on the output from LASSO to further reduce the number of predictors in our Cox regression survival models. LASSO predictors were generated based on finding the best linear combination of immune cell types, which predict disease status. Although LASSO is a useful tool for prediction of disease status, it can result in overfitting for individual level variable evaluation, which we address by using backward selection, where predictors were retained in our regression model if they both minimized the Akaike Information Criterion (AIC) and were found to be significant predictors of overall survival. To further evaluate the survival model resulting from backward selection, we ran a sensitivity analysis on all M0 tumors, including stage III tumors, which were not used to develop our immune infiltration based survival model.
We applied the CIBERSORT deconvolution algorithm for additional confirmation of the immune cell types present in the TME.26 As CIBERSORT examines only 22 immune cell types, we did not implement LASSO on CIBERSORT outputs. CIBERSORT nicely complements the BASE algorithm, as it provides estimates of immune cell proportions in the TME as opposed to degree of immune infiltration provided by BASE, which does not allow for comparisons of immune infiltration levels between different immune cell types but allows comparison of immune infiltration scores between samples. CIBERSORT was implemented on filtered (p-value for Pearson correlation coefficient (R) < 0.05) immune cell fractions for each individual. Similar to BASE, all regression and Cox survival models using CIBERSORT were adjusted for age and sex.
Gene set enrichment analysis
We performed Gene Set Enrichment Analysis (GSEA)27 using RNA-seq data and examined the human oncogenic (MSigDB Version 6.1 C6) and immunological (MSigDB Version 6.1 C7)28,29 gene set signatures. Based on the oncogenic and immune signature profiles in RCC, we specifically examined senescence and exhaustion pathways from MSigDB C2, and compared them between M0 and M1 samples. We also performed single-sample GSEA (ssGSEA)30,31 for specific pathways, as the ssGSEA scores served as inputs for generalized linear regression and Cox regression models.
Immune checkpoint and immune activation markers
For all gene expression analyses, log10-normalized expression was used. Expression of key immunomodulatory factors was compared between M0 and M1 tumors using Wilcoxon-rank sum tests. Additionally, we assessed the association between the expression of key immune checkpoint molecules and immune infiltration scores using regression modeling, which adjusts for age, sex, and tumor purity. Here, the ABSOLUTE algorithm, which requires copy number data, was used to determine tumor purity.32 Last, associations between immune infiltrates and senescence enrichment were assessed using Spearman rank correlation.
Regression modeling and use of interaction terms
We implemented regression modeling in order to elucidate predictors of disease aggressiveness by determining the association of specific co-variates, such as senescence enrichment, with expression of immunomodulatory molecules. In our regression models we include an interaction term to determine if the association between senescence enrichment and expression of immunomodulatory molecules varies based on if the tumor is metastatic or not (Table 2). We also implemented logistic regression modeling with interaction terms to assess if the association between immunotherapy outcome and expression of immunomodulatory molecules is affected by tumor senescence enrichment (Table 3). In other words, we asked: does the synergy between tumor senescence and immune activity influence response to immune checkpoint blockade therapy? Last, we implemented regression modeling with interaction terms to determine if the synergy between tumor senescence and immune activity influences percent change in tumor size from baseline in the immunotherapy cohorts (Table 4).
Table 2.
TP53-inactivation induced senescence differentially influences the tumor microenvironment in M0 and M1 tumors. We developed age and sex adjusted regression models using single-sample GSEA (ssGSEA) enrichment scores from TCGA RCC tumors, M0 (n = 297) and M1 (n = 79), to determine whether the TP53-inactivation induced senescence pathway, SENESCENCE_TP53_TARGETS_UP, associates with VHL-induced hypoxia and expression of key immune markers linking cellular senescence and immune infiltration. In models 1–4, we show the association between senescence enrichment and cGAS (), STING (), PRF1 , and GZMA expression by regressing SENESCENCE_TP53_TARGETS_UP enrichment scores on the log10 normalized gene expression of these markers. In model 5 we show the association between senescence enrichment and hypoxia (MANIA_HYPOXIA_VHL_UP) enrichment in M0 and M1 RCC tumors. Our regression models adjust for age and sex and specifically examine if there is an interaction between SENESCENCE_TP53_TARGETS_UP enrichment and metastasis status in order to determine if tumor senescence activity associates with markers of inflammation differently based on M0 or M1 status.
| Model | Outcome | Variable | SE | p-value | |
|---|---|---|---|---|---|
| cGAS | 1.365 | 0.3475 | 0.000102 | ||
| −2.714 | 0.7275 | 0.000221 | |||
| STING | 1.660 | 0.2838 | 1.08*10−08 | ||
| −1.551 | 0.5942 | 0.00941 | |||
| PRF1 | 2.397 | 0.5247 | 6.66*10−06 | ||
| −3.236 | 1.098 | 0.00342 | |||
| GZMA | 2.054 | 0.6228 | 0.00106 | ||
| −2.930 | 1.303 | 0.0252 | |||
| 0.7909 | 0.09713 | 5.97*10−15 | |||
| −0.5865 | 0.2033 | 0.00415 |
whereis the expected normalized gene expression of cGAS (MB21D1), STING, PRF1, or GZMA, is SENESCENCE_TP53_TARGETS_UP pathway enrichment scores, is the expected pathway enrichment score of , the MANIA_HYPOXIA_VHL_TARGETS_UP pathway, is metastasis status, is age, and is sex.
Note: SE is standard error
Table 3.
Predictive effect of senescence immune interactions on immunotherapy response. Logistic regression modeling was implemented on the Miao et al., 2018 immunotherapy dataset (n = 33), which consists of the discovery (n = 16), and validation (n = 17) cohorts, to determine if senescence immune interactions were predictive of response to immune checkpoint inhibitors. We specifically focused on immunological factors that interact with senescence enrichment to predict immunotherapy response. Best RECIST score was binned as a binary variable – Response (CR/PR = 1) and Disease (PD/SD = 0) and this was the main outcome. We also compared the performance of our senescence-immune interaction predictors to the immunophenotype score (IPS) . All models were adjusted for age and cohort status (discovery vs. validation) and were compared to the null model (), which also adjusts for age, and cohort status.
| Model | Predictors | SE | p-value | AIC | Model p-value | |
|---|---|---|---|---|---|---|
| −53.80 | 28.43 | 0.0580 | ||||
| −101.5 | 50.67 | 0.0451 | ||||
| 157.7 | 80.61 | 0.0504 | ||||
| 43.83 | 0.01266 | |||||
| −29.07 | 12.68 | 0.0219 | ||||
| −91.35 | 39.09 | 0.0195 | ||||
| 146.1 | 62.69 | 0.0198 | ||||
| 41.19 | 0.00371 | |||||
| −32.00 | 19.05 | 0.0930 | ||||
| −18.97 | 10.37 | 0.0675 | ||||
| 29.72 | 16.19 | 0.0665 | ||||
| 48.09 | 0.08661 | |||||
| −24.5609 | 15.0025 | 0.101 | ||||
| −26.028 | 15.197 | 0.0868 | ||||
| 39.661 | 22.965 | 0.0842 | ||||
| 48.77 | 0.1168 | |||||
| −100.901 | 45.30 | 0.0259 | ||||
| −130.121 | 59.72 | 0.0294 | ||||
| 206.31 | 91.424 | 0.0294 | ||||
| 40.97 | 0.00335 | |||||
| 0.4435 | 0.4275 | 0.299 | ||||
| 49.52 | 0.2846 |
where is assosciated with response, is age, is cohort status (discovery vs. validation), is SENESCENCE_TARGETS_UP pathway enrichment score, is MANIA_HYPOXIA_VHL_TARGETS_UP pathway enrichment score, are normalized gene expression values for the cGAS (MB21D1), PDCD1, GZMA, and PRF1 genes respectively, and is the immunophenotype score (IPS). All models were compared with the to determine significance.
Note: SE is standard error and AIC is Akaike Information Criterion
Table 4.
Predictive effect of senescence immune interactions on percent change in tumor size from baseline. Regression modeling was implemented on the Miao et al., 2018 immunotherapy dataset (n = 33), which consists of the discovery (n = 16), and validation (n = 17) cohorts, to determine if enrichment of the TP53-inactivation induced senescence pathway, SENESCENCE_TP53_TARGETS_UP, interacts with immune markers to influence percent change in tumor size from baseline in patients who received immune checkpoint blockade therapy. We assessed if the interaction between SENESCENCE_TP53_TARGETS_UP pathway enrichment and cGAS , PDCD1 , GZMA , PRF1, or GZMB expression influenced change in tumor size from baseline. We compared the performance of our senescence immune interaction predictors to the immunophenotype score (IPS) . All regression models were adjusted for age and cohort status and were compared to the null model, which also adjusts for age and cohort status to determine model significance.
| Model | Predictors | SE | p-value | AIC | Model p-value | ||
|---|---|---|---|---|---|---|---|
| 839.8 | 383.1 | 0.0392 | |||||
| −1316 | 534.9 | 0.0222 | |||||
| 293.0 | 0.341 | 0.0340 | |||||
| 398.7 | 205.1 | 0.0648 | |||||
| −630.1 | 304.8 | 0.0507 | |||||
| 296.5 | 0.252 | 0.172 | |||||
| 225.5 | 112.5 | 0.0575 | |||||
| −384.0 | 169.07 | 0.0333 | |||||
| 294.1 | 0.314 | 0.059 | |||||
| 399.2 | 209.9 | 0.0704 | |||||
| −465.0 | 242.7 | 0.0684 | |||||
| 294.5 | 0.304 | 0.0719 | |||||
| 425.5 | 234.5 | 0.0833 | |||||
| −777.0 | 383.3 | 0.0549 | |||||
| 295.8 | 0.271 | 0.127 | |||||
| −17.21 | 8.586 | 0.0564 | |||||
| 293.9 | 0.214 | 0.0449 |
where is the percent change in tumor size from baseline, is age, is cohort status, is SENESCENCE_TP53_TARGETS_UP pathway enrichment scores, and are normalized gene expression values for the cGAS (MB21D1), PDCD1, GZMA, PRF1, and GZMB genes respectively. Lastly, is the immunophenotype score (IPS). All models were compared with the model to determine significance
Note: SE is standard error and AIC is Akaike Information Criterion, and is the R-Squared value for regression the model.
BAF/PBAF subunit mutation analysis
BAF/PBAF is a chromatin remodeling complex regulating expression including selected immune cell types through effects on the SWI/SNF family.33 Mutation status for each BAF/PBAF subunit gene was obtained by examining publicly available somatic variant calls in the mutation annotation format (MAF) files synthesized from whole exome seq data for each RCC sample in TCGA. MAF files for each individual were converted into a binary matrix, where a 1 for a gene indicates the presence of a mutation in the .MAF file while a 0 indicates no mutation. Using our binary matrices, we specifically examined mutation status (presence or absence) in 12 BAF/PBAF subunits, which are known to be commonly mutated in cancers and were found to have mutations in RCC.34 These 12 subunits are: ARID2, BCL7A, BRD7, PBRM1, PHF10, SARC4A, SARCB1, SARCC2, SARCC1, SARCCA2, SARCD3, SS18. We also examined if having a mutation in any one of the BAF/PBAF subunits associates with senescence activity by pooling mutation status information on the 12 BAF/PBAF subunits and examining its association with senescence.
Validation of tumor senescence and immune interactions in melanoma
We further demonstrate the value of incorporating senescence immune interactions in predicting outcome from immunotherapy in melanoma, which is known to be highly immunogenic and often responsive to checkpoint inhibition therapy. We validated our RCC findings using TCGA melanoma (denoted SKCM) RNA-seq and clinical data (n = 469),17 as well as one RNA-seq dataset published by Hugo et al., 2016 on metastatic melanoma pre-treatment tumor lesions from patients treated with anti-PD-1 (n = 26).35 For the Hugo et al., 2016 dataset, we converted all fragments per kilobase of transcript per million (FPKM) expression values into transcripts per million (TPM) using the equation: in order to maintain consistency with the Miao et al., 2018 dataset.
Comparison with immunophenotype score (IPS) in RCC and melanoma
To evaluate the performance of our senescence immune interaction predictors in predicting outcomes from checkpoint blockade therapy, we compared our predictors with the immunophenotype score (IPS), which characterizes tumor immunogenicity.36 All predictors, including IPS, were incorporated as covariates into logistic regression models which adjust for age and sex or cohort status (discovery vs. validation for RCC).
Results
M1 RCC tumors are inflamed compared to M0
RCC M1 tumors have high CD4 + T cell-2 and CD8 + T cell-1 infiltrates (see Table S1 for description of BASE immune cell types) while M0 tumors exhibit stronger macrophage, dendritic cells (DC), monocyte, and B cell infiltrates (Figure 2A). Of the 11 immune cell types predictive of metastasis based on penalized regression, three cell types, including activated DC and CD8 + T cell-1, interact with metastasis status to influence survival (Figure 2C, Fig. S5). Individual level evaluation of immune predictors using backward selection shows three immune cell types, CD8 + T cell-1, CD4 + T cell-2, and activated DC (DC Active), are predictive of survival in an age and sex-adjusted cox regression model (Figure 2B). CD8 + T cell-1 and CD4 + T cell-2 infiltrates negatively associate with survival, while DC Active infiltration is associated with improved overall survival. Sensitivity analysis in all M0 tumors, including stage III, which were excluded in our initial analysis to determine immune predictors, shows CD4 + T cell-2 infiltration is predictive of overall survival in M0 RCC patients. Based on these results, we focused on CD8 + T cell-1, CD4 + T cell-2, and activated DC (DC Active), as CIBERSORT and GSEA (Fig. S3-S4, Table S2-S4) analyses have also confirmed their association with metastasis status and survival (Figure 2B, Table S2). Specifically, CIBERSORT and GSEA confirm that the CD4 + T cell-2 signature in BASE is detecting CD4+ memory T cells, while the activated DC and CD8 + T cell signatures in BASE are indeed detecting activated DCs and CD8 + T cells respectively (Fig. S3-S5, Table S3-S5).
Figure 2.
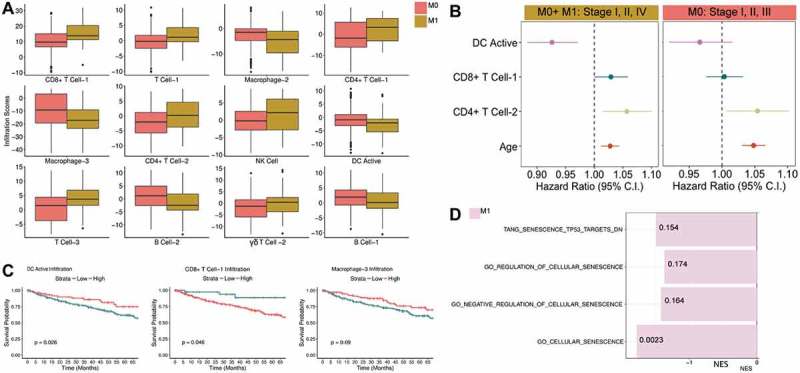
Renal clear cell carcinoma tumor immune microenvironment characterization A) Shown are 12 out of 13 BASE-generated immune cell types found to be predictors of metastasis status for RCC based on penalized regression modeling. B) Results from backward selection applied to penalized regression predictors show three immune cell types, CD8 + T cell-1, DC Active (Activated DC), and CD4+ Memory T cell are significant predictors of overall survival in an age and sex adjusted cox proportional hazards model. Sensitivity analysis shows the effect of immune predictors on overall survival in all M0 tumors (stage I/II, n = 297, stage III, n = 123). C) Immune scores for three predictors, CD8 + T cell-1, CD4 + T cell-2, and Macrophage-3, which interact with metastasis status in regression models of survival, were binned as high or low. The effect of these three immune infiltration scores on survival is shown. D) We performed GSEA analysis for all senescence pathways in the MSigDB database. The normalized enrichment score (NES) is displayed on the x-axis. Pathways shown are significant for M1 post-FDR correction (Q-values are shown with Q < 0.2).
Due to the increased immune activity observed in M1 tumors compared to M0, we examined the expression of immune checkpoint and activation markers, and found M1 tumors exhibit increased expression of both immune checkpoint and activation markers (Fig. S6). Moreover, the positive association between immune checkpoint marker expression (TIGIT, CD38, CTLA4, PDCD1) and CD8 + T cell-1 infiltration is significantly stronger in M1 tumors compared to M0 (Fig. S7, Table S6). These findings align with previous work which also demonstrates CD8 + T cells with increased expression of immune checkpoint markers in the absence of active DCs are associated with a worse prognosis.8 In order to better characterize T cell dysfunction in M1 tumors based on bulk tumor gene expression, we examined pathways defining four possible T cell states in the bulk M1 tumor, senescence, anergy, stem-ness, and exhaustion, and examined their association with T cell infiltration. We found no differences in the expression of T cell-specific senescence and anergy markers/pathways between M0 and M1 tumors. Based on these results, we next assessed tumor pathways, such as tumor senescence, which can lead to tumor inflammation and immune dysfunction.
We find M1 RCC tumors are highly senescent with four out of eight senescence pathways enriched in M1 tumors (Figure 2D) compared to M0. Moreover, high oncogenic activity was observed in M1 tumors (Table S7) including a TP53 inactivation oncogenic pathway which associates with increased senescence activity (Fig. S8). This suggested to us that the tumor itself rather than surrounding immune cells is senescent. Differential expression of genes in the TP53-inactivation induced senescence gene sets was also examined to confirm differential enrichment (Fig. S9). Therefore, we focused on senescence pathways induced by TP53 inactivation as mutations in TP53 are in the top 10 most common mutations found in RCC with a mutation frequency of 2.88% based on TCGA mutation analysis,37 and unlike other senescence pathways, we can say with greater confidence that TP53-inactivation induced senescence activity derives from the tumor itself rather than surrounding non-tumor cells. Despite the relatively low TP53 mutation rate in RCC, TP53 mutations are associated with poor prognosis,38 and expression of the KIF2C (kinesin family member 2C) protein, which regulates cellular senescence of human primary cells via a p53-dependent pathway, is higher in TP53 mutated cancers,39 including RCC.40 Given the increased senescence and inflammation signatures observed in M1 tumors, we wanted to determine if senescence activity associates with inflammation and T cell checkpoint marker expression and if these associations are different between M1 and M0 tumors.
Tumor senescence associates with inflammation inducers differently in M0 tumors compared to M1
In order to determine if the association between TP53-inactivation induced senescence enrichment and immune cytolytic activity varies based on tumor aggressiveness, we implemented multivariate regression modeling. In other words, we wanted to know if the effect of senescence enrichment on expression of key molecules and pathways depends on whether senescence enrichment is observed in M0 or M1 tumors. Specifically, we examined hypoxia pathways, which are known to induce inflammation (Table 2, Table S10), and the expression of the cystolic DNA sensors cGAS and STING, which directly link senescence activity with immunity. We also examined the expression of PRF1 and GZMA, which are known predictive markers of immune cytolytic activity across multiple cancer types as demonstrated by Rooney et al.41 Hypoxia (MANIA_HYPOXIA_VHL_UP)42 and senescence (SENESCENCE_TP53_TARGETS_UP) enrichment associate differently depending on if the tumor is metastatic or not (Table 2, Table S10). Similarly, the metastasis state of the tumor appears to modify the association between senescence enrichment and expression of the cystolic DNA sensors, cGAS and STING as well as expression of the immune cytolytic markers, GZMA and PRF1 (Table 2, Table S10). Based on these results, we explored if considering the combined effect of senescence and inflammation was predictive of prognosis in individuals who received checkpoint blockade therapy.
Combined effect of tumor senescence and inflammation influence response to checkpoint blockade therapy
We implemented regression modeling to determine if the interactions between senescence pathway activity (SENESCENCE_TP53_TARGETS_UP) and specific immune markers, such as PDCD1, MB21D1 (cGAS), GZMA, and PRF1 influence response to checkpoint blockade therapy as measured by percent change in tumor size from baseline and best RECIST score (Table 3–4). The molecules PDCD1, cGAS, and GZMA, PRF1, and GZMB were specifically examined because cGAS is known to induce SASP in tumor cells, PDCD1 is targeted by anti-PD-1 therapy and is expressed by both exhausted and activated T cells, and expression of GZMA, PRF1, and GZMB, is indicative of cytolytic immune activity. Tumors with increased expression of immuno-modulatory molecules but low SENESCENCE_TP53_TARGETS_UP enrichment are associated with poor response to checkpoint blockade therapy, while tumors with both increased SENESCENCE_TP53_TARGETS_UP enrichment and increased expression of immunomodulatory molecules are associated with good response to checkpoint blockade therapy. These findings are highlighted in Table 3 (M1-M5), which shows senescence enrichment alone and expression of immunomodulatory molecules alone are associated with poor response to treatment ( is negative), while the interaction between senescence enrichment and immunomodulatory molecules is associated with good response to treatment ( is positive). This suggests high SENESCENCE_TP53_TARGETS_UP activity coupled with immune activation is predictive of positive response from checkpoint blockade therapy in RCC. To further evaluate these results, we examined the association of senescence immune interactions with percent change in tumor size from baseline in the immunotherapy cohort and observed similar results (Table 4). Moreover, we compared the performance of our senescence immune interaction predictors with the immunophenotype score (IPS), which characterizes tumor immunogenicity. Overall, senescence immune interactions were superior predictors of checkpoint blockade response in RCC than IPS, as assessed by the AIC and values of the regression models (Table 3–4).
Next, we examined the relationship between senescence and overall survival in patients who either did or did not receive checkpoint blockade therapy. We hypothesized that senescence activity alone may negatively influence survival, while senescence activity coupled with increased immune activation, such as through checkpoint blockade therapy, may improve overall survival. Therefore, senescence activity in individuals treated with checkpoint blockade therapy should display a positive association with survival compared to individuals who did not receive immunotherapy as checkpoint blockade therapy boosts immune activation.
High tumor senescence activity associates with improved overall survival in individuals who have received checkpoint blockade therapy, but it associates with worse overall survival in individuals who have not received checkpoint blockade therapy
We examined the role of TP53-inactivation induced senescence activity on survival in individuals who did (Miao et al. dataset) and did not (TCGA cohort) receive immunotherapy. We computed senescence enrichment scores for all M0 and M1 samples and segregated tumors into high and low scoring based on the median for the distribution. In the TCGA cohort, the SENESCENCE_TP53_TARGETS_DN pathway was predictive of overall survival (Figure 3A) such that increased SENESCENCE_TP53_TARGETS_DN enrichment is associated with worse survival even after adjusting for metastasis status (Table S8).
Figure 3.
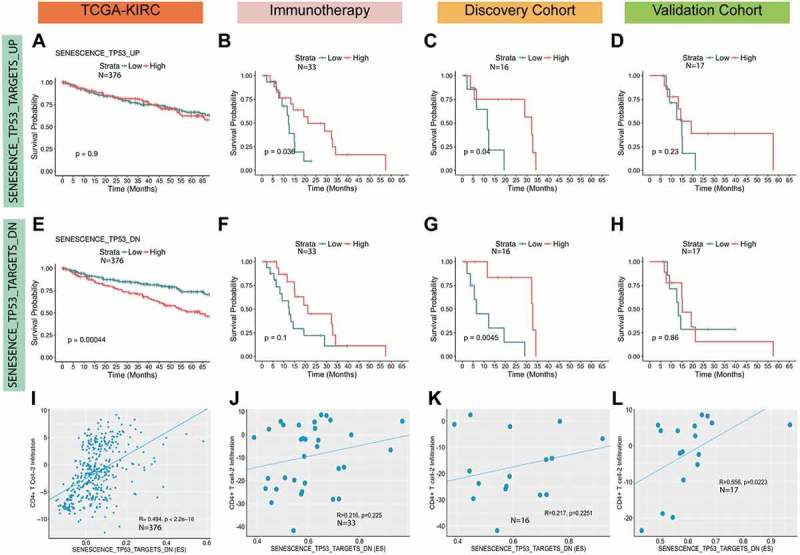
TP53-inactivation induced senescence pathways correlate with survival and T cell infiltration. The effect of high vs. low single-sample gene set enrichment (ssGSEA) scores for the SENESCENCE_TP53_TARGETS_UP (A) and SENESCENCE_TP53_TARGETS_DN (E) pathways in the TCGA RCC (M0 and M1) cohort was examined using Kaplan-Meier (KM) survival curves. High vs. low ssGSEA scores for SENESCENCE_TP53_TARGETS_UP (B) and SENESCENCE_TP53_TARGETS_DN (F) in all individuals who received immunotherapy in the Miao et al., 2018 dataset is also shown as well as KM curves comparing high and low ssGSEA scores for SENESCENCE_TP53_TARGETS_UP (C, D) and SENESCENCE_TP53_TARGETS_DN (G, H) in the discovery and the validation cohorts. The cut-offs for high and low scores was set at the median for the score distribution in each cohort. We also examined the association of SENESCENCE_TP53_TARGETS_DN enrichment with CD4 + T cell-2 infiltration scores in all cohorts (I-L) using the spearman rank correlation test.
In the immunotherapy cohorts, and in the discovery cohort specifically, increased enrichment of the SENESCENCE_TP53_TARGETS_UP pathway was predictive of survival (Figure 3B-C), while the SENESCENCE_TP53_TARGETS_DN pathway was only predictive of overall survival in the discovery cohort (Figure 3G). In the validation cohort alone, we did not find any differences in survival due to high vs. low senescence enrichment. Furthermore, Cox regression, which adjusts for age, sex, and cohort status shows increased SENESCENCE_TP53_TARGETS_DN enrichment associates with improved survival in the immunotherapy cohorts (Table S9). Of note, we did not find any differences between the discovery and validation cohorts with respect to SENESCENCE_TP53_TARGETS_UP43 or SENESCENCE_TP53_TARGETS_DN43 activity (Fig. S2).
Increased senescence enrichment is predictive of worse overall survival in the TCGA dataset. However, in the immunotherapy cohorts and in the discovery cohort specifically, we find increased senescence enrichment improves survival. This reversal in the association between survival and senescence activity based on checkpoint blockade therapy matches our hypothesis, which states senescence activity alone may negatively influence survival, while senescence activity coupled with immune activation, such as via checkpoint blockade therapy, may improve overall survival.
Validation of senescence immune predictors in melanoma
To validate the senescence immune findings from RCC, we evaluated our senescence immune predictors on TCGA SKCM tumors and on the pre-treatment metastatic melanoma anti-PD-1 therapy dataset published by Hugo et al., 2016.35 We show that senescence SENESCENCE_TP53_TARGETS_DN enrichment alone is both a strong predictor of survival and immunotherapy response in metastatic melanoma and it is also a better predictor of immunotherapy response than IPS. However, our senescence immune interactions did not predict immunotherapy response in metastatic melanoma with the exception of STING and SENESCENCE_TP53_TARGETS_UP (see Fig. S11, Table S11).
SENESCENCE_TP53_TARGETS_DN activity is negatively associated with mutations in the BAF/PBAF subunits
As mutations in the PBRM1 gene are associated with T cell-mediated tumor killing,44 delayed onset of tumor senescence,45 and benefit from immunotherapy, we wondered if PBRM1 mutations are associated with senescence enrichment. Using regression modeling, we found a negative association between SENESCENCE_TP53_TARGETS_UP enrichment and PBRM1 mutation allele frequency in the RCC immunotherapy dataset (Fig. S12E). Next, we looked through TCGA RCC mutation data to determine if mutations in BAF/PBAF subunits, including PBRM1, are associated with reduced senescence activity. We found having a mutation in any of the 12 commonly mutated BAF/PBAF subunits associates with reduced senescence activity (Fig. S12A). Moreover, we found three BAF/PBAF subunits, PBRM1, SS18, and BRD7, which independently associate with reduced senescence enrichment (Fig. S12 B-D). Our findings lead us to hypothesize that PBAF/BAF subunit mutations may influence immunotherapy outcomes via their effects on tumor senescence.
Senescence activity correlates with CD4 + T cell-2 infiltration
We find both immune infiltration and senescence activity vary with metastasis status. Therefore, we examined if immune cell types predictive of disease aggressiveness (i.e. metastasis) also correlate with TP53-inactivation induced senescence activity. We found CD4 + T cell-2 infiltration positively correlates with the SENESCENCE_TP53_TARGETS_UP enrichment in the TCGA (= ~ 0.49) and validation (= ~ 0.55) cohorts, but not in the discovery cohort (Figure 3I-L). In addition, we examined the effect of immune infiltration on overall survival in the immunotherapy cohorts, which revealed CD8 + T cell-1 infiltration may be associated with worse survival (trending towards significance p = 0.06) while CD4+ Tcell-2 and activated DC infiltration do not affect overall survival (Figure S5).
Discussion
Our first goal was to determine predictors of disease aggressiveness by comparing M0 and M1 tumors. We found metastatic tumors (M1) are inflamed and senescent compared to non-metastatic (M0) tumors, and they also exhibit increased expression of immune checkpoint markers, which was consistently associated with CD8 + T cell-1 infiltration. Furthermore, we show CD8 + T cell-1 infiltration continues to be associated with poor survival even in individuals treated with checkpoint blockade therapy (Figure S5). These findings align with previous studies, which show CD8 + T cells with increased expression of immune checkpoint markers are associated with poor survival. In addition, M1 tumors potentially show uncoupling of TP53-inactivation induced senescence activity and its downstream pro-inflammatory signals. Our focused analysis of the Miao et al. data suggests this may have implications for response to immunotherapy and overall survival (Figure 1B, Fig. S11, Table S11). Increased activation of oncogenic pathways (Table S7) in M1 tumors may be driving oncogene-induced cellular senescence activity,13 which can paradoxically result in the recruitment of immune cells through the release of cytokines while also promoting pro-tumorigenic effects such as the activation of EMT and hypoxia pathways10,13 (Figure 1B). Notably, we also found strong correlations between EMT, hypoxia and senescence pathways in both the RCC TCGA and the immunotherapy datasets (Fig. S13). As such, our TCGA analysis suggests senescence activity could be developed as a marker of metastasis risk. However, additional in vivo experimental studies should be conducted to support these findings.
Figure 1.
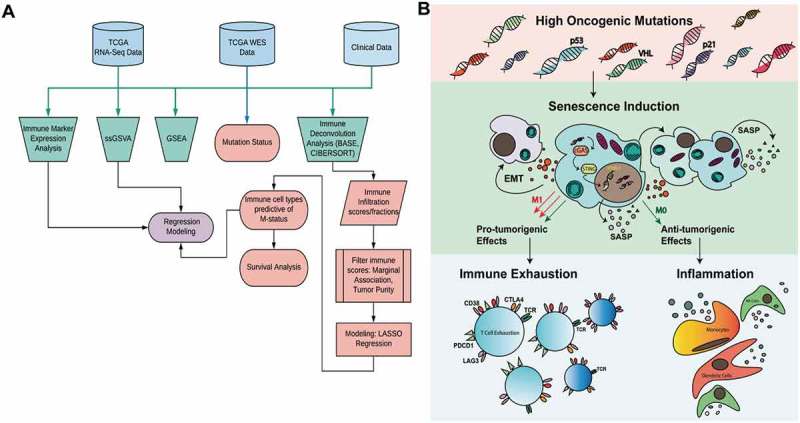
Outline of methods and relationship between senescence and immune cells in the tumor microenvironment. A) TCGA renal clear cell carcinoma RNA-seq and whole exome sequencing data were used in combination with tumor staging and survival information to discern differences between metastatic (M1-stage IV, n = 79) and non-metastatic (M0-stage II/III, n = 297) tumors. Five different analyses were performed to determine immune infiltration, pathway activity, and mutation status. Outputs from these analyses were used in regression models to characterize and assess the role of the tumor microenvironment in metastatic disease. B) A diagram outlining a potential mechanism by which high oncogenic activity can lead to senescence induction, which has both pro and anti-tumorigenic effects.
Previous studies have demonstrated the role of cGAS-STING signaling in linking tumor senescence with innate immunity. For example, cytoplasmic chromatin is also known to trigger inflammation in senescence and cancer by activating the innate DNA sensing pathway cGAS-STING.46 Moreover, cGAS is both a key sensor of cystolic DNA and is essential for DNA-damage induced cellular senescence and SASP, which not only defines senescence, but also leads to the expression of inflammatory cytokines and the activation of innate immunity.12 cGAS-STING activity coupled with anti-PD-1 therapy has been shown to result in the greatest reduction in tumor burden in mouse models of non-small cell lung cancer, and this is thought to result from activation of dendritic cells, and increased antigen presentation to CD8 + T cells.47 We outline a potential mechanism by which senescence may influence the TME in Figure1B. Based on our results, it appears senescence activity alone is associated with poor survival in RCC, while senescence activity in individuals who receive immunotherapy is associated with improved survival. This suggests senescence coupled with activation of immunity via cGAS-STING signaling is predictive of checkpoint blockade therapy response in RCC as demonstrated from our predictive regression models (Table 3–4). Moreover, we also showed that senescence activity is a strong predictor of survival and anti-PD-1 response in metastatic melanoma (Table S11, Fig. S11). However, we do note that senescence immune interactions behave differently in melanoma than in RCC and these effects may be tumor type or tumor immune landscape specific. Additional in vivo studies should be performed to test the role of senescence immune modulation in multiple cancer types.
Lastly, we examined the relationship between senescence and mutations in PBRM1, and, more broadly, in BAF/PBAF subunits. PBRM1 encodes a subunit of the SWI/SNF chromatin complex, is a regulator of p53, and is required for the induction of replicative senescence.45 Moreover, PBRM1 knockdown is associated with a delay in the onset of senescence activity. Our findings match the literature as individuals with a PBRM1 mutation tend to have reduced senescence activity. We also showed mutations in other BAF/PBAF subunits associate with reduced senescence activity (Fig S12A-D). However, the exact mechanism by which PBRM1 or BAF/PBAF subunit mutations influence senescence remains unclear.
In conclusion, our assessment of TME predictors of disease aggressiveness and subsequent analyses on immunotherapy datasets led us to hypothesize a potential mechanism linking senescence with tumor inflammation in RCC, which we have outlined in Figure 1B. We hope this hypothesis generating research allows for future studies to better characterize the role of senescence in influencing the TME and its potential implications in immunotherapy treatment.
Limitations and future directions
It should be noted that it is difficult to assess which specific cell types exhibit senescence from a bulk tumor analysis. The lack of increased expression of immune senescence specific markers, such as CD57, KLRG-1, and CD160, suggested tumor cells and not immune cells in the RCC M1 TME undergo senescence. However, examination of these markers alone is insufficient for determining which cell types activate cellular senescence pathways. Single-cell sequencing would better allow us to elucidate the role of senescence in tumors and or immune cells. Furthermore, experimental evidence is needed to validate our computational findings on the possible role of different immune cell subsets in influencing the TME, as a major drawback of computational methods is that they are correlative and not causative, and therefore can be used only for hypothesis generating purposes. Additionally, while LASSO deals with co-linear predictors, it picks the most predictive of the co-linear predictors, which is statistically appropriate, but may not reflect the underlying biology. Although we have outlined a potential mechanism to help explain differences in response to checkpoint blockade therapy in RCC, due to the correlative nature of our study, our findings on the role of cellular senescence and innate immunity in influencing treatment outcome should also be considered as hypothesis generating. However, future studies on determining predictors of immunotherapy outcome should consider incorporating information on tumor senescence.
Funding Statement
This work was supported by the HHS | NIH | U.S. National Library of Medicine (NLM) [1K01LM012426]; HHS | NIH | U.S. National Library of Medicine (NLM) [5T32LM012204-03].
Acknowledgments
The authors are grateful for the financial support from research grants, (1) 5T32LM12204-2 NIH-NLM, (2) 1K01LM012426 NIH-NLM. We would also like to thank Dr. Frederick S. Varn and Dr. Elizer Van Allen for their comments and support on this manuscript.
Conflict of Interest
There are no conflicts of interest for all authors
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be access on the publisher’s website.
References
- 1.Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V. Renal cell carcinoma. Nat Rev Dis Prim. 2017;3:17009. doi: 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meskawi M, Sun M, Trinh Q-D, Bianchi M, Hansen J, Tian Z, Rink M, Ismail S, Shariat SF, Montorsi F, et al. A review of integrated staging systems for renal cell carcinoma. Eur Urol. 2012;62(2):303–314. doi: 10.1016/j.eururo.2012.04.049. [DOI] [PubMed] [Google Scholar]
- 3.Weinstock M, McDermott D.. Targeting PD-1/PD-L1 in the treatment of metastatic renal cell carcinoma. Ther Adv Urol. 2015;7(6):365–377. doi: 10.1177/1756287215597647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choueiri TK, Fishman MN, Escudier B, McDermott DF, Drake CG, Kluger H, Stadler WM, Perez-Gracia JL, McNeel DG, Curti B,et al. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma. Clin Cancer Res. 2016;22(22):5461–5471. doi: 10.1158/1078-0432.CCR-15-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bossé D, Wankowicz SM, Cullen D, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science (80-). 2018;359(6377):801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster WS, Lohse CM, Thompson RH, Dong H, Frigola X, Dicks DL, Sengupta S, Frank I, Leibovich BC, Blute ML, et al. Mononuclear cell infiltration in clear-cell renal cell carcinoma independently predicts patient survival. Cancer. 2006;107(1):46–53. doi: 10.1002/cncr.21951. [DOI] [PubMed] [Google Scholar]
- 7.Geissler K, Fornara P, Lautenschläger C, Holzhausen H-J, Seliger B, Riemann D. Immune signature of tumor infiltrating immune cells in renal cancer. Oncoimmunology. 2015;4(1):e985082. doi: 10.4161/2162402X.2014.985082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giraldo NA, Becht E, Pages F, Skliris G, Verkarre V, Vano Y, Mejean A, Saint-Aubert N, Lacroix L, Natario I, et al. Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res. 2015;21(13):3031–3040. doi: 10.1158/1078-0432.CCR-14-2926. [DOI] [PubMed] [Google Scholar]
- 9.Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007;117(11):3421–3426. doi: 10.1172/JCI32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker DJ, Alimirah F, van Deursen JM, Campisi J, Hildesheim J. Oncogenic senescence: a multi-functional perspective. Oncotarget. 2017;8(16):27661–27672. doi: 10.18632/oncotarget.15742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74(3):665–674. doi: 10.1158/0008-5472.CAN-13-0992. [DOI] [PubMed] [Google Scholar]
- 12.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci. 2017;114(23):E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davalos AR, Coppe J-P, Campisi J, Desprez P-Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29(2):273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–1070. doi: 10.1038/ncb3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen K, Liu J, Cao X. cGAS-STING pathway in senescence-related inflammation. Natl Sci Rev. December 2017. doi: 10.1093/nsr/nwx146. [DOI] [Google Scholar]
- 16.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network, Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, et al. Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R Foundation for Statistical Computing R: A language and environment for statistical computing. Vienna, Austria: Accessed December6, 2017 https://www.r-project.org/. [Google Scholar]
- 19.Wickham H. ggplot2. Cham: Springer International Publishing; 2016. doi: 10.1007/978-3-319-24277-4. [DOI] [Google Scholar]
- 20.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 21.Varn FS, Andrews EH, Mullins DW, Cheng C. Integrative analysis of breast cancer reveals prognostic haematopoietic activity and patient-specific immune response profiles. Nat Commun. 2016;7:10248. doi: 10.1038/ncomms10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heng TSP, Painter MW, Immunological Genome Project Consortium, Elpek K, Lukacs-Kornek V, Mauermann N, Turley SJ, Koller D, Kim FS, Wagers AJ, et al. The immunological genome project: networks of gene expression in immune cells. Nat Immunol. 2008;9(10):1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 23.Varn FS, Wang Y, Mullins DW, Fiering S, Cheng C. Systematic pan-cancer analysis reveals immune cell interactions in the tumor microenvironment. Cancer Res. 2017;77(6):1271–1282. doi: 10.1158/0008-5472.CAN-16-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16(1):64. doi: 10.1186/s13059-015-0620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J R Stat Soc Ser B (Statistical Methodol). 2011;73(3):273–282. doi: 10.1111/j.1467-9868.2011.00771.x. [DOI] [Google Scholar]
- 26.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Godec J, Tan Y, Liberzon A, Tamayo P, Bhattacharya S, Butte AJ, Mesirov JP, Haining WN. Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity. 2016;44(1):194–206. doi: 10.1016/j.immuni.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14(1):7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, . Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, Parsons R, Zhao K, Wang W. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005;19(14):1662–1667. doi: 10.1101/gad.1323805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1(5):e1500447. doi: 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hack H, Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18(1):248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 37.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Peng S, Jiang N, Wang A, Liu S, Xie H, Linpei G, Cai Q, Niu Y. Prognostic and clinicopathological value of p53 expression in renal cell carcinoma: a meta-analysis. Oncotarget. 2017;8(60):102361–102370. doi: 10.18632/oncotarget.21971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gwon M-R, Cho JH, Kim J-R. Mitotic centromere-associated kinase (MCAK/Kif2C) regulates cellular senescence in human primary cells through a p53-dependent pathway. FEBS Lett. 2012;586(23):4148–4156. doi: 10.1016/j.febslet.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Sun Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget. 2017;8(1):624–643. doi: 10.18632/oncotarget.13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maina EN, Morris MR, Zatyka M, Raval RR, Banks RE, Richards FM, Johnson CM, Maher ER. Identification of novel VHL target genes and relationship to hypoxic response pathways. Oncogene. 2005;24(28):4549–4558. doi: 10.1038/sj.onc.1208649. [DOI] [PubMed] [Google Scholar]
- 43.Tang X, Milyavsky M, Goldfinger N, Rotter V. Amyloid-β precursor-like protein APLP1 is a novel p53 transcriptional target gene that augments neuroblastoma cell death upon genotoxic stress. Oncogene. 2007;26(52):7302–7312. doi: 10.1038/sj.onc.1210542. [DOI] [PubMed] [Google Scholar]
- 44.Pan D, Kobayashi A, Jiang P, de Andrade LF, En Tay R, Luoma A, Tsoucas D, Qiu X, Lim K, Rao P, et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science (80-). 2018;359(6377):770–775. doi: 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burrows AE, Smogorzewska A, Elledge SJ. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci. 2010;107(32):14280–14285. doi: 10.1073/pnas.1009559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;2017. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci. 2017;114(7):1637–1642. doi: 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.