

Abstract
The IκB kinase (IKK) is considered to control gene expression primarily through activation of the transcription factor NF‐κB. However, we show here that IKK additionally regulates gene expression on post‐transcriptional level. IKK interacted with several mRNA‐binding proteins, including a Processing (P) body scaffold protein, termed enhancer of decapping 4 (EDC4). IKK bound to and phosphorylated EDC4 in a stimulus‐sensitive manner, leading to co‐recruitment of P body components, mRNA decapping proteins 1a and 2 (DCP1a and DCP2) and to an increase in P body numbers. Using RNA sequencing, we identified scores of transcripts whose stability was regulated via the IKK‐EDC4 axis. Strikingly, in the absence of stimulus, IKK‐EDC4 promoted destabilization of pro‐inflammatory cytokines and regulators of apoptosis. Our findings expand the reach of IKK beyond its canonical role as a regulator of transcription.
Keywords: EDC4, IKK, P bodies, post‐transcriptional regulation, RNA stability
Subject Categories: Immunology, RNA Biology, Signal Transduction
Introduction
The IKK complex is a key mediator of cellular response to numerous stimuli, including DNA damage, bacterial and viral antigens, cytokines and oxidative stress (Hayden & Ghosh, 2008; Oeckinghaus et al, 2011; Hinz & Scheidereit, 2014). It consists of the catalytic subunits, IKKα and IKKβ, and the regulatory subunit IKKγ (NEMO). IKK is believed to exert its function primarily through activation of the downstream transcription factor, NF‐κB. In response to stimulus, IKK phosphorylates IκB proteins, leading to their proteolysis and liberation of NF‐κB. The latter drives the expression of numerous genes regulating cell proliferation and differentiation, apoptosis and inflammation (Karin & Ben‐Neriah, 2000; Hayden & Ghosh, 2008). Constituting the largest fraction of transcriptional targets of NF‐κB are inflammatory cytokines and chemokines that are key modulators of the immune response. Strength and duration of their expression are further regulated by mRNA stability, determined by intrinsic cis elements, frequently located in the 3′ untranslated region (UTR), and by recruited trans factors, including RNA‐binding proteins (RBPs; Hao & Baltimore, 2009; Schoenberg & Maquat, 2012). Many mammalian mRNAs contain AU‐rich elements (AREs) that generally render the transcript less stable and more prone to decay.
RNA‐binding proteins can promote rapid degradation of their target mRNA in either the 3′ to 5′ or in the 5′ to 3′ direction. The latter requires the removal of the 5′ cap structure by DCP1a and DCP2, followed by exonucleolytic digestion by exoribonuclease 1 (XRN1; Parker & Sheth, 2007). EDC4 is essential for the decapping process as it provides a scaffold for DCP1a, DCP2 and XRN1 (Jonas & Izaurralde, 2013; Chang et al, 2014). In addition, EDC4 is required for the assembly of the RBPs into higher order complexes, known as processing bodies (P bodies; Yu et al, 2005; Decker et al, 2007; Parker & Sheth, 2007). P bodies belong to a group of non‐membrane‐bound organelles, containing both proteins and RNA (Braun et al, 2012). mRNAs might be targeted to P bodies for degradation (Brengues et al, 2005; Teixeira et al, 2005). Other studies have shown that mRNAs are stored in P bodies and can be released to enter translation (Brengues et al, 2005; Bhattacharyya et al, 2006) and that a vast majority is protected from 5′ decay (Kedersha et al, 2005). In addition, a recent genome‐wide study revealed that decay or stabilization of P‐body‐associated mRNAs is controlled in a context‐dependent manner and in response to stress (Teixeira et al, 2005). These multiple functions of P bodies may thus strongly impact the timing and amplitude of cellular responses to exogenous stimuli.
Here, we show a direct link between IKK and change in RNA stability of scores of transcripts. We identified the P body scaffold protein EDC4 as a novel interaction partner of the IKK complex. IKKβ phosphorylated EDC4 when activated by TNFα, IL‐1β, or genotoxic or oxidative stress. Phosphorylation of EDC4 was crucial for its interaction with other RBPs, and subsequent formation of higher order complexes, reflected in an increase in detectable P bodies. RNA‐Seq analysis revealed post‐transcriptional regulation of hundreds of mRNAs by the IKK‐EDC4 axis in response to stress. Formerly, IKK‐dependent gene regulation was regarded to occur via activation of the transcription factor NF‐κB. Our data provide evidence for a global mechanism and an extensive NF‐κB‐independent function of the IKK complex in the regulation of mRNA stability.
Results
IKK complex interacts with P body scaffold protein EDC4
We identified novel interaction partners of IKKγ by co‐immunoprecipitation from SILAC‐labelled human osteosarcoma cells treated with ionizing irradiation (IR; Fig 1A, Appendix Fig S1A and Table EV1). Gene Ontology (GO) term analysis and clustering of the proteins with increased association with IKKγ after IR stimulation showed an expected enrichment for the biological processes “response to DNA damage stimulus”, “post‐translational protein modification”, “regulation of apoptosis”, but interestingly, also for “mRNA metabolic process” (Appendix Fig S1B). The latter group comprised numerous RNA‐binding proteins, exoribonuclease 3′‐5′ (DIS3), RNase inhibitor 1 (RNH1), and P body proteins Poly(RC) binding protein 1 (PCBP1), and EDC4, among others (Appendix Fig S1A). EDC4 is essential for P body formation and was therefore selected for further analysis.
Figure 1. EDC4 interacts with IKK in a stimulus‐sensitive manner.
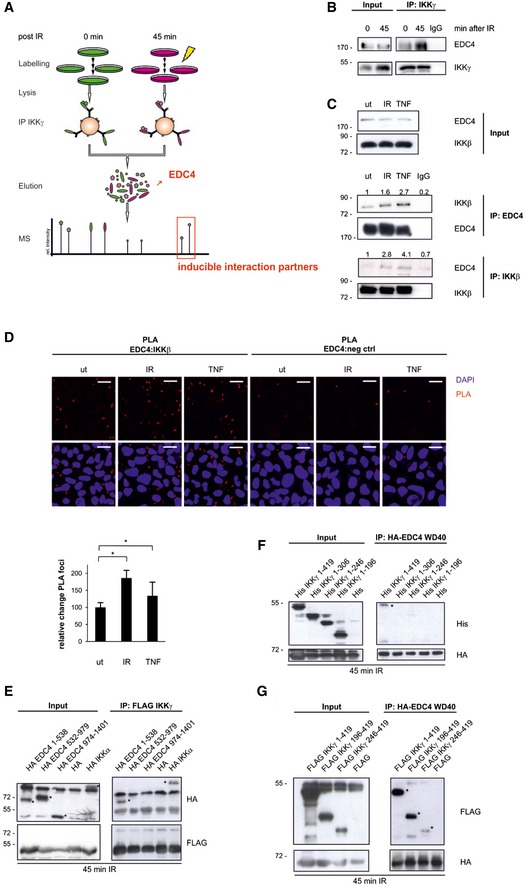
- Schematic diagram of the SILAC screen.
- Immunoprecipitation of endogenous IKKγ from cytoplasmic cell lysates of unstimulated (ut) or irradiated U2‐OS cells followed by Western blot (WB) of IKKγ and EDC4.
- Immunoprecipitation of endogenous EDC4 or IKKβ from unstimulated (ut), irradiated (20 Gy; 45 min post‐stimulus) or TNFα‐treated (10 ng/ml; 15 min) U2‐OS cells, WB of EDC4 or IKKβ. Fold changes of co‐precipitated proteins are indicated. Results are representative for three experiments.
- Proximity ligation assay (PLA) of unstimulated (ut), irradiated (20 Gy: 45 min post‐stimulus) or TNFα‐treated (10 ng/ml; 45 min post‐stimulus) U2‐OS cells; IKKβ‐EDC4 interaction (red), nucleus (blue). As a negative control, PLA was performed without primary IKKβ antibody. Scale bar: 25 μm. Bottom panel: relative quantitation of independent experiments (n = 2) by ImageJ software, at least 500 cells per experiment ± s.d. unpaired t‐test, *P < 0.05.
- Co‐expression of full‐length FLAG‐IKKγ with HA‐tagged EDC4 sub‐regions, wild‐type HA‐IKKα (positive control) or empty vector (negative control) in HEK293 cells, followed by immunoprecipitation of FLAG‐IKKγ and WB of FLAG and HA. Immunoprecipitation was performed with anti‐FLAG sepharose from whole‐cell lysates of irradiated (10 Gy) cells. Left panel: input, right panel: FLAG‐IP. Asterisks denote specific bands. Note that the three EDC4 fragments reveal an aberrant migration relative to calculated molecular weight in SDS–PAGE (Braun et al, 2012).
- HEK293 cells expressing N‐terminally His‐tagged IKKγ deletion constructs (as shown in Appendix Fig S1H) or empty His‐vector together with HA‐tagged EDC4 WD40 domain (HA‐EDC41–538). Immunoprecipitates from whole‐cell lysates of irradiated cells with anti‐HA sepharose were analysed by WB for His and HA. Left panel, input; right panel, HA‐IP. Asterisks, specific bands.
- Co‐immunoprecipitation as in (F) with C‐terminally FLAG‐tagged IKKγ.
Source data are available online for this figure.
Co‐immunoprecipitation of endogenous IKK and EDC4 confirmed the increased interaction in response to irradiation (Fig 1A and B, Appendix Fig S1A). To determine whether EDC4 is a substrate of the IKK complex, interaction of EDC4 with IKKβ was analysed. IKKβ is activated by DNA damage and also other canonical IKK stimuli, including TNFα. Reciprocal co‐immunoprecipitation of endogenous IKKβ and EDC4 showed a basal interaction that was enhanced in response to IR and to TNFα (Fig 1C). To rule out the possibility that this interaction was mediated through RNA, reciprocal co‐immunoprecipitations were repeated in the presence of RNase A and RNase T1 (Appendix Fig S1C). In situ analysis by proximity ligation assays (PLAs) revealed a direct and an IR‐ or TNFα‐enhanced interaction between EDC4 and IKKβ (Fig 1D). These findings suggest that IKK‐EDC4 interaction is not restricted to DNA damage signalling, but can be induced by different stimuli that activate IKK. We further expanded our analysis to other components of P bodies and to the IKK cascade and showed DNA damage‐induced interaction of IKKγ with P body marker DDX6 (Appendix Fig S1D) and of EDC4 with TRAF6 (TNF receptor‐associated factor 6), which is an essential component of the cytoplasmic IKK signalling cascade (Appendix Fig S1E; Hinz et al, 2010).
We next asked which protein domains are required for the IKKγ‐EDC4 interaction (Fig 1E–G, Appendix Fig S1F–H). The EDC4 protein contains an N‐terminal WD40 domain and a C‐terminal α‐helical domain connected by a serine‐rich linker region (Appendix Fig S1G). In vitro co‐IP assays showed that the N‐terminal WD40 domain is necessary and sufficient for interaction with full‐length IKKγ (Appendix Fig S1F). Precipitation of sub‐regions of EDC4 from irradiated cells confirmed that the WD40 domain of EDC4 interacts with IKKγ (Fig 1E).
We further showed that the leucine zipper and zinc‐finger motifs in IKKγ are required for binding to EDC4 (Fig 1F and G, Appendix Fig S1H). Collectively, these results demonstrate that cellular stress promotes interaction between EDC4 and the IKK complex.
The IKK complex phosphorylates EDC4 on serines in the WD40 domain and in the serine‐rich linker
To identify IKK substrate sites in EDC4, we performed in vitro kinase assays with endogenous IKK and the purified recombinant sub‐regions of EDC4. IR, TNFα and IL‐1β all led to inducible phosphorylation of EDC4 in the WD40 domain and the serine‐rich linker region, indicating that canonical IKKγ/IKKβ signalling mediates phosphorylation of EDC4 (Fig 2A).
Figure 2. IKK phosphorylates EDC4.
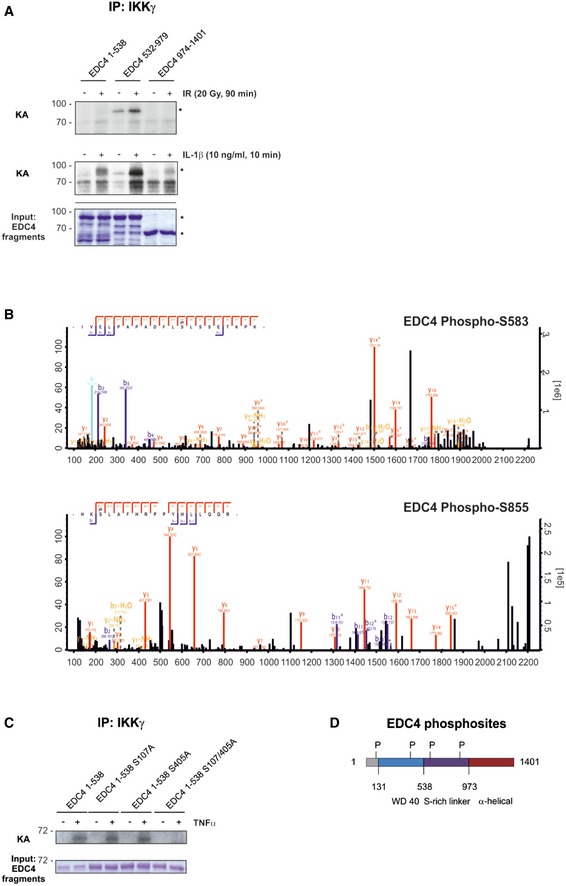
- In vitro kinase assay (KA) using endogenous IKKβ from cells, unstimulated or stimulated with IR (20 Gy, 45 min; top panel) or IL‐1β (10 ng/ml, 10 min; bottom panel) with purified GST‐EDC4 domains as indicated. Lower panel: cold KA as above, Coomassie blue staining. Asterisks denote specific bands.
- IKK phosphosite identification in EDC4 by mass spectrometry (see Table EV2 for MS data). Endogenous IKK purified from unstimulated or TNFα‐treated (10 ng/ml, 15 min) U2‐OS cells by immunoprecipitation of IKKγ was used in a cold KA with recombinant EDC4 sub‐regions, followed by MS analysis. Top, MS spectrum for phospho‐serine 583. Bottom, MS spectrum for phospho‐serine 855.
- In vitro KA of IKKβ (as in A) from TNFα‐stimulated cells with purified recombinant Strep‐EDC4 WD40 domain (EDC4 1–538) and point mutants for IKK phosphosites, S107A, S405A and S107/405A. Below: cold kinase assay.
- Diagram of EDC4 indicating IKKβ‐phosphorylated serines.
Source data are available online for this figure.
Subsequent MS analysis revealed serines 583 and 855 in the serine‐rich linker region of EDC4 as IKK phosphosites (Fig 2B, Table EV2). Because of limited sequence coverage by MS, we searched the dbPTM database and found serines 107 and 405 as further potential IKK phosphorylation sites. Combined substitution of serines 107 and 405 with alanines, but not of each alone, resulted in complete loss of IKK‐mediated phosphorylation (Fig 2C). These data confirmed that IKK phosphorylates EDC4 at two serines in the WD40 domain of EDC4 (Ser107 and Ser405) and at two serines in the serine‐rich linker region of EDC4 (Ser583 and Ser855; Fig 2D). To confirm that phosphorylation of EDC4 by IKK occurs in cells, we additionally performed MS analysis on endogenous, immunoprecipitated EDC4 in IKKβ or EDC4 CRISPR knockout cell lines or control cells (Appendix Fig S2A–C, Table EV3). TNFα stimulation resulted in phosphorylation of EDC4 at Ser583 in an IKK‐dependent manner. This site, along with serines 107 and 855, was likewise reported as phosphosite of EDC4 by the PhosphoSitePlus.org database (Appendix Fig S2D). In summary, we demonstrated that EDC4 is an IKK substrate in cells.
Phosphorylation of EDC4 by IKK enhances formation of P bodies
EDC4 is essential for the assembly of RBP complexes and RNA, whose microscopically detectable foci are referred to as P bodies (Yu et al, 2005). P bodies are detectable in eukaryotic cells under normal cellular conditions, but increase in size and number in response to exogenous and endogenous stress (Kedersha et al, 2005; Teixeira et al, 2005). Furthermore, P body size and number are proportional to and dependent on the pool of translationally silenced mRNAs (Parker & Sheth, 2007). Therefore, we analysed changes in P body formation following DNA damage. The number of P bodies was significantly increased after irradiation, as seen by immunofluorescence staining of cells with P body marker DDX6 in different human primary and cancer cell lines (Fig 3A and Appendix Fig S3A and B). Furthermore, both IKKβ and IKKγ co‐localized to the P body foci and the number of foci containing EDC4 and IKKβ or IKKγ increased after stimulation (Appendix Fig S3C and D). Since IKK directly phosphorylates EDC4, we tested whether other known IKK stimuli would also affect P body formation. Indeed, TNFα, IL‐1β or hydrogen peroxide‐induced T‐loop phosphorylation of IKKβ, as expected, and led to an increase in P body numbers (Appendix Fig S3E–J). Depletion of IKKβ or IKKγ attenuated P body induction in stimulated cells (Fig 3B and Appendix Fig S3K–N). Similarly, an IKK inhibitor abrogated P body induction in response to DNA damage (Appendix Fig S3O).
Figure 3. Phosphorylation of EDC4 by IKK promotes P body formation.
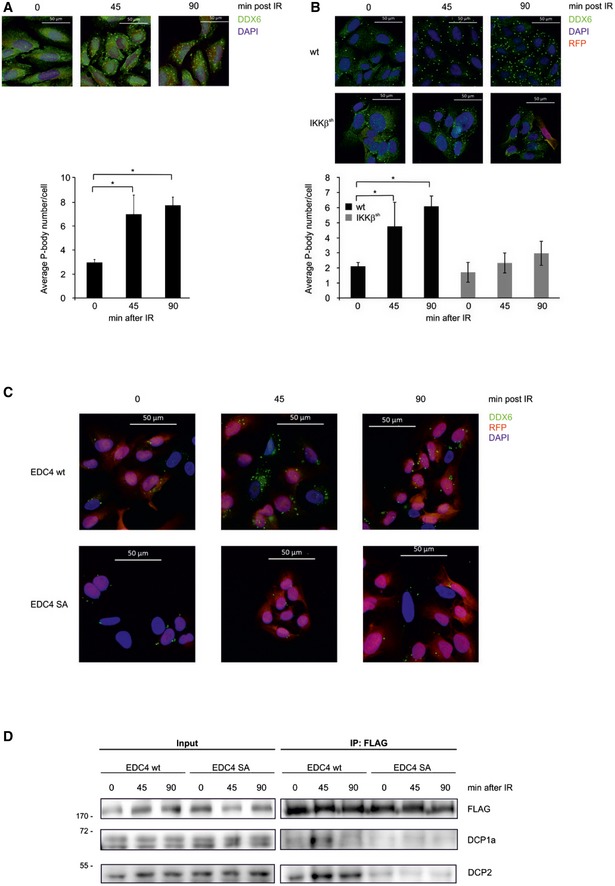
- Fluorescence microscopy using anti‐DDX6 antibody (green) of untreated U2‐OS cells, or 45 or 90 min post‐irradiation (IR). Nuclei stained with DAPI (blue), scale bar 50 μm. Bottom panel: quantification of P‐body foci from independent experiments (n = 3) by ImageJ software, 100 cells per experiment ± s.d. unpaired t‐test, *P < 0.01. Equivalent results were obtained with staining for P body components EDC4, DCP1a and DCP2 (not shown).
- As described in panel (A), except in clonal U2‐OS cells stably expressing pTRIPZ RFP‐coupled shIKKβ. Cells were treated with doxycycline (IKKβsh) or left untreated (wt) and analysed 45 or 90 min after IR. Bottom panel: P body quantification as in (A).
- Fluorescence microscopy using EGFP‐tagged EDC4 (green) and DAPI in clonal U2‐OS cells stably expressing pTRIPZ RFP‐coupled shEDC4 and reconstituted with either wild‐type EGFP‐tagged EDC4 or EGFP‐tagged EDC4 phospho‐deficient Ser107/405/583/855Ala mutant (SA). Cells were pre‐treated with doxycycline to deplete endogenous EDC4 prior to IR.
- Immunoprecipitation of clonal U2‐OS cells stably expressing pTRIPZ RFP‐coupled shEDC4 to deplete endogenous EDC4 and reconstituted with either shRNA‐resistant wild‐type FLAG‐tagged EDC4 or FLAG‐tagged EDC4 phospho‐deficient mutant (SA). Endogenous EDC4 was depleted with doxycycline treatment prior to IR and co‐immunoprecipitation with anti‐FLAG sepharose. IP lysates were analysed by Western blot with anti‐FLAG, anti‐DCP1a and anti‐DCP2 antibodies. Left: input. Right: FLAG‐IP.
Source data are available online for this figure.
Depletion of EDC4 confirmed its essential role in P body formation (Appendix Fig S3P–S; Yu et al, 2005). CRISPR‐mediated knockout of IKKβ or EDC4 confirmed the requirement of these two factors for the induction of P body formation in response to IR and TNFα (Appendix Fig S3S). Of note, extended absence of IKKβ in the CRISPR‐mediated knockout cells led to a higher basal number of P bodies. But neither IR nor TNFα led to a further increase in foci number (Appendix Fig S3S, right panel), as also seen with IKKβ or IKKγ siRNA‐mediated depletion or treatment with an IKK inhibitor. We reconstituted EDC4‐depleted cells with EDC4 wild‐type (wt) or with non‐phosphorylatable EDC4 SA mutant to determine whether phosphorylation of EDC4 by IKK was necessary for P body induction. Reintroduction of wt EDC4 rescued formation of P bodies and their stimulus‐dependent increase. In contrast, introduction of the EDC4 SA mutant led to a low basal level of P body formation and could not restore their amplification (Fig 3C). Moreover, the induced interaction of P body components DCP1a and DCP2 was only observed with the wt EDC4, but not with the EDC4 SA mutant (Fig 3D). Further factors required for mRNA stabilization and storage are likely assembled in the same manner. Taken together, these findings suggest that phosphorylation of EDC4 by IKK promotes assembly of P bodies in response to diverse stimuli.
P bodies share many components with cytoplasmic stress granules (SGs; Kedersha et al, 2005). To determine whether SGs might be connected to the IKK‐EDC4 response, we analysed whether SG formation would be affected by agents that activate IKK (Appendix Fig S4A). Only hydrogen peroxide treatment, as expected, but not TNFα, IL‐1β or IR, led to significant SG formation at analysed time points. Thus, IKK signalling significantly affects the formation of P bodies but not of SGs (Appendix Fig S4B).
IKK‐EDC4 axis differentially regulates stability of hundreds of transcripts
To identify transcripts post‐transcriptionally regulated by IKK or EDC4 in unstimulated cells or in response to DNA damage, we performed actinomycin D chase experiments with wild‐type, EDC4‐ or IKKβ‐depleted cells followed by RNA‐Seq (Fig 4). We used an early IR time point where we detected induction of P bodies (see above), but no significant transcriptional response via the IKK‐NF‐κB axis (Fig 4A, Tables EV4 and EV5 and Appendix Fig S5A and B). Transcript expression (due to changes in transcription, co‐transcriptional regulation and post‐transcriptional stabilization or degradation) was affected by DNA damage in both positive and negative manner (Appendix Fig S5A and B). Expression of genes responsible for cell proliferation and differentiation was negatively regulated (Appendix Fig S5C). A significant number of transcripts were, however, controlled solely post‐transcriptionally (Appendix Fig S5D). The functional cluster “Transcription from RNA Polymerase II promoter” was attenuated both through altered expression and through destabilization (Appendix Fig S5C and D). Additionally, irradiation increased the stability of inflammatory and cytokine responses (Appendix Fig S5D–F).
Figure 4. EDC4 and IKK regulate stability of multiple transcripts.
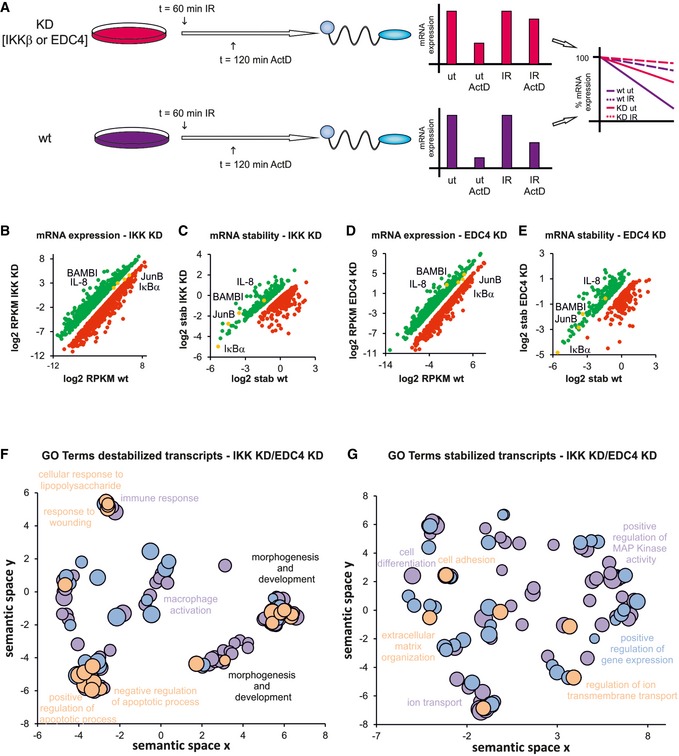
- Schematic diagram of ActD chase experiments to determine stability of transcripts. pTRIPZ constructs encoding shIKKβ or shEDC4 (as above) were treated with dox to induce knockdown (KD, IKKβsh, EDC4sh) or left untreated (wt). ActD treatment, 120 min, IR at 60 min prior to harvest. RNA was analysed by qRT–PCR. Bar charts represent raw data. Line graphs represent normalized stability, were IR or unstimulated samples were set at 100% and ActD‐treated samples represent residual expression.
- Graph showing mRNAs with increased (green) or decreased (red) expression in IKK‐depleted cells (see A) relative to wt. mRNA expression was measured by RNA‐Seq, and significance was calculated for two corresponding conditions and cut‐off set for P‐value < 0.1 and fold change > 2 (Table EV4A)
- Graph illustrating mRNAs with increased (green) or decreased (red) stability in IKK‐depleted cells (see A) relative to wt. mRNA stability was determined by RNA‐Seq (Table EV4B).
- Analysis of mRNA expression in EDC4‐depleted cells relative to wt as in (A; Table EV4C).
- Analysis of mRNA stability in EDC4‐depleted cells relative to wt as in (B; Table EV4D).
- GO terms of transcripts which are destabilized by IKK or EDC4 using REVIGO for removal of redundancy of GO terms (Supek et al, 2011). Scatterplot clustering of groups with functionally similar GO terms. Each circle represents a GO term; size indicative of gene target numbers; the x‐axis and y‐axis are semantic coordinates given by REVIGO; violet: EDC4‐depleted, blue: IKK‐depleted, orange: terms identical for both EDC4 and IKK‐depleted samples; P‐value < 0.05 (Table EV4E). Note that semantic co‐positioning in the plot indicates functional similarity of GO terms.
- Visualization of GO terms of mRNAs that are stabilized by IKK or EDC4, as in (F; Table EV4F).
A significant proportion of mRNAs whose expression changed in an IKKβ‐ or EDC4‐dependent manner (Fig 4B and D) were regulated via stabilization of their transcripts (Fig 4C and E). Overall, IKKβ or EDC4 regulated the stability of a large number of transcripts, respectively (Fig 4A–E and Table EV4), a third of which showed regulation by both IKK and EDC4 (Table EV4). In addition to the IKK‐EDC4 axis, both factors also independently regulated mRNA stability (Fig 4B–E, Table EV4). This likely involves other RBPs that interact with IKK (Appendix Fig S1A).
Transcripts destabilized via the IKKβ‐EDC4 axis were enriched for GO terms “response to wounding”, “negative regulation of apoptotic process” and “cellular response to lipopolysaccharide” and included numerous NF‐κB targets, which were regulated under these unstimulated conditions in an NF‐κB‐independent manner (Fig 4F, Tables EV4 and EV6). In unstimulated cells, stabilized targets showed enrichment for GO terms “cell adhesion” and “extracellular matrix organization” and “regulation of ion transmembrane transport” (Fig 4G and Table EV4).
Transcripts which were destabilized via the IKK‐EDC4 axis included IL‐8, CSF2, CCL5, CEBPβ and KLF10, while selectively stabilized transcripts comprised CCL7, CCL11, FOXA1, IRF7 and p63 (Table EV4).
IL‐8 mRNA is one of the first identified transcripts whose stability is regulated by P bodies following IL‐1β stimulation and whose transcription is regulated by NF‐κB (Rzeczkowski et al, 2011). We therefore used IL‐8 as a bona fide target of IKK and EDC4 for an in‐depth analysis on the impact on mRNA stability. The stability of IL‐8 mRNA increased after IR treatment compared to its baseline stability in wild‐type cells (Appendix Fig S6A and B).
We first compared exon–exon with exon–intron spanning IL‐8 transcripts (Appendix Fig S6B) and did not detect any unprocessed exon–intron pre‐mRNA for IL‐8 in ActD‐treated cells, confirming complete inhibition of transcription. Therefore, irradiation increased IL‐8 mRNA expression through stabilization of pre‐existing transcripts. Stabilization of IL‐8 mRNA by DNA damage was confirmed in several cell types (Appendix Fig S6C–E) and using the other IKK‐activating stimuli, TNFα, IL‐1β or hydrogen peroxide (Appendix Fig S6F and G).
With RNA‐Seq, we have identified numerous pro‐inflammatory cytokines that are destabilized via the IKK‐EDC4 axis in the absence of stimulus and whose activity shows no further increase in response to IR (Fig 4). We used IL‐8 to analyse how IKK and EDC4, and the phosphorylation state of the latter, regulate post‐transcriptional expression of IL‐8 mRNA. Stable depletion of IKKβ or EDC4 in unstimulated cells resulted in an increase in IL‐8 mRNA due to enhanced stability. Irradiation did not lead to further stabilization (Appendix Fig S6H and I).
To determine whether mRNA stability of IL‐8 depends on the phosphorylation state of EDC4, EDC4‐depleted cells were reconstituted with the wt EDC4 or the phospho‐deficient EDC4 SA mutant. Reconstitution with the wt EDC4 but not EDC4 SA rescued stabilization of IL‐8 mRNA in irradiated cells (Appendix Fig S6J, middle panel). Expression and stability of IL‐8 mRNA in cells expressing EDC4 SA resembled that of EDC4 and IKKβ knockdown cells (Appendix Fig S6H–J, lower panel). These results indicate that EDC4 suppressed the expression of IL‐8 in unstimulated cells. Upon phosphorylation of EDC4 by IKK, degradation of IL‐8 mRNA was abrogated, leading to increased stability of the transcript. Taken together, these data indicate that IKK regulates mRNA stability of IL‐8 via phosphorylation of EDC4.
We additionally validated other targets shown by RNA‐Seq to be regulated via the IKK‐EDC4 axis. Thus, we performed actinomycin D chase experiments on JunB (transcriptional target of NF‐κB), BAMBI (NF‐κB‐independent target) and IκBα (NF‐κB target that did not show stability regulation by the IKK‐EDC4 axis; Appendix Fig S7A–E). To confirm that IKK and EDC4 indeed act along the same axis on these targets, we repeated the experiments with co‐knockdown of EDC4 and IKKβ (Appendix Fig S7E). No additive effects of IKK and EDC4 depletion were observed, suggesting that IKK and EDC4 function through a common axis. RNA transcripts of JunB and BAMBI showed strong regulation by IKK‐EDC4, whereas IκBα mRNA did not show alteration in its stability. These data confirmed that RNA stability of transcripts was controlled irrespective of their regulation by transcription (Appendix Fig S7A–E).
A large fraction of transcripts was already regulated at a basal level by EDC4 and IKKβ (Table EV4, Fig 4C and E). This group contained numerous cytokines and chemokines (Table EV4). It was previously shown that mRNAs of cytokines are enriched for AU‐rich elements in their 3′ UTRs (Hao & Baltimore, 2009). AREs are known to regulate RNA stability in both positive and negative manner (Blanco et al, 2014; Zid & O'Shea, 2014; Halstead et al, 2015). To determine whether AREs in the 3′ UTR would be overrepresented in the above‐mentioned groups of transcripts, we analysed the occurrence of the annotated AREs, using 1,000 randomly selected 3′ UTRs as control (Table EV7). We found that transcripts that were destabilized by IKKβ and EDC4 at basal level showed a significant enrichment for AREs. Many of these targets were cytokines and chemokines including IL‐1α, IL‐8, IL‐15 and IL‐17D (Table EV8). Of the ARE‐rich transcripts whose basal stability was negatively regulated, 138 were co‐regulated/destabilized by both IKKβ and EDC4 (Tables EV4 and EV8). We confirmed the impact of IKKβ and EDC4 on ARE‐containing mRNAs by expression of a luciferase reporter containing 0, 5 or 7 repeats of an ARE motif in wt, IKKβ‐ or EDC4‐depleted cells (Appendix Fig S8A). Loss of EDC4 or IKKβ or irradiation of wild‐type cells led to increased expression of the luciferase reporter, suggesting that both proteins can act on transcript stability through ARE motifs.
DNA damage leads to activation of a number of kinases. Some, including p38, JNK and ERK1/2, have been linked to post‐transcriptional regulation of ARE‐containing transcripts by phosphorylation of RBPs or P body components (Reinhardt et al, 2010; Nagashima et al, 2015). We inhibited p38, JNK and ERK1/2 to determine whether they also play a role in mRNA stabilization (Appendix Fig S8B). Analysis of transcript stability of different target mRNAs revealed divergent functions of the investigated kinases: while IL‐8 mRNA was stabilized in response to IR, pre‐treatment with p38, JNK or MEK inhibitors led to a reduction in IR‐induced stabilization. The stability of IL‐8 mRNA in unstimulated cells was not affected, confirming that the three kinases do affect IL‐8 mRNA stability following DNA damage, but in a manner opposed to IKK‐EDC4. Analysis of additional transcripts revealed that the target of IKK‐EDC4, BAMBI, was not regulated by the three kinases. TNFAIP3, which was not affected by depletion of IKK or of EDC4, was positively regulated by JNK in IR‐treated cells (Appendix Fig S8C). These findings show that kinases other than IKK are additionally involved in mRNA regulation following DNA damage, but with great divergence in targets and effects, supporting the notion that stress signalling leads to modulation of P body assembly and function via various kinase pathways. Therefore, P bodies may function as hubs of signal integration leading to custom regulation of individual targets in response to stress.
We suggest a mechanism whereby IKK phosphorylates EDC4 in response to cellular stress, resulting in differential regulation of stability of scores of transcripts (Fig 5). About 80% of genes whose stability was regulated by IKK are not known as NF‐κB target genes (Fig 5, Table EV6). Therefore, this regulation not only precedes transcription, mediated by the IKK‐NF‐κB axis, but also affects a much larger number of targets.
Figure 5. Post‐transcriptional gene regulation by IKK .
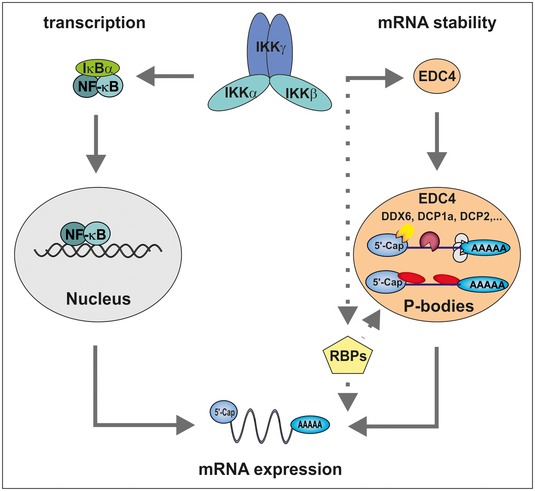
Proposed model for IKK in the regulation of mRNA stability versus transcription. IKK phosphorylates IκB proteins, promoting their degradation. Subsequently, NF‐κB translocates to the nucleus where it promotes transcription of dozens of target genes. Concurrently, IKK interacts with and phosphorylates EDC4, leading to an increase in P bodies. Recruited mRNAs are either degraded or stored in a translationally repressed state. This results in differential regulation of stability of hundreds of transcripts. In addition, IKK regulates various other RBPs contributing to the post‐transcriptional regulation of mRNA levels.
Discussion
IκB kinase is known as a regulator of transcription, mainly through activation of NF‐κB. Here, we demonstrate that activated IKK induces P body formation, thereby expanding its function in the control of gene expression to regulation of mRNA stability.
Recent reports link the modulation of single components of the IKK‐NF‐κB pathway at the RNA level by post‐transcriptional regulation (Glasmacher et al, 2010; Iwasaki et al, 2011; Murakawa et al, 2015). In this work, we reveal an NF‐κB‐independent function of the IKK complex in the global regulation of mRNA stability.
We identified the P body scaffold protein EDC4 as a novel interaction partner and substrate of IKKγ and β, respectively. Importantly, we showed that a fraction of IKK co‐localizes with EDC4 to the P body foci. On activation by TNFα, IL‐1β or ionizing irradiation, IKK phosphorylates EDC4, promoting its association with DCP1a/DCP2 and assembly of P bodies, resulting in stimulus‐dependent post‐transcriptional regulation of distinct sets of transcripts.
The observation that IKK regulates mRNA stability in the absence of activating stimuli deserves a special consideration. Surprisingly, RNA stability of a similar number of distinct mRNAs was controlled by IKK in unstimulated and in stimulated cells. Although part of this regulation may be due to low‐level basal catalytic activity of IKKβ, another possibility is a structural role of physical interaction of IKKγ and β with P body components and/or other RBPs.
Similar to IKK, EDC4 regulates a vast number of transcripts in unstimulated cells. Transcripts controlled by EDC4 showed significant enrichment for GO terms encompassing inflammation and immune response. Our data indicate that EDC4 and IKK silence noise in gene expression via destabilization of mRNAs encoding cytokines or proteins regulating apoptosis that are produced in low quantities by basal transcription. This dual facet of IKK, as a post‐transcriptional attenuator and a transcriptional activator of inflammatory cytokines, would allow for a swift and efficient response to stimuli.
Reports describing murine and human phenotypes arising from mutations or loss of the decapping components are lacking, despite clear evidence from Arabidopsis and Drosophila of their importance in development (Xu et al, 2006; Weil et al, 2012). We speculate that mutations in the decapping machinery and specifically in EDC4 would contribute to inflammatory and autoimmune disorders, characterized by increased cytokine/chemokine production.
The IKK‐NF‐κB axis is known as a crucial inducer of cytokine expression. However, studies using IKKγ‐deficient mice demonstrated that these succumb to cutaneous inflammation driven by inflammatory cytokines in the absence of activated NF‐κB (Makris et al, 2000). These observations could be explained by a contribution of altered transcript stability due to the lack of IKKγ. Indeed, we observed increased expression of cytokine mRNAs in IKK‐depleted cells, indicating that IKK can also function as a negative regulator of cytokine expression through regulation of mRNA stability.
Many cytokines, for example IL‐8, IL‐1α, CCL5 and CSF2 as well as chemokines and other pro‐inflammatory signalling molecules that were destabilized in unstimulated cells in IKK‐ or EDC4‐dependent manner, were stabilized via the IKK‐EDC4 axis in response to stimulus. The mechanism of this change in regulation via IKK and EDC4 is not yet understood. The fate of a specific mRNA might be determined by the degree of EDC4 phosphorylation, dependent on the status of IKK activity. Furthermore, additional, post‐translational modifications of P body components (Rzeczkowski et al, 2011; Arribas‐Layton et al, 2016; Tenekeci et al, 2016) might be involved. Taken together, IKK emerges as a pivotal regulator of gene expression, with the number of transcripts regulated by IKK through changes in stability clearly exceeding the transcriptome regulated by the IKK‐NF‐κB axis (Fig 5). Additionally, post‐transcriptional regulation of mRNA by IKK affords a faster and more flexible regulation of gene expression: swift response to a stimulus can be achieved by selective stabilization or destabilization of targets, bypassing the need for complex transcription and RNA‐processing events.
What determines whether a transcript is degraded or stabilized is an outstanding question. Previous reports evoke cis elements, including AREs (Bakheet et al, 2001) and co‐recruited effectors, including the mRNA decapping proteins DCP1a and DCP2 (Kafasla et al, 2014). We found that transcripts which were destabilized by both EDC4 and IKKβ were enriched for AREs. Many of these ARE‐rich transcripts, including IL‐8, MMP7 and BMP4, encode secreted factors or cytokines. Therefore, we speculate that in the absence of stimulus, ARE‐rich transcripts are targeted for clearance via the IKK‐EDC4 axis. DNA damage leads to a switch in regulation of these transcripts, allowing their accumulation. A rapid change in the compositional state, and therefore function, of P bodies likely contributes to the observed switch in post‐transcriptional regulation (Mitchell & Parker, 2014; Banani et al, 2016). In response to stress, different kinases may dictate composition of P bodies through changes in assembly and subcellular localization of RBPs and P body proteins or through alteration of their affinity for interaction partners or mRNA. As we have shown here, IKK‐mediated phosphorylation of EDC4 regulates its association with DCP1a and DCP2 in response to DNA damage, promoting the formation of P‐bodies. Similarly, phosphorylation of DCP1a by JNK or ERK was shown to alter its localization to P‐bodies (Rzeczkowski et al, 2011; Aizer et al, 2013; Tenekeci et al, 2016). Post‐translational modifications might not only affect interaction among constitutive components of these cellular bodies, but also change recruitment of RBPs and therefore of their target mRNAs. Intriguing examples for the dynamic, kinase‐mediated control of RBPs are TTP and RC3H1 (roquin) both of which localize to P bodies upon specific stimuli (Fenger‐Gron et al, 2005; Franks & Lykke‐Andersen, 2007; Mino et al, 2015), adding another layer of regulation to the IKK‐NF‐κB signalling pathway through regulating stability of its key players (Navarro et al, 2011; Tiedje et al, 2014, 2016; Mino et al, 2015; Murakawa et al, 2015).
In summary, we propose that EDC4 as part of P bodies serves as a signalling node that integrates signals from different kinases, including IKK. Integration of numerous signals would then result in multifaceted regulation of mRNA stability of select targets that surpasses regulation by transcription alone.
Materials and Methods
Cell culture
U2‐OS, HEK293 and HepG2 cells were cultured in DMEM (Gibco) supplemented with 10% FCS (Gibco) and 1× penicillin/streptomycin (Gibco). Early passage BJ cells were obtained from ATCC (Manassas, USA) and were cultured with MEM (Gibco) supplemented with 10% FCS and 1× penicillin/streptomycin. Irradiation was carried out with STS OB 29 with a 137Cs source using 20 Gy, unless indicated otherwise. U2‐OS cells bearing dox‐inducible shRNA were cultured in medium containing tet‐free serum (Clontech). Expression of shRNA was induced with 2 μg/ml doxycycline for 7 days. Controls were handled equally without the addition of dox. Scrambled shRNA was used as controls to confirm that doxycycline does not affect gene expression under the conditions used. CRISPR‐Cas9 was performed as described previously (Joung et al, 2017), using the following guide RNA sequences: EDC4 (fwd: 5′ CACCGCTGCGCGAGCATCGACATCG 3′, rev: 5′ AAAC CGATGTCGATGCTCGCGCAG C 3′), IKKβ (5′ GATTTGGAAATGTCATCCGA 3′). Guide RNAs against EDC4 or IKKβ were cloned into lenti‐CRISPRv2 vector (Addgene plasmid #52961). Empty CRISPRv2 vector was used to generate the control cell line. CRISPR lentivirus was used to transfect U2‐OS cells, followed by selection with puromycin. Monoclonal cell lines were generated and used for experiments.
RNA analysis
RNA extraction
RNA was extracted using TRIzol reagent according to manufacturer's instructions (Thermo Fisher, Carlsbad, USA). In brief, cells were harvested in 1 ml TRIzol, followed by addition of 200 μl chloroform. After phase separation, total RNA was precipitated from the aqueous layer with isopropanol. The RNA pellet was washed twice with 75% RNase‐free ethanol, air‐dried and resuspended in nuclease‐free water.
cDNA synthesis
iScript cDNA synthesis kit from Promega was used according to manufacturers’ instructions.
qRT–PCR
qRT–PCR was performed as described previously (de Oliveira et al, 2016). Primers for qRT–PCR analysis were designed according to MIQE guidelines (Minimum Information for Publication of qRT‐PCR Experiments) using NCBI primer blast, choosing a melting temperature of 62°C. The DNA folding tool from the mfold server (http://unafold.rna.albany.edu/?q=mfold/DNA-Folding-Form) was used to exclude primers forming secondary structures. By using three serial dilutions of cDNA, primer efficiencies were determined, only primers with efficiencies varying around 100% were used for analysis. To determine fold induction of target genes, two reference genes with an m value lower than 0.5 were used (RPL13A, HPRT1).
RNA‐Seq
Stranded mRNA sequencing libraries were prepared with 500 ng total RNA according to manufacturer's protocol (Illumina). The libraries were sequenced in 1 × 100 + 7 manner on HiSeq 2000 platform (Illumina).
RNA‐Seq raw data processing: Raw RNA‐Seq data quality was confirmed using FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Minor adaptor contamination and low‐quality bases were filtered using Trimmomatic (v.0.33; Bolger et al, 2014) using the settings “ILLUMINACLIP:$adaptors:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36” where $adaptors is a compilation of adapter sequences released by Illumina (http://support.illumina.com/). The filtered reads were aligned against the hg19 genome build of Homo sapiens using the STAR aligner (v.2.4.2a; Dobin et al, 2013). Using the Rsubread package (Liao et al, 2013), gene expression levels were quantified based on short read counts mapping to the genes according to ENSEMBL v75 genome annotation (Cunningham et al, 2015; settings: strand‐specific: no; multimapping reads: not counted; multi‐overlapping reads: not counted). For each sample, between ~78 and ~82% of the aligned reads could be assigned to gene features. The raw read counts obtained for each gene using Rsubread were normalized to RPKM values, which were obtained by the formula “(109 × C)/(N × L)”, where C represents number of reads mapping to a gene, N represents total mapped reads from the sample, and L represents total exon length in base pairs of the gene.
RNA‐Seq data were analysed for RNA stability: basal stability is defined as the residual expression of mRNA after actinomycin D treatment in unstimulated cells and was calculated according to the following formula: basal stability = ut ActD/ut. IKKβ or EDC4 dependence is assessed by comparing basal stability in knockdown cells versus wt cells. Induced stability is defined as the residual expression of mRNA after actinomycin D treatment in irradiated cells, according to the formula: induced stability = IR ActD/IR. IKKβ or EDC4 dependence is assessed by comparing induced stability in knockdown cells versus wt cells (lower panel, right). Calculations refer to Tables EV4 and EV5 and graphs Fig 4C and E–G, and Appendix Fig S5B and D.
GO terms were obtained using DAVID https://david.ncifcrf.gov version 6.8, and redundant GO terms were removed and summarized using REVIGO (Supek et al, 2011).
AU‐rich element (ARE) search and comparison
Nine different types of ARE motifs were downloaded from AREsite database (Gruber et al, 2011). In order to find out whether a group of genes (selected based on deregulation status in different mutant/treated backgrounds) have an enrichment of ARE sequences, first all the transcripts of the genes in the group of interest were found. Then, the number of non‐overlapping ARE motif matches (by simple pattern matching) in the exonic three prime UTR regions (according to the ENSEMBL v75 UTR annotations) was counted for each transcript and normalized per 1,000 base pairs of searched sequence. As a control group, 1,000 genes were randomly drawn and the ARE motif matches were counted in three prime UTRs of the transcripts of the control group and normalized. Finally, the normalized frequency of ARE motif matches was compared using Wilcoxon rank‐sum test to test whether the gene group of interest has a higher frequency of ARE motif instances in the transcript three prime UTR regions compared to the background distribution.
Protein analysis
Nuclear/cytoplasmic fractionation
For subcellular fractionation, plates were washed with buffer A [10 mM Tris–HCl (pH 7.9); 1.5 mM MgCl2; 10 mM KCl] and harvested by scraping in 500 μl buffer A including protease and phosphatase inhibitors [0.5 mM DTT; 0.4 mM Pefabloc; complete protease inhibitor cocktail (Roche); 10 mM NaF; 8 mM β‐glycerophosphate; 0.2 mM Na3VO4]. Following incubation on ice, NP‐40 was added to a final concentration of 0.5%. Samples were vortexed, centrifuged and supernatants containing the cytoplasmic fraction were transferred to new tubes. The pellet was washed with buffer A and resuspended in three volumes buffer C [20 mM Tris–HCl (pH 7.9); 25% glycerol; 0.42 M NaCl; 1.5 mM MgCl2; 0.2 mM EDTA; 0.5 mM DTT; protease inhibitor cocktail; 10 mM NaF; 20 mM β‐glycerophosphate; 300 μM Na3VO4]. Before insoluble nuclear debris was pelleted by centrifugation, lysates were adjusted to a concentration of 150 mM NaCl by addition of buffer A. The supernatants containing the nuclear fraction were transferred to new reaction tubes.
Blots were analysed with Fusion Solo (Vilber Lourmat) and quantitated (Fusion Capt V16.05a).
Electrophoretic mobility shift assays (EMSA)
EMSA was performed with nuclear fractions or whole‐cell extracts (WCE). Whole‐cell extract was prepared using Baeuerle buffer (20 mM HEPES pH 7.9; 350 mM NaCl; 20% glycerol; 1 mM MgCl2; 0.5 mM EDTA; 0.1 mM EGTA; 1% NP‐40), including complete protease inhibitor cocktail (Roche), 10 mM NaF, 8 mM β‐glycerophosphate, 0.2 mM Na3VO4, 1 mM DTT.
In vitro kinase assay
The potential substrate sub‐regions were recombinantly expressed and purified. Endogenous IKK complex was immunoprecipitated from kinase IP lysates [50 mM HEPES (pH 7.5); 150 mM NaCl; 1.5 mM MgCl2; 1 mM EDTA; 1% Triton X‐100; 10% glycerol; complete protease inhibitor cocktail (Roche), 10 mM NaF, 8 mM β‐glycerophosphate, 0.2 mM Na3VO4, 1 mM DTT; calyculin A] of untreated or stimulated cells with an IKKγ antibody. The purified sub‐regions were incubated with the IKK complex in the presence of [γ‐32P] ATP. A purified IκBα fragment (aa 1–54) was used as positive control. Samples were boiled and subjected to SDS–PAGE and autoradiography.
Samples for MS analysis of EDC4 phosphosites were prepared as described above. Kinase assay was performed in the presence of cold ATP. Samples were boiled and subjected to MS analysis.
For analysis of phosphorylation of endogenous EDC4, EDC4 was immunoprecipitated from MS IP lysates [150 mM NaCl, 50 mM Tris pH 7.5, 1% IGPAL‐CA630, 0.5% deoxycholate, 0.1% sodiumdodecylsulfate, 5% glycerol, MS‐SAFE Protease and Phosphatase Inhibitor (SIGMA)] of U2‐OS CRISPR cells [wt control, EDC4 knockout (as negative control) or IKKβ knockout]. Proteins were digested on‐beads with trypsin as described (Hubner et al, 2010) and desalted using the stage tip protocol (Rappsilber et al, 2003). Peptides were separated on an EASY‐nLC 1200 system (Thermo Fisher Scientific), packed in‐house (75 μm × 20 cm, 1.9 μm C18 resin, ReproSil‐Pur C18‐AQ material, Dr Maisch GmbH) and coupled to a Q Exactive Plus instrument (Thermo Fisher Scientific) applying a flow rate of 250 nl/min. Peptides were separated using a 1‐h gradient ramping from 2 to 30% LC solvent B (0.1% formic acid, 90% acetonitrile) in LC solvent A (0.1% formic acid, 3% acetonitrile). MS acquisition took place in data‐dependent mode using the Top20 peaks for MS2 fragmentation at a resolution of 70,000 for MS1 and 15,000 for MS2 and a maximum injection time of 100 ms. Database search was performed using MaxQuant version 1.5.2.8 (Cox & Mann, 2008) with oxidized methionine, acetylated N‐termini and phosphorylation on serine, threonine and tyrosine as variable modifications. Carbamidomethylation was used as a fixed modification and search occurred against a human UniProt database (2018‐05) with a peptide and protein FDR cut‐off of 1% but without using a site‐FDR cut‐off while the match between runs option was turned on.
Immunoprecipitation
For immunoprecipitation of endogenous protein complexes, cells were harvested in ice‐cold CHAPS buffer [Tris–HCl pH 7.4, 110 mM NaCl and 50 mM EDTA, supplemented with 1% CHAPS complete protease inhibitor cocktail (Roche), 10 mM NaF, 8 mM β‐glycerophosphate, 0.2 mM Na3VO4, 1 mM DTT], incubated with primary antibodies overnight and 1 h with G Fast Flow Sepharose Beads (GE Healthcare, Little Chalfont, UK). Samples were boiled and subjected to SDS–PAGE.
To analyse protein–protein interaction between recombinant proteins in vitro, purified GST‐tagged full‐length IKKγ and Strep‐tagged EDC4 sub‐regions were used. Strep‐tagged IKKγ was used as a positive control. The purified recombinant proteins were mixed in 400 μl IP buffer (150 mM NaCl; 20 mM Tris–HCl pH 7.9; 10% glycerol; 0.1% NP‐40; 0.4 mM Pefabloc; 1 mM DTT) and incubated for 90 min. GST‐IKKγ was precipitated using glutathione sepharose (GE Healthcare). IPs were washed five times with IP buffer, boiled and analysed by SDS–PAGE.
Immunofluorescence
Immunofluorescence was performed as described previously (Prox et al, 2012), with the following antibodies: EDC4 (F‐2, Santa Cruz; Cell Signaling), DDX6 (Bethyl) and G3BP1 (H‐10, Santa Cruz). Briefly, 2 × 105 cells were seeded in six‐well plates on sterilized cover slips 48 h before experiment. Following treatment, cells were fixed in 4% PFA for 10–15 min, quenched with 0.12% glycine in 0.2% saponin (in PBS) and blocked with 10% FCS in 0.2% saponin (in PBS). Samples were incubated in 0.2% saponin containing the primary antibodies overnight at 4°C. Cover slips were washed five times in 0.2% saponin before incubation in 0.2% saponin containing the secondary antibodies for 1 h at room temperature. Cover slips were washed five times in 0.2% saponin and ddH2O and fixed on glass slides using mowiol supplemented with DAPI.
DuoLink (Merck, Kenilworth, USA) proximity ligation assay (PLA) was performed according to the manufacturer's protocol; U2‐OS cells were seeded on sterilized cover slips 48 h before experiment. Cells were fixed in 4% PFA and quenched with 0.12% glycine in 0.2% saponin (in PBS) as described above. For blocking, cells were incubated for 1 h at 37°C in the provided blocking solution. Incubation with primary antibodies (EDC4 F1, Santa Cruz, IKKγ FL‐419, Santa Cruz, both diluted 1:200) was done overnight at 4°C.
SILAC‐MS analysis
For co‐immunoprecipitation of cytoplasmic IKK complexes, cells were lysed with a hypoosmotic lysis buffer [10 mM Tris–HCl (pH 7.9); 1.5 mM MgCl2; 10 mM KCl] using glass homogenizers. Prior to co‐IP, IKKγ FL‐419 (Santa Cruz Biotechnology, Santa Cruz, USA) was crosslinked to Protein G‐coated Dynabeads (Invitrogen, Carlsbad, USA). Precipitates were washed five times with IP wash buffer (100 mM Tris–HCl pH 7.9, 150 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, supplemented with 0.4 mM Pefabloc). For mass spectrometry, precipitates were eluted at 37°C with 6 M urea/2 M thiourea. Proteins in 6 M urea/2 M thiourea buffer derived from cytoplasmic IKKγ immunoprecipitation experiments with U2‐OS cells were treated with tris(2‐carboxyethyl)phosphine and chloroacetamide to reduce and alkylate sulfhydryl groups, followed by an enzymatic digest with endopeptidases LysC and trypsin. After a solid‐phase extraction and desalting, peptides were eluted, lyophilized and reconstituted in a 0.1% formic acid/3% ACN solution. Peptides were separated on a reversed‐phase column (20 cm length, 75 μm ID, 3 μm Dr. Maisch C18) with a gradient from 3 to 36% ACN in 120 min and measured in data‐dependent acquisition mode using an Orbitrap Q Exactive instrument (Thermo). The raw files were analysed with MaxQuant 1.2.2.5. Cut‐off for induced Interaction partners of IKKγ was set to at least twofold change, resulting in values for H/L ratio, normalized to IKKγ values, of at least 2 in the forward experiment and 0.5 in the reverse experiment. For presentation in the heatmap, gitools‐2.3.0 was used.
Antibodies and reagents
Reagents were added as follows: doxycycline hydrochloride (2 μg/ml, Sigma), actinomycin D (10 ng/ml, Sigma), IL‐1β (10 ng/ml, Enzo), hydrogen peroxide (100 μM, Roth) and TNFα (10 ng/ml, Enzo).
Antibodies were as follows: p65 (C‐20), IκBα (C‐21), IKKγ (FL‐419), EDC4 (F‐1), hDcp1a (D‐13), hDCP2 (V‐25), PARP‐1 (F‐2), TRAF6 (H‐274) and G3BP1 (H‐10) were obtained from Santa Cruz Biotechnology (Santa Cruz, USA); DCP1a, DCP2 and DXX6 were from Bethyl Laboratories (Montgomery, USA); EDC4, IKKβ, p65‐S538 (Cell Signaling Technology, Denver, USA) and IKKγ (Cl54; BD Bioscience, Franklin Lakes, USA).
Transfection/transduction
Plasmid and siRNA transfections were carried out as described previously (Stilmann et al, 2009) using siRNA against IKKγ (GGAAGAGCCAACUGUGUGA), IKKβ (CUUAGAUACCUUCAUGAAA) and EDC4 (GGCUCUGGUUAAUGGCAAA) or against IKKγ (GGAAGAGCCAACUGUGUGA) and IKKγ (siRNA ID S16186 from Thermo Scientific) or Xrn1 (siRNA ID s29015 from Thermo Scientific). Scrambled control siRNA (AllStars) was obtained from Qiagen.
Full‐length EDC4 plasmid was obtained from Harvard plasmID database, and mammalian expression plasmids of HA‐tagged EDC4 1–538, 532–979 and 974–1,401 were a gift of E. Izaurralde (Braun et al, 2012). For bacterial expression and purification of recombinant proteins, sequences were cloned into pASK‐IBA17+ expression plasmid and pGEX‐6P‐3 expression plasmid. Point mutants were generated using the site‐directed mutagenesis kit from Qiagen and cloned into mammalian FLAG and EGFP vectors. pTRIPZ constructs for EDC4, IKKβ and scrambled (Dharmacon, Lafayette, USA) were transfected into HEK293T cells and supernatant used for transduction as per manufacturer's instruction (http://dharmacon.gelifesciences.com/uploadedfiles/resources/ptripz-inducible-lentiviral-manual.pdf). Clonal selection was performed using puromycin.
pEZX‐MT01 constructs were a gift by Antje Hirsekorn from the Ohler laboratory (pEZX‐MT01 vector from GeneCopoeia, 3′ UTR sequence for 5× ARE: 5′ ATTTATTTATTTATTTATTTGTTTGTTTCACTGGCTCTGAGGCCAGTGAAGTTTTTTGCCCAACTGGAATTTAAAAGATGTGTGTCT 3′, and for 7× ARE: 5′ ATTTATTTATTTATTTATTTATTTATTTCACTGGCTCTGAGGCCAGTGAAGTTTTTTGCCCAACTGGAATTTAAAAGATGTGTGTCT 3′).
Data availability
Accession numbers for RNA‐Seq, SILAC‐MS and phosphosite analysis are E‐MTAB‐7199, MSV000082975 and MSV000082890, respectively.
Author contributions
Conceptualization, NM, MK and CS; formal analysis, OP, WS, BU and GD, PM; investigation, NM, MK, PB, ABT, MH and BP; resources, AA, GD, WC and PM. Writing–original draft, MK and NM; writing–review and editing, NM, MK and CS, funding acquisition, CS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We are grateful to Sabine Jungmann and Lynn Krueger for excellent technical assistance. We would like to thank Markus Landthaler for HEK cells bearing inducible HA‐DDX6 vector, Elisa Izaurralde for HA‐EDC4 constructs and Antje Hirsekorn/Uwe Ohler laboratory, for pEZX‐MT01 plasmids. We thank Jana Wolf and Mireya Plass Pórtulas for helpful discussion and suggestions. This work was supported in part by funding from German Federal Ministry of Education and Research (BMBF) as part of the RNA Bioinformatics Center of the German Network for Bioinformatics Infrastructure (de.NBI) [031 A538C RBC (de.NBI)] to BU, from DFG (HI 1549/1‐1 and SCHE 277/8‐1) to MH and CS and from BMBF, CancerSys (ProSiTu) to CS.
The EMBO Journal (2018) 37: e98658
See also: ND Perkins (December 2018)
References
- Aizer A, Kafri P, Kalo A, Shav‐Tal Y (2013) The P body protein Dcp1a is hyper‐phosphorylated during mitosis. PLoS One 8: e49783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribas‐Layton M, Dennis J, Bennett EJ, Damgaard CK, Lykke‐Andersen J (2016) The C‐terminal RGG domain of human Lsm4 promotes processing body formation stimulated by arginine dimethylation. Mol Cell Biol 36: 2226–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS (2001) ARED: human AU‐rich element‐containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res 29: 246–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, Rosen MK (2016) Compositional control of phase‐separated cellular bodies. Cell 166: 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W (2006) Stress‐induced reversal of microRNA repression and mRNA P‐body localization in human cells. Cold Spring Harb Symp Quant Biol 71: 513–521 [DOI] [PubMed] [Google Scholar]
- Blanco FF, Sanduja S, Deane NG, Blackshear PJ, Dixon DA (2014) Transforming growth factor beta regulates P‐body formation through induction of the mRNA decay factor tristetraprolin. Mol Cell Biol 34: 180–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JE, Truffault V, Boland A, Huntzinger E, Chang CT, Haas G, Weichenrieder O, Coles M, Izaurralde E (2012) A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5′ exonucleolytic degradation. Nat Struct Mol Biol 19: 1324–1331 [DOI] [PubMed] [Google Scholar]
- Brengues M, Teixeira D, Parker R (2005) Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 310: 486–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CT, Bercovich N, Loh B, Jonas S, Izaurralde E (2014) The activation of the decapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1. Nucleic Acids Res 42: 5217–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho‐Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kahari AK, Keenan S et al (2015) Ensembl 2015. Nucleic Acids Res 43: D662–D669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Teixeira D, Parker R (2007) Edc3p and a glutamine/asparagine‐rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae . J Cell Biol 179: 437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger‐Gron M, Fillman C, Norrild B, Lykke‐Andersen J (2005) Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell 20: 905–915 [DOI] [PubMed] [Google Scholar]
- Franks TM, Lykke‐Andersen J (2007) TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU‐rich elements. Genes Dev 21: 719–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, Kremmer E, Wang X, Heissmeyer V (2010) Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA‐independent post‐transcriptional repression. Nat Immunol 11: 725–733 [DOI] [PubMed] [Google Scholar]
- Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL (2011) AREsite: a database for the comprehensive investigation of AU‐rich elements. Nucleic Acids Res 39: D66–D69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead JM, Lionnet T, Wilbertz JH, Wippich F, Ephrussi A, Singer RH, Chao JA (2015) Translation. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 347: 1367–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Baltimore D (2009) The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol 10: 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2008) Shared principles in NF‐kappaB signaling. Cell 132: 344–362 [DOI] [PubMed] [Google Scholar]
- Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C (2010) A cytoplasmic ATM‐TRAF6‐cIAP1 module links nuclear DNA damage signaling to ubiquitin‐mediated NF‐kappaB activation. Mol Cell 40: 63–74 [DOI] [PubMed] [Google Scholar]
- Hinz M, Scheidereit C (2014) The IkappaB kinase complex in NF‐kappaB regulation and beyond. EMBO Rep 15: 46–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M (2010) Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol 189: 739–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H, Takeuchi O, Teraguchi S, Matsushita K, Uehata T, Kuniyoshi K, Satoh T, Saitoh T, Matsushita M, Standley DM, Akira S (2011) The IkappaB kinase complex regulates the stability of cytokine‐encoding mRNA induced by TLR‐IL‐1R by controlling degradation of regnase‐1. Nat Immunol 12: 1167–1175 [DOI] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E (2013) The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev 27: 2628–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F (2017) Genome‐scale CRISPR‐Cas9 knockout and transcriptional activation screening. Nat Protoc 12: 828–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafasla P, Skliris A, Kontoyiannis DL (2014) Post‐transcriptional coordination of immunological responses by RNA‐binding proteins. Nat Immunol 15: 492–502 [DOI] [PubMed] [Google Scholar]
- Karin M, Ben‐Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF‐[kappa]B activity. Annu Rev Immunol 18: 621–663 [DOI] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke‐Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169: 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed‐and‐vote. Nucleic Acids Res 41: e108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris C, Godfrey VL, Krahn‐Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M (2000) Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X‐linked disorder incontinentia pigmenti. Mol Cell 5: 969–979 [DOI] [PubMed] [Google Scholar]
- Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, Uehata T, Tartey S, Akira S, Suzuki Y, Vinuesa CG, Ohler U, Standley DM, Landthaler M, Fujiwara T, Takeuchi O (2015) Regnase‐1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell 161: 1058–1073 [DOI] [PubMed] [Google Scholar]
- Mitchell SF, Parker R (2014) Principles and properties of eukaryotic mRNPs. Mol Cell 54: 547–558 [DOI] [PubMed] [Google Scholar]
- Murakawa Y, Hinz M, Mothes J, Schuetz A, Uhl M, Wyler E, Yasuda T, Mastrobuoni G, Friedel CC, Dolken L, Kempa S, Schmidt‐Supprian M, Bluthgen N, Backofen R, Heinemann U, Wolf J, Scheidereit C, Landthaler M (2015) RC3H1 post‐transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF‐kappaB pathway. Nat Commun 6: 7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima T, Inoue N, Yumoto N, Saeki Y, Magi S, Volinsky N, Sorkin A, Kholodenko BN, Okada‐Hatakeyama M (2015) Feedforward regulation of mRNA stability by prolonged extracellular signal‐regulated kinase activity. FEBS J 282: 613–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro MN, Goebel J, Feijoo‐Carnero C, Morrice N, Cantrell DA (2011) Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat Immunol 12: 352–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S (2011) Crosstalk in NF‐kappaB signaling pathways. Nat Immunol 12: 695–708 [DOI] [PubMed] [Google Scholar]
- de Oliveira KA, Kaergel E, Heinig M, Fontaine JF, Patone G, Muro EM, Mathas S, Hummel M, Andrade‐Navarro MA, Hubner N, Scheidereit C (2016) A roadmap of constitutive NF‐kappaB activity in Hodgkin lymphoma: dominant roles of p50 and p52 revealed by genome‐wide analyses. Genome Med 8: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Sheth U (2007) P bodies and the control of mRNA translation and degradation. Mol Cell 25: 635–646 [DOI] [PubMed] [Google Scholar]
- Prox J, Willenbrock M, Weber S, Lehmann T, Schmidt‐Arras D, Schwanbeck R, Saftig P, Schwake M (2012) Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell Mol Life Sci 69: 2919–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA, Yaffe MB (2010) DNA damage activates a spatially distinct late cytoplasmic cell‐cycle checkpoint network controlled by MK2‐mediated RNA stabilization. Mol Cell 40: 34–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzeczkowski K, Beuerlein K, Muller H, Dittrich‐Breiholz O, Schneider H, Kettner‐Buhrow D, Holtmann H, Kracht M (2011) c‐Jun N‐terminal kinase phosphorylates DCP1a to control formation of P bodies. J Cell Biol 194: 581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE (2012) Regulation of cytoplasmic mRNA decay. Nat Rev Genet 13: 246–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C (2009) A nuclear poly(ADP‐ribose)‐dependent signalosome confers DNA damage‐induced IkappaB kinase activation. Mol Cell 36: 365–378 [DOI] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, Smuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D, Sheth U, Valencia‐Sanchez MA, Brengues M, Parker R (2005) Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 11: 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenekeci U, Poppe M, Beuerlein K, Buro C, Muller H, Weiser H, Kettner‐Buhrow D, Porada K, Newel D, Xu M, Chen ZJ, Busch J, Schmitz ML, Kracht M (2016) K63‐Ubiquitylation and TRAF6 pathways regulate mammalian P‐body formation and mRNA decapping. Mol Cell 62: 943–957 [DOI] [PubMed] [Google Scholar]
- Tiedje C, Holtmann H, Gaestel M (2014) The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J Interferon Cytokine Res 34: 220–232 [DOI] [PubMed] [Google Scholar]
- Tiedje C, Diaz‐Munoz MD, Trulley P, Ahlfors H, Laass K, Blackshear PJ, Turner M, Gaestel M (2016) The RNA‐binding protein TTP is a global post‐transcriptional regulator of feedback control in inflammation. Nucleic Acids Res 44: 7418–7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil TT, Parton RM, Herpers B, Soetaert J, Veenendaal T, Xanthakis D, Dobbie IM, Halstead JM, Hayashi R, Rabouille C, Davis I (2012) Drosophila patterning is established by differential association of mRNAs with P bodies. Nat Cell Biol 14: 1305–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yang JY, Niu QW, Chua NH (2006) Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. Plant Cell 18: 3386–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Yang WH, Gulick T, Bloch KD, Bloch DB (2005) Ge‐1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA 11: 1795–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, O'Shea EK (2014) Promoter sequences direct cytoplasmic localization and translation of mRNAs during starvation in yeast. Nature 514: 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Data Availability Statement
Accession numbers for RNA‐Seq, SILAC‐MS and phosphosite analysis are E‐MTAB‐7199, MSV000082975 and MSV000082890, respectively.