

Abstract
Type 2 diabetes mellitus (DM) is a lifelong metabolic disease, characterized by hyperglycaemia which gradually leads to the development and progression of vascular complications. It is recognized as a global burden disease, with substantial consequences on human health (fatality) as well as on health-care system costs. This review focuses on the topic of historical discovery and understanding the complexity of the disease in the field of pathophysiology, as well as development of the pharmacotherapy beyond insulin. The complex interplay of insulin secretion and insulin resistance developed from previously known “ominous triumvirate” to “ominous octet” indicate the implication of multiple organs in glucose metabolism. The pharmacological approach has progressed from biguanides to a wide spectrum of medications that seem to provide a beneficial effect on the cardiovascular system. Despite this, we are still not achieving the target treatment goals. Thus, the future should bring novel antidiabetic drug classes capable of acting on several levels simultaneously. In conclusion, given the raising burden of type 2 DM, the best present strategy that could contribute the most to the reduction of morbidity and mortality should be focused on primary prevention.
Keywords: Type 2 diabetes mellitus, Physical activity, Hyperglycaemia, Insulin resistance, Hypoglycaemic agents
Core tip: Type 2 diabetes mellitus (DM) is a global burden disease and one of the leading all-cause mortality causes due to cardiovascular (CV) complications. The rapid raise in the understanding of its pathogenesis resulted in treatment approach options beyond insulin that also provide beneficial CV effect. We discuss this scientific pathological and pharmacological development through a comprehensive historical approach. The wide spectrum of therapeutic agents currently used in type 2 DM treatment result in a CV mortality reduction which is not exclusively in correlation with glucose-lowering potency but is linked to its mechanism of action.
INTRODUCTION
Diabetes mellitus (DM) is chronic, lifelong progressive metabolic disease characterized by hyperglycaemia due to absolute or relative insulinopaenia. There are several different types of DM and each are caused by a complex interplay between genetic predisposition and environmental factors. The metabolic dysregulation that contributes to hyperglycaemia includes diminished insulin secretion, impaired glucose utilization or increased glucose production, and eventually causes pathophysiological changes in multiple organs and organ systems[1]. Despite all the scientific advances in the field of pathophysiology, diagnosis and treatment, the prevalence of DM has shown a dramatic rise over the past 200 years. Nowadays, DM represents a global burden disease with fatal consequences on human health, and significant impact on health-care system costs. It is estimated that in 2017, there were 451 million people (ages 18-99 years) with diabetes worldwide[2], and this number is expected to rise, mostly due to type 2 DM. Thus, we review herein the longitudinal history and therapeutic approaches beyond insulin for type 2 DM.
HISTORY AND CLASSIFICATION OF DM
The first documented symptoms of DM were recorded by ancient physicians in 1552 B.C. in a 3rd Dynasty Egyptian papyrus, being described as a rare mysterious disease characterized by excessive urination which leads to emaciation and death[3,4]. Around year 150 of the new era, the term “diabetes mellitus” meaning “honey” and “siphon” was introduced by an ancient Greek physician Aretaeus, reflecting the sweet urine taste in affected individuals[5]. However, its recognition as what is called a “clinical entity” - a condition that has separate and distinct existence from any known underlying cause or specific treatment option - occurred in an 1822 publication in the New England Journal of Medicine and Surgery[6].
The idea beyond this disease was not clarified until 1889, when Josph von Mering and Oskar Minkowski found that pancreatomy performed on a dog resulted in fatal diabetes[7,8]. In 1910, Edward Albert Sharpey-Schafer hypothesized that this might be due to the lack of a single pancreatic chemical, which he called “insulin”[6]. His hypothesis was confirmed by the discovery of insulin in 1921 by Frederick Banting and Charles Best[9,10]. After initially reversing diabetes in a dog using an extract from pancreatic islets of a healthy dog, together with James Collip and John Macleod they purified the hormone from bovine pancreas and used it to treat diabetes in humans[11].
From that point of time, DM has represented a fertile ground for scientific research[12]. Since 1923, 10 scientists have received a Nobel prize for diabetes-related investigation[6]. Over the past two centuries it became clear that DM does not represent a unique clinical condition with a common pathophysiological background. Namely, insulin transformed the lives of children and young adults with diabetes but had limited impact upon the survival of those diagnosed at the age of 50[6].
The classification of DM is primarily based on the pathogenic process that results in hyperglycaemia. In brief, it is now well known that severe insulin deficiency accounts for about 10% of all DM cases and is characterized by selective autoimmune destruction of insulin producing pancreatic β-cells, which are classified as type 1 DM, usually occurring in younger, lean individuals[1]. The majority of patients, however, belong to the group with insulin resistance as the core pathophysiological disorder rather than insulin deficiency[1], classified as type 2 DM. This type of DM is phenotypically often accompanied by central obesity, hypertension and dyslipidaemia.
The different pathophysiological background of hyperglycaemia was first proposed by Himsworth[13] in the year 1936, who tested the ability of injected insulin to clear an oral glucose load from the circulation. He concluded that there were insulin sensitive patients whose diabetes was due to insulin deficiency and insulin insensitive patients whose diabetes was due to resistance to insulin. These findings were strengthened by the research of Claude Bernard, who showed that blood glucose is also regulated by the non-glucose precursors driven by the liver[14-16], which probably represent the core of diabetes classification that was, however, not adopted until the 1970s[17] under the terms “maturity onset diabetes” and “non-insulin-dependent diabetes” (NIDDM). Those were abandoned in favour of the newer terminology between 1980 and the 1990s. The categorization of DM involving two principal groups, namely type 1 and type 2 DM, raises its own concerns, especially in terms of type 2 DM because of diversity in clinical presentation and the natural course of the disease requiring an individual therapeutic approach[18].
PATOPHYSIOLOGY OF TYPE 2 DM: WHAT HAVE WE LEARNED IN THE LAST CENTURY?
Type 2 DM (formerly known as NIDDM) is a common metabolic disorder characterized by insulin resistance, relative impairment in insulin secretion, and certain degree of genetic predisposition, the prevalence of which markedly rises with the degree of obesity[1]. It is often accompanied by hypertension and dyslipidaemia: high serum low density lipoprotein concentrations and low serum high density lipoprotein concentrations that increase cardiovascular (CV) risk. The constellation of these clinical conditions is referred to as metabolic syndrome. Although, the risk factors associated with this type of DM were observed as early on as the 1920s and the term “metabolic syndrome” was coined in the 1950s when the French physician Jean Vague noticed that upper body obesity seemed to be associated with an increased risk for the conditions of atherosclerosis, diabetes, kidney stones and gout[19]. He also noticed that these patients show significant improvement in their diabetes, high blood cholesterol and high triglycerides following a low-calorie and low-carbohydrate diet[20]. The term became commonly used in the 1970s, and in the 1988, Gerald Reaven hypothesized that insulin resistance could be the underlying factor linking this constellation of abnormalities, which he went on to name “syndrome X”[21,22].
Indeed, it is now well known that type 2 DM usually presents with varying degrees of insulin resistance, consequent relative insulin deficiency, and hyperglycaemia which further impair pancreatic β-cell function, resulting in a vicious cycle of metabolic state worsening[23]. In addition, it is now well known that the majority of type 2 DM patients have genetic risk for its development. Its importance is supported primarily due to the observation that normoglycaemic offspring of type 2 DM parents have reduced non-oxidative glucose metabolism associated with increased muscle intracellular lipid content and reduced muscle glycogen synthesis[1]. This is due to a complex interaction among many genes and environmental factors (i.e., a complex polygenic interplay which finally results in insulin resistance, namely decreased insulin sensitivity) that represents a core pathophysiological factor in type 2 DM development.
The exact molecular mechanism leading to insulin resistance has not been elucidated so far. Although the amount of insulin receptor expression on target tissues is diminished due to insulin’s cellular internalization and reduced tyrosine kinase activity, the different expression probably represents the secondary and not the primary defect. It is considered that the post-receptor alterations in insulin receptor substrate-1 (IRS-1), regulating phosphorylation and dephosphorylation, might play a predominant role in this condition. Precisely, there is an imbalance between IRS-1 tyrosine and serine phosphorylation (Figure 1)[24]. Diminished IRS-1 tyrosine phosphorylation results in reduced translocation of glucose transporter type 4 (GLUT-4) to the plasma membrane, which enables glucose influx into the cells. Simultaneously, enhanced IRS-1 serine phosphorylation activates mitogen-activated proteins, whose action is not involved in metabolic but in mitotic insulin activity and proinflammatory pathways activation which result in intramitochondrial stress and further enhance insulin resistance. It is also implicated in the diabetes-related micro- and macrovascular complications’ development. In brief, insulin resistance consists of two tightly coupled mechanisms: lack of suppression of glucose production and lack of glucose uptake by peripheral tissues, primarily muscles. Skeletal muscles usually utilize more than 80% of the circulating glucose in the presence of circulating insulin, while in the condition of insulin resistance this effect is diminished[19-22,24,25].
Figure 1.
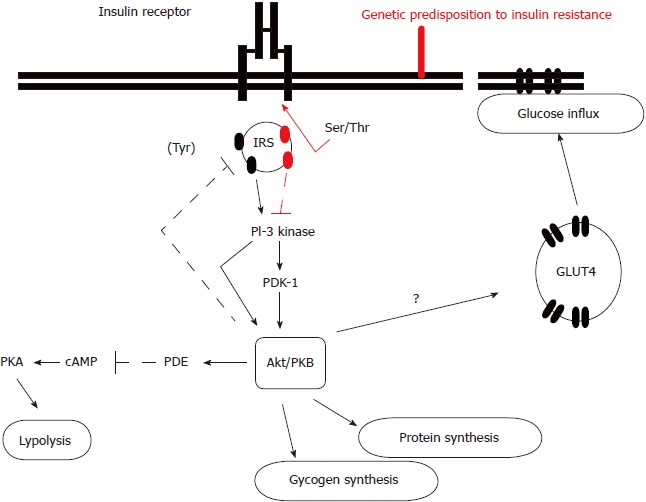
The molecular mechanism of insulin resistance. In insulin resistance, the binding of insulin to its receptor does not result in serine phosphorylation of insulin receptor substrate-1 and activation of the cascade of intracellular substrates’ activation which result in glucose influx, glucagon and protein synthesis, and lipolysis inhibition. IRS: Insulin receptor substrate; Ser/Thr: Serine/threonine protein kinase; Tyr: Tyrosine kinase; PI-3: Phosphatidylinositol 3; PDK-1: Phosphoinositide-dependent protein kinase-1; Akt/PBK: AKT serine/threonine kinase 1 (protein kinase B family); PDE: Phosphodiesterase; cAMP: Cyclic adenosine monophosphate; PKA: Protein kinase A; GLUT4: Glucose transporter type 4.
Furthermore, during the overnight fast, there is a substantial (1.8-2.0 mg/kg per min) glucose production, essential to meet the needs of the brain and other neural tissues whose uptake accounts for 50%-60% of total glucose disposal and is insulin independent. In euglycaemic individuals, the hepatic glucose production is suppressed following the glucose influx into the portal vein due to rise in insulin and inhibition in glucagon release[1]. In type 2 DM, this mechanism is diminished, which then results in both fasting as well as postprandial hyperglycaemia. The mechanisms involved in hepatic glucose production include hyperglucagonaemia, increased levels of circulating glucose precursors, free fatty acid oxidation, enhanced sensitivity to glucagon and decreased sensitivity to insulin[1,24].
A gradual increase in insulin resistance requires a notable higher amount of insulin in order to overcome hyperglycaemia. Consequently, as pancreatic β-cells start to release insulin from its secretory granules, a higher amount of amylin appears in higher concentration in circulation but also in pancreas itself[1,23]. Circulating amylin decreases glucose uptake in peripheral tissues, i.e., enhances insulin resistance, while the pancreatic amylin further decreases pancreatic insulin secretion contributing to the hyperglycaemic state in both cases[1]. Thus, this complex interplay of insulin secretion and insulin resistance in the liver and the skeletal muscle, also known as “ominous triumvirate”[1], represented the first proposed fundamental mechanism of type 2 DM development and progression over the last two decades, i.e., ever since it was established by a prospective study carried out by Jallut et al[26] in 1990 and later supported by many prospective studies carried out in diverse ethnic populations[27,28].
However, since 1990 and until 2009, due to technological and pharmacological advances, there was a growing body of literature suggesting that the “ominous triumvirate” was not the sole pathophysiological disturbance in type 2 DM. An exponential growth of experimental and clinical studies suggested the possible implications of other organs: primarily pancreatic glucagon producing cells (α-cells), visceral adipose tissue, gut, kidneys and central nervous system. This was finally defined by DeFronzo[29] in 2009 under the term “ominous octet”. As it gradually became evident that persons affected by type 2 DM have mostly a specific adipose tissue topography, i.e., that visceral obesity is often accompanying type 2 DM, the two might be considered a part of this pathogenic process. It has been shown that they are also insulin resistant, resulting in the antilipolytic effect and thus leading to daylong elevation in the plasma free-fatty acid concentration[30-36], which not only further disrupts pancreatic β-cell function[37,38] but also promotes hepatic and muscle insulin resistance[39-41] and stimulates gluconeogenesis[42,43]. Moreover, the visceral adipocytes have a secretory capacity for a number of biological active products, including the adipokines, namely adiponectin, leptin, resistin, tumour necrosis factor-α, interleukine-1β, plasminogen activator inhibitor-1, retinol binding protein 4, etc., which are all associated with the function of intermediary metabolism[30,44].
After the basic elucidation of these so-called “dysharmonius quartet”, at the beginning of the 2000s, the concept of type 2 DM pathophysiology was further expanded to include the gastrointestinal tissues as the fifth member of the “quintessential quintet”. Even back in 1932, it was observed that the administration of an extract from the upper intestine could produce a fall in blood glucose, and the presumed hormone was named “incretin”[45]. Moreover, it was noticed that oral glucose administration resulted in a much greater insulin secretion, as compared to intravenous glucose infusion in a concentration that mimicked orally-absorbed plasma glucose concentration[46-48]; the phenomena was called “incretin effect”. The discovery of two gastrointestinal peptides - glucose-dependent insulinotrophic peptide secreted by the K-cells of the more proximal small intestine, and glucagon like peptide-1 (GLP-1) secreted by the L-cells of the distal small intestine - probably mediate > 99% of this “incretin effect”. They have been shown to delay gastric emptying, stimulate insulin and suppress glucagon secretion in a glucose-dependent manner. In fact, the diminished “incretin effect” has been repeatedly shown in type 2 DM patients[49-52].
The sixth member of the “octet” is the pancreatic α-cell. We have already mentioned glucagon, i.e., the lack of glucagon suppression in the context of hepatic insulin resistance as well as in the incretion part, so it does not come as surprise that the higher fasting plasma glucose has been documented in several clinical studies in type 2 DM individuals back from the 1970s[53-58]. It has been demonstrated that higher concentrations of fasting glucagon closely correlate with the increase in fasting hepatic glucose production as described in detail earlier in the paper. Furthermore, the authors showed a simultaneous decrease following somatostatin infusion. Thus, it is clear that hyperglucagonemia merits being considered as one of the key features in the pathogenesis type 2 DM.
In addition to muscle, liver, pancreatic α-cell and β-cells, adipocytes and the gut, the kidney is one of the two most recent members implicated in the pathophysiology of type 2 DM. Although excessive urination was one of the first described characteristics of DM[3-5] and the effort of its reduction was one of the first therapeutic approaches to DM while the sweet urine taste represented one of the first diagnostic approaches for DM, its pivotal role in type 2 DM pathogenesis was only recently explained. Physiologically, a total of approximately 162 g of glucose is filtered by kidneys on daily basis, and 90% of that amount is reabsorbed by the high capacity of sodium-glucose-transporter-2 (SGLT2) in the renal proximal tubule, while the remaining 10% of the filtered glucose is reabsorbed by the SGLT1 transporter in the descending part of the proximal tubule[59]. The result is that no glucose appears in the urine in healthy individuals. During the last two decades it became evident that in both type 1 and type 2 DM the maximal renal tubular reabsorptive capacity is increased[60,61]. This mechanism is due to markedly increased levels of SGLT2 mRNA and protein itself in the proximal renal tubular cells in type 2 DM[62]. In conclusion, the overactivation of an adaptive response by the kidney to conserve glucose seems to play an important role in hyperglycaemia development in type 2 DM.
Finally, the last component of the “ominous octet” is the central nervous system. Following functional magnetic resonance imaging investigations, it became evident that certain hypothalamic areas, precisely those which are involved in appetite regulation, show diminished inhibitory response in insulin-resistant type 2 DM individuals compared to normal glucose tolerant subjects, even though the plasma insulin response was noted as markedly increased in the obese group. This indicates that besides peripheral tissues, the insulin resistance, a common feature of all type 2 DM subjects, exists is the central nervous system.
Therefore, it is evident that the last few decades provided better and comprehensive knowledge on the pathophysiology of type 2 DM, which certainly had repercussion on the pharmacological treatment approach.
LONGITUDINAL HISTORY OF TREATMENT OPTIONS IN TYPE 2 DM
From the very beginning, even back in the times when frequent and excessive urination was considered a hallmark of DM, physicians tried to understand how it could be managed. They initially used the methods that could decrease this process, and there are documents reporting that horseback riding often was one of the first proposed methods[5]. Centuries later, Rollo[63] established the link between consumed food and the amount of glucose in the urine. He observed that carbohydrates increased glucose levels, while animal products’ consumption resulted in less glucose[3,4,64]. Thus, he proposed that DM treatment should be based on a high fat and protein rich diet with low carbohydrates. This modification of diet became the recommended treatment for diabetes until the discovery of insulin[65].
Since the first proposal in 1877, a series of modifications and even individualized approaches, such as “oat-cure”, “potato therapy” and the “starvation diet”, persisted until 1916. This was the year when a Boston scientist, Elliot Joslin, was established as one of the leading diabetes experts upon creating The Treatment of Diabetes Mellitus textbook, wherein he reported that fasting diet combined with regular but moderate physical activity could significantly reduce the risk of death in diabetes[66]. Although the diet and physical activity concept underwent numerous changes, especially over the last two decades, they still represent two official fundamental treatment approaches for type 2 DM, beyond pharmacological treatment[67], and probably will remain so in the future.
The pharmacological treatment options in type 2 DM, however, did not occur until after the 1900s, and in the century of their development resulted in a wide spectrum of insulin and non-insulin hypoglycaemic agents experiencing an exponential growth. Although the discovery of insulin is considered the beginning of the pharmacological “DM-treatment” era, it is less known that the discovery and the use of oral hypoglycaemic agents (OHL) started 60 years ago, with an OHL class of biguanides that now represent the basic, first-line pleiotropic agent known as metformin[67]. Namely, during the medieval times, the French lilac plant (Galega officinalis) was used to relieve DM symptoms in Southern and Eastern Europe[68]. At the beginning of the 20th century, the anti-hyperglycaemic compound of the plant, guanidine, was isolated, synthesized and named Synthalin[69]. The further development of Synthalin and other guanidine homologs was stopped since they were hepatotoxic, and completely ended with the discovery and global use of insulin[70]. However, a resurgence of interest in the biguanides occurred several decades later, as the pathophysiology of DM as well as the difference in its clinical presentation (i.e., before-mentioned classification) became clearer. Although metformin was introduced in 1959, its wide clinical use started two decades after and it was not approved in the United States until 1990[68-71]; meanwhile, other agents from this class - phenformin and buformin - resulted in a significant number of lactic acidosis, which led to their withdrawal[70]. Nowadays, metformin represents the only clinically significant biguanide whose primary mechanism of action is the ability to reduce hepatic gluconeogenesis and glycogenolysis due to enhancement of insulin resistance[72].
The sulfonylureas (SUs) represent the second and latest class of OHL, the discovery of which was triggered by the observation of an accidental hypoglycaemic effect. All the other OHL classes developed rapidly during the last two to three decades, accompanied by a deeper understanding of the complex pathophysiology of type 2 DM at the molecular level. The history of SUs starts in 1937, with the observation of hypoglycaemic activity of the synthetic sulphur compounds[73] and which was further confirmed by hypoglycaemia occurrence in typhoid patients treated with antibiotic para-amino-sulfonamide-isopropyl-thiodiazole[74]. In 1946, Auguste Loubatieres confirmed that aryl SU compounds stimulated release of insulin and, therefore, they require the remaining pancreatic β-cell function to achieve the effect[73,74]. Soon after, in the 1950s, the first SU - tolbutamide - was marketed in Germany[74], followed by the introduction of the other first-generation SU agents-chlorpropamide, acetohexamide, and tolazamide. The next advancement in SU therapy was the release of the more potent second-generation agents, back in 1984 - glipizide, glyburide, and gliklazide - and the third-generation agent glimepiride which was released in 1995[73-75]. Nowadays, the SUs are widely used, since they are generally safe, inexpensive, and relatively predictable, with hypoglycaemia as the primarily use-limiting side effect. In addition, due to research advances in seeking an agent that could be used with less fear of hypoglycaemia, the class of meglitinides was released on the market in 2000[76,77]. The meglitinides act similar to SUs, i.e., they enhance insulin secretion but their effect is diminished at low glucose concentrations. Thus, they might cause hypoglycaemia but less frequently and less severe than the “conventional” SUs[74].
The class of thiazolidinediones (TZDs) was initially introduced to the market in the middle of 1990. As a peroxisome proliferator–activated receptor-γ drug class activator, they were recognized soon after the discovery that the activation of this precise cell surface receptor enhances skeletal muscle insulin sensitivity and reduces hepatic glucose production[31,78]. This class of agents was thought to possess a similar but more durable effect than metformin. Troglitazone was the first TZD approved by the United States’ Federal Drug Administration (FDA), and was soon accompanied by pioglitazone and rosiglitazone[79]. Trosiglitazone was removed from the market 4 years after its release, in 2000. This was due to the FDA having received reports of 63 hepatic failure cases with lethal outcome in patients treated with troglitazone[80]. Two other TZDs, rosiglitazone and pioglitazone, have been linked to fluid retention, which limited their use in patients with congestive heart failure. Pioglitazone has been shown to have a potentially modest beneficial impact on CV disease but has also been associated with a possible increase in the incidence of bladder cancer[79]. However, the wide use of rosiglitazone was soon associated with an increased risk of myocardial infarction, which led to its temporary market restriction, i.e., it remained available only in the United States. However, in November 2013, there was a change in position based on the findings of the large “Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes” (RECORD) study, demonstrating that individuals treated with rosiglitazone did not have an elevated risk of myocardial infarction compared to those taking other antihyperglycemic agents[81,82]. Despite this, pioglitazone remains the most used TZD at the present time, taking its place in all the available diabetes management guidelines as the first to second option, but one cannot speculate whether rosiglitazone will dominate the market in the near future.
Before the endocrine role of gut itself in the “incretin effect” became a clear and important pathophysiological player in type 2 DM development (which then led to introduction of so called “incretin” group of anti-hyperglycaemic agents), a class of drugs targeting gut, precisely small intestine α-glucosidase and thereby decreasing glucose absorption, was introduced into clinical practice[74-78]. The first drug in this category of α-glucosidase inhibitors that reached the market was acarbose, back in 1995, followed a year later by miglitol. Despite the logical background of drug development, neither drug has ever been used widely, probably because of the modest impact on A1C and their gastrointestinal side effects. However, following the discovery and elucidation of the incretin-insulin pathway, the “incretin based” class of agents became an intriguing and rapid growing area of research and development in the pharmaceutical industry.
At first, researchers became interested in the development of dipeptidyl-peptidase inhibitors - agents that could be taken orally and would prolong the circulating half-life of endogenous incretins[45-47,74]. First among these was sitagliptin, in 2006[83]; this was soon followed by saxagliptin, linagliptin, vildagliptin and alogliptin, comprising a separate OHL class, the “gliptines”. Obviously, these drugs showed good results in the post-market clinical trials, as nowadays they are recommended even as second-line diabetes therapy[67]. Parallel to those, GLP-1 analogues were developed. The first analogue, exenatide was produced from exendin-4, which was isolated from the salivary gland venom of the Gila monster (Heloderma suspectum)[45-47,74]. This drug become available for clinical use in 2005[74]. A second GLP-1 receptor agonist, liraglutide, was approved in 2010. Soon after, i.e., over the last 8 years, the market of GLP-1 analogues has grown exponentially, starting with the once-weekly form of exenatide, dulaglutide, lixisenatide and albiglutide, to lately with semaglutide[74]. Although these drugs major advantage is weight loss, they also show cardio- and neuro-protective effects[67,84,85]. However, the exact mechanism of these post-market results remains to be elucidated.
And finally, after the “ominous octet” was completed with the elucidation of overactivation or renal tubular glucose reabsorptive capacity[59-62] due to a markedly increased level of SGLT2 in the proximal renal tubular cells[86], the selective SGLT-2 inhibitors were developed. More precisely, when SGLT-2 is antagonized, the excess of glucose in the renal tubules is not reabsorbed but instead secreted in the urine. Canagliflozin was the first SGLT-2 inhibitor to be approved by the FDA, in March 2013[87], which was followed by dapagliflozin in early 2014 and finally by empagliflozin.
Thus, here we described a wide spectrum of non-insulin therapeutic agents currently used in type 2 DM treatment. The official recommendations on their efficacy and safety, indications and contraindications, and effective combinations change almost on a yearly basis; yet, simultaneously we are experiencing an increase in diabetes-related complications, especially those leading to CV death[2,67]. This would, however, come as surprise if we kept in mind the results from the “Action to Control Cardiovascular Risk in Diabetes” trial[81] which clearly demonstrated that aggressive glycaemic control does not reduce the risk of CV death, despite the reduction in myocardial infarction.
NON-INSULIN THERAPEUTIC APPROACH AND CV RISK
As aforementioned in detail, due to the complex pathophysiological background of type 2 DM with the wide spectrum of CV risk factors that coexist in addition to the hyperglycaemia itself. It is important to emphasize here that the promising effects in terms of CV risk reduction were first published back in the late 90’s, as an observation in the “STOP-NIDDM Trial” (an international study on the efficacy of an alpha-glucosidase inhibitor to prevent type 2 DM in a population with impaired glucose tolerance)[88]. The primary outcome of the study that clearly demonstrated that acarbose can prevent or delay the progression of impaired glucose tolerance to type 2 DM was recently confirmed in the “Acarbose Cardiovascular Evaluation” (ACE) trial[89], but it showed no effect in CV risk reduction. Thus, the “Empagliflozin Cardiovascular Outcome Event Trial in type 2 DM Patients–Removing Excess Glucose” (EMPA-REG OUTCOME) was the first clinical study that demonstrated superiority of a glucose lowering agent, in CV disease, heart failure, and renal and mortality endpoints compared to placebo[90]. Given the glycated haemoglobin (HbA1c) reduction of 0.45%, blood pressure in approximately 5/2 mmHg, and reduction in body weight by approximately 2%, the question arose: Could these results have been predicted based on what we knew about the mode of action?
Thus, shortly after the EMPA-REG OUTCOME trial was published, Ferrannini et al[90] developed a so-called “thrifty substrate” hypothesis which posits that in conditions of mild but persistent hyperketonaemia, β-hydroxybutyrate is taken up by the heart and oxidized into fatty acids. This selection of substrates improves the transduction process of oxygen consumption into work efficiency in the myocardiocytes. In addition, the enhanced oxygen release to the myocardium through haemoconcentration-driven by the diuresis[91,92] might affect a powerful synergy with the substrate shift. The rationale for this hypothesis came from experimental studies in diet-induced obese rats treated with dapagliflozin[93], ipragliflozin[94], or tofogliflozin[95] that demonstrated accelerated lipolysis and increased circulating ketone body levels, especially in the fasting state or when animals were fed in pairs. In addition, this was also confirmed in patients with type 2 DM following a 4-wk course of treatment with 25 mg of empagliflozin[96]. Concomitantly, both fasting and post-meal plasma β-hydroxybutyrate concentrations were increased 2-fold to 3-fold and these changes were similar in time course, though attenuated in extent, in a group of non-diabetic volunteers receiving the drug. This hypothesis of the thrifty substrate might also explain the similar outcome obtained by the “Combined results from the Canagliflozin Cardiovascular Assessment Study” (CANVAS)[97]. Nevertheless, the post-market period has been too short to support any general conclusion at this time.
And finally, some of the GLP-1 agonists, i.e., liraglutde[98] and semaglutide[99] have shown a significant relative risk reduction compared with placebo for the 3-point major adverse CV event primary outcome and relative risk in CV as well as in all-cause mortality, having low-to-moderate between-trial statistical heterogeneity. However, the concerns about their potential in pancreatic cell proliferation observed in experimental studies has not been elucidated so far, due to the relatively short post-market period[100].
CONCLUSION
In this narrative review, we described the rapid development of the pathophysiology type 2 DM concept accompanied by lifesaving treatment options beyond insulin that have dramatically enhanced the quality of life and life expectancy of affected individuals. The nephroprotective effects of angiotensin-receptor blockade, angiotensin-converting enzyme inhibition and protein restriction have been shown[99-104], while laser photocoagulation has preserved the vision of millions of patients with diabetic retinopathy[105]. The target hyperglycaemic agents’ development has resulted in better glycaemic control, which has increased the focus of their potential in the context of development of diabetic complication preventive strategies.
It is now known that better gluco-regulation results in development and progression of microvascular complications, according to the large, population-based studies of Diabetes Control and Complications Trial[106] as well as United Kingdom Prospective Diabetes Study[107]. Additionally, the follow-up ACCORD study has showed reduced myocardial infarction with improved glycaemic control, but it didn’t provide an all-cause CV mortality rate. This finding raised awareness that an exclusively glucose-centric approach to diabetes will most likely not lead to reduction in all-cause CV disease mortality[98]. This finding was further strengthened by the Steno-2 trial[108,109], which demonstrated up to 50% CVD mortality with a multifactorial, instead of gluco-centric, control.
Thus, the present position statement for type 2 DM treatment comprises the simultaneous approach to control of glucose along with lipids, blood pressure and obesity[109,110]. Since obesity accompanies more than 80% of the type 2 DM population and contributes to other targeted factors’ improvement, its treatment strategy should be a priority in the comprehensive assessment of diabetes care. According to the American Diabetes Association Standards of Care from 2016, bariatric surgery should be considered in obesity management, in addition to behaviour modification and pharmacotherapy[111].
However, despite all the knowledge and all the pharmaceutic agents that are available, there will always be a need for more effective treatment options in order to affect the disease in an even more precise pathophysiological pathway, in the near future. For instance, we can expect a completely novel antidiabetic drug class-oxidative phosphorylation blockers, currently represented by imeglimin[112]. The underlying mechanism of action of this drug class consists of balancing bioenergetics in mitochondria and consequent insulin resistance to result in balance of insulin secretion and utilization as well as hepatic gluconeogenesis suppression. Thus, it is important to emphasize that this is not the sole promising novel drug class. This is indicated by findings from the research on the first-in-class drug, an adenosine monophosphate (AMP)-activated protein kinase activator, targeting one of the key players in the process of energy balance preservation, especially during caloric disturbances[113], and, finally, findings on the first monoclonal antibody, bimagrumab, that blocks the myostatin type II receptor, which results in fat reduction[114].
In conclusion, given the raising burden of type 2 DM and CV mortality due to diabetes, despite all of the therapeutic options that are available or will become available in due time, we should be focused on primary prevention, i.e., targeting preventive public health policies and in the rigorous evidence-based initiatives to introduce dietary products that will address metabolic disturbances.
ACKNOWLEDGMENTS
The authors acknowledge Dr. Lora Stanka Kirigin Biloš, a native English speaker for assistance on the English language editing process.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest.
Manuscript source: Invited manuscript
Peer-review started: August 29, 2018
First decision: October 5, 2018
Article in press: November 26, 2018
Specialty type: Endocrinology and metabolism
Country of origin: Croatia
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Barzilay J, Parikh M S- Editor: Ma RY L- Editor: A E- Editor: Song H
Contributor Information
Kristina Blaslov, Department of Endocrinology, Diabetes and Metabolic Diseases Mladen Sekso, University Hospital Center Sestre Milosrdnice, Zagreb 10000, Croatia. kblaslov@gmail.com.
Fran Stjepan Naranđa, School of Medicine, University of Zagreb, Zagreb 10000, Croatia.
Ivan Kruljac, Department of Endocrinology, Diabetes and Metabolic Diseases Mladen Sekso, University Hospital Center Sestre Milosrdnice, Zagreb 10000, Croatia.
Ivana Pavlić Renar, School of Medicine, University of Zagreb, Zagreb 10000, Croatia.
References
- 1.Powers AC. Diabetes Mellitus. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, et al., editors. Harrison’s Principles of Internal Medicine, 18th ed. New York: McGraw-Hill; 2012. [Google Scholar]
- 2.International Diabetes Federation. IDF Diabetes Atlas, 8th ed. Brussels: International Diabetes Federation; 2017. [Google Scholar]
- 3.Guthrie DW, Humphreys SS. Diabetes urine testing: an historical perspective. Diabetes Educ. 1988;14:521–526. doi: 10.1177/014572178801400615. [DOI] [PubMed] [Google Scholar]
- 4.Eknoyan G, Nagy J. A history of diabetes mellitus or how a disease of the kidneys evolved into a kidney disease. Adv Chronic Kidney Dis. 2005;12:223–229. doi: 10.1053/j.ackd.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Reece E. The history of diabetes mellitus. In: Reece E, Coustan D, et al., editors. Diabetes Mellitus in Pregnancy. New York: Churchill Livingstone; 1995. [Google Scholar]
- 6.Polonsky KS. The past 200 years in diabetes. N Engl J Med. 2012;367:1332–1340. doi: 10.1056/NEJMra1110560. [DOI] [PubMed] [Google Scholar]
- 7.von Mering J, Minkowski O. Diabetes mellitus nach Pankreas extirpation. Arch Exp Pathol Pharmacol. 1890;26:371–387. [Google Scholar]
- 8.Brogard JM, Vetter T, Blickle JF. Discovery of pancreatic diabetes in Strasbourg. Diabete Metab. 1992;18:104–114. [PubMed] [Google Scholar]
- 9.Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc J. 1922;12:141–146. [PMC free article] [PubMed] [Google Scholar]
- 10.Bliss M. The discovery of insulin: the inside story. Publ Am Inst Hist Pharm. 1997;16:93–99. [PubMed] [Google Scholar]
- 11.Geyelin HR, Harrop G, Murray MF, Corwin E. The use of insulin in juvenile diabetes. J Metabolic Res. 1922;2:767–792. [Google Scholar]
- 12.Dublin LI. The facts of life from birth to death. Macmillan: New York; 1951. [Google Scholar]
- 13.Himsworth HP. Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. Diabet Med. 2011;28:1440–1444. doi: 10.1111/j.1464-5491.2011.3508.x. [DOI] [PubMed] [Google Scholar]
- 14.Cori CF, Cori GT. Carbohydrate metabolism. Annu Rev Biochem. 1946;15:193–218. doi: 10.1146/annurev.bi.15.070146.001205. [DOI] [PubMed] [Google Scholar]
- 15.Houssay BA, Smyth FS, Foglia VG, Houssay AB. Comparative diabetogenic action of the hypophysis from various animals. J Exp Med. 1942;75:93–106. doi: 10.1084/jem.75.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer EH. Phosphorylase and the origin of reversible protein phosphorylation. Biol Chem. 2010;391:131–137. doi: 10.1515/bc.2010.011. [DOI] [PubMed] [Google Scholar]
- 17.From the NIH: Successful diet and exercise therapy is conducted in Vermont for “diabesity”. JAMA. 1980;243:519–520. [PubMed] [Google Scholar]
- 18.Gale EA. Is type 2 diabetes a category error? Lancet. 2013;381:1956–1957. doi: 10.1016/S0140-6736(12)62207-7. [DOI] [PubMed] [Google Scholar]
- 19.NHS. 2018. Metabolic syndrome. Accessed August 27. Available from: https://www.nhs.uk/conditions/metabolic-syndrome/ [Google Scholar]
- 20.The Globe diabetes community. 2018. Metabolic syndrome. Accessed August 27. Available from: https://www.diabetes.co.uk/diabetes-and-metabolic-syndrome.html. [Google Scholar]
- 21.BBC. 2018. Metabolic syndrome. Accessed August 27. Available from: http://www.bbc.co.uk/radio4/science/casenotes_20080122.shtml. [Google Scholar]
- 22.International Diabetes Federation. 2018. Guidelines. Accessed August 27. Available from: https://www.idf.org/e-library/guidelines.html. [Google Scholar]
- 23.Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. beta-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab. 2005;90:493–500. doi: 10.1210/jc.2004-1133. [DOI] [PubMed] [Google Scholar]
- 24.Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98:2133–2223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 26.Jallut D, Golay A, Munger R, Frascarolo P, Schutz Y, Jéquier E, Felber JP. Impaired glucose tolerance and diabetes in obesity: a 6-year follow-up study of glucose metabolism. Metabolism. 1990;39:1068–1075. doi: 10.1016/0026-0495(90)90168-c. [DOI] [PubMed] [Google Scholar]
- 27.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 28.Levy J, Atkinson AB, Bell PM, McCance DR, Hadden DR. Beta-cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: the 10-year follow-up of the Belfast Diet Study. Diabet Med. 1998;15:290–296. doi: 10.1002/(SICI)1096-9136(199804)15:4<290::AID-DIA570>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 29.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, DeFronzo RA. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest. 1989;84:205–213. doi: 10.1172/JCI114142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groop LC, Saloranta C, Shank M, Bonadonna RC, Ferrannini E, DeFronzo RA. The role of free fatty acid metabolism in the pathogenesis of insulin resistance in obesity and noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1991;72:96–107. doi: 10.1210/jcem-72-1-96. [DOI] [PubMed] [Google Scholar]
- 32.Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab. 2004;89:463–478. doi: 10.1210/jc.2003-030723. [DOI] [PubMed] [Google Scholar]
- 33.Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. 2013;9:191–200. doi: 10.5114/aoms.2013.33181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonadonna RC, De Fronzo RA. Glucose metabolism in obesity and type 2 diabetes. Diabete Metab. 1991;17:112–135. [PubMed] [Google Scholar]
- 35.DeFronzo RA. Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int J Clin Pract Suppl. 2004:9–21. doi: 10.1111/j.1368-504x.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 36.Fraze E, Donner CC, Swislocki AL, Chiou YA, Chen YD, Reaven GM. Ambient plasma free fatty acid concentrations in noninsulin-dependent diabetes mellitus: evidence for insulin resistance. J Clin Endocrinol Metab. 1985;61:807–811. doi: 10.1210/jcem-61-5-807. [DOI] [PubMed] [Google Scholar]
- 37.Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L, DeFronzo R, Cusi K. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–2474. doi: 10.2337/diabetes.52.10.2461. [DOI] [PubMed] [Google Scholar]
- 38.Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF. Prolonged elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese nondiabetic humans but not in individuals with type 2 diabetes. Diabetes. 2000;49:399–408. doi: 10.2337/diabetes.49.3.399. [DOI] [PubMed] [Google Scholar]
- 39.Thiébaud D, DeFronzo RA, Jacot E, Golay A, Acheson K, Maeder E, Jéquier E, Felber JP. Effect of long chain triglyceride infusion on glucose metabolism in man. Metabolism. 1982;31:1128–1136. doi: 10.1016/0026-0495(82)90163-9. [DOI] [PubMed] [Google Scholar]
- 40.Felber JP, Vannotti A. Effects of fat infusion on glucose tolerance and insulin plasma levels. Med Exp Int J Exp Med. 1964;10:153–156. doi: 10.1159/000135410. [DOI] [PubMed] [Google Scholar]
- 41.Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williamson JR, Kreisberg RA, Felts PW. Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc Natl Acad Sci USA. 1966;56:247–254. doi: 10.1073/pnas.56.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bevilacqua S, Bonadonna R, Buzzigoli G, Boni C, Ciociaro D, Maccari F, Giorico MA, Ferrannini E. Acute elevation of free fatty acid levels leads to hepatic insulin resistance in obese subjects. Metabolism. 1987;36:502–506. doi: 10.1016/0026-0495(87)90051-5. [DOI] [PubMed] [Google Scholar]
- 44.Bays HE, González-Campoy JM, Bray GA, Kitabchi AE, Bergman DA, Schorr AB, Rodbard HW, Henry RR. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev Cardiovasc Ther. 2008;6:343–368. doi: 10.1586/14779072.6.3.343. [DOI] [PubMed] [Google Scholar]
- 45.Holst JJ. Glucagon-like peptide-1: from extract to agent. The Claude Bernard Lecture, 2005. Diabetologia. 2006;49:253–260. doi: 10.1007/s00125-005-0107-1. [DOI] [PubMed] [Google Scholar]
- 46.Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3:153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 48.Meier JJ, Nauck MA. Incretins and the development of type 2 diabetes. Curr Diab Rep. 2006;6:194–201. doi: 10.1007/s11892-006-0034-7. [DOI] [PubMed] [Google Scholar]
- 49.Vilsbøll T, Holst JJ. Incretins, insulin secretion and Type 2 diabetes mellitus. Diabetologia. 2004;47:357–366. doi: 10.1007/s00125-004-1342-6. [DOI] [PubMed] [Google Scholar]
- 50.Gutniak M, Orskov C, Holst JJ, Ahrén B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. 1992;326:1316–1322. doi: 10.1056/NEJM199205143262003. [DOI] [PubMed] [Google Scholar]
- 51.Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care. 1992;15:270–276. doi: 10.2337/diacare.15.2.270. [DOI] [PubMed] [Google Scholar]
- 52.Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 53.Consoli A, Nurjhan N, Reilly JJ Jr, Bier DM, Gerich JE. Mechanism of increased gluconeogenesis in noninsulin-dependent diabetes mellitus. Role of alterations in systemic, hepatic, and muscle lactate and alanine metabolism. J Clin Invest. 1990;86:2038–2045. doi: 10.1172/JCI114940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsuda M, Defronzo RA, Glass L, Consoli A, Giordano M, Bressler P, Delprato S. Glucagon dose-response curve for hepatic glucose production and glucose disposal in type 2 diabetic patients and normal individuals. Metabolism. 2002;51:1111–1119. doi: 10.1053/meta.2002.34700. [DOI] [PubMed] [Google Scholar]
- 55.Unger RH, Aguilar-Parada E, Müller WA, Eisentraut AM. Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest. 1970;49:837–848. doi: 10.1172/JCI106297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64:106–110. doi: 10.1210/jcem-64-1-106. [DOI] [PubMed] [Google Scholar]
- 57.Wise JK, Hendler R, Felig P. Evaluation of alpha-cell function by infusion of alanine in normal, diabetic and obese subjects. N Engl J Med. 1973;288:487–490. doi: 10.1056/NEJM197303082881003. [DOI] [PubMed] [Google Scholar]
- 58.Boden G, Soriano M, Hoeldtke RD, Owen OE. Counterregulatory hormone release and glucose recovery after hypoglycemia in non-insulin-dependent diabetic patients. Diabetes. 1983;32:1055–1059. doi: 10.2337/diab.32.11.1055. [DOI] [PubMed] [Google Scholar]
- 59.Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782–790. doi: 10.4158/EP.14.6.782. [DOI] [PubMed] [Google Scholar]
- 60.Noonan WT, Shapiro VM, Banks RO. Renal glucose reabsorption during hypertonic glucose infusion in female streptozotocin-induced diabetic rats. Life Sci. 2001;68:2967–2977. doi: 10.1016/s0024-3205(01)01090-6. [DOI] [PubMed] [Google Scholar]
- 61.Kamran M, Peterson RG, Dominguez JH. Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. J Am Soc Nephrol. 1997;8:943–948. doi: 10.1681/ASN.V86943. [DOI] [PubMed] [Google Scholar]
- 62.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54:3427–3434. doi: 10.2337/diabetes.54.12.3427. [DOI] [PubMed] [Google Scholar]
- 63.Bernard MC. Lecons sur Le Diabete et La Glycogenese Animale. Paris: Bailliereet Fils. 1877 [Google Scholar]
- 64.Papaspyros NS. The History of Diabetes Mellitus, 2nd ed. Stuttgart: Georg Thieme Verlag; 1964. [Google Scholar]
- 65.Allan FN. Diabetes before and after insulin. Med Hist. 1972;16:266–273. doi: 10.1017/s0025727300017750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joslin EP. The Treatment of Diabetes Mellitus. Can Med Assoc J. 1916;6:673–684. [PMC free article] [PubMed] [Google Scholar]
- 67.Introduction: Standards of Medical Care in Diabetes-2018. Diabetes Care. 2018;41:S1–S2. doi: 10.2337/dc18-Sint01. [DOI] [PubMed] [Google Scholar]
- 68.White JR, Campbell RK, White JR. Overview of the medications used to treat type 2 diabetes. In: White JR, Campbell RK, et al., editors. Medications for the Treatment of Diabetes. Alexandria: American Diabetes Association; 2008. pp. 5–15. [Google Scholar]
- 69.Frank E, Nothnamm M, Wagner A. Über synthetische dargestellte Korper mit Insulinartiger Wirkung auf den normallen und diabetisched Organismus. Klin Wchnschr. 1926;5:2011–2107. [Google Scholar]
- 70.Jarvis B, Elkinson S. Agents in development for type 2 diabetes. Drugs R D. 1999;2:95–99. doi: 10.2165/00126839-199902020-00002. [DOI] [PubMed] [Google Scholar]
- 71.Alberti KGMM, Zimmet P, Defronzo RA: International Textbook of Diabetes Mellitus, 2nd ed. New York: John Wiley & Sons. 1997 [Google Scholar]
- 72.Rodbard HW, Blonde L, Braithwaite SS, Brett EM, Cobin RH, Handelsman Y, Hellman R, Jellinger PS, Jovanovic LG, Levy P, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13 Suppl 1:1–68. doi: 10.4158/EP.13.S1.1. [DOI] [PubMed] [Google Scholar]
- 73.Levine R. Sulfonylureas: background and development of the field. Diabetes Care. 1984;7 Suppl 1:3–7. [PubMed] [Google Scholar]
- 74.Quianzon CC, Cheikh IE. History of current non-insulin medications for diabetes mellitus. J Community Hosp Intern Med Perspect. 2012:2. doi: 10.3402/jchimp.v2i3.19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Galloway JA, Diabetes Mellitus, 9th ed. Indianapolis: Eli Lilly and Company. 1988 [Google Scholar]
- 76.US Food and Drug Administration. 2018. Prandin. Accessed August 27. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails. [Google Scholar]
- 77.US Food and Drug Administration. 2018. Precose. Accessed August 27. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails. [Google Scholar]
- 78.Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R, Matthews DR; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD) Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetes Care. 2012;35:1364–1379. doi: 10.2337/dc12-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kendall DM. Thiazolidinediones: the case for early use. Diabetes Care. 2006;29:154–157. doi: 10.2337/diacare.29.1.154. [DOI] [PubMed] [Google Scholar]
- 80.US Food and Drug Administration. Troglitazone: presentation to advisory committee. Accessed August 27, 2018. Available from: http://www.fda.gov/ohrms/dockets/ac/00/slides/3615s1a.PPT. [Google Scholar]
- 81.ACCORD Study Group, Gerstein HC, Miller ME, Genuth S, Ismail-Beigi F, Buse JB, Goff DC Jr, Probstfield JL, Cushman WC, Ginsberg HN, Bigger JT, Grimm RH Jr, Byington RP, Rosenberg YD, Friedewald WT. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med. 2011;364:818–828. doi: 10.1056/NEJMoa1006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nature world news. 2018. FDA eases restrictions on Glaxo diabetes drug Avandia. Accessed August 27. Available from: http://www.natureworldnews.com/articles/5075/20131126/fda-eases-restrictions-glaxos-drug.htm. [Google Scholar]
- 83.Neumiller JJ. Differential chemistry (structure), mechanism of action, and pharmacology of GLP-1 receptor agonists and DPP-4 inhibitors. J Am Pharm Assoc (2003) 2009;49 Suppl 1:S16–S29. doi: 10.1331/JAPhA.2009.09078. [DOI] [PubMed] [Google Scholar]
- 84.Hausenloy DJ, Yellon DM. GLP-1 therapy: beyond glucose control. Circ Heart Fail. 2008;1:147–149. doi: 10.1161/CIRCHEARTFAILURE.108.810887. [DOI] [PubMed] [Google Scholar]
- 85.MacDonald PE, El-Kholy W, Riedel MJ, Salapatek AM, Light PE, Wheeler MB. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes. 2002;51 Suppl 3:S434–S442. doi: 10.2337/diabetes.51.2007.s434. [DOI] [PubMed] [Google Scholar]
- 86.Forbes. 2018. FDA approves first SGLT2 inhibitor for diabetes. Accessed August 27. Available from: http://www.forbes.com/sites/larryhusten/2013/03/29/fda-approves-first-sglt2-inhibitor-for-diabetes. [Google Scholar]
- 87.Chiasson JL, Gomis R, Hanefeld M, Josse RG, Karasik A, Laakso M. The STOP-NIDDM Trial: an international study on the efficacy of an alpha-glucosidase inhibitor to prevent type 2 diabetes in a population with impaired glucose tolerance: rationale, design, and preliminary screening data. Study to Prevent Non-Insulin-Dependent Diabetes Mellitus. Diabetes Care. 1998;21:1720–1725. doi: 10.2337/diacare.21.10.1720. [DOI] [PubMed] [Google Scholar]
- 88.Holman RR, Coleman RL, Chan JCN, Chiasson JL, Feng H, Ge J, Gerstein HC, Gray R, Huo Y, Lang Z, et al. Effects of acarbose on cardiovascular and diabetes outcomes in patients with coronary heart disease and impaired glucose tolerance (ACE): a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017;5:877–886. doi: 10.1016/S2213-8587(17)30309-1. [DOI] [PubMed] [Google Scholar]
- 89.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373:2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 90.Ferrannini E, Mark M, Mayoux E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care. 2016;39:1108–1114. doi: 10.2337/dc16-0330. [DOI] [PubMed] [Google Scholar]
- 91.McMurray J. EMPA-REG - the “diuretic hypothesis”. J Diabetes Complications. 2016;30:3–4. doi: 10.1016/j.jdiacomp.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 92.Devenny JJ, Godonis HE, Harvey SJ, Rooney S, Cullen MJ, Pelleymounter MA. Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obesity (Silver Spring) 2012;20:1645–1652. doi: 10.1038/oby.2012.59. [DOI] [PubMed] [Google Scholar]
- 93.Yokono M, Takasu T, Hayashizaki Y, Mitsuoka K, Kihara R, Muramatsu Y, Miyoshi S, Tahara A, Kurosaki E, Li Q, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol. 2014;727:66–74. doi: 10.1016/j.ejphar.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 94.Suzuki M, Takeda M, Kito A, Fukazawa M, Yata T, Yamamoto M, Nagata T, Fukuzawa T, Yamane M, Honda K, et al. Tofogliflozin, a sodium/glucose cotransporter 2 inhibitor, attenuates body weight gain and fat accumulation in diabetic and obese animal models. Nutr Diabetes. 2014;4:e125. doi: 10.1038/nutd.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, Mari A, Pieber TR, Muscelli E. Shift to Fatty Substrate Utilization in Response to Sodium-Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients With Type 2 Diabetes. Diabetes. 2016;65:1190–1195. doi: 10.2337/db15-1356. [DOI] [PubMed] [Google Scholar]
- 96.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR; CANVAS Program Collaborative Group. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med. 2017;377:644–657. [Google Scholar]
- 97.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2016;375:311–322. doi: 10.1056/NEJMoa1603827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375:1834–1844. doi: 10.1056/NEJMoa1607141. [DOI] [PubMed] [Google Scholar]
- 99.Azoulay L, Filion KB, Platt RW, Dahl M, Dormuth CR, Clemens KK, Durand M, Juurlink DN, Targownik LE, Turin TC, et al. Incretin based drugs and the risk of pancreatic cancer: international multicentre cohort study. BMJ. 2016;352:i581. doi: 10.1136/bmj.i581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barnett AH, Bain SC, Bouter P, Karlberg B, Madsbad S, Jervell J, Mustonen J; Diabetics Exposed to Telmisartan and Enalapril Study Group. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med. 2004;351:1952–1961. doi: 10.1056/NEJMoa042274. [DOI] [PubMed] [Google Scholar]
- 101.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 102.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 103.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 104.Parving HH, Lehnert H, Bröchner-Mortensen J, Gomis R, Andersen S, Arner P; Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345:870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- 105.Klahr S, Levey AS, Beck GJ, Caggiula AW, Hunsicker L, Kusek JW, Striker G. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. N Engl J Med. 1994;330:877–884. doi: 10.1056/NEJM199403313301301. [DOI] [PubMed] [Google Scholar]
- 106.Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 107.Diabetes Control and Complications Trial Research Group, Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, Davis M, Rand L, Siebert C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 108.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 109.Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med. 2003;348:383–393. doi: 10.1056/NEJMoa021778. [DOI] [PubMed] [Google Scholar]
- 110.Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580–591. doi: 10.1056/NEJMoa0706245. [DOI] [PubMed] [Google Scholar]
- 111.Standards of Medical Care in Diabetes-2016: Summary of Revisions. Diabetes Care. 2016;39 Suppl 1:S4–S5. doi: 10.2337/dc16-S003. [DOI] [PubMed] [Google Scholar]
- 112.Vuylsteke V, Chastain LM, Maggu GA, Brown C. Imeglimin: A Potential New Multi-Target Drug for Type 2 Diabetes. Drugs R D. 2015;15:227–232. doi: 10.1007/s40268-015-0099-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim KH, Lee IS, Park JY, Kim Y, An EJ, Jang HJ. Cucurbitacin B Induces Hypoglycemic Effect in Diabetic Mice by Regulation of AMP-Activated Protein Kinase Alpha and Glucagon-Like Peptide-1 via Bitter Taste Receptor Signaling. Front Pharmacol. 2018;9:1071. doi: 10.3389/fphar.2018.01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Garito T, Roubenoff R, Hompesch M, Morrow L, Gomez K, Rooks D, Meyers C, Buchsbaum MS, Neelakantham S, Swan T, et al. Bimagrumab improves body composition and insulin sensitivity in insulin-resistant individuals. Diabetes Obes Metab. 2018;20:94–102. doi: 10.1111/dom.13042. [DOI] [PubMed] [Google Scholar]