

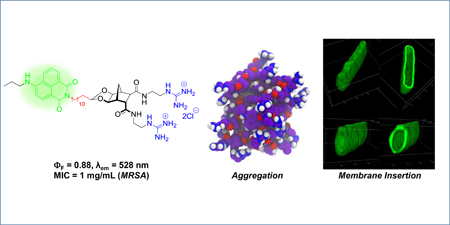
Abstract
The design, synthesis and evaluation of a small series of potent amphiphilic norbornane antibacterial agents has been performed (compound 10 MIC = 0.25 μg/mL against MRSA). Molecular modelling indicates rapid aggregation of this class of antibacterial agent prior to membrane association and insertion. Two fluorescent analogues (compound 29 with 4-amino-naphthalimide and 34 with 4-nitrobenz-2-oxa-1,3-diazole fluorophores) with good activity (MIC = 0.5 μg/mL against MRSA) were also constructed and confocal microscopy studies indicate that the primary site of interaction for this family of compounds is the bacterial membrane.
Keywords: Antimicrobial, antibacterial, amphiphilic, fluorescence, naphthalimide, norbornane, microscopy
Graphical Abstract
1. Introduction
There exists an urgent need to combat the emergence of multi-drug resistant (MDR) bacteria.[1–7] Positive steps have been taken to resolve the problem of bacterial resistance, and this issue has received significant media attention and high-priority recognition by leading health organisations including WHO, British Health and the IDSA.[5] A number of initiatives have been aimed at enticing pharmaceutical companies to re-establish antibacterial product development.[8] Nevertheless, the inevitability of bacterial resistance to any newly approved drug reinforces the need to have new classes of antibacterial agents in the development pipeline; and particularly new drugs that limit the potential for bacterial resistance.[9, 10]
Cationic antimicrobial peptides (CAMPs) are naturally occurring compounds that exhibit activity against most microbes including bacteria.[11, 12] The ability of CAMPs to adopt or be preorganised in a structurally amphiphilic manner, allows them to interact and ultimately perturb the bacterial membrane.[13, 14] Given their longstanding success in nature, it is widely thought that the development of bacterial resistance to these agents should be limited.[15, 16] The synthesis of peptidomimetics that also possess well-defined amphiphilic structure is therefore a logical step in the pursuit of new antibacterial agents and a number of research groups are currently active in this field.[17–20]
The rigid norbornane scaffold has previously been used to prepare preorganised anion hosts[21, 22] and in turn facially amphiphilic cationic peptidomimetics,[23–25] including bisethers (such as 1, Figure 1)[24] and norbornane acetals (such as 2),[23] with both classes active against Gram-positive and Gram-negative bacterial strains. The acetal family were the more potent with structure-activity relationship (SAR) studies indicating that a dicationic charge and a long hydrophobic component were essential for activity.[23]
Figure 1:
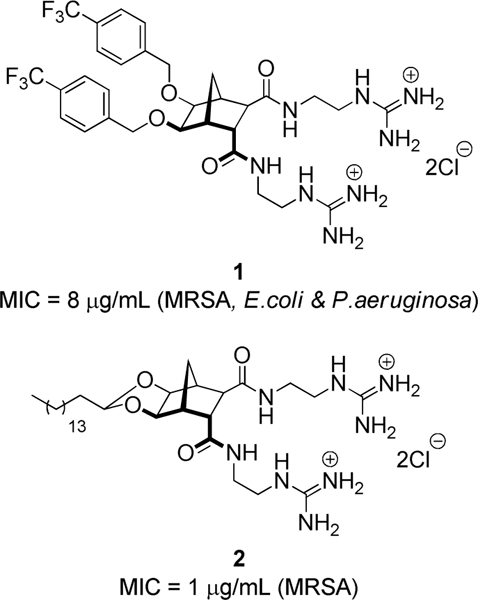
Previously reported norbornane-based antibacterial agents.[23, 24]
In order to further probe the SAR of these compounds, we envisaged analogues of bisguanidinium 2 where the guanidine groups were further modified with, for example, a hydrophobic group. While a bisphenylguanidine norbornane has been previously reported,[25] the isolation of this product proved troublesome and as such we sought to devise a simpler and more robust synthetic route.
While it was originally hypothesised that the mode of action (MOA) of this norbornane class of compounds involved the membrane as the primary target, no experiments have been performed to establish this fact. Moreover, although the majority of CAMPs are membrane active there are many examples in which these peptides also have an intracellular target.[26] A combination of cellular targets is thought to lead to greater antibacterial activity as well as increasing the barrier to evolved resistance.[26]
Fluorescence microscopy has been used to help elucidate antibacterial MOA of several peptidomimetic compounds,[27, 28] and compounds that aren’t inherently fluorescent can often be tagged with a small organic fluorophore with little impact on biological activity.[29] A plethora of small organic fluorescent compounds are available—including the 4-amino-1,8-naphthalimide[30] and 4-nitrobenz-2-oxa-1,3-diazole (NBD) fluorophores[31]—and these have been successfully used to provide an insight into important biological processes. Both of these fluorophores exhibit large Stokes shifts, high quantum yields and typically emit in the green portion of the UV-visible spectrum (495–570 nm).[32, 33]
This study outlines the synthesis and evaluation of six new norbornane-based antibacterial agents, including the most potent to date, and two fluorescent analogues that also possess excellent antibacterial activity. A combination of molecular modelling and fluorescence microscopy has been used to provide an insight into the MOA of this class of compounds.
2. Results and Discussion
2.1. Chemistry
Our preferred reagent for introducing the guanidine group was isothiourea 3[34, 35] and this compound was used here as the initial starting point to generate further functionalised guanidinylating agents. Unfortunately, the direct alkylation of 3 using NaH and BnBr gave 4 in poor yield. Neverthless, using Mitsunobu conditions (BnOH, PPh3, DIAD) benzylation was successful and 4 was isolated in excellent yield (95%, Scheme 1). Heating bisamines 5 and 8[23] with guanidinylating agent 4 and Et3N in CH2Cl2 gave the Boc-protected guanidines 6 and 9 respectively. The Boc-groups were then cleaved using methanolic HCl (generated from AcCl in MeOH) to give the desired guanidine hydrochloride salts 7 and 10.
Scheme 1:
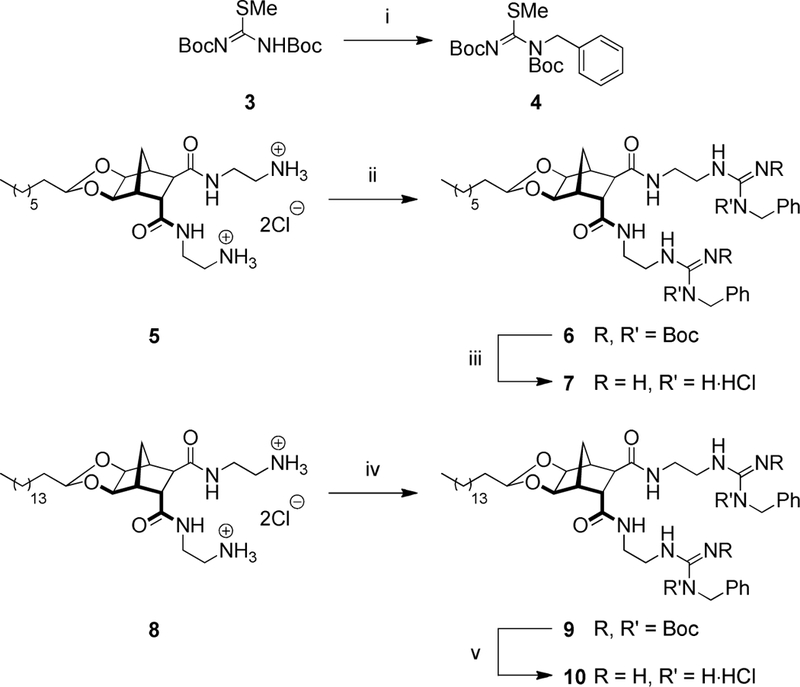
Reagents and conditions: (i) BnOH, DIAD, Ph3P, THF, 66 °C, 16 h, 95%; (ii) 4, Et3N, CH2Cl2, 40 °C, 2 d, 30%; (iii) AcCl, MeOH, 21 °C, 24 h, 93%; (iv) 4, Et3N, CH2Cl2, 80 °C, 2 d, 14%; (v) AcCl, MeOH, 21 °C, 24 h, 74%;
It is known that the anion recognition group is important to the success of many antimicrobial peptides (AMPs) due to their ability to interact with negatively charged phosphates on the outer bacterial membrane.[36–38] To date, we have investigated a number of different anion recognition groups with varying degrees of success.[23] A guanidiniocarbonylpyrrole (GCP, 11) moiety has been reported by the Schmuck group[39] and has been shown to interact with anions that are part of large biological macromolecules.[40–42] In light of this work, we furnished bisamine 5 with the GCP group over two steps to give acyl guanidinium 13 (Scheme 2).
Scheme 2:
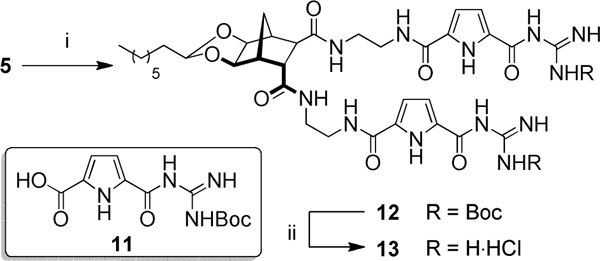
Reagents and conditions: (i) 11, PyBOP, DMAP, DMF, 80 °C, 16 h, 42%; (ii) AcCl, MeOH, 21 °C, 24 h, 99%.
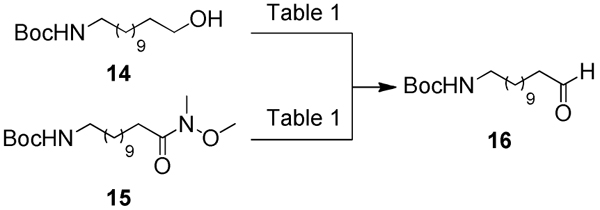
In order to further functionalise the hydrophobic component of the norbornane acetals, N-Boc-protected dodecanal 16 was desired. The synthesis of aldehyde 16 has been previously reported by Zeiler and co-workers where the oxidation of 14 was achieved using Swern conditions in 73% yield.[43] Unfortunately in our hands, and despite altering reaction conditions, including the amount of oxalyl chloride, temperature and reaction duration, aldehyde 16 could only be isolated in a 35% yield after chromatographic purification (Table 1, entries 1–3). Additional studies using 1H NMR indicated that aldehyde 16 degraded quickly; with the resonance assigned to the aldehyde (δ = 9.76 ppm) decreasing in size and relative integration over time. As such, another approach that could generate 16 rapidly, in high yields and without the need for a laborious column chromatography step was pursued.
Table 1:
Optimised synthesis of aldehyde 16.
 |
| Entry | S.M.a | Redox agent (equiv.) |
Temp. (°C) |
Time (h) |
Yield (%) |
|---|---|---|---|---|---|
| 1b | 14 | (COCl2)2 (1.2)c | 21 | 24 | 35 |
| 2 | 14 | (COCl2)2 (1.2)c | –78 | 4 | 18 |
| 3 | 14 | (COCl2)2 (2.4)c | –78 | 3 | 20 |
| 4 | 14 | PCC (1.5) | 21 | 3 | 6 |
| 5 | 14 | PCC (3.0) | 21 | 3 | 79 |
| 6 | 14 | TEMPO (0.2) | 21 | 48 | 58 |
| 7 | 15 | LiAlH4 (1.5) | –78 | 2 | 95 |
S.M. = starting material
Stirred at –78 °C for 3 h before warming to 21 °C and stirred for a further 21 h
DMSO (5.0 equiv.) used
Pyridinium chlorochromate (PCC) has been used extensively for the oxidation of primary alcohols to aldehydes.[44–46] Initially following the procedure described by Sanders and co-workers,[47] treating alcohol 14 with PCC (1.5 equiv.) gave aldehyde 16 in poor yield (6%), again, after chromatographic purification (Table 1, entry 4). However, using a larger excess of PCC (3.0 equiv.) the alcohol was consumed after 3 hours and 16 was isolated in 79% yield, but again chromatographic purification was required (Table 1, entry 5). Oxidation of 14 using TEMPO (0.2 equiv.) and (diacetoxyiodo)benzene, [48] afforded aldehyde 16 in 58% yield after a column chromatography step (Table 1, entry 6). By far the most successful approach was reduction of Weinreb amide 15 using LiAlH4 to give aldehyde 16 in 95% yield after only two hours and with no requirement for chromatographic purification (Table 1, entry 7). Maintaining the reaction temperature at –78 °C prevented the formation of side-products and spectroscopically pure 16 was isolated after a simple extractive work-up.
Freshly prepared aldehyde 16 was reacted with diol 17 using previously established methodology[23] to give acetal 18 in a 78% yield (Scheme 3). The attachment of two guanidinium groups to the norbornane scaffold, using freshly prepared 2-[2,3-Bis(tert-butoxycarbonyl)guanidino]ethylamine (22),[34] was carried out over three steps.[24] Briefly, hydrolysis of the methyl esters gave dicarboxylic acid 19 in 88% yield. Standard EDCI/HOBt coupling conditions were employed to attach two units of 22 to the norbornane scaffold. Global deprotection using methanolic HCl gave the desired product 21 as the trihydrochloride salt.
Scheme 3:
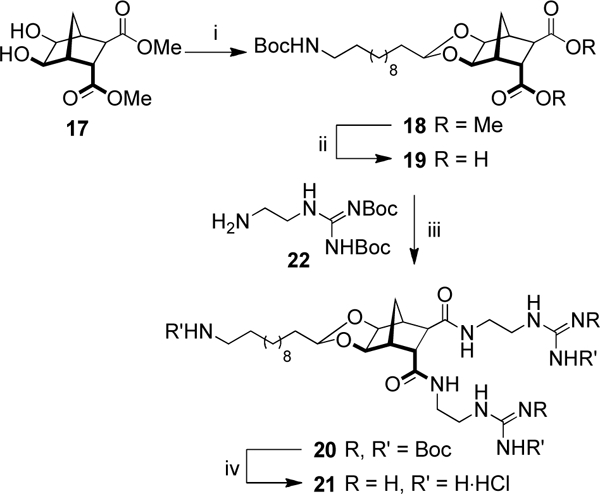
(i) 16, TsOH·H2O, MgSO4, PhMe, 110 °C, 3 h, 78%; (ii) NaOH, THF/H2O, 21 °C, 16 h, 88%; (iii) 22, EDCI, HOBt, CHCl3, 50 °C, 30 min, 32%; (iv) AcCl, MeOH, 21 °C, 2 d, 99%.
In order to attach a fluorescent tag to the norbornane framework, Boc-protected amine 18 was deprotected to give amine 23 using the same methanolic HCl procedure described earlier. Condensation between amine 23 and 4-bromo-1,8-naphthalic anhydride 24 afforded the corresponding 4-bromo-1,8-naphthalimide 25 in near-quantitative yield (99%, Scheme 4). It should be noted that a trace amount of starting material 24 remained (as evidenced by 1H NMR spectroscopy) but this was eventually removed using column chromatography in the next step. While amination of the 4-bromo position of naphthalimides can be achieved using thermal SNAr conditions, concomitant amide formation could also occur at the methyl ester sites. As such, the method reported by Fleming and co-workers was followed to convert bromide 25 to the fluorescent 4-aminopropyl-1,8-naphthalimide using propylamine, in the presence of Pd2(dba)3 CHCl3, xantphos and Cs2CO3 (3.0 equiv.) in PhCH3 at 70 °C.[49] After chromatographic purification, fluorescent norbornane 26 was isolated in excellent yield (88%). The attachment of two guanidinium groups to the norbornane scaffold was achieved over three steps, following the previously described methodology, to give naphthalimide 29 (Scheme 4).
Scheme 4:
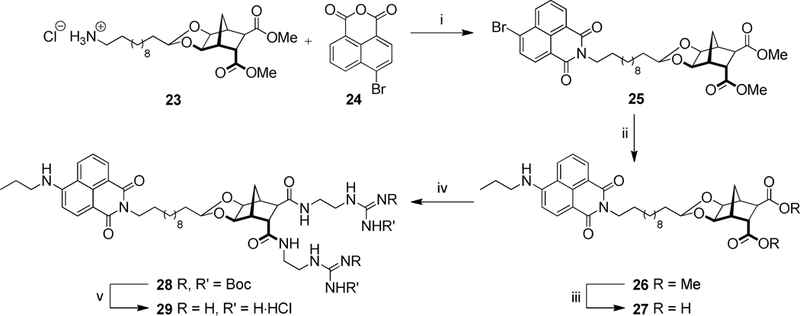
Reagents and conditions: (i) Et3N, EtOH, MW: 100 °C, 45 min, 99%; (ii) Propylamine, Pd2(dba)3CHCl3, Xantphos, Cs2CO3, PhMe, 70 °C, 24 h, 88%; (iii) NaOH, THF/H2O, 21 °C, 16 h, 88%; (iv) 22, EDCI, HOBt, DMF, 21 °C, 2 d, 82%; (v) AcCl, MeOH, 21 °C, 2 d, 97%.
A similar protocol was used to construct the NBD analogue (Scheme 5). Aromatic substitution of commercially available NBC-Cl (30) with amine 23 proceeded smoothly to give the NBD-norbornane conjugate (31). After hydrolysis, amide coupling and Boc-deprotection, the desired NBD analogue (34) was isolated as the dihydrochloride salt.
Scheme 5:
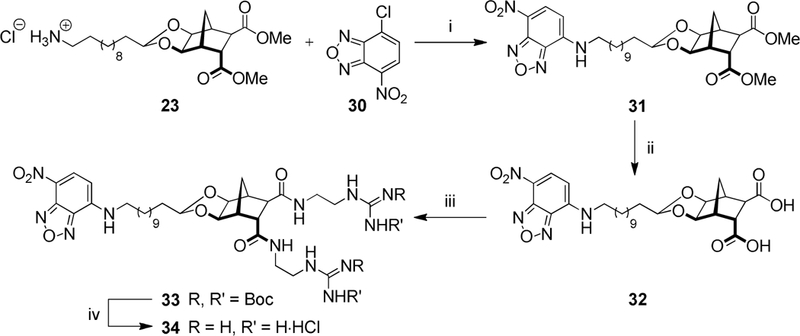
Reagents and conditions: (i) Et3N, MeOH, 21 °C, 24 h, 68%; (ii) NaOH, THF/H2O, 21 °C, 16 h, 78%; (iii) 22, EDCI, HOBt, DMF, 21 °C, 3 d, 65%; (iv) AcCl, MeOH, 21 °C, 2 d, 95%.
2.2. Photophysical properties
All fluorescent analogues displayed properties typical of their respective fluorophores. Naphthalimide derivatives 28 and 29 exhibited absorption maxima at ~440 nm (Table 2) with corresponding emission maxima of ~530 nm in DMSO and 549 nm in H2O with Stokes shifts in the range of 84–100 nm. Quantum yields for 28 and 29 in DMSO were high (0.89 and 0.88, respectively) whereas the quantum yield for 29 in H2O was significantly reduced (Φf = 0.16). Similarly, the NBD analogues also displayed characteristic fluorescent behaviour in DMSO (λabs ~ 475 nm, λex = 538 nm, Φf = 0.55) In H2O a slight bathochromic shift in emission and decreased quantum yield was observed (λex = 550 nm, Φf = 0.02). Nonetheless, the properties observed for 29 and 34 in H2O were amenable for fluorescence microscopy.
Table 2:
Photophysical properties
| Compound | Solvent | λabs (nm) |
λem (nm) |
Stokes shift (nm) |
Φf |
|---|---|---|---|---|---|
| 28 | DMSO | 442 | 526 | 84 | 0.89 |
| 29 | DMSO | 437 | 528 | 91 | 0.88 |
| 29 | H2O | 449 | 549 | 100 | 0.16 |
| 33 | DMSO | 475 | 538 | 63 | 0.55 |
| 34 | DMSO | 472 | 538 | 66 | 0.55 |
| 34 | H2O | 475 | 550 | 75 | 0.02 |
2.3. Antimicrobial activity
The six new compounds were evaluated against a range of Gram-negative and Gram-positive bacteria. The principal means of assessing activity was by broth-micro dilution (BMD) assay, to ascertain the minimum inhibitory concentration (MIC), of each compound. For some analogues, disk diffusion assay (Kirby-Bauer), was also performed (see ESI for results). The new compounds exhibited excellent activity against a range of Gram-positive bacterial strains with MIC values comparable to Vancomycin (Table 3). The analogues containing the benzyl substituted guanidine groups (7, and 10) were the most active, with MIC values as low as 0.25 μg/mL; an improvement in activity compared to their previously synthesised non-substituted counterparts (no activity observed and one MIC value of 1μg/mL, respectively).[23] The tricationic analogue (21) only showed moderate activity (MIC = 8–16 μg/mL) despite the presence of an extra charged moiety; reinforcing the notion that structural amphiphilicity is required for antibacterial activity. Importantly, both naphthalimide 29 and NBD 34 were active (MIC value for both = 1 μg/mL against MRSA); comparable to the previously reported non-fluorescent norbornane 2 (MIC value of 2 μg/mL against MRSA).[23] This result suggests that the attachment of the largely hydrophobic fluorescent tag has minimal impact on the antibacterial activity of these compounds. Unfortunately, the GCP analogue (13) failed to show any activity at all. When these compounds were evaluated against Gram-negative bacterial strains only moderate or weak activity was noted, the best result was obtained for benzylguanidine 7 and NBD 34; both with MIC values of 8 μg/mL against Escherichia coli (Table 3). Given that a number of groups have previously demonstrated the antifungal properties of CAMPs,[50, 51] compounds 7, 29 and 34 were also evaluated against pathogenic fungal strains (Table S3). All compounds exhibited activity, highlighted by an MIC of 0.25 μg/mL for naphthalimide 29 against Cryptococcus neoformans; considerably more potent than the clinically used antifungal agent Fluconazole (MIC = 8 μg/mL, Table S3).
Table 3:
MIC values (μg/mL)
| Bacterial strain | 7 | 10 | 13 | 21 | 29 | 34 | COLa | VANb |
|---|---|---|---|---|---|---|---|---|
| A. baumannii ATCC 19606 | >32 | >128 | >128 | >32 | >32 | 32 | 0.06 | NTc |
| P. aeruginosa ATCC 27853 | 32 | >128 | >128 | >32 | >32 | 32 | 0.25 | NT |
| K. pneumoniae ATCC 700603 | >32 | >128 | >128 | >32 | >32 | >32 | 0.03 | NT |
| E. coli ATCC 25922 | 8 | >128 | >128 | >32 | >32 | 8 | 0.06 | NT |
| S. aureus ATCC 43300 | 2 | 0.25 | >128 | 16 | 1 | 1 | NT | 1 |
| S. aureus NARSA NRS17 | 1 | 0.25 | >128 | 16 | 4 | 1 | NT | 4 |
| S. aureus NARSA NRS1 | 2 | 0.5 | >128 | 8 | 4 | 4 | NT | 4 |
| S. aureus NARSA VRS 10 | 1 | 0.5 | 128 | 8 | 2 | 0.5 | NT | >64 |
| S. pneumoniae ATCC 7000677 | 2 | 1 | >128 | 8 | 1 | 1 | NT | 1 |
COL = Colistin sulphate
VAN = Vancomycin
NT = Not tested
The cytotoxicity of most compounds when tested against human embryonic kidney cells (HEK293, Table 4) was modest, with CC50 values >50 μg/mL. While the highly active benzylguanidine 10 did show considerable toxicity the CC50 was still an order of magnitude above its MIC. Pleasingly, benzylgaunidine 7 and fluorescent analogues 29 and 34, each of which exhibited excellent antibacterial activity against Gram-positive bacteria (MIC 1, 2 and 0.5 μg/mL respectively), showed CC50 values >50 μg/mL.
Table 4:
Cytotoxicity (CC50) against HEK293 cells (μg/mL)
| 7 | 10 | 13 | 21 | 29 | 34 | TAMa | |
|---|---|---|---|---|---|---|---|
| CC50 | >50 | 7.8 | >100 | >50 | >50 | >50 | 13 |
TAM = Tamoxifen
2.4. Molecular modelling
Molecular dynamics simulations were performed to identify potential interactions between the compounds and a model of a cell membrane from Gram-negative bacteria (see experimental for details regarding model membrane construction). The Gram-positive cell wall is comprised of a thick peptidoglycan layer whilst Gram-negative bacteria have a comparatively thin peptidoglycan layer within their cellular envelope (up to 90% versus 10%, respectively).[52, 53] Molecular modelling that incorporates peptidoglycans presents a significant challenge as the complete assembled structure of peptidoglycans is not yet fully understood.[54] As such, a Gram-negative bacterial cell membrane was chosen for this study to circumvent complications associated with peptidoglycans. The compounds modelled included the previously reported norbornanes bisguanidine (2) and bisamine (8)[23] as well as naphthalimide (29) and benzylguanidine (7). Two sets of simulations were performed; one set to determine if these compounds self-aggregate, and the second set to determine how they interact with the model bacterial cell wall.
Self-association was readily apparent and all compounds were found to aggregate to form clusters (Figure 2), a logical outcome given the amphiphilic nature of the compounds. The aggregation occurred relatively quickly, on the sub-microsecond timescale, and as such it is likely that, in vivo, a level of aggregation occurs prior to insertion into the lipid bilayer. Dimerisation, and even oligomerisation, to form the active antibacterial agent, is a feature of a number of antibiotics including Vancomycin,[55] Daptomycin[56] and a variety of cationic antimicrobial peptidomimetics.[57]
Figure 2:
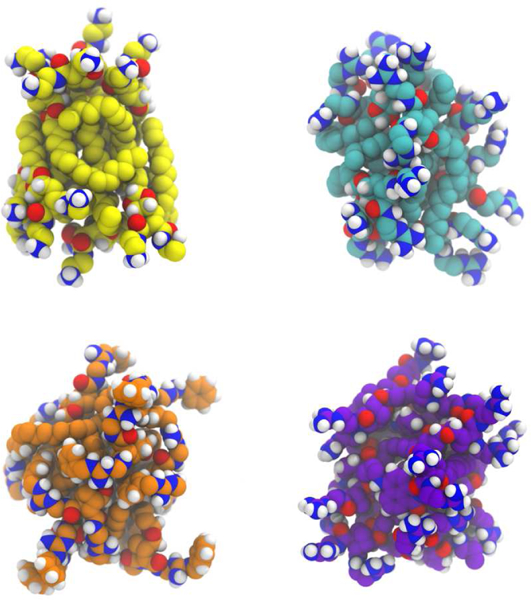
A snapshot of the assembled ten molecule aggregates. Yellow, cyan, orange and purple represent the carbon atoms of the bisamine (8), bisguanidine (2), benzylguanidine (7) and naphthalimide (29) species respectively. Nitrogen is coloured blue, oxygen = red, hydrogen = white). Water molecules and Cl- have been omitted for clarity.
Additional computational studies were performed on bisguanidine (2), chosen as a representative example, to probe how this class of compounds interacts with bacterial cell membranes. An aggregate consisting of 28 molecules of 2 (see experimental section for the construction of this aggregate) was inserted into the aqueous (extracellular) portion of a Gram-negative bacterial inner membrane model (Figure 3, LHS). The 228 aggregate was partially absorbed into the membrane in each of the three replicate simulations and full absorption was expected were the simulations to be continued. The incorporation of the 228 aggregate into the membrane occurred in a series of steps. First, the aggregate contacted the membrane. The aggregate stayed attached at the same position of the membrane due to strong electrostatic interactions between the positively charged 228 aggregate and the negatively charged lipid molecules (DMPG). The aggregate maintained its integrity for a few hundred nanoseconds until individual molecules of 2 were drawn into the membrane (Figure 3, Centre). The aggregate then slowly broke down with additional individual molecules diffusing into the membrane (Figure 3, RHS). While no distinct pore formation was observed, some water molecules did penetrate into the membrane (see video in supplementary material). These water molecules appeared to be stabilised inside the membrane by interactions with the charged guanidine moieties of 2. In one replicate a significant amount of membrane curvature was evident when the intact aggregate contacted the membrane (see Figure S50). In all three replicates the interaction between the 228 aggregate and the model membrane followed an identical pattern that indicates this class of compound aggregates quickly in vivo prior to inserting into to the membrane, before slowly breaking apart. This series of events would likely decrease membrane stability, increase membrane permeability and ultimately lead to cell lysis—a known mode of action for CAMPs and mimics such as Colistin.[37]
Figure 3:

Simulation snapshots highlighting the insertion/absorption of bisguanidine aggregate (228) into a model membrane. LHS: Simulation begins with the aggregate deliberately positioned in the “extracellular environment” above the model membrane. Centre: membrane contact established and lipid absorption underway. RHS: simulation end with aggregate embedded in the membrane and some dispersion evident. Lipid molecules are coloured grey (PPoPE) and red (DMPG), 2 is coloured by element (carbon = cyan, nitrogen = blue, oxygen = red and hydrogen = white). Water molecules, Na+ and Cl- have been omitted for clarity. A video of the membrane insertion process is available in the electronic supplementary information.
2.5. Fluorescence microscopy
Confocal microscopy was performed to assess localisation of fluorescent compounds 29 and 34 in Gram-positive and Gram-negative bacteria. Two control fluorophores were also assessed, naphthalimide 35 and NBD 36 (Figure 4, see ESI for the full synthesis and characterisation of these controls).
Figure 4:
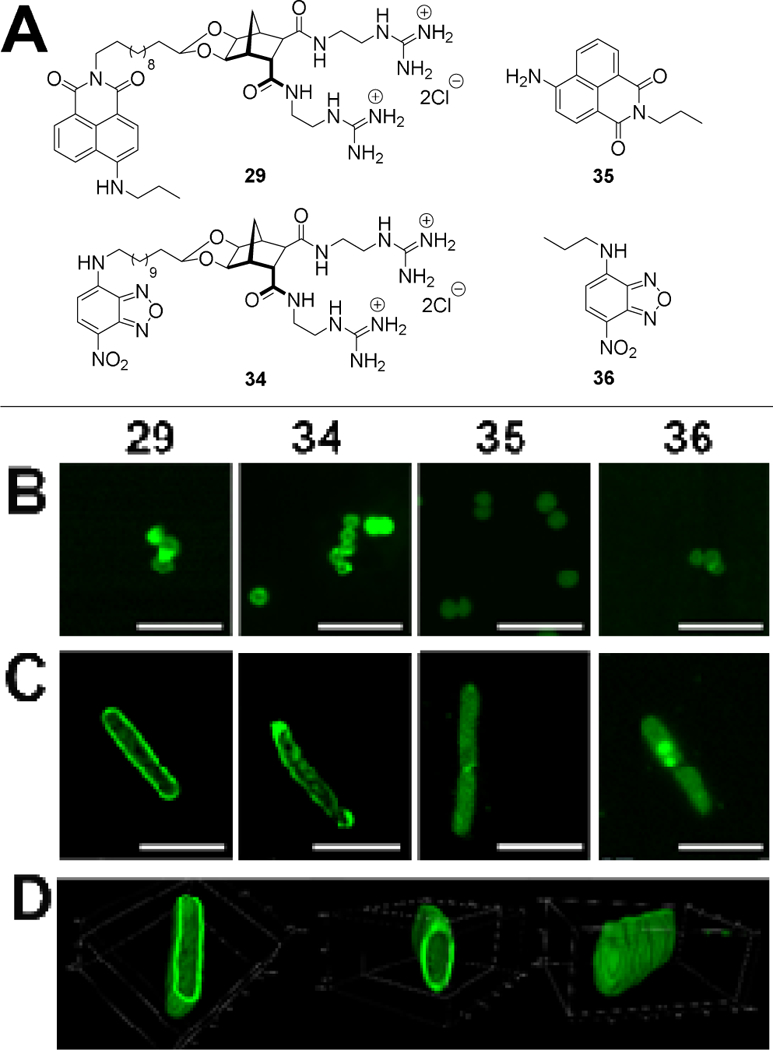
(A) Structure of compounds used in microscopy studies: Norbornanes 29 and 34 plus controls 35 and 36. (B–C) Localisation of compounds in bacteria. Confocal micrographs showing localisation of compounds in (B) S. aureus MRSA and (C) E. coli. Scale bar = 5 μm. (D) 3D rendering showing cell wall localisation of compound 29 in E. coli. Box dimensions x:y:z 5.4 μm:4.0 μm:2.3 μm.
Bacteria were incubated for 20 minutes with 10 μg/mL of the fluorescent compound. In Gram-positive S. aureus the staining pattern for both compounds 29 and 34 was similar with fluorescence observed throughout the cell but the intensity was greatest at the cell periphery (Figure 4B). Naphthalimide 35 localised throughout the cell and NBD 36 localised to the cell periphery; similar to norbornane compounds 29 and 34, however the overall fluorescence was considerably less intense for the control compounds 35 and 36. This decreased intensity is likely a result of weaker interactions with the bacterial membrane as both control compounds 35 and 36 lack the guanidine groups that act as anion recognition moieties targeting the phosphate groups present in the bacterial outer membrane.
When Gram-negative E. coli was incubated with 29 and 34, a similar distribution within the cells was observed (Figure 4C). Once again, greater localisation occurred at the cell periphery, with distinct regions of higher concentration particularly evident when three dimensional images were taken (Figure 4D, also see ESI for associated video). The simple organic fluorophores used as controls (35 and 36) occasionally localised to similar regions, but with less consistency and intensity, than compounds 29 and 34, again highlighting the role of the positively charged groups present on the norbornane-based compounds for cell wall interactions.
The use of molecular modelling and fluorescence microscopy indicate that this class of antibacterial agent likely aggregates rapidly prior to interacting with the bacterial cell membrane. Both the modelling outcomes and the fluorescence microscopy studies using naphthalimide 29 and NBD 34 clearly show the norbornane-based compounds localised on the bacterial membrane highlighting the important role of the cationic groups. Fluorescence microscopy experiments showed that naphthalimide 29 interacts with the membrane for both S. aureus and E. coli, however cell penetration was only observed in S. aureus; a potential cause for the increased antibacterial activity demonstrated by these compounds against Gram-positive bacterial species. Indeed, many AMPs such as the bacteriocin family, which includes colicins and lantibiotics,[58] demonstrate strain-specific antibacterial activity, despite relying on a mode of action that primarily involves membrane interactions as the first step.[59, 60] With this in mind and comparing the outcomes to those of CAMPs in the literature,[61, 62] it is suggested that these compounds function as mimetics of cationic antimicrobial peptides.
3. Conclusions
In summary, a series of norbornane-based CAMP mimics were synthesised—two dibenzylguanidine substituted (7 and 10), one guanidiniocarbonylpyrrole-functionalised (13), one tricationic (21) and two fluorescently tagged, using the 4-amino naphthalimide and NBD fluorophores (29 and 34, respectively). Incorporation of the dibenzylguanidine groups enhanced the antibacterial activity of the compounds, albeit in the case of 10 some cytotoxicity was observed. The importance of retaining a structurally amphiphilic topology was reinforced as non-amphiphilic tricationic analogue 21 was considerably less active than its amphiphilic diatonic counterparts. Antibacterial activity of the fluorescent analogues was comparable to the previously described norbornane acetal 2 with each compound active against several strains of Gram-positive bacteria (MIC values as low as 0.5 μg/mL). Additionally, NBD 34 exhibited activity against several Gram-negative bacterial strains. This structurally amphiphilic class of low-molecular weight CAMP mimics are readily synthesised and possess promising antimicrobial activity.
4. Experimental
4.1. Chemistry
The following compounds and their respective precursors were prepared using literature methods and full reaction details can be found in the supplementary information; 3,[34] 5,[23] 8,[23] 14,[63] 17,[24] 22,[35] 35[64, 65] and 36.[66] Compound 11 was kindly donated by the Schmuck research group and its synthesis has been previously reported.[39]
4.1.1. N,N’-Bis(tert-butoxycarbonyl)-N-benzyl-S-methylisothiourea (4).[67]
To a stirring solution of methylisothiourea 3 (1.04 g, 3.58 mmol) in anhydrous THF (12.8 mL) at ambient temperature under an inert atmosphere, was added Ph3P (1.50 g, 5.73 mmol) followed by BnOH (490 μL, 4.65 mmol). The homogenous solution was cooled to 0 °C, before Diisopropyl azodicarboxylate (DIAD, 1.1 mL, 5.37 mmol) was added slowly. The yellow solution was warmed to ambient temperature and then heated to 66 °C for 16 h. The reaction mixture was concentrated under reduced pressure and the crude material was diluted with H2O (10 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic phases were washed with brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo to give a yellow oil which was stirred in pet. spirits (20 mL) at ambient temperature for 1 h. The resulting white precipitate was collected using vacuum filtration and discarded. The remaining filtrate was purified using column chromatography (10% EtOAc in pet. spirits) to give a light green oil (1.29 g, 95%). Rf = 0.38 (10% EtOAc in pet. spirits). 1H NMR (270 MHz, CDCl3) δ 7.36–7.24 (5H, m, ArH), 4.78 (2H, s, CH2), 2.28 (3H, s, CH3), 1.52 (9H, s, t-Bu), 1.39 (9H, s, t-Bu). 13C NMR (67.5 MHz, CDC13) δ 163.3, 158.1, 152.1, 137.5, 128.7, 128.5, 127.8, 127.6, 127.4 82.8, 81.9, 52.6, 28.2, 28.1, 15.7. HRMS (ESI, m/z) for C19H28N2O4S [M + Na]+ calc. 403.1662; found 403.1664.
4.1.2. tert-Butyl-[12-(methoxy[methyl]amino)-12-oxododecyl]carbamate (15).
A mixture of carboxylic acid 40 (5.46 g, 17.30 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI, 4.02 g, 20.94 mmol), 1-hydroxybenzotriazole (HOBt, 241 mg, 1.78 mmol), N,O-dimethylhydroxylamine hydrochloride (2.05 g, 20.97 mmol), CHCl3 (80 mL) and N,N-diisopropylethylamine (DIPEA, 15.4 mL, 86.5 mmol) was stirred at 65 °C for 24 h. The reaction was cooled to ambient temperature and the mixture was washed with 0.1 M HCl (40 mL), sat. NaHCO3 (40 mL), brine (40 mL), then dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using column chromatography (20–50% EtOAc in pet. spirits) to afford the title compound (5.40 g, 87%) as a colourless viscous oil. Rf = 0.56 (50% EtOAc in pet. spirits). 1H NMR (400 MHz, CDCl3) δ 4.51 (1H, br s, NH), 3.67 (3H, s, OCH3), 3.17 (3H, s, NCH3), 3.09–3.08 (2H, m, NHCH2), 2.40 (2H, t, J = 7.5 Hz, CH2CO), 1.65–1.57 (2H, m, CH2), 1.46–1.39 (9H, m, t-Bu), 1.29–1.26 (16H, m, 8 × CH2). 13C NMR (100 MHz, CDCl3) δ 174.9, 156.1, 79.2, 61.3, 40.8, 32.3, 32.0, 30.2, 29.64, 29.63, 29.58, 29.57, 29.5, 29.4, 28.6, 26.9, 24.8. HRMS (ESI, m/z) for C19H38N2O4 [M + H]+ calc. 359.2904; found 359.2904.
4.1.3. N-(tert-Butoxycarbonyl)-12-aminododecanal (16).
Method A[43]
To the stirring solution of oxalyl chloride (140 μL, 1.60 mmol) in anhydrous CH2Cl2 (8 mL) at –78 °C, was added DMSO (230 μL, 3.19 mmol), alcohol 14 (400 mg, 1.33 mmol) in anhydrous CH2Cl2 (1.5 mL) and Et3N (1 mL, 7.18 mmol) successively. Stirring was maintained under an inert atmosphere at –78 °C for 3 h before being warmed to ambient temperature and stirred for a further 16 h. The reaction was diluted with H2O (20 mL) and extracted with Et2O (2 × 20 mL). The combined organic phase was washed with brine (20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using column chromatography (10% EtOAc in pet. spirits) to give the title compound (140 mg, 35%) as a white solid.
Method B[47]
Alcohol 14 (1.04 g, 3.43 mmol) was added to a stirring mixture of neutral alumina (5.09 g, 50.0 mmol) and pyridinium chlorochromate (PCC, 2.18 g, 10.1 mmol) in CH2Cl2 (33 mL) and stirring was maintained at ambient temperature for 3 h. The solid material was removed by filtration (rinsing with CH2Cl2) and the filtrate was concentrated in vacuo. The crude material was purified using column chromatography (20% EtOAc in pet. spirits) to give the product (808 mg, 79%) as a white solid.
Method C[48]
Alcohol 14 (209 mg, 0.693 mmol) was stirred in CH2Cl2 (1.4 mL) at ambient temperature before (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO, 13 mg, 0.083 mmol) and (diacetoxyiodo)benzene (246 mg, 0.762 mmol) were added. The pale orange solution was stirred at ambient temperature for a further for 24 h before more TEMPO (13 mg, 0.083 mmol) was added. Stirring was maintained for another 24 h and the reaction was then quenched with sat. Na2SO3 (8 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic phase was washed with sat. NaHCO3 (5 mL), brine (5 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using column chromatography (20% EtOAc in pet. spirits) to give the title compound (112 mg, 58%) as a white solid.
Method D[68]
A stirring solution of Weinreb amide 15 (994 mg, 2.77 mmol) in anhydrous THF (30 mL) was added LiAlH4 (4.2 mL, 4.16 mmol, 1 M solution in Et2O), at –78 °C under an inert atmosphere. Stirring was maintained for 2 h before the reaction was quenched with 1 M HCl (20 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic phase was washed with brine (30 mL), dried (MgSO4), filtered and concentrated in vacuo to give a white solid (790 mg, 95%) that did not require further purification. Rf = 0.45 (20% EtOAc in pet. spirits). m.p. 51–53 °C (lit. 51 °C).[43] 1H NMR (500 MHz, CDCl3) δ 9.76 (1H, t, J = 1.8 Hz, CHO), 4.48 (1H, br s, NH), 3.09 (2H, t, J = 7.0 Hz, CH2NHBoc), 2.41 (2H, dt, J = 7.4, 1.8 Hz, CH2CHO), 1.65–1.59 (2H, m, CH2), 1.47–1.44 (11H, m, CH2, t-Bu), 1.29–1.26 (14H, m, 7 × CH2). 13C NMR (125 MHz, CDCl3) δ 203.1, 156.2, 79.2, 44.1, 40.8, 30.2, 29.63, 29.60, 29.51, 29.46, 29.4, 29.3, 28.6, 26.9, 22.2. HRMS (ESI, m/z) for C17H33NO3 [M + Na]+ calc. 322.2353; found 322.2353.
4.1.4. 8-endo-9-exo-Di[2ʹ-(2ʺ,3ʺ-bis-tert-butoxycarbonyl-3ʺ-benzylguanidino)ethylcarbamoyl]-4-heptyl-3,5-dioxatricyclo [5.2.1.02,6] decane (6).
A mixture of diamine 5 (80 mg, 0.165 mmol) and Et3N (60 μL, 0.41 mmol) in CH2Cl2 (300 μL) was stirred for 30 min at ambient temperature before a solution of methylisothiourea 510 (130 mg, 0.341 mmol) in CH2Cl2 (900 μL) was added in one portion and the reaction was heated to 40 °C for 2 d. The reaction mixture was diluted with CH2Cl2 (10 mL), transferred to a separatory funnel and washed with H2O (2 × 5 mL), brine (5 mL), dried (MgSO4) and filtered. The solvent was removed in vacuo and the crude material was purified using flash column chromatography (20–70% EtOAc in pet. spirits) to give the title compound (53 mg, 30%) as a colourless viscous oil. Rf = 0.46 (70% EtOAc in pet spirits). 1H NMR (500 MHz, CDCl3) δ 7.33–7.27 (10H, m, ArH), 4.79 (4H, br s, ArCH2), 4.52 (1H, br s, H4), 4.10 (1H, br s, H6), 3.92 (1H, d, J = 5.5 Hz, H2), 3.33–3.21 (8H, m, 4 × NHCH2), 2.87 (1H, app. t, J = 5.1 Hz, H8), 2.61 (1H, br s, H9), 2.57 (1H, br s, H7), 2.32 (1H, d, J = 5.9 Hz, H1), 1.76 (1H, d, J = 9.9 Hz, H10a), 1.61–1.25 (49H, m, 6 × CH2, 4 × t-Bu, H10s), 0.87 (3H, t, J = 6.8 Hz, CH3). 13C NMR (125 MHz, CDCl3) δ 174.3, 172.6, 153.5 (2 × C), 138.0, 137.8, 128.7 (6 × C), 128.3, 128.2, 128.0, 127.8, 104.0, 83.3, 83.1, 81.5, 79.7 (2 × C), 78.7, 51.4, 51.3, 47.8, 44.6, 44.3, 43.2, 43.0, 39.6, 38.8, 33.0, 32.5, 31.9, 29.8, 29.6, 29.3, 28.33, 28.32, 28.21, 28.17, 24.3, 22.7, 14.2. HRMS (ESI, m/z) for C57H84N8O12 [M + H]+ calc. 1075.6438; found 1075.6429.
4.1.5. 4-Heptyl-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-di[carboxamidoethyl-(3″- benzyl)guanidine] hydrogen chloride (7).
To a stirring solution of Boc-protected guanidine 6 (40 mg, 0.04 mmol) and MeOH (370 μL) was added dropwise AcCl (70 μL, 0.987 mmol), and the reaction was stirred for 24 h at ambient temperature. The reaction was concentrated in vacuo and co-evaporated with MeOH (2 × 0.5 mL), to afford the title compound (25 mg, 93%) as a white solid. 1H NMR (270 MHz, CD3OD) δ 7.40–7.30 (10H, m, ArH), 4.61 (1H, t, J = 4.6 Hz, H4), 4.42–4.41 (4H, m, 2 × ArCH2), 4.03 (1H, d, J = 5.4 Hz, H2), 3.98 (1H, d, J = 5.4 Hz, H6), 3.41–3.34 (8H, m, 4 × NHCH2), 3.21 (1H, app. t, J = 4.8 Hz, H8), 2.62–2.61 (2H, m, H1, H7), 2.45 (1H, br s, H9), 1.72 (1H, d, J = 10.2 Hz, H10a), 1.60–1.56 (2H, m, CH2), 1.45 (1H, d, J = 10.2 Hz, H10s), 1.39–1.29 (10H, m, 5 × CH2), 0.89 (3H, t, J = 6.8 Hz, CH3). 13C NMR (67.5 MHz, CD3OD) δ 176.7, 174.7, 157.7 (2 × C), 129.9 (4 × C), 129.0 (4 × C), 128.5 (2 × C), 128.4 (2 × C), 105.1, 82.8, 79.9, 47.7, 47.0, 46.2, 46.1, 45.1, 44.9, 42.4 (2 × C), 39.7, 39.6, 33.9, 32.9, 32.7, 30.6, 30.3, 25.2, 23.7, 14.4. HRMS (ESI, m/z) for C37H54N8O4 [M + 2H]2+ calc. 338.2207; found 338.2208.
4.1.6. 8-endo-9-exo-Di[2′-(2″,3″-bis-tert-butoxycarbonyl-3″-benzylguanidino)ethylcarbamoyl]-4-pentadecyl-3,5-dioxatricyclo [5.2.1.02,6] decane (9).
A mixture of diamine 8 (284 mg, 0.476 mmol) and Et3N (30 μL, 0.21 mmol) in CH2Cl2 (2.5 mL) was stirred for 30 min at ambient temperature in a pressure vessel, before a solution of methylisothiourea 4 (431 mg, 1.13 mmol) in CH2Cl2 (2.5 mL) was added in one portion and the reaction was heated to 80 °C for 2 d. The reaction mixture was diluted with CH2Cl2 (10 mL), transferred to a separatory funnel and washed with H2O (2 × 10 mL), brine (10 mL), dried (MgSO4) and filtered. The solvent was removed in vacuo and the crude material was purified using flash column chromatography (50–70% EtOAc in pet. spirits) to give the title compound (83 mg, 14%) as a colourless viscous oil. Rf = 0.65 (70% EtOAc in pet spirits). 1H NMR (270 MHz, CDCl3) δ 7.35–7.26 (10H, m, ArH), 4.78 (4H, br s, ArCH2), 4.52 (1H, br s, H4), 4.10 (1H, d, J = 9.8 Hz, H6), 3.92 (1H, d, J = 5.5 Hz, H2), 3.31–3.14 (8H, m, 4 × NHCH2), 2.87 (1H, app. t, J = 5.1 Hz, H8), 2.61 (1H, br s, H9), 2.57 (1H, br s, H7), 2.32 (1H, d, J = 5.8 Hz, H1), 1.75 (1H, d, J = 9.8 Hz, H10a), 1.61–1.25 (61H, m, 12 × CH2, 4 × t-Bu, H10s), 0.87 (3H, t, J = 7.0 Hz, CH3). 13C NMR (67.5 MHz, CDCl3) δ 174.3, 172.6, 153.4 (2 × C), 138.0, 137.9, 128.7 (6 × C), 128.3 (2 × C), 127.8 (2 × C), 104.0, 83.3, 83.1, 81.5, 79.7, 78.8, 77.3, 51.4, 51.3, 47.8, 44.7, 44.3, 43.2, 43.0, 39.7, 38.8, 33.0, 32.5, 32.0, 29.80, 29.77, 29.71, 29.67, 29.5, 28.34 (3 × C), 28.32 (3 × C), 28.21, 28.17, 24.3, 22.8, 14.3.
4.1.7. 4-Pentadecyl-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-di[carboxamidoethyl-(3″-benzyl)guanidine] hydrogen chloride (10).
To a stirring solution of Boc-protected guanidine 9 (101 mg, 0.085 mmol) and MeOH (5 mL) was added dropwise AcCl (250 μL, 3.52 mmol), and the reaction was stirred for 24 h at ambient temperature. The reaction was concentrated in vacuo and co-evaporated with MeOH (2 × 0.5 mL), to afford the title compound (54 mg, 74%) as a white solid. m.p. 174–180 °C (slow decomposition). 1H NMR (270 MHz, CD3OD) δ 7.38–7.31 (10H, m, ArH), 4.61 (1H, t, J = 4.6 Hz, H4), 4.47–4.41 (4H, m, 2 × ArCH2), 4.04 (1H, d, J = 5.6 Hz, H2), 3.98 (1H, d, J = 5.6 Hz, H6), 3.40–3.30 (8H, m, 4 × NHCH2), 3.21 (1H, app. t, J = 4.8 Hz, H8), 2.65–2.61 (2H, m, H1, H7), 2.46 (1H, br s, H9), 1.72 (1H, d, J = 10.2 Hz, H10a), 1.60–1.55 (2H, m, CH2), 1.45 (1H, d, J = 10.2 Hz, H10s), 1.36–1.22 (24H, m, 12 × CH2), 0.90 (3H, t, J = 6.8 Hz, CH3). 13C NMR (67.5 MHz, CD3OD) δ 176.7, 174.7, 157.7 (2 × C), 137.6, 129.9 (3 × C), 129.0 (4 × C), 128.5 (2 × C), 128.4 (2 × C), 105.1, 82.8, 80.0, 47.7, 46.9, 46.2, 46.1, 45.2, 44.9, 42.4 (2 × C), 39.7, 39.6, 33.9, 33.1, 32.7, 30.77, 30.75, 30.7 (2 × C), 30.64 (2 × C), 30.62, 30.5, 25.2, 23.7, 14.4. HRMS (ESI, m/z) for C45H70N8O4 [M + 2H]2+ calc. 394.2833; found 394.2842.
4.1.8. 8-endo-9-exo-Di[2′-(5″-acetamido-1H-pyrrole-2″-N-tert-butoxycarbonyl-guanidiniocarbonyl)ethylcarbamoyl]-4-heptyl-3,5-dioxatricyclo[5.2.1.02,6]decane (12).
A solution of GCP 11 (104 mg, 0.116 mmol), (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP, 163 mg, 0.313 mmol), 4-(dimethylamino)pyridine (DMAP, 8 mg, 0.07 mmol) and DMF (0.5 mL) was stirred at ambient temperature. To the homogenous yellow solution was added Et3N (90 μL, 0.646 mmol) and the solution gradually turned to red and then eventually bright orange. To the stirred solution was added diamine 5 (56 mg, 0.116 mmol) and the biphasic mixture was stirred at 80 °C for 16 h. The reaction was quenched with H2O (2 mL) and the white precipitate was collected using vacuum filtration. The crude material was stirred in 10% pet. spirits in CH2Cl2 for 1 h, cooled and then collected using vacuum filtration to afford the desired product (47 mg, 42%) as an off-white solid. Rf = 0.41 (89% CH2Cl2, 10% MeOH, 1% NH4OH). m.p. 217–252 °C (slow decomposition). 1H NMR (500 MHz, DMSO-d6) δ 11.30–10.88 (4H, br s, 4 × NH), 9.34 (2H, br s, 2 × NH), 8.58 (2H, br s, 2 × NH), 8.40 (2H, br s, 2 × NH), 8.12–8.10 (2H, m, 2 × NH), 6.80–6.75 (4H, m, 4 × CH), 4.44 (1H, t, J = 4.3 Hz, H4), 3.84 (1H, d, J = 5.6 Hz, H6), 3.79 (1H, d, J = 5.6 Hz, H2), 3.36–3.27 (8H, m, 4 × CH2), 3.14–3.09 (1H, m, H8), 2.50 (2H, m, H1, H7), 2.29 (1H, br s, H9), 1.45 (19H, br s, t-Bu, H10a), 1.21–1.08 (13H, m, 6 × CH2, H10s), 0.85 (3H, t, J = 6.6 Hz, CH3). 13C NMR (125 MHz, DMSO-d6) δ 173.1 (2 × C), 171.2 (2 × C), 159.8 (4 × C), 158.5 (2 × C), 129.7 (2 × C), 113.7 (2 × C), 111.7 (2 × C), 111.6 (2 × C), 102.9, 81.0, 78.2, 46.3, 44.3, 43.2, 42.7, 38.7 (2 × C), 38.4 (2 × C), 32.4, 31.2, 31.0, 29.0, 28.7, 27.8, 23.8, 22.1, 14.0.
4.1.9. 4-Heptyl-3,5-dioxatricydo[5.2.1.02,6]decane-8-endo-9-exo-di[carboxamidoethyl-(5″-acetamido-1-H-pyrrole-2″)-guamdiniocarbonyl] hydrogen chloride (13).
To a stirring solution of Boc-protected guanidine 12 (93 mg, 0.096 mmol) and MeOH (2 mL) was added dropwise AcCl (280 μL, 3.94 mmol), and the reaction was stirred for 24 h at ambient temperature. The reaction was concentrated in vacuo and co-evaporated with MeOH (2 × 0.5 mL), to afford the title compound (79 mg, 99%) as a brown solid. m.p. 206–209 °C. :H NMR (500 MHz, DMSO-d6) δ 12.34–12.31 (2H, m, 2 × NH), 11.95–11.94 (2H, m, 2 × NH), 8.63–8.43 (8H, m, 8 × NH), 8.17–8.14 (2H, m, 2 × NH), 7.50 (2H, br s, 2 × CH), 6.84 (2H, br s, 2 × CH), 4.49 (1H, t, J = 4.7 Hz, H4), 3.87 (1H, d, J = 4.9 Hz, H6), 3.82 (1H, d, J = 4.9 Hz, H2), 3.75–3.63 (2H, br s, 2 × NH), 3.28–3.13 (8H, m, 4 × CH2), 3.09 (1H, app. t, J = 4.8 Hz, H8), 2.50 (2H, m, H1, H7), 2.30 (1H, br s, H9), 1.50–1.45 (3H, m, CH2, H10a), 1.28–1.10 (11H, m, 5 × CH2, H10s), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (125 MHz, DMSO-d6) δ 173.1, 171.2, 159.6 (2 × C), 159.3 (2 × C), 155.4 (2 × C), 132.90, 132.87, 125.4 (2 × C), 115.9 (2 × C), 112.4 (2 × C), 102.9, 81.0, 78.2, 46.3, 44.4, 43.2, 42.7, 38.72, 38.71, 38.5, 38.3, 32.4, 31.2, 31.0, 28.9, 28.6, 23.8, 22.1, 14.0. HRMS (ESI, m/z) for C35H52N12O8 [M + 2H]2+ calc. 384.2010; found 384.2019.
4.1.10. Dimethyl 4-[11′-(tert-butoxycarbonylammo)undecyl]-3,5-dioxatricydo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxylate (18).
A stirring suspension of diol 17 (323 mg, 1.32 mmol), TsOH H2O (13 mg, 0.07 mmol), MgSO4 (166 mg, 1.38 mmol) and PhMe (2.2 mL) was treated with aldehyde 16 (587 mg, 1.96 mmol) and heated to 110 °C for 3 h. Solid MgSO4 was removed by filtration and the filtrate was diluted with EtOAc (20 mL), washed with H2O (2 × 10 mL), brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using column chromatography (20% EtOAc in pet. spirits) to afford the title compound (542 mg, 78%) as a colourless oil. Rf = 0.44 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 4.65 (1H, t, J = 5.2 Hz, H4), 4.50 (1H, br s, NH), 4.02 (1H, d, J = 5.6 Hz, H2), 3.89 (1H, d, J = 5.6 Hz, H6), 3.70–3.69 (6H, m, 2 × Me), 3.22 (1H, app. t, J = 4.8 Hz, H8), 3.08 (2H, app. t, J = 7.0 Hz, CH2NHBoc), 2.71 (1H, d, J = 4.8 Hz, H9), 2.64–2.63 (2H, m, H1, H7), 1.77 (1H, dd, J = 10.9, 1.4 Hz, H10a), 1.64–1.60 (2H, m, CH2), 1.43 (9H, s, t-Bu), 1.39–1.24 (19H, m, 9 × CH2, H10s). 13C NMR (125 MHz, CDCl3) δ 174.1, 172.9, 156.1, 104.3, 81.4, 79.1, 78.9, 52.5, 52.3, 45.4, 45.1, 43.8, 43.4, 40.8, 32.9, 31.7, 30.2, 29.82, 29.78, 29.64, 29.59, 29.5, 29.4, 28.6, 26.9, 24.3. HRMS (ESI, m/z) for C28H47NO8 [M + Na]+ calc. 548.3194; found 548.3171.
4.1.11. 4-[11′-(tert-butoxycarbonylamino)undecyl]-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxylic acid (19).
To the stirred solution of the ester 18 (27 mg, 0.05 mmol) in THF (210 μL), 2M NaOH (110 μL) was added and the reaction was stirred for 16 h at ambient temperature. The reaction was concentrated under reduced pressure and all organic-soluble impurities were extracted with CH2Cl2 (2 × 5 mL). The aqueous solution was acidified with sat. KH2PO4 (pH = 5), extracted with EtOAc (3 × 8 mL), dried (MgSO4) and filtered to give a colourless oil (22 mg, 88%). 1H NMR (500 MHz, CDCl3) δ 4.68 (1H, t, J = 4.9 Hz, H4), 4.59 (1H, br s, NH), 4.04 (2H, s, H2, H6), 3.27 (1H, app. t, J = 5.2 Hz, H8), 3.07 (2H, br s, NHCH2), 2.78 (1H, d, J = 4.3 Hz, H1), 2.73 (1H, br s, H7), 2.65 (1H, d, J = 5.2 Hz, H9), 1.82 (1H, d, J = 10.2 Hz, H10a), 1.66–1.62 (2H, m, CH2), 1.51–1.25 (28H, m, 9 × CH2, t-Bu, H10s). 13C NMR (125 MHz, CDCl3) δ 178.4, 177.2, 104.4, 81.4, 79.5, 78.8, 45.3, 45.2, 43.7, 43.4, 40.8, 32.7, 31.9, 30.1, 29.8, 29.63, 29.56, 29.5 (2 × C), 29.4, 28.6, 26.9, 24.2. HRMS (ESI, m/z) for C26H43NO8 [M + H]+ calc. 498.3061; found 498.3054.
4.1.12. 8-endo-9-exo-Di[2′-(2″,3″-bis-tert-butoxycarbonylguanidino)ethylcarbamoyl]-4-[11′-(tert- butoxycarbonylamino)undecyl]-3,5-dioxatricyclo[5.2.1.02,6] decane (20).
A MW vial was charged with diacid 19 (21 mg, 0.004 mmol), EDCI (24 mg, 0.126 mmol), HOBt (1 mg, 0.01 mmol) and anhydrous CHCl3 (210 μL) and was stirred at ambient temperature for 30 min. Amine 22 (38 mg, 0.13 mmol) was then added and the reaction was irradiated for 30 min at 50 °C. The resulting homogenous clear liquid was diluted with CHCl3 (10 mL), washed with H2O (2 × 5 mL), brine (5 mL), dried (MgSO4), filtered, and concentrated in vacuo to afford a colourless oil that was purified using flash column chromatography (20% EtOAc in pet. spirits-EtOAc) to give the title compound (14 mg, 32%) as a colourless oil. Rf = 0.75 (EtOAc). 1H NMR (500 MHz, CDCl3) δ 11.47–11.44 (2H, m, 2 × NH), 8.67 (1H, br s, NH), 8.56 (1H, br s, NH), 8.04 (1H, t, J = 4.1 Hz, NH), 6.92 (1H, br s, NH), 4.60 (1H, t, J = 4.8 Hz, H4), 4.50 (1H, br s, NH), 4.02 (1H, d, J = 5.7 Hz, H6), 3.95 (1H, d, J = 5.7 Hz, H2), 3.60–3.56 (4H, m, 2 × NHCH2), 3.45–3.35 (4H, m, 2 × NHCH2), 3.11–3.07 (2H, m, BocNHCH2), 2.94 (1H, app. t, J = 5.3 Hz, H8), 2.70 (1H, d, J = 4.1 Hz, H1), 2.57 (1H, br s, H7), 2.45 (1H, d, J = 5.3 Hz, H9), 1.77 (1H, d, J = 9.7 Hz, H10a), 1.62–1.57 (2H, m, CH2), 1.50–1.43 (50H, m, 2 × CH2, 5 × t-Bu, H10s), 1.36–1.24 (14H, m, 7 × CH2). 13C NMR (125 MHz, CDCl3) δ 174.2, 172.1, 163.1, 162.7, 157.8, 157.0, 156.1, 153.2, 153.1, 104.1, 83.8, 83.6, 81.7, 80.1, 80.0, 79.1, 79.0, 47.8, 44.4, 44.2, 43.0, 42.1, 40.8, 40.4, 40.1, 40.0, 33.0, 32.5, 32.1, 30.2, 29.8, 29.7, 29.6, 29.4, 28.6, 28.38, 28.37, 28.20, 28.19, 27.0, 24.3, 22.8. HRMS (ESI, m/z) for C52H91N9O14 [M + H]+ calc. 1066.6758; found 1066.6775.
4.1.13. 4-[11′-(Amino)undecyl]-3,5-dioxatricyclo[5.2.1.02’6]decane-8-endo-9-exo-dicarboxamidoethylguanidine hydrogen chloride (21).
To a stirring solution of Boc-protected guanidine 20 (14 mg, 0.01 mmol) and MeOH (1 mL) was added dropwise AcCl (30 μL, 0.422 mmol), and the reaction was stirred for 48 h at ambient temperature. The reaction was concentrated in vacuo and co-evaporated with MeOH (2 × 0.5 mL), to afford the title compound (9 mg, 99%) as a colourless oil. 1H NMR (500 MHz, CD3OD) 5 4.66 (1H, t, J = 4.6 Hz, H4), 4.05 (1H, d, J = 5.5 Hz, H6), 4.00 (1H, d, J = 5.5 Hz, H2), 3.37–3.26 (4H, m, 2 × NHCH2), 3.21 (1H, app. t, J = 5.0 Hz, H8), 2.96–2.85 (6H, m, 3 × NHCH2), 2.62–2.61 (2H, m, H1, H7), 2.45 (1H, br s,, H9), 1.73 (1H, d, J = 10.3 Hz, H10a), 1.64–1.58 (4H, m, 2 × CH2), 1.48 (1H, d, J = 10.3 Hz, H10s), 1.40–1.29 (16H, m, 8 × CH2). 13C NMR (125 MHz, CD3OD) δ 176.5, 174.6, 158.9 (2 × C), 105.1, 82.8, 80.0, 47.7, 47.0, 45.1, 44.9, 41.93, 41.88, 40.0, 39.7, 39.6, 33.9, 32.7, 30.8, 30.70, 30.67, 30.6, 30.5, 30.3, 29.2, 27.5, 25.2. HRMS (ESI, m/z) for C27H51N9O4 [M + 3H]3+ calc. 189.4761; found 189.4766.
4.1.14. Dimethyl 4-(11′-aminoundecanyl)-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxylate hydrogen chloride (23).
To a stirring solution of Boc-protected amine 18 (511 mg, 0.97 mmol) and MeOH (10 mL) was added dropwise AcCl (700 μL, 9.85 mmol), and the reaction was stirred for 24 h at ambient temperature. The reaction mixture was concentrated in vacuo, co-evaporated with MeOH (2 × 0.5 mL) and the title compound was isolated (444 mg, 99%) as an off-white sticky residue. 1H NMR (500 MHz, CD3OD) δ 4.66 (1H, t, J = 4.8 Hz, H4), 4.00 (1H, d, J = 5.6 Hz, H2), 3.90 (1H, d, J = 5.6 Hz, H6), 3.71 (3H, s, Me), 3.70 (3H, s, Me), 3.21 (1H, app. t, J = 5.0 Hz, H8), 2.91 (2H, app. t, J = 7.6 Hz, CH2NH3), 2.64–2.62 (2H, m, H1, H7), 2.56 (1H, br s, H9), 1.74 (1H, dd, J = 10.7, 1.5 Hz, H10a), 1.67–1.58 (4H, m, 2 × CH2), 1.41–1.29 (17H, m, 8 × CH2, H10s). 13C NMR (125 MHz, CD3OD) δ 175.4, 174.0, 105.2, 82.5, 80.0, 52.8, 52.6, 46.6, 46.2, 44.9, 44.5, 40.8, 33.8, 32.4, 30.62, 30.57 (3 × C), 30.5, 30.2, 28.6, 27.4, 25.2. HRMS (ESI, m/z) for C23H39NO6 [M + H]+ calc. 426.2850; found 426.2847.
4.1.15. Dimethyl 4-[11′-(6″-bromo-1″,3″-dioxo-1H-benzo[de]isoquinoline-2(3H))undecyl]-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxylate (25).
A 10 mL microwave vial was charged with 4-bromo-1,8-naphthalic anhydride (264 mg, 0.953 mmol), norbornane hydrochloride 23 (444 mg, 0.961 mmol), Et3N (140 μL, 1.00 mmol) and EtOH (3 mL) and heated to 100 °C using microwave irradiation for 45 min. The orange slurry was diluted with H2O (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic phase was washed with 0.1 M HCl (30 mL), sat. NaHCO3 (30 mL), brine (30 mL), dried (MgSO4), filtered and concentrated in vacuo to give the title compound (649 mg, 99%) as an orange oil. 1H NMR (270 MHz, CDCl3) δ 8.64 (1H, dd, J = 7.4, 1.2 Hz, H9″), 8.55 (1H, dd, J = 8.6, 1.2 Hz, H7″), 8.40 (1H, d, J = 7.8 Hz, H4″), 8.02 (1H, d, J = 7.8 Hz, H5″), 7.83 (1H, dd, J = 8.6, 7.4 Hz, H8″), 4.64 (1H, t, J = 4.9 Hz, H4), 4.18–4.12 (2H, m, H11′), 4.02 (1H, d, J = 5.5 Hz, H2), 3.89 (1H, d, J = 5.5 Hz, H6), 3.70 (6H, s, 2 × Me), 3.23 (1H, app. t, J = 4.9 Hz, H8), 2.71 (1H, d, J = 4.9 Hz, H9), 2.65–2.64 (2H, m, H1, H7), 1.78 (1H, dd, J = 11.0, 1.5 Hz, H10s), 1.74–1.58 (4H, m, H1′, H10′), 1.42–1.25 (17H, m, 8 × CH2, H10s). 13C NMR (67.5 MHz, CDCl3) δ 174.1, 172.9, 163.74, 163.73, 133.3, 132.1, 131.3, 131.2, 130.8, 130.3, 129.1, 128.2, 123.3, 122.4, 104.3, 81.4, 78.9, 52.5, 52.3, 45.4, 45.2, 43.8, 43.4, 40.7, 32.9, 31.8, 29.7 (2 × C), 29.62, 29.60, 29.5 (2 × C), 28.2, 27.2, 24.3. HRMS (ESI, m/z) for C35H4279BrNO8 [M + Na]+ calc. 706.1986; found 706.1972.
4.1.16. Dimethyl 4-[11′-(6″-propylamino-1″,3″-dioxo-1H-benzo[de]isoquinolme-2(3H))undecyl]-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxylate (26).
A stirring solution of 4-bromo naphthalimide norbornane 25 (28 mg, 0.041 mmol), Pd2(dba)3CHCl3 (2 mg, 0.002 mmol), Xantphos (1 mg, 0.002 mmol), propylamine (5 drops) and Cs2CO3 (40 mg, 0.123 mmol) in PhMe (410 μL) was heated at 70 °C for 24 h. The solvent was removed in vacuo and the resulting residue was loaded onto SiO2 and purified using column chromatography (2% MeOH in 1:4 EtOAc in pet. spirits) to give the title compound (23 mg, 88%) as a yellow oil. Rf = 0.19 (2% MeOH in 1:4 EtOAc in pet. spirits). 1H NMR (400 MHz, CDCl3) δ 8.57 (1H, dd, J= 7.4, 1.1 Hz, H9″), 8.45 (1H, d, J = 8.5 Hz, H4″), 8.08 (1H, dd, J = 8.4, 1.1 Hz, H7″), 7.61 (1H, dd, J = 8.4, 7.4 Hz, H8″), 6.72 (1H, d, J = 8.5 Hz, H5″), 5.26–5.25 (1H, m, NH), 4.65 (1H, t, J = 4.9 Hz, H4), 4.16–4.12 (2H, m, H11′), 4.02 (1H, d, J = 5.7 Hz, H2), 3.90 (1H, dd, J = 5.7, 1.4 Hz, H6), 3.70 (3H, s, Me), 3.69 (3H, s, Me), 3.40–3.35 (2H, m, NHCH2), 3.22 (1H, app. t, J = 5.1 Hz, H8), 2.72 (1H, d, J = 4.9 Hz, H9), 2.65–2.64 (2H, m, H1, H7), 1.88–1.76 (3H, m, CH2CH3, H10a), 1.74–1.67 (2H, m, H10′), 1.64–1.59 (2H, m, H1′), 1.43–1.24 (17H, m, 8 × CH2, H10s), 1.11 (3H, t, J = 7.4 Hz, CH3). 13C NMR (100 MHz, CDCl3) δ 174.1, 172.9, 164.8, 164.3, 149.5, 134.6, 131.2, 130.8, 125.8, 124.8, 123.4, 120.3, 110.5, 104.5, 104.4, 81.4, 78.9, 52.5, 52.3, 45.6, 45.4, 45.2, 43.8, 43.4, 40.4, 32.9, 31.8, 29.69, 29.67 (2 × C), 29.6 (2 × C), 29.5, 28.4, 27.3, 24.4, 22.4, 11.8. HRMS (ESI, m/z) for C38H50N2O8 [M + H]+ calc. 663.3640; found 663.3648.
4.1.17. 4-[11′-(6″-Propylamino-1″,3″-dioxo-1H-benzo[de]isoquinoline-2(3H))undecyl]-3,5-dioxatricyclo[5.2.1.02,6] decane-8-endo-9-exo-dicarboxylic acid (27).
A biphasic solution of ester 26 (434 mg, 0.655 mmol) in 2 M NaOH/THF (1:4, 7 mL) was stirred at ambient temperature for 16 h. The reaction mixture was extracted with CH2Cl2 (2 × 15 mL) and the isolated aqueous phase was acidified to pH = 1 using 2 M HCl and extracted with EtOAc (3 × 15 mL). The combined organic phase was washed with brine (15 mL), dried (MgSO4), filtered and concentrated in vacuo to afford the title compound (366 mg, 88%) as an orange solid. m.p. 94–95 °C. 1H NMR (500 MHz, CDCl3) δ 8.56 (1H, dd, J = 7.5, 1.1 Hz, H9″), 8.44 (1H, d, J = 8.5 Hz, H4″), 8.08 (1H, dd, J = 8.5, 1.1 Hz, H7″), 7.58 (1H, dd, J = 8.5, 7.5 Hz, H8″), 6.69 (1H, d, J = 8.5 Hz, H5″), 4.68 (1H, t, J = 4.8 Hz, H4), 4.14–4.11 (2H, m, H11′), 4.05 (2H, br s, H2, H6), 3.37 (2H, app. t, J = 7.3 Hz, NHCH2), 3.28 (1H, app. t, J= 5.2 Hz, H8), 2.79 (1H, d, J = 4.3 Hz, H7), 2.73 (1H, br s, H1), 2.66 (1H, d, J = 5.2 Hz, H9), 1.85–1.81 (3H, m, CH2, H10a), 1.72–1.59 (4H, m, 2 × CH2), 1.39–1.21 (17H, m, 8 × CH2, H10s), 1.10 (3H, t, J = 7.4 Hz, CH3). 13C NMR (125 MHz, CDCl3) 5 178.1, 176.9, 164.9, 164.5, 149.7, 134.8, 131.4, 129.9, 126.0, 124.8, 123.2, 120.2, 110.1, 104.5, 104.4, 81.3, 78.8, 45.6, 45.2, 45.1, 43.7, 43.4, 40.5, 32.7, 31.9, 29.8, 29.64, 29.58, 29.55, 29.4 (2 × C), 28.4, 27.3, 24.2, 22.4, 11.8. HRMS (ESI, m/z) for C36H46N2O8 [M – H]– calc. 633.3181; found 633.3190.
4.1.18. 8-endo-9-exo-Di[2′-(2″,3″-bis-tert-butoxycarbonylguamdmo)ethylcarbamoyl]-4-[11′-(6″-propylamino-1″,3″-dioxo-1H-benzo[de]isoqumolme-2(3H))undecyl]-3,5-dioxatricyclo[5.2.1.02,6] decane (28).
A solution of diacid 27 (285 mg, 0.449 mmol), EDCI (260 mg, 1.35 mmol), HOBt (6 mg, 0.05 mmol), aminoethylguanidine 22 (423 mg, 1.40 mmol) and anhydrous DMF (6 mL) was stirred at ambient temperature for 2 d under an inert atmosphere. The reaction mixture was diluted with EtOAc (25 mL) and washed with H2O (15 mL), brine (3 × 15 mL), dried (MgSO4), filtered and concentrated in vacuo to give a yellow oil. The crude material was purified using column chromatography (EtOAc) to afford the title compound (444 mg, 82%) as a yellow solid. Rf = 0.50 (EtOAc). m.p. 98–99 °C. Φf = 0.89 (DMSO). 1H NMR (500 MHz, CDCl3) δ 11.48 (1H, br s, NH), 11.45 (1H, br s, NH), 8.63 (1H, t, J = 6.1 Hz, NH), 8.57 (1H, dd, J = 7.4, 1.0 Hz, H9″), 8.51 (1H, t, J = 5.8 Hz, NH), 8.45 (1H, d, J = 8.5 Hz, H4″), 8.09 (1H, dd, J = 8.5, 1.0 Hz, H7″), 8.01 (1H, t, J = 4.1 Hz, NH), 7.61 (1H, dd, J = 8.5, 7.4 Hz, H8″), 6.86 (1H, t, J= 5.3 Hz, NH), 6.72 (1H, d, J = 8.5 Hz, H5″), 5.29 (1H, t, J = 5.0 Hz, NH), 4.60 (1H, t, J = 4.9 Hz, H4), 4.15–4.12 (2H, m, H11′), 4.02 (1H, d, J = 5.5 Hz, H6), 3.94 (1H, d, J = 5.5 Hz, H2), 3.60–3.52 (4H, m, 2 × NHCH2), 3.44–3.36 (6H, m, 3 × NHCH2), 2.94 (1H, app. t, J = 5.2 Hz, H8), 2.78 (1H, br s, H7), 2.70 (1H, d, J = 4.1 Hz, H1), 2.44 (1H, d, J = 5.2 Hz, H9), 1.87–1.80 (2H, m, CH2), 1.77 (1H, d, J = 9.5 Hz, H10a), 1.72–1.67 (2H, m, CH2), 1.62–1.57 (2H, m, CH2), 1.50–1.48 (37H, m, 4 × t-Bu, H10s), 1.42–1.24 (16H, m, 8 × CH2), 1.10 (3H, t, J = 7.4 Hz, CH3). 13C NMR (125 MHz, CDCl3) δ 174.2, 172.1, 164.8, 164.3, 163.5, 163.0, 157.9, 157.1, 153.2, 149.5, 134.6, 131.2, 129.9, 125.8, 124.8, 123.4, 120.3, 110.5, 104.5, 104.2, 83.7, 83.4, 81.6, 79.9, 79.7, 79.0, 47.8, 45.6, 44.30, 44.25, 42.3, 40.4, 40.2, 40.04, 39.97, 33.0, 32.5, 29.74, 29.71, 29.69, 29.66, 29.6, 28.40, 28.38, 28.3, 28.20, 28.19, 27.3, 24.4, 22.4, 11.8. HRMS (ESI, m/z) for C62H94N10O14 [M + H]+ calc. 1203.7024; found 1203.7040.
4.1.19. 4-[11′-(6″-Propylamino-1″,3″-dioxo-1H-benzo[de]isoquinoline-2(3H))undecyl]-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxamidoethylguanidine hydrogen chloride (29).
To a stirring solution of Boc-protected guanidine 28 (87 mg, 0.072 mmol) and MeOH (720 μL) was added dropwise AcCl (200 μL, 2.81 mmol), and the reaction was stirred for 2 d at ambient temperature. The reaction mixture was concentrated in vacuo, co-evaporated with MeOH (2 × 0.5 mL) and the title compound was isolated (61 mg, 97%) as an off-white sticky residue. m.p. 122–159 °C (slow decomposition). Φf = 0.88 (DMSO) and 0.16 (H2O). 1H NMR (500 MHz, CD3OD) δ 8.55 (1H, dd, J = 8.5, 1.1 Hz, H9″), 8.50 (1H, dd, J = 7.5, 1.1 Hz, H7″), 8.35 (1H, d, J = 8.7 Hz, H4″), 7.63 (1H, dd, J = 8.5, 7.5 Hz, H8″), 6.78 (1H, d, J = 8.7 Hz, H5″), 4.65 (1H, t, J = 4.7 Hz, H4), 4.12–4.09 (2H, m, H11′), 4.05 (1H, d, J = 5.7 Hz, H6), 3.99 (1H, d, J = 5.7 Hz, H2), 3.43–3.28 (10H, m, 5 × NHCH2), 3.23 (1H, app. t, J = 5.1 Hz, H8), 2.62–2.61 (2H, m, H1, H7), 2.44 (1H, br s, H9), 1.85–1.78 (2H, m, CH2), 1.73 (1H, d, J= 10.0 Hz, H10a), 1.70–1.66 (2H, m, CH2), 1.61–1.57 (2H, m, CH2), 1.47 (1H, d, J= 10.0 Hz, H10s), 1.40–1.28 (16H, m, 8 × CH2), 1.07 (3H, t, J = 7.4 Hz, CH3). 13C NMR (125 MHz, CD3OD) δ 176.5, 174.6, 166.3, 165.8, 158.9 (2 × C), 152.8, 136.1, 132.2, 131.3, 129.4, 125.4, 123.4, 121.9, 109.1, 105.11, 105.09, 82.8, 79.9, 47.8, 46.8, 46.2, 45.1, 44.9, 42.0, 41.9, 41.0, 39.7, 39.6, 33.9, 32.7, 30.6 (4 × C), 30.5, 30.4, 29.1, 28.1, 25.2, 22.7, 11.9. HRMS (ESI, m/z) for C42H62N10O6 [M + 2H]2+ calc. 402.2500; found 402.2506.
4.1.20. Dimethyl 4-[11′-(7″-nitrobenzo[c][1,2,5]oxadiazol-4″-amino)undecyl]-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxylate (31).
A solution of norbornane hydrochloride 23 (371 mg, 0.803 mmol), 4-nitrobenz-2-oxa-1,3-diazole chloride (160 mg, 0.803 mmol) and Et3N (170 μL, 1.22 mmol) in MeOH (8 mL) was stirred at ambient temperature for 24 h. The reaction mixture was concentrated under reduced pressure then diluted with EtOAc (20 mL). The organic phase was washed with 0.1 M HCl (10 mL), sat. NaHCO3 (10 mL), brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo. The orange residue was purified using column chromatography (10% EtOAc in CH2Cl2) to give the title compound (320 mg, 68%) as a dark orange viscous oil. Rf = 0.9 (10% EtOAc in CH2Cl2). :H NMR (500 MHz, CDCl3) δ 8.48 (1H, d, J = 8.6 Hz, H6″), 6.33 (1H, t, J = 4.7 Hz, NH), 6.17 (1H, d, J = 8.6 Hz, H5″), 4.65 (1H, t, J = 4.8 Hz, H4), 4.02 (1H, d, J = 5.6 Hz, H6), 3.94 (1H, d, J = 5.6 Hz, H2), 3.70 (3H, s, Me), 3.69 (3H, s, Me), 3.50–3.46, (2H, m, H11′), 3.22 (1H, app. t, J = 5.2 Hz, H8), 2.71 (1H, d, J = 4.5 Hz, H7), 2.65–2.64 (2H, m, H1, H9), 1.83–1.75 (3H, m, H10′, H10a), 1.64–1.60 (2H, m, H1′), 1.48–1.42 (2H, m, H9′), 1.39–1.26 (15H, m, H10s, 7 × CH2). 13C NMR (125 MHz, CDCl3) δ 174.1, 172.9, 144.4, 144.1, 144.0, 136.7, 123.9, 104.3, 98.6, 81.3, 78.9, 52.5, 52.3, 45.4, 45.1, 44.1, 43.7, 43.4, 32.9, 31.7, 29.60, 29.57, 29.5 (3 × C), 29.3, 28.6, 27.1, 24.3. HRMS (ESI, m/z) for C29H40N4O9 [M + H]+ calc. 589.2868; found 589.2883.
4.1.21. 4-[11′-(7″-Nitrobenzo[c] [1,2,5]oxadiazol-4″-amino)undecyl]-3,5-dioxatricydo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxylic acid (32).
A biphasic solution of ester 31 (259 mg, 0.440 mmol) in 2 M NaOH/THF (1:4, 3 mL) was stirred at ambient temperature for 16 h. The reaction mixture was extracted with CH2Cl2 (2 × 5 mL) and the aqueous phase was isolated and acidified to pH = 1 using 2 M HCl and then extracted with EtOAc (3 × 15 mL). The combined organic phase was washed with brine (15 mL), dried (MgSO4), filtered and concentrated in vacuo to afford the title compound (193 mg, 78%) as an orange solid. m.p. 98–99 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.55 (1H, t, J = 5.3 Hz, NH), 8.50 (1H, d, J = 9.1 Hz, H6″), 6.40 (1H, d, J = 9.1 Hz, H5″), 4.61 (1H, t, J = 4.8 Hz, H4), 3.96 (1H, d, J = 5.8 Hz, H6), 3.88 (1H, d, J = 5.8 Hz, H2), 3.45–3.44, (2H, m, H11′), 3.00 (1H, app. t, J = 5.5 Hz, H8), 2.52 (1H, d, J = 4.6 Hz, H7), 2.45 (1H, br s, H1), 2.40 (1H, d, J = 5.5 Hz, H9), 1.70–1.63 (2H, m, H10′), 1.59 (1H, d, J = 9.6 Hz, H10a), 1.54–1.49 (2H, m, H1’), 1.37–1.19 (17H, m, H10s, 8 × CH2). 13C NMR (100 MHz, DMSO-d6) δ 174.6, 173.3, 145.2, 144.4, 144.2, 138.0, 120.5, 103.2, 99.1, 80.7, 78.2, 45.0, 44.4, 43.4, 43.2, 42.6, 32.3, 31.2, 28.9 (5 × C), 28.7, 27.6, 26.4, 23.7. HRMS (ESI, m/z) for C27H36N4O9 [M – H]– calc. 559.2410; found 559.2424.
4.1.22. 8-endo-9-exo-Di[2″-(2″,3″-bis-tert-butoxycarbonylguanidino)ethykarbamoyl]-4-[11′-(7″-mtrobenzo[c][1,2,5]oxadiazol-4″-amino)undecyl]-3,5-dioxatricydo[5.2.1.02,6]decane (33).
A solution of diacid 32 (200 mg, 0.357 mmol), EDCI (222 mg, 1.16 mmol), HOBt (5 mg, 0.04 mmol), aminoethylguanidine 22 (339 mg, 1.12 mmol) and anhydrous DMF (5.1 mL) was stirred at ambient temperature for 3 d under an inert atmosphere. The reaction mixture was diluted with EtOAc (25 mL) and washed with H2O (15 mL), brine (3 × 15 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using flash column chromatography (50% EtOAc in CH2Cl2) and the title compound (263 mg, 65%) was isolated as an orange solid. Rf = 0.3 (50% EtOAc in CH2Cl2). m.p. 117–124 °C (slow decomposition). Φf = 0.55 (DMSO). 1H NMR (500 MHz, CDCl3) δ 11.48 (1H, br s, NH), 11.44 (1H, br s, NH), 8.64 (1H, t, J = 6.1 Hz, NH), 8.51–8.49 (2H, m, H6″, NH), 8.03 (1H, t, J = 4.1 Hz, NH), 6.87 (1H, t, J = 5.2 Hz, NH), 6.32 (1H, t, J = 5.2 Hz, NH), 6.17 (1H, d, J = 8.7 Hz, H5″), 4.61 (1H, t, J = 4.7 Hz, H4), 4.04 (1H, d, J = 5.6 Hz, H6), 3.95 (1H, d, J = 5.6 Hz, H2), 3.60–3.35 (10H, m, 5 × NHCH2), 2.93 (1H, app. t, J = 5.2 Hz, H8), 2.70 (1H, d, J = 4.1 Hz, H1), 2.58 (1H, br s, H7), 2.45 (1H, d, J = 5.2 Hz, H9), 1.83–1.76 (3H, m, H10′, H10a), 1.61–1.58 (2H, m, H1′), 1.53–1.43 (37H, m, H10s, 4 × t-Bu), 1.38–1.27 (16H, m, 8 × CH2). 13C NMR (125 MHz, CDCl3) 5 174.2, 172.0, 163.5, 163.0, 157.9, 157.1, 153.2 (2 × C), 144.4, 144.04, 144.00, 136.6, 124.1, 104.1, 98.6, 83.7, 83.4, 81.7, 79.9, 79.7, 79.0, 47.9, 44.3, 44.2, 44.1, 43.0, 42.3, 40.2, 40.1, 40.0, 32.9, 32.5, 29.61, 29.56, 29.54, 29.51, 29.48, 29.3, 28.7, 28.41, 28.39, 28.21, 28.20, 27.1, 24.2. HRMS (ESI, m/z) for C53H84N12O15 [M + H]+ calc. 1129.6252; found 1129.6265.
4.1.23. 4-[11′-(7″-Nitrobenzo[c][1,2,5]oxadiazol-4″-amino)undecyl]-3,5-dioxatricyclo[5.2.1.02,6]decane-8-endo-9-exo-dicarboxamidoethylguanidine hydrogen chloride (34).
To a stirring solution of Boc-protected guanidine 33 (89 mg, 0.079 mmol) and MeOH (790 μL) was added dropwise AcCl (230 μL, 3.23 mmol), and the reaction was stirred for 2 d at ambient temperature. The reaction mixture was concentrated in vacuo, co-evaporated with MeOH (2 × 0.5 mL) and the title compound was isolated (60 mg, 95%) as an orange solid. m.p. 121–124 °C. Φf = 0.55 (DMSO) and 0.02 (H2O). 1H NMR (500 MHz, CD3OD) δ 8.53 (1H, d, J = 8.8 Hz, H6″), 6.35 (1H, d, J = 8.8 Hz, H5″), 4.64 (1H, t, J = 4.7 Hz, H4), 4.04 (1H, d, J = 5.6 Hz, H6), 3.99 (1H, d, J = 5.6 Hz, H2), 3.55–3.52 (2H, m, H11′), 3.40–3.29, (8H, m, 4 × CH2NH), 3.22 (1H, app. t, J= 5.0 Hz, H8), 2.62–2.61 (2H, m, H1, H7), 2.44 (1H, br s, H9), 1.80–1.75 (2H, m, H10′), 1.72 (1H, d, J = 10.4 Hz, H10a), 1.59–1.55 (2H, m, H1’), 1.48–1.28 (17H, m, H10s, 8 × CH2). 13C NMR (125 MHz, CD3OD) δ 176.5, 174.6, 158.8 (2 × C), 146.7, 145.9, 145.6, 138.6, 122.9, 105.1, 99.5, 82.8, 79.9, 47.7, 46.9, 45.1, 44.9, 44.7, 42.0, 41.9, 39.7, 39.6, 33.9, 32.7, 30.61, 30.57, 30.51 (2 × C), 30.49, 30.3, 29.2, 28.0, 25.2. HRMS (ESI, m/z) for C33H52N12O7 [M + 2H]2+ calc. 365.2114; found 365.2115.
4.2. Disk Diffusion Assay.
A stock solution of 10 mg/mL was made for each compound under observation using DMSO as a solvent. Each stock solution was then diluted 1:2 to bring the concentration to 5 mg/mL in DMSO. The diluted solutions were then filter-sterilised using a 0.2-μm nylon filter, and 10 μL of the 5 mg/mL stock was pipetted onto a blank disk (i.e. 50 μg/disk; Oxoid Limited, Hampshire, UK). Suspensions of all bacterial isolates were adjusted to a 0.5 McFarland standard (in 0.9% NaCl) before they were swabbed onto nutrient agar plates. The controls used were a 10 μg Colistin disk (sulphate, Oxoid), 10 μL of DMSO and a plate swabbed with saline from the dispenser used.
4.3. Minimum Inhibitory Concentration (MIC) determination.
Bacteria were obtained either from American Type Culture Collection (ATCC; Manassas, VA, USA) or Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) as listed in Table S1. Bacteria were cultured in Nutrient broth (NB; Bacto Laboratories, catalogue No. 234000) or Mueller-Hinton broth (MHB; Bacto Laboratories, catalogue No. 211443) at 37 °C overnight with shaking (~180 RPM). A sample of each culture was diluted 50-fold in fresh MHB and incubated at 37 °C for 1.5–3 h with shaking (~180 RPM). Compound stock solutions were prepared as 10 mg/mL in DMSO and Colistin was dissolved in milli-Q water at 5.12 mg/mL. The compounds, at twice the final desired concentration, were serially diluted 2-fold across the wells of 96–well plates (Non-Binding Surface (NBS), Corning, catalogue No. 3641). Mid-log phase bacterial cultures (after 1.5–3 h incubation) were diluted to give a final cell density of 5×105 colony forming units (CFU)/mL when adding 50 μL to each well giving a final compound concentration range of 32 μg/mL to 0.015 μg/mL (DMSO ≤ 1%). MICs were determined visually after 18–20 h of incubation at 37 °C, with the MIC defined as the lowest compound concentration at which no bacterial growth was visible. Determined MIC values are the result of two independent experiments of n = 2, giving a final dataset of n = 4.
4.4. Cytotoxicity evaluation.
HEK293 (ATCC CRL-1573, human embryonic kidney) cells were seeded as 3000 cells per well in a 384-well plate in DMEM medium (GIBCO-Invitrogen #11995–073), in which 10% of FBS was added. Cells were incubated for 24 h at 37 °C, 5% CO2 to allow cells to attach to the plates. A concentration series of compounds was then added into each well. The cells were incubated with the compounds for 24 h at 37 °C, 5% CO2. After the incubation, 10 μM resazurin (dissolved in PBS) was added to each well. The plates were then incubated for 2 h at 37 °C, 5% CO2. The fluorescence intensity was read using Polarstar Omega with excitation/emission 560/590. The data was analysed by Prism software. Results are presented as the average percentage of control ± SD for each set of duplicate wells.
4.5. Microscopy.
Escherichia coli (ATCC© 25922™, USA) and Staphylococcus aureus (Staphylococcus aureus subsp. aureus Rosenbach (ATCC® 43300™ MRSA), USA) were grown in 50 mL of Luria-Bertani (LB) broth at 37 °C with gentle shaking overnight. S. aureus and E. coli were incubated with for 20 min at 37 °C with 10 μg/mL of fluorescent compound (29, 34, 35 or 36) in LB broth. For time dependent assessment, E. coli were incubated with 10 μg/mL of compound 34 for 5 min, 30 min, 1 h, 2 h, 4 h and 18 h. S. aureus were fixed in 4% paraformaldehyde for 20 min before being wet mounted onto slides for visualisation. Imaging was performed on a Nikon A1+ confocal microscope (Nikon, Japan) using a 60× object with excitation at 488 nm and emissions detected at 500–550 nm by a GaAsP PMT detector. 3D reconstruction and rendering were performed using NIS elements software (Nikon, Japan).
4.6. Modelling.
GROMACS version 2016.1 molecular dynamics package[69] in conjunction with the GROMOS 54A7 force field[70] was used for all MD simulations. Water was represented explicitly using the simple point charge (SPC) model.[71] Parameters for norbornane molecules prepared in the study were determined by the Automated Topology Builder.[72] Each system was simulated under periodic boundary conditions in a rectangular simulation box with a timestep of 2 fs. The temperature of the system was maintained by coupling each component of the system to an external temperature bath at 310 K with a coupling constant of τT = 0.1 ps using a velocity rescaling thermostat. The pressure was maintained at 1 bar by weakly coupling the system to a semi-isotropic pressure bath using an isothermal compressibility of 4.5 × 10–5 bar–1 and a coupling constant of τP = 0.5 ps. During the simulations, the length of all bonds in all non-water molecules were constrained using the LINCS algorithm.[73] The SETTLE algorithm[74] was used to constrain the geometry of water molecules. Electrostatic interactions were calculated using particle mesh Ewald summation and nonbonded interactions were calculated with a cut-off of 1.0 nm. Both were updated each timestep. All images and videos were prepared in VMD.[74]
Aggregation simulations were conducted on bisamine (8), bisguanidine (2), benzylguanidine (7) and naphthalimide (29) species. Each simulation system consisted of 10 molecules of norbornane based compound, 20 chloride ions and 15,000 water molecules. Solute molecules were randomly positioned in a 9 × 9 × 9 nm box, to which water was then added. 1000 steps of energy minimisation, then 500 ps under an NPT ensemble was simulated for each species. Finally 300 ns, 400 ns, 200 ns, 200 ns long simulations was conducted for 8, 2, 7 and 29 respectively using an NVT ensemble until a single aggregate formed. To construct the aggregate for use in the bilayer simulations, the same procedure was followed on a system comprising of 200 molecules of 2, 400 chloride ions and 30,000 water molecules in a 15 × 15 × 15 box. The NVT ensemble was simulated for 100 ns.
The starting coordinates of the model membrane used in the bilayer simulations were taken from Anandan et al.[75]. This represents the inner membrane of a Gram-negative bacteria. The outer membrane was not modelled as the structure of the peptidoglycan layer remains elusive. The model membrane is composed of 80% (408 molecules) of 1-palmitoyl-2-palmitoleyl-sn-glycero-3-phophoethanolamine (PPoPE) and 20% (104 molecules) of 1,2-dimyristoyl-sn-glycero-3-phospho-(1’-rac)-glycerol (DMPG). The membrane in Anandan et al. contained both D and L-enantiomers of the DMPG head group; this was corrected in this study so that only the naturally occurring L-enantiomer was modelled. The model also contained 31,457 water molecules, 260 sodium ions and 156 chloride ions. After 5000 steps of energy minimisation, the system was equilibration using an NPT ensemble for 100 ns.
The largest aggregate of 2 (28 molecules), was placed with random orientation in the water surrounding the model membrane. The addition of more water molecules was necessary to accommodate the aggregate; ca. 11,500 additional water molecules were added, expanding the system size in the direction normal to the bilayer plane. Three replicates were then energy minimised for 10000 steps each, then were simulated using an NPT ensemble. The first replicate was simulated for 780 ns; the second and third replicates were simulated for 500 ns.
Supplementary Material
Highlights.
Antibacterial norbornane based amphiphiles with MIC as low as 0.25 μg/mL.
Fluorescent microscopy of fluorophore-tagged analogues shows membrane localisation.
Molecular modelling indicates rapid aggregation prior to membrane insertion.
Acknowledgements
F.M.P., S.M.H., & T.D.A. thank the ARC (DP140100227) and the Strategic Research Centre for Chemistry and Biotechnology (Deakin University) for financial support and a top-up scholarship for S.M.H. The authors would also like to thank the Australian Research Council for funding Deakin University’s Magnetic Resonance Facility through LIEF grant LE110100141. C.A.B was funded by a Future Industry Accelerator Research and Development Voucher Scheme. J.L. & R.L.N. are supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI098771). J.L. is an Australian NHMRC Senior Research Fellow, while M.A.C. is an Australian NHMRC Principal Research Fellow. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. The MIC and cytotoxicity assays were performed in collaboration with CO-ADD (Community for Open Antimicrobial Drug Discovery, co-add.org)[76] funded by the Wellcome Trust (UK) and The University of Queensland (Australia).
Footnotes
Supporting Information Available: Synthetic procedures for all known compounds and copies of NMR spectra (1H and 13C) for all new compounds. All bacterial strains tested, and all cytotoxicity data. Additional molecular modelling images. Fluorescence microscopy and molecular modelling videos can be supplied upon request. This material is available free of charge via the Internet at http://
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Walsh CT, Wencewicz TA, Prospects for new antibiotics: a molecule-centered perspective, J. Antibiot, 67 (2014) 7–22. [DOI] [PubMed] [Google Scholar]
- [2].Butler MS, Blaskovich MA, Cooper MA, Antibiotics in the clinical pipeline in 2013, J. Antibiot, 66 (2013) 571–591. [DOI] [PubMed] [Google Scholar]
- [3].Boucher HW, Talbot GH, Benjamin DK, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D, 10 × ‘20 Progress—Development of new drugs active against Gram-negative Bacilli: an update from the Infectious Diseases Society of America, Clin. Infect. Dis, 56 (2013) 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schwarz S, Johnson AP, Transferable resistance to colistin: a new but old threat, J. Antimicrob. Chemother, 71 (2016) 2066–2070. [DOI] [PubMed] [Google Scholar]
- [5].Antimicrobial resistance: global report on surveillance, World Health Organization, (2014).
- [6].Shore CK, Coukell A, Roadmap for antibiotic discovery, Nat Microbiol, 1 (2016) 16083. [DOI] [PubMed] [Google Scholar]
- [7].McGuinness WA, Malachowa N, DeLeo FR, Focus: infectious diseases: vancomycin resistance in staphylococcus aureus, YJBM, 90 (2017) 269. [PMC free article] [PubMed] [Google Scholar]
- [8].National Action Plan for Combating Antibiotic-Resistant Bacteria, Centres for Disease Control, Washington, (2015).
- [9].Fisher JF, Mobashery S, Endless resistance. Endless antibiotics?, MedChemComm, 7 (2016) 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bush K, Investigational agents for the treatment of Gram-negative bacterial infections: a reality check, ACS Infect. Dis, 1 (2015) 509–511. [DOI] [PubMed] [Google Scholar]
- [11].Jenssen H, Hamill P, Hancock REW, Peptide antimicrobial agents, Clin. Microbiol. Rev, 19 (2006) 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hancock RE, Sahl H-G, Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies, Nat. Biotechnol, 24 (2006) 1551–1557. [DOI] [PubMed] [Google Scholar]
- [13].Ding B, Yin N, Liu Y, Cardenas-Garcia J, Evanson R, Orsak T, Fan M, Turin G, Savage PB, Origins of cell selectivity of cationic steroid antibiotics, J. Am. Chem. Soc, 126 (2004) 13642–13648. [DOI] [PubMed] [Google Scholar]
- [14].Chou H-T, Wen H-W, Kuo T-Y, Lin C-C, Chen W-J, Interaction of cationic antimicrobial peptides with phospholipid vesicles and their antibacterial activity, Peptides, 31 (2010) 1811–1820. [DOI] [PubMed] [Google Scholar]
- [15].Isaksson J, Brandsdal BO, Engqvist M, Flaten GE, Svendsen JSM, Stensen W, A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption, J. Med. Chem, 54 (2011) 5786–5795. [DOI] [PubMed] [Google Scholar]
- [16].Perron GG, Zasloff M, Bell G, Experimental evolution of resistance to an antimicrobial peptide, Proc. R. Soc. B, 273 (2006) 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen X, Dings RP, Nesmelova I, Debbert S, Haseman JR, Maxwell J, Hoye TR, Mayo KH, Topomimetics of amphipathic β-sheet and helix-forming bactericidal peptides neutralize lipopolysaccharide endotoxins, J. Med. Chem, 49 (2006) 7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thaker HD, Sgolastra F, Clements D, Scott RW, Tew GN, Synthetic mimics of antimicrobial peptides from triaryl scaffolds, J. Med. Chem, 54 (2011) 2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bremner JB, Keller PA, Pyne SG, Boyle TP, Brkic Z, David DM, Garas A, Morgan J, Robertson M, Somphol K, Binaphthyl-based dicationic peptoids with therapeutic potential, Angew. Chem. Int. Ed, 49 (2010) 537–540. [DOI] [PubMed] [Google Scholar]
- [20].Robertson M, Bremner JB, Coates J, Deadman J, Keller PA, Pyne SG, Somphol K, Rhodes DI, Synthesis and antibacterial activity of C2-symmetric binaphthyl scaffolded amino acid derivatives, Euro. J. Med. Chem, 46 (2011) 4201–4211. [DOI] [PubMed] [Google Scholar]
- [21].Lowe AJ, Dyson GA, Pfeffer FM, Factors influencing anion binding stoichiometry: the subtle influence of electronic effects, Euro. J. Org. Chem, 2008 (2008) 1559–1567. [Google Scholar]
- [22].Lowe AJ, Dyson GA, Pfeffer FM, Steric and electronic factors influencing recognition by a simple, charge neutral norbornene based anion receptor, Org. Biomol. Chem, 5 (2007) 1343–1346. [DOI] [PubMed] [Google Scholar]
- [23].Hickey SM, Ashton TD, Khosa SK, Robson RN, White JM, Li J, Nation RL, Yu HY, Elliott AG, Butler MS, Huang JX, Cooper MA, Pfeffer FM, Synthesis and evaluation of cationic norbornanes as peptidomimetic antibacterial agents, Org. Biomol. Chem, 13 (2015) 6225–6241. [DOI] [PubMed] [Google Scholar]
- [24].Hickey SM, Ashton TD, White JM, Li J, Nation RL, Yu HY, Elliott AG, Butler MS, Huang JX, Cooper MA, Pfeffer FM, Synthesis of norbornane bisether antibiotics via silver-mediated alkylation, RSC Advances, 5 (2015) 28582–28596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Henderson LC, Li J, Nation RL, Velkov T, Pfeffer FM, Developing an anion host for lipid A binding and antibacterial activity, Chem. Commun, 46 (2010) 3197–3199. [DOI] [PubMed] [Google Scholar]
- [26].Hale JDF, Hancock REW, Alternative mechanisms of action of cationic antimicrobial peptides on bacteria, Expert Rev. Anti. Infec. Ther, 5 (2007) 951–959. [DOI] [PubMed] [Google Scholar]
- [27].Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RLA, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Käch A, Eberl L, Riedel K, DeMarco SJ, Robinson JA, Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa, Science, 327 (2010) 1010–1013. [DOI] [PubMed] [Google Scholar]
- [28].Hasper HE, Kramer NE, Smith JL, Hillman J, Zachariah C, Kuipers OP, De Kruijff B, Breukink E, An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II, Science, 313 (2006) 1636–1637. [DOI] [PubMed] [Google Scholar]
- [29].Stewart PS, Davison WM, Steenbergen JN, Daptomycin rapidly penetrates a Staphylococcus epidermidis biofilm, Antimicrob. Agents Chemother, 53 (2009) 3505–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Duke RM, Veale EB, Pfeffer FM, Kruger PE, Gunnlaugsson T, Colorimetric and fluorescent anion sensors: an overview of recent developments in the use of 1,8-naphthalimide-based chemosensors, Chem. Soc. Rev, 39 (2010) 3936–3953. [DOI] [PubMed] [Google Scholar]
- [31].Chattopadhyay A, Chemistry and biology of N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-labeled lipids: fluorescent probes of biological and model membranes, Chem. Phys. Lipids, 53 (1990) 1–15. [DOI] [PubMed] [Google Scholar]
- [32].Bruno TJ, Svoronos PD, CRC handbook of fundamental spectroscopic correlation charts, CRC Press, 2005. [Google Scholar]
- [33].Lavis LD, Raines RT, Bright ideas for chemical biology, ACS Chem. Biol, 3 (2008) 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hickey SM, Ashton TD, Khosa SK, Pfeffer FM, An optimised synthesis of 2-[2, 3-Bis (tert-butoxycarbonyl) guanidino] ethylamine, Synlett, 23 (2012) 1779–1782. [Google Scholar]
- [35].Hickey SM, Ashton TD, Pfeffer FM, Facile synthesis of guanidine functionalised building blocks, Asian J. Org. Chem, 4 (2015) 320–326. [Google Scholar]
- [36].Peschel A, How do bacteria resist human antimicrobial peptides?, Trends Microbiol, 10 (2002) 179–186. [DOI] [PubMed] [Google Scholar]
- [37].Warren HS, Kania SA, Siber G, Binding and neutralization of bacterial lipopolysaccharide by colistin nonapeptide, Antimicrob. Agents Chemother, 28 (1985) 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brogden KA, Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?, Nat. Rev. Microbiol, 3 (2005) 238–250. [DOI] [PubMed] [Google Scholar]
- [39].Schmuck C, Bickert V, Merschky M, Geiger L, Rupprecht D, Dudaczek J, Wich P, Rehm T, Machon U, A facile and efficient multi-gram synthesis of N-protected 5-(guanidinocarbonyl)-1H- pyrrole-2-carboxylic acids, Euro. J. Org. Chem, 2008 (2008) 324–329. [Google Scholar]
- [40].Schmuck C, Carboxylate binding by 2-(guanidiniocarbonyl)pyrrole receptors in aqueous solvents: improving the binding properties of guanidinium cations through additional hydrogen bonds, Chem-Eur. J, 6 (2000) 709–718. [DOI] [PubMed] [Google Scholar]
- [41].Schmuck C, Geiger L, Design and synthesis of a new class of arginine analogues with an improved anion binding site in the side chain, Chem. Commun, (2005) 772–774. [DOI] [PubMed] [Google Scholar]
- [42].Schmuck C, Heil M, Scheiber J, Baumann K, Charge interactions do the job: a combined statistical and combinatorial approach to finding artificial receptors for binding tetrapeptides in water, Angew. Chem. Int. Ed, 44 (2005) 7208–7212. [DOI] [PubMed] [Google Scholar]
- [43].Zeiler E, Korotkov VS, Lorenz-Baath K, Böttcher T, Sieber SA, Development and characterization of improved β-lactone-based anti-virulence drugs targeting ClpP, Bioorgan. Med. Chem, 20 (2012) 583–591. [DOI] [PubMed] [Google Scholar]
- [44].Agarwal S, Tiwari HP, Sharma JP, Pyridinium chlorochromate: an improved method for its synthesis and use of anhydrous acetic acid as catalyst for oxidation reactions, Tetrahedron, 46 (1990) 4417–4420. [Google Scholar]
- [45].Corey EJ, Suggs JW, Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds, Tetrahedron Lett, 16 (1975) 2647–2650. [Google Scholar]
- [46].Hunsen M, Pyridinium chlorochromate catalyzed oxidation of alcohols to aldehydes and ketones with periodic acid, Tetrahedron Lett, 46 (2005) 1651–1653. [Google Scholar]
- [47].Sanders TC, Seto CT, 4-Heterocyclohexanone-based inhibitors of the serine protease plasmin, J. Med. Chem, 42 (1999) 2969–2976. [DOI] [PubMed] [Google Scholar]
- [48].Amatore M, Beeson TD, Brown SP, MacMillan DWC, Enantioselective linchpin catalysis by SOMO catalysis: an approach to the asymmetric a-chlorination of aldehydes and terminal epoxide formation, Angew. Chem. Int. Ed, 48 (2009) 5121–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fleming CL, Ashton TD, Pfeffer FM , Synthesis of 4-amino substituted 1,8-naphthalimide derivatives using palladium-mediated amination, Dyes and Pigments, 109 (2014) 135–143. [Google Scholar]
- [50].Findlay B, Zhanel GG, Schweizer F, Cationic amphiphiles, a new generation of antimicrobials inspired by the natural antimicrobial peptide scaffold, Antimicrob. Agents Chemother, 54 (2010) 4049–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Makovitzki A, Avrahami D, Shai Y, Ultrashort antibacterial and antifungal lipopeptides, P. Nat. A. Sci, 103 (2006) 15997–16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ruiz N, Kahne D, Silhavy TJ, Advances in understanding bacterial outer-membrane biogenesis, Nat. Rev. Microbiol, 4 (2006) 57. [DOI] [PubMed] [Google Scholar]
- [53].Dalrymple OK, Stefanakos E, Trotz MA, Goswami DY, A review of the mechanisms and modeling of photocatalytic disinfection, Appl. Catal. B-Environ, 98 (2010) 27–38. [Google Scholar]
- [54].Hong X, Hopfinger AJ, Construction, molecular modeling, and simulation of Mycobacterium tuberculosis cell walls, Biomacromolecules, 5 (2004) 1052–1065. [DOI] [PubMed] [Google Scholar]
- [55].Beauregard DA, Williams DH, Gwynn MN, Knowles DJ, Dimerization and membrane anchors in extracellular targeting of vancomycin group antibiotics, Antimicrob. Agents Chemother, 39 (1995) 781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Straus SK, Hancock REW, Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides, Biochim. Biophys. Acta, 1758 (2006) 1215–1223. [DOI] [PubMed] [Google Scholar]
- [57].Marquette A, Mason AJ, Bechinger B, Aggregation and membrane permeabilizing properties of designed histidine-containing cationic linear peptide antibiotics, J.Pept. Sci, 14 (2008) 488–495. [DOI] [PubMed] [Google Scholar]
- [58].Parisien A, Allain B, Zhang J, Mandeville R, Lan C, Novel alternatives to antibiotics: bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides, J. Appl. Microbiol, 104 (2008) 1–13. [DOI] [PubMed] [Google Scholar]
- [59].Park Y, Hahm K, Antimicrobial peptides (AMPs): peptide structure and mode of action, J. Biochem. Mol. Biol, 38 (2005) 507–516. [DOI] [PubMed] [Google Scholar]
- [60].Yang L, Harroun TA, Weiss TM, Ding L, Huang HW, Barrel-stave model or toroidal model? A case study on melittin pores, Biophys. J, 81 (2001) 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Aron Z, Opperman TJ, The hydrophobic trap—the Achilles heel of RND efflux pumps, Res. Microbiol. (In Press), (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Opperman T, Nguyen S, Recent advances toward a molecular mechanism of efflux pump inhibition, Front. Micribiol, 6 (2015) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Forbes CC, DiVittorio KM, Smith BD, Bolaamphiphiles promote phospholipid translocation across vesicle membranes, J. Am. Chem. Soc, 128 (2006) 9211–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Xuan W, Pan R, Cao Y, Liu K, Wang W, A fluorescent probe capable of detecting H2S at submicromolar concentrations in cells, Chem. Commun, 48 (2012) 10669–10671. [DOI] [PubMed] [Google Scholar]
- [65].Hearn KN, Nalder TD, Cox RP, Maynard HD, Bell TDM, Pfeffer FM, Ashton TD, Modular synthesis of 4-aminocarbonyl substituted 1,8-naphthalimides and application in single molecule fluorescence detection, Chem. Commun, 53 (2017) 12298–12301. [DOI] [PubMed] [Google Scholar]
- [66].Buchanan MK, Needham CN, Neill NE, White MC, Kelly CB, Mastro-Kishton K, Chauvigne-Hines LM, Goodwin TJ, McIver AL, Bartolotti LJ, Frampton AR, Bourdelais AJ, Varadarajan S, Glycoconjugated site-selective DNA-methylating agent targeting glucose transporters on glioma cells, Biochemistry, 56 (2017) 421–440. [DOI] [PubMed] [Google Scholar]
- [67].Black SL, Chauvignac C, Grundt P, Miller CN, Wood S, Traynor JR, Lewis JW, Husbands SM, Guanidino N-substituted and N,N-disubstituted derivatives of the κ-opioid antagonist GNTI, J. Med. Chem, 46 (2003) 5505–5511. [DOI] [PubMed] [Google Scholar]
- [68].Halim M, Tremblay MS, Jockusch S, Turro NJ, Sames D, Transposing molecular fluorescent switches into the near-IR: development of luminogenic reporter substrates for redox metabolism, J. Am. Chem. Soc, 129 (2007) 7704–7705. [DOI] [PubMed] [Google Scholar]
- [69].Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 1 (2015) 19–25. [Google Scholar]
- [70].Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF, Definition and testing of the GROMOS force-field versions 54A7 and 54B7, Eur. Biophys. J, 40 (2011) 843. [DOI] [PubMed] [Google Scholar]
- [71].Hermans J, Berendsen HJ, Van Gunsteren WF, Postma JP, A consistent empirical potential for water-protein interactions, Biopolymers, 23 (1984) 1513–1518. [Google Scholar]
- [72].Malde AK, Zuo L, Breeze M, Stroet M, Poger D, Nair PC, Oostenbrink C, Mark AE, An automated force field topology builder (ATB) and repository: version 1.0, J. Chem. Theory Comput, 7 (2011) 4026–4037. [DOI] [PubMed] [Google Scholar]
- [73].Hess B, Bekker H, Berendsen HJ, Fraaije JG, LINCS: a linear constraint solver for molecular simulations, J. Comput. Chem, 18 (1997) 1463–1472. [Google Scholar]
- [74].Miyamoto S, Kollman PA, Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models, J. Comput. Chem, 13 (1992) 952–962. [Google Scholar]
- [75].Anandan A, Evans GL, Condic-Jurkic K, O’Mara ML, John CM, Phillips NJ, Jarvis GA, Wills SS, Stubbs KA, Moraes I, Structure of a lipid A phosphoethanolamine transferase suggests how conformational changes govern substrate binding, P. Nat. A. Sci, 114 (2017) 2218–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Blaskovich MA, Zuegg J, Elliott AG, Cooper MA, Helping chemists discover new antibiotics, ACS Infect. Dis, 1 (2015) 285–287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.