

Abstract
Using molecular dynamics simulations and electronic structure theory, we shed light on the charge dynamics that causes the differential interaction of tumor suppressor protein p53 with the p21 and Gadd45 genes in response to oxidative stress. We show that the sequence dependence of this selectivity results from competing charge transfer to the protein and through the DNA, with implications on the use of genome editing tools to influence the p53 regulatory function.
Graphical Abstract
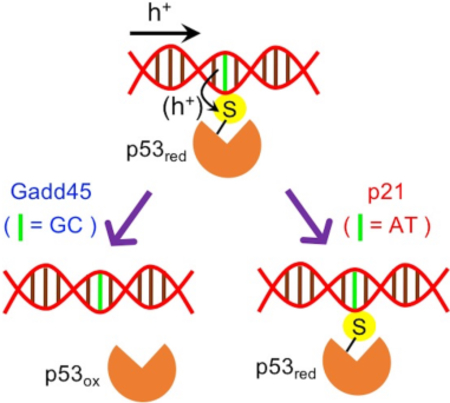
The tumor suppressor protein p53 prevents tumor formation by sensing various cellular stresses and activating or repressing a myriad of genes. p21WAF1/Cip1 and Gadd45 are two target genes that are expressed when the Cys-rich core domain of p53 binds to their sequence-specific DNA response elements at the promoters. p21 mediates the p53-dependent G1 growth phase arrest by hindering the activities of cyclin-dependent kinases,1 inhibiting DNA replication, and interfering with DNA polymerase activity.2 The activated Gadd45 recruits nucleotide and/or base excision repair factors to gene-specific loci,3 promoting DNA repair4 and demethylation.3
Some of the cellular stresses sensed by p53 include reactive oxygen species, which serve as signaling molecules to regulate biological and physiological processes.5 In many proteins, redox signaling can involve H2O2-mediated oxidation of Cys residues,5 as well as DNA-mediated signaling initiated by base oxidation.6 Therefore, it is important to understand how p53 differentiates target genes for the redox regulation of metabolic and signaling pathways under oxidative stress.
Experimental studies7–9 have shown that, under conditions of DNA oxidation, the core DNA-binding domain of p53 dissociates more easily from the Gadd45 consensus sequence than from the p21 sequence. The dissociation is expected to occur because of hole transfer (HT) from the oxidized DNA to a proximal Cys residue of p53. In fact, the HT oxidizes Cys to a cysteinyl radical cation that can form a disulfide bond with a neighboring Cys residue. This disulfide bond formation in turn attenuates p53-DNA binding, thus causing the DNA dissociation.10 Interestingly, the preferred dissociation of the Gadd45 DNA sequence from p53 is also consistent with the differential expression of p53-inducible genes in human ML-1 myeloblastic leukemia cells after DNA damage by ionizing radiation.11 As apoptosis can serve as a fail-safe mechanism to prevent uncontrolled cellular proliferation under persistent oxidative stress,12 p53 reacts by underexpressing Gadd45 (which hampers inefficacious DNA repair) and continuing to express p21 (which promotes G1 cell cycle arrest and eventual cellular apoptosis13). This differential response of p53 to the oxidized Gadd45 and p21 DNA sequences needs to be explained in terms of structure, free energy landscape,9 and dynamical aspects of the p53-DNA charge transfer (CT) that are studied here.
Structurally, the key underlying feature of p53-DNA binding is the ability of p53 to bind to a consensus sequence located within the downstream gene promoter.14 This DNA sequence consists of two decameric palindromic sequences spaced 0 to 21 base pairs (bps) apart.15, 16 Crystal structures show that four p53 proteins self-assemble on two DNA palindromic sequences (which thus represent the two half-sites for protein binding) to form a tetramer.17 One half-site bound to two p53 proteins is shown in Fig. 1a, where a key C277 residue is nestled into the major groove and forms H-bonds with the proximal purines.18 Indeed, it was demonstrated that modification of C277 at the DNA-binding interface can regulate p53 cellular activity in response to environmental redox signals.19 Selective alkylation of C277 by 2-sulfonylpyrimidines helps to stabilize p53 mutants and is especially potent at killing cancer cells.20
Fig. 1.

Protein-DNA models used in the molecular dynamics (MD) simulation of the p53 protein complexed with the Gadd45 and p21 half-site sequences. a) Two p53 proteins (labeled ‘top’ and ‘bottom’) bound to a half-site DNA sequence (PDB ID 2ADY). b) Original DNA sequence (in blue) in the 2ADY structure and bp replacements producing the p21 and Gadd45 model sequences (see Fig. 3 of Ref. 12). The DNA decamer usually consists of the sequence indicated in purple, where R denotes a purine nucleobase (G or A), W is either A or T, and Y is a pyrimidine nucleobase (C or T). The yellow and orange circles indicate the locations of the DNA binding to the key Cys residue (C277) in the top and bottom protein, respectively. Key differences between the Gadd45 and p21 bp sequences in the proximity of the C277 residues are at positions R3 and Y1.
Near each redox-active C277 residue, the Gadd45 and p21 consensus sequences (see Fig. 1b) only differ by one bp, which is guanine-cytosine (GC) in Gadd45 and adenine-thymine (AT) in p21. This difference plays a critical role in the interpretation of the differential response of p53 to the presence of an electron hole, produced by oxidative stress, in the p21 and Gadd45 sequences. In fact, since (i) the oxidation potential landscape of DNA favors hole residence on guanines and (ii) the oxidation potentials of G and Cys are 1.29 V and 0.9 V, respectively (thus enabling energetically downhill HT to Cys), the presence of G near C277 drives DNA-to-p53 HT, with consequent disulfide bond formation and DNA-protein dissociation.9, 18 Yet, kinetic validation of this mechanism has not been provided. From a dynamic perspective, the probability of HT to Cys depends on two factors: how frequently the low-redox potential nucleobase aligns with the DNA major groove-p53 interface,9 and the competition between the time scales of the HT through the bp at position R3 or Y1 and the HT to Cys. Here, we used MD simulations to investigate the first factor. Then, we performed electronic structure and kinetic analysis on MD snapshots to study the second factor, which touches unresolved issues in the understanding of DNA charge transport, including the localization/delocalization of the transferring charge.21 The G proximal to C277 can speed up22 the charge transit through the DNA in Gadd45 compared to p21. This faster charge conduction competes with the possibility that the hole transiently localized on G transfers to Cys. Some models of thermally induced hole hopping through DNA22 predict that, in p21 DNA, the hole should jump between G bases at positions R1 and C (we focus the analysis on this part of the palindromic sequence) via superexchange. However, can molecular motion bring one of the two G bases sufficiently close to C277 to enable direct HT? May HT from one of the G bases to Cys compete with superexchange between the guanines separated by two bps (Fig. 1b)? Depending on the DNA-p53 conformation and on the polarization of the local environment, may an A nucleobase be transiently occupied by the hole and transfer it to the Cys? In this case, A-to-Cys HT would be even more downhill, thermodynamically, than G-to-Cys HT. Considering the strong hole localization in DNA that emerges from previous studies,23 may the HT through DNA be treated as sequential hopping between bps? These questions highlight the complications of establishing a relation between DNA sequence and p53 response to DNA oxidation in a dynamic perspective. Here, we explain this relation by comparative analysis of the HT steps to C277 and through the DNA, after calculating the parameters that describe all relevant CT steps within the framework of Marcus theory.24 We find, in terms of CT dynamics, that a single bp difference in the p21 and Gadd45 sequences can modulate the HT from the oxidized DNA to C277 and thus can influence the functionally relevant dissociation of the p53-DNA complex, in good agreement with experimental results.8, 9, 18
The systems studied comprise two p53 proteins (the ‘top’ and ‘bottom’ molecules in Fig. 1) bound to the Gadd45 or the p21 sequence. To build these systems, we made substitutions in the original DNA sequence from the PDB structure 2ADY17 to obtain models of the p21 and p53 sequences that match the sequences used in the experiments of Ref. 8 in the Cys binding regions (Fig. 1b). The conformational dynamics of the resulting protein-DNA complexes was simulated using classical MD (see details in ESI), with MD production runs of 40 ns. Both systems were well equilibrated after 10 ns, and the RMSDs (Fig. S1) show similar structural fluctuations for the human p53-Gadd45 DNA and p53-p21 DNA complexes.
We extracted one MD snapshot every 2.5 ns in the 10–40 ns time window to calculate the CT parameters involved in the HT steps illustrated in Fig. 2. The rates associated with these steps were described using the nonadiabatic rate expression24
| (1) |
The CT parameters in eqn. (1) are the electronic coupling, VIF, between the initial (I) and final (F) diabatic (localized) electronic states, the reaction free energy ΔG°, and the reorganization energy λ. We calculated VIF for portions of the p53-DNA interface containing the methanethiol moiety and the relevant bp, using a DFT implementation (with the M11 functional25 and the 6–31g** basis set) of the formula26–28
| (2) |
(a and b are the coefficients of the ground state expansion on the diabatic states, SIF is the overlap of these states, and ΔEIF is their energy difference; ΔEv is the vertical energy gap between the adiabatic ground and first-excited states). The ΔG° values were estimated as differences between the experimental oxidation potentials of the charge donor and acceptor. The reorganization energies were evaluated combining Marcus’ expression for λ24 with DFT analysis (see ESI).
Fig. 2.

HT models for the contacts of p53 with (a) Gadd45 and (b) p21 DNA sequences. The arrows denote the possible HT steps. The HT rate constant values (in s–1) refer to the top (black) and bottom (purple) contacts. We assume that protein and DNA unbind after HT to Cys. Thus, the HT steps from Cys are not shown. The DNA sequence runs from the 5’ (left) to the 3’ (right) end in both top and bottom contact models.
The mean-square electronic coupling over the selected MD snapshots, , for each redox pair in Fig. 2 is reported in Table 1. The Gadd45 DNA sequence has similar coupling to the top and bottom proteins, as is expected from the similarity of the two p53-DNA contacts. In contrast, the p21 model sequence is more strongly coupled to the Cys in the bottom protein than to the one in the top protein. This difference may also be a consequence of our modeling. The bp triplet near the upper Cys is one bp away from the edge of the DNA model sequences, while two extra bps were maintained on the side of the bp triplet in contact with the lower Cys (cf. Fig. 1 and 2). This choice was made to minimize the changes to the actual DNA sequence that is in complex with the p53 proteins in the 2ADY structure, and to explore the extension of the DNA sequence on the sides of the contact bp triplet needed to assure the structural stability of the p53-DNA contact. Our findings indicate that including only one extra bp in the DNA sequence complexed with the proteins is a borderline modeling choice that was sufficient to constrain the DNA-protein contact geometry in the case of Gadd45 but failed to do so in the case of p21. Table 2 shows that the ranges of bp-Cys distances (see Section S3) for the three bps in Fig. 2b, at the interfaces of p21 DNA model with the top and bottom proteins. These distances correlate very well with the couplings in Table 1, thus also supporting the robustness of our method for calculating couplings. We note that, although the DNA structural fluctuations can be reduced by the presence of the proteins, the average values of the electronic couplings between adjacent bps are similar to the expectations29 for free DNA. For the p21 DNA-bottom p53 contact, e.g., we obtain 〈VIF〉 values of 43, 2, 58 and 13 meV for AT1-AT2, GC-CG, AT-GC and AT2-CG, respectively.
Table 1.
Values of mean-square electronic coupling, , in eV2 and coherence parameter, C, for each redox pair in Fig. 2, at the top and bottom p53-Gadd45 DNA and p53-p21 DNA contacts.
| DNA | protein | redox pair | 〈VIF2〉 | C |
|---|---|---|---|---|
| Gadd45 gene model | Top | AT-Cys | 2.32 × 10−5 | 0.34 |
| GC-Cys | 2.39 × 10−3 | 0.19 | ||
| CG-Cys | 4.56 × 10−5 | 0.36 | ||
| AT-GC | 2.92 × 10−3 | 0.56 | ||
| GC-CG | 9.16 × 10−6 | 0.33 | ||
| Bottom | AT-Cys | 5.30 × 10−5 | 0.32 | |
| GC-Cys | 7.75 × 10−3 | 0.35 | ||
| CG-Cys | 2.50 × 10−4 | 0.50 | ||
| AT-GC | 6.21 × 10−3 | 0.54 | ||
| GC-CG | 1.64 × 10−5 | 0.26 | ||
| p21 gene model | Top | AT1-Cys | 1.08 × 10−6 | 0.48 |
| AT2-Cys | 6.34 × 10−9 | 0.33 | ||
| CG-Cys | 6.45 × 10−7 | 0.52 | ||
| AT1-AT2 | 1.82 × 10−3 | 0.66 | ||
| AT2-CG | 2.00 × 10−3 | 0.38 | ||
| Bottom | AT1-Cys | 4.29 × 10−5 | 0.17 | |
| AT2-Cys | 1.52 × 10−5 | 0.32 | ||
| CG-Cys | 1.39 × 10−4 | 0.39 | ||
| AT1-AT2 | 2.75 × 10−3 | 0.66 | ||
| AT2-CG | 2.36 × 10−4 | 0.76 | ||
Table 2.
Ranges of distances (in Å) spanned by the indicated bp-Cys pairs, over the selected MD snapshots, at the DNA contacts with the top and bottom proteins.
| p21 DNA | AT1-Cys | AT2-Cys | CG-Cys |
|---|---|---|---|
| top | 7.37–9.34 | 9.29–11.66 | 8.50–10.28 |
| bottom | 5.70–8.43 | 7.55–9.82 | 5.73–9.61 |
Table 1 also shows the values of the coherence parameter30 , which is a descriptor of the coupling sensitivity to structural fluctuations: C ≅ 1 for very rigid systems; C ≅ 0 for very flexible systems with strong dependence of the coupling on the atomic conformation. The C values in Table 1 indicate that the electronic couplings are rather sensitive to the fluctuations of the protein-DNA contact regions. Moreover, a comparison of the bottom panel in Table 1 with Table 2 shows that the C value need not correlate strictly with the p53-DNA distance in this complex system. The closer contact between the p21 sequence and the bottom protein, compared to the top protein, seems to enable a broader range of coupling values that depend on subtleties of the contact (including the bp-Cys distance, the molecular orientations, etc.), thus leading to smaller C values.
Irrespective of whether we consider the top or bottom DNA-protein contact in the system of Fig. 1a, Table 1 shows that the intermediate bp in the contact bp triplet is much more strongly coupled to Cys, and more weakly coupled to the next bp, in the protein complex with the Gadd45 sequence than in the complex with the p21 sequence. The relative differences in the electronic couplings are accentuated in the HT rates (Fig. 2), favoring a slower transit of the charge across the bp triplet and its transition to C277 in the Gadd45 system compared to p21. In particular, in the complex of p53 with the Gadd45 sequence, the CT rates computed for HT to CG and C277 are approximately in the ranges 103-104 s–1 and 107-108 s–1, respectively, while the ranges 105-107 s–1 and 103-107 s–1 were computed for the p21 system. Note that the predicted HT is faster between AT2 and CG than between GC and CG because of both a larger electronic coupling and a negative reaction free energy (see Section S4). Thus, considering the connections among Cys oxidation, disulfide bridge formation, and protein dissociation from DNA, the HT rates in Fig. 2 demonstrate, in kinetic terms, the higher propensity of p53 to dissociate from the Gadd45 DNA sequence than from the p21 sequence in the presence of an electron hole generated by oxidative stress. This conclusion can be clearly stated without constructing a specific kinetic model. Constructing such a model accurately would require taking into account the probability for the hole to tunnel through both AT1 and AT2, rather than transiently residing in AT2, in the system of Fig. 2b, while the transferring charge is expected to transiently localize on the guanine close to C277 (Fig. 2a) in the Gadd45 sequence. Therefore, the use of a detailed kinetic model would further strengthen the conclusions of this study. Importantly, our MD simulations find that the three bps in the HT models of Fig. 2 remain close to the two C277 residues during the evolution of the protein-DNA complex. Therefore, HT to the protein from purine-containing bps outside of the contact bp triplet could not compete with HT by superexchange through AT1–AT2 and thus favor HT to the protein in the complex with p21 DNA.
In conclusion, this study provides an explanation, in terms of protein-DNA CT dynamics, of the experimental finding7–9 that DNA-to-protein HT can cause the selective binding of tumor suppressor protein p53 to p21 DNA, in contrast to its dissociation from Gadd45 DNA, under oxidative stress. We show that the intimate relationship between p53 activation/function (contributing to genome stability) and the cell redox environment is a matter of kinetic competition between HT through DNA and HT from the DNA to residue C277, which enables the sequence selectivity of the p53 binding to different promoters.
The implications of our findings may be extended to related proteins that contain redox-active residues in their DNA-binding domains and should be considered in strategies to prevent p53 aggregation by the formation of disulfide bridges.31 As well, the kinetic competition of HT processes at the p53-DNA interface described here suggests a potential role for genome engineering tools such as CRISPR-Cas932, 33 to edit the bp sequence precisely and hence to modulate the relative efficiency of the competing CT pathways determining the p53 regulatory function.
Supplementary Material
Acknowledgements
We acknowledge use of the Duke Compute Cluster and support of our research by the National Institutes of Health (Grant GM-48043).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R, Khokhar AR and Kuang J, Oncogene, 2005, 24, 2929–2943. [DOI] [PubMed] [Google Scholar]
- 2.Abbas T and Dutta A, Nat. Rev. Cancer, 2009, 9, 400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niehrs C and Schäfer A, Trends Cell Biol., 2012, 22, 220–227. [DOI] [PubMed] [Google Scholar]
- 4.Barreto G, Schäfer A, Marhold J, Stach D, Swaminathan SK, Handa V, Döderlein G, Maltry N, Wu W, Lyko F and Niehrs C, Nature, 2007, 445, 671–675. [DOI] [PubMed] [Google Scholar]
- 5.Schieber M and Chandel Navdeep S., Curr. Biol, 2014, 24, R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genereux JC, Boal AK and Barton JK, J. Am. Chem. Soc, 2010, 132, 891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buzek J, Latonen L, Kurki S, Peltonen K and Laiho M, Nucleic Acids Res., 2002, 30, 2340–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augustyn KE, Merino EJ and Barton JK, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 18907–18912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer KN and Barton JK, Biochemistry, 2014, 53, 3467–3475. [DOI] [PubMed] [Google Scholar]
- 10.Takada T and Barton JK, J. Am. Chem. Soc, 2005, 127, 12204–12205. [DOI] [PubMed] [Google Scholar]
- 11.Chin PL, Momand J and Pfeifer GP, Oncogene, 1997, 15, 87–99. [DOI] [PubMed] [Google Scholar]
- 12.Kannan K and Jain SK, Pathophysiology, 2000, 7, 153–163. [DOI] [PubMed] [Google Scholar]
- 13.Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novák B and Bakal C, Nat. Commun, 2017, 8, 14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW and Vogelstein B, Nat. Genet, 1992, 1, 45–49. [DOI] [PubMed] [Google Scholar]
- 15.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and Vogelstein B, Cell, 1993, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- 16.Kearns S, Lurz R, Orlova EV and Okorokov AL, Nucleic Acids Res, 2016, 44, 6185–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE and Shakked Z, Mol. Cell, 2006, 22, 741–753. [DOI] [PubMed] [Google Scholar]
- 18.Schaefer KN, Geil WM, Sweredoski MJ, Moradian A, Hess S and Barton JK, Biochemistry, 2015, 54, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaar JL, Basse N, Joerger AC, Stephens E, Rutherford TJ and Fersht AR, Protein Sci, 2010, 19, 2267–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bauer MR, Joerger AC and Fersht AR, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, E5271–E5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renaud N, Berlin YA, Lewis FD and Ratner MA, J. Am. Chem. Soc, 2013, 135, 3953–3963. [DOI] [PubMed] [Google Scholar]
- 22.Bixon M and Jortner J, J. Am. Chem. Soc, 2001, 123, 12556–12567. [DOI] [PubMed] [Google Scholar]
- 23.Uskov DB and Burin AL, Phys Rev B, 2008, 78, 073106. [Google Scholar]
- 24.Kuznetsov AM and Ulstrup J, Electron Transfer in Chemistry and Biology: An Introduction to the Theory, John Wiley & Sons, New York, 1999. [Google Scholar]
- 25.Peverati R and Truhlar DG, J. Phys. Chem. Lett, 2011, 2, 2810–2817. [Google Scholar]
- 26.Migliore A, J. Chem. Phys, 2009, 131, 114113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Migliore A, Chem J Theory Comput, 2011, 7, 1712–1725. [DOI] [PubMed] [Google Scholar]
- 28.Teo RD, Terai K, Migliore A and Beratan DN, Phys. Chem. Chem. Phys, 2018, 20, 26063–26067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voityuk AA, J. Chem. Phys, 2008, 128, 115101. [DOI] [PubMed] [Google Scholar]
- 30.Balabin IA and Onuchic JN, Science, 2000, 290, 114–117. [DOI] [PubMed] [Google Scholar]
- 31.Wiman KG, Oncogene, 2010, 29, 4245. [DOI] [PubMed] [Google Scholar]
- 32.Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA and Zhang F, Science, 2013, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE and Church GM, Science, 2013, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.