

In this study, Janssen et al. investigated whether differential histone-modifying activities are required for DSB repair in euchromatin and heterochromatin domains. They used locus-specific DSB induction in Drosophila animal tissues and cultured cells to more deeply interrogate the impact of dKDM4A on chromatin changes, temporal progression, and pathway utilization during DSB repair and found that distinct pre-existing chromatin states require specialized epigenetic alterations to ensure proper DSB repair.
Keywords: Drosophila, H3K56me3, H3K9me3, dKDM4A, double-strand breaks, euchromatin, heterochromatin, histone demethylation, homologous recombination, repair pathway choice
Abstract
Repair of DNA double-strand breaks (DSBs) must be orchestrated properly within diverse chromatin domains in order to maintain genetic stability. Euchromatin and heterochromatin domains display major differences in histone modifications, biophysical properties, and spatiotemporal dynamics of DSB repair. However, it is unclear whether differential histone-modifying activities are required for DSB repair in these distinct domains. We showed previously that the Drosophila melanogaster KDM4A (dKDM4A) histone demethylase is required for heterochromatic DSB mobility. Here we used locus-specific DSB induction in Drosophila animal tissues and cultured cells to more deeply interrogate the impact of dKDM4A on chromatin changes, temporal progression, and pathway utilization during DSB repair. We found that dKDM4A promotes the demethylation of heterochromatin-associated histone marks at DSBs in heterochromatin but not euchromatin. Most importantly, we demonstrate that dKDM4A is required to complete DSB repair in a timely manner and regulate the relative utilization of homologous recombination (HR) and nonhomologous end-joining (NHEJ) repair pathways but exclusively for heterochromatic DSBs. We conclude that the temporal kinetics and pathway utilization during heterochromatic DSB repair depend on dKDM4A-dependent demethylation of heterochromatic histone marks. Thus, distinct pre-existing chromatin states require specialized epigenetic alterations to ensure proper DSB repair.
One of the most harmful DNA lesions is a double-strand break (DSB), the improper repair of which can lead to formation of aberrant chromosomes linked to cancer and developmental diseases (Roukos and Misteli 2014). At DSBs, the severed strands of the DNA helix can be repaired by a variety of mechanisms, but the two major DSB repair pathways are nonhomologous end joining (NHEJ) and homologous recombination (HR). NHEJ repairs DNA by ligating both ends of the DSB together, often resulting in small insertions and deletions (indels) at the break site. HR repair involves more extensive processing of the DSB site, in which 5′-to-3′ end resection of the DSB ends produces ssDNA that invades and perfectly copies homologous sequences to repair the DSB site (Ciccia and Elledge 2010). Less understood is how these pathways efficiently repair DSBs within the wide spectrum of chromatin environments in the nucleus.
Constitutive heterochromatin is riddled with repetitive DNA sequences, associated with transcriptional repression, and predominantly localizes to pericentromeric and telomeric regions of individual chromosomes. These domains are enriched for histone H3 Lys9 dimethylation and trimethylation (H3K9me2/me3) and its cognate “reader,” heterochromatin protein 1 (HP1) (Allshire and Madhani 2018). Other histone modifications, such as H3K56me2/me3 (Jack et al. 2013), are also highly enriched in heterochromatin.
Erroneous recombination between a DSB and the highly abundant homologous repetitive sequences in heterochromatin located on nonhomologous chromosomes can lead to harmful aberrant chromosomal structures, such as dicentrics and acentrics. Heterochromatic DSBs strikingly move outside the heterochromatin domain during repair, which likely prevents erroneous recombination and instead promotes “safe” repair from sister chromatids or homologous chromosomes (Chiolo et al. 2011; Janssen et al. 2016, 2018). Despite observing similar repair kinetics and pathway utilization for DSBs induced in euchromatin and heterochromatin in Drosophila (Janssen et al. 2016), stark differences in chromatin composition, biophysical properties (Larson et al. 2017; Strom et al. 2017), and DSB dynamics (Chiolo et al. 2011) suggest that different chromatin-modifying activities are required to repair euchromatic and heterochromatic DSBs. Several heterochromatin-enriched proteins, such as Kap1 (Goodarzi et al. 2008), SMC5/6, and HP1a (Chiolo et al. 2011), are required to ensure proper DSB repair in heterochromatin, yet the impact of histone-modifying activities remains unclear.
We previously identified the Drosophila histone demethylase dKDM4A (dJMJD2) (Lloret-Llinares et al. 2008) as a heterochromatin-enriched protein required for normal heterochromatin structure and function (Colmenares et al. 2017). dKDM4A's enzymatic activity is not required for heterochromatin maintenance but is important for movement of DSBs to the heterochromatin periphery (Colmenares et al. 2017). However, whether this reflects a more general role of dKDM4A in DSB repair in both euchromatin and heterochromatin remains unknown. dKDM4A directly binds HP1a (Lin et al. 2008) and is part of the jumonji family of Fe(II)- and α-ketoglutarate-dependent lysine demethylases (Whetstine et al. 2006). This demethylase family plays important roles in mammalian DNA damage repair (Mallette et al. 2012; Khoury-Haddad et al. 2014; Awwad and Ayoub 2015) and development (Nottke et al. 2009), and its members are often overexpressed in cancer (Black et al. 2013). dKDM4A is structurally most homologous to human KDM4D, since both contain the enzymatic JmjN and JmjC domains but lack the PHD and Tudor domains found in human KDM4A-C (Lloret-Llinares et al. 2008). dKDM4A demethylates H3K36me2/me3 in vivo as well as in vitro (Lin et al. 2008; Lloret-Llinares et al. 2008; Crona et al. 2013). However, dKDM4A depletion results in no observable changes in H3K36me3 levels in heterochromatin, indicating that dKDM4A-mediated demethylation of H3K36 is specific for euchromatic sites (Colmenares et al. 2017). Loss of dKDM4A also results in increased levels of two heterochromatic methyl marks in vivo (H3K9me2/me3 and H3K56me2/me3) (Lloret-Llinares et al. 2008; Tsurumi et al. 2013; Colmenares et al. 2017), but demethylation of these marks by dKDM4A has not been observed in vitro (Lin et al. 2008). Thus, the impact of dKDM4A on H3K9me3 and H3K56me3 demethylation in vivo may be indirect or requires additional factors not present in purified systems.
Using our previously developed single-DSB systems in Drosophila animals (Janssen et al. 2016), we report here that pericentromeric heterochromatin, but not euchromatin, exhibits dKDM4A-dependent demethylation of H3K9me3 and H3K56me3 specifically at sites of DSBs. In addition, dKDM4A loss in Drosophila animals does not affect euchromatic DSB repair, whereas completion of heterochromatic DSB repair is significantly delayed. Strikingly, sequence analysis of DSB repair products, synthetic lethality assays, and live imaging of HR repair proteins after CRISPR/Cas9-mediated cleavage of heterochromatic repeats reveals that depletion of dKDM4A results in a heterochromatin-specific increase in the ratio of HR:NHEJ repair events, concomitant with increased HR protein recruitment to DSBs. This relative increase in HR protein recruitment can be rescued by transiently inhibiting H3K9 methyltransferase activity during DSB induction, indicating that dKDM4A functions to counter H3K9me2/me3 at the DSB site. Together, our results establish that DSBs in heterochromatin, but not in euchromatin, require specific DSB-induced dKDM4A-dependent chromatin changes to promote timely DSB repair progression and NHEJ repair pathway choice.
Results
dKDM4A promotes monomethylation of H3K9 and H3K56 (H3K9me1 and H3K56me1) specifically at heterochromatic DSBs
To analyze the specific chromatin changes that occur at either euchromatic or heterochromatic DSBs, we used our previously developed single-break systems (DR-white) in Drosophila animals (Janssen et al. 2016). DR-white is integrated at specific euchromatic and heterochromatic sites and contains a recognition site for the DSB-inducing I-SceI endonuclease (Fig. 1A; Do et al. 2014).
Figure 1.

Increased H3K9me1 and H3K56me1 at heterochromatic DSBs is dependent on dKDM4A. (A) Schematic overview of the DR-white single-DSB system (Janssen et al. 2016) integrated in seven different fly lines in either euchromatic sites (EC; black; 1x chromosome 2 [2eu_1] or 1x chromosome 3 [3eu_1]) or heterochromatic sites (HC; blue; 2x chromosome 2 [2het_1 and 2het_2], and 3x chromosome 3 [3het_1, 3het_2, and 3het_3]). The I-SceI cut site in the DR-white construct is targeted for induction of a single DSB by the I-SceI endonuclease. (B) Schematic of ChIP (chromatin immunoprecipitation) experiments (results are shown in C–F). Third instar larvae with DR-white integrations in the presence or absence (control) of the hsp.I-SceI transgene were heat-shocked for 1 h at 37°C. Chromatin was prepared from larvae harvested 6–9 h after heat shock and subjected to ChIP and quantitative PCR (ChIP-qPCR) using a primer set 1.4 kb downstream from the I-SceI cut site. Relative enrichment over input was calculated for each ChIP sample. The relative increase after DSB induction (fold change) equals ChIP enrichment levels in hsp.I-SceI-expressing larvae (+DSB) divided by ChIP levels in larvae not expressing hsp.I-SceI (no DSB). (C–F) ChIP-qPCR analysis of the fold change in H3K9me1 (C,E) and H3K56me1 (D,F) levels observed upon single DSB induction at DR-white sites in euchromatin (black) and heterochromatin (blue) in wild-type (+) or homozygous dKDM4A deletion mutant (ΔdKDM4A) larvae. The dotted line indicates “no change” in the level of the respective marks between samples with and without a DSB. (n.s.) P-value ≥ 0.05; (*) P-value < 0.05; (**) P-value < 0.01, t-test, unpaired. Averages are shown for n ≥ 5 samples per condition +SEM.
We first focused on assessing changes in histone modifications enriched in heterochromatin (H3K9me2/me3 and H3K56me2/me3) (Jack et al. 2013; Allshire and Madhani 2018). ChIP-qPCR (chromatin immunoprecipitation [ChIP] followed by quantitative PCR [qPCR]) was performed on extracts from third instar larval tissue following heat shock-inducible expression of I-SceI (Fig. 1B). We observed an increase in H3K9me3 at both euchromatic and heterochromatic DBS sites (Supplemental Fig. S1A), which reproduces the H3K9me3 enrichments found by others at both euchromatic and heterochromatic DSB sites (Ayrapetov et al. 2014; Tsouroula et al. 2016). All antibodies used for ChIP in this study show selectivity for their respective modification when compared with other histone H3 methylation marks (Supplemental Fig. S2). However, using stringent validation methods (see the Materials and Methods; Shah et al. 2018), we were not able to identify any H3K9me3 antibodies that displayed high ChIP efficiency and selectivity (based on testing of 13 H3K9me3 antibodies) (Supplemental Fig. S2A; M. Keogh [Epicypher], pers. comm.). Nevertheless, both H3K9me3 antibodies used in this study do show higher specificity for H3K9me3 over other H3 methylation marks (Supplemental Fig. S2), and others have observed increased H3K9me3 levels at both euchromatic and heterochromatic DSBs (Ayrapetov et al. 2014; Tsouroula et al. 2016), supporting the validity of the results. The increased H3K9me3 levels at DSBs in mammals have been attributed to the methyltransferase activity of Suv39h1 (Ayrapetov et al. 2014). In addition, we identified an increase in H3K56me2 at the majority of euchromatic and heterochromatic DSBs (Supplemental Fig. S1B). However, the relative increases in H3K9me3 and H3K56me2 were similar for euchromatic and heterochromatic DSBs (Supplemental Fig. S1A,B); thus, these modifications increase at DSBs regardless of the pre-existing chromatin environment.
In contrast, ChIP-qPCR analyses revealed that H3K9me1 and H3K56me1 levels only increased at heterochromatic DSBs (1.2-fold to 1.5-fold and 1.3-fold to 1.6-fold compared with undamaged sites, respectively), suggesting that specific chromatin-modifying activities could play a role at heterochromatic DSBs (Fig. 1C,D; see Supplemental Fig. S1C,D for relative enrichment over input). Importantly, the H3K9me1 antibodies used in this study showed high selectivity and efficiency (Supplemental Fig. S2B). dKDM4A promotes H3K9me2/me3 and H3K56me2/me3 demethylation in vivo (Lloret-Llinares et al. 2008; Tsurumi et al. 2013; Colmenares et al. 2017), and we previously observed dKDM4A-dependent reductions in bulk H3K56me3 levels after irradiation of cultured Drosophila cells (Colmenares et al. 2017). Thus, we hypothesized that dKDM4A could be responsible for the increased H3K9me1 and H3K56me1 levels after single-DSB induction. Indeed, dKDM4A deletion mutants (ΔdKDM4A) (Crona et al. 2013) exhibited a significant decrease in the accumulation of H3K9me1 at heterochromatic DSBs—from 1.2-fold to 1.6-fold enrichment in wild type to no enrichment in ΔdKDM4A (Fig. 1E). Similarly, H3K56me1 at heterochromatic DSB sites decreased significantly in the absence of dKDM4A (Fig. 1F), whereas H3K9me1 and H3K56me1 levels at euchromatic sites were unaffected (Fig. 1E,F). Loss of dKDM4A did not alter the levels of the DSB-associated phosphorylation of H2Av (γH2Av, equivalent to γH2AX in mammals), one of the first markers for DNA damage (Supplemental Fig. S1E). Importantly, depletion of histone H3 (observed previously in budding yeast) (Hauer et al. 2017) was not visible at heterochromatic DSBs compared with undamaged DNA in the presence or absence of dKDM4A (Supplemental Fig. S1F), indicating that loss of histone H3 at these single DSBs is unlikely to play a role in the observed modification changes. Thus, the observed decreases in H3K9me1 and H3K56me1 upon dKDM4A depletion are not due to a defect in upstream DNA damage signaling pathways or loss of H3 at DSBs.
In other systems, increased H3K36me3 levels are associated with changes in DSB repair pathway choice (Aymard et al. 2014; Jha et al. 2014), and dKDM4A can demethylate H3K36me3 in vitro (Lin et al. 2008). However, our previous analysis demonstrated that dKDM4A depletion had no effect on the normally very low levels of H3K36me3 in bulk heterochromatin (Colmenares et al. 2017). Indeed, direct assessment by ChIP-qPCR analysis using validated antibodies (Supplemental Fig. S2C,D) revealed no dKDM4A-dependent changes in the levels of H3K36me3 or H3K36me1 at euchromatic and heterochromatic DR-white insertions in the presence (Supplemental Fig. S1G,H) or absence (Supplemental Fig. S3A) of DSBs. Thus, we found no evidence for dKDM4A-mediated H3K36 demethylation at heterochromatic DSBs in Drosophila, although transient dKDM4A-independent changes in H3K36 methylation status cannot be ruled out (Supplemental Fig. S1G,H). Finally, H3K9me1/me3 and H3K56me1/me2 levels at undamaged euchromatic and heterochromatic sites were not affected by the absence of dKDM4A, indicating that changes in H3K9 and H3K56 methylation specifically result from DSBs at DR-white integrations (Supplemental Fig. S3B,C).
Our results indicate that increased H3K9/K56 methylation occurs at both euchromatic and heterochromatic DSBs (Supplemental Fig. S1A). In contrast, dKDM4A-dependent demethylation of H3K9me2/me3 and H3K56me2/me3 occurs only at heterochromatic DSBs, not euchromatic DSBs, and thereby results in increased enrichments for H3K9me1 and H3K56me1 (Fig. 1C–F).
dKDM4A ensures timely DSB repair in heterochromatin, not euchromatin
The dKDM4A-dependent histone methylation changes observed only at heterochromatic DSBs led us to hypothesize that this epigenetic modifier may specifically impact repair of damaged DNA in heterochromatin. Therefore, we analyzed DSB repair kinetics in wild-type and ΔdKDM4A larval tissue by timing the appearance and disappearance of DNA damage foci, visualized via live imaging of fluorescently tagged Mu2 (MDC1 in mammals), a γH2Av-binding protein. Analysis was performed on third instar larval wing and leg discs containing a heterochromatic or euchromatic DR-white insertion and trimethoprim-inducible ecDHFR-I-SceI expression (Fig. 2; Janssen et al. 2016). Strikingly, loss of dKDM4A resulted in a significant delay in Mu2 disappearance at heterochromatic DSBs when compared with wild-type tissue (Fig. 2A,B). Fifty percent of Mu2 foci at single heterochromatic DBSs in wild-type tissue disappeared ∼60 min after appearance (Fig. 2B, light-blue line), compared with ∼190 min in homozygous ΔdKDM4A tissues (Fig. 2B, dark-blue line). This significant delay in repair kinetics (approximately three times) in ΔdKDM4A tissue was confirmed with a second heterochromatic DR-white insertion (Supplemental Fig. S4A). Importantly, the ΔdKDM4A repair delay was rescued by expression of a dKDM4A wild-type transgene (Fig. 2B, red line), ruling out effects of unrelated mutations present in the ΔdKDM4A animals. In contrast, the kinetics of DSB repair at a euchromatic DR-white insertion were not significantly affected by the presence (Fig. 2C, grey line) or absence (Fig. 2C, black line) of dKDM4A. We conclude that dKDM4A is required for timely DSB repair specifically in heterochromatin and not in euchromatin. The delay is likely restricted to heterochromatic DSBs (Fig. 2B,C; Supplemental Fig. S4A) because dKDM4A is enriched in heterochromatin through direct binding to HP1a. Indeed, in contrast to a wild-type transgene, a dKDM4A transgene with a mutation in the PxVxL motif (V423A) that abrogates HP1 binding (Colmenares et al. 2017) is unable to rescue DSB relocalization defects upon loss of endogenous dKDM4A in cells in culture (Supplemental Fig. S4B). This indicates that the specificity of dKDM4A for heterochromatin repair is dependent on its heterochromatin-specific enrichment.
Figure 2.

Timely repair of heterochromatic DSBs depends on the histone demethylase dKDM4A. (A) Induced expression of ecDHFR-I-SceI (stabilized by the presence of trimethoprim) (Janssen et al. 2016) and eYFP-tagged Mu2 (DSB protein) in DR-white larval tissue allows visualization and tracking of DSBs over time. Representative images of Mu2-eYFP dynamics at heterochromatic (HC; 3het_3) DSBs in larval cells in the presence (wild type; top) or absence (ΔdKDM4A; homozygous mutant; bottom) of endogenous dKDM4A. (B,C) Time-lapse analysis of single Mu2-YFP focus disappearance (minutes from appearance, as depicted in A) in third instar larval discs with an insertion of DR-white in either heterochromatic sites (HC; 3het_3; blue; B) or euchromatic sites (EC; 3eu_1; black; C). Cells were imaged in wild-type (light line), dKDM4A homozygous mutant (ΔdKDM4A; dark line), or dKDM4A homozygous mutant larval tissue in the presence of a wild-type dKDM4A transgene (ΔdKDM4A + dKDM4A-WT; red line). The time point of Mu2 focus appearance was set at t = 0 for each individual focus. (n) The number of cells with a single Mu2 focus imaged for the indicated conditions. (n.s.) P-value ≥ 0.1; (**) P-value < 0.0001, log-rank (Mantel-Cox) test.
dKDM4A mutants depend on intact HR repair, but not NHEJ, for their viability
We found previously that loss of dKDM4A resulted in synthetic lethality when combined with mutations in the DNA damage response kinase ATR or Mu2, demonstrating that dKDM4A mutant flies depend on canonical DNA damage repair signaling for their survival (Colmenares et al. 2017). To get more insight into the role of dKDM4A in heterochromatin DSB repair, we determined whether survival of dKDM4A mutant flies depends on the presence of proteins required for specific DSB repair pathways. Interestingly, we found that homozygous dKDM4A mutants display significantly elevated levels of synthetic lethality (relative to a wild-type control) in combination with loss or depletion of HR-specific repair proteins required for resection (Tosca/Exo1, dCtIP, or dBLM) or homology search and strand invasion (dRad51) (Fig. 3A). The dKDM4A mutant viability ranged from 0% in the absence of CtIP to 79% in the presence of one of the dRad51 mutants. In contrast, dKDM4A mutants were mostly viable and fertile when combined with depletion for NHEJ repair proteins, including dKu70 and dKu80 RNAi or a ligase 4 mutant (126%, 91%, and 88% viability, respectively, when compared with single mutants for dKDM4A or the repair protein) (Fig. 3A). We conclude that survival of dKDM4A mutant flies specifically depends on the presence of HR repair proteins and not NHEJ proteins.
Figure 3.

Loss of dKDM4A leads to increased usage of HR at heterochromatic DSBs. (A) Quantification of the number of offspring resulting from crosses between dKDM4A mutant flies and the indicated repair mutants. (n.s.) P-value ≥ 0.05; (*) P-value < 0.05; (**) P-value < 0.01, unpaired t-test. Numbers indicate the percentages of offspring. (B,C) DR-white PCR products from larval genomic DNA with the indicated genotypes were analyzed using Sanger sequencing following trimethoprim-induced stabilization of ecDHFR-I-SceI throughout larval development (∼4–5 d). Sequence analysis was performed using TIDE (tracking of indels by decomposition) (Brinkman et al. 2014) to determine the percentage of HR (intact white gene) and NHEJ (small indels) products relative to the total amount of repair products identified. (n.s.) P-value ≥ 0.05; (*) P-value < 0.05; (**) P-value < 0.01, unpaired t-test. Averages + SEM are plotted for n ≥ 5 larvae per condition.
dKDM4A loss results in increased HR usage at heterochromatic DSBs
The dKDM4A-dependent changes in chromatin and repair kinetics specific to heterochromatic DSBs and the dependency of dKDM4A mutant flies on an intact HR repair pathway suggest that dKDM4A could regulate DSB repair pathway utilization only in heterochromatin. We specifically hypothesized that dKDM4A could be required for proper NHEJ repair of heterochromatin breaks and that its loss results in an increased dependency on HR repair. To test this hypothesis, we sequenced repair products from euchromatic and heterochromatic DR-white insertion sites after I-SceI expression in third instar larvae (Fig. 3B; Janssen et al. 2016). Strikingly, loss of dKDM4A significantly increased the proportion of HR repair products only at heterochromatic DSBs, compared with wild-type larval tissue, with a concomitant decrease in NHEJ repair products (Fig. 3B). For example, HR levels increased from 35% in wild type to 48% in dKDM4A mutant larvae at the 2het_2 heterochromatic DR-white insertion (Fig. 3B). This increase in HR repair was independent of changes in the total number of identified repair products (Supplemental Fig. S5A) or increased background mutation levels (Supplemental Fig. S5B), indicating that the increase in HR truly reflects changes in repair pathway use. Interestingly, in line with the absence of a defect in Mu2 focus kinetics at euchromatic breaks (Fig. 2C), DSB repair pathway choice remained unaffected in the presence or absence of dKDM4A at two euchromatic DR-white insertion sites (Fig. 3B). Importantly, increased HR repair at heterochromatic DSBs was rescued by introducing a wild-type dKDM4A transgene (Fig. 3C) and was reproduced in the presence of a different dKDM4A deletion mutation (ΔdKDM4A) (Supplemental Fig. S5C).
Cell cycle stage is a major determinant of DSB repair pathway choice (Chapman et al. 2012), with NHEJ dominating in the G1 phase and HR dominating in the S and G2 phases. To rule out the possibility that altered repair pathway usage in dKDM4A mutants reflects changes in cell cycle progression, we performed cell cycle analysis using the Fly-FUCCI system (Zielke et al. 2014). There were no significant cell cycle changes in dKDM4A mutant flies compared with wild type (Supplemental Fig. S5D), indicating that the observed increase in HR at heterochromatic DSBs in dKDM4A mutants is not due to increased time in S or G2. Pathway utilization changes were also not due to altered expression of repair genes, since RNA sequence analysis did not reveal any significant changes in the levels of HR or NHEJ genes in the absence of dKDM4A in both flies and cell culture (Supplemental Fig. S5E). Altogether, we conclude that dKDM4A promotes timely DSB repair and regulates pathway utilization in heterochromatin but does not impact repair of euchromatic DSBs.
Importantly, the increased HR levels upon dKDM4A depletion are not simply due to loss of canonical heterochromatin properties, since HP1a depletion results in a decrease in HR levels at both euchromatic and heterochromatic DSBs (Supplemental Fig. S6A), similar to what has been described previously in mammalian cells (Baldeyron et al. 2011). In addition, loss of the second KDM4 family member in Drosophila, dKDM4B, which promotes H3K9me3 demethylation in vivo (Lloret-Llinares et al. 2008; Tsurumi et al. 2013), also resulted in decreased levels of HR at both euchromatic and heterochromatic DSBs in third instar larvae (Supplemental Fig. S6B). Thus, HP1a and dKDM4B appear to play a more general role in promoting HR in both euchromatic and heterochromatic DSB repair, in contrast to the impact of dKDM4A specifically on heterochromatin DSB repair. Finally, in contrast to dKDM4A mutant flies, we found that dKDM4B mutant flies are not synthetically lethal with ATR (mei-41), the major DNA damage checkpoint kinase in flies (Supplemental Fig. S6C), indicating that dKDM4A plays a more prominent role in repair than dKDM4B.
dKDM4A depletion results in increased HR protein localization at heterochromatic DSBs
The fact that HR levels increase in the absence of dKDM4A indicates that dKDM4A normally inhibits HR protein recruitment and/or promotes NHEJ at heterochromatic DSBs. To directly test the hypothesis that dKDM4A impacts recruitment of HR proteins to heterochromatic DSBs, we developed an inducible CRISPR/Cas9 system in Drosophila cultured cells that generates DSBs in the 11- to 12-base-pair (bp) dodeca satellite tandem repeats (Fig. 4A, left) located in the chromosome 3 pericentromere (Abad et al. 1992). Generating a Cas9 fusion with the ecDHFR degradation domain (Cho et al. 2013) results in induction of DSBs at dodeca repeats upon addition of the stabilizing agent trimethoprim and dodeca single-guide RNA (sgRNA) expression. Dodeca fluorescence in situ hybridization (dodeca-FISH) combined with γH2Av immunofluorescence (IF) staining confirmed specific DSB induction at dodeca repeats upon ecDHFR-Cas9 induction in the absence or presence of dKDM4A (Supplemental Fig. S7A). The level of γH2Av intensity at dodeca DSBs did not differ between control and dKDM4A-depleted cells, indicating that loss of dKDM4A did not affect the efficiency of ecDHFR-Cas9-induced DSBs (Supplemental Fig. S7B).
Figure 4.

Increased HR protein localization at heterochromatic breaks in the absence of dKDM4A. (A, left) Trimethoprim addition stabilizes ecDHFR-Cas9. Concomitant expression of dodeca sgRNA with ecDHFR-Cas9 results in DSB induction at heterochromatic dodeca repeats in the pericentromeric heterochromatin of chromosome 3. (Right) Representative images of time-lapse movies of S2 cells stably expressing HP1a (red) and Tosca (HR protein; green) transiently transfected with fluorescently tagged inducible Cas9 (ecDHFR-Cas9; blue) and dodeca sgRNA. (B) Quantification of the number of cells with Tosca localization at dodeca repeats upon trimethoprim addition in yellow (control; black) or dKDM4A-depleted (gray) cells. Trimethoprim was added to the culture medium 10 min prior to imaging. Analysis was limited to cells that had visible Cas9 protein induction. (**) P-value < 0.01, unpaired t-test. Averages + SD are plotted for n = 3 experiments per condition. (C) Quantification of the level of Tosca protein present at Cas9-induced heterochromatic DSBs. Analysis was limited to cells that had visible HR protein localization to Cas9-induced DSBs, and nuclear background signal was subtracted. Each dot indicates one dodeca “focus” with Tosca (top) localization. (*) P-value < 0.05, unpaired t-test. n ≥ 46 cells per condition. The horizontal line indicates average + SEM. (D) Representative images of cells expressing CtIP-YFP (HR protein; green) and Cerulean-HP1a (heterochromatin protein; red) 30 min following 5 Gy of irradiation. Light-gray arrows point to CtIP foci in euchromatin, and blue arrows point to heterochromatic CtIP foci. (E,F) Quantification of images as in D of CtIP foci localization following 5 Gy of irradiation. The number of CtIP foci present within euchromatin (outside the HP1a domain; “eu”; light-gray arrows; E) or heterochromatin (within the HP1a domain; “het”; blue arrows; F) were quantified by live analysis of cells transfected with yellow RNAi (control; n = 35 cells) or dKDM4A RNAi (n = 61 cells). Averages are shown ±SEM. (*) P-value < 0.05, unpaired t-test. (G) Quantification of the level of CtIP-YFP protein present at ionizing radiation (IR)-induced heterochromatic (het) and euchromatic (eu) DSBs in cells transfected with yellow (control) or dKDM4A dsRNAs and imaged as in D. Analysis was performed 30 min after 5 Gy of IR, and nuclear CtIP background signal was subtracted. Each dot indicates one CtIP “focus.” (n.s.) P-value ≥ 0.05; (**) P-value < 0.001, unpaired t-test. n ≥ 74 cells per condition. The horizontal line indicates average + SEM. (H) Quantification of the number of cells with Tosca-YFP localization to dodeca repeats upon stabilization of Cas9 (through trimethoprim addition) in yellow (control) or dKDM4A-depleted cells in the presence (gray) or absence (black) of 0.5 µM Su(var)3-9 inhibitor chaetocin. Trimethoprim and chaetocin were added 10 min prior to the start of the movie. Analysis was limited to cells that had visible Cas9 induction. (n.s.) P-value ≥ 0.05; (*) P-value < 0.05, unpaired t-test. Averages + SD are plotted for n = 3 experiments per condition.
We next assessed HR repair protein recruitment to dodeca DSBs by live analysis of ecDHFR-Cas9, HP1a (to visualize the heterochromatin domain), and Tosca (Exo1 homolog; HR protein required for resection) (Fig. 4A, right; Chiolo et al. 2011). In line with our observations that loss of dKDM4A in mutant larval discs results in increased HR products only from heterochromatic DSBs (Fig. 3B), we observed that dKDM4A depletion resulted in a significant increase in cells displaying Tosca recruitment to dodeca DSBs compared with wild-type controls (from 57% to 83%) (Fig. 4B). In addition, we observed a significant increase in Tosca levels at the dodeca DSBs (Fig. 4C). We found a similar increase in the recruitment of CtIP, another HR end resection protein, to dodeca DSBs in the absence of dKDM4A (Supplemental Fig. S7C–E), indicating that dKDM4A loss has a general effect on early HR protein recruitment. Thus, loss of dKDM4A increases not only the number of cells that recruit HR proteins to DSBs (Fig. 4B; Supplemental Fig. S7D) but also the actual number of Tosca and CtIP molecules at heterochromatic DSBs (Fig. 4C; Supplemental Fig. S7E). dKDM4A was efficiently depleted in S2 cells (Supplemental Fig. S7F), and we did not observe significant cell cycle changes in S2 cells in the presence or absence of dKDM4A, ruling out a role for cell cycle-induced changes in HR protein recruitment (Supplemental Fig. S7G).
To determine whether the dKDM4A-dependent changes in HR protein recruitment were specific for DSBs in heterochromatin, we performed live analysis following irradiation of S2 cells, which allows a direct comparison between euchromatic and heterochromatic CtIP recruitment (Fig. 4D). We did not observe any significant changes in the kinetics of CtIP foci in euchromatin in the absence of dKDM4A (Fig. 4D,E), whereas the number of CtIP foci in heterochromatin remained significantly higher when compared with control cells even at 70 min after irradiation (Fig. 4D,F). Similar to what we observed at ecDHFR-Cas9-induced DSBs, increased intensity of irradiation-induced CtIP foci was evident in heterochromatin, but not in euchromatin of dKDM4A-depleted cells, when compared with control irradiated cells (Fig. 4D,G).
Overall, these results demonstrate that dKDM4A normally inhibits recruitment of early HR proteins (e.g., end resection) to DSBs but only within heterochromatin. We showed previously that early HR events occur within the heterochromatin domain, whereas late HR events (e.g., Rad51-dependent homology search) happen only after DSBs relocalize outside the heterochromatin domain (Chiolo et al. 2011). It is possible that dKDM4A normally also prevents aberrant localization of late HR proteins within the heterochromatin domain, as observed previously for the Su(var)3-9 histone methyltransferase and HP1a (Chiolo et al. 2011). Therefore, we tested the effect of dKDM4A depletion on Rad51 localization following irradiation of Drosophila cells. Notably, Rad51 does not localize to DSBs within the heterochromatin domain in the presence or absence of dKDM4A (Supplemental Fig. S7H). We therefore conclude that elevated HR usage upon dKDM4A depletion is due to increased recruitment of early HR resection proteins to heterochromatic DSBs and does not result from aberrant Rad51 recruitment to DSBs inside the heterochromatin domain.
dKDM4A counteracts heterochromatic methyltransferase activity at DSBs
Together, our results suggest that the inability to demethylate heterochromatic marks at DSBs upon dKDM4A depletion results in increased HR usage. This also suggests that the increases in HR upon loss of dKDM4A should be rescued (reduced to normal levels) by inhibiting H3K9me3 methyltransferase activity. Since prolonged inhibition of H3K9 methyltransferase activity will result in reduced HP1a and dKDM4A localization to heterochromatin (Colmenares et al. 2017), we only transiently inhibited H3K9 methyltransferases upon Cas9-dependent DSB induction using the small molecule inhibitor chaetocin (Greiner et al. 2005). In line with previous data that implicate H3K9 dimethyltransferase and trimethyltransferase activity in promoting repair and HR at euchromatic DSBs through methylation of H3K9 (Ayrapetov et al. 2014; Alagoz et al. 2015), we found that transient chaetocin treatment of control RNAi cells reduced the number of cells with Tosca-positive dodeca DSBs from 47% to 30% (Fig. 4H). Strikingly, loss of H3K9 methyltransferase activity completely rescued the increased Tosca recruitment induced by dKDM4A depletion (Fig. 4H). The number of dKDM4A-depleted cells with visible Tosca localization at dodeca DSBs was reduced from 74% to 39% upon chaetocin treatment. These findings suggest that H3K9 methyltransferase activity is required for HR protein recruitment and is normally counteracted by dKDM4A demethylation to reduce HR protein recruitment and promote NHEJ in heterochromatin. Since we only transiently inhibited H3K9 methyltransferase activity during break induction, these results also suggest that newly deposited H3K9 me2/me3 promotes HR protein recruitment at heterochromatic DSBs. This is in line with our H3K9me3 ChIP data (Supplemental Fig. S1A) as well as transient H3K9me3 increases observed at euchromatic DSBs (Ayrapetov et al. 2014). Importantly, short-term chaetocin treatment did not reduce the number of γH2Av foci at dodeca DSBs (Supplemental Fig. S8A) or lead to defects in cell cycle progression (Supplemental Fig. S8B), ruling out the possibility that reduced Cas9 efficiency or cell cycle-related changes were responsible for the observed reduction in Tosca recruitment.
In sum, we conclude that loss of dKDM4A-mediated demethylation of heterochromatic histone marks results in increased recruitment of early HR repair proteins and a higher frequency of HR repair but only at heterochromatic DSBs.
Discussion
The effects of the local chromatin landscape on DSB repair remain poorly understood. Different chromatin environments drive a variety of nuclear functions, and each is associated with specific molecular and biophysical properties. Thus, different chromatin domains could require distinct epigenetic changes to ensure proper access to the DNA damage repair machinery. To address this question, we induced single DSBs (I-SceI) in Drosophila animals as well as multiple DSBs (CRISPR/Cas9 or irradiation) in cell culture to assess the role of the histone demethylase dKDM4A in DSB repair in two distinct chromatin domains: heterochromatin and euchromatin.
This study extends our previous demonstration that dKDM4A is important for heterochromatic DSB movement (Colmenares et al. 2017) by revealing that heterochromatic, but not euchromatic, DSBs require dKDM4A for timely repair and repair pathway choice (Fig. 5). Without dKDM4A, DSB foci persist, are delayed in their relocalization outside the heterochromatin domain, and become heavily dependent on the HR pathway for repair and organismal survival. Our findings also suggest that direct or indirect demethylation by dKDM4A of the heterochromatin-enriched marks H3K9me3 and H3K56me3 into a monomethylated state at the DSB site mediates the repair process. This is consistent with increased H3K9me3 and H3K56me3 levels observed in vivo upon loss of dKDM4A (Lloret-Llinares et al. 2008; Tsurumi et al. 2013; Colmenares et al. 2017) but reveals a new target site; namely, heterochromatic DSBs. Unlike the mammalian orthologs (Whetstine et al. 2006), purified dKDM4A does not promote H3K9me3 demethylation in vitro (Lin et al. 2008), and in vitro demethylation of H3K56me3 remains untested. It is possible that dKDM4A directly demethylates H3K9me2/3 and/or K56me2/3 in vivo, but the recombinant dKDM4A used in the in vitro assays lacks specific DNA damaged-induced post-translational modifications or cofactors. Regardless of whether dKDM4A directly or indirectly promotes H3K9/K56me2/3 demethylation, our results clearly demonstrate that the heterochromatin landscape undergoes dKDM4A-dependent changes necessary to promote efficient DSB repair and modulate pathway utilization.
Figure 5.
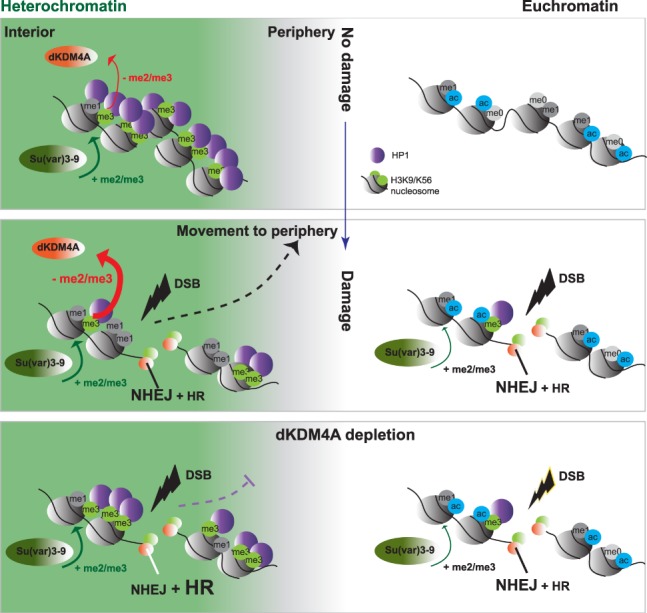
Model for the role of dKDM4A in heterochromatin DSB repair. (Top) Heterochromatin (green) and euchromatin (white) are two distinct chromatin environments within the nucleus, in which heterochromatin is characterized by H3K9me2/me3 and H3K56me2/me3. In contrast, euchromatin is characterized by histone modifications associated with increased transcriptional activity, such as acetylation. (Middle) DSBs in euchromatin and heterochromatin are repaired by NHEJ or HR. Regions surrounding the DSBs in both chromatin domains undergo an increase in H3K9me2/me3 and H3K56me2/me3. However, only heterochromatic DSBs move to the heterochromatin periphery, and the highly abundant heterochromatin marks are demethylated. This DSB movement and histone demethylation depend on the activity of dKDM4A and promote timely DSB repair. (Bottom) In the absence of dKDM4A demethylase activity, heterochromatic DSBs cannot be demethylated, which results in defective or delayed movement of the DSBs to the heterochromatin periphery, delayed DSB repair, and an increase in the usage of the HR repair pathway.
H3K9me2/3 normally recruits HP1a, which is thought to mediate heterochromatin compaction and specific biophysical properties (Eissenberg and Elgin 2000; Larson et al. 2017; Strom et al. 2017). The DSB-associated dKDM4A-dependent demethylation of H3K9me2/3 could therefore lead to localized loss of heterochromatin structure and increased accessibility to specific repair proteins, resulting in timely DSB repair (Fig. 2). The contribution of the newly discovered H3K56me3 modification to heterochromatin structure is less well understood (Jack et al. 2013; Colmenares et al. 2017). However, the previously identified reduction of H3K56me3 during heterochromatin replication (Jack et al. 2013) as well as a role for H3K56me1 in facilitating replication through its binding to PCNA (Yu et al. 2012) suggest that demethylation of H3K56me3 could result in increased accessibility of heterochromatin to the replication machinery. We therefore hypothesize that dKDM4A-dependent demethylation of heterochromatic substrates specifically facilitates DSB repair completion and pathway utilization through three non-mutually exclusive mechanisms: (1) restructuring of the local heterochromatin landscape into a repair-competent state, (2) recruitment of specific DNA repair pathway components such as NHEJ, and (3) mobilization of the DSB to the heterochromatin periphery to prevent aberrant recombination and improve access to the repair machinery.
In the first model, the DNA damage-activated demethylation of H3K9me3 and H3K56me3 by dKDM4A (Fig. 1C–F) facilitates DSB repair by increasing the accessibility of otherwise repressive heterochromatin. We demonstrated previously that the frequencies of HR and NHEJ repair pathway usage are highly similar in euchromatin and heterochromatin domains (Janssen et al. 2016) despite major differences in molecular composition and biophysical structure. Results presented here, however, indicate that heterochromatic DSBs require specific dKDM4A-dependent chromatin changes to achieve the same relative usage of NHEJ and HR pathways as euchromatic DSBs (Fig. 3). We propose that modifier-dependent generation of “repair-competent” chromatin structures more closely resemble euchromatin, which generally has low levels of H3K9me3 and H3K56me3 and high levels of H3K9me1 and H3K56me1 (Supplemental Fig. S3B,C; Ho et al. 2014). Conversion of heterochromatin into a more euchromatin-like composition provides a logical explanation for previous surprising observations that euchromatic and heterochromatic DSBs normally display very similar repair timing and pathway usage (Janssen et al. 2016). Future investigations into the chromatin composition of DNA repair sites in a variety of pre-existing chromatin domains will be important to identify “repair-competent” chromatin states.
Second, the increased frequency of HR repair and decreased frequency of NHEJ repair in the absence of dKDM4A (Fig. 3B) suggest that different repair pathways may be promoted or inhibited by specific histone modifications and associated proteins. Indeed, a recent study described the induction of specific chromatin changes associated with HR or NHEJ repair in euchromatic regions (Clouaire et al. 2018). Chromatin modifications have been implicated in the recruitment of specific repair proteins. For example, H3K36me3 has been suggested to recruit CtIP through its reader protein, LEDGF, at DSBs in actively transcribed euchromatin (Daugaard et al. 2012; Aymard et al. 2014), whereas demethylation of H3K4me3 allows the recruitment of the ZMYND8/NuRD complex to promote HR (Gong et al. 2017). In our study, we found that the absence of dKDM4A-mediated demethylation of H3K9me3 and H3K56me3 is associated with higher levels of HR protein recruitment and HR repair products (Figs. 3B, 4). This indicates that HR proteins prefer or tolerate the presence of H3K9me3 and H3K56me3 at DSB sites or that NHEJ proteins prefer H3K9me1 and H3K56me1 chromatin. Consistent with these observations, dKDM4A mutants depend on HR proteins for their viability (Fig. 3A), indicating that in the absence of H3K9me3 and H3K56me3 demethylation, DSBs depend on repair by HR.
Transient increases in the heterochromatin mark H3K9me3, observed at both euchromatic and heterochromatic DSBs (Supplemental Fig. S1A,B; Ayrapetov et al. 2014; Tsouroula et al. 2016), can potentially recruit or maintain HP1a at the DSB site. In mammals, this in turn appears to promote HR by repositioning 53BP1 away from the DSB (Baldeyron et al. 2011; Alagoz et al. 2015) as well as through direct binding of HP1a to HR proteins (Wu et al. 2015). It is therefore possible that dKDM4A-mediated demethylation of the highly abundant H3K9me3 mark normally promotes NHEJ by reducing HP1a at the break site, thereby lowering the affinity of HR proteins to heterochromatic DSBs. Consistent with this hypothesis, inhibition of H3K9me2/3 rescued the increased HR protein recruitment to heterochromatic DSBs in dKDM4A-depleted cells, revealing the dependency of HR protein recruitment on (de novo) H3K9me2/me3 at the DSB site (Fig. 4H). In contrast to heterochromatin, euchromatic DSBs have relatively low H3K9/K56me2/3 levels (Supplemental Fig. S3B,C) and may not need active demethylation to continue NHEJ repair. Alternatively, euchromatic DSBs may be dependent on another yet unidentified demethylase to remove damage-induced increases in H3K9me2/me3 and H3K56me2/me3 (Supplemental Fig. S1A,B). Whether HR and NHEJ proteins bind to specific methyl marks and differ between euchromatic and heterochromatic DSBs needs to be directly tested in future in vitro binding studies.
In the third model, demethylation of H3K9me3 and H3K56me3 may promote DNA repair by facilitating the mobilization of DSBs outside the heterochromatin domain and, ultimately, to the nuclear periphery (Chiolo et al. 2011; Colmenares et al. 2017). dKDM4A-dependent removal of heterochromatin marks could locally reduce levels of proteins (e.g., HP1a) (Chiolo et al. 2011) that normally promote inclusion in the phase-separated heterochromatin (Larson et al. 2017; Strom et al. 2017). This demethylation could result in incompatibility with the biophysical properties (e.g., liquid phase separation) of heterochromatin, causing either active expulsion or passive separation of DSBs from the heterochromatin domain. In the absence of dKDM4A, DSBs will therefore remain “phase-compatible” and stay within heterochromatin. We showed previously that early HR events, such as resection, normally occur within heterochromatin but that later HR events, such as Rad51 recruitment, only occur once DSBs leave the heterochromatin domain (Chiolo et al. 2011) and associate with the nuclear periphery (Ryu et al. 2015). It is therefore possible that the delayed DSB repair (Fig. 2) and increased HR usage in the absence of dKDM4A (Figs. 3B, 4) results primarily from delayed DSB relocalization (Colmenares et al. 2017), which could promote HR through the accumulation of resection proteins (Fig. 4B–G; Supplemental Fig. S7C–E). Consequently, elevated resection frequencies (or lengths) would inhibit NHEJ-mediated processing of DSBs. We hypothesize that these resected DSBs eventually do move outside the heterochromatin domain to promote Rad51 loading and completion of HR repair (Supplemental Fig. S7H).
dKDM4A mutant flies are only ∼70% viable when compared with wild-type flies (Colmenares et al. 2017), suggesting that repair deficiency due to loss of dKDM4A is detrimental to organismal health in the absence of induced DNA damage. Whether delayed DSB repair and increased usage of HR induced by dKMD4A loss results in increased aberrant recombination among heterochromatic repeats, as observed upon loss of HP1a, Su(var)3-9, or the SMC5/6 complex (Peng and Karpen 2007; Chiolo et al. 2011), remains to be determined. In cancer cells, where various members of the KDM4 family are overexpressed and drive tumorigenesis (Black et al. 2012), the effect of KDM4 on DNA repair in different chromatin environments remains unknown (Mallette et al. 2012; Khoury-Haddad et al. 2014). Our results emphasize the importance of studying the effects of KDM4 family members on genetic instability in human diseases and raise the possibility that KDM4 overexpression in cancer cells perturbs DNA repair pathways specifically in repetitive regions, which are highly dependent on controlled modulation of the heterochromatin landscape (Janssen et al. 2018).
In conclusion, this study demonstrates that heterochromatin and euchromatin require different chromatin-modifying activities to ensure efficient DSB repair and regulate repair pathway utilization. Many studies of DNA damage-associated chromatin proteins and modifiers do not incorporate key information about modifications present before damage induction and how they change during different stages of repair. This makes it hard to draw conclusions about the roles of chromatin proteins at DSBs in different chromatin domains. Future endeavors in other organisms would benefit from using locus-specific and chromatin domain-specific DNA damage induction; single-DSB systems provide a clean, physiologically relevant way to analyze repair mechanisms and avoid the toxicity and possibly unrelated chromatin changes associated with high levels of DSBs. This is crucial to generate a deeper understanding of how different pre-existing chromatin states—and associated chromatin changes—impact DNA damage repair and maintain genetic stability and, ultimately, how they impact organismal health and disease.
Materials and methods
Constructs
HP1a, dKDM4A, ecDHFR-Cas9, Tosca, and CtIP were cloned into pCOPIA vectors containing N-terminal YFP, GFP, mCherry, or Cerulean epitope tags. Dodeca sgRNA (for sequence, see Supplemental Table S1) was cloned into the pU6-3-sgRNA vector (Addgene, 45946).
Fly lines and genetic assays
Flies were grown at room temperature on standard medium, except where otherwise noted. A list of all fly lines used is in Supplemental Table S1. Parental flies used for viability assays were comprised of double mutants heterozygous for dKDM4A and a repair gene or heterozygous dKDM4A mutants with either an RNAi construct or a GAL4 gene driven by an Actin5C promoter. The percentage viability of the progeny was calculated using the ratio of adult dKDM4A mutant homozygotes to dKDM4A mutant heterozygotes (over balancer), normalized to the ratio of wild-type homozygotes to heterozygotes. Due to the subviability of dKDM4A mutant flies, ratios obtained from double mutants of dKDM4A and another gene were normalized to ratios for the dKDM4A mutant alone and adjusted to set the dKDM4A mutant at 100% to facilitate comparisons. All genotypes analyzed were quantified from five to 15 crosses and conducted in a y w genetic background.
DR-white repair product analysis
Quantification of somatic repair products in DR-white, I-SceI larvae was performed by inducing I-SceI expression in larvae, as described previously (Janssen et al. 2016). The upstream white[I-SceI-cut] gene in the DR-white construct was amplified using a primer pair specific for the DR-white construct (Supplemental Table S1) with Phire polymerase (Phire Animal Tissue Direct kit, Thermo Fisher) for 30 cycles. PCR products were treated with 0.5 µL of ExoSAP-IT (Affymetrix) and subsequently sequenced by Genewiz with a DR-white Sanger sequencing primer (Supplemental Table S1). Analysis of Sanger sequences was performed using the TIDE (tracking of indels by decomposition) algorithm (http://tide.nki.nl), a computational protocol designed by the laboratory of Dr. Bas van Steensel and published previously (Brinkman et al. 2014). HR products were identified as conversions of the I-SceI recognition site to the wild-type white sequence (essentially a 23-bp deletion). Indels of up to 25 bp were categorized as NHEJ products, with the exception of 23-bp deletion products, which were categorized as HR products.
Cell culture and manipulation
Stable lines were generated by cotransfection of expression constructs with pCOPIA_Hygro (Life Technologies) using the DOTAP liposomal transfection reagent (Roche) and selection for hygromycin resistance at 100 µg/mL (Life Technologies). Transient transfections were conducted using the TransIT-2020 reagent (Mirus), and live imaging was performed 72 h later. For RNAi experiments, dsRNA was generated from MEGAScript T7 transcription kit (Life Technologies) and PCR products containing T7 promoter sequences and the target regions (Supplemental Table S1). Tissue culture cells were transfected with 5–10 µg of dsRNA for 5 d with DOTAP reagent (Roche). Irradiation experiments were conducted by exposing cells to 5 Gy of X-rays from a 130-kV Faxitron TRX5200. Trimethoprim was from Sigma (T7883), and chaetocin was from Cayman Chemical (13156).
EdU, FISH, and IF staining
For all IF stainings, tissue culture cells were fixed on slides with 3.6% paraformaldehyde for 5 min and permeabilized with 0.4% Triton-X in PBS. For EdU labeling, cells were incubated with 10 µM EdU for 30 min and subsequently fixed as described above. EdU visualization was performed according to the Click-iT EdU Alexa fluor 488 Thermo Fisher protocol (C10337). Following fixation, IF was performed. Cells were incubated for 1 h at room temperature in PBS and 0.4% Triton-X/5% milk blocking solution. Primary antibodies were incubated overnight at 4°C in the same blocking solution. Secondary antibody incubations were performed for 2 h at room temperature in PBS and 0.4% Triton. Nuclei were counterstained with DAPI and mounted in Prolong Gold anti-fade (Life Technologies).
For IF-FISH, the IF protocol was performed as described above, except that primary antibody incubation was performed for 2 h, and the secondary antibody incubation was performed for 1 h, all at room temperature. Slides were subsequently washed three times in PBS and 0.1% Triton and post-fixed for 10 min with 3.6% formaldehyde. FISH was conducted by stepwise heating of samples from 37°C to 70°C in 2× sodium citrate buffer with 0.1% Tween-20 and 50% formamide followed by incubation with 250 ng of heat-denatured dodeca BNA probes (Integrated DNA Technologies) (for sequence, see Supplemental Table S1) in 50% formamide, 2× sodium citrate buffer, and 10% dextran sulfate overnight at 37°C.
Imaging
Images of cultured cells or wing discs were acquired using a 60× oil immersion objective (NA 1.40) on a DeltaVision microscope (DeltaVision Spectris; Applied Precision, LLC), and images were deconvolved using SoftWoRx (Applied Precision, LLC). Time-lapse images were acquired once every 10 min. Image analysis and focus tracking of deconvolved images were performed manually using Fiji image analysis software.
For live Mu2 foci tracking and Fly-FUCCI cell cycle analysis, third instar wing discs were dissected and placed on a slide in 10 µL of Schneider S1 medium supplemented with 10% FBS and covered with a 22-mm × 22-mm #1.5 coverslip (VWR) as described previously (Lerit et al. 2014). To induce ecDHFR-I-SceI (in larval tissue) or ecDHFR-Cas9 (in S2 cells), 400 µM or 10 µM trimethoprim, respectively, was added 10 min prior to imaging.
Chromatin preparation and qPCR
Third instar larvae (with [+DSB] and without [control] hsp.I-SceI transgene) were heat-shocked for 1 h at 37°C. Larvae were snap-frozen 6–9 h after heat shock in liquid nitrogen and kept at −80°C until chromatin preparation. For the preparation of chromatin, 30–40 larvae were subjected to homogenization, fixation, and sonication following the modENCODE protocol (http://www.modencode.org). ChIP was performed as described elsewhere (O'Geen et al. 2011) by using 5–10 µg of antibody and 2–4 µg of chromatin. Enrichment for histone modifications was quantified by qPCR using Absolute Blue qPCR SYBR low ROX mix (Thermo Fisher Scientific) and primers specific for the 3xP3 locus in the DR-white construct as well as the yellow gene as an internal control. qPCR was performed on the 7500 Fast real-time PCR system (Applied Biosystems). Primer sequences are in Supplemental Table S1. The fold enrichment of the specific histone mark upon DSB induction was calculated by dividing the relative increase over input for the heat-shocked sample containing hsp.I-SceI by the relative increase over input for the heat-shocked sample that did not contain hsp.I-SceI.
We used the Epicypher SNAP-ChIP K-MetStat panel (19-1001) to validate the specificity of the antibodies used for ChIP (Supplemental Fig. S2). The K-Met Stat panel contained individually barcoded nucleosomes associated with a specific histone H3 methylation mark (me0, H3K4me1/me2/me3, H3K9me1/me2/me3, H3K27me1/me2/me3, and H3K36me1/me2/me3). Following the sonication step, 0,4 µL of 0.6 nM K-MetStat stock was added per 2 µg of chromatin. ChIP was subsequently performed, and the enrichment for each modified nucleosome was determined by qPCR using primers specific for the individual barcoded nucleosomes. The specificity of all antibodies used for ChIP in this study was validated, with the exception of H3K56 antibodies due to unavailability of H3K56-modified nucleosomes in the K-MetStat panel.
Antibodies
The antibodies used for ChIP-qPCR were anti-H3K9me1 (EpiCypher, 13-0014; Abcam, 8896), H3K9me3 (Abcam, 8898; Active Motif, 39765), H3K56me1 (Abcam, 66857), H3K56me2 (Active Motif, 39277), H3K36me3 (Abcam, 9050; Cell Signaling Technology, 4909S), H3K36me1 (Abcam, 176920), Histone H3 (Abcam, ab1791), and γH2Av (mouse; Hybridoma Bank, UNC93-5.2.1). The primary antibodies used for IF were anti-γH2Av (mouse; Hybridoma Bank, UNC93-5.2.1), anti-Rad51 (a kind gift from J. Kadonaga), and anti-Cyclin A (mouse; 1:10; Developmental Studies Hybridoma Bank, A12). The secondary antibodies used for IF were Alexa 488/568/647 goat anti-mouse (1:1000; Thermo Fisher Scientific).
RT-qPCR
RNA was isolated by homogenizing two to three larvae in 200 µL of Trizol and incubating at room temperature for 5 min. Following addition of 40 µL of chloroform, lysates were shaken, incubated for 2–3 min at room temperature, and centrifuged at 12,000g for 15 min at 4°C. The aqueous phase was transferred to a new tube, and RNA was precipitated with 100 µL of isopropanol and 5 µg of glycogen. Following 10 min of incubation at room temperature, samples were centrifuged at 12,000g for 10 min at 4°C. The RNA was washed and centrifuged once in the presence of ice-cold 70% ethanol. The resulting RNA pellet was resuspended in 50 µL of dH2O. cDNA was synthesized using SuperScript III (Invitrogen) and oligo dT primers (IDT) following standard cDNA synthesis protocol (Invitrogen). qPCR was subsequently performed on the cDNA with gene-specific primers on the 7500 Fast real-time PCR system (Applied Biosystems). Primer sequences are in Supplemental Table S1.
Supplementary Material
Acknowledgments
Special thanks to the Karpen, Dr. Priscilla Cooper, and Dr. Sue Celniker laboratories as well as Dr. Or Gozani and Dr. Tie-Mei Li for their invaluable input during laboratory meetings and project design. These studies were supported by National Institutes of Health (NIH) grants R01 GM086613 and R01 GM117420 to G.H.K., Dutch Cancer Society (KWF) post-doctoral fellowship 2013-5854 to A.J., and an Innovative Genomics Institute grant to A.J., S.U.C., and G.H.K.
Author contributions: A.J. and S.U.C. performed most experiments and analysis. T.L. performed experiments in Figure 3, B and C, and Supplemental Figure S3, A–C. A.J., S.U.C., and G.H.K. contributed to project planning, experimental design, interpretations of results, and manuscript preparation.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.317537.118.
References
- Abad JP, Carmena M, Baars S, Saunders RD, Glover DM, Ludena P, Sentis C, Tyler-Smith C, Villasante A. 1992. Dodeca satellite: a conserved G+C-rich satellite from the centromeric heterochromatin of Drosophila melanogaster. Proc Natl Acad Sci 89: 4663–4667. 10.1073/pnas.89.10.4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagoz M, Katsuki Y, Ogiwara H, Ogi T, Shibata A, Kakarougkas A, Jeggo P. 2015. SETDB1, HP1 and SUV39 promote repositioning of 53BP1 to extend resection during homologous recombination in G2 cells. Nucleic Acids Res 43: 7931–7944. 10.1093/nar/gkv722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, Madhani HD. 2018. Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19: 229–244. 10.1038/nrm.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awwad SW, Ayoub N. 2015. Overexpression of KDM4 lysine demethylases disrupts the integrity of the DNA mismatch repair pathway. Biol Open 4: 498–504. 10.1242/bio.201410991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. 2014. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21: 366–374. 10.1038/nsmb.2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. 2014. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci 111: 9169–9174. 10.1073/pnas.1403565111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldeyron C, Soria G, Roche D, Cook AJL, Almouzni G. 2011. HP1α recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol 193: 81–95. 10.1083/jcb.201101030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JC, Van Rechem C, Whetstine JR. 2012. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 48: 491–507. 10.1016/j.molcel.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, et al. 2013. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154: 541–555. 10.1016/j.cell.2013.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B. 2014. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42: e168 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MR, Boulton SJ. 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47: 497–510. 10.1016/j.molcel.2012.07.029 [DOI] [PubMed] [Google Scholar]
- Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. 2011. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144: 732–744. 10.1016/j.cell.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho U, Zimmerman SM, Chen LC, Owen E, Kim JV, Kim SK, Wandless TJ. 2013. Rapid and tunable control of protein stability in Caenorhabditis elegans using a small molecule. PLoS One 8: e72393 10.1371/journal.pone.0072393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T, Rocher V, Lashgari A, Arnould C, Aguirrebengoa M, Biernacka A, Skrzypczak M, Aymard F, Fongang B, Dojer N, et al. 2018. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol Cell 72: 250–262.e6. 10.1016/j.molcel.2018.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmenares SU, Swenson JM, Langley SA, Kennedy C, Costes SV, Karpen GH. 2017. Drosophila histone demethylase KDM4A has enzymatic and non-enzymatic roles in controlling heterochromatin integrity. Dev Cell 42: 156–169.e5. 10.1016/j.devcel.2017.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crona F, Dahlberg O, Lundberg LE, Larsson J, Mannervik M. 2013. Gene regulation by the lysine demethylase KDM4A in Drosophila. Dev Biol 373: 453–463. 10.1016/j.ydbio.2012.11.011 [DOI] [PubMed] [Google Scholar]
- Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sorensen CS, Petersen NH, Sorensen PH, Lukas C, Bartek J, et al. 2012. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol 19: 803–810. 10.1038/nsmb.2314 [DOI] [PubMed] [Google Scholar]
- Do AT, Brooks JT, Le Neveu MK, LaRocque JR. 2014. Double-strand break repair assays determine pathway choice and structure of gene conversion events in Drosophila melanogaster. G3 4: 425–432. 10.1534/g3.113.010074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissenberg JC, Elgin SC. 2000. The HP1 protein family: getting a grip on chromatin. Curr Opin Genet Dev 10: 204–210. 10.1016/S0959-437X(00)00058-7 [DOI] [PubMed] [Google Scholar]
- Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM. 2017. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J Cell Biol 216: 1959–1974. 10.1083/jcb.201611135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, Jeggo PA. 2008. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 31: 167–177. 10.1016/j.molcel.2008.05.017 [DOI] [PubMed] [Google Scholar]
- Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. 2005. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol 1: 143–145. 10.1038/nchembio721 [DOI] [PubMed] [Google Scholar]
- Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen-Hughes T, et al. 2017. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24: 99–107. 10.1038/nsmb.3347 [DOI] [PubMed] [Google Scholar]
- Ho JWK, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn K-A, Minoda A, Tolstorukov MY, Appert A, et al. 2014. Comparative analysis of metazoan chromatin organization. Nature 512: 449–452. 10.1038/nature13415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack AP, Bussemer S, Hahn M, Punzeler S, Snyder M, Wells M, Csankovszki G, Solovei I, Schotta G, Hake SB. 2013. H3K56me3 is a novel, conserved heterochromatic mark that largely but not completely overlaps with H3K9me3 in both regulation and localization. PLoS One 8: e51765 10.1371/journal.pone.0051765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Breuer GA, Brinkman EK, van der Meulen AI, Borden SV, van Steensel B, Bindra RS, LaRocque JR, Karpen GH. 2016. A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev 30: 1645–1657. 10.1101/gad.283028.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Colmenares SU, Karpen GH. 2018. Heterochromatin: guardian of the genome. Annu Rev Cell Dev Biol 34: 265–288. 10.1146/annurev-cellbio-100617-062653 [DOI] [PubMed] [Google Scholar]
- Jha DK, Pfister SX, Humphrey TC, Strahl BD. 2014. SET-ting the stage for DNA repair. Nat Struct Mol Biol 21: 655–657. 10.1038/nsmb.2866 [DOI] [PubMed] [Google Scholar]
- Khoury-Haddad H, Guttmann-Raviv N, Ipenberg I, Huggins D, Jeyasekharan AD, Ayoub N. 2014. PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proc Natl Acad Sci 111: E728–737. 10.1073/pnas.1317585111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ. 2017. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547: 236–240. 10.1038/nature22822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerit DA, Plevock KM, Rusan NM. 2014. Live imaging of Drosophila larval neuroblasts. J Vis Exp 10.3791/51756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-H, Li B, Swanson S, Zhang Y, Florens L, Washburn MP, Abmayr SM, Workman JL. 2008. Heterochromatin protein 1a stimulates histone H3 lysine 36 demethylation by the Drosophila KDM4A demethylase. Mol Cell 32: 696–706. 10.1016/j.molcel.2008.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret-Llinares M, Carré C, Vaquero A, de Olano N, Azorín F. 2008. Characterization of Drosophila melanogaster JmjC+N histone demethylases. Nucleic Acids Res 36: 2852–2863. 10.1093/nar/gkn098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. 2012. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J 31: 1865–1878. 10.1038/emboj.2012.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottke A, Colaiacovo MP, Shi Y. 2009. Developmental roles of the histone lysine demethylases. Development 136: 879–889. 10.1242/dev.020966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Geen H, Echipare L, Farnham PJ. 2011. Using ChIP-seq technology to generate high-resolution profiles of histone modifications. Methods Mol Biol 791: 265–286. 10.1007/978-1-61779-316-5_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JC, Karpen GH. 2007. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat Cell Biol 9: 25–35. 10.1038/ncb1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukos V, Misteli T. 2014. The biogenesis of chromosome translocations. Nat Cell Biol 16: 293–300. 10.1038/ncb2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, Karpen GH, Chiolo I. 2015. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat Cell Biol 17: 1401–1411. 10.1038/ncb3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RN, Grzybowski AT, Cornett EM, Johnstone AL, Dickson BM, Boone BA, Cheek MA, Cowles MW, Maryanski D, Meiners MJ, et al. 2018. Examining the roles of H3K4 methylation states with systematically characterized antibodies. Mol Cell 72: 162–177.e7. 10.1016/j.molcel.2018.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH. 2017. Phase separation drives heterochromatin domain formation. Nature 547: 241–245. 10.1038/nature22989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsouroula K, Furst A, Rogier M, Heyer V, Maglott-Roth A, Ferrand A, Reina-San-Martin B, Soutoglou E. 2016. Temporal and spatial uncoupling of DNA double strand break repair pathways within mammalian heterochromatin. Mol Cell 63: 293–305. 10.1016/j.molcel.2016.06.002 [DOI] [PubMed] [Google Scholar]
- Tsurumi A, Dutta P, Shang R, Yan SJ, Li WX. 2013. Drosophila Kdm4 demethylases in histone H3 lysine 9 demethylation and ecdysteroid signaling. Sci Rep 3: 2894 10.1038/srep02894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, et al. 2006. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125: 467–481. 10.1016/j.cell.2006.03.028 [DOI] [PubMed] [Google Scholar]
- Wu W, Nishikawa H, Fukuda T, Vittal V, Asano M, Miyoshi Y, Klevit RE, Ohta T. 2015. Interaction of BARD1 and HP1 is required for BRCA1 retention at sites of DNA damage. Cancer Res 75: 1311-1321. 10.1158/0008-5472.CAN-14-2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Song C, Zhang Q, DiMaggio PA, Garcia BA, York A, Carey MF, Grunstein M. 2012. Histone H3 lysine 56 methylation regulates DNA replication through its interaction with PCNA. Mol Cell 46: 7–17. 10.1016/j.molcel.2012.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke N, Korzelius J, van Straaten M, Bender K, Schuhknecht GF, Dutta D, Xiang J, Edgar BA. 2014. Fly-FUCCI: a versatile tool for studying cell proliferation in complex tissues. Cell Rep 7: 588–598. 10.1016/j.celrep.2014.03.020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.