

Abstract
Paclitaxel is a widely used chemotherapy drug, but development of resistance leads to treatment failure. Tumor cells that are treated with a sublethal dose of paclitaxel for a long period of time show the epithelial‐mesenchymal transition (EMT) phenotype, which leads to metastasis and resistance. All‐trans retinoic acid (ATRA) is always used in combination with paclitaxel and can reverse EMT in many types of cancer cells. The ability of ATRA to reverse EMT in chemoresistant cells is still unknown. In the present study, the ability of ATRA to reverse EMT in paclitaxel‐resistant cells was investigated. Three colorectal cancer cell lines, HCT116, LoVo and CT26, were treated with sublethal doses of paclitaxel to create resistant cell lines. Western blotting, immunocytochemistry, and “parachute” dye‐coupling assays showed that ATRA reverses EMT, inhibits nuclear factor kappa B (NF‐κΒ), and upregulates gap junctions in paclitaxel‐resistant cells. Scratch wound‐healing and Transwell assays showed that ATRA decreases the migration and invasion abilities of paclitaxel‐resistant cells. In addition, the CT26 cell line was used in the Balb/c pulmonary metastasis model to show that ATRA reduces metastasis of paclitaxel‐resistant cells in vivo. Given these data, ATRA may reverse EMT by inhibiting NF‐κΒ and upregulating gap junctions in paclitaxel‐resistant cells.
Keywords: all‐trans‐retinoic acid, epithelial‐mesenchymal transition, gap junction, NF‐κΒ, paclitaxel
1. INTRODUCTION
Long‐term chemotherapy treatment often leads to drug resistance and metastasis. Paclitaxel is one of the most effective chemotherapy drugs, but many patients develop resistance to it during treatment. Paclitaxel‐resistant cells have a high metastatic ability.1 Paclitaxel stabilizes microtubules, blocks the cell cycle at the G2/M phase, and activates proapoptotic signaling to induce cell death.1 As a mitotic inhibitor, paclitaxel has an obvious anti‐proliferative effect; however, there is growing evidence that epithelial‐mesenchymal transition (EMT) is associated with acquired resistance to paclitaxel.2, 3 During EMT, epithelial cells lose their polarized organization and intercellular junctions, undergo changes in phenotype and cytoskeletal organization, and acquire mesenchymal characteristics. Together, these changes increase metastatic ability of the cells.4 Long‐term exposure to sublethal doses of paclitaxel induces EMT.3 EMT promotes scattering and local dissemination from the tumor surface and tends to be resistant to anoikis, which further contributes to metastasis.5 Hence, targeting EMT is a plausible strategy to combat and manage metastasis induced by paclitaxel resistance.
For this reason, many agents have been used in combination with paclitaxel to challenge resistance.6 The combination of all‐trans‐retinoic acid (ATRA) and paclitaxel has been used to treat a variety of cancers for more than 10 years. This combination causes more apoptosis and less invasion than paclitaxel alone.7, 8 ATRA is the predominant natural metabolite of vitamin A and is involved in many important biological processes, such as morphogenesis, vision, growth, metabolism, differentiation, and cellular homeostasis.9 ATRA is also used in both chemopreventive and therapeutic schemes. As an anti‐cancer agent, ATRA inhibits carcinogenesis by three mechanisms: arresting further growth of abnormal cells, inducing differentiation of abnormal cells to stop proliferation, and inducing apoptosis.10 ATRA reverses EMT in many types of cancer cells by restoring the expression of E‐cadherin, downregulating the expression of vimentin, and increasing cell‐to‐cell interactions.11 It has also been reported that ATRA inhibits cancer cell invasion and metastasis in a variety of cancer types.12, 13 Whether ATRA can be used as a single agent to reverse EMT in chemoresistant cancer cells is still unknown. The mechanism by which ATRA inhibits the EMT caused by chemotherapeutic drugs also needs to be investigated.
Therefore, it was hypothesized that ATRA could reverse EMT in paclitaxel‐resistant colorectal cancer cells in vitro and in vivo. In this study, three colorectal cancer cell lines, HCT116, LoVo, and CT26, were used to investigate the potential reverse effect of ATRA on EMT.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
All cell culture media, trypsin and antibiotics were purchased from Gibco (Grand Island, NY, USA), and FBS was purchased from HyClone (Logan, UT, USA). Mouse anti‐E‐cadherin antibody, mouse anti‐N‐cadherin antibody, mouse anti‐nuclear factor kappa B (anti‐NF‐κΒ) p65 antibody, mouse anti‐fibronectin antibody, mouse anti‐β‐actin antibody, mouse anti‐connexin 32 antibody, and mouse anti‐snail antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti‐MMP‐9 antibody, rabbit anti‐IKKα antibody, rabbit anti‐IκΒα antibody, rabbit anti‐vimentin antibody, rabbit anti‐β‐catenin antibody, rabbit anti‐connexin 43 antibody, rabbit anti‐connexin 26 antibody, rabbit anti‐retinoic acid receptor α antibody, rabbit anti‐Histone H3 antibody, goat anti‐rabbit IgG‐peroxidase, goat anti‐mouse IgG‐peroxidase, goat anti‐mouse IgG FITC, goat anti‐rabbit IgG FITC, goat anti‐mouse IgG TRITC, DAPI, ATRA, paclitaxel, Calcein‐AM, CM‐Dil and MTT were purchased from Sigma‐Aldrich (St Louis, MO, USA). Immobilon membranes were purchased from Merck Millipore (Bedford, MA, USA). ECL Plus substrate, RIPA lysis buffer and bicinchoninic acid reagents were purchased from CWBio (Beijing, China). Nuclear and cytoplasmic protein extraction kit and crystal violet were purchased from Beyotime (Shanghai, China). pTARGET Mammalian Expression Vector system was purchased from Promega Corporation (Madison, WI, USA). Lipofectamine reagent was purchased from Invitrogen (Waltham, MA, USA).
2.2. Cell lines
Cell lines HCT116, LoVo, and CT26 were purchased from the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai Shi, China). HCT116 and LoVo cell lines are human colorectal cancer cell lines. CT26 is a murine colon adenocarcinoma cell line derived from N‐nitroso‐N‐methylurethane‐treated Balb/c mice. All cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin.
2.3. Paclitaxel and ATRA treatment
Paclitaxel‐resistant cell lines were developed by long‐term, low‐dose treatment of all three cell lines with paclitaxel. Initially, the cells were treated for 24 hours with varying concentrations of paclitaxel (LoVo, 5 nmol/L; HCT116, 4 nmol/L; CT26, 2 nmol/L). The cells were cultured in medium without paclitaxel to recover for about 2 weeks and then treated with higher concentrations of paclitaxel. Cells that could stably grow and proliferate in high concentrations of paclitaxel were established. The paclitaxel‐resistant cells were named “P”. These resistant cells and their parental cells were both treated with media containing 1 μmol/L ATRA for 48 hours and named “PR” and “R”, respectively. The effect of 24‐hour treatment with paclitaxel on cell growth was measured by MTT assay. The cell growth inhibition rate was calculated by defining the absorption of cells not treated with paclitaxel (control) as 100%. Measurements were carried out in triplicate. These cells were used in subsequent assays.
2.4. Connexin 43 transfection
Connexin 43 (Cx43) was transfected to all paclitaxel‐resistant cells. The method was carried out as previously described.14 Briefly, cDNA of the human Cx43 coding region was inserted into pTARGET vector. Constructed expression vector was transfected to cells by lipofection. Cx43 expression in transfected cells is identified by western blotting and immunocytochemistry. GJ function is also measured by “Parachute” dye‐coupling assay. These transfected cells were named “C”. The details of transfection are described in Document S1.
2.5. Western blotting
For detection of IKKα, IκΒα, NF‐κΒ and retinoic acid receptor α (RARα), cytosolic and nuclear proteins were isolated from harvested cells using a nuclear and cytoplasmic protein extraction kit (Beyotime, Shanghai, China) according to the manufacturer's protocol.
For detection of Cx43, connexin 32 (Cx32), connexin 26 (Cx26), E‐cadherin, N‐cadherin, fibronectin, MMP‐9, snail, vimentin, and β‐catenin, 106 cells were lysed in 500 mL RIPA lysis buffer supplemented with protease inhibitors for 20 minutes at 4°C. The lysates were centrifuged, and bicinchoninic acid reagents were used to quantitate the protein content.
Western blotting was carried out as previously described.15 Briefly, protein samples were suspended in SDS loading buffer. After boiling, 10 μg protein was separated by SDS‐PAGE and then transferred to immobilon membranes using the semi‐dry blotting method. The membranes were probed with antibodies using standard techniques. Finally, the protein bands were visualized using ECL Plus and exposed film. Each assay was carried out in triplicate.
2.6. Immunocytochemistry analysis
Immunofluorescence staining was used to analyze Cx43, E‐cadherin, and vimentin expression, and NF‐κΒ activation. The procedure was carried out as previously described.15 In brief, cells were washed gently with PBS, fixed in 10% formalin at room temperature for 20 minutes, treated with 0.5% Triton X‐100 for 5 minutes at 4°C and then blocked with 5% normal goat serum overnight at 4°C. Slides were incubated with primary antibody for 1 hour at 37°C. After washing with PBS, the slides were incubated with FITC‐ and/or TRITC‐conjugated secondary antibody and DAPI for 1 hour at 37°C. Subsequently, the slides were washed with PBS to remove antibody and sealed. The slides were photographed immediately using a fluorescence microscope (Olympus, Tokyo, Japan).
2.7. “Parachute” dye‐coupling assay
Gap junction (GJ) function was examined using the “Parachute” dye‐coupling assay as described by Wang et al.6 Cells were seeded in six‐well plates and grown to 80%‐100% confluency. CM‐Dil and Calcein‐AM were used to determine GJ function. CM‐Dil is a red fluorescent dye that cannot spread to coupled cells. Calcein‐AM is a green fluorescent dye that can spread to adjacent cells by GJ. Calcein‐AM (10 μg/mL) and CM‐Dil (5 μg/mL) were used to stain each well of donor cells for 30 minutes at 37°C. Donor cells were then trypsinized and seeded onto a monolayer of receiver cells grown in another well. The ratio of donor to receiver cells was 1:150. GJ formed between donor and receiver cells after 4 hours of incubation at 37°C. Fluorescence was detected using a fluorescence microscope. Red fluorescence of CM‐Dil indicated the location of donor cells. The green fluorescence of Calcein‐AM was used to calculate the number of illuminant receiver cells around each donor cell. This value was used to evaluate GJ function.
2.8. Scratch wound‐healing assay
To determine the migratory ability of the cells, the classical scratch wound‐healing assay was carried out as previously described.15 Briefly, cells were cultured until they formed a monolayer in six‐well plates. Cells were serum‐starved overnight, and an artificial scratch wound was created by a 10‐μL pipette tip. The plates were washed with PBS to remove cell debris and then maintained in serum‐free culture at 37°C, 5% CO2. Migration photos were captured at 0, 12, 24, and 48 hours after scratching. Experiments were repeated independently in triplicate. The following equation was used to calculate percent wound closure: percent wound closure (%) = [1 − (L t/L 0)] × 100, where L t represents width of scratch at time t and L 0 represents initial width of scratch.
2.9. Cell invasion assay
Cell invasion assays were carried out as previously described15 using 24‐well Matrigel‐coated chambers (6.5 mm in diameter, 8 μm pore‐size, 100 μg/cm2 Matrigel, Corning, Tewksbury, MA, USA). Briefly, cells were grown until they were subconfluent, then serum‐starved for 24 hours. Cells were detached using trypsin, and 2 × 105 cells were added to the upper Transwell chamber in 500 μL serum‐free media. To the lower chamber, 750 μL media with 10% FBS was added. All conditions were repeated in triplicate. After 24‐hour incubation at 37°C and 5% CO2, cells that had not migrated were removed using a cotton swab and cells that had migrated were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 30 minutes. Images of three different fields were captured for each membrane.
2.10. Experimental pulmonary metastasis and treatment
CT26, CT26‐P or CT26‐C cells (2 × 105/0.2 mL) were trypsinized and injected into the tail vein of Balb/c mice (6‐week‐old, female) to establish a model for metastatic lung tumors. ATRA was dissolved in 5% HCO‐60 solution and prepared for dosing (0.585 mg/kg) in accordance with the report by Suzuki et al.16 ATRA was injected into the tail vein of the CT26 or CT26‐P group daily for 7 days following tumor cell injection. Mice were killed 2 weeks after tumor cell injection, and tumor nodules on the surface of the lungs were counted. Survival time was compared among groups. All animal experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978).
2.11. Statistical analysis
All data represent mean ± standard deviation. Statistical analysis was carried out by Student's t test using SPSS software. P value <0.05 was considered statistically significant.
3. RESULTS
3.1. Establishment and phenotype of paclitaxel‐resistant cell lines
After continuous treatment with increasing concentrations of paclitaxel, resistant cells were established. The cobblestone morphology of the HCT116‐P and LoVo‐P cells changed to spindle and fiber shapes (Figure 1), which is typical of the fibroblastic phenotype. As seen in Figure 1, ATRA treatment and Cx43 transfection can partially reverse the fibroblastic phenotype of HCT116‐P and LoVo‐P cells to the epithelial phenotype. Paclitaxel treatment of the mesenchymal cells CT26‐P caused some morphological changes, which were reversed by ATRA treatment and Cx43 transfection, although the changes were not significant. Paclitaxel IC50 for the cells were determined using the MTT assay. Results indicated that long‐term sublethal dosage of paclitaxel significantly increases the IC50. Paclitaxel‐resistant cells lost most of their resistance after they were treated with ATRA or transfected by Cx43, but ATRA treatment did not impact the IC50.
Figure 1.
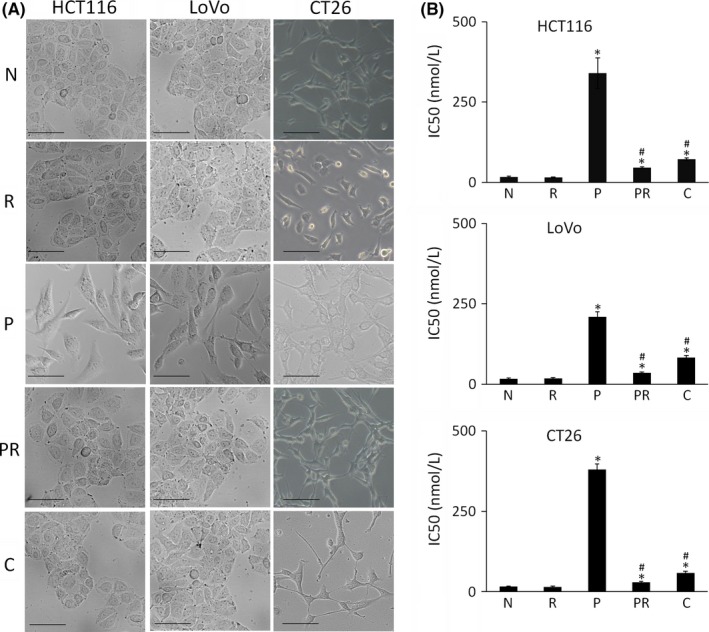
Morphological change and paclitaxel IC50 of colorectal cancer cells. A, Morphological changes in three colorectal cancer cells lines (HCT116, LoVo, CT26) following treatment as indicated. “N” are parental cells, “R” are parental cells treated with all‐trans‐retinoic acid (ATRA) for 48 h, “P” are paclitaxel‐resistant cells, “PR” are paclitaxel‐resistant cells treated with ATRA for 48 h, and “C” are Cx43 transfected paclitaxel‐resistant cells. Scale bars, 20 μm. B Paclitaxel IC50 of the cells. “R”, “P”, “PR” and “C” were compared with “N” using Student's t test. “PR” and “C” were also compared with “P” using Student's t test. *P < .05 represents a significant difference compared to “N”, and #P < .05 represents a significant difference compared to “P”
3.2. Connexin 43 transfection increased Cx43 expression and GJ function significantly in paclitaxel‐resistant cells
Connexin 43 transfection obviously increased Cx43 expression in paclitaxel‐resistant cells (Figure 2). Most Cx43 were located on the membrane (Figure S2A‐C), which increased GJ function significantly in these cells (Figure S2D,E).
Figure 2.
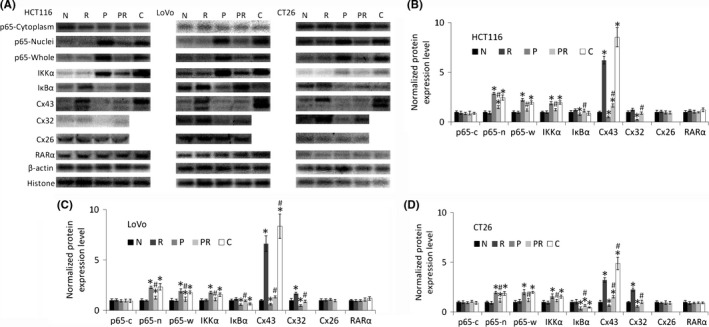
Western blot analysis of nuclear factor kappa B (NF‐κΒ) p65, IKKα, IκΒα, Cx43, Cx32, Cx26 and RARα expression in colorectal cancer cells. p65 expression was measured in the cytoplasmic, nucleolar, and whole‐cell lysates to determine whether NF‐κΒ translocation to the nucleus is induced by paclitaxel and all‐trans‐retinoic acid (ATRA). “N” are parental cells, “R” are parental cells treated with ATRA for 48 h, “P” are paclitaxel‐resistant cells, “PR” are paclitaxel‐resistant cells treated with ATRA for 48 h, and “C” are Cx43 transfected paclitaxel‐resistant cells. A, Western blot images. β‐Actin is used for cytoplasmic loading control, including p65‐cytoplasm, p65‐whole, IKKα, IκΒα, Cx43, Cx32, and Cx26. Histone H3 is used for nucleolar loading control of p65‐nuclei and RARα. B‐D, Bar diagrams of densitometric analysis where “c” represents cytoplasmic, “n” represents “nucleolar”, and “w” represents whole‐cell lysates. “R”, “P”, “PR” and “C” were compared to “N” using Student's t test. “PR” and “C” were also compared with “P” using Student's t test. *P < .05 represents a significant difference from “N”, and #P < .05 represents a significant difference from “P”. Cx, connexin; RARα, retinoic acid receptor α
3.3. High levels of NF‐κΒ activation in paclitaxel‐resistant cells can be inhibited with ATRA treatment
Nuclear factor kappa B is a proinflammatory cytokine that promotes survival, adhesion, and invasion of cancer cells.17 As a key transcriptional regulator of extracellular proteases, NF‐κΒ activation also contributes to EMT, which is important for cancer metastasis.18
Nuclear factor kappa B activation was assessed by assaying for the expression and location of the NF‐κΒ p65 subunit. Under normal conditions, NF‐κΒ binds to an inhibitor of NF‐κΒ (IκΒ) to form an inactive compound in the plasma. Many chemotherapeutic drugs, including paclitaxel, can induce IκΒ degradation to activate NF‐κΒ in cancer cells.19 When external stimulus causes NF‐κΒ to dissociate from IκΒ, NF‐κΒ is activated and transfers to the nucleus. This process is mediated mainly by the activation of IκΒ kinase (IKK). IKK is composed of a regulatory protein (IKKγ) and a heterodimer of IKKα and IKKβ. As IKK and IκΒ are cytoplasmic proteins, they can only be detected in cytosolic extractions.20
Western blotting (Figure 2) and immunocytochemistry (Figure S1) both show that all paclitaxel‐resistant cells had relatively high expression of NF‐κΒ in their nuclei. The assays also showed that expression of IKKα increased and IκΒα decreased in paclitaxel‐resistant cells. These expression changes were reversed with ATRA treatment. The results suggest that ATRA may prevent the IκΒ degradation and increased expression of IKK induced by paclitaxel. It has been reported that ATRA inhibits NF‐κΒ activation in many cell types.21, 22 In this study, treatment with ATRA was found to reduce NF‐κΒ expression in the nuclei of resistant cells, but not in those of parental colorectal cancer cells. Furthermore, Cx43 transfection does not obviously reduce NF‐κΒ expression in the nuclei of resistant cells. RARα expression is not significantly affected by long‐time paclitaxel treatment, so ATRA can play an effective role in paclitaxel‐resistant cells.
3.4. All‐trans retinoic acid treatment and Cx43 increase the function of gap junctions in paclitaxel‐resistant cells
Gap junctions are important channels that connect adjacent cells or the extracellular environment. GJ inhibit EMT, invasion, and metastasis.23 GJ are composed of connexins, a protein family encoded by a group of tumor suppressor genes considered as targets for chemotherapy. Cx43 is the most widely expressed connexin in a variety of tissues and is a structural component of GJ.23 Some reports have shown that paclitaxel inhibits GJ function and Cx43 expression to decrease cytotoxicity of itself.6, 14 Cx43 as well as other GJ molecules, such as Cx32 and Cx26, has been shown to inhibit primary tumor cell progression. Their expression decreases after some chemotherapeutic agents.24, 25 Furthermore, ATRA has been reported as a GJ potentiator.26, 27 As such, the ability of ATRA to upregulate GJ function in paclitaxel‐resistant colorectal cancer cells was investigated.
Figure 2 and Figure S2 show that paclitaxel‐resistant cells had lower Cx43 and Cx32 expression and GJ function. Treatment with ATRA increased Cx43 and Cx32 expression and GJ function in both parental and paclitaxel‐resistant cells. ATRA treatment increased Cx43 expression in cell membranes, even those of paclitaxel‐resistant cells (Figure S2A‐C). However, neither ATRA nor paclitaxel had obvious effects on Cx26 expression.
3.5. All‐trans retinoic acid and Cx43 both decrease mesenchymal marker expression and increase epithelial marker expression in paclitaxel‐resistant cells
Long‐term paclitaxel treatment was shown to induce treatment resistance and EMT.28 Figure 3 and Figure S3 show that long‐term paclitaxel treatment induced EMT in all three colorectal cancer cell lines. Specifically, compared to their parental cells, paclitaxel‐resistant cells had higher expression of many EMT markers, including fibronectin, MMP‐9, N‐cadherin, snail, vimentin, and β‐catenin as well as lower expression of E‐cadherin. Treatment with ATRA or Cx43 transfection reversed these changes in all three paclitaxel‐resistant cells lines, suggesting that ATRA and Cx43 may reverse the EMT in paclitaxel‐resistant cells.
Figure 3.
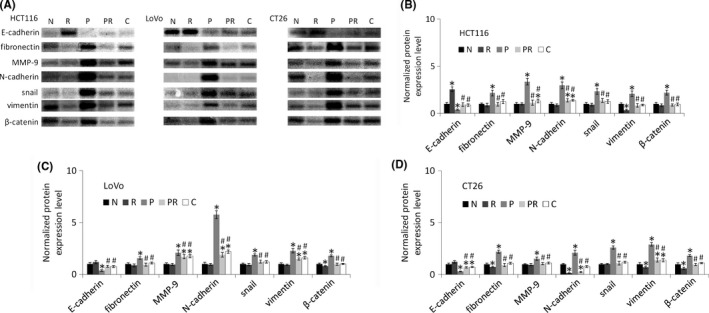
Western blot analysis of E‐cadherin, fibronectin, MMP‐9, N‐cadherin, snail, vimentin, and catenin in colorectal cancer cell lines. “N” are parental cells, “R” are parental cells treated with all‐trans‐retinoic acid (ATRA) for 48 h, “P” are paclitaxel‐resistant cells, “PR” are paclitaxel‐resistant cells treated with ATRA for 48 h, and “C” are Cx43 transfected paclitaxel‐resistant cells. A, Western blot images. β‐Actin in Figure 2 was used as the loading control for all proteins in this figure. B‐D, Bar diagrams of densitometric analysis. “R”, “P”, “PR” and “C” were compared with “N” using Student's t test. “PR” and “C” were compared with “P” using Student's t test. *P < .05 represents a significant difference from “N”, and #P < .05 represents a significant difference from “P”
3.6. All‐trans retinoic acid and Cx43 both reduce the migration and invasion abilities of paclitaxel‐resistant cells
All‐trans retinoic acid can reduce the migration and invasion abilities of many types of cancer cells, such as pancreatic and colorectal.29, 30 To investigate whether ATRA reduces the migration and invasion abilities of paclitaxel‐resistant cells, scratch wound‐healing and Transwell invasion assays were carried out. As shown in Figure 4, all three paclitaxel‐resistant cells had higher migration abilities than their parental cells. ATRA and Cx43 transfection can both reduce the migration ability of these resistant cells. In addition, in the wound‐healing assay, cell density did not change after 48 hours of treatment with ATRA. This lack of change indicates that reduced cell migration is mainly attributed to the decreased migratory capability of cells, rather than a decrease in the number of cells resulting from apoptosis. As shown in Figure 5, the Transwell invasion assay showed that ATRA and Cx43 transfection both reduced the invasive ability of paclitaxel‐resistant cells. In addition, ATRA also reduced the migration and invasion abilities of parental cells.
Figure 4.
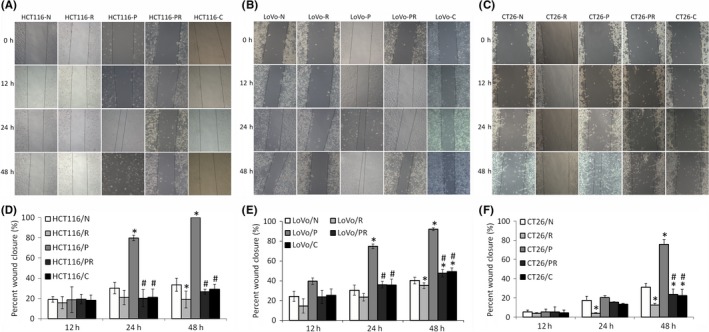
Migration of colorectal cancer cells as measured by scratch wound‐healing assay (original magnification 100×). “N” are parental cells, “R” are parental cells treated with all‐trans‐retinoic acid (ATRA) for 48 h, “P” are paclitaxel‐resistant cells, “PR” are paclitaxel‐resistant cells treated with ATRA for 48 h, and “C” are Cx43 transfected paclitaxel‐resistant cells. A, Migration image of HCT116. B, Migration image of LoVo. C, Migration image of CT26. D‐F, Bar diagrams of scratch wound‐healing assays of HCT116, LoVo and CT26. “R”, “P”, “PR” and “C” were compared to “N” using Student's t test. “PR” and “C” were compared with “P” using Student's t test. *P < .05 represents a significant difference from “N” and #P < .05 represents a significant difference from “P”
Figure 5.
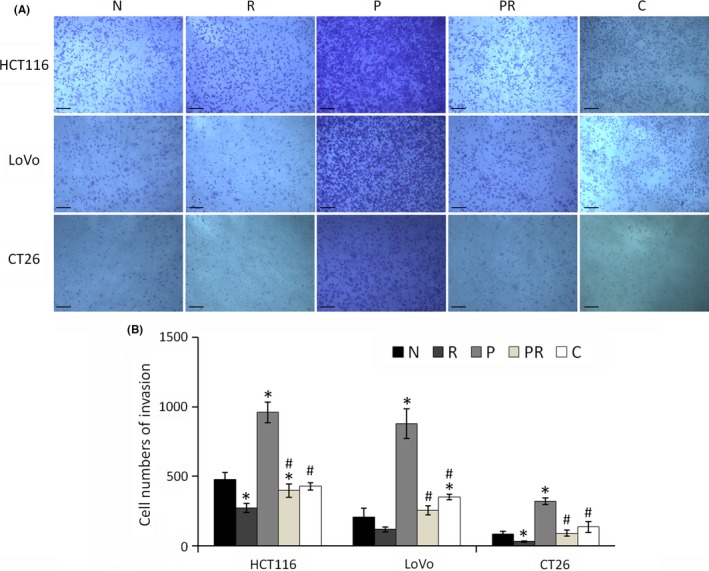
Invasion of colorectal cancer cells as measured by Transwell assay. “N” are parental cells, “R” are parental cells were treated with all‐trans‐retinoic acid (ATRA) for 48 h, “P” are paclitaxel‐resistant cells, “PR” are paclitaxel‐resistant cells treated with ATRA for 48 h, and “C” are Cx43 transfected paclitaxel‐resistant cells. A, Image of Transwell assay. Scale bars, 100 μm. B, Bar diagrams of invasion. “R”, “P”, “PR” and “C” were compared to “N” using Student's t test. “PR” and “C” were compared with “P” using Student's t test. *P < .05 represents a significant difference from “N”, and #P < .05 represents a significant difference from “P”
3.7. All‐trans retinoic acid and Cx43 reduce metastasis of paclitaxel‐resistant cells in vivo
Ability of ATRA and Cx43 transfection to reduce metastasis of paclitaxel‐resistant CT26 cells was evaluated using a mouse pulmonary metastasis model (Figure 6 and Figure S4). Higher numbers of cancer nodules in the lungs of mice injected with CT26‐P cells compared to mice injected with CT26 cells indicates that paclitaxel‐resistant cells have a higher potential to metastasize than parental cells. Treatment with ATRA of mice injected with CT26 and CT26‐P decreased the number of cancer nodules, suggesting that ATRA inhibits metastasis of both paclitaxel‐resistant and parental colorectal cancer cells. Mice injected with CT26‐C also had fewer cancer nodules, suggesting that Cx43 transfection inhibits metastasis. Figure 6C shows the survival curves of all treatment five groups. All the mice ultimately died because of pulmonary metastasis. The mice injected with CT26‐P cells had the shortest survival time. However, mice treated with ATRA, whether injected with CT26 or CT26‐P cells, survived significantly longer than their untreated counterparts. Treatment of mice with ATRA (0.585 mg/kg) only resulted in survival beyond 60 days. In addition, mice injected with CT26‐C cells had longer survival time than those injected with CT26‐P cells.
Figure 6.
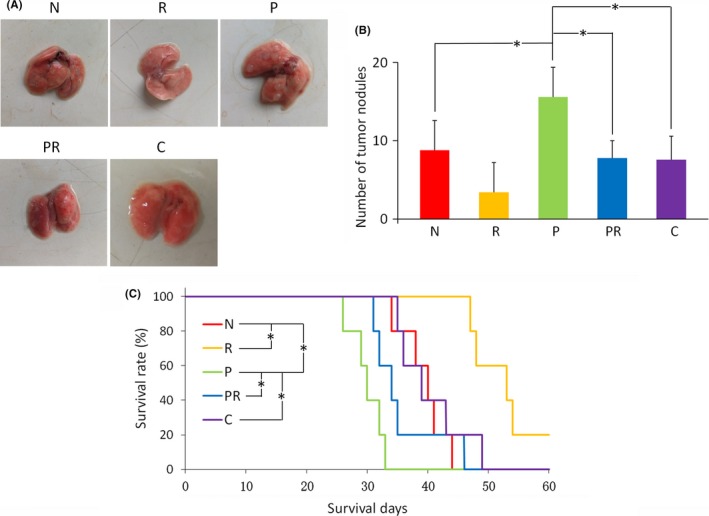
Pulmonary metastases and the survival curves of mice, n (per group) = 5. “N” are mice injected with CT26 cells, “R” are mice of group “N” treated with all‐trans‐retinoic acid (ATRA), “P” are mice injected with CT26‐P cells, “PR” are mice of group “P” treated with ATRA, and “C” are mice injected with CT26‐C cells. A, Typical morphology of the lungs of each treatment group. B, Number of tumor nodules for each treatment group. *P < .05 represents a significant difference. C, Survival curves of the mice. *P < .05 represents a significant difference
4. DISCUSSION
Tumor cells undergo a series of metastatic steps to become malignant and invasive. EMT is an important process involved in these steps and results in morphological changes. During EMT, cells lose epithelial properties, such as expression of E‐cadherin, and acquire mesenchymal characteristics, which increase the motility and invasion abilities of cells. According to several studies, there is a close link between EMT and resistance to several chemotherapeutic agents.3, 4 These agents may alter the apoptotic process and lead to increased expression of EMT‐inducible factors, resulting in failure of therapy. In the present study, the three colorectal cancer cell lines, LoVo, HCT116 and CT26, also showed more mesenchymal characteristics when they acquired resistance to paclitaxel.
It has been reported that ATRA inhibits the activation of NF‐κΒ and upregulates the expression of Cx43, which are both involved in the EMT process.17, 20, 21, 22, 24, 25 Paclitaxel‐resistant cells often have high levels of NF‐κΒ activation.31 Expression of NF‐κΒ is sufficient to induce EMT and has been known to upregulate expression of snail and downregulate expression of E‐cadherin.32 Our results show that ATRA can inhibit NF‐κΒ by decreasing the activity of IKK. This may lead to the re‐expression of E‐cadherin in paclitaxel‐resistant cells following ATRA treatment.
Generally, cancer cells lose their GJ function when they acquire chemoresistance.33 Re‐expression of Cx43 can enhance the cytotoxicity of paclitaxel.14 As a structural component of GJ, Cx43 is responsible for the transfer of water‐soluble molecules directly from one cell to another without passing through the membrane. Enhancement of the cytotoxic effects of Cx43 is attributed to the increased transfer of drugs from one cell to another. Connexins are known tumor suppressors and were recently found to also inhibit metastasis.34, 35 EMT is a process by which cells gradually lose contact. GJ composed of Cx43 provide an intercellular channel which can also form a hemichannel to exchange materials with the extracellular matrix. For example, Cx43 reduces EMT marker expression in endometrial stromal cells and promotes the switch from N‐ to E‐cadherin in glioma stem cells.34, 35 In this study, ATRA was found to upregulate Cx43 expression and GJ function in all three paclitaxel‐resistant colorectal cancer cell lines. Cx43 transfection to paclitaxel‐resistant cells also reversed their EMT phenotype. These results suggest that ATRA may reverse EMT by upregulating Cx43 expression. Upregulation of Cx43 can increase the interaction between cells and the external environment, which may offset the loss of intercellular communication induced by EMT. We also found that Cx32 expression decreases in paclitaxel‐resistant cells and ATRA upregulates it. However, Cx26 is not affected by ATRA and paclitaxel. Cx32 is considered a tumor suppressor. It can inhibit invasiveness and growth of cancer cells and impede tumor progression.36 These results suggested that other GJ molecules may also contribute to the EMT reverse. In vitro results confirm that ATRA treatment reversed EMT phenotypes in paclitaxel‐resistant cells. Scratch wound‐healing and Transwell assays also indicate that ATRA reduces the migration and invasion abilities of paclitaxel‐resistant cells. Furthermore, ATRA was found to reduce the migration and invasion abilities of parental CT26 cells as well as reverse EMT, which may be because CT26 is a mesenchymal cell line.
ATRA and Cx43 transfection reduces the expression of fibronectin, MMP‐9, N‐cadherin, snail, vimentin, and β‐catenin and increases the expression of E‐cadherin in paclitaxel‐resistant cells and parental CT26 cells, showing that ATRA reverses EMT in mesenchymal cells and Cx43 contributes to this effect. Fibronectin, a mesenchymal marker, is an extracellular matrix protein. E‐cadherin is the main cell‐to‐cell and cell‐to‐matrix adhesion protein. N‐cadherin and vimentin are both mesenchymal cell‐related proteins. “Cadherin switch”, which refers to the downregulation of E‐cadherin and upregulation of N‐cadherin, is a common feature of EMT. NF‐κΒ activation may induce snail expression, and repression of E‐cadherin expression by the transcription factor snail is a central event during the loss of epithelial phenotype.32 Like snail, β‐catenin is another transcription factor underlying the specific gene program of EMT. It can repress the transcription of E‐cadherin and enhance the expression of MMP, such as MMP‐9.37
Last, in vivo results show that pulmonary metastasis is inhibited by ATRA treatment and Cx43 transfection inhibits metastasis of CT26‐P cells. Although CT26 is a mesenchymal cell line, several mesenchymal markers were upregulated when paclitaxel resistance was acquired. Invasion, migration, and metastatic abilities also increased in the paclitaxel‐resistant cells. CT26 is commonly used in the Balb/c pulmonary metastasis model.38 In vivo results show that ATRA inhibits pulmonary metastasis induced by paclitaxel‐resistant CT26 cells, which suggests that ATRA could be used to reverse EMT in chemoresistant cells.
In summary, paclitaxel is a standard treatment for various cancers but it can increase the motility and invasion of cancer cells. ATRA is commonly used in combination with paclitaxel to improve patient outcomes.39 It has been reported that this combination is effective for inhibition of the invasion of tumor cells.8 In our study, it was confirmed that, as a single agent, ATRA reverses the upregulation of EMT in paclitaxel‐resistant colorectal cancer cell lines. ATRA was found to increase Cx43 expression and GJ function and inhibit NF‐κΒ, which may all be involved in reversing EMT. The in vitro and in vivo results confirmed that ATRA reduces the invasion, migration, and metastatic abilities of paclitaxel‐resistant colorectal cancer cells. This study provides data that can be used for the development of new therapeutic approaches for the treatment of chemorefractory tumors in the future.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was partially supported by the National Basic Research Program of China Grant number 2016YFC0102404, and Science and Technology Fund of Tianjin Municipal Health Bureau under grant number 2015KY30, and the Natural Science Foundation of China Grant numbers 11774256, 61505109, and National Key Scientific Research Instrument Development Project Grant number 61427819, and Tianjin 131 Innovative Talent Training Project in 2018, and the State Key Laboratory of Medicinal Chemical Biology of Nankai University.
Shi G, Zheng X, Wu X, Wang S, Wang Y, Xing F. All‐trans retinoic acid reverses epithelial‐mesenchymal transition in paclitaxel‐resistant cells by inhibiting nuclear factor kappa B and upregulating gap junctions. Cancer Sci. 2019;110:379–388. 10.1111/cas.13855
Guiling Shi and Xiaoli Zheng contributed equally to this work.
Contributor Information
Yijia Wang, Email: yijiawang_1980@163.com.
Fei Xing, Email: xingfei@sdut.edu.cn.
REFERENCES
- 1. Wang TH, Wanf HS, Soong YK. Paclitaxel‐induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88(11):2619‐2628. [DOI] [PubMed] [Google Scholar]
- 2. Du F, Wu X, Liu Y, et al. Acquisition of paclitaxel resistance via PI3K‐dependent epithelial‐mesenchymal transition in A2780 human ovarian cancer cells. Oncol Rep. 2013;30(3):1113‐1118. [DOI] [PubMed] [Google Scholar]
- 3. Kajiyama H, Shibata K, Terauchi M, et al. Chemoresistance to paclitaxel induces epithelial‐mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31(2):277‐283. [PubMed] [Google Scholar]
- 4. Tiwari N, Gheldof A, Tatari M, Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol. 2012;22(3):194‐207. [DOI] [PubMed] [Google Scholar]
- 5. Zhou Z, Zhang L, Xie B, et al. FOXC2 promotes chemoresistance in nasopharyngeal carcinomas via induction of epithelial mesenchymal transition. Cancer Lett. 2015;363(2):137‐145. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y, Zhang C, Zhang S, et al. Kanglaite sensitizes colorectal cancer cells to Taxol via NF‐κΒ inhibition and connexin 43 upregulation. Sci Rep. 2017;7(1):1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karmakar S, Banik NL, Ray SK. Combination of all‐trans retinoic acid and paclitaxel‐induced differentiation and apoptosis in human glioblastoma U87MG xenografts in nude mice. Cancer. 2010;112(3):596‐607. [DOI] [PubMed] [Google Scholar]
- 8. Hong GY, Jeong YI, Sang JL, Lee E, Oh JS, Lee HC. Combination of paclitaxel‐ and retinoic acid‐incorporated nanoparticles for the treatment of CT‐26 colon carcinoma. Arch Pharmacal Res. 2011;34(3):407‐417. [DOI] [PubMed] [Google Scholar]
- 9. Ross SA, Mccaffery PJ, Drager UC, De Luca LM. Retinoids in embryonal development. Physiol Rev. 2000;80(3):1021. [DOI] [PubMed] [Google Scholar]
- 10. Hansen LA, Sigman CC, Andreola F, Ross SA, Kelloff GJ, De Luca LM. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis. 2000;21(7):1271‐1279. [PubMed] [Google Scholar]
- 11. Cui J, Gong M, He Y, Li Q, He T, Bi Y. All‐trans retinoic acid inhibits proliferation, migration, invasion and induces differentiation of hepa1‐6 cells through reversing EMT in vitro. Int J Oncol. 2015;48(1):349. [DOI] [PubMed] [Google Scholar]
- 12. Lan L, Cui D, Luo Y, et al. Inhibitory effects of retinoic acid on invasiveness of human thyroid carcinoma cell lines in vitro. J Endocrinol Invest. 2009;32(9):731‐738. [DOI] [PubMed] [Google Scholar]
- 13. Liu H, Zang C, Fenner MH, Possinger K, Elstner E. PPARγ Ligands and ATRA inhibit the invasion of human breast cancer cells in vitro. Breast Cancer Res Treat. 2003;79(1):63‐74. [DOI] [PubMed] [Google Scholar]
- 14. Wang SQ, Zhang SW, Zhao ZY, Zhang CZ, Yang XY, Wang YJ. Connexin 43 enhances paclitaxel cytotoxicity in colorectal cancer cell lines. Exp Ther Med. 2017;14:1212‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi G, Zheng X, Zhang S, et al. Kanglaite inhibits EMT caused by TNF‐α via NF‐κΒ inhibition in colorectal cancer cells. Oncotarget. 2018;9(6):6771‐6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suzuki S, Kawakami S, Chansri N, Yamashita F, Hashida M. Inhibition of pulmonary metastasis in mice by all‐trans retinoic acid incorporated in cationic liposomes. J Controlled Release. 2006;116(1):58‐63. [DOI] [PubMed] [Google Scholar]
- 17. Sutcliffe AM, Clarke DL, Bradbury DA, Corbett LM, Patel JA, Knox AJ. Transcriptional regulation of monocyte chemotactic protein‐1 release by endothelin‐1 in human airway smooth muscle cells involves NF‐κB and AP‐1. Br J Pharmacol. 2010;157(3):436‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Q, Helfand BT, Jang TL, et al. Nuclear factor‐κB‐mediated transforming growth factor‐β‐induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clin Cancer Res. 2009;15(10):3557‐3567. [DOI] [PubMed] [Google Scholar]
- 19. Gupta SC, Kannappan R, Reuter S, Kim JH, Aggarwal BB. Chemosensitization of tumors by resveratrol. Ann N Y Acad Sci. 2011;1215(1):150‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayden MS, Ghosh S. Signaling to NF‐κB. Genes Dev. 2004;18(18):2195‐2224. [DOI] [PubMed] [Google Scholar]
- 21. Karkeni E, Bonnet L, Astier J, et al. All‐trans‐retinoic acid represses chemokine expression in adipocytes and adipose tissue by inhibiting NF‐κB signaling. J Nutr Biochem. 2017;42:101‐107. [DOI] [PubMed] [Google Scholar]
- 22. Hong K, Zhang Y, Guo Y, et al. All‐trans retinoic acid attenuates experimental colitis through inhibition of NF‐κB signaling. Immunol Lett. 2014;162(1):34‐40. [DOI] [PubMed] [Google Scholar]
- 23. Yamasaki H, Naus CCG. COMMENTARY: role of connexin genes in growth control. Carcinogenesis. 1996;17(6):1199. [DOI] [PubMed] [Google Scholar]
- 24. Tang N, Wang Q, Wu D, Zhang S, Zhang Y, Tao L. Differential effects of paclitaxel and docetaxel on gap junctions affects their cytotoxicities in transfected HeLa cells. Mol Med Rep. 2013;8:638‐644. [DOI] [PubMed] [Google Scholar]
- 25. Teleki I, Krenacs T, Szasz MA, et al. The potential prognostic value of connexin 26 and 46 expression in neoadjuvant‐treated breast cancer. BMC Cancer. 2013;13:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y, Zhang S, Zhang C, et al. Investigation of an SPR biosensor for determining the influence of connexin 43 expression on the cytotoxicity of cisplatin. Analyst. 2016;141(11):3411‐3420. [DOI] [PubMed] [Google Scholar]
- 27. Liu Y, Wen Q, Chen X, et al. All‐trans retinoic acid arrests cell cycle in leukemic bone marrow stromal cells by increasing intercellular communication through connexin 43‐mediated gap junction. J Hematol Oncol. 2015;8(1):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang JM, Yang GY, Medina DJ, Vassil AD, Liao J, Hait WN. Treatment of multidrug resistant (MDR1) murine leukemia with P‐glycoprotein substrates accelerates the course of the disease. Biochem Biophys Res Commun. 1999;266(1):167‐173. [DOI] [PubMed] [Google Scholar]
- 29. Adachi Y, Itoh F, Yamamoto H, et al. Retinoic acids reduce matrilysin (matrix metalloproteinase 7) and inhibit tumor cell invasion in human colon cancer. Tumor Biol. 2001;22(4):247‐253. [DOI] [PubMed] [Google Scholar]
- 30. Guan J, Zhang H, Wen Z, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL‐6 in cancer associated fibroblast cells. Cancer Lett. 2014;345(1):132‐139. [DOI] [PubMed] [Google Scholar]
- 31. In LL, Azmi MN, Ibrahim H, Awang K, Nagoor NH. 1'S‐1’‐acetoxyeugenol acetate: a novel phenylpropanoid from Alpinia conchigera enhances the apoptotic effects of paclitaxel in MCF‐7 cells through NF‐κB inactivation. Anticancer Drugs. 2011;22(5):424‐434. [DOI] [PubMed] [Google Scholar]
- 32. Julien S, Puig I, Caretti E, et al. Activation of NF‐kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26(53):7445‐7456. [DOI] [PubMed] [Google Scholar]
- 33. Su M, Zhang Q. Deficiency of gap junction composed of connexin43 contributes to oxaliplatin resistance in colon cancer cells. Oncol Lett. 2017;14(3):3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu J, Berga SL, Johnston‐Macananny EB, et al. Endometrial stromal decidualization responds reversibly to hormone stimulation and withdrawal. Endocrinology. 2016;157(6):2432‐2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gangoso E, Thirant C, Chneiweiss H, Medina JM, Tabernero A. A cell‐penetrating peptide based on the interaction between c‐Src and connexin43 reverses glioma stem cell phenotype. Cell Death Dis. 2014;5(5):e1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hada S, Sato H, Virgona N, et al. Connexin 32 expression reduces malignant phenotype in human A549 adenocarcinoma cells: implication of Src involvement. Oncol Rep. 2006;16(5):1149‐1154. [PubMed] [Google Scholar]
- 37. Oral D, Erkekoglu P, Kocer‐Gumusel B, Chao MW. Epithelial‐mesenchymal transition: a special focus on phthalates and bisphenol A. J Environ Pathol Tox. 2016;35(1):43. [DOI] [PubMed] [Google Scholar]
- 38. Weng YL, Liao HF, Li AF, Chang JC, Chiou RY. Oral administration of resveratrol in suppression of pulmonary metastasis of BALB/c mice challenged with CT26 colorectal adenocarcinoma cells. Mol Nutr Food Res. 2010;54(2):259‐267. [DOI] [PubMed] [Google Scholar]
- 39. Arrieta O, González‐De la Rosa Claudia H, Aréchaga‐Ocampo E, et al. Randomized phase II trial of All‐trans‐retinoic acid with chemotherapy based on paclitaxel and cisplatin as first‐line treatment in patients with advanced non‐small‐cell lung cancer. J Clin Oncol. 2010;28(21):3463‐3471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials