

Abstract
Inhibition of the hypoxia-inducible factor 1α (HIF-1α) pathway by disrupting its association with the transcriptional coactivator p300 inhibits angiogenesis and tumor development. Development of HIF-1α/p300 inhibitors has been hampered by preclinical toxicity; therefore, we aimed to identify novel HIF-1α/p300 inhibitors. Using a cell-free assay designed to test compounds that block HIF-1α/p300 binding, 170 298 crude natural product extracts and prefractionated samples were screened, identifying 25 active extracts. One of these extracts, originating from the marine sponge Latrunculia sp., afforded six pyrroloiminoquinone alkaloids that were identified as positive hits (IC50 values: 1–35 μM). Luciferase assays confirmed inhibition of HIF-1α transcriptional activity by discorhabdin B (l) and its dimer (2), 3-dihydrodiscorhabdin C (3), makaluvamine F (5), discorhabdin H (8), discorhabdin L (9), and discorhabdin W (11) in HCT 116 colon cancer cells (0.1–10 μM, p < 0.05). Except for 11, all of these compounds also reduced HIF-1α transcriptional activity in LNCaP prostate cancer cells (0.1–10 μM, p < 0.05). These effects occurred at noncytotoxic concentrations (<50% cell death) under hypoxic conditions. At the downstream HIF-1α target level, compound 8 (0.5 μM) significantly decreased VEGF secretion in LNCaP cells (p < 0.05). In COLO 205 colon cancer cells no activity was shown in the luciferase or cytotoxicity assays. Pyrroloiminoquinone alkaloids are a novel class of HIF-1α inhibitors, which interrupt the protein−protein interaction between HIF-1α and p300 and consequently reduce HIF-related transcription.
Graphical Abstract
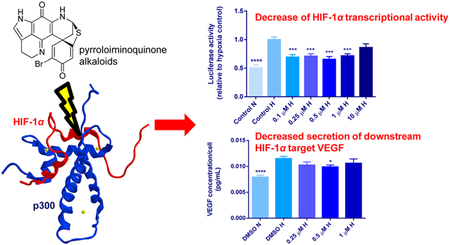
Hypoxia, a general feature of solid tumors, plays a critical role in various cellular processes including angiogenesis, cancer development, and progression. Cellular responses to hypoxia are orchestrated by activation of the heterodimeric transcription factor hypoxia-inducible factor 1 (HIF-1). HIF-1 is composed of an O2-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit. Upon stabilization by hypoxia, the HIF-1α subunit accumulates and dimerizes with HIF-1β. The HIF-1α/β complex binds to cognate hypoxia response elements (HREs) and activates transcription of many HIF-1 target genes, such as vascular endothelial growth factor (VEGF), glucose transporter 1, enolase-1 (ENO1), and lactate dehydrogenase A (LDHA).1 In this process, the transcriptional coactivators p300 and CREB-binding protein (CBP) bind to HIF-1α to stabilize the HIF-1α/β-HRE complex.1
In many human tumors HIF-1α is overexpressed, including colon, breast, lung, and prostate carcinomas.2 In certain cancer types, including colon cancer,3 expression of HIF-1α is associated with VEGF expression and microvessel density.4 Direct inhibition of HIF-1α, e.g., by disrupting the binding between HIF-1α and p300, is therefore an interesting and widely studied drug target in cancer research.5–9 Previously, our laboratory generated a high-throughput screening (HTS) assay that can be used to identify small-molecule inhibitors of HIF-1α through inhibiting the binding interaction between the C-terminal transactivation domain (CTAD) of HIF-1α and the cysteine histidine-rich domain 1 (CH1) domain of p300.9 We subsequently showed that the natural compounds gliotoxin, chaetocin, and chetomin (CTM), members of the epidithiodiketopiperazine (ETP) family, were able to disrupt the HIF-1α/p300 complex in multiple cancer cell lines, downregulated the expression of HIF-1α target genes, inhibited angiogenesis in vitro, and inhibited tumor growth in vivo.6,9 However, clinical application of ETPs is limited by their toxicity.6
Therefore, our objective was to identify more specific and less toxic HIF-1α inhibitors by using the HTS assay we developed to screen for compounds and natural product extracts that can disrupt the HIF-1α/p300 complex. Our recently described HIF-1α/p300 assay10 enabled us to perform a high-throughput screening of extracts from the National Cancer Institute’s Natural Products Repository, which led to the discovery of pyrroloiminoquinone alkaloids including discorhabdin and makaluvamine alkaloids originating from a Latrunculia sp. sponge as potential HIF-1α/p300 inhibitors. These natural products comprise a large and well-studied family of marine-derived alkaloids that contain a tricyclic pyrroloiminoquinone core structure11,12 and have been reported to exhibit many potent biological activities, including cytotoxic, antiviral, antimalarial, and immunomodulatory effects.12 However, the molecular mechanisms or target of these compounds remain to be elucidated. Here we describe the identification of pyrroloiminoquinone alkaloids as HIF-1α/p300 inhibitors, followed by their biological effects in several cancer cell lines.
RESULTS AND DISCUSSION
HIF-1 α/p300 Screening Results.
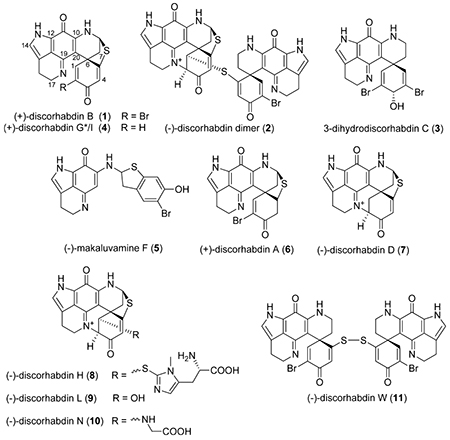
A total of 170 298 crude natural product extracts and prefractionated extract samples were screened in the HIF-1α/p300 assay at a concentration of 10 μg/mL. Reconfirmation studies of the initial hits identified 25 extracts as potential projects for bioassay-guided natural product isolation efforts. One of these extracts (NSC# C005119), sourced from an Australian Latrunculia sp. marine sponge, exhibited robust activity in the screening assay, and it was selected for a detailed chemical study. Fractionation of the extract by repeated chromatography on diol, Sephadex LH-20, and C18 media ultimately led to the isolation of a family of pyrroloiminoquinone alkaloids, namely, (+)-discorhabdin B (l),13 a (−)-discorhabdin B dimer (2),14 3-dihydrodiscorhabdin C (3),15 (+)-discorhabdin G*/I (4),16,17 and (−)-makaluvamine F (5).18 These compounds were all previously isolated from marine sponges, with the exception of the dimeric compound 2, which was shown to form as a consequence of long-term storage of discorhabdin B (l).14 Structural assignments and purity of all of the isolated natural products were confirmed by LC-MS and by comparison of their spectroscopic data (HRMS, 1H NMR, 13C NMR, αD), with the appropriate literature values.
To enlarge the panel of compounds for comparative testing, samples of (+)-discorhabdin A (6), (−)-discorhabdin D (7), (−)-discorhabdin H (8), (−)-discorhabdin L (9), (−)-discorhabdin N (10), and (−)-discorhabdin W (11), previously reported from New Zealand specimens of Latrunculia sp.,17 were included in the study. Spectroscopic analysis of all the compounds used in this study confirmed they were ≥95% pure (Supporting Information). Compounds 1, 2, 3, 5, 8, and 9 were identified as the most potent HIF-1α/p300 inhibitors in the screening assay (IC50 values shown in Table 1) and were evaluated further for biological activity in cell-based assays. Discorhabdin W (11) showed no activity in the screening assay and was therefore included as a negative control in subsequent experiments.
Table 1.
IC50 Values of the Most Potent Compounds in the HIF-1α/p300 Screening Assay
| compound | IC50 value (μM) ± SD |
|---|---|
| chetomin | 1.9 ± 0.5 (positive control) |
| 1 | 3.7 ± 1.8 |
| 2 | 2.4 ± 0.1 |
| 3 | 3S.2 ± 1S.6 |
| 5 | 8.3 ± 0.2 |
| 8 | 2.2 ± 0.6 |
| 9 | 0.73 ± 0.18 |
| 11 | >100 (negative control) |
Cytotoxicity in Human Cancer Cell Lines.
Our previous studies with inhibitors of HIF-1α/p300 demonstrated sensitivity in HCT 116 and prostatic carcinoma cell lines.6,9 Therefore, we tested the cytotoxicity of compounds 1, 2, 3, 5, 8, 9, and 11 in HCT 116 and LNCaP cells under hypoxic conditions (0.5−1% O2). Tirapazamine (TPZ), previously shown to be an HIF-1α inhibitor,19 was used as a positive control in all CCK-8 assays since TPZ increases chemosensitivity in many oxygen-deprived human cancer cell lines.20 As shown in Table 2, most compounds were well tolerated in HCT 116 cells at concentrations up to 10 μM (18 h) or 50 μM (48 h). Interestingly, pyrroloiminoquinone alkaloids (including compounds 3 and 4) have previously been shown to be highly cytotoxic in HCT 116 cells, with IC50 values in the lower micromolar range.21 Most likely, the difference in cytotoxicity is due to a longer treatment time (72 h) performed under normoxic conditions in the study by Antunes et al.21 The hypoxic conditions in our cytotoxicity experiments could also explain the difference in drug sensitivity, as certain drugs are more cytotoxic in normoxia than in hypoxia.20 In fact, the observed limited cytotoxicity under hypoxia suggests that oxygen could mediate the cytotoxicity of pyrroloiminoquinone alkaloids, since their reduction potentials fall within biologically relevant reduction ranges,18,22 suggesting that these compounds could couple with oxygen to generate reactive oxygen species. In LNCaP cells discorhabdin and makaluvamine alkaloids were also well tolerated (Table 2), as shown by minimal cytotoxicity at the maximum test concentration of 10 μM. Also in colorectal COLO 205 cells the majority of the tested compounds (2, 5, 8, 11) did not cause substantial cell death up to concentrations of 10 μM (Table 2).
Table 2.
Cytotoxicity Results of Pyrroloiminoquinone Alkaloids in the CCK-8 Cytotoxicity Assay under Hypoxic Conditions in Three Cancer Cell Lines
| ICSO value (μM) ± SEMa | ||||
|---|---|---|---|---|
| compound | HCT 116 (18 h, 0.5% O2) | HCT 116 (48 h, 0.5% O2) | LNCaP (18 h, 1% O2) | COLO 205 (48 h, 0.5% O2) |
| tirapazamine (positive control) | 72.7 ± 6.9 | 11.9 ± 1.3 | -c | 47.7 ± 12.2d |
| 1 | 3.1 ± 1.4 | NA | 7.0 ± 1.3 | 1.8 ± 1.5 |
| 2 | >10 | 26.8 ± 5.4b | >10 | >10 |
| 3 | 8.6 ± 0.3b | NA | >10 | 5.1 ± 1.1 |
| 5 | 7.2 ± 1.9 | NA | >10 | >10 |
| 8 | >10 | >50 | >10 | >10 |
| 9 | 6.2 ± 2.2 | >50 | 8.3 ± 2.5 | 7.6 ± 1.2 |
| 11 | >10 | NA | >10 | >10 |
IC50 values are presented as the average of triplicate experiments ± SEM (n = 3 or 4).
Outlier excluded (duplicate experiment).
IC50 not calculated: only one concentration tested (25 μM).
IC50 based on quintuplicate experiments (n = 3 or 4).
Biological Activity I: Inhibition of HIF-1α Activity in an HIF Reporter Gene Assay.
A dual luciferase reporter assay was carried out to determine the inhibitory effects of compounds 1,2, 3, 5, 8, 9, and 11 on the transcriptional activity of HIF-1α. Hereto HCT 116, LNCaP, and COLO 205 cells were transfected with an HIF-1 reporter plasmid containing a hypoxia response element that mediates HIF-1-dependent gene transcription. In HCT 116 cells, all compounds significantly decreased HIF-1α transcriptional activity at one or more concentrations (Figure 1). As shown by the greatest effect at the lowest test concentration of 0.1 μM, compound 2 appeared to be the most potent at decreasing reporter activity. Compounds 1, 2, 3, 8, 9, and 10 were active at concentrations that were not notably cytotoxic (<50% cell death) in the cytotoxicity assay (Figure 1), indicating that the decrease in the HRE-mediated reporter activity was not caused by nonspecific cell death. Surprisingly, compound 11, which was inactive in the protein−protein assay, reduced HIF reporter activity in HCT 116 cells. None of the tested alkaloids (0.1−10 μM) significantly decreased the hypoxia-induced luciferase signal in COLO 205 cells (Supporting Information). It has been shown previously that the hypoxic induction of HIF-1α is differentially regulated in different colon cancer cells. As such, anti-VEGF antibodies that significantly inhibited the hypoxic induction of HIF-1α in HCT 116 cells were not effective in COLO 205 cells.23 Indeed, in our studies HCT 116 cells were more sensitive to inhibition of HIF-1α activity by pyrroloiminoquinone alkaloids. The differential effects of these compounds on HIF-1α transcriptional activity in COLO 205 vs HCT 116 colon cancer cells demonstrate that discorhabdin and makaluvamine alkaloids exhibit cell-type specificity similar to anti-VEGF antibodies. Most compounds also reduced luciferase activity at one or more concentrations in LNCaP cells (Figure 2). In more detail, the following compounds were effective at nontoxic concentrations (based on cytotoxicity results presented in Table 2): 1 (1 μM), 2 (1 μM), 3 (1 μM), 5 (0.1, 1 μM), 8 (0.1 μM), and 9 (1 μM). Compound 11 did not show activity in LNCaP cells. The results of the HIF reporter assay in HCT 116 and LNCaP cells are similar to previous reports that drugs inactive in cytotoxicity assays (e.g., thalidomide, lenalidomide) may still have clinical activity through mechanisms on the immune system or on the tumor microenvironment that may not be detectable in a cytotoxic cell line screen.24 Interestingly, in the HIF reporter assay several compounds (2 and 8 in HCT 116 and LNCaP cells and 9 in HCT 116 cells) displayed a U-shaped dose−response: their activity at lower concentrations was reduced at higher concentrations. U-shaped dose−response curves are often observed with antiangiogenic agents,25 as exemplified by the effects of the angiogenesis inhibitor endostatin on HIF-1α gene expression in human endothelial cells26 and on tumor growth in tumor-bearing mice.27,28 In addition, other angiogenesis inhibitors (e.g., interferon-α,29 rosiglitazone,30 angiostatin31) exhibited similar U-shaped dose−efficacy curves in vivo. Various other mechanisms of action have been reported for pyrroloiminoquinone alkaloids. For example, the electrophilic activity of compound 1 has been suggested to potentiate its cytotoxic effects.14 We, however, do not expect that electrophilic reactivity is the underlying mechanism for disrupting the HIF-1α/p300 binding, because of the presence of a positively charged zinc ion between the C-TAD of HIF-1α and CH1 of p300.9 In fact, ejection of this zinc ion was shown to be the mechanism of action of ETPs.9 Whether pyrroloiminoquinone alkaloids share the same mechanism of action has yet to be determined. Moreover, despite lacking the electrophilic Δ1,2 unsaturation, compounds 8 and 9 still showed activity in our HIF reporter assay in both HCT 116 and LNCaP cells. Furthermore, makaluvamines, such as compound 5, have been shown to inhibit DNA topoisomerase II,18 which is involved in the translation of HIF-1α.32 Clearly, these compounds may have multiple mechanisms of action, as shown by our discovery of compound 5 as HIF-1α/p300 inhibitor. Wada et al. tested a range of discorhabdin A (compound 6) analogues in a panel of cytotoxicity and mechanism-based assays;33 however they could not conclude anything definitive regarding the molecular mechanism or targets of these compounds. All of the assays were run under oxic conditions, whereas the assays in our study were run under hypoxic conditions. As such, we demonstrated the significance of oxygen in mediating the cytotoxic effects of discorhabdins. Several discorhabdins have been tested for antitumor activity in vivo, but discorhabdin D (compound 7) is the only reported discorhabdin with such activity.34 Despite showing cytotoxicity to cancer cells in vitro, several discorhabdins (A, B, C,34 P, and discorhabdin analogue 54a33) did not reduce tumor growth in xenograft models. This lack of activity is likely due to instability of the compounds in vivo, since the latter two compounds either were not detected or were barely detectable in blood after administration.33 On the other hand, the iminoquinone structure of the active discorhabdin D has been reported to be more stable due to cross-linking between the nitrogen atom of the iminoquinone and the α-carbon of the spiroenone.33 On the basis of this finding, compounds 2, 8, and 9 also have stable iminoquinone structures and are therefore most likely to be stable in vivo. Therefore, these compounds were selected for further testing in downstream assays.
Figure 1.

HIF reporter assay results of pyrroloiminoquinone alkaloids in HCT 116 cells after 18 h treatment under hypoxic conditions (0.5% O2). Reporter activity values of pyrroloiminoquinone alkaloids are normalized to hypoxia control (control H) and are expressed as the average of triplicate experiments ±SEM (n = 3 or 4). For the positive control chetomin results are average values ±SEM from 5 to 6 replicate experiments (n = 4). Data were analyzed using the Wilcoxon rank sum test with Hochberg’s method for adjusting p values for multiple comparisons (reference group: control H). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. N = normoxia; H = hypoxia.
Figure 2.

HIF reporter assay results of pyrroloiminoquinone alkaloids in LNCaP cells after 18 h treatment under hypoxic conditions (1% O2). Reporter activity values of pyrroloiminoquinone alkaloids are normalized to hypoxia control (control H) and are expressed as the average of triplicate experiments ±SEM (n = 3 or 4). For the positive control chetomin results are average values ±SEM from 5 to 6 replicate experiments (n = 4). Data were analyzed using the Wilcoxon rank sum test with Hochberg’s method for adjusting p values for multiple comparisons (reference group: control H). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. N = normoxia; H = hypoxia.
Biological Activity II: Effects on Downstream HIF-1α Target Gene.
Since VEGF is one of the downstream targets of HIF-1α, we conducted VEGF ELISA assays in LNCaP and HCT 116 cells under hypoxic conditions. LNCaP cells were treated for 18 h with compounds 2, 8, and 9 within the concentration range that significantly reduced HIF-1 transcriptional activity in the HIF reporter gene assay (Figure 2). As shown in Figure 3A–C, a significant decrease of VEGF secretion was observed for compound 8 (0.5 μM), while compound 9 caused a borderline significant decrease of VEGF concentrations (0.25–1 μM). The effects of the test compounds were cell-type specific, as shown by the VEGF ELISA results in HCT 116 cells. After 48 h treatment with compounds 2, 8, and 9 at concentrations with activity in the HIF reporter assay, none of these compounds significantly reduced hypoxia-induced VEGF secretion after comparison with the hypoxia control (Figure 3D,E; data not shown). However, we did observe a statistically nonsignificant trend in the decrease of VEGF secretion by compound 2 (0.1–0.25 μM) after both 18 h (data not shown) and 48 h treatments (Figure 3D). Discrepancies between HIF activity and effects on HIF downstream targets have been previously reported for HIF inhibitors.35 In our experience, HIF-1α/p300 inhibitors have been shown to have differential effects on downstream HIF targets that are cell-type specific.6 For example, gliotoxin lowered ENO1, VEGF, and LDHA mRNA expression in PC3 prostate cancer cells but exhibited no effects in HCT 116 cells. Previous studies by McKee et al. on HIF-2 inhibitors with demonstrated HIF reporter activity also produced variable results with affecting downstream VEGF protein levels as well as other downstream targets that were cell-line-dependent.35 This suggests that direct HIF inhibitors may exhibit more than one mechanism of action that would subsequently affect downstream target genes, and these effects may be cell-type-specific. Considering the differential effects of HIF-1α/p300 inhibitors on downstream HIF target genes,6 ongoing and future studies will elucidate the effects of our compounds on the expression of other HIF-1 downstream targets at the post-transcriptional and post-translational level.
Figure 3.

VEGF ELISA results of compounds 2, 8, 9, and chetomin in LNCaP (A−C) and HCT 116 cells (D−F) after 18 h (LNCaP) or 48 h (HCT 116) treatment under hypoxic conditions (0.5−1% O2). Results are expressed as VEGF concentration per cell (pg/mL) and are expressed as the average of triplicate (LNCaP) or quadruplicate (HCT 116) experiments ± SEM (n = 3). Data were analyzed using either the exact Wilcoxon rank sum test (LNCaP) or the stratified Wilcoxon rank sum test (HCT 116) with Hochberg’s method for adjusting p values for multiple comparisons (reference group: control H). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. N = normoxia; H = hypoxia.
In summary, for the first time we describe the novel role of pyrroloiminoquinone alkaloids as potential HIF-1α/p300 inhibitors. Five discorhabdins and one makaluvamine alkaloid that disrupted the HIF-1α/p300 interaction in a cell-free protein−protein assay were tested in COLO 205, HCT 116, and LNCaP cell lines. The activity of the compounds was confirmed in HCT 116 and LNCaP cells (but not in COLO 205 cells): the vast majority of the compounds inhibited the transcriptional activity of HIF-1α (Table 3). Compounds 1, 2, 3, 5, 8, and 9 were active in both cell lines and are therefore the most interesting compounds for further evaluation. Promisingly, results of the reporter gene assay were confirmed by inhibitory activity on VEGF secretion by compound 8 and to a lesser extent by compound 9 in LNCaP cells. Future preclinical studies will characterize the molecular mechanism of action of pyrroloiminoquinone alkaloids, their effects on angiogenesis and tumor growth, and toxicity profile in order to determine their potential as novel HIF-1α/p300 inhibitors.
Table 3.
Concentrations of Pyrroloiminoquinone Alkaloids Causing Significant Inhibition of Luciferase Activity in COLO 205, HCT 116, and LNCaP Cells (p < 0.05)
| compound | COLO 205 | HCT 116 | LNCaP |
|---|---|---|---|
| 1 | no effect | 0.1,0.25,0.5, 1, 10a μM | 1, 10a μM |
| 2 | no effect | 0.1, 0.25, 0.5, 1 μM | 1 μM |
| 3 | no effect | 10 μM | 1, 10a μM |
| 5 | no effect | 0.25, 1 μM | 0.1, 1, 10 μM |
| 8 | no effect | 0.5, 1 μM | 0.1 μM |
| 9 | no effect | 0.1, 0.25, 0.5, 1, 10a μM | 1, 10a μM |
| 11 | no effect | 0.1, 0.25,0.5, 1, 10 μM | no effect |
>50% cell death in CCK-8 assay.
EXPERIMENTAL SECTION
General Experimental Procedures.
TPZ, CTM, and dimethyl sulfoxide (DMSO) originated from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS) and charcoal/dextran-treated FBS were purchased from Atlanta Biologicals (Lawrenceville, GA, USA). Pen Strep (100 U/mL penicillin +100 μg/mL streptomycin), Opti-MEM, McCoy’s 5A medium, Lipofectamine 2000 and Lipofectamine 3000 transfection reagent, and 0.25% trypsin-EDTA were purchased from Life Technologies (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Dulbecco’s phosphate-buffered saline (DPBS) was purchased from Lonza Ltd. (Basel, Switzerland).
HIF-1α/p300 Screening Assay.
The screening of potential HIF-1α/p300 inhibitors was carried out using a cell-free protein−protein assay. The basic principle of this assay is to determine HIF-1α/p300 disruption by measuring displacement of the HIF-1α binding domain of p300 (CH1) from the p300binding domain of HIF-1α (C-TAD) that is immobilized on streptavidin-coated multiwell plates.10,36 Details of this assay were as described previously.10
Isolation of Pyrroloiminoquinone Alkaloids.
A 2.0 g aliquot of the Latrunculia sp. extract (NSC# C005119) was dissolved in MeOH− CH2O2 (1:1), adsorbed onto4gof diol solid support (YMC Diol 120Å, 5−50 mm) and then placed on top of a prepacked column of diol media (17.1 g). The column was eluted with 120 mL volumes of hexane− CH2O2 (9:1), CH2O2−EtOAc (20:1), 100% EtOAc, EtOAc−MeOH (5:1), and 100% MeOH. The HIF-1α/p300 inhibitory activity was concentrated in the fractions eluted with EtOAc−MeOH (198 mg) and MeOH (913 mg). These fractions were combined and then chromatographed on Sephadex LH-20 elutedwith CH2Q2−MeOH (1:1). A dark green band was collected (472 mg) from the LH-20 column, and after solvent removal it was separated by C18 reversed-phase chromatography using a step-gradient elution with 100% H2O, H2O−MeOH (3:1), H2O−MeOH (1:1) H2O−MeOH (1:3), 100% MeOH, MeOH−CH2Cl2 (1:1), and 100% CH2Cl2. The material eluted from C18 with H2O− MeOH (1:1 and 1:3) was combined (60 mg) and rechromatographed on diol eluting with EtOAc−MeOH + 0.05% TFA (5:1), EtOAc−MeOH + 0.05% TFA (3:1), EtOAc−MeOH + 0.05% TFA (1:1), 100% MeOH, and MeOH−CH2Cl2 (1:1). The active diol fractions were purified by repeated C18 HPLC using a CH3CN−H2O + 0.05% TFA gradient from 20% CH3CN to 100% CH3CN over 40 min to provide 5.1 mg of (+)-discorhabdin B (1), 4.0 mg of (−)-discorhabdin B dimer (2), 3.9 mg of 3-dihydrodiscorhabdin C (3), 2.3 mg of (+)-discorhabdin G*/I (4), and 7.8 mg of (−)-makaluvamine F (5).
Cell Lines.
COLO 205 cells (ATCC) were cultured in RPMI-1640 medium (ATCC), HCT 116 cells (ATCC) were grown in McCoy’s 5A medium (Gibco), and phenol-free RPMI-1640 medium (Gibco) was used for culturing of LNCaP cells (ATCC). Maintenance medium for each cell line was supplemented with 10% FBS and 1% Pen Strep. Cells were cultured in 5% CO2 and 95% air at 37 °C. For hypoxia experiments cells were placed in a hypoxia chamber (BioSpherix) set at 0.5% O2 for COLO 205 and HCT 116 cells or at 1% O2 for LNCaP cells (37 °C).
Cytotoxicity Assay.
Cells (in 100 μL of maintenance medium) were seeded into 96-well plates at a concentration of 15 000 (COLO 205, LNCaP) or 10 000 (HCT 116) cells/well. Twenty-four hours later, cells were treated with fresh medium containing either pyrroloiminoquinone alkaloid, positive control TPZ, or DMSO (0.5%, v/v) followed by 18 h (HCT 116, LNCaP) or 48 h (HCT 116, COLO 205) incubation under hypoxic conditions. Each concentration was tested in quadruplicate, and experiments were performed in triplicate. The tested concentration ranges were 0.001−10, 0.05−50, and 0.1 − 10 μM for COLO 205, HCT 116, and LNCaP cells, respectively. The next morning 10 μL of CCK-8 (Dojindo Molecular Technologies, Inc.) was added to each well. After incubation for 2 h at 37 °C, absorbance was read at 450 nm on a SpectraMax M2 fluorescence plate reader (Molecular Devices). The fluorescent signal of each sample was normalized to the average signal of the DMSO-treated controls to calculate percent cell viability.
HIF Reporter Assay.
In order to investigate inhibition of HIF-1α transcriptional activity by discorhabdin and makaluvamine alkaloids, a dual luciferase reporter assay (Promega) was carried out. Hereto, COLO 205 and HCT 116 cells were seeded into 96-well plates for 24 h at a concentration of 30 000 and 50 000 cells/well, respectively. Subsequently, cells were transiently cotransfected with 100 ng of pGL4.42[luc2P/HRE/Hygro] plasmid (Promega) containing four copies of the HRE sequence fused to a firefly luciferase reporter and 10 ng of pRL-TK (Renilla control reporter) plasmid using Lipofect-amine 2000 according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were treated with medium containing 0.1% FBS with 100 μL of pyrroloiminoquinone alkaloids (0.01−10 μM) or the positive controls CTM (0.5 μM) and TPZ (25 μM). Each concentration was tested in quadruplicate, and experiments were performed in triplicate. Plates were then incubated under normoxic or hypoxic (0.5% O2) conditions for 18 h. Cells were lysed, and luminescence was measured on a GloMax 96 microplate luminometer with dual injectors (Promega) according to the Dual-Luciferase Reporter Assay System manual (Promega). Luciferase activity in each well was analyzed by normalizing the HRE-luciferase signal to the pRL-TK luciferase signal. Subsequently, the fold-induction of each sample was calculated by dividing its normalized luciferase signal by the average HRE:TK ratio of the hypoxic controls (DMSO).
For LNCaPs, the seeding density was 15 000 cells/well and cells were cotransfected with pRL-TK-HRE plasmid (10 ng of pGL4.42[luc2P/HRE/Hygro] plasmid, 1 ng of pRL-TK plasmid), with Lipofectamine 3000 (0.3 μL/well) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were treated with phenol-free RPMI-1640 medium containing 0.1% charcoal-stripped FBS with pyrroloiminoquinone alkaloids (0.1,1,10 μM) and incubated for 18 h in the hypoxia chamber (1% O2). Other procedures in these cells were similar as described earlier for COLO 205 and HCT 116 cells.
VEGF ELISA.
HCT 116 cells were seeded into 96-well plates at a concentration of 25 000 and 15 000 cells/well, respectively. After an overnight incubation at 37 °C, maintenance medium was replaced with 100 of medium (5% charcoal-stripped FBS for HCT 116 cells; 0.1% charcoal-stripped FBS for LNCaP cells) containing pyrroloiminoquinone alkaloids (compound 2, 8, or 9) or control (DMSO or CTM). HCT 116 cells were treated at the following drug concentrations: 0.1, 0.25, and 0.5 μM for compound 2, 0.25, 0.5, and 1 μM for compound 8, 0.1, 0.25, and 0.5 μM for compound 9, and 0.5 μM for CTM. LNCaP cells were treated with these compounds at 0.25, 0.5, and 1 μM, while CTM was added at 0.25 μM. After incubation for 48 h at 0.5% O2 (HCT 116) or 18 h at 1% O2 (LNCaP), supernatant was diluted 4-fold (HCT 116) or 2-fold (LNCaP), and 200 μL of sample was used for VEGF quantification with the Quantikine VEGF ELISA kit (R&D Systems, Inc.). In addition, cell viability was determined using the CCK-8 assay to calculate VEGF secretion per cell.
Statistical Analyses.
The exact Wilcoxon rank sum test was applied to each comparison of one concentration of a compound vs the hypoxic control in the luciferase and VEGF ELISA (LNCaP) data. Results of the VEGF ELISA experiments in HCT 116 cells were analyzed with the stratified Wilcoxon rank sum test with experiment as the stratification factor to compare the effects of a compound vs the hypoxic control. For both the luciferase and the VEGF ELISA data Hochberg’s method was used for correcting the p values for the multiple concentrations tested. Results were considered statistically significant if p < 0.05 after correction.
Supplementary Material
ACKNOWLEDGMENTS
Thanks to the Natural Products Support Group at NCI-Frederick for extract preparation, and S. Tarasov and M. Dyba (Biophysics Resource Core, Structural Biophysics Laboratory, CCR) and H. Bokesch (MTL) for assistance with high-resolution mass spectrometry. We thank Dr. M. Stoffels for advice. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. This project was also funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.5b00846.
1H and 13C NMR spectra for compounds 1–11 (S1–S20); HIF reporter assay results of pyrroloiminoquinone alkaloids in COLO 205 cells (S21) (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Semenza GL Nat. Rev. Cancer 2003, 3, 721–732. [DOI] [PubMed] [Google Scholar]
- (2).Zhong H; De Marzo AM; Laughner E; Lim M; Hilton DA; Zagzag D; Buechler P; Isaacs WB; Semenza GL; Simons JW Cancer Res. 1999, 59, 5830–5835. [PubMed] [Google Scholar]
- (3).Kuwai T; Kitadai Y; Tanaka S; Onogawa S; Matsutani N; Kaio E; Ito M; Chayama K Int. J. Cancer 2003, 105, 176–181. [DOI] [PubMed] [Google Scholar]
- (4).Hirota K; Semenza GL Crit. Rev. Oncol. Hematol 2006, 59, 15–26. [DOI] [PubMed] [Google Scholar]
- (5).Burslem GM; Kyle HF; Breeze AL; Edwards TA; Nelson A; Warriner SL; Wilson AJ ChemBioChem 2014, 15, 1083–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Reece KM; Richardson ED; Cook KM; Campbell TJ; Pisle ST; Holly AJ; Venzon DJ; Liewehr DJ; Chau CH; Price DK; Figg WD Mol. Cancer 2014, 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Na YR; Han KC; Park H; Yang EG Biochem. Biophys. Res. Commun 2013, 434, 879–884. [DOI] [PubMed] [Google Scholar]
- (8).Kwon HS; Kim DR; Yang EG; Park YK; Ahn HC; Min SJ; Ahn DR Bioorg. Med. Chem. Lett 2012, 22, 5249–5252. [DOI] [PubMed] [Google Scholar]
- (9).Cook KM; Hilton ST; Mecinovic J; Motherwell WB; Figg WD; Schofield CJ J. Biol. Chem 2009, 284, 26831–26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chan ST; Patel PR; Ransom TR; Henrich CJ; McKee TC; Goey AK; Cook KM; Figg WD; McMahon JB; Schnermann MJ; Gustafson KR J.Am. Chem. Soc 2015, 137, 5569–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Antunes EM; Copp BR; Davies-Coleman MT; Samaai T Nat. Prod. Rep 2005, 22, 62–72. [DOI] [PubMed] [Google Scholar]
- (12).Hu JF; Fan H; Xiong J; Wu SB Chem. Rev 2011, 111, 5465–5491. [DOI] [PubMed] [Google Scholar]
- (13).Perry NB; Blunt JW; Munro MH Tetrahedron 1988, 44, 1727–1734. [Google Scholar]
- (14).Lam CF; Grkovic T; Pearce AN; Copp BR Org. Biomol. Chem 2012, 10, 3092–3097. [DOI] [PubMed] [Google Scholar]
- (15).Copp BR; Fulton KF; Perry NB; Blunt JW; Munro MH J. Org. Chem 1994, 59, 8233–8238. [Google Scholar]
- (16).Reyes F; Martin R; Rueda A; Fernandez R; Montalvo D; Gomez C; Sanchez-Puelles JM J. Nat. Prod 2004, 67, 463–465. [DOI] [PubMed] [Google Scholar]
- (17).Grkovic T; Pearce AN; Munro MH; Blunt JW; Davies-Coleman MT; Copp BR J. Nat. Prod 2010, 73, 1686–1693. [DOI] [PubMed] [Google Scholar]
- (18).Radisky DC; Radisky ES; Barrows LR; Copp BR; Kramer RA; Ireland CM J. Am. Chem. Soc 1993, 115, 1632–1638. [Google Scholar]
- (19).Zhang J; Cao J; Weng Q; Wu R; Yan Y; Jing H; Zhu H; He Q; Yang B PLoS One 2010, 5, e13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Strese S; Fryknas M; Larsson R; Gullbo J BMC Cancer 2013, 13, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Antunes EM; Beukes DR; Kelly M; Samaai T; Barrows LR; Marshall KM; Sincich C; Davies-Coleman MT J. Nat. Prod 2004, 67, 1268–1276. [DOI] [PubMed] [Google Scholar]
- (22).Antunes EM; Maree SE; Nyokong T; Davies-Coleman MT; Maree MD J. Chem. Res 2005, 2005, 780–783. [Google Scholar]
- (23).Calvani M; Trisciuoglio D; Bergamaschi C; Shoemaker RH; Melillo G Cancer Res. 2008, 68, 285–291. [DOI] [PubMed] [Google Scholar]
- (24).Holbeck SL; Collins JM; Doroshow JH Mol. Cancer Ther. 9, 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Reynolds AR Dose-Response 2009, 8, 253–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Abdollahi A; Hahnfeldt P; Maercker C; Grone HJ; Debus J; Ansorge W; Folkman J; Hlatky L; Huber PE Mol. Cell 2004, 13, 649–663. [DOI] [PubMed] [Google Scholar]
- (27).Celik I; Surucu O; Dietz C; Heymach JV; Force J; Hoschele I; Becker CM; Folkman J; Kisker O Cancer Res. 2005, 65, 11044–11050. [DOI] [PubMed] [Google Scholar]
- (28).Tjin Tham Sjin RM; Naspinski J; Birsner AE; Li C; Chan R; Lo KM; Gillies S; Zurakowski D; Folkman J; Samulski J; Javaherian K Cancer Gene Ther. 2006, 13, 619–627. [DOI] [PubMed] [Google Scholar]
- (29).Slaton JW; Perrotte P; Inoue K; Dinney CP; Fidler IJ Clin. Cancer Res. 1999, 5, 2726–2734. [PubMed] [Google Scholar]
- (30).Panigrahy D; Singer S; Shen LQ; Butterfield CE; Freedman DA; Chen EJ; Moses MA; Kilroy S; Duensing S; Fletcher C; Fletcher JA; Hlatky L; Hahnfeldt P; Folkman J; Kaipainen AJ Clin. Invest 2002, 110, 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lalani AS; Chang B; Lin J; Case SS; Luan B; Wu-Prior WW; VanRoey M; Jooss K Mol. Ther 2004, 9, 56–66. [DOI] [PubMed] [Google Scholar]
- (32).Creighton-Gutteridge M; Cardellina JH 2nd; Stephen AG; Rapisarda A; Uranchimeg B; Hite K; Denny WA; Shoemaker RH; Melillo G Clin. Cancer Res. 2007, 13, 1010–1018. [DOI] [PubMed] [Google Scholar]
- (33).Wada Y; Harayama Y; Kamimura D; Yoshida M; Shibata T; Fujiwara K; Morimoto K; Fujioka H; Kita Y Org. Biomol. Chem 9, 4959–4976. [DOI] [PubMed] [Google Scholar]
- (34).Perry NB; Blunt JW; Munro MH J. Org. Chem 1988, 53, 4127–4128. [Google Scholar]
- (35).McKee TC; Rabe D; Bokesch HR; Grkovic T; Whitson EL; Diyabalanage T; Van Wyk AW; Marcum SR; Gardella RS; Gustafson KR; Linehan WM; McMahon JB; Bottaro DP J. Nat. Prod 2012, 75, 1632–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kung AL; Zabludoff SD; France DS; Freedman SJ; Tanner EA; Vieira A; Cornell-Kennon S; Lee J; Wang B; Wang J; Memmert K; Naegeli HU; Petersen F; Eck MJ; Bair KW; Wood AW; Livingston DM Cancer Cell 2004, 6, 33–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.