

Abstract
The μ-opioid receptor (MOR) agonist morphine is commonly used for pain management, but it has severe adverse effects and produces analgesic tolerance. Thus, alternative ways of stimulating MOR activity are needed. We found that MrgC11, a sensory neuron-specific G protein-coupled receptor, may form heteromeric complexes with MOR. Peptide-mediated activation of MrgC11 enhanced MOR recycling by inducing coendocytosis and sorting of MOR for membrane reinsertion. MrgC11 activation also inhibited the coupling of MOR to β-arrestin-2 and enhanced the morphine-dependent inhibition of cAMP production. Intrathecal coadministration of a low dose of an MrgC agonist potentiated acute morphine analgesia and reduced chronic morphine tolerance in wild-type mice but not in Mrg-cluster knockout (Mrg KO) mice. BAM22, a bivalent agonist of MrgC and opioid receptors, enhanced the interaction between MrgC11 and MOR and produced stronger analgesia than did the individual monovalent agonists. Morphine-induced neuronal and pain inhibition was reduced in Mrg KO mice compared to that in wild-type mice. Our results uncover MrgC11-MOR interactions that lead to positive functional modulation of MOR. MrgC shares genetic homogeneity and functional similarity with human MrgXI. Thus, harnessing this positive modulation of MOR function by Mrg signaling may enhance morphine analgesia in a sensory neuron- specific fashion to limit central side effects.
INTRODUCTION
Agonists to the μ-opioid receptor (MOR), including morphine, remain the primary analgesic options for treating severe acute and chronic pain. Unfortunately, because they bind to MORs expressed throughout the central nervous system (CNS), dose-limiting central adverse effects, addiction, and the development of analgesic tolerance present substantial barriers to their clinical use (1). In addition, the opioid epidemic with abuse and accidental death from overdose has been escalating. Guanine nucleotide-binding protein (G protein)- coupled receptors (GPCRs), such as adrenergic and δ-opioid receptors (DORs), may interact with MORs and affect morphine analgesia profoundly (2–4). However, these receptors are also widely distributed in the CNS, and their ligands are associated with central side effects. A substantial portion of morphine analgesia is mediated by MORs that are expressed on primary sensory neurons in the dorsal root ganglion (DRG) and trigeminal ganglion (5). Therefore, selectively modulating MOR activity in peripheral afferent neurons would be a promising strategy to enhance morphine analgesia while limiting central side effects.
GPCRs can form receptor homomers and heteromers, which may show different signaling and biologic effects (3). The fact that MOR tends to form heteromeric complexes with other GPCR family members that enable their functional interactions may offer an opportunity to identify new drug targets for pain inhibition that cause fewer side effects than conventional opioid analgesics (3, 6–8). However, a primary sensory neuron-specific GPCR partner that enhances MOR activity and morphine analgesia has not been identified. One possibility is subtype C of the Mas-related GPCR family (MrgC; mouse MrgC11 and rat rMrgC), which is expressed mostly in small-diameter primary sensory neurons but not in the CNS (9, 10). MrgCs have gained attention for their association with pain and itch (9, 11, 12). Studies suggest that activation of MrgCs on the central terminals of DRG neurons by intrathecal drug administration inhibits pain in various rodent models (11, 13,14). By conducting the first comprehensive examination of physical and functional interplay between mouse MrgC11 and MOR, we tested the hypotheses that the two GPCRs form heteromeric complexes and that MrgC11 may positively modulate MOR function.
RESULTS
MrgC11 may physically interact with MOR at the cellular level
MrgC11 is expressed exclusively in primary sensory neurons in the DRG and trigeminal ganglion and is distributed in small-diameter, likely nociceptive, neurons (9, 10). Because MrgC11 may contribute to pain inhibition at the central terminals of primary sensory neurons similar to the function of MORs (11, 13, 14), we investigated the potential interaction of the two receptors. In an immunofluorescence staining study, immunoactivity for MrgC and that for MOR were highly colocalized in mouse (74%) and rat (90%) DRG neurons (Fig. 1A). Colocalization of MrgC and MOR immunoreactivity was also identified in mouse spinal cord (fig. S1). The specificity of MrgC antibody toward Mrg family members was confirmed in previous studies, which showed an absence of MrgC immunoreactivity in the spinal cord and DRG of Mrg knockout (KO) mice (15, 16). Furthermore, MrgC is expressed specifically on sensory neurons in the peripheral nervous system, not in neurons or glial cells of the CNS (9, 15,16). The specificity of monoclonal antibody toward MOR was confirmed here in conditional MOR KO mice (fig. S2), which were generated by crossing Pirt-Cre mice with Oprm1fl/fl mice to exclusively delete MOR expression in primary sensory neurons.
Fig. 1. Interaction between MrgC11 and MOR.
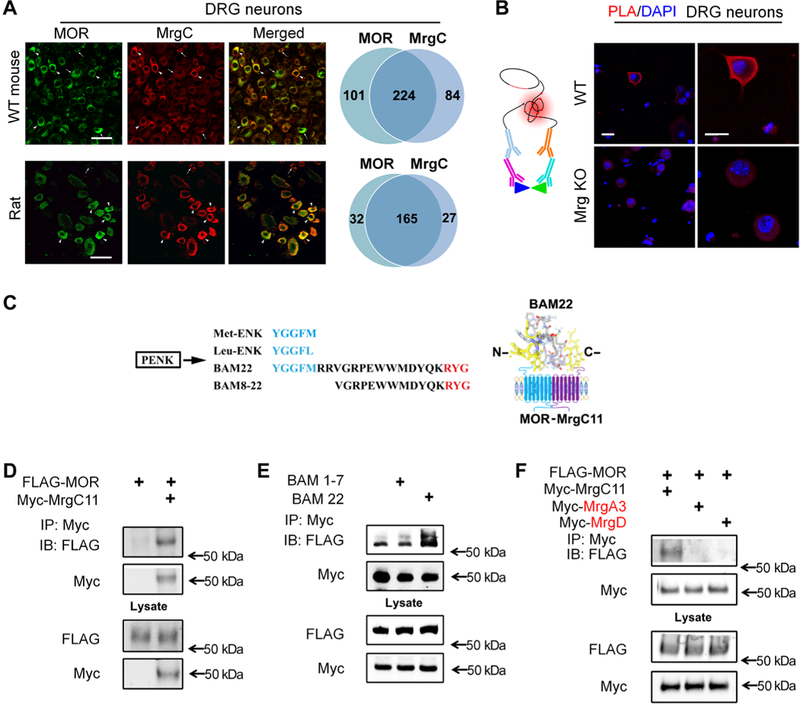
(A) Immunostaining for MrgC and MOR in DRG neurons of wild-type (WT) mice and rats (n = 3 animals per group). Arrowheads, (strongly) double-labeled cells; arrows, single-labeled cells. Right: Venn diagrams portray cells with strong coexpression. Scale bars, 50 mm. (B) Left: Schematic shows the main principles of the PLA. Right: PLA signal (red) for colocalization of MrgC and MOR antibodies in DRG neurons from WT and Mrg KO mice. DAPI (4’,6-diamidino-2-phenylindole) (blue) counterstained the nuclei. Images are representative of four experiments. Scale bars, 20 mm. (C) Left: Amino acid sequences of Met-ENK, Leu-ENK, BAM22, and BAM8–22 from the same precursor, proenkephalin (PENK), in which the consensus sequence (blue) binds the opioid receptor in the N terminus and the three-amino acid sequence (red) binds MrgC in the C terminus. Right: Diagram showing how BAM22 binds to both MOR and MrgCII. (D) Immunoprecipitation (IP) of Myc and immunoblotting (IB) of solubilized protein extracts derived from HEK293T cells transfected with Myc-MrgC11 and FLAG-MOR. (E) As described in (D), cells cotransfected with Myc-MrgC11 and FLAG-MOR were treated with bath application of full-length BAM22 or BAM1–7. (F) As described in (D), from cells cotransfected with FLAG-MOR and Myc-MrgC11, Myc-MrgA3, or Myc-MrgD. (D to F) Data are representative of three to four experiments.
Next, we directly interrogated whether MrgC11 and MOR form a complex in a heterologous expression system by conducting co-IP experiments in HEK293T cells cotransfected with FLAG-MOR and Myc-MrgC11. In addition, we examined the effect of BAM22, a cleavage product of proenkephalin A that has not only a canonical opioid receptor binding YGGFM (Met-enkephalin, BAM1–7) motif in its N terminus but also a RYG motif at its C terminus that specifically binds to MrgC (Fig. 1C) (10). Therefore, BAM22 serves as a unique bivalent ligand for both MrgC and opioid receptors, whereas BAM8– 22 activates MrgC only. Myc-MrgC11 coeluted from affinity resin in the presence of FLAG-MOR, and anti-FLAG antibody immunoprecipitated a band that corresponded to MrgC11 (Fig. 1D). In contrast, the specific bands were not observed in the precipitates prepared from cells expressing Myc-MrgC11 alone. BAM22 enhanced MrgC11 and MOR interaction in HEK293T cells (Fig. 1E), but BAM1–7 (BAM without the MrgC binding site) did not. This finding suggests that the full-length bivalent agonist BAM22 may act as a bridge to facilitate MrgC11 association with MOR. MOR appears to physically interact specifically with MrgC11 because we did not detect FLAG-MOR immunoreactivity either in Myc-MrgA3 precipitates of HEK293T cells that coexpressed Myc-MrgA3 and FLAG-MOR or in cells that coexpressed Myc-MrgD and FLAG-MOR (Fig. 1F). Together, these results provide further evidence that MrgC11 resides close to MOR in the native environment of DRG neurons and forms heteromeric complexes that could render functional interaction.
We used these antibodies in a proximity ligation assay (PLA) to examine the proximity of the two GPCRs in individual mouse DRG neurons. PLA is a relatively sensitive technique that detects colocalization of proteins in individual cells, which may be inferred as interaction, and reveals cellular and molecular architecture (17–19). PLA signal, located mostly near the cell membrane, was detected only in DRG neurons of wild-type mice but not in Mrg KO mice (Fig. 1B). Because possible nonselective binding of MrgC and MOR antibodies may confound PLA findings, we further carried out PLA in human embryonic kidney (HEK) 293T cells transfected with plasmids expressing FLAG-MOR, Myc-MrgC11, or both. FLAG-tagged MOR and Myc-tagged MrgC11 were then detected by anti-FLAG and anti-Myc antibodies. PLA signal appeared only in cells that were transfected with both receptors (fig. S3). Since PLA can detect proteins that are within 40 nm of one another (18,19), these findings suggest that MrgC11 and MOR may reside in close proximity in DRG neurons and in HEK293T cells cotransfected with both receptors.
C-terminal domains of MrgC11 and MOR are important to their physical interaction
We next searched for the MrgC11 domain that mediates its physical interaction with MOR. Such a site would be important for developing candidate drugs to modulate their interactions. Computational analysis and receptor crystal structure studies have indicated that transmembrane domain 5 (TM5), TM6, and the TM1-TM2-helix8 interface in GPCRs may be involved in receptor oligomerization (20). By comparing the MrgC11 protein sequence with the GPCRs that can form heteromers with MOR, we predicted that TM2 of MrgC11 may be important to MrgC11-MOR interaction. Therefore, we initially constructed a mutated MrgC11 (MrgC11-TM2TM6) in which the predicted TM2 (MrgC11-TM2) was substituted by TM6 (MrgC11-TM6). Unexpectedly, co-IP showed that the mutated MrgC11 still interacted with MOR in cotransfected HEK293T cells (Fig. 2A), suggesting that TM2 is not important to MrgC11-MOR interaction.
Fig. 2. MrgC11 interacts with MOR through the CTD.
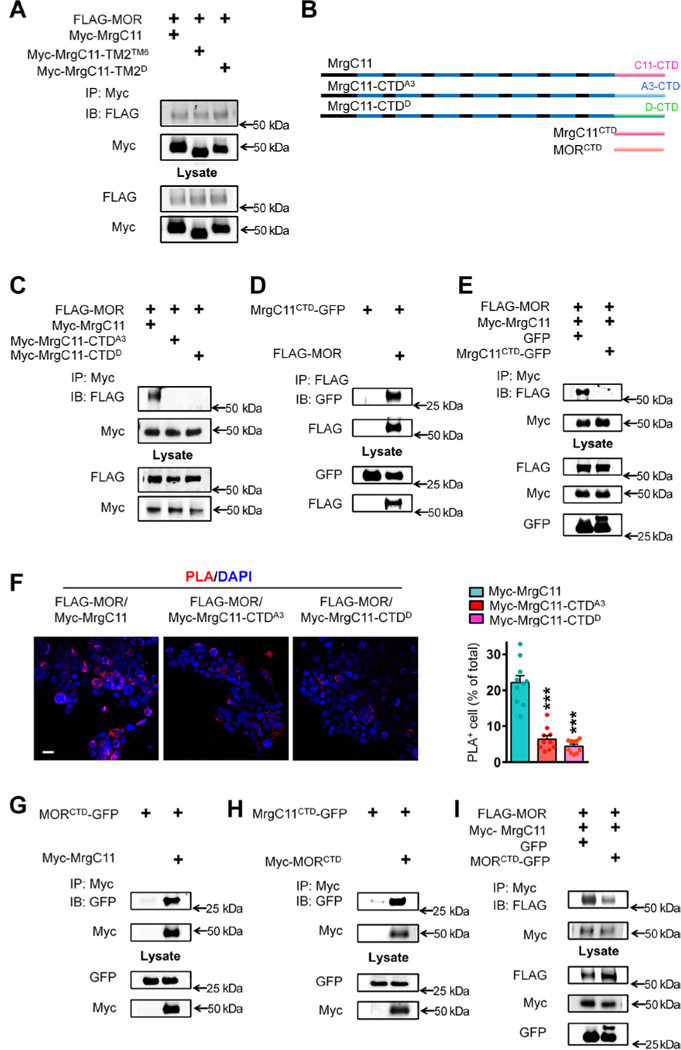
(A) Co-IP to examine the interaction between FLAG-MOR and WT Myc-MrgC11 or two mutant MrgC11s in HEK293T cells. In two mutant MrgC11s, the second TM2 region was swapped with TM6 of MrgC11 (Myc-MrgC11-TM2TM6) or TM2 of MrgD (Myc-MrgC11-TM2D). (B) The diagram shows WT MrgC11; mutant MrgC11s in which the CTD was substituted with that of MrgA3 (MrgC11-CTDA3) or MrgD (MrgC11-CTDD); MrgC11 CTD (MrgC11 CTD); and MOR CTD (MORCTD). (C) Co-IP to examine the interaction between FLAG-MOR and WT Myc-MrgC11, MrgC11-CTDA3, and MrgC11-CTDD mutants in transfected cells. (D) Co-IP to examine the interaction between FLAG-MOR and CTD of MrgC11 (MrgC11CTD-GFP). (E) Co-IP to examine MrgC11-MOR interaction in the presence of MrgC11CID-GFP. (F) Left: PLA of HEK293T cells cotransfected with FLAG-MOR and WT Myc-MrgC11, mutant MrgC11-CTDA3, or MrgC11-CTDD. Scale bar, 20 μm. Right: Quantification of PLA signal (n = 10 experiments per group). (G and H) Co-IP to examine the interaction between MORCTD-GFP and WT Myc-MrgC11 (G) and between Myc-MORCTD and MrgC11CTD-GFP (H). (I) Co-IP to examine whether expression of MORctd-GFP reduces MOR and MrgC11 interaction. (F) ***P < 0.001 versus Myc-MrgC11 by one-way analysis of variance (ANOVA) and Bonferroni post hoc test. Values are mean ± SEM. Data in (A), (C) to (E), and (G) to (I) are representative of three to four experiments.
We then examined whether the C-terminal domain (CTD) of MrgC11 is critical to receptor interaction. Because MrgA3 and MrgD did not interact with MOR (Fig. 1F), we constructed a mutated MrgC11 in which the predicted MrgC11-CTD was substituted with the CTD of MrgA3 or MrgD (Fig. 2B). Co-IP revealed that MrgC11-MOR interaction was nearly lost in HEK293T cells cotransfected with FLAG-MOR and mutated MrgC11-CTDA3 or MrgC11-CTDD (Fig. 2C). To further examine the role of the CTD in MrgC11 and MOR interaction, we constructed a plasmid expressing a fusion protein that contains MrgC11-CTD with green fluorescent protein (GFP) fused at the C terminus (MrgC11CTD-GFP). MrgC11CTD-GFP interacted directly with MORs in cotransfected cells (Fig. 2D). Furthermore, MrgC11CTD-GFP may compete with full-length MrgC11 for interaction with MOR because it reduced heteromer formation between FLAG-MOR and Myc-MrgC11 (Fig. 2E). In line with the co-IP findings, PLA showed that the associations between MOR and the two mutated MrgC11s were substantially weaker than that between MOR and wild-type MrgC11 (Fig. 2F) and that interaction between wild-type MrgC11 and MOR could be reduced by MrgC11CTD- GFP (fig. S4). Collectively, these findings confirm that the CTD of MrgC11 is indispensable to its physical interaction with MOR.
Co-IP studies showed that the CTD is also critical for MOR interaction with MrgC11. First, we constructed a plasmid expressing a fusion protein that contains MOR-CTD with GFP fused at the C terminus (MORCTD-GFP), which directly interacted with MrgC11 in cotransfected cells (Fig. 2G). Second, Myc-MORCTD also interacted with MrgC11CTD-GFP (Fig. 2H). Finally, MORCTD-GFP competed with MOR for interaction with MrgC11 and reduced heteromer formation between FLAG-MOR and Myc-MrgC11 (Fig. 2I).
Next, we investigated whether heteromer formation also occurs between MOR and MrgX1, the human homolog to MrgC that shares genetic homogeneity and functional similarity (9,10,21). PLA signals appeared in HEK293T cells that were cotransfected with FLAG- MOR and Myc-MrgX1 (fig. S5A). Furthermore, co-IP experiments showed that Myc-MrgX1 coeluted from affinity resin in the presence of FLAG-MOR and that anti-FLAG antibody immunoprecipitated a band that corresponded to MrgX1 (fig. S5B), suggesting that MOR and MrgX1 may also interact at the cellular level.
MrgC11 activation induces coendocytosis of MOR and targets MOR into the recycling pathway
Heteromer formation between MORs and other GPCRs enables functional interaction and modulation of receptor trafficking properties (2, 3, 6). How the formation of MrgC11-MOR complex affects MOR trafficking remains unclear. Hence, we examined the endocytosis of MOR in HEK293T cells cotransfected with Myc-MrgC11 and FLAG-MOR. The Myc and FLAG peptide tags were located at the N termini of MrgC11 and MOR, respectively, and were exposed to the extracellular space after the receptors were inserted into the plasma membrane (Fig. 3A). We prelabeled the Myc-MrgC11 and FLAG- MOR on the surface of live cells using mouse anti-Myc and rabbit anti-FLAG antibodies. Under control conditions, the prelabeled MrgC11 and MOR were located mostly on the cell surface. After a 45-min stimulation with the MrgC-selective agonists BAM8–22 (5 μM) and JHU58 (5 μM) (13, 14), the prelabeled MrgC11 and MOR were both internalized and redistributed into intracellular vesicle-like structures (Fig. 3A). In contrast, no marked co-internalization of MrgC11 and MOR was observed after applying morphine (5 μM for 45 min), an MOR agonist known to induce minimal endocytosis of MOR. However, cotreatment of morphine with a lower dose of BAM8–22 (1 μM) induced significant co-internalization of both receptors (Fig. 3A, and B). The internalized receptors were largely colocalized in the same intracellular compartments. These findings provide direct evidence that MrgC11 activation induces and facilitates the co-internalization and coendocytosis of MrgC11 and MOR.
Fig. 3. MrgC11 activation leads to co-internalization and targeting of surface MORs into the recycling pathway.
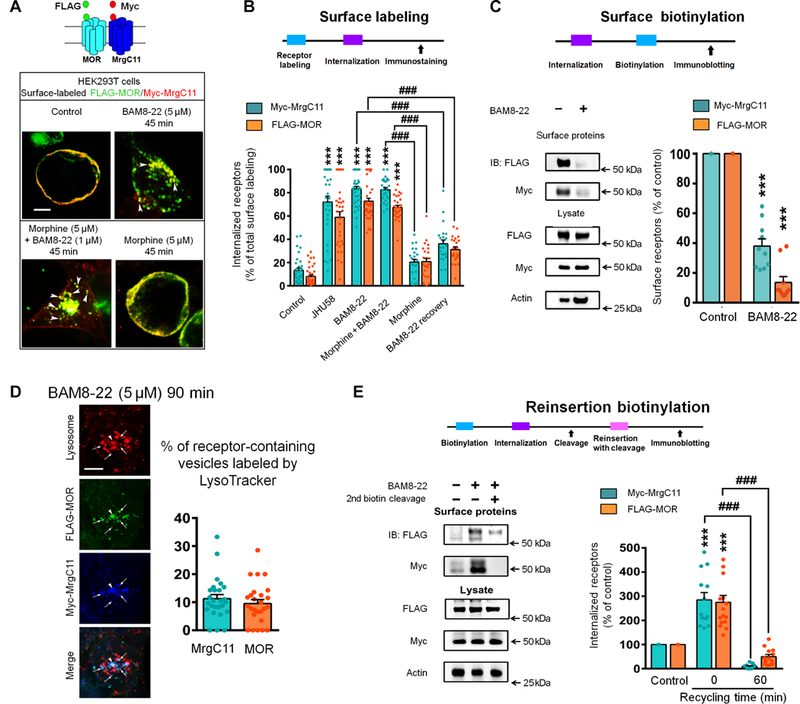
(A) Diagram depicts FLAG-MOR and Myc-MrgC11 at the cell surface. Representative images show co-internalization of FLAG-MOR and Myc-MrgC11 in HEK293T cells 45 min after administration of BAM8–22 (5 mM), morphine (5 mM), or both. Arrowheads, internalized receptors. Scale bar, 10 mm. Data are representative of three experiments. (B) Experimental design and quantitative data show the ratio of internalized MrgC11 and MOR versus total surface immunostaining (n = 23 to 40 cells). (C) Immunoblotting and quantitative data of MOR and MrgC11 on the cell surface before and after BAM8–22 (5 mM, n = 11 per group). (D) Left: Representative images show that MORs (arrows) were co-internalized with MrgC11 after BAM8–22 exposure and were not sorted into lysosome-like compartments labeled by LysoTracker (arrowheads). Right: Quantification of receptor-containing vesicles labeled by LysoTracker (n = 26 per group). Scale bar, 10 mm. (E) Left: Immunoblotting showed a change of biotinylated receptors after a second biotin cleavage, providing a measure of receptor recycling. Right: Recycling was quantified by comparing the internalized MrgC11 and MOR at 0 min and 60 min of recycling time (n = 11 per group). (B, C, and E) Two-way ANOVA and Bonferroni post hoc test. ***P < 0.001 versus control; ###P < 0.001. Values are mean ± SEM.
We then confirmed MrgC11 activation-induced co-internalization of MOR by using immunoblotting and quantified the receptors present on the cell surface. In HEK293T cells cotransfected with Myc- MrgC11 and FLAG-MOR, we biotinylated the proteins that remained on the cell surface after BAM8–22 treatment and precipitated them with immobilized streptavidin. We found that BAM8–22, which only binds MrgC, markedly reduced MOR and MrgC11 on the cell surface (Fig. 3C). Another MrgC agonist, JHU58 (5 μM) (13, 14), induced robust co-internalization of MrgC11 and MOR in mouse DRG neurons that were cotransfected with Myc-MrgC11 and FLAG- MOR (fig. S6A). In addition, the coexpression of MrgC11CTD-GFP, which interacts directly with MORs and hence competes with full- length MrgC11 for interaction (Fig. 2D, and E), significantly attenuated BAM8–22-induced MOR and MrgC11 co-internalization (fig. S6B). These findings suggest that physical interaction between MrgC11 and MOR is essential for receptor co-internalization.
GPCRs activated by selective agonists can be driven into divergent lysosomal and recycling pathways, resulting in different postendocytic fates and differences in cell signaling strength (3, 7,22). Hence, understanding the postendocytic fate of MOR after co-internalization with MrgC11 is important for predicting functional outcome. By co- staining Myc-MrgC11 with a LysoTracker probe in MrgC11-expressing HEK293T cells, we showed that only a small amount of MrgC11 was redistributed in lysosome-like compartments labeled by the LysoTracker probe after 90-min treatment with BAM8–22 and JHU-58 (5 μM; fig. S7A). Few MORs were present in the lysosome after BAM8–22 treatment (Fig. 3D). Thus, internalized MrgC11 and MOR were not sorted into the degradation pathway. To directly test whether the internalized receptors are reinserted back into the plasma membrane, we performed biotinylation assays of receptor recycling in HEK293T cells (Fig. 3E). MOR and MrgC11 underwent very weak endocytosis under basal conditions (Fig. 3A), but application of BAM8–22 (5 μM) caused robust co-internalization of surface MOR and MrgC11 (Fig. 3B, and C, and fig. S7, B and C). Internalized MORs and MrgC11s were mostly reinserted into the plasma membrane after 60 min, with only 18% of the internalized MORs and 3% of the internalized MrgC11s retained intracellularly (Fig. 3E). Together, these findings suggest an endocytic sorting of both MOR and MrgC11 through recycling pathways after MrgC11 activation.
MrgC11 agonism alters acute morphine inhibition of cAMP production
To complement the findings of physical interaction, we next examined whether MrgC11 agonism enhances acute morphine-induced inhibition of cyclic adenosine 3’,5’-monophosphate (cAMP) production, which occurs through MOR signaling coupled to the G; pathway. In HEK293T cells that coexpressed MOR, MrgC11, and a cAMP biosensor construct, acute treatment with morphine dose dependently decreased forskolin-induced intracellular cAMP production (Fig. 4A), but BAM8–22 by itself had little effect (Fig. 4B). Notably, coapplication of BAM8–22 (10−6 M) significantly potentiated the inhibition of cAMP by low doses of morphine (10−7 and 10−8 M). This finding cannot be attributed to additive effect (Fig. 4C, and D). Furthermore, this enhancement by BAM8–22 did not occur in cells lacking MrgC11 expression (Fig. 4D), suggesting MrgC11 involvement.
Fig. 4. MrgC agonist potentiates acute inhibition of cAMP production by morphine.
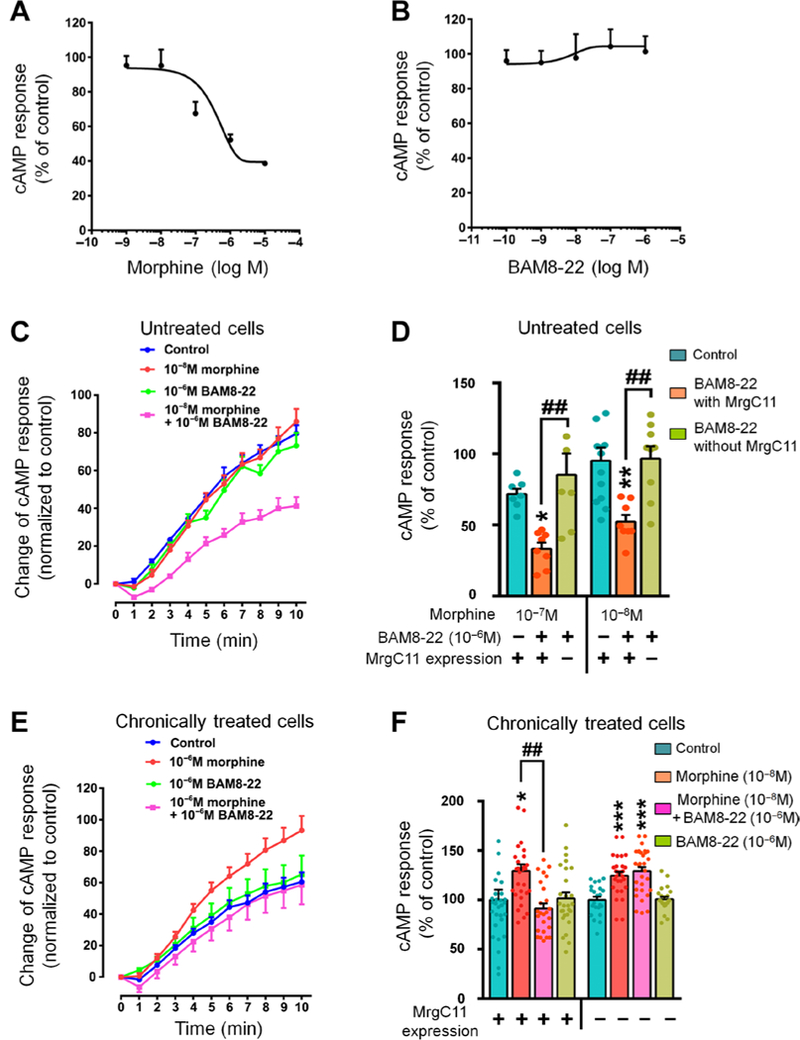
(A) Dose-response curve of morphine to inhibit forskolin-induced cAMP production in HEK293T cells coexpressing MOR, MrgCII, and cAMP biosensor construct (n = 4 experiments). (B) Dose-response curve of BAM8–22 effect on forskolin-induced cAMP production (n = 3 experiments). (C) Time course of forskolin-induced cAMP production in response to different drug treatments in HEK293T cells (n = 6 to 8 experiments). (D) Quantification of forskolin-induced cAMP production after different drug treatments (n = 7 to 12 experiments). (E) Time course of effects of different drug treatments on superactivation of the cAMP pathway in HEK293T cells after withdrawal from 14-hour chronic morphine (10−6 M) or vehicle treatment (n = 25 to 27 experiments). (F) Quantification of different drug treatments on chronic morphine- induced cAMP superactivation in cells transfected with MOR alone and in cells cotransfected with MOR and MrgC11 (n = 25 to 29 experiments). (D and F) One-way ANOVA with Bonferroni post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control (blue bar); ##P < 0.01 versus the indicated group. All values are mean ± SEM.
In contrast to the inhibition of cAMP by acute morphine, chronic morphine treatment of cells produces a compensatory up-regulation of adenylyl cyclase (23–25). Thus, we next examined whether BAM8–22 alleviates the paradoxical cAMP “superactivation” after chronic morphine treatment. HEK293T cells transfected with plasmids expressing FLAG-MOR, Myc-MrgC11, and a cAMP biosensor were treated chronically with morphine. Consistent with previous findings (25), chronic morphine treatment led to increased cAMP production (superactivation; Fig. 4E). Pretreatment with BAM8–22 blocked chronic morphine-induced cAMP superactivation in cells that expressed both MrgC11 and MOR but not in cells without MrgC11 (Fig. 4F). Therefore, activation of MrgC11 may enhance acute morphine inhibition of cAMP but reduce superactivation of cAMP after chronic morphine treatment.
MrgC11 activation reduces MOR and β-arrestin-2 coupling
Previous studies suggested that MOR coupling to the β-arrestin-2 pathway may be associated with the development of some morphine side effects (for example, respiratory depression and gastrointestinal dysfunction) and analgesic tolerance (26–28). To examine whether MrgC11 activation affects MOR and β-arrestin-2 coupling, we used a NanoBiT complementation assay (Promega) to monitor β-arrestin recruitment (29). In NanoBiT titration experiments, stimulation of MOR with morphine or DAMGO [(D-Ala2, N-MePhe4, Gly- ol)-enkephalin] for 20 min dose-dependently increased the NanoLuc luminescence, which serves to indicate the recruitment of β-arrestin-2 to MOR (Fig. 5A, and B). In line with previous findings (27, 30, 31), morphine induced much less β-arrestin-2 recruitment than did DAMGO (Fig. 5B), and MrgC11 showed little β-arrestin-2 coupling either before or after activation by BAM8–22 (Fig. 5C, and D). The CTD of MOR also showed substantial β-arrestin coupling (Fig. 5E). Furthermore, MOR and β-arrestin-2 coupling was significantly reduced by the expression of Myc-MORCTD (Fig. 5F), suggesting that the CTD of MOR is important to β-arrestin recruitment.
Fig. 5. MrgC11-MOR interaction reduces MOR and β-arrestin-2 coupling.
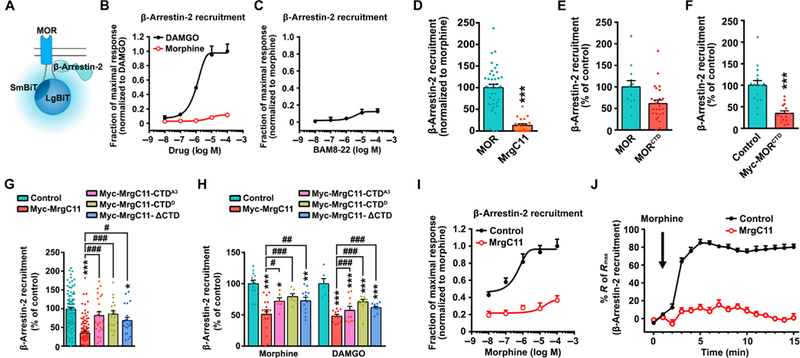
(A) Schematic representation of β-arrestin recruitment measured by NanoBiT complementation assay. (B) Dose-response curves of morphine and DAMGO for stimulation of MOR/β-arrestin-2 coupling in HEK293T cells coexpressing MOR-SmBiT and LgBiT-β-arrestin-2 (n = 3 to 4 experiments). (C) Dose-response curves of BAM8–22 for inducing MrgC11/β-arrestin-2 coupling (n = 3 to 4). (D) MrgC11/β-arrestin coupling (n = 20) versus MOR/β-arrestin coupling (n = 40). E) Recruitment of β-arrestin to MOR (n = 12) and its CTD (n = 25). (F) Effects of expression of Myc-MORCTD on MOR/β-arrestin coupling (n = 14 to 16). (G) Effects of coexpression of WT and mutant MrgC11s on morphine-induced MOR/β-arrestin-2 (n = 21 to 89). (H) Effects of expression of MrgC11 and MrgC11 C-terminal mutants on morphine- and DAMGO-induced β-arrestin-2 recruitment (n = 10 to 23). (I) Dose-response curves and (J) time course of morphine (100 μM) for stimulation of MOR/β-arrestin-2 coupling in cells transfected with MrgC11 or control plasmid (n = 8 to 12 per group). (D to F) ***P < 0.001 by Student’s t test. (G and H) *P < 0.05, **P < 0.01, and ***P < 0.001 versus control; #P < 0.05, ##P < 0.01, and ###P < 0.001 by one-way ANOVA with Bonferroni post hoc test. Values are mean ± SEM.
In the resting state, MOR showed significantly lower affinity to β-arrestin-2 in cells transfected with MrgC11 than in cells transfected with control plasmid. Furthermore, MrgC11 with mutated CTDs (CTDD and CTDA3) that do not interact with MOR failed to inhibit MOR and β-arrestin-2 coupling (Fig. 5G). In the activated state, both morphine and DAMGO induced significantly less β-arrestin-2 recruitment to MOR in cells coexpressing wild-type MrgC11 but not in cells coexpressing MrgC11 with mutated CTDs (Fig. 5H).
To better determine the ability of MrgC11 activation to interfere with MOR and β-arrestin-2 coupling, we further established dose- response functions for morphine-induced recruitment of β-arrestin-2 to MOR in the absence or presence of MrgC11. The expression of MrgC11 produced a rightward shift in the dose-response curve (Fig. 5I). Morphine-induced recruitment of β-arrestin-2 was pronounced and rapidly increased to a plateau level in cells expressing the control plasmid. However, this effect was largely abolished in cells expressing wild-type MrgC11 (Fig. 5J). These findings suggest that MrgC11 activation may reduce the coupling of MOR to β-arrestin-2, possibly because of its interaction with MOR at the CTD, which may bias MOR signaling away from the β-arrestin-2 pathway.
Cotreatment with MrgC agonist enhances morphine analgesia
Physical interaction with other GPCRs can have a positive or negative effect on MOR signaling and function. After demonstrating the positive functional modulation of MOR by MrgC11 activation at the cellular level (Figs. 2 to 5), we next examined the effect of MrgC11- MOR interaction on morphine analgesia in vivo. In the tail immersion test, 1 nmol BAM8–22 (by intrathecal injection), which is below its effective dose for pain inhibition, markedly enhanced the inhibition of heat nociception by a low dose of morphine [1.3 nmol, intrathecally (i.t.)] in wild-type mice but not in Mrg KO mice (Fig. 6A). BAM22, a full-length bivalent peptide ligand that binds both MrgC11 and opioid receptors, induced robust heat antinociception in wild-type mice at 1.3 nmol, a dose at which BAM8–22 and morphine would be ineffective (blue; Fig. 6A). In line with these findings, a low dose of BAM8–22 (1 nmol, i.t.) also potentiated the inhibition of heat nociception induced by a subeffective dose of morphine (1.3 nmol, i.t.) in both the hot plate test and Hargreaves test (fig. S8). We conducted isobolographical analysis to further determine whether intrathecal coadministration of BAM8–22 and morphine induces additive or synergistic antiheat nociception. The dose-response curves for morphine, BAM8–22, and combined drug treatment (1:1 ratio) were established for the tail immersion test (Fig. 6B). The parameters were calculated by fitting nonlinear least-squares curves, and the half-maximal effective dose (ED50) was projected by yielding a minimum residual sum of squares of deviations from the theoretical curve (32, 33). The experimental ED50 of combined drug treatment was 0.6 nmol [95% confidence interval (CI), 0.3 to 1.2 nmol], which was much lower than the ED50 of morphine (3.2 nmol; 95% CI, 1.4 to 7.4 nmol) or of BAM8–22 (4.1 nmol; 95% CI, 2.0 to 8.5 nmol). The experimental ED50 of the combined treatment (red dot) fell outside the 95% CIs of the theoretical additive ED50 (Fig. 6C, black line), indicating a synergistic action (34,35). Calcium-imaging assays also showed that acute cotreatment of DRG neurons with a subeffective dose of morphine (0.2 μM) and a low dose of BAM8–22 (0.2 μM) significantly inhibited the increase in intracellular Ca2+ concentration ([Ca2+];) evoked by capsaicin (0.3 μM) in wild-type mice but not in Mrg KO mice (Fig. 6D). BAM8–22 at this dose did not induce [Ca2+] response in DRG neurons (fig. S9).
Fig. 6. Intrathecal BAM8-22 potentiates morphine analgesia.
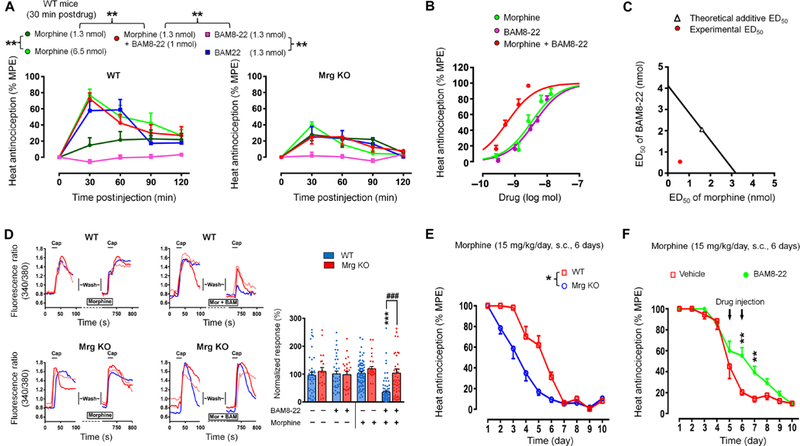
(A) Time course of pain inhibition after intrathecal drug administration in tail immersion test (52°C) of WT (n = 9 to 12 per group) and Mrg KO mice (n = 7 to 9 per group). BAM22, a dual agonist to MrgC and opioid receptor. (B) Dose-response curves of pain inhibition [% maximum possible effect (MPE) at 30 min postdrug] by intrathecal morphine, BAM8-22, and combined (1:1 ratio, n = 8 to 9 per group). (C) Isobolographical analysis. The experimental ED50 of the combined drug treatment (0.6 nmol; red dot) falls outside the 95% confidence limits of the theoretic additive ED50 (black line). (D) Effects of coapplication of BAM8-22 (0.2 μM) with a subeffective dose of morphine (0.2 μM) on capsaicin (0.3 μM)–induced [Ca2+]i increase in WT and Mrg KO DRG neurons (n = 21 to 79). (E) Time course of changes of the analgesic effect of morphine after repeated treatments [15 mg/kg, subcutaneously (s.c.)] in WT (n = 7) and Mrg KO (n = 9) mice. (F) Effects of BAM8-22 (delivered in two 5 nmol intrathecal injections/day on tolerance days 5 and 6; n = 8) and vehicle (n = 7) on the development of morphine tolerance in WT mice. *P < 0.05, **P < 0.01,and ***P < 0.001; ###P < 0.001 by two-way mixed model ANOVA and Bonferroni post hoc test. Values are mean ± SEM.
In a chronic morphine tolerance study, the pain-inhibiting effect of morphine (10 mg/kg) in the tail immersion test decreased quickly from day 2 in Mrg KO mice and completely vanished at 5 to 6 days after repetitive subcutaneous administrations. However, in wild-type mice, the decrease in morphine analgesia began at a later date (day 4) with the same treatment (Fig. 6E, and F). After 10 days of chronic morphine administration, cumulative dose-response curves showed a significant 3.6-fold shift of the morphine ED50 to the right in wildtype mice, whereas the potency of morphine in Mrg KO mice showed an even greater reduction (4.6-fold; fig. S10A and table S1). Thus, morphine tolerance was facilitated in Mrg KO mice, as compared to that in wild-type mice. Administration of BAM8–22 (5 nmol, i.t.) to wild-type mice before the daily morphine injection on days 5 and 6 increased the analgesic effect of morphine on days 6 and 7 compared to that in vehicle-treated mice (Fig. 6F and fig. S10B). Together, these findings suggest that activation of MrgC11 may counteract the development of morphine analgesic tolerance.
Intrathecal coadministration of BAM8–22 (1 nmol) and morphine (1.3 nmol) in wild-type mice did not produce well-known morphine side effects such as changes in exploration and locomotor activity, which we assessed in the open-field test (fig. S11A). Total distance traveled and time spent in the center zone (in 10 min) were similar in mice that received intrathecal coadministration of morphine and BAM8–22 and those that received saline (fig. S11, B and C). In contrast, the total distance traveled was significantly increased, and the time spent in the center significantly decreased after systemic morphine (5 mg/kg, s.c.) administration.
Morphine-induced neuronal inhibition and analgesia are reduced in Mrg KO mice
To complement our findings from the agonist study in wild-type mice, we also compared morphine analgesia in wild-type and Mrg KO mice. In the tail immersion test (52°C), hot plate test, and Hargreaves test, the baseline (meaning predrug) response latencies to noxious heat stimuli were all similar in naїve Mrg KO and wild-type mice, suggesting normal heat nociception (Fig. 7A). However, morphine (2.5 mg, i.t.)-induced heat antinociception, as indicated by an increase in tail-flick latency and paw withdrawal latency (Fig. 7B, to D), was significantly lower in Mrg KO mice than in wild-type mice. In an inflammatory pain model, both Mrg KO and wild-type mice developed mechanical and heat hypersensitivities on the ipsilateral hind paw at 2 to 3 days after intraplantar injection of complete Freund’s adjuvant, as indicated by increased paw withdrawal frequency to von Frey stimulation and decreased paw withdrawal latency to noxious heat stimulation, respectively (Fig. 7E, to G). Although 2.5 mg of morphine (by intrathecal injection) normalized mechanical (Fig. 7E) and heat hypersensitivity (Fig. 7F, and G) in wild-type mice at 30 min after injection, the effect was significantly reduced in Mrg KO mice. The inhibition of neuropathic pain-related behavior by intrathecal morphine was also reduced in Mrg KO mice (Fig. 7H, and I). Both wild-type and Mrg KO mice developed mechanical and heat hypersensitivity on the ipsilateral hind paw at 1 to 2 weeks after chronic constriction injury (CCI) of the sciatic nerve. Morphine (2.5 mg, i.t.) attenuated mechanical and heat hypersensitivity in wild-type mice at 30 min after injection but was significantly less effective in Mrg KO mice. The reduced morphine analgesia in Mrg KO mice was not due to a decrease in MOR expression in the DRG and spinal cord (fig. S12).
Fig. 7. Intrathecal morphine-induced pain inhibition is reduced in Mrg KO mice.
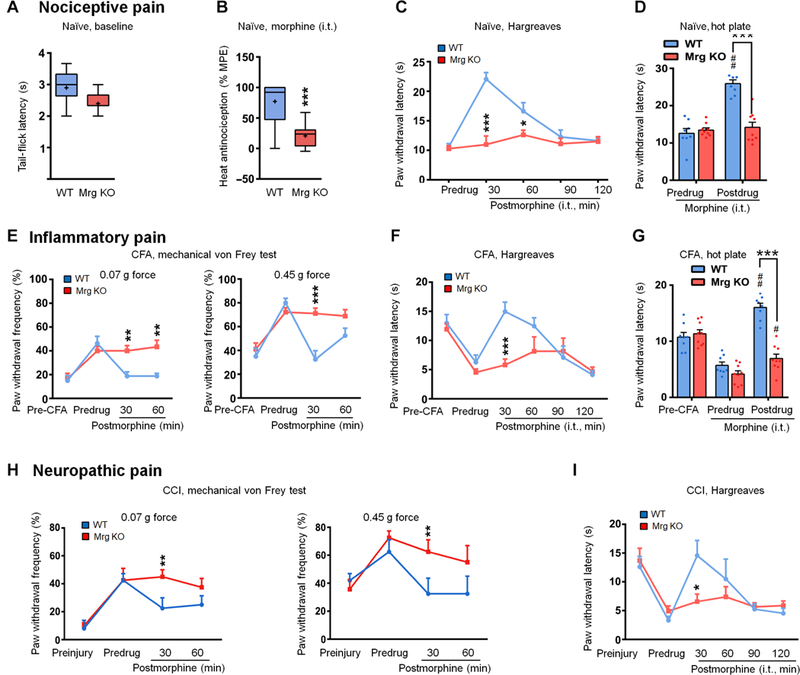
(A) Tail-flick latencies of naïve WT (n = 19) and Mrg KO mice (n = 11) in the tail immersion test (52°C). (B) Intrathecal morphine-induced (7.8 nmol, 5 ml, i.t.) heat antinociception in naïve WT (n = 19) and Mrg KO (n = 11) mice. Box-and-whisker plots show median (horizontal line), mean (+), interquartile range (box), and maximum and minimum values (“whiskers”). ***P < 0.001 by Mann-Whitney U test. (C and D) Morphine- induced heat antinociception (7.8 nmol, 5 ml) in naïve WT (n = 8) and Mrg KO (n = 9) mice in Hargreaves test (C) and hot plate test (D). (E) At 2 to 3 days after intraplantar injection of complete Freund’s adjuvant (CFA), effects of morphine (7.8 nmol, i.t.) on the increased paw withdrawal frequency to mechanical stimulation in WT mice (n = 8) and Mrg KO mice (n = 9). (F and G) Morphine (7.8 nmol, i.t.) inhibition of inflammatory heat hypersensitivity in CFA-treated WT and Mrg KO mice in the Hargreaves test (F) and hot plate test (G). (H) At 1 to 2 weeks after CCI, effects of morphine (7.8 nmol, i.t.) on the increased paw withdrawal frequency to mechanical stimulation in WT and Mrg KO mice (n = 6 per group). (I) Effects of morphine (7.8 nmol, i.t.) on heat hypersensitivity in CCI-WT and CCI-Mrg KO mice. (C to I) Values are mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 versus WT or indicated group; #P < 0.05 and ##P < 0.01 versus predrug by two-way mixed model ANOVA with Bonferroni post hoc test.
Next, using calcium imaging, we evaluated the involvement of MrgC11 in regulation of MOR activity in native DRG neurons. Morphine attenuates intracellular Ca2+ mobilization through activation of MOR, which inhibits voltage-dependent calcium channels (8, 36). Morphine dose-dependently reduced capsaicin (0.3 μM)- evoked increases in [Ca2+]i in DRG neurons from wild-type mice. However, this inhibition of Ca2+ response by morphine was significantly decreased in DRG neurons from Mrg KO mice (Fig. 8A). Because changes in MOR-mediated inhibition in the soma of DRG neurons may not reflect changes in their central terminals, we further conducted patch-clamp recording of evoked excitatory postsynaptic currents (eEPSCs) from dorsal horn neurons that may receive monosynaptic and polysynaptic inputs from nociceptive afferent neurons (Fig. 8B). In wild-type mice, eEPSCs were recorded from substantia gelatinosa (SG; lamina II) neurons in spinal cord slices (L4-L5 segments) (13). High-intensity paired-pulse stimulation (500 μA, 0.1 ms, 400 ms apart, three tests per min) was applied at the dorsal root to activate high-threshold afferent fibers (C fibers).
Fig. 8. Hypothetical model illustrating MrgC11 regulation of MOR activity.
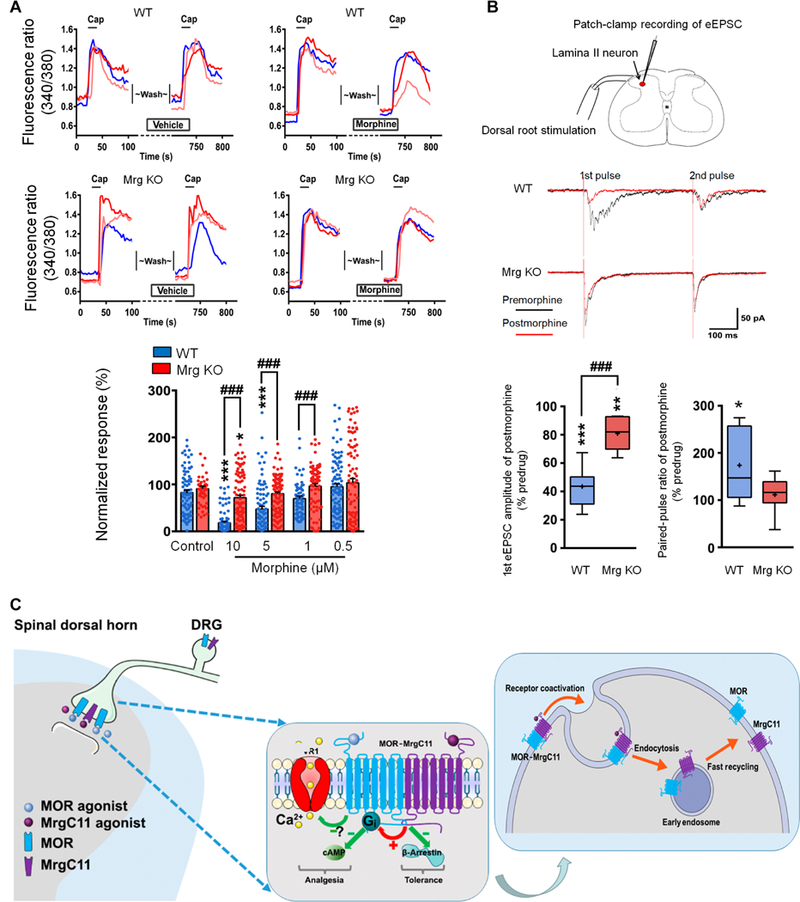
(A) Representative traces and quantification show morphine (5 μM) inhibition of capsaicin (Cap; 0.3 μM)–induced [Ca2+]i increase in WT and Mrg KO DRG neurons (n = 54 to 117). Values are mean ± SEM. *P < 0.05 and ***P < 0.001 versus control; ###P < 0.001 by two-way mixed model ANOVA with Bonferroni post hoc test. (B) Top: Patch-clamp recording in a lamina II neuron and representative traces of eEPSCs to paired-pulse test stimulation (500 μA, 0.1 ms, 400-ms interval) before and 5 min after morphine (1 μM). Bottom: Quantification of morphine inhibition of eEPSCs and changes in pairedpulse ratio (n = 8 per group). Box-and-whisker plots show median (horizontal line), mean (+), interquartile range (box), and maximum and minimum values (whiskers). *P < 0.05, **P < 0.01, and ***P < 0.001 versus predrug; ###P < 0.001 by Mann-Whitney U test. (C) Hypothetical model by which MrgC11 regulates MOR activity. Left: Coactivation of MrgC11 and MOR facilitates Gi-coupled regulation of cAMP and shifts the MOR response to agonist away from β-arrestin-2 signaling pathways that mediate morphine tolerance toward the Gi signaling pathways mediating a therapeutic response. Right: Activation of MrgC11 in the MOR-MrgC11 complex leads to receptor coendocytosis. The internalized receptors are sorted into recycling endosomes.
Paired-pulse stimulation is often used to examine short-term neuronal plasticity, which may reflect the probability of neurotransmitter release from presynaptic terminals (14, 37, 38). Accordingly, we calculated the paired-pulse ratio (PPR) of eEPSCs to determine whether a decrease of presynaptic release of neurotransmitters contributes to drug-induced inhibition of eEPSCs. The PPR is defined as the peak amplitude of the second eEPSC (P2) over the first eEP- SC (P1) evoked by two pulses. At baseline (predrug), the PPR was <1 in both naïve wild-type and naïve Mrg KO mice. A value less than 1 suggests paired-pulse depression, which may result when fewer presynaptic vesicles are available to release neurotransmitter in the second pulse than in the first pulse (37, 38). This observation is consistent with previous findings in spinal cord tissue using a similar paired-pulse stimulation protocol (14, 39,40). Bath application of 1 μM morphine strongly inhibited eEPSCs to the first pulse in SG neurons from wild-type mice, which was associated with an increase in PPR (Fig. 8B). This result suggests that inhibition of eEPSCs by morphine may involve a reduction in presynaptic excitatory neurotransmitter release (14, 37, 38). However, the inhibitory effect of morphine on eEPSCs was significantly reduced in Mrg KO mice. Together, these loss-of-function studies suggest that endogenous Mrg signaling may play an important role in morphine analgesia and in MOR-mediated neuronal inhibition under physiologic conditions. Additional tests are needed to determine whether the recorded lamina II neurons receive monosynaptic and/or polysynaptic inputs from primary afferents.
DISCUSSION
Our study revealed that MrgC11 and MOR interact physically at the cellular level and lead to functional consequences. Activation of MrgC11 promoted coendocytosis of MORs targeted into the recycling pathway, reduced the coupling of MOR to β-arrestin-2, and enhanced morphine inhibition of cAMP production. MrgC agonist potentiated acute morphine analgesia and reduced chronic morphine tolerance, further illustrating a positive functional modulation of MOR by MrgC11 signaling in vivo. Notably, Mrg KO mice exhibited compromised morphine analgesia, as compared to wild-type mice. Our study reveals a previously unrecognized essential role of MrgC11 in modulating MOR activity and implies that MrgC11 may represent a primary sensory neuron-specific partner GPCR that modulates MOR activity and morphine analgesia.
PLA and co-IP studies suggested that MrgC-enhanced MOR activity may arise from physical interplay between the two proteins through a critical interface on their CTDs. MrgC11 activation led to the co-internalization of MrgC11 and MOR and hence may alter the trafficking properties of MOR (2, 6,41). Internalized GPCRs are either degraded in lysosomes or resensitized and recycled (7, 22, 41). Moreover, heteromers and homomers may traffic via different postinternalization pathways (2, 42). We found that the internalized MOR was not redistributed in lysosome-like compartments. Therefore, the formation of MrgC11-MOR complex through oligomerization may facilitate MOR endocytosis and sorting into the recycling pathway for reinsertion into the plasma membrane. It is possible that MOR activity may also be modulated by MrgC11 signaling through other intracellular mechanisms, such as increased coupling of MOR to the Gi pathway, which would modulate G protein-coupled receptor kinase and β-arrestin-mediated MOR desensitization (26, 28,31). It remains to be determined how MrgC11 and MOR interaction disrupts the coupling of MOR to β-arrestin-2.
Our results further provide converging biologic and pharmacologic evidence for positive functional modulation of MOR by MrgC11 agonism. Inhibition of cAMP production contributes to morphine analgesia (30, 43), but the paradoxical increase in adenylyl cyclase activity (superactivation) by chronic morphine treatment indicates cellular withdrawal (25, 44). Notably, BAM8–22 enhanced acute morphine-induced inhibition of cAMP production and reduced chronic morphine-induced cAMP superactivation, which may be ameliorated by increased MOR endocytosis and recycling (7, 25). Accordingly, facilitation of MOR endocytosis with MrgC agonist may reduce protracted MOR desensitization and cellular adaptations that are associated with morphine tolerance. Intrathecal coadministration of a low dose of BAM8–22 increased morphine-induced antinociception and limited morphine tolerance. Furthermore, BAM8– 22 enhanced morphine inhibition of the capsaicin-evoked [Ca2+]i increase in wild-type DRG neurons but not in Mrg KO DRG neurons. These effects contrast with those from interactions between MOR and DOR (6, 42), which target MOR into the degradation pathway (2).
Intrathecal MrgC agonist inhibits pain in animal models without notable side effects (11–13, 45–47). BAM8–22, in combination with morphine, did not elicit signs of discomfort after intrathecal administration. However, subcutaneous injection of BAM8–22 induces scratching behavior, suggesting that MrgC may function in sensory processing of itch at peripheral terminals in the skin (12, 48,49). The different effects of MrgC agonists may be due to peripheral and spinal receptors being coupled to different signaling pathways (49–51). Activation of MrgC and MrgX1 inhibits high voltage-activated calcium currents, which reduces neurotransmitter release and attenuate spinal nociceptive transmission (13, 14, 21, 52). In contrast, BAM8–22 in the periphery activates the Gq pathway to open TRPA1 channels to evoke itch (12, 48, 49). The phenomenon of one receptor signaling through two opposing pathways is also seen for serotonin receptors (53, 54).
Activation of both MrgC11 and MOR by BAM22 may contribute to its greater analgesic effect. Bivalent endogenous ligands are rare, but another example is the endothelin-1 (ET-1) peptide (55). Like the ET-1 peptide, which facilitates and stabilizes the heterodimeric conformation, BAM22 may promote the formation of hetero-oligomers by MrgC11 and MOR. Engineered bivalent ligands have been generated to improve drug efficacy by targeting homo-oligomers or heterooligomers. The affinity and efficacy of the dimeric ligands depend on the length of the spacer that links the molecules. On the basis of the PEP-FOLD de novo peptide structure prediction method, the distance between the two pharmacophores in BAM22 is 15 to 20 Å, a length that may enable BAM22 to bridge neighboring MrgC11 and MOR effectively.
We also present evidence that pain inhibition by morphine is partially dependent on endogenous Mrg signaling, as suggested by findings in Mrg KO mice. Furthermore, acute morphine analgesia was enhanced, and chronic morphine tolerance was attenuated by cotreatment with a low dose of MrgC agonist. Additional studies using MrgC11 KO mice will help further validate the role of MrgC11 in morphine analgesia. Among the 12 functional Mrgs that were deleted in Mrg KO mice, MrgC11 is the only receptor for BAM8–22 (12, 13, 56). Accordingly, the effects of BAM8–22 on morphine analgesia are likely to be mediated by MrgC11 in wild-type mice.
The escalation of opioid abuse, addiction, and accidental death associated with the current opioid epidemic has been striking. A significant proportion of morphine analgesia is mediated by MORs on primary sensory neurons and their central terminals in the spinal cord (5, 57). Our findings suggest that receptor oligomerization may allow MrgC to positively modulate MOR function (Fig. 8C). MrgC shares genetic homogeneity with human MrgX1, and the sequence of BAM peptides is conserved from rodent to human (9, 10,21, 58). Similar to MrgC, MrgX1 activation also inhibits high voltage-activated calcium current and induces pain inhibition (14, 21). Our PLA and co-IP studies suggested that MrgX1 also interacts with MOR at the cellular level. Nevertheless, physical and functional MOR-MrgX1 interactions require further investigation. The recent development of MrgX1 transgenic mice has made it possible to test MrgX1 ligands in conventional mouse models (52). Because MrgC11-MOR interaction occurs only in primary sensory neurons, harnessing this unprecedented positive modulation of MOR function by Mrg signaling may enhance morphine analgesia and reduce tolerance in a primary sensory neuron-specific fashion. Such drugs should limit the side effects of ipioids by reducing the required dose of morphine and minimize central adverse effects because they may not enhance MOR activation in the brain.
MATERIALS AND METHODS
Mice
For all animal experiments, male mice (20 to 30 g) were used at 2 to 3 months of age. Mice were housed in groups of three to five on a standard 12-hour light/12-hour dark cycle, and all behavioral experiments were performed during the light cycle. No animals were excluded from the study, and randomization of experimental groups was not required. The experimenter was blind to animal genotypes during behavioral testing. Before all behavioral tests, mice were habituated to behavior rooms for 30 min. Mice that were tested on multiple behavioral paradigms were given a resting period of at least 1 week between experiments. All animal work was approved by the Animal Care and Use Committee of Johns Hopkins University School of Medicine.
Mrg KO mice were generated as previously described (12). They were backcrossed to C57BL/6 mice for a minimum of nine generations before homozygous Mrg KO mice were generated. Mrg KO mice have a deletion of 845 kb in chromosome 7, which contains 12 intact Mrg genes, including MrgC11 (11, 12).
To generate MrgA3-eGFP-WT mice, a mouse BAC clone (RP23– 311C15) containing the entire MrgA3 gene was purchased from the Children’s Hospital Oakland Research Institute. The BAC clone was modified by using homologous recombination in bacteria to generate the MrgA3 GFP-Cre transgenic line (16).
Oprm1fl/fl mice have a floxed MOR-encoding gene in which exon 2 and exon 3 are flanked by two loxP sites (59). To delete MORs specifically from primary sensory neurons, we crossed the Oprm1fl/fl with Pirt-Cre mice in which the Cre recombinase is under control of the Pirt promoter and expressed exclusively in >95% of all DRG neurons (60, 61).
Plasmid construction
All primers and oligonucleotides used for the construction of plasmids are listed in table S2. The complementary DNAs of mouse MrgC11, MOR1, MrgA3, and MrgD were cloned into a pCMV-HA/Myc plasmid (Clontech), and the epitope was positioned at the N termini of receptors. The Myc-MrgC11-CTDA3, Myc-MrgC11-CTDD, Myc- MrgC11-DCTD, MrgC11-TM2TM6, and MrgC11-TM2D were constructed by two-step polymerase chain reaction (PCR) with the Q5 mutagenesis kit (New England Biolabs). The C-terminal regions of MrgC11 and MOR were first amplified by PCR (primers in table S3) and then cloned into the eGFP-N3 vector (Clontech) or a pCMV-Myc plasmid. The mouse MrgC11, MOR1, and β-arrestin-2 were inserted into a NanoBiT PPI plasmid (Clontech). The small subunit (SmBiT) was positioned at the C termini of receptors, and the large subunit (LgBiT) was positioned at the N terminus of β-arrestin-2.
Cell culture and transfection
HEK293T cells were cultured in growth medium that consisted of 90% Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) (Invit- rogen) at 37°C in the presence of 95% O2 and 5% CO2. Using Lipo- fectamine 2000 (Thermo Fisher Scientific), we transfected the cells with 2 to 4 μg of plasmid/35-mm dish or 0.1 to 0.2 μg of plasmid/96-well micropolate well and cultured them for 1 to 2 days before experiments. We routinely assessed the cells with DAPI staining to confirm the absence of mycoplasma.
Cultures of dissociated DRG neurons
DRG from all spinal levels of 4-week-old mice or rats were collected in cold DH10 [90% DMEM/F-12, 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) (Invitrogen)] and treated with enzyme solution [dispase (5 mg/ml) and collagenase type I (1 mg/ml) in Hanks’ balanced salt solution without Ca2+ or Mg2+ (Invitrogen)] at 37°C. After trituration and centrifugation, cells were resuspended in DH10, plated on glass coverslips coated with poly-D-lysine (0.5 mg/ml; Biomedical Technologies Inc.) and laminin (10 μg/ml; Invitrogen), cultured in an incubator (95% O2 and 5% CO2) at 37°C, and used within 24 hours.
Immunohistochemistry
HEK293T cells cotransfected with Myc-MrgC11 and FLAG-MOR expression plasmids were preincubated with mouse anti-Myc antibody (1:100; M4439, Sigma-Aldrich), rabbit anti-FLAG antibody (1:100; F7425, Sigma-Aldrich), and/or LysoTracker Red DND-99 (1:500; Thermo Fisher Scientific) for 30 min at 37°C. Cells were then treated with 5 μM BAM 8–22, JHU58, morphine, or DAMGO for 30 or 90 min. Cells were fixed with 4% paraformaldehyde and 0.2% picric acid and then immunostained. wild-type mice and Mrg-cluster gene KO (Mrg KO) mice were fixed in 4% paraformaldehyde and 0.2% picric acid. Sections of L4-L5 DRG and spinal segments were cut on a cryostat and immunostained with custom-made primary rabbit polyclonal MrgC antibody (1:500; Proteintech Group Inc.) (15, 16) or a mixture of the MrgC antiserum and guinea pig anti-MOR antibody (1:400; GP10106, Neuromics). For secondary antibodies, we used Alexa 568-conjugated (Thermo Fisher Scientific) or Alexa 488-conjugated (A11008, Molecular Probes) goat antibody to rabbit, Alexa 488- conjugated goat antibody to mouse (A10667, Molecular Probes), Cy3-conj ugated goat antibody to guinea pig (A11073, Jackson Laboratory), and Alexa 488-conjugated goat antibody to mouse immunoglobulin G1 (A21124, Molecular Probes). All secondary antibodies were diluted 1:100 in blocking solution. The total number of neurons in each section was determined by counting both labeled and unlabeled cell bodies. Tissues from different experimental groups were processed together.
Proximity ligation assay
PLA was performed with Duolink reagents (Sigma-Aldrich). The HEK293T cells and dissociated DRG neurons were cultured as described above. The next day, cells were fixed with 4% paraformaldehyde at room temperature and then blocked and incubated with primary antibodies (1:200; rabbit anti-MrgC, Proteintech Group Inc.; 1:200; mouse anti-MOR, mAb63256, Abcam) overnight at 4°C. After incubation, the coverslips were washed with 0.1% Triton X-100 in phosphate-buffered saline (PBS) and then incubated with secondary antibodies (anti-mouse MINUS probe and anti-rabbit PLUS probe) for 2 hours at room temperature. Coverslips were then washed with 0.1% Triton X-100 in PBS and transferred to wash buffer A (Sigma- Aldrich). The PLA reaction was performed according to the manufacturer’s instructions in 20 μl of reaction solution per coverslip in a humidified chamber. Coverslips were mounted in mounting medium (Sigma-Aldrich) and analyzed on a confocal microscope (Leica).
Cell surface biotinylation and immunoblotting
The transfected HEK293T cell cultures were incubated in extracellular solution (ECS; 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 10 mM Hepes, and 10 mM sucrose) or ECS containing different drugs with or without Sulfo-NHS-LC-Biotin (0.5 mg/ml; Pierce) for 45 min at 37°C and then in PBS (Ca2+/Mg2+) containing biotin for 45 min at 4°C. After the reaction was stopped, cells were lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer [50 mM tris (pH 7.5), 150 mM NaCl, 10% glycerol, 0.1% Triton X-100, and bovine serum albumin (0.5 mg/ml)]. The lysates were then precipitated with streptavidin. Samples were subjected to SDS- polyacrylamide gel electrophoresis, transferred to membranes, and probed with the indicated antibodies: FLAG (1:2000; Cell Signaling Technology) and Myc (1:4000; Cell Signaling Technology). Protein bands were detected with enhanced chemiluminescence (Bio-Rad). Western blots were imaged with the ImageQuant LAS 4000 (GE Healthcare Life Sciences) and analyzed with ImageJ 1.46a software.
Biotinylation assay of receptor endocytosis and recycling with cleavable biotin reagent
For the biotinylation assay of receptor endocytosis, the transfected HEK293T cell cultures were biotinylated with Sulfo-NHS-SS-Biotin (1.5 mg/ml; Pierce). Drugs were present throughout all steps and incubations except for the 4°C biotinylation reaction. Cells were then incubated at either 4°C to block membrane trafficking or 37°C for various times to allow endocytosis to occur. The remaining surface biotin was then cleaved by reducing its disulfide linkage with glutathione cleavage buffer (50 mM glutathione in 75 mM NaCl and 10 mM EDTA containing 1% bovine serum albumin and 0.075 M NaOH) twice for 15 min each at 4°C. Cell membranes were prepared, and biotinylated proteins were precipitated. The recycling of MOR and MrgC11 receptors was measured biochemically by the loss of internalized receptors specifically labeled with cleavable (disulfide-linked) biotin. The transfected HEK293T cell cultures were treated with pharmacologic agents and surface-biotinylated as described above and then transferred to 37°C for 30 min to allow endocytosis to occur. Cells were cooled to 4°C to stop membrane trafficking, and the remaining surface biotin was quantitatively cleaved with glutathione. We returned cultures to serum-free growth medium containing 50 mM glutathione at 37°C (or 4°C) for various times to allow internalized receptors to recycle before the cells were cooled to 4°C and incubated with glutathione cleavage buffer. Residual biotinylated (internalized) receptors were isolated and detected by immunoblot analysis. Western blots were imaged with the ImageQuant LAS 4000 (GE Healthcare Life Sciences) and analyzed with ImageJ 1.46a software.
Immunoprecipitation
Cells and tissue were lysed in ice-cold RIPA buffer. The suspended lysate was immunoprecipitated at room temperature for 10 min with protein G Dynabeads (Thermo Fisher Scientific) that had been preincubated for 10 min with 0.5 to 2 μg of mouse anti-Myc (Sigma- Aldrich) or anti-FLAG antibody (Sigma-Aldrich) at room temperature. Immunoprecipitates were collected and aspirated. The Dynabeads were then resuspended in RIPA buffer, washed, and incubated in SDS sample buffer for 10 min at 70°C. The precipitate was immuno- blotted as described above. Antibodies against FLAG (1:2000; Cell Signaling Technology) and Myc (1:4000; Cell Signaling Technology) were used.
Real-time measurement of cAMP dynamics
Briefly, HEK293T cells were plated into 96-well plates and then transfected with the biosensor (Promega), MOR, and/or MrgC11 plasmids. For acute drug experiments in naive cells, the cells were pre-equilibrated with 2% GloSensor cAMP reagent (Promega) in CO2-independent medium for 2 hours at room temperature, beginning 48 hours after transfection. The cells were then preincubated with the test compound for 10 min before addition of forskolin (15 μM) alone or mixed with other test compounds. Luminescence was quantified on a FlexStation reader (Molecular Devices) for 10 min after the addition of forskolin. For chronic drug experiments, cells were exposed to the drug for 14 hours, rinsed three times in drug- free medium to initiate a withdrawal phase, and then given 15 μM forskolin. Luminescence signal was measured during the following 10 min. For data expressed as cAMP fold of baseline, relative luminescent units (RLUs) were measured for 1000 ms at baseline before the test drug incubation, and all subsequent RLUs were then divided by this baseline. The cAMP fold change was calculated by dividing the maximum RLU during the 10-min period by the baseline average RLU and then normalizing to the control group. For data expressed as cAMP response change, the RLU change was calculated by subtracting the RLU during the 10-min test period from baseline RLU and normalizing it to the maximum RLU change (calculated by subtracting the maximum RLU during the 10-min test period from baseline RLU in the control group). Each data point was a minimum of three wells per experimental replicate.
β-Arrestin recruitment assays
β-Arrestin recruitment was measured by the NanoBiT complementation assay (Promega). Assays were conducted exactly as instructed by the manufacturer (29). Briefly, HEK293T cells were plated into 96-well plates (Corning) at 2 × 104 cells per well and then cotransfected the next day with the different NanoBiT plasmids (1:1 ratio of interacting pairs). For MOR/β-arrestin and MrgC11/β-arrestin, we used mouse MOR or MrgC11 containing C-terminal SmBiT- tagged (MOR-SmBiT or MrgC11-SmBiT) and LgBiT-tagged N-terminal β-arrestin-2 (LgBiT-β-arrestin-2). At 24 hours after transfection, growth medium was exchanged with Opti-MEM I, and Nano-Glo Live Cell Substrate was added. Cells were then incubated for 15 min at 37°C, and luminescence was measured with a FlexStation reader (Molecular Devices) at 37°C. After thermal equilibration, morphine or DAMGO in Opti-MEM I was injected, and measurements were continued. To measure MOR-mediated β-arrestin recruitment in the presence or absence of MrgC11 or MrgC11 mutant coexpression, we cotransfected HEK293T cells with MOR-SmBiT, LgBiT-β- arrestin-2, and MrgC11 in a 16:16:1 ratio. We substituted pCMV-Myc for MrgC11 to maintain the same concentration of DNA transfected. At least 24 hours after transfection, luminescence was measured on a FlexStation reader (Molecular Devices), as described above. Data were normalized to that of DAMGO or morphine and analyzed with the sigmoidal dose-response function of GraphPad Prism 6.0.
Calcium imaging
Intracellular calcium was measured as previously described (16) by an experimenter blind to genotype. Briefly, neurons were loaded with Fura-2-acetomethoxyl ester (Thermo Fisher Scientific) for 30 min in the dark at 22°C. After being washed, cells were imaged at 340- and 380-nm excitation for detection of intracellular free calcium.
Whole-cell voltage-clamp recordings from spinal cord slices
We used procedures as described in our previous studies (14, 39). Briefly, the lumbosacral segment of spinal cord was removed rapidly from wild-type or Mrg KO mice and placed in ice-cold, low-sodium Krebs solution (95 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4-H2O, 6 mM MgCl2, 1.5 mM CaCl2, 25 mM glucose, 50 mM sucrose, and 1 mM kynurenic acid) saturated with 95% O2/5% CO2. For electrophysiology recording, spinal cord slices were stabilized with a grid (ALA Scientific) and submerged in a low-volume recording chamber (SD Instruments) that was perfused with room temperature Krebs solution (125 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4-H2O, 1 mM MgCl2, 2 mM CaCl2, and 25 mM glucose, with 10 μM GABAA receptor blocker bicuculline and 1 μM glycinergic receptor blocker strychnine) bubbled with a continuous flow of 95% O2/5% CO2. Whole-cell patch-clamp recording of lamina II cells was carried out under oblique illumination with an Olympus fixed-stage microscope system (BX51). Data were acquired with pCLAMP 10 software (Molecular Devices) and a MultiClamp amplifier. Using a puller (P1000), we fabricated thin-walled glass pipettes (World Precision Instruments) that had a resistance of 3 to 6 megohm and were filled with internal solution (120 mM K-gluconate, 20 mM KCl, 2 mM MgCl2, 0.5 mM EGTA, 2 mM Na2-adenosine triphosphate, 0.5 mM Na2-guanosine triphosphate, and 20 mM Hepes). The cells were voltage-clamped at −70 mV. Membrane current signals were sampled at 10 kHz and low-pass-filtered at 2 kHz. Larger-bore pipettes filled with Krebs solution were used for dorsal root stimulation. To evoke EPSCs, we delivered paired-pulse test stimulation to the dorsal root consisting of two synaptic volleys (500 μA, 0.1 ms) 400 ms apart at a frequency of 0.05 Hz to activate high-threshold afferent fibers (C fibers), followed by a 0.1-ms, 5-mV depolarizing pulse [to measure series resistance (Rseries) and input resistance (Rinput)]. We monitored Rseries and Rinput and discarded cells if either of these values changed by more than 20%. The amplitudes of the first (P1) and the second (P2) EPSC were measured, and the PPR was calculated as PPR = P2/P1.
Behavioral studies
An experimenter blinded to genotype carried out all behavioral tests, and all protocols were approved by the Animal Care and Use Committee of Johns Hopkins University School of Medicine. Drugs were suspended in 0.9% saline to the desired working concentration and injected into the intrathecal space of mice under brief isoflurane (1.5%) anesthesia. A 30-gauge, 0.5-inch needle connected to a 50-μl syringe was inserted into one side of the L5 or L6 spinous process and moved carefully forward to the intervertebral space. A tail flick indicated that the tip of the needle was inserted into the subarachnoid space. The needle was removed after administration of drug solution. One day before behavioral testing, mice were acclimated to the environment for 1 hour.
Mechanical sensitivity was assessed with the von Frey test by the frequency method. Two calibrated von Frey monofilaments (low force, 0.07 g; high force, 0.45 g) were used. Each von Frey filament was applied perpendicularly to the plantar side of each hind paw for ~1 s; the stimulation was repeated 10 times to both hind paws. The occurrence of paw withdrawal in each of these 10 trials was expressed as a percent response frequency: paw withdrawal frequency = (number of paw withdrawals/10 trials) x 100%.
For the Hargreaves test, mice were placed under a transparent plastic box (4.5 cm x 5 cm x 10 cm) on a glass floor. Infrared light was delivered through the glass floor to the hind paw. After acclimatization sessions, the latency for the animal to withdraw its hind paw was measured.
For the hot plate latency test, a rectangular metal surface was heated to 55°C. The antinociceptive response was the latency observed from the time the rat was placed on the heated surface until the first overt behavioral sign of nociception such as (i) licking of a hind paw, (ii) vocalization, or (iii) an escape response. The timer was stopped by a foot-operated pedal, and the rat was immediately removed from the hot plate. A maximum hot plate latency of 30 s was used to prevent tissue damage to the rat’s paws.
Antinociception was assessed with the water immersion tail-flick test in 52°C water. The latency to the first sign of a rapid tail flick was taken as the behavioral endpoint. Each mouse was tested for baseline latency. The test compound dissolved in 5 μl of vehicle was administrated via lumbar puncture. A maximum score was assigned (100%) to animals that did not respond within 10 s. Antinociception was calculated by the formula: % maximum possible effect = 100 x (test latency - baseline latency)/(10 - baseline latency). Single-dose data were analyzed with one-way ANOVA and t test for group comparisons (mean ± SEM). Groups of mice were used to determine ED50 values of morphine analgesia after intrathecal or subcutaneous administration of morphine. Analgesia was defined as a doubling or greater of the baseline latency for an individual mouse. ED50 values for morphine and 95% confidence limits were determined with the Bliss program. For chronic morphine treatment, ED50 was determined 10 days after daily morphine administration (5 mg/kg, s.c.) with a cumulative dose-response paradigm.
The open-field test was used to assess the effect of drug administration on spontaneous exploration and locomotor activity of animals. Animals were placed in an open field chamber (40 cm x 40 cm plastic box with a wall height of 33 cm) for 10 min. We analyzed total distance traveled, time spent in the center, mean travel speed, and number of internal periphery crossings in video recordings using SMART 3 software (Panlab Harvard Apparatus).
Data analysis
All statistical analysis was carried out with GraphPad Prism software or IBM SPSS Statistics 21. We performed a normality test on the data by using the Shapiro-Wilk test. Data that followed a normal distribution are means ± SEM. Comparisons of normally distributed data consisting of two groups were made by Student’s t tests. Comparisons of data in three or more groups were made by one-way ANOVA, followed by the Bonferroni post hoc test. Comparisons of two or more factors across multiple groups were made by two-way ANOVA, followed by the Bonferroni post hoc test. Data that did not follow a normal distribution were analyzed with nonparametric ANOVA (Friedman and Kruskal-Wallis) and expressed as medians. Post hoc tests (Wilcoxon matched-pairs test and Mann-Whitney U test) were used to analyze specific data points. Statistical significance was set at P < 0.05 in all tests. Sample sizes were determined in each study based on a statistical power analysis (power = 0.80, α = 0.05 (two-sided)] or previous similar studies (2, 4, 11, 13,16, 62–64). Representative data are from experiments that were replicated biologically at least three times with similar results. All behavioral experiments consisted of a minimum of six animals per group. For the dose- response curve analysis, the parameters, minimum or maximum effect (Emax), ED50, and Hill coefficient (n) were calculated by fitting nonlinear least-squares curves, according to previous studies (32, 33).
For the drug interaction analysis, we calculated the value of the theoretic additive ED50 according to the previously described method (34, 35), and the drug interactions are presented in the conventional isobolography.
Supplementary Material
Acknowledgments:
We thank C. F. Levine (scientific editor, Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University) for editing the manuscript.
Funding: This study was supported by NIH grants NS70814 (to Y.G.), NS54791 (to X.D.), NS099879 (to Y.G.), and NS26363 (to S.N.R.) and by the Johns Hopkins Blaustein Pain Research Fund (to Y.G.). X.D. is an investigator of the Howard Hughes Medical Institute. This work was facilitated by the Pain Research Core funded by the Blaustein Fund and the Neurosurgery Pain Research Institute at Johns Hopkins University.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice ASC, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M, Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 14, 162–173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He S-Q, Zhang Z-N, Guan J-S, Liu H-R, Zhao B, Wang H-B, Li Q, Yang H, Luo J, Li Z-Y, Wang Q, Lu Y-J, Bao L, Zhang X, Facilitation of μ-opioid receptor activity by preventing 5-opioid receptor-mediated codegradation. Neuron 69, 120–131 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KDG, Pin J-P, Volkow ND, Waldhoer M, Woods AS, Franco R, Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 5, 131–134 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA, A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc. Natl. Acad. Sci. U.S.A. 101, 5135–5139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein C, Schäfer M, Machelska H, Attacking pain at its source: New perspectives on opioids. Nat. Med. 9, 1003–1008 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Gomes I, Fujita W, Gupta A, Saldanha SA, Negri A, Pinello CE, Eberhart C, Roberts E, Filizola M, Hodder P, Devi LA, Identification of a m-5 opioid receptor heteromer-biased agonist with antinociceptive activity. Proc. Natl. Acad. Sci. U.S.A. 110, 12072–12077 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He L, Fong J, von Zastrow M, Whistler JL, Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell 108, 271–282 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Jordan BA, Devi LA, G-protein-coupled receptor heterodimerization modulates receptor function. Nature 399, 697–700 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong X, Han S-K, Zylka MJ, Simon MI, Anderson DJ, A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106, 619–632 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Lembo PMC, Grazzini E, Groblewski T, O’Donnell D, Roy M-O, Zhang J, Hoffert C, Cao J, Schmidt R, Pelletier M, Labarre M, Gosselin M, Fortin Y, Banville D, Shen SH, Strom P, Payza K, Dray A, Walker P, Ahmad S, Proenkephalin A gene products activate a new family of sensory neuron—Specific GPCRs. Nat. Neurosci. 5, 201–209 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Guan Y, Liu Q, Tang Z, Raja SN, Anderson DJ, Dong X, Mas-related G-protein- coupled receptors inhibit pathological pain in mice. Proc. Natl. Acad. Sci. U.S.A. 107, 15933–15938 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng H-J, Geng Y, Undem BJ, Kollarik M, Chen Z-F, Anderson DJ, Dong X, Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell 139, 1353–1365 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He S-Q, Li Z, Chu Y-X, Han L, Xu Q, Li M, Yang F, Liu Q, Tang Z, Wang Y, Hin N, Tsukamoto T, Slusher B, Tiwari V, Shechter R, Wei F, Raja SN, Dong X, Guan Y, MrgC agonism at central terminals of primary sensory neurons inhibits neuropathic pain. Pain 155, 534–544 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, He S-Q, Xu Q, Yang F, Tiwari V, Liu Q, Tang Z, Han L, Chu Y-X, Wang Y, Hin N, Tsukamoto T, Slusher B, Guan X, Wei F, Raja SN, Dong X, Guan Y, Activation of MrgC receptor inhibits N-type calcium channels in small-diameter primary sensory neurons in mice. Pain 155, 1613–1621 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He S-Q, Han L, Li Z, Xu Q, Tiwari V, Yang F, Guan X, Wang Y, Raja SN, Dong X, Guan Y, Temporal changes in MrgC expression after spinal nerve injury. Neuroscience 261, 43–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han L, Ma C, Liu Q, Weng H-J, Cui Y, Tang Z, Kim Y, Nie H, Qu L, Patel KN, Li Z, McNeil B, He S, Guan Y, Xiao B, LaMotte RH, Dong X, A subpopulation of nociceptors specifically linked to itch. Nat. Neurosci. 16, 174–182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Söderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius K-J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L-G, Landegren U, Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Nahalkova J, Novel protein-protein interactions of TPPII, p53, and SIRT7. Mol. Cell. Biochem. 409, 13–22 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Thompson CM, Bloom LR, Ogiue-Ikeda M, Machida K, SH2-PLA: A sensitive in-solution approach for quantification of modular domain binding by proximity ligation and real-time PCR. BMC Biotechnol. 15, 60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S, Crystal structure of the m-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Ikeda SR, Modulation of ion channels and synaptic transmission by a human sensory neuron-specific G-protein-coupled receptor, SNSR4/mrgX1, heterologously expressed in cultured rat neurons. J. Neurosci. 24, 5044–5053 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg L-O, Wundrack N, Beyer A, Grecksch G, Hollt V, Receptor endocytosis counteracts the development of opioid tolerance. Mol. Pharmacol. 67, 280–287 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Sharma SK, Klee WA, Nirenberg M, Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc. Natl. Acad. Sci. U.S.A. 72, 3092–3096 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siuda ER, Copits BA, Schmidt MJ, Baird MA, Al-Hasani R, Planer WJ, Funderburk SC, McCall JG, Gereau IV RW, Bruchas MR, Spatiotemporal control of opioid signaling and behavior. Neuron 86, 923–935 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn AK, Whistler JL, Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron 32, 829–839 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin F-T, Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Bohn LM, Gainetdinov RR, Lin F-T, Lefkowitz RJ, Caron MG, μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Dang VC, Chieng B, Azriel Y, Christie MJ, Cellular morphine tolerance produced by ßarrestin-2-dependent impairment of μ-opioid receptor resensitization. J. Neurosci. 31, 7122–7130 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, Machleidt T, Kirkland TA, Wood MG, Eggers CT, Encell LP, Wood KV, NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang X-P, Sassano MF, Giguere PM, Löber S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK, Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pierce KL, Lefkowitz RJ, Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2, 727–733 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Gong N, Wang Y-C, Wang H-L, Ma A-N, Hashimoto K, Wang Y-X, Interactions of the potent D-amino acid oxidase inhibitor CBIO with morphine in pain and tolerance to analgesia. Neuropharmacology 63, 460–468 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Huang J, Cai Q, Chen Y, Hong Y, Treatment with ketanserin produces opioid-mediated hypoalgesia in the late phase of carrageenan-induced inflammatory hyperalgesia in rats. Brain Res. 1303, 39–47 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Tallarida RJ, Stone DJ Jr., Raffa RB, Efficient designs for studying synergistic drug combinations. Life Sci. 61, PL417–PL425 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Tallarida RJ, Porreca F, Cowan A, Statistical analysis of drug-drug and site-site interactions with isobolograms. Life Sci. 45, 947–961 (1989). [DOI] [PubMed] [Google Scholar]
- 36.Schroeder JE, McCleskey EW, Inhibition of Ca2+ currents by a mu-opioid in a defined subset of rat sensory neurons. J. Neurosci. 13, 867–873 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu XY, Liu ZC, Sun YG, Ross M, Kim S, Tsai FF, Li QF, Jeffry J, Kim JY, Loh HH, Chen ZF, Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell 147, 447–458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fioravante D, Regehr WG, Short-term forms of presynaptic plasticity. Curr. Opin. Neurobiol. 21, 269–274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sdrulla AD, Xu Q, He SQ, Tiwari V, Yang F, Zhang C, Shu B, Shechter R, Raja SN, Wang Y, Dong X, Guan Y, Electrical stimulation of low-threshold afferent fibers induces a prolonged synaptic depression in lamina II dorsal horn neurons to high-threshold afferent inputs in mice. Pain 156, 1008–1017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morisset V, Urban L, Cannabinoid-induced presynaptic inhibition of glutamatergic EPSCs in substantia gelatinosa neurons of the rat spinal cord. J. Neurophysiol. 86, 40–48 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Lipscombe D, Raingo J, Internalizing channels: A mechanism to control pain? Nat. Neurosci. 9, 8–10 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Fujita W, Gomes I, Devi LA, Heteromers of μ-δ opioid receptors: New pharmacology and novel therapeutic possibilities. Br. J. Pharmacol. 172, 375–387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ingram SL, Williams JT, Opioid inhibition of Ih via adenylyl cyclase. Neuron 13, 179–186 (1994). [DOI] [PubMed] [Google Scholar]
- 44.Williams JT, Christie MJ, Manzoni O, Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev. 81, 299–343 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Hong Y, Dai P, Jiang J, Zeng X, Dual effects of intrathecal BAM22 on nociceptive responses in acute and persistent pain-potential function of a novel receptor. Br. J. Pharmacol. 141, 423–430 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen T, Hu Z, Quirion R, Hong Y, Modulation of NMDA receptors by intrathecal administration of the sensory neuron-specific receptor agonist BAM8–22. Neuropharmacology 54, 796–803 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Zeng X, Huang H, Hong Y, Effects of intrathecal BAM22 on noxious stimulus-evoked c-fos expression in the rat spinal dorsal horn. Brain Res. 1028, 170–179 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Sikand P, Dong X, LaMotte RH, BAM8–22 peptide produces itch and nociceptive sensations in humans independent of histamine release. J. Neurosci. 31, 7563–7567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.S. R. Wilson, K. A. Gerhold, A. Bifolck-Fisher, Q. Liu, K. N. Patel, X. Dong, D. M. Bautista, TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor- mediated itch. Nat. Neurosci. 14, 595–602 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Yang F-C, Okuda T, Dong X, Zylka MJ, Chen C-L, Anderson DJ, Kuner R, Ma Q, Mechanisms of compartmentalized expression of Mrg class G-protein-coupled sensory receptors. J. Neurosci. 28, 125–132 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delmas P, Abogadie FC, Buckley NJ, Brown DA, Calcium channel gating and modulation by transmitters depend on cellular compartmentalization. Nat. Neurosci. 3, 670–678 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Li Z, Tseng P-Y, Tiwari V, Xu Q, He S-Q, Wang Y, Zheng Q, Han L, Wu Z, Blobaum AL, Cui Y, Tiwari V, Sun S, Cheng Y, Huang-Lionnet JHY, Geng Y, Xiao B, Peng J, Hopkins C, Raja SN, Guan Y, Dong X, Targeting human Mas-related G protein-coupled receptor X1 to inhibit persistent pain. Proc. Natl. Acad. Sci. U.S.A. 114, E1996–E2005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crozier RA, Ajit SK, Kaftan EJ, Pausch MH, MrgD activation inhibits KCNQ/M- currents and contributes to enhanced neuronal excitability. J. Neurosci. 27, 4492–4496 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rau KK, McIlwrath SL, Wang H, Lawson JJ, Jankowski MP, Zylka MJ, Anderson DJ, Koerber HR, Mrgprd enhances excitability in specific populations of cutaneous murine polymodal nociceptors. J. Neurosci. 29, 8612–8619 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Houde M, Desbiens L, D’Orléans-Juste P, Endothelin-1: Biosynthesis, Signaling and Vasoreactivity. Adv. Pharmacol. 77, 143–175 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Li Z, He SQ, Tseng PY, Xu Q, Tiwari V, Yang F, Shu B, Zhang T, Tang Z, Raja SN, Wang Y, Dong X, Guan Y, The inhibition of high-voltage-activated calcium current by activation of MrgC11 involves phospholipase C-dependent mechanisms. Neuroscience 300, 393–403 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guan Y, Borzan J, Meyer RA, Raja SN, Windup in dorsal horn neurons is modulated by endogenous spinal μ-opioid mechanisms. J. Neurosci. 26, 4298–4307 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zylka MJ, Dong X, Southwell AL, Anderson DJ, Atypical expansion in mice of the sensory neuron-specific Mrg G protein-coupled receptor family. Proc. Natl. Acad. Sci. U.S.A. 100, 10043–10048 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weibel R, Reiss D, Karchewski L, Gardon O, Matifas A, Filliol D, Becker JA, Wood JN, Kieffer BL, Gaveriaux-Ruff C, Mu opioid receptors on primary afferent nav1.8 neurons contribute to opiate-induced analgesia: Insight from conditional knockout mice. PLOS ONE 8, e74706 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim AY, Tang Z, Liu Q, Patel KN, Maag D, Geng Y, Dong X, Pirt, a phosphoinositide- binding protein, functions as a regulatory subunit of TRPV1. Cell 133, 475–485 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim YS, Anderson M, Park K, Zheng Q, Agarwal A, Gong C, Saijilafu L Young S He PC Lavinka F Zhou D Bergles M Hanani Y Guan DC Spray X. Dong, Coupled activation of primary sensory neurons contributes to chronic pain. Neuron 91, 1085–1096 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He S-Q, Yang F, Perez FM, Xu Q, Shechter R, Cheong Y-K, Carteret AF, Dong X, Sweitzer SM, Raja SN, Guan Y, Tolerance develops to the antiallodynic effects of the peripherally acting opioid loperamide hydrochloride in nerve-injured rats. Pain 154, 2477–2486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rios C, Gomes I, Devi LA, μ opioid and CB1 cannabinoid receptor interactions: Reciprocal inhibition of receptor signaling and neuritogenesis. Br. J. Pharmacol. 148, 387–395 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jordan BA, Trapaidze N, Gomes I, Nivarthi R, Devi LA, Oligomerization of opioid receptors with β2-adrenergic receptors: A role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. U.S.A. 98, 343–348 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.