

Abstract
Mutations spawn genetic variation which, in turn, fuels evolution. Hence, experimental investigations into the rate and fitness effects of spontaneous mutations are central to the study of evolution. Mutation accumulation (MA) experiments have served as a cornerstone for furthering our understanding of spontaneous mutations for four decades. In the pregenomic era, phenotypic measurements of fitness-related traits in MA lines were used to indirectly estimate key mutational parameters, such as the genomic mutation rate, new mutational variance per generation, and the average fitness effect of mutations. Rapidly emerging next-generating sequencing technology has supplanted this phenotype-dependent approach, enabling direct empirical estimates of the mutation rate and a more nuanced understanding of the relative contributions of different classes of mutations to the standing genetic variation. Whole-genome sequencing of MA lines bears immense potential to provide a unified account of the evolutionary process at multiple levels—the genetic basis of variation, and the evolutionary dynamics of mutations under the forces of selection and drift. In this review, we have attempted to synthesize key insights into the spontaneous mutation process that are rapidly emerging from the partnering of classical MA experiments with high-throughput sequencing, with particular emphasis on the spontaneous rates and molecular properties of different mutational classes in nuclear and mitochondrial genomes of diverse taxa, the contribution of mutations to the evolution of gene expression, and the rate and stability of transgenerational epigenetic modifications. Future advances in sequencing technologies will enable greater species representation to further refine our understanding of mutational parameters and their functional consequences.
Keywords: effective population size, genetic drift, mutation rate, mutation accumulation, next-generation sequencing, whole-genome sequencing, RNA-Seq
Introduction
Darwin’s theory of evolution by natural selection is inextricably dependent on the presence of heritable variation among individuals within a population. For evolutionary change to occur, there must exist genetic variation that enables the spread of one genotype in lieu of another genotype via the action of major evolutionary forces, such as natural selection or random genetic drift. Indeed, this relationship is embodied in Fisher’s fundamental theorem of natural selection (Fisher 1930) which mathematically demonstrates a correlation between the amount of genetic variation in a population and the rate of evolutionary change by natural selection. Mutation, as the evolutionary force that induces this genetic variation, therefore occupies a central place in evolutionary biology. However, the majority of spontaneous mutations have detrimental effects on organismal fitness (Muller 1950). The rate and fitness effects of new mutations impinge on a multitude of evolutionary and biological phenomena, including but not limited to the maintenance of genetic variation (Lynch and Walsh 1998; Charlesworth and Hughes 1999), the contribution to quantitative trait variation (Caballero and Keightley 1994; Azevedo et al. 2002), the evolution of sex, mating systems and recombination (Pamilo et al. 1987; Kondrashov 1988; Charlesworth 1990; Peck et al. 1997; Otto and Michalakis 1998; Neiman et al. 2010), inbreeding depression (Charlesworth D and Charlesworth B 1987; Charlesworth et al. 1990; Deng and Lynch 1996), the evolution of senescence (Hamilton 1966; Partridge and Barton 1993; Charlesworth and Hughes 1996), the persistence of gene duplicates (Li 1980; Walsh 1995; Force et al. 1999), and the evolution of ploidy level (Kondrashov and Crow 1991; Perrot et al. 1991). Lastly, there has been much interest in the consequences of spontaneous mutations for the maintenance of numerous threatened populations of plants and animals at small population sizes (Lynch and Gabriel 1990; Gabriel et al. 1993; Lande 1994; Lynch et al. 1995a, 1995b; Katju et al. 2018).
Given the centrality of mutations in genetics and evolution, significant effort has been expended in gaining insights into the rate and molecular properties of newly originating mutations. The evolutionary fate of mutations in a population depends on the rate at which they originate as well as the combined action of evolutionary forces, such as natural selection and genetic drift (Kimura 1983; Ohta 1992; Yampolsky and Stoltzfus 2001; Charlesworth 2009; Halligan and Keightley 2009). A key challenge in mutation research is owing to a paradox regarding the nature of mutations. While mutational variation is requisite for adaptive evolution, the vast majority of mutations leading to a change in phenotype usually have detrimental or deleterious effects on the fitness of the carrier (Keightley and Eyre-Walker 1999; Drake 2006). Hence, wild or natural populations under intense selection offer extremely limited opportunities to conduct a comprehensive analysis of newly originating mutations given that the majority are rapidly eradicated via selection in a short evolutionary period. Mutation accumulation (MA hereafter) experiments, theoretically considered by Muller in the 1920s (1928) but experimentally pioneered by Mukai and Ohnishi (Mukai 1964; Mukai et al. 1972; Ohnishi 1977a, 1977b, 1977c), have served as an exemplar approach to estimate key mutational parameters from phenotypic data in the pregenomic era. The underlying principle behind MA experiments is straightforward: Multiple replicate lines derived from an inbred ancestral stock population are allowed to evolve independently of one another under conditions of extreme bottlenecking each generation. In species where selfing is the primary mode of reproduction (e.g., Saccharomyces cerevisiae, Chlamydomonas reinhardtii, Caenorhabditis elegans and Caenorhabditisbriggsae, Daphnia, and Arabidopsis), Ne is kept constant at one individual per generation. For obligate outcrossing species such as Drosophila, each new generation has a sibling mating pair as the founders. This regime of selfing or inbreeding dictates that newly arising mutations, if not lost via drift, are rapidly driven to homozygosity in diploid species. In microbial systems, single-cell bottlenecks can be created via restreaking of colonies (Andersson and Hughes 1996; Kibota and Lynch 1996) or single cell dilution (Krasovec et al. 2016). The repeated bottlenecks severely diminish the efficacy of natural selection, promoting evolutionary divergence due to the accumulation of mutations by random genetic drift (fig. 1). Where possible, excess individuals descended from the same ancestral genotype/line as the experimental lines are cryopreserved in a presumably inert, unevolving state for subsequent phenotypic or molecular comparisons with experimental lines subjected to multiple MA generations. Hence, MA studies circumvent the challenges of studying mutations in natural populations where strong selection may purge the very mutational variants of interest.
Fig. 1.

—Schematic of a classical MA experiment. For simplicity, the figure depicts a single chromosome pair in a selfing diploid species. Multiple MA lines (n), all descended from a common ancestral progenitor line, are independently maintained for t generations under an experimental regime of consecutive bottlenecks that drastically reduces the efficacy of selection, thereby enabling the accumulation of spontaneous mutations within experimental lines under the influence of genetic drift. Excess individuals descended from the progenitor line are preserved where possible, to serve as ancestral controls for phenotypic, molecular and/or genomic comparisons with the evolved MA lines bearing new mutations. New spontaneous mutations, denoted by colored lines on chromosomes, initially exist in a heterozygous form but can be lost due to genetic drift (not shown for simplicity) or rapidly become homozygous due to the inbreeding/selfing regime imposed in MA experiments. Following t generations, MA lines are expected to have diverged phenotypically due to the accumulation of varying mutation loads (both with respect to the total number and types of mutations) owing to the stochastic nature of the spontaneous mutation process, culminating in an increase in phenotypic between-line variance. Adapted from Halligan and Keightley (2009).
Under the assumption that the majority of newly occurring mutations have deleterious fitness effects, an expected signature of MA studies is an average fitness decline of the experimental lines and an increase in among-line variance with additional generations of bottlenecking. As the vast majority of mutations occur and become fixed/lost spontaneously under the experimental regime of MA studies, they represent an ideal and relatively unbiased sample set for investigating the rates, fitness effects, and other properties of spontaneous mutations. The fitness effect of a mutation can range continuously from lethal to deleterious to neutral to beneficial. Loss or fixation of mutations and their consequences for population fitness depend upon the selection coefficients (s) associated with individual mutations and the effective population size, Ne. For sexually reproducing diploids, the dynamics of mutations with |s| ≪ 1/2Ne and |s| ≫ 1/2Ne are dictated by drift and selection, respectively (Kimura 1962, 1983). Similarly, for haploid species, the dynamics of mutations with |s| ≪ 1/Ne and |s| ≫ 1/Ne are dictated by drift and selection, respectively. Deleterious mutations with extremely large effects are unlikely to pose a long-term threat to population fitness as they are rapidly eradicated via selection and unlikely to reach fixation; those with extremely small or no effects would be effectively neutral. Although the long-term consequence of a mutation is dependent on the effective size of a population, the prevailing opinion is that the most detrimental class of mutations influencing long-term population fitness includes mutations with intermediate selection coefficients (Ohta 1992). Such mutations would be eradicated via purifying selection at high Ne, but can behave in an effectively neutral manner and reach fixation by genetic drift under low Ne conditions although they may not be neutral with respect to absolute fitness (Lynch et al. 1999). Therefore, small populations subjected to attenuated selection and an increased magnitude of genetic drift can potentially accumulate mutations with extremely large effects in addition to ones with moderate to very slight effects. It should be mentioned that while the majority of MA experiments display a pattern of average fitness decline, it is not universally observed as some experimental lines may maintain ancestral fitness levels despite an extended MA regime (Hall et al. 2013; Dillon and Cooper 2016; Krasovec et al. 2017). A lack of fitness decline could be owing to the stochastic accumulation of mutations in some lines but not others, a load of neutral to near-neutral mutations with minimal contribution to phenotypic evolution, or the choice of a trait lacking a substantial fitness component in the benign MA experimental conditions.
Since the initial experiments of Mukai and Ohnishi, many MA studies (both spontaneous and mutagen-induced) have been conducted in a diverse set of organisms, from viruses to multicellular eukaryotes (reviewed by Halligan and Keightley 2009). In a period spanning approximately three decades (mid-1960s to late 1990s), most of our insights into the basic fundamental properties of new genetic variation stemming from spontaneous mutations have been gleaned from phenotypic analyses of these time- and labor-intensive MA experiments. The MA experiments from this period provided indirect estimates of key mutational parameters for life-history or quantitative traits, such as the haploid genome-wide mutation rate per generation (U), the average selection coefficient of mutations [E(a)], the degree of dominance of new mutations, the nature of epistatic interactions between mutations, and their environmental context-dependence, among others (see Halligan and Keightley 2009). For example, phenotypic estimates of U in eukaryotes ranged widely (>700-fold) from 0.00065 to 0.47 per genome per generation, likely reflecting differences in experimental conditions and the nature of the fitness-trait measured (Mukai 1964; Mukai et al. 1972; Houle et al. 1992; Keightley and Caballero 1997; García-Dorado et al. 1998; Fry et al. 1999; Vassilieva et al. 2000; Ávila and Garcia-Dorado 2002; Charlesworth et al. 2004; Joseph and Hall 2004; Baer et al. 2005; Schoen 2005). Another intriguing result from phenotypic analyses of MA studies is that assays under competitive or stress conditions tend to yield higher estimates of U (Fry et al. 1999; Gong et al. 2005) relative to benign assays suggesting that phenotypic data from MA studies under benign conditions can detect causal mutations only if they are of moderate to large effects. If phenotypic assays consistently underestimate U relative to direct molecular approaches, this points to the possibility of a large fraction of cryptic new mutations with very mild deleterious effects on fitness or some unknown fraction of mutations that behave neutrally under benign conditions but may be deleterious in the wild. Together, this vast range in values of U from phenotypic assays of MA lines and discrepancies in U estimates from benign versus competitive phenotypic assays underscores the idea that our ability to infer U is limited by experimental resolution and simplifying assumptions implicit in the analytical approach (e.g., equal fitness effects of new mutations).
The advent of the genomic revolution since the late 1990s has led to a burgeoning of studies directly employing whole-genome sequencing (WGS) technology to directly estimate the mutation rate in MA lines of diverse species. Direct WGS approaches, currently utilizing next- or second-generation (Illumina/Solexa, 454 Pyrosequencing, SOLiD/Applied Biosystems, Ion Torrent) and third-generation sequencing technologies (PacBio), offer both short (25–200 bp) and long (up to 10 kb) DNA sequences (reads) that are generated using a massively parallel, automated approach. Short reads of the genomes of MA lines (MA-WGS, henceforth) and the ancestral control are then assembled using a published reference genome. MA-WGS approaches offer considerable advantages in furthering our understanding of the spontaneous mutation process. First, they yield a direct empirical estimate of the genome-wide spontaneous mutation rate inclusive of 1) mutations leading to phenotypic changes, 2) previously undetected cryptic neutral or nearly neutral mutations with no discernible effect on phenotype, and 3) cryptic deleterious mutations with no fitness effects under benign laboratory conditions while engendering phenotypic effects under wild or stringent conditions. A second important consideration is that MA-WGS studies enable direct estimation of the spontaneous mutation rates of different classes of mutations, such as base substitutions, short insertion and deletion events, inversions, and copy-number changes. Third, MA-WGS approaches enable estimation of mutation rates in nuclear versus organellar genomes (mitochondrial, chloroplast) of eukaryotic species. Fourth, MA-WGS permits more nuanced investigations into the heterogeneity of rates and properties of spontaneous mutations occurring in 1) different genomic regions (interchromosomal, and intrachromosomal regions such as arms, cores, and tips), 2) genomic regions that may be under differing selective constraints such as exonic regions under more stringent selection versus intergenic and intron regions that may evolve in a more neutral fashion overall, and 3) differential mutability and selective constraints at specific sites within exonic, intronic, and intergenic regions. Lastly, high-throughput RNA-sequencing technology has the potential to usher in the first genome-wide insights into the transcriptional and functional consequences of different mutational classes, in conjunction with the role of environmental conditions and differing developmental stages in dictating the realized phenotype.
There have been several excellent reviews of MA experiments and their evolutionary implications (García-Dorado et al. 1999; Keightley and Eyre-Walker 1999; Lynch et al. 1999; Halligan and Keightley 2009) based on phenotypic measurements of MA lines. However, the last decade has seen a rapid emergence of studies partnering classical MA experiments with modern next-generation sequencing technology to generate direct molecular estimates of the spontaneous mutation rates pertaining to different classes of mutations and in different genomic regions with initial forays into the use of transcriptomics to investigate the effects of mutation on gene expression divergence. In this review, we summarize the findings of these MA-WGS studies and discuss their influence on our current understanding of the spontaneous mutation process in diverse organisms. We have largely limited our discussion to spontaneous MA experiments using high-throughput genomic approaches, but have included earlier genome-wide studies of MA lines using Sanger sequencing approaches where relevant. We have reviewed and synthesized the results of spontaneous MA-WGS studies to compare spontaneous mutation rates and the spectrum of mutations across prokaryotes, unicellular eukaryotes, and multicellular eukaryotes to determine both taxa-specific and broadly shared features across these diverse organisms. We additionally review in detail the mutation process in one organellar genome, namely the mitochondrial DNA (mtDNA) of eukaryotes. Our analysis further delves into the comparison of phenotypic versus direct molecular estimates of the genomic mutation rate U and offers explanations for the observed discrepancy that exists between the two estimates. The evolution of mutation rates as a function of genome size and effective population size (Ne) is further explored though a thorough treatment of the subject is provided in preceding reviews (Baer et al. 2007; Lynch 2010a; Lynch et al. 2016). Lastly, we provide the first comprehensive review of transcriptional and epigenetic changes due to mutation, as gleaned from MA-WGS studies.
Mutational Landscape in Prokaryotic Genomes
MA experiments in prokaryotes typically involve picking and streaking colonies on agar. Each time a colony is restreaked, the population of cells in the colony is passed through a bottleneck of a single cell. After 20–30 generations, the number of cells per colony can be in the range of 106–109 but the Ne remains small because of the repeated single-cell bottlenecks, or roughly half the number of generations of growth in the colony. Experiments in Salmonella typhimurium and Escherichiacoli showed that there is an average decrease in growth rates associated with repeated single-cell bottlenecks and a divergence in growth rates between lines, both hallmarks of MA (Andersson and Hughes 1996; Kibota and Lynch 1996). Furthermore, multiple lines of evidence suggest that selection is negligible in MA studies of prokaryotes, and that the rates and patterns of mutations in prokaryotic genomes have not been biased by selection during repeated colony restreaking.
Spontaneous Rates of Base Substitutions
In prokaryotes, the spontaneous rate of base substitution, μbs, ranges ∼300-fold, from 7.9 × 10−11 to 2.34 × 10−8/site/generation (table 1) with a median rate of 3.28 × 10−10. Although the sample size is still fairly limited, the species that have been analyzed thus far range broadly in genome size, number of chromosomes and G+C-content. These include Mesoplasma florum with a genome size of only 780 kb and a G+C-content of 27%, Mycobacterium smegmatis with a genome size of 7 Mb and G+C-content of 67%, Burkholderia cenocepacia with a genome size of 8 Mb and G+C-content of 67% and three chromosomes (most prokaryotes have only one circular chromosome), and Deinococcus radiodurans, famous for being the world’s most extremophile bacterium according to the Guinness Book of World Records. In addition to these, there are mutation rate measurements from more traditionally studied bacteria, such as Bacillus subtilis, E.coli (several strains), Pseudomonas sp. aeruginosa, and Salmonella typhimurium.
Table 1.
Estimates of Spontaneous Nuclear Base Substitution and Small Insertion–Deletion (Indels) Mutation Rates from MA Experiments Using High-Throughput Sequencing Approaches
| Species | Kingdom | Group | Ne | Average MA Gens. | μtotal (/site/gen) | μbs (/site/gen) | μindel (/site/gen) | Ratio μbs:μindel | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Prokaryotes | |||||||||
| Bacillus subtilis | Bacteria | Terrabacteria | — | 5,645 | — | 3.28 × 10−10 | — | — | Sung et al. (2015) |
| Burkholderia cenocepacia | Bacteria | Proteobacteria | — | 5,554 | 1.50 × 10−10 | 1.33 × 10−10 | 1.68 × 10−11 | 8:1 | Dillon et al. (2015) |
| Deinococcus radiodurans | Bacteria | Terrabacteria | — | 5,961 | 5.21 × 10−10 | 4.99 × 10−10 | 2.17 × 10−11 | 23:1 | Long et al. (2015) |
| Escherichia coli K12 | Bacteria | Proteobacteria | — | 6,000 | 2.38 × 10−10 | 2.20 × 10−10 | 1.81 × 10−11 | 12:1 | Lee et al. (2012) |
| Escherichia coli K12 | Bacteria | Proteobacteria | — | 6,114 | — | 3.12 × 10−10 | 3.12 × 10−11 | 10:1 | Foster et al. (2015) |
| Mesoplasma florum L1 | Bacteria | Terrabacteria | — | 2,351 | 1.16 × 10−8 | 9.78 × 10−9 | 1.85 × 10−9 | 5:1 | Sung, Ackerman, et al. (2012); Sung et al. (2015) |
| Mycobacterium smegmatisa | Bacteria | Terrabacteria | — | ∼49,000 | 6.54 × 10−10 | 5.27 × 10−10 | 1.27 × 10−10 | 4:1 | Kucukyildirim et al. (2016) |
| Pseudomonas aeruginosa | Bacteria | Proteobacteria | — | ∼2,500 | 9.30 × 10−11 | 7. 90 × 10−11 | 1.44 × 10−11 | 5:1 | Dettman et al. (2016) |
| Pseudomonas fluorescensa | Bacteria | Proteobacteria | — | 5,240 | 2.51 × 10−8 | 2.34 × 10−8 | 1.65 × 10−9 | 14:1 | Long et al. (2015) |
| Salmonella typhimurium LT2 | Bacteria | Proteobacteria | — | 5,000 | — | 7.00 × 10−10 | — | — | Lind and Andersson (2008) |
| Vibrio cholerae 2740–80 | Bacteria | Proteobacteria | — | 6,453 | 1.24 × 10−10 | 1.07 × 10−10 | 1.71 × 10−11 | 6:1 | Dillon et al. (2017) |
| Vibrio fischeri ES114 | Bacteria | Proteobacteria | — | 5,187 | 2.64 × 10−10 | 2.07 × 10−10 | 5.68 × 10−11 | 4:1 | Dillon et al. (2017) |
| Unicellular eukaryotes | |||||||||
| Bathycoccus prasinos | Eukaryota | Plants | 8.5 | 4,994 | 4.39 × 10−10 | 3.02 × 10−10 | 1.37 × 10−10 | 2:1 | Krasovec et al. (2017) |
| Chlamydomonas reinhardtii | Eukaryota | Plants | — | 1,730 | 1.11 × 10−10 | 6.76 × 10−11 | 4.36 × 10−11 | 2:1 | Sung, Ackerman, et al. (2012) |
| Chlamydomonas reinhardtii | Eukaryota | Plants | 6.5 | 940 | 1.15 × 10−9 | 9.63 × 10−10 | 1.90 × 10−10 | 5:1 | Ness et al. (2015) |
| Dictyostelium discoideum | Eukaryota | Protists | — | 1,000 | — | 2.90 × 10−11 | — | — | Saxer et al. (2012) |
| Micromonas pusilla | Eukaryota | Plants | 6 | 4,145 | 9.76 × 10−10 | 8.15 × 10−10 | 1.61 × 10−10 | 5:1 | Krasovec et al. (2017) |
| Ostreococcus mediterraneus | Eukaryota | Plants | 7 | 8,379 | 5.92 × 10−10 | 4.92 × 10−10 | 1.00 × 10−10 | 5:1 | Krasovec et al. (2017) |
| Ostreococcus tauri | Eukaryota | Plants | 8.5 | 17,250 | 4.79 × 10−10 | 4.19 × 10−10 | 6.00 × 10−11 | 7:1 | Krasovec et al. (2017) |
| Paramecium tetraurelia | Eukaryota | Protists | — | 3,300 | 2.33 × 10−11 | 1.94 × 10−11 | 3.87 × 10−12 | 5:1 | Sung, Tucker, et al. (2012) |
| Saccharomyces cerevisiae | Eukaryota | Fungi | 10 | 4,800 | 3.50 × 10−10 | 3.30 × 10−10 | 2.00 × 10−11 | 17:1 | Lynch et al. (2008) |
| Saccharomyces cerevisiae | Eukaryota | Fungi | — | 1,740 | 2.90 × 10−10 | 2.90 × 10−10 | 0 | — | Nishant et al. (2010) |
| Saccharomyces cerevisiae | Eukaryota | Fungi | — | 2,500 | 3.60 × 10−10 | 3.60 × 10−10 | 0 | — | Serero et al. (2014) |
| Saccharomyces cerevisiae | Eukaryota | Fungi | 10 | 2,062 | 1.72 × 10−10 | 1.67 × 10−10 | 5.03 × 10−12 | 33:1 | Zhu et al. (2014) |
| Schizosaccharomyces pombe | Eukaryota | Fungi | — | 1,700 | 2.73 × 10−10 | 2.13 × 10−10 | 6.00 × 10−11 | 4:1 | Farlow et al. (2015) |
| Schizosaccharomyces pombe | Eukaryota | Fungi | 10.3 | 1,952 | 3.40 × 10−10 | 1.70 × 10−10 | 1.70 × 10−10 | 1:1 | Behringer and Hall (2016) |
| Tetrahymena thermophila | Eukaryota | Protists | — | 1,000 | — | 7.61 × 10−12 | — | — | Long et al. (2016) |
| Multicellular eukaryotes | |||||||||
| Arabidopsis thaliana | Eukaryota | Plants | 1 | 30 | 8.40 × 10−9 | 7.10 × 10−9 | 1.30 × 10−9 | 5:1 | Ossowski et al. (2010) |
| Caenorhabditis briggsae | Eukaryota | Metazoa | 1 | 250 | — | 1.33 × 10−9 | — | — | Denver et al. (2012) |
| Caenorhabditis elegans | Eukaryota | Metazoa | 1 | 250 | — | 2.10 × 10−9 | — | — | Denver et al. (2009) |
| Caenorhabditis elegans | Eukaryota | Metazoa | 1 | 250 | — | 1.45 × 10−9 | — | — | Denver et al. (2012) |
| Daphnia pulex | Eukaryota | Metazoa | 1 | 128 | — | 3.80 × 10−9 | — | — | Keith et al. (2016) |
| Daphnia pulex | Eukaryota | Metazoa | 1 | 82 | — | 2.30 × 10−9 | — | — | Flynn et al. (2017) |
| Drosophila melanogaster | Eukaryota | Metazoa | 2 | 262 | 4.83 × 10−9 | 3.46 × 10−9 | 1.37 × 10−9 | 3:1 | Keightley et al. (2009) |
| Drosophila melanogaster | Eukaryota | Metazoa | 2 | 149 | 5.94 × 10−9 | 5.49 × 10−9 | 4.50 × 10−10 | 12:1 | Schrider et al. (2013) |
| Drosophila melanogasterb | Eukaryota | Metazoa | — | 60 | 6.00 × 10−9 | 5.21 × 10−9 | 7.90 × 10−10 | 7:1 | Huang et al. (2016) |
| Drosophila melanogaster | Eukaryota | Metazoa | 2 | 52 | 6.37 × 10−9 | 6.03 × 10−9 | 3.38 × 10−10 | 18:1 | Sharp and Agrawal (2016) |
| Drosophila melanogaster | Eukaryota | Metazoa | 2 | 36–53 | — | 4.90 × 10−9 | — | — | Assaf et al. (2017) |
| Mus musculus | Eukaryota | Metazoa | — | 20–21 | 5.71 × 10−9 | 5.40 × 10−9 | 3.10 × 10−10 | 17:1 | Uchimura et al. (2015) |
| Pristionchus pacificus | Eukaryota | Metazoa | 1 | 142 | — | 2.00 × 10−9 | — | — | Weller et al. (2014) |
Naturally occurring mutator strain.
Autosomal mutation rate only.
The mutation rates measured by sequencing MA lines can differ significantly from previous published estimates using single indicator loci. For example, the MA-WGS estimate of the mutation rate for Salmonella typhimurium is 7 × 10−10/site/generation (Lind and Andersson 2008) whereas a reporter locus approach using various reversion mutations in lacZ constructs yielded a mutation rate of 9 × 10−11/site/generation (Hudson et al. 2003). Likewise, the first MA-WGS-based rates for E. coli (Lee et al. 2012) were roughly one-third of previously accepted estimates using reporter genes (Drake 1991). The discrepancies between MA measurements of mutation rates and reporter loci can have several causes. First, the growth conditions of bacteria during MA and in classical mutation rate experiments are different. In a traditional mutation rate experiment, a large number of independent liquid cultures are plated on selective medium which reveals the phenotypes of the mutant cell; whereas in MA experiments, the bacteria grow in colonies on a plate. The difference between growth in liquid versus solid medium could well contribute to discrepancies between mutations rates. Furthermore, a reporter locus may not be representative of the genome as a whole. In addition, classical mutation rate experiments depend on the phenotypes of reporter loci. In some cases where the mutation rate estimate is based on the reversion of a mutant gene, the original mutation may be leaky. Cells with the leaky mutations can, in some cases, pass through additional generations on a selective medium, and accrue additional mutations that were absent in the original culture. This in turn would result in an overestimation of the mutation rate. Alternatively, the mutant phenotype that is being screened may need time to develop, resulting in a phenotypic lag. A good example of this was provided in experiments that compared mutation rate measurements from WGS versus estimates from resistance to rifampicin and nalidixic acid (Lee et al. 2012). The mutation rates based on the frequency of antibiotic resistant colonies were much lower, presumably because mutants take time, even a few generations, to fully develop resistance.
Rates of Small Insertions and Deletions
Small insertion and deletion events (indels, henceforth) refer to the insertions or deletions of a small number of nucleotide bases, typically 50 bp or less. Variation between species with regard to published small indel rates can be problematic because of the use of different criteria to estimate these rates by different research groups. The small indel rates have been based on indels of <5 nt, <10 or even <146 bp (Lee et al. 2012; Dettman et al. 2016). Furthermore, the identification of indels in short-read alignments is beset with difficulties. One concern regarding the analysis of indels is that different studies do not use the same pipeline for variant calling, and this variability in indel calling methods frequently yields different results (O’Rawe et al. 2013; Hasan et al. 2015). Although most analyses of MA lines use Sanger sequencing on a sample of variants to estimate the proportion of false positives, false negatives can also impact the results and different variant-calling methods may have their own intrinsic biases in calling indels, contributing to the variation among different studies (Hasan et al. 2015).
The spontaneous mutation rate for small indel events, μindel, in ten prokaryotic species ranges ∼128-fold, from 1.44 × 10−11 to 1.85 × 10−9/site/generation, with P.aeruginosa and Mesoplasma florum displaying the lowest and highest rate, respectively (table 1). Despite these limitations and differences in methodologies for indel variant-calling, it seems clear that small indels are less frequent than base substitutions in each of the ten species of bacteria listed in table 1. The ratio of base substitutions to indels ranges from four in Mycobacterium smegmatis and Vibrio fischeri to 23 in Deinococcus radiodurans. Indels occur most frequently in simple sequence repeats, and the indel rate is correlated with both the number of repeats and the length of the repeat motif (Lee et al. 2012; Long et al. 2015; Dettman et al. 2016; Dillon et al. 2017). The majority of MA experiments in prokaryotes have found a deletion bias, with small deletions being more frequent than small insertions (table 2). Similar results have been obtained previously, for example, by analyzing insertions and deletions in bacterial pseudogenes (Mira et al. 2001). However, mismatch-repair deficient strains of bacteria can have a radically altered spectrum of indel mutations. These include an insertion bias in the naturally occurring mutator strain of Mycobacterium smegmatis and a stronger bias toward single nucleotide indels (Long et al. 2015; Kucukyildirim et al. 2016; Dillon et al. 2017).
Table 2.
Properties and Mutation Bias of Spontaneous Base Substitutions and Small Indels Observed via High-Throughput Sequencing of MA Lines
| Species | AT Biasa | Ts:Tv Mutation Bias | RatioNonsyn:Syn | Ratio of Insertions to Deletions | Reference |
|---|---|---|---|---|---|
| Prokaryotes | |||||
| Bacillus subtilis NCIB3610 | 0.60 | 6:1 | 3:1 | — | Sung et al. (2015) |
| Burkholderia cenocepacia | 0.83 | 2:1 | 3:1 | 0.94 | Dillon et al. (2015) |
| Deinococcus radiodurans | 0.49 | 3:1 | 3:1 | 1.11 | Long et al. (2015) |
| Escherichia coli K12 substr. MG1655 | 1.24 | 3:1 | 2:1 | 0.40 | Lee et al. (2012) |
| Escherichia coli ED1a | 2.09 | 3:1 | 3:1 | 0.19 | Foster et al. (2015) |
| Escherichia coli IAI1 | 2.04 | 2:1 | 2:1 | 0.19 | Foster et al. (2015) |
| Mesoplasma florum L1 | 15.97 | 3:1 | 6:1 | 0.98 | Sung, Ackerman, et al. (2012) |
| Mycobacterium smegmatisb | 0.73 | 3:1 | 2:1 | 2.14 | Kucukyildirim et al. (2016) |
| Vibrio cholerae 2740–80 | 2.71 | 3:1 | 2:1 | 0.29 | Dillon et al. (2017) |
| Vibrio fischeri ES114 | 4.26 | 2:1 | 5:1 | 0.58 | Dillon et al. (2017) |
| Unicellular eukaryotes | |||||
| Bathycoccus prasinos | 2.89 | 1:1 | 2:1 | 1.00 | Krasovec et al. (2017) |
| Chlamydomonas reinhardtii | 1.10 | 1:1 | — | 1.60 | Sung, Ackerman, et al. (2012) |
| Chlamydomonas reinhardtii | 2.88 | 2:1 | 2:1 | 0.84 | Ness et al. (2015) |
| Micromonas pusilla | 1.00 | 2:1 | 3:1 | 0.17 | Krasovec et al. (2017) |
| Ostreococcus mediterraneus | 1.31 | 3:1 | 4:1 | 0.38 | Krasovec et al. (2017) |
| Ostreococcus tauri | 1.74 | 7:1 | 2:1 | 0.63 | Krasovec et al. (2017) |
| Paramecium tetraurelia | 12.86 | 1:1 | 2:1 | _ (5:0) | Sung, Tucker, et al. (2012) |
| Saccharomyces cerevisiae | 3.96 | 1:1 | 3:1 | _ (0:1) | Lynch et al. (2008) |
| Saccharomyces cerevisiae | 2.23 | 2:1 | 3:1 | 0.45 | Zhu et al. (2014) |
| Schizosaccharomyces pombe | 2.65 | 2:1 | 3:1 | 6.00 | Farlow et al. (2015) |
| Schizosaccharomyces pombe | 2.97 | 1:1 | 2:1 | 6.13 | Behringer and Hall (2016) |
| Tetrahymena thermophila | 10.04 | 3:1 | 2:1 | — | Long et al. (2016) |
| Multicellular eukaryotes | |||||
| Arabidopsis thaliana | 6.09 | 5:1 | 3:1 | 0.50 | Ossowski et al. (2010) |
| Caenorhabditis elegans | 2.24 | 1:1 | 2:1 | — | Denver et al. (2009) |
| Daphnia pulex | 2.69 | 3:1 | — | — | Keith et al. (2016) |
| Drosophila melanogaster | 2.08 | 2:1 | 2:1 | 0.17 | Keightley et al. (2009) |
| Drosophila melanogaster | 4.33 | 6:1 | 9:1 | 0.20 | Schrider et al. (2013) |
| Drosophila melanogaster | 2.85 | 2:1 | 3:1 | 0.33 | Huang et al. (2016) |
| Drosophila melanogaster | 3.84 | 2:1 | 3:1 | 0.32 | Sharp and Agrawal (2016) |
| Drosophila melanogaster | 3.12 | 2:1 | — | — | Assaf et al. (2017) |
| Pristionchus pacificus | 5.16 | 2:1 | 3:1 | — | Weller et al. (2014) |
Note.—Ts and Tv refer to transitions and transversions, respectively. Nonsyn and Syn refer to nonsynonymous and synonymous substitutions in protein-coding genes, respectively.
Weighted by genomic nucleotide composition.
Naturally occurring mutator strain.
Local Context-Dependence of Spontaneous Mutations
Neighboring Bases
The importance of base composition of neighboring bases for mutation rates was first suggested by Seymour Benzer as a part of his classic work on the fine structure of genes (Benzer 1961). It has long been known that certain combinations of nucleotides can be either underrepresented or overrepresented. In principle, such deviations from random expectations can result from either context-dependent mutation rates or selection for or against certain sequence motifs in genomes. Although many MA studies lack a sufficient number of mutations to test whether the rates of particular nucleotide substitutions are influenced by the identity of neighboring nucleotides, several experiments with bacteria, both wild-type and DNA-repair deficient, have provided evidence for strong context-dependence. The results from MA experiments have uncovered both general trends and species-specific patterns of context-dependent mutations. As an example of a general trend, YR (pyrimidine–purine) and RY dimers have higher mutation rates than YY and RR dimers (Sung et al. 2015). Focal nucleotides with G or C on their 5′ or 3′ side have higher mutation rates than those bearing A or T on their 5′ or 3′ side in Bacillus subtilis, E. coli, Deinococcus radiodurans, and Pseudomonas fluorescens but not in M. florum (Lee at al. 2012; Sung et al. 2015). Mismatch-repair-deficient strains such as E. coli mutL and Bacillus subtilis have similar context dependence as their wild-type counterparts. Incorporating additional 5′ and 3′ neighboring bases to the analysis (5-mers and 7-mers) does not have a significant effect, suggesting that the context-dependence is due to the immediately adjacent nucleotides (Sung et al. 2015).
Computer simulations have revealed that the observed frequency of nucleotide triplets in the genome of M. florum was strongly correlated with the equilibrium frequency of triplets using its context-dependent mutation rates, but the frequency of triplets in E. coli and Bacillus subtilis exhibited no such correlation (Sung et al. 2015). Mesoplasmaflorum has a smaller Ne than either E. coli and Bacillus subtilis, which fits the prediction that the base composition of species with small Ne should resemble the context-dependent mutational equilibrium more than species with larger Ne (Sung et al. 2015).
Chromatin Organization
Additional local structural characteristics of bacterial chromosomes can also influence their mutation rates. In mismatch-repair-deficient E. coli, the density of mutations across the genome is nonrandom and increases and decreases in a wave-like function with distance from the origin of replication (Foster et al. 2013). The mutation rates were positively correlated with the degree of predicted superhelicity.
Nuclear Mutations in Eukaryotic Genomes
Base Substitutions
Direct genome-wide estimates of the spontaneous base substitution rate, μbs, have been generated for ten unicellular and eight multicellular eukaryotic species (table 1). The subset of unicellular eukaryotic species includes five algae, two fungi, and three protists. Spontaneous rates of nuclear base substitutions in unicellular eukaryotes range from 7.61 × 10−12 to 8.15 × 10−10/site/generation, representing a ∼100-fold difference among the ten species, with a median μbs of 2.94 × 10−10/site/generation. The robustness of these estimates can be indirectly verified for three species, the algae C. reinhardtii and the fungal species S. cerevisiae and Schizosaccharomyespombe, wherein different researchers have generated mutation rates from independent MA experiments varying in time span (MA generations) and sequencing platform. These independent estimates of the mutation rate differ by ∼3-fold for C. reinhardtii (Ness et al. 2012; Sung, Ackerman, et al. 2012), ∼2-fold for S. cerevisiae (Lynch et al. 2008; Nishant et al. 2010; Serero et al. 2014; Zhu et al. 2014), and only ∼1.25-fold for Schizosaccharomyes pombe (Farlow et al. 2015; Behringer and Hall 2016). The average μbs for the algal, fungal and protist species are 5.09 × 10−10, 2.39 × 10−10 and 1.87 × 10−11, respectively. The extremely small sample size of the data set and the biased species representation preclude robust statistical testing, but the data suggest that the wide range in overall mutation rates reported for unicellular eukaryotes stems largely from the extremely low mutation rates observed in protists (Saxer et al. 2012; Sung, Ackerman, et al. 2012; Long et al. 2016). Indeed, the ciliate Tetrahymena thermophila (Long et al. 2016) currently has the lowest base substitution rate observed for any species tested in an MA setting, across both prokaryotes and eukaryotes. Given that protists do not represent a natural clade or a formal taxon, additional species testing is required to determine the cause(s) of and extent to which substitution rates may be constrained among various clades within this paraphyletic group.
With respect to multicellular eukaryotes, genome-wide rates of spontaneous base substitution are known via MA experiments in one plant species and seven metazoans (table 1, and references therein). Estimates of μbs for multicellular eukaryotes range from 1.33 to 7.1 × 10−9/site/generation, with the nematode Caenorhabditis. briggsae and the angiosperm Arabidopsisthaliana representing the lower and upper ends of the rate spectrum, respectively. The median μbs is 2.53 × 10−9/site/generation. The range of base substitution rates in multicellular eukaryotes is ∼5-fold, far narrower than the ∼100-fold difference observed for unicellular eukaryotes. If only metazoans are considered, the difference in base substitution rates contracts further, to a 4-fold difference. The nematodes, Caenorhabditis elegans, Caenorhabditis briggsae, and Pristionchus pacificus, exhibit an average base substitution rate of 1.7 × 10−9. The five independent estimates of the mutation rate for Drosophilamelanogaster differ by ∼2-fold with an average rate of 5.02 × 10−9. The microcrustacean, Daphnia pulex, falls in the middle of the metazoan spectrum, with an average rate of 3.05 × 10−9. Additional MA experiments in plants will be required to address whether the A. thaliana rate is representative of the taxon, and is, on average, higher than that of metazoans. The median μbs of unicellular eukaryotes is more similar to that of prokaryotes (∼1.1-fold difference) relative to multicellular eukaryotes (∼9-fold difference) and may be due to larger effective population sizes of unicellular eukaryotes and greater intensity of selection on the evolution of the mutation rate (see section on the Sources of Variation in Mutation Rates).
Small Indel Events
Direct genome-wide estimates of the small indel rate, μindel, have been generated for nine unicellular and three multicellular eukaryotic species (table 1). The subset of unicellular eukaryotic species includes five algae, two fungi, and one ciliate. In unicellular eukaryotes, μindel ranges from 3.87 × 10−12 to 1.61 × 10−10/site/generation, representing a ∼40-fold difference among the eight species, with a median μindel of 8.82 × 10−11/site/generation. Average μindel values for the algal, fungal and protist species are 1.07 × 10−10, 6.06 × 10−11 and 3.87 × 10−12, respectively. The data set for small indel rates in multicellular eukaryotes is more limited, with one estimate for Arabidopsis (1.3 × 10−9/site/generation), four independent estimates for D. melanogaster (average 7.4 × 10−10/site/generation), and one estimate for Mus musculus (3.1 × 10−9/site/generation). The average small indel rate is ∼1 order of magnitude greater in multicellular eukaryotes (1.13 × 10−9/site/generation) relative to unicellular eukaryotes (8.24 × 10−11/site/generation). If all 12 species of eukaryotes are pooled together, the μindel ranges from 3.87 × 10−12 to 1.3 × 10−9/site/generation, representing a ∼340-fold difference among them, and with a median μindel of 1.16 × 10−10/site/generation.
The small sample size of the data set and biased species representation preclude robust statistical testing, but the data are suggestive of some trends. For each of the 12 eukaryotic species, small indels are, on average, less frequent than base substitutions (table 1, and references therein), recapitulating the pattern observed in prokaryotes. With the exception of Schizosaccharomyes pombe (Behringer and Hall 2016), the ratio of base substitutions to indels ranges from 2 in the algal species Bathycoccus prasinos (Krasovec et al. 2017) and C. reinhardtii to 33 in one estimate for S. cerevisiae (Zhu et al. 2014). Additionally, the size of small deletions is frequently greater than that of small insertions (Ness et al. 2015; Krasovec et al. 2017). Arabidopsis and Drosophila display a deletion bias as is observed in the majority of MA experiments with prokaryotes. However, there are also notable exceptions to the rule of a deletion bias. Two independent MA experiments with Schizosaccharomyes pombe found that insertions were six times more common than deletions (Farlow et al. 2015; Behringer and Hall 2016). There were also instances of discordant results within the same species. Experiments with genetically divergent lines of C. reinhardtii have found significant variation in mutation rates, including indel rates (table 2). The most extensive MA experiment in C. reinhardtii found that deletions were more common than insertions and that deletions were, on average, larger than insertions (Ness et al. 2015). However, there was considerable variation between lines, which also includes variation in the patterns of indel mutations. One line in particular displayed an excess of 9-bp deletions that were not associated with any particular sequence motifs. After removing the disproportionately large number of 9-bp deletions from this line, the average frequency of deletions was not significantly different from the average frequency of insertions, but the average length of deletions was still greater than the average length of insertions.
Mutational Spectra of Nuclear Changes
All eukaryotic genomes analyzed to date have a strong A/T mutation bias (table 2). The data are consistent with a substantial contribution from oxidative damage resulting in 5-hydroxyuracil from oxidative deamination of 5-methylcytosine and C:G→T:A transitions, and 8-oxoguanine resulting in G:C→ T:A transversions (Duncan and Miller 1980; Grollman and Moriya 1993). Not only are these major sources of mutation in eukaryotes, but also a major source of mutation rate variation within species. MA experiments in D. melanogaster uncovered genetic variation in mutation rate that was primarily due to high levels of C:G→T:A transitions in one line (Schrider et al. 2013). In light of these results, it is possible to calculate the expected equilibrium base composition at silent sites and compare it with the observed. Thus far, it appears that the G+C-content in silent sites of genomes is higher than expected based on mutation pressure alone. GC-biased gene conversion is one possible neutral mechanism for increasing G+C-content (Duret and Galtier 2009), but it is not clear whether it is sufficient to counter the pervasive erosion of G+C by spontaneous mutations (Weller et al. 2014; Keith et al. 2016).
Copy-Number Changes (Large Duplications and Deletions)
The importance of gene duplications in the evolution of life has long been recognized (Ohno 1970). More recently, a technological revolution in genomics has revealed both a rich history of past gene duplications written in sequenced genomes (reviewed by Katju 2012) and an abundance of gene copy-number variation (CNV) caused by duplications and deletions in natural populations (reviewed by Katju and Bergthorsson 2013; Bergthorsson and Katju 2016). The frequency of duplications in populations is determined by the rate of spontaneous duplications and their preservation or elimination by natural selection and genetic drift. By comparing the rate and spectrum of spontaneous gene duplication with the rate of fixation of duplications in genomes and their distribution in natural populations, we gain valuable insight into the relative roles that the duplication rate, selection, and genetic drift play in determining the fate of duplications in natural populations and as a source of evolutionary novelties.
Using a combination of oligonucleotide array comparative genomic hybridization (oaCGH) and pulsed-field gel electrophoresis, Lynch et al. (2008) analyzed eight S. cerevisiae MA lines that were passaged through 200 single-cell bottlenecks and ∼4,800 generations. The spontaneous duplication and deletion rates were measured to be 3.4 × 10−6 and 2.1 × 10−6/gene/generation, respectively. An earlier study involving the analysis of ten MA lines of Caenorhabditis elegans by oaCGH provided the first empirical, genome-wide estimates of the spontaneous rate of duplication rate in a multicellular eukaryote (Lipinski et al. 2011). The duplication rate was found to be 3.4 × 10−7 per gene/generation when all gene duplications were included (complete and partial genes). When only completely duplicated genes were considered, the duplication rate was 1.25 × 10−7/gene/generation. Paired-end sequencing of D. melanogaster MA lines found that the duplication rate was similar to that in Caenorhabditis elegans: 3.75 × 10−7 duplications/gene/generation for partial or complete duplications and 1.25 × 10−7/gene/generation if only complete duplications were considered (Schrider et al. 2013). The spontaneous gene duplication rate for single-copy genes in Daphnia pulex is 3.27 × 10−5 (Keith et al. 2016), an order of magnitude higher than the oaCGH-based estimate in Caenorhabditis elegans (Lipinski et al. 2011) or D. melanogaster (Schrider et al. 2013). Recently, Konrad et al. (2018) used Illumina sequencing and a modified oaCGH approach on a different set of Caenorhabditis elegans MA lines to generate a μduplication estimate of 2.9 × 10−5 which is very similar to that for Daphnia (Keith et al. 2016) and almost 2 orders of magnitude greater than the preceding estimate for Caenorhabditis elegans (Lipinski et al. 2011). MA experiments in Salmonella estimated the deletion rate to be 5 × 10−7 (Nilsson et al. 2005). The same MA experiments that measured the gene duplication rates in eukaryotes also measured the deletion rates. The gene deletion rates for S. cerevisiae (Lynch et al. 2008), Caenorhabditis elegans (Konrad et al. 2018), D. melanogaster (Schrider et al. 2013) and Daphnia pulex (Keith et al. 2016) were estimated to be 2.1 × 10−6, 0.5 × 10−5, 9.37 × 10−7 and 3.71 × 10−5/gene/generation, respectively. Empirical, genome-wide estimates of the spontaneous duplication and deletion rate from MA experiments are presented in table 3.
Table 3.
Rates of Copy-Number Change (Gene Duplications and Deletions) per Gene per Generation Estimated from Empirical Genome-Wide Analyses of Mutation Accumulation Experiments Using High-Throughput Approaches
| Species | μduplication | μdeletion | μcopy-number | Reference |
|---|---|---|---|---|
| Prokaryotes | ||||
| Salmonella typhimurium LT2 | — | 5.0 × 10−7 | — | Nilsson et al. (2005) |
| Unicellular eukaryotes | ||||
| Saccharomyces cerevisiae | 3.4 × 10−6 | 2.1 × 10−6 | 5.5 × 10−6 | Lynch et al. (2008) |
| Multicellular eukaryotes | ||||
| Caenorhabditis elegans | 3.4 × 10−7 | 2.2 × 10−7 | 5.6 × 10−7 | Lipinski et al. (2011) |
| Caenorhabditis elegans | 2.9 × 10−5 | 0.5 × 10−5 | 3.4 × 10−5 | Konrad et al. (2018) |
| Daphnia pulexa | 2.3 × 10−5 | 2.9 × 10−5 | 5.2 × 10−5 | Keith et al. (2016) |
| Drosophila melanogaster | 3.7 × 10−7 | 9.4 × 10−7 | 1.3 × 10−6 | Schrider et al. (2013) |
Note.—The spontaneous rate of gene duplication and deletion are denoted by μduplication and μdeletion, respectively. μcopy-number denotes the combined rate of copy-number change by either gene duplication or deletion.
Averaged across asexual and cyclical lines for single-copy genes only.
Comparisons of duplication and deletion rates from MA experiments to the patterns of gene acquisition and loss in 1) sequenced genomes, and 2) natural populations have been used to make inferences about selection operating on CNVs. The probability that a gene is duplicated or deleted in any one generation is an order of magnitude greater than the base substitution rate. This observation regarding the high rate of spontaneous gene duplications and deletions speaks to their importance in introducing genetic variation, and this is corroborated by multiple studies showing abundant CNV in natural populations. Second, the rates of spontaneous gene duplication are orders of magnitude higher than the rates of gene duplications estimated from the age distribution of gene duplicates in sequenced genomes. If natural selection eradicates some fraction of gene duplicates in their infancy before they accrue any nucleotide substitutions, the age distribution of extant gene duplicates within a genome will result in an underestimate of the spontaneous duplication rate. The observation that empirical measures of the gene duplication and deletion rates from MA experiments are orders of magnitude higher than those from bioinformatic analysis of sequenced genomes is best explained by the loss of the vast majority of young CNVs by natural selection in the latter (Lipinski et al. 2011; Schrider et al. 2013).
The duplication/deletion rates in MA lines have been compared with natural polymorphism in the same species to make inferences about natural selection on CNVs. For Daphnia pulex, the observed number of base pairs in CNVs is close to 19-fold lower than expected from the rate and size distribution of copy-number changes in MA experiments (Keith et al. 2016). The results suggest that most large CNVs are deleterious and purged from Daphnia pulex populations by purifying selection. Furthermore, comparisons of the duplication/deletion rates in MA lines with CNVs in natural populations of D. melanogaster concluded that 99% of all new CNVs were deleterious, and moreover, that CNVs were 10-fold more likely to be removed by natural selection than amino acid replacement substitutions (Schrider et al. 2013).
Rate and Spectrum of Mutations in Eukaryotic Mitochondrial Genomes
Introduction
Since the ancient evolutionary event wherein an α-proteobacterium took up residence in a eukaryotic host cell and evolved to become the modern-day energy workhorse of eukaryotic cells now known as mitochondria, most of its independent function and genetic material has been lost or transferred to the host nucleus. Modern mitochondria retain a fraction of their ancestral genome to manufacture the components required for ATP production. The biology and transmission genetics of mtDNA is an unorthodox one, with additional and striking taxa-specific differences. The mutation rate of animal mitochondria exceeds that of their host’s nuclear genome by an order of magnitude or more (Brown et al. 1982), and mitochondrial mutations are increasingly being associated with a variety of human diseases (Wallace and Chalkia 2013; Wallace 2015). The rapid rate of molecular evolution also renders metazoan mitochondria an amenable tool in evolutionary studies, as a marker for determining relationships between closely related populations or species and in studies of contemporary geographic distributions of organisms (Avise 2000). In contrast, plant mitochondrial genomes possess extremely low rates of sequence evolution relative to the nuclear genome (Wolfe et al. 1987) and have been gainfully employed in investigating deeper phylogenetic relationships (Bowe et al. 2000). A similarly wide diversity in pattern is displayed in the inheritance of mtDNA across taxa (reviewed by White et al. 2008). In the majority of instances, mtDNA is inherited uniparentally through the maternal germline. However, even in species with a predominantly maternal transmission pattern, biparental inheritance of mtDNA can occur at low frequencies via paternal leakage (Neale et al. 1989; Kondo et al. 1990; Gyllensten et al. 1991; Kvist et al. 2003; Ballard and Whitlock 2004; Barr et al. 2005; McCauley et al. 2005; White et al. 2008). Doubly uniparental inheritance of mtDNA, wherein female offspring inherit maternal mtDNA and male offspring inherit the mtDNA of both parents, is observed in several bivalve families (Zouros et al. 1994; Skibinski et al. 1994; reviewed by Breton et al. 2007). At the other end of the spectrum, a few plant species including cucumbers and some conifers (Havey 1997; Neale et al. 1989) are reported to have a predominantly paternal mode of mtDNA transmission.
An early and long-held assumption in the study of mitochondria was that individuals only possessed one mtDNA haplotype, often referred to as homoplasmy (Birky 2001). A state of homoplasmy necessitates that mtDNA molecules are essentially nonrecombining. This presumed lack of recombination in mtDNA came with the implicit assumption that existing variation was generated by mutational changes alone, thereby establishing it as the molecular marker of choice for delineating evolutionary change in populations and species and dating evolutionary events. The last two decades have demonstrated that the population structure of mitochondria is far more complex and is best described as a nested hierarchy of populations, with multiple mtDNA molecules per mitochondria, multiple mitochondria per oocyte, multiple oocytes per females, and so forth (Rand 2001). Newly arising mtDNA mutations create a heterogeneous population of mutant and wild-type mtDNA molecules, generating a state known as heteroplasmy. Heteroplasmy can be regarded as an intermediate polymorphic stage following the origin of new mitochondrial alleles via mutation and preceding their ultimate fixation or loss within the nested population hierarchy of mitochondria. The frequency of these heteroplasmic alleles can shift during meiotic and mitotic events, due to both random genetic drift as well as natural selection (Rand 2001; Wallace 2015). A state of heteroplasmy can also enable the formation of novel recombinant mtDNA molecules. Although the extent to which this occurs is still under vigorous debate (Kraytsberg et al. 2004; reviewed by Barr et al. 2005; Hagström et al. 2014), there is clear evidence for recombination in fungal (Taylor 1986; MacAlpine et al. 1998; Birky 2001), plant (Lonsdale et al. 1988; Remacle et al. 1995; Städler and Delph 2002; Bergthorsson et al. 2003), and animal (Passamonti et al. 2003; Ladoukakis and Eyre-Walker 2004; reviewed by Piganeau et al. 2004) mitochondria. The existence of even rare recombination in mitochondrial genomes can impede the accumulation of deleterious mutations (Charlesworth et al. 1993; Neiman and Taylor 2009).
Both traditional Sanger and massively parallel sequencing technologies have facilitated direct molecular analyses of MA lines to generate genome-wide estimates of the rate and spectrum of spontaneous mitochondrial mutations in eight unicellular/multicellular eukaryote species (table 4). Of these nine studies, five have utilized next-generation sequencing technology (Haag-Liautard et al. 2008; Lynch et al. 2008; Saxer et al. 2012; Sung, Tucker, et al. 2012; Konrad et al. 2017). Caenorhabditiselegans mtDNA evolution has been studied independently in two different sets of MA lines (Denver et al. 2000; Konrad et al. 2017) and with different sequencing platforms (Sanger vs. next-generation Illumina sequencing), thereby providing some insight into the relative performance of each platform. While metazoan mtDNA genomes have been better represented among the multicellular eukaryotes, to date we have no insight into genome-wide rates and spectrum of mtDNA in plants, despite MA experiments in A.thaliana (Schultz et al. 1999; Shaw et al. 2000) and in the genus Amsinckia (Schoen 2005). The mutational dynamics of plant mtDNA genomes are expected to exhibit a sharp contrast to their metazoan counterparts given that plant mtDNA has an extremely low mutation rate (Wolfe et al. 1987). However, analysis of the mutational process in the mtDNA genomes of land plants may not be biologically feasible for the reasons of extremely low mutation rates, lengthier generation times, large genome size, and the repetitive base content of the genomes. Mitochondrial genomes of algal MA lines (e.g., Krasovec et al. 2016) may offer a more feasible option given their smaller genome size, and amenability to MA experiments.
Table 4.
Estimates of Spontaneous Mitochondrial Mutation Rates and Spectra Derived from Mutation Accumulation Experiments in Eight Eukaryotic Species Using Traditional (Sanger) or High-Throughput Sequencing Approaches
| Species | μtotal | μbs | μindel | Ratio of Indel:Single-base Substitutions | A/T Content of mtDNA Genome (%) | Base Changes Increasing A/T Content (%) | mtDNA Ne | Reference |
|---|---|---|---|---|---|---|---|---|
| Unicellular eukaryotes | ||||||||
| Dictyostelium discoideuma | 0.7 × 10−8 | — | — | — | — | — | — | Saxer et al. (2012) |
| Paramecium tetraureliaa | — | 6.96 × 10−8 | — | — | — | — | — | Sung, Tucker, et al. (2012) |
| Saccharomyces cerevisiaea | 2.0 × 10−8 | 1.22 × 10−8 | 0.75 × 10−8 | 0.61 | 84 | 33 | — | Lynch et al. (2008) |
| Multicellular eukaryotes | ||||||||
| Caenorhabditis briggsae | — | 7.20 × 10−8 | — | — | 76 | 87 | — | Howe et al. (2010) |
| Caenorhabditis elegans | 16.0 × 10−8 | 9.70 × 10−8 | 6.30 × 10−8 | 0.65 | 76 | 29 | — | Denver et al. (2000) |
| Caenorhabditis elegansa | 10.5 × 10−8 | 4.32 × 10−8 | 6.14 × 10−8 | 1.42 | 76 | 89 | 62–100 | Konrad et al. (2017) |
| Drosophila melanogastera | 7.8 × 10−8 | 6.20 × 10−8 | 1.60 × 10−8 | 0.26 | 82 | 86 | 13–42 | Haag-Liautard et al. (2008) |
| Daphnia pulex | 15.5 × 10−8 | 3.15 × 10−8 | 12.35 × 10−8 | 3.92 | 62 | 60 | 5–10 | Xu et al. (2012) |
| Pristionchus pacificus | 7.6 × 10−8 | 4.50 × 10−8 | 3.20 × 10−8 | 0.71 | 76 | 57 | — | Molnar et al. (2011) |
High-throughput or next-generation sequencing platform.
Overall Rate of Spontaneous Mutation in mtDNA Genomes
The overall, genome-wide rate of spontaneous mtDNA mutations (/site/generation), μtotal, is currently available for six taxonomically diverse species (two unicellular and four multicellular eukaryotes) (table 4). The empirical estimates for μtotal include both base substitutions and indel events and range ∼23-fold, from 7 × 10−9 to 1.6 × 10−7/site/generation (table 4). If only multicellular eukaryotes are considered, the range in mutation rates is considerably narrower, varying only ∼2-fold from 7.6 × 10−8 to 16 × 10−8/site/generation. Likewise, there is a ∼3-fold difference in the overall mtDNA mutation rate for the two unicellular eukaryotes, S. cerevisiae and Dictyosteliumdiscoideum, although it should be noted that the base substitution rate in Paramecium tetraurelia is significantly higher than these overall mutation rates and comparable to those generated for metazoan species. Hence, the sample size is extremely limited and the rate estimates too variable for unicellular eukaryotes to enable a broad generalization of their rates of mtDNA evolution with reference to each other as well as to their multicellular counterparts. In general, overall mtDNA mutation rates are consistently higher in metazoans but the mechanistic reason(s) for this difference is obscure.
Spontaneous Rate of Base Substitutions in mtDNA Genomes
Direct empirical estimates of the spontaneous mtDNA base substitution rate, μbs, from Sanger or high-throughput sequencing of MA lines are currently available for seven species (two unicellular and five multicellular eukaryotes, respectively). Estimates of μbs for the unicellular eukaryotes S. cerevisiae and the protist Paramecium tetraurelia differ ∼6× (1.22 × 10−8 versus 6.96 × 10−8 base substitutions/nucleotide site/generation, respectively) (table 4). For the five multicellular eukaryotes, the spontaneous mtDNA base substitution rate is surprisingly consistent, varying ∼3× with a range of 3.15 × 10−8 to 9.7 × 10−8 base substitutions/nucleotide site/generation with the rate in Daphnia pulex representing the lower end of the spectrum (table 4). The paucity of estimates for unicellular eukaryotic species precludes a meaningful comparison and potential insights into how they may differ from multicellular species.
Spontaneous Rate of Indel Events in mtDNA Genomes
There exists a slightly greater disparity in the spontaneous mutation rate for indel events, μindel (table 4) relative to μbs. μindel estimates from five eukaryotes (one unicellular, four multicellular) range ∼16×, from 0.75 × 10−7 to 1.23 × 10−7 changes/site/generation, with S. cerevisiae and Daphnia pulex displaying the lowest and highest rate, respectively. μindel estimates exceed μbs for Daphnia pulex (Xu et al. 2012) and Caenorhabditis elegans (Konrad et al. 2017), but the converse is observed for D. melanogaster, S. cerevisiae, D. melanogaster, and Pristionchus pacificus (Haag-Liautard et al. 2008; Lynch et al. 2008; Molnar et al. 2011). This is reflected in the ratio of indel to single-base substitutions which ranges from 0.61 to 3.92 (table 4). Hence, no discernible pattern can be ascribed to the frequency of indel events among taxonomic groups given the extremely limited sample size in the case of unicellular eukaryotes and the fact that metazoan species have indel rates that either exceed or are lesser than their base substitution rates. However, in general, species-specific μindel estimates appear to be quite similar to their μbs counterparts, with the exception of Daphnia pulex.
Mutational Spectrum of Base Substitutions in mtDNA Genomes
In general, metazoan mitochondrial genomes tend to be A+T-biased (Castellana et al. 2011, and references therein), although there are some notable exceptions. What factors dictate the extant base composition of a mitochondrial genome? The simplest model posits that the prevalent base composition is due to mutational input. In terms of the A+T-rich mtDNA genomes, the observed skew in base composition is therefore owing to a strong, biased mutation pressure toward A/T base substitutions. An alternative competing hypothesis posits that the observed base composition in mtDNA genomes reflects the influence of countering selective forces to maintain an optimum equilibrium. Hence, in the case of the A+T-rich mtDNA genomes, it is possible that spontaneous G/C base substitutions arise more frequently but are subsequently eradicated via purifying selection to enhance an A+T skew in base composition. An analysis of the spectrum of new spontaneous base substitutions in the mtDNA genomes of long-term MA lines can help distinguish between these two competing hypotheses. In this kind of analyses, third codon positions and intergenic regions are less likely to be under selection and are hence preferable to first and second codon positions in detecting the cumulative effects of prevalent mutation biases in the genome. Genome-wide analyses of spontaneous mitochondrial mutations in MA lines first conducted in Caenorhabditis elegans using a direct sequencing approach (Denver et al. 2000) reported a strongly biased mutation pressure toward G/C changes. Given that the Caenorhabditis elegans mtDNA genome has a 76% A+T-content, Denver et al. (2000) therefore argued for a dominant role of selection in shaping the base composition of the mtDNA genome. Similar to the pattern observed in Caenorhabditis elegans by Denver et al. (2000), Lynch et al. (2008) concluded a G/C mutation bias in S. cerevisiae. The conclusions from subsequent mtDNA analysis of MA lines of other multicellular eukaryotic species have been at odds with the pattern first observed in Caenorhabditis elegans (Denver et al. 2000) and S. cerevisiae (Lynch et al. 2008). A strong G/C → A/T mutation bias has been reported in both D. melanogaster (Haag-Liautard et al. 2008) and the nematode Pristionchus pacificus (Molnar et al. 2011). Likewise, a strong bias toward A/T mtDNA mutations was also reported in a study that employed Sanger sequencing of Caenorhabditis briggsae MA lines (Howe et al. 2010). These contrasting patterns of mtDNA base substitution bias in otherwise A+T-rich mtDNA genomes were referred to as a “muddle of mutation across taxa” (Montooth and Rand 2008). A recent study investigating the spontaneous mtDNA mutation process via Illumina paired-end sequencing in an independent set of long-term Caenorhabditis elegans MA lines provides evidence for an extremely strong G/C → A/T mutation bias with 89% of new spontaneous point mutations resulting in an increased A+T-content (Konrad et al. 2017). This finding contradicts those of Denver et al. (2000) and underscores the contribution of a strongly biased A/T mutation pressure leading to the skewed base composition observed in mtDNA genomes of all multicellular eukaryotes studied to date via MA experiments (table 4). A general conclusion regarding the role of mutation biases versus selection in dictating base composition of the mtDNA genomes of unicellular eukaryotes is currently lacking. Further in-depth analyses of the mtDNA mutational spectrum of additional unicellular eukaryotic species such as Dictyostelium discoideum and Paramecium tetraurelia are much needed to offer a comparative genomic perspective regards any notable differences among diverse unicellular eukaryotes themselves and in relation to their multicellular counterparts.
The Emerging Pervasiveness of Heteroplasmy
The advent of next-generation sequencing technology has significantly transformed our understanding and ubiquity of mitochondrial heteroplasmy by enabling the detection of extremely rare mtDNA variants that typically remain undetected via other approaches. Heteroplasmies represent an intermediate polymorphic step in the trajectory of mtDNA variants, from their origin as a single copy to ultimate fixation in an individual or cell type. The identification and extent of heteroplasmy has important implications for the evolution of mitochondrial genomes, including the effective population size of mtDNA, the influence of genetic drift versus selection in dictating their future evolutionary dynamics, and the opportunities they may create for recombination events in a supposedly linked genome thought to be vulnerable to Muller’s Ratchet (Li et al. 2010).
Studies using a Sanger sequencing approach in Caenorhabditis briggsae, Pristionchus pacificus, and Daphnia pulex were able to detect mtDNA variants ranging in frequencies from 0.22 to fixation, although there appears to be a significant difference among the studies as well with respect to the range of detectable frequencies of mtDNA variants (table 5). In general, the majority of mtDNA mutations (75–100%) detected via Sanger sequencing tend to exist in high frequencies of >0.5 within an individual. High-throughput sequencing approaches far exceed the capacity of Sanger technology in the detection of mtDNA heteroplasmies given that the vast majority of mutations detected in D. melanogaster (Haag-Liautard et al. 2008) and Caenorhabditis elegans (Konrad et al. 2017) MA lines occur in a heteroplasmic condition. Pyrosequencing, as was conducted in the fly MA lines, offered greater sensitivity relative to the Sanger approach in that only 50% of the mtDNA variants detected occurred in >0.5 frequency (Haag-Liautard et al. 2008). In contrast, a recent study in Caenorhabditis elegans employing Illumina, paired-end sequencing technology found that only 30% of detected mtDNA mutations occurred in frequencies >0.5 (Konrad et al. 2017). Next-generation sequencing enabled the accurate detection of extremely rare variants in the Caenorhabditis elegans mtDNA genome with frequencies as low as 0.01. Indeed, Konrad et al.’s (2017)Caenorhabditis elegans study found that the median frequency of the detected mtDNA variants in MA lines was 0.18 which is considerably lower than that found in the remainder four multicellular eukaryotes (0.53–1.0; table 5), with only 2% of all mtDNA mutations having reached fixation within 35 MA lines after 300–400 MA generations. Together, these findings are a significant departure from the initial notion that individuals are generally homoplasmic (Birky 2001), that is, they only carry one mtDNA haplotype. In addition, Konrad et al. (2017) also assessed mtDNA variants in 38 Caenorhabditis elegans natural isolates and observed a bimodal distribution with variants present in either high or low frequency, and disproportionately fewer variants in intermediate frequencies. Heteroplasmic variants in natural isolates tend to be present in low frequencies in contrast to a more uniform distribution of heteroplasmic variants under genetic drift conditions in the N = 1 MA lines, suggesting a role for natural selection in the suppression of intracellular frequencies of potentially deleterious variants in the wild (Konrad et al. 2017).
Table 5.
Distribution and Frequencies of Heteroplasmic mtDNA Mutations Identified in Mutation Accumulation Lines of Five Eukaryotic Species Using Differing Sequencing Technologies
| Species | Sequencing Technology | Frequency Range of mtDNA Variants | Median Frequency | % Fixed Mutations (Frequency = 1) | % Mutations with >0.5 Frequency | Reference |
|---|---|---|---|---|---|---|
| Drosophila melanogaster | Pyrosequencing | 0.06–1.0 | 0.53 | 20 | 50 | Haag-Liautard et al. (2008) |
| Caenorhabditis briggsae | Sanger | 0.51–1.0 | 0.93 | 47 | 100 | Howe et al. (2010) |
| Pristionchus pacificus | Sanger | 0.30–1.0 | 1.00 | 75 | 75 | Molnar et al. (2011) |
| Daphnia pulex | Sanger | 0.22–1.0 | 1.00 | 61 | 78 | Xu et al. (2012) |
| Caenorhabditis elegansa | Illumina, paired-end | 0.01–1.0 | 0.18 | 2 | 30 | Konrad et al. (2017) |
mtDNA mutations across all MA lines comprising three differing population size treatments.
Mitochondrial Effective Population Size, Ne[mtDNA]
Mitochondria are subjected to selection and genetic drift not only in a population of individuals but also in populations of mitochondria within the cells of individuals (Rand 2001). A new mtDNA variant arising via mutation in the germline is initially present as one unique haplotype in the extant population of mitochondrial genomes within a cell of an individual. The presence of this new mtDNA haplotype engenders a heteroplasmic state wherein the cytoplasm now comprises an aggregate of different mitochondrial haplotypes. The time (number of generations) it takes to realize the evolutionary fate of this new mtDNA mutant, eventual loss or fixation within the cytoplasm, will be determined by the forces of selection and/or genetic drift as well as the effective population size of extant mtDNA molecules in the cell. This mitochondrial effective population size, Ne[mtDNA], is defined as the “effective number of maternal mitochondria transmitted to progeny” (Haag-Liautard et al. 2008). If the new mtDNA variant is neutral with respect to fitness, then under the neutral theory of molecular evolution (Kimura and Ohta 1969), its persistence as a neutral polymorphism is critically dependent on the effective population size of mtDNA molecules. Because the mitochondrial population size within a cell can vary significantly across different developmental stages and tissue types, and the observation that mtDNA haplotype frequencies can dramatically shift within as little as one generation from mother to offspring, there is widespread acceptance for the existence of a mitochondrial bottleneck in the host germ line (Bergstrom and Pritchard 1998; White et al. 2008). While bottlenecks in population genetics are typically equated with loss of genetic diversity and enhanced stochasticity due to the influence of genetic drift, it has been cogently argued that mitochondrial bottlenecks, while accelerating the rate of genetic load within some lineages, can actually serve to facilitate selection among lineages and serve as a brake for mutational degradation via Muller’s Ratchet (Bergstrom and Pritchard 1998). The frequency distribution of new mtDNA variants detected in MA studies can serve as a powerful means to quantify the Ne[mtDNA] if heteroplasmies are evident, as was done by Haag-Liautard et al. (2008) using a maximum-likelihood approach in their study of mtDNA evolution in D. melanogaster MA lines. This approach has since been applied to generate estimates of Ne[mtDNA] from MA studies of Daphnia pulex (Xu et al. 2012) and Caenorhabditis elegans (Konrad et al. 2017) (table 5). Ne[mtDNA] is estimated to be 5–10 copies for Daphnia pulex (Xu et al. 2012), 13–42 for D. melanogaster (Haag-Liautard et al. 2008), and 62–100 for Caenorhabditis elegans (Konrad et al. 2017) (table 5). The 10-fold difference in the range of these estimates most likely stems from the use of different sequencing technologies utilized by these studies given their differing degrees of sensitivity in the detection of heteroplasmies, which in turn directly influences the estimation of Ne[mtDNA]. It is likely that all of these estimates of Ne[mtDNA] are in fact conservative, given that extremely low-frequency variants were likely excluded in the data set of identifiable mtDNA mutations, either because of a detection bias or confounded with false-positive calls.
Degree of Congruence between Genome-Wide Mutation Rates as Estimated from Phenotypic Assays versus High-Throughput Data
MA experiments were originally designed to estimate the rate of deleterious mutations that affected a particular phenotype. Initially, the phenotype of the greatest interest was some proxy estimate of fitness, such as the number of viable offspring, but in principle it can be used to estimate the mutation rate that impacts any other physical or behavioral trait. Naturally, the molecular mutation rates are expected to be much greater than the phenotypic mutation rates as only a small fraction of mutations will significantly impact any given phenotype. Furthermore, there may exist a cryptic class of mutations with small fitness effects which are undetectable in phenotypic assays under benign laboratory conditions, thereby leading to an underestimation of phenotypically based genomic mutation rates (Davies et al. 1999; Halligan and Keightley 2009). Figure 2 compares indirect phenotypic estimates of U with direct molecular estimates from MA-WGS studies. Direct molecular estimates of U can exceed phenotypic estimates of U by up to 5,000-fold. The average discrepancy between direct molecular and phenotypic estimates of U is ∼125-fold. Two striking exceptions to this rule are phenotypic-based mutation rates in two species of protists, T. thermophila and Dictyostelium discoideum. These species have extraordinarily low nuclear mutation rates, at least based on single nucleotide polymorphisms whereas their phenotypic rates are within the normal range found for other taxa. The reasons for this are not clear. However, it is possible that other classes of mutations such as mtDNA variants, small indels, structural variants, or copy-number changes can account for some of this discrepancy, as well as transgenerational epigenetic changes. Because some copy-number changes can be quite large and span multiple loci, they have the potential to change the expression of many genes simultaneously and thereby exert disproportionately large effects on a phenotype. Additionally, transgenerational epigenetic changes may be of importance in some taxa. Another notable pattern in Figure 2 is that there can be considerable intraspecific variation in the phenotypic estimates of U depending on the fitness trait assayed. Drosophilamelanogaster and A. thaliana represent the most extreme examples within this data set wherein the range in phenotypic estimates of U exceeds 300-fold.
Fig. 2.

—Phenotypic estimates of the genome-wide mutation rate, U, as a function of the direct molecular estimates of the genome-wide nucleotide mutation rate, Ubs, generated from whole-genome sequence data. U is represented as the number of mutations per genome per generation. For direct molecular estimates of the U from MA-WGS studies, the base substitution rate was utilized as it was the most readily available across different MA studies for different species. Multiple data points for a species represent phenotypic estimates of U for different fitness traits assayed. For species with multiple molecular estimates of U from WGS data, the average rate was used. The dashed red line represents a hypothetical one-to-one relationship between phenotypic and molecular estimates of U. With the exception of the two protist species Dictyostelium discoideum and T. thermophila, direct molecular estimates of U can be up to several orders of magnitude higher than their counterparts from Bateman–Mukai or maximum likelihood analyses of phenotypic data. Prokaryotic species are denoted by circles. Unicellular and multicellular eukaryotes are denoted by triangles and squares, respectively. All plotted data are presented in supplementary table S1, Supplementary Material online.
Sources of Variation in Mutation Rates
A major goal of investigations into mutation rate variation is to identify fundamental principles that govern the evolution of the mutation rate across all domains of life. Is there an optimal mutation rate that balances the need for removing deleterious mutations with a need for introducing new beneficial mutations? Do sex and recombination influence mutation rate evolution? Do larger genomes demand greater fidelity of DNA replication?
Drake’s Rule and the Drift-Barrier Hypothesis
In a classic analysis of mutation rates across several microbial genomes, John Drake described an inverse linear relationship between genome size and mutation rate in DNA-based microbes (Drake 1991). Remarkably, the number of mutations per genome per generation appeared to be constant (0.003) over several orders of magnitude difference in both genome size and the per-nucleotide mutation rate. The relationship between genome size and mutation rate was taken to suggest that selection operates on minimizing the deleterious mutation rate per genome, and that the mutation rate is the product of a tradeoff between reducing the mutation rate by more accurate replication and repair, and the physiological cost of higher replication fidelity. This original study by Drake (1991) comprised a small sample size with only four species of bacteriophage and three cellular organisms, and was based on mutations in reporter loci.
MA-WGS studies in the genomic era in diverse species have demonstrated that spontaneous base substitution rates can vary over 4 orders of magnitude, from 10−12 to 10−8 per site per generation (table 1). A reevaluation of the relationship between genome size and the genome-wide mutation rate from MA experiments shows that the inverse relationship may still hold, but only among microbes (prokaryotes and unicellular eukaryotes) (fig. 3A). In striking contrast, the mutation rate scales positively with genome size in the case of multicellular eukaryotes (fig. 3A; Lynch 2010a). In prokaryotes, which typically possess sparse intergenic DNA, few pseudogenes and no spliceosomal introns, the fraction of the genome that is under selection may be adequately approximated by the size of the genome. In contrast, for multicellular eukaryotes with a substantial fraction of disposable genomic DNA, the coding part of the genome has been used as a proxy for the fraction of the genome that is presumably under selection, and is therefore a target for deleterious mutations. Employing only the coding portion of multicellular eukaryotic genomes as an independent variable significantly improves the fit with mutation rate (Sung, Ackerman, et al. 2012). However, the mutation rates of microbes and multicellular eukaryotes correlate with effective population size, Ne, in a broadly similar manner, eliminating the need to find different causal explanations for the evolution of mutation rates for these groups (fig. 3B and C). The relationship between Ne and the mutation rate is predicted by the drift-barrier hypothesis, which states that the limits to the evolution of improved replication fidelity are determined by a combination of diminishing benefits of further improvement in fidelity and genetic drift in finite populations (Lynch 2010a; Sung, Ackerman, et al. 2012). According to the drift-barrier hypothesis, the main obstacle to reducing the mutation rate in the wild does not arise from trade-offs with the physiological cost of increased fidelity, although such trade-offs may exist. Rather, the obstacle to reducing the mutation rate results in part from the limits, set by Ne, to the efficacy of natural selection in removing deleterious mutations that increase the mutation rate. Additionally, genetic drift in finite populations limits the efficacy of selection in fixing much rarer beneficial mutations that reduce the mutation rate. Consequently, selection in very large populations can attain (and maintain) greater improvement in replication fidelity relative to smaller effective populations (Lynch 2010b; Lynch et al. 2016). The drift-barrier hypothesis does not deny the importance of the size of the mutational target, the part of the genome that is under selection, as an important determinant in the evolution of mutation rate. However, the primary contributing factor is still Ne which determines the contribution of genetic drift to the evolution of mutation rate. As such, it is currently the best explanation for the large-scale patterns in mutation rate variation across genomes across all domains of life, including viruses.
Fig. 3.
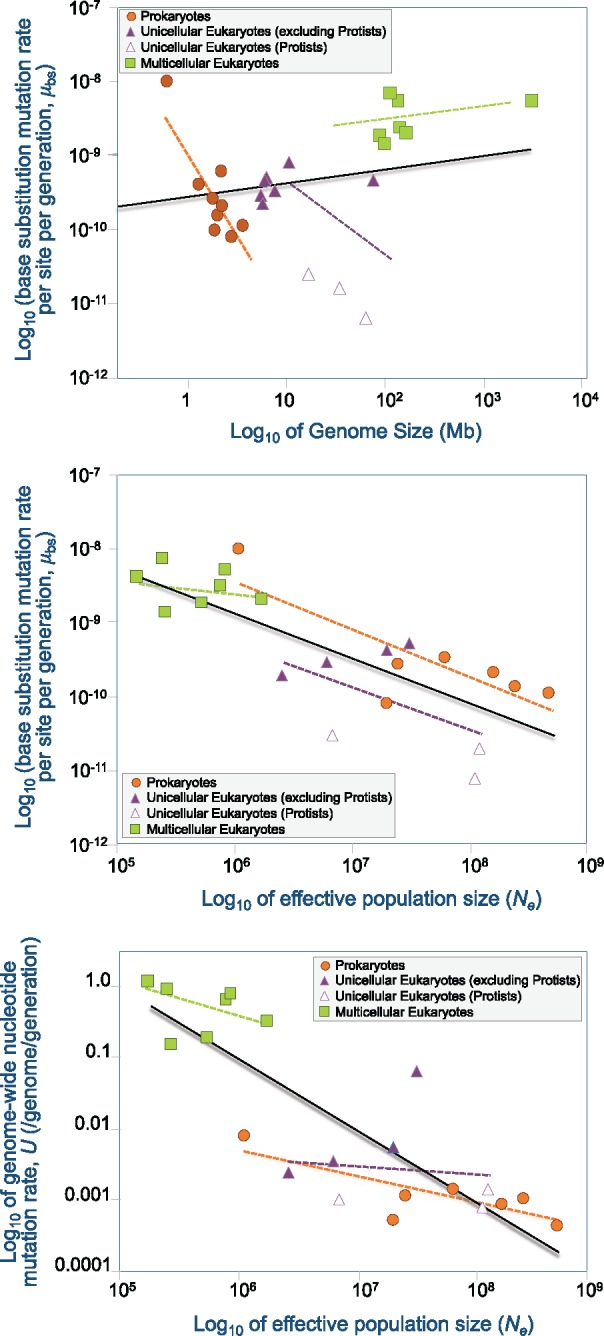
—Relationship between spontaneous mutation rates from MA-WGS studies, genome size and effective population size (Ne). Prokaryote, unicellular and multicellular eukaryotes species are represented by orange circles, purple triangles, and green squares, respectively. Three protists (the ciliates Paramecium tetraurelia and T. thermophila, and the social amoeba Dictyostelium discoideum) are represented in open triangles. The solid black lines are representative of the entire data set comprising prokaryote, unicellular eukaryotes, and multicellular eukaryotes. Dashed orange, purple, and green lines are representative of prokaryotes, unicellular eukaryotes, and multicellular eukaryotes, respectively. All plotted data are presented in supplementary table S2, Supplementary Material online. (A) Base substitution mutation rate per nucleotide site per generation, μbs, as a function of genome size. The mutation rate is inversely correlated with genome size in prokaryotes (r = −0.90, P = 0.009, n = 9). (B) Base substitution mutation rate per nucleotide site per generation, μbs, as a function of effective population size, Ne. μbs is inversely correlated with Ne across all taxa (r = −0.78, P = 3E-05, n = 21) and within prokaryotes (r = −0.81, P = 0.028, n = 7). (C) Genome-wide mutation rate per genome per generation, U, as a function of effective population size, Ne. U is inversely correlated with Ne across all taxa (r = −0.83, P < 10−5, n = 21) and within prokaryotes (r = −0.80, P = 0.031, n = 7).
Base Composition Bias
There exists immense variation in the base composition of genomes. Among the prokaryotes, for instance, G+C-content can vary from 16.5% in Carsonella ruddii (Nakabachi et al. 2006) to 75% in Anaeromyxobacter dehalogenans (Sanford et al. 2002). Base composition within prokaryotic genomes can also vary locally. For example, regions or genes that were recently acquired by horizontal gene transfer can differ significantly from the average base composition of the genome (Lawrence and Ochman 1997). Furthermore, the two strands of the bacterial chromosome can have different compositional biases that are associated with leading and lagging strand replication (Lobry 1996).
The diversity in G+C-content, both within and between genomes, has engendered both neutral- (mutation bias or GC-biased gene conversion) and selection-based hypotheses for their origin. Perhaps, the simplest explanation for the immense variation in G+C-content between and within genomes is that they reflect prevailing mutation biases, or mutation pressure. Freese (1962) and Sueoka (1962) proposed that the G+C-content of genomes represents the equilibrium state of the rate of mutations from G/C → A/T and A/T → G/C. In this view, the amino acid composition of proteins imposes constraints on the otherwise neutral evolution of G+C-content, and hence only the G+C-content at silent sites is expected to reach equilibrium from mutation pressure alone (Sueoka 1988). Deviations from the expected equilibrium have in turn been viewed as evidence of selection on G+C-content, or evidence for other processes that influence G+C-content, such as GC-biased gene conversion.
Base substitution patterns in genomes have been analyzed by mutation experiments employing reporter loci, polymorphisms in natural populations, and MA experiments. Reporter loci have the disadvantage of being confined to a single or few locations in the genome, as well as the possibility that the phenotypes for different mutations may not all take the same time to develop, thereby potentially biasing the results. Polymorphisms in natural populations may have been subject to natural selection, and MA experiments are typically performed in a single or few environments and may not reflect the variation in mutation patterns found in the wild. The A/T mutation bias in prokaryotes ranges from ∼0.6 to 16 in MA experiments. The majority of MA experiments with wild-type bacteria have found a mutation bias toward higher A/T content. MA experiments with E. coli found that in wild-type strains, G/C → A/T mutations occur at rates 1.24 − 2× greater than A/T → G/C mutations. All else being equal, an A/T bias predicts that silent sites should be A/T-rich. Instead, silent sites in E. coli tend to be slightly G/C-rich. Some species with relatively G/C-rich genomes, such as B.cenocepacia (66.8% G/C), Mycobacterium smegmatis (65.6% G/C), and Deinococcus radiodurans (67% G/C), do indeed display a mutation bias toward a higher G/C-content (Dillon et al. 2015; Kucukyildirim et al. 2016). On the opposite end of the A/T mutation bias spectrum is Mesoplasma florum with an A/T bias of ∼16.0 (Sung, Ackerman, et al. 2012).
Indel rates can disproportionally affect repeats based on G+C-content. In mismatch-repair-deficient lines of P. aeruginosa, indels occurred primarily in homopolymeric runs of G/C base pairs (Dettman et al. 2016). Furthermore, there was an evident strand bias in the indel rate as indels were more common with a G in the lagging strand template compared with the leading strand. In B. cenocepacia, G/C base pairs were deleted more frequently than A/T base pairs without a commeasurable increase in G/C base pair insertions compared with A/T insertions. This bias toward deletions in G/C base pairs would contribute to an increase in the A+T-content of genomes in the absence of opposing selective mechanisms for increasing or maintaining high G+C-content.
Unsurprisingly, there is a correlation between the predicted and the observed G+C-content of prokaryotic genomes. However, the observed G+C-contents tend to be greater than predicted by mutation pressure alone. This difference reflects, among other things, the constraints that the genetic code places on the base composition of genome. Amino acids with high G/C codons are required for protein function in genomes regardless of the mutation bias, and this sets limits to the degree to which nucleotide composition of the genome reflects the prevailing mutation biases. In addition, selection on silent sites and G/C-biased gene conversion also contribute to the deviation of the observed from the expected base composition of genomes. Furthermore, the deviations from the equilibrium G+C-content (GCeq) can also contribute to the variation in mutation rates. The higher G+C-content of genomes compared with their GCeq is predicted to result in higher mutation rates relative to genomes at GCeq (Krasovec et al. 2017). The contribution of elevated G+C-content to mutation rates can be substantial, and there exists a significant correlation between the observed deviation from the GCeq of genomes and their mutation rate (Krasovec et al. 2017).
Leading/Lagging Strand Differences in Mutation Rates
Differences in the replication of the leading and lagging strands can lead to differences in the rates and spectrum of mutations, depending on which strand of the DNA molecule is being used as a template (Wu and Maeda 1987). The consequences of leading/lagging strand asymmetry in mutation rates are easiest to detect in prokaryotes, which have a conserved single origin of replication (Wu 1991; Lobry 1996). Assuming that there are no differences in mutational biases between the two DNA strands, the intrastrand frequencies of any base and its complementary base should be equal (A=T, C=G). Deviation from this parity rule can result from selection or differences in mutation rates between the two strands (Sueoka 1995). Bacterial genomes frequently display asymmetry in intrastrand base frequencies which switch signs at the origin of replication. For example, there may be an excess of G relative to C on one side of the origin of replication on a particular DNA strand which changes to an excess of C relative to G on the other side of the replication origin on the same DNA strand (Lobry 1996). MA experiments in E. coli have found significant leading/lagging strand differences for specific mutation rates. For instance, A/T → G/C transitions were more frequent when A is on the lagging strand template and T is on the leading strand template. Likewise, G/C → A/T transitions were more frequent when C was on the lagging strand template and G was on the leading strand template (Lee et al. 2012; Shewaramani et al. 2017). Moreover, context-specific mutation rates also display strand bias (Sung et al. 2015). In contrast, no leading/lagging strand differences in mutation rates were detected in Salmonella typhimurium (Lind and Andersson 2008).
Location within a Genome
Various genomic features, such as G+C-content, recombination rate, and the timing of replication of different chromosomes or chromosomal regions, have the potential to influence the frequencies and types of mutations. Nucleotide polymorphism in natural populations is correlated with recombination frequency, which is usually attributed to natural selection and not differences in mutation rates (Begun and Aquadro 1992; Cutter and Choi 2010; McGaugh et al. 2012). However, mutation rates are correlated with recombination rate in diverse taxa, including humans, Arabidopsis, honey bees, and Caenorhabditis elegans (Arbeithuber et al. 2015; Francioli et al. 2015; Yang et al. 2015; Konrad et al. 2018; Smith et al. 2018). In Caenorhabditis elegans, novel gene copy-number changes occur more frequently in the chromosome arms with higher recombination rates, compared with the cores with lower recombination rates (Konrad et al. 2018). Similarly, in honey bees, more mutations occurred in the vicinity of crossovers than expected by chance (Yang et al. 2015).
The change in the nucleotide pool during replication has been suggested to influence mutation rates and the mutation spectrum as a function of replication timing (Wolfe et al. 1989; Gu and Li 1994). The potential for replication timing to introduce intragenomic variation in mutation rate has also been investigated in families and in MA experiments with mixed results. In human families, there was a positive correlation between replication timing and mutation rate, suggesting that late-replicating regions have higher mutation rates than early-replicating regions in some studies (Francioli et al. 2015; Jónsson et al. 2017; Smith et al. 2018). However, the late-replication contribution was confounded with father’s age as young fathers contributed more to the late replication effect in one of the studies (Francioli et al. 2015). In contrast, another study of human families reached the contrasting conclusion that early replicating genes have higher mutation rates (Wong et al. 2016; Smith et al. 2018).
Burkholderia cenocepacia, a Gram-negative bacterium, contains three chromosomes bearing significant differences in the rates and spectra of mutations (Dillon et al. 2015). The highest and lowest base substitution rates were observed on chromosomes I and II, respectively, which is opposite to the rate of evolution of the genes on these chromosomes. Furthermore, the spontaneous rate of G/C → T/A transversions was highest on chromosomes III, whereas the rate of A/T → C/G transversions was highest on chromosomes I. However, dividing the genome into early and late replicating regions did not clarify whether these differences in mutation rate and spectrum between chromosomes could be attributed to the timing of replication.
Rate of Transcription and Its Effects on Mutation Rate
Analyses of the effects of transcription on mutation rates have reached divergent conclusions, even in the same species (e.g., Martincorena et al. 2012; Chen and Zhang 2013). Some experiments have suggested that high levels of transcription increase mutation rates (Klapacz and Bhagwat 2002; Hudson et al. 2003; Kim and Jinks-Robertson 2012; Alexander et al. 2013). MA experiments with Salmonella typhimurium appear to confirm this relationship as highly expressed genes with high codon adaptation index (CAI) were hit with significantly more mutations than expected by chance (Lind and Andersson 2008). In B. cenocepacia, a Gram-negative bacterium with three chromosomes, the largest chromosome (chromosome I) which harbors a disproportionately larger fraction of essential and highly expressed genes also exhibits the highest mutation rate of the genome’s three chromosomes (Dillon et al. 2015). The high mutation rate in chromosome I stands in contrast with the slower rate of molecular evolution of genes on this chromosome. Although consistent with mutagenic consequences of transcription, the difference in mutation rate between different chromosomes could also be the consequence of early versus late replication of different chromosomes (Dillon et al. 2015). In contrast, experiments in E. coli mutL mutants found a negative correlation between CAI and the number of mutations, which suggests that gene expression may not increase the mutation rate in E. coli (Lee et al. 2012). In these cases, the rate of transcription was inferred indirectly from CAI or location in the genome. Analysis of MA in microalgae found that transcript abundance was negatively associated with mutations in intergenic regions, implicating transcription-coupled repair in reducing the mutation rate (Krasovec et al. 2017). However, this association was not detected in coding sequences of the same species (Krasovec et al. 2017). The relative contributions of transcription-coupled repair and transcription-associated mutagenesis to the mutation rate seem to vary between species and between regions of the genome, although in microbes, transcription appears to cause a slight increase in their mutation rates (Lynch et al. 2016).
Intraspecific Variation in Mutation Rates
MA studies with different strains within species have also found that there can be significant intraspecific variation in the mutation rate. The intraspecific variation in mutation rate at a genome-wide level was elegantly demonstrated in MA experiments with C. reinhardtii which found a 7-fold difference in mutation rate between six genetically diverse strains (Ness et al. 2015). The causes of intraspecific variation in mutation rates are still not well understood. It has been shown that mutator alleles can increase in frequency during adaptation to novel environments, and it is possible that some intraspecific variation arises from transient alleles increasing the mutation rate due to selection (Sniegowski et al. 1997; Taddei et al. 1997; Raynes et al. 2011). However, variation in mutation rate is also expected from mutation–selection balance of novel detrimental mutations that increase the mutation rate.
Paternal Contribution to Variation in Mutation Rates
Haldane (1935) suggested that mutation rates could be higher in males than in females. This hypothesis is supported by considerable evidence amassed from comparing variation and divergence in the sex chromosomes relative to the autosomes (Miyata et al. 1987; Ellegren 2007; Wilson Sayres and Makova 2011). WGS analysis of the frequency of spontaneous mutations in human families has provided direct estimates of the relative paternal and maternal contributions to mutation rates and moreover found a strong correlation with paternal age (Kong et al. 2012; Francioli et al. 2015; Jonsson et al. 2017). The male contribution to mutation rate is primarily due to the greater number of cell divisions in the male germline than in the female germline and not due to a higher mutation rate per cell division in males (Link et al. 2017). It appears that the age of the father contributes significantly to the variation in mutation among humans and may, in fact, explain most of the variation in mutation rates in human families (Kong et al. 2012; Jonsson et al. 2017). This association with paternal age has also been observed in chimpanzees (Venn et al. 2014). The strong male contribution to mutation frequency would also contribute to interspecific variation in mutation rate as, all else being equal, species with older breeding males should have higher per generation mutation rates relative to species with young breeding males. An analysis of new mutations in a family of collared flycatchers found only slightly more mutations attributable to males than females, as well as an overall lower mutation rate compared with humans (Smeds et al. 2016). The authors speculated that lower mutation rates in birds and mice compared with humans and chimpanzees can in part be explained by paternal mutations (Smeds et al. 2016).
Transcriptional Consequences of Spontaneous Mutations
The first progression toward understanding the eventual phenotypic consequences of mutation is to determine the influence of mutations on the evolution of gene expression. Alterations in the expression profiles of both protein-coding and regulatory genes can effect morphological change, with a growing body of evidence implicating a strong role for regulatory changes in the process that was previously obscured (Beldade et al. 2002; Wittkopp et al. 2003; Wray et al. 2003; Abzhanov et al. 2004; Shapiro et al. 2004; Fay and Wittkopp 2008; Romero et al. 2012). The genomics revolution has facilitated the development of technologies capable of generating a transcriptome, namely the quantification of an entire set of transcripts in a cell specific to a particular environmental condition and unique developmental stage of an organism. The transcripts under study are not restricted to mRNAs; indeed, a major goal of transcriptomics is enable analysis of all flavors of transcripts additionally encompassing noncoding RNA and small RNAs (Wang et al. 2009). In the late 1990s and early 2000s, hybridization-based approaches involving custom-made or commercial microarrays initially served as the method of choice for investigating patterns of global gene expression. However, a major limitation of microarray technology is its dependence on an a priori known genome sequence to facilitate probe design, which certainly played a role in restricting initial transcriptome analysis to that of a handful of model species. Microarray technology has further limitations, namely 1) greater noise in a data set stemming from high background levels due to cross-hybridization which can lead to spurious correlations (Okoniewski and Miller 2006), 2) limits to range of detection due to background and saturation of signals, and 3) challenges associated with the comparison of expression profiles across different sets of experiments (Wang et al. 2009). Commencing in 2008, the high-throughput, sequence-based approach of RNA-Seq has revolutionized the field of transcriptomics given 1) its nonreliance on existing genomic sequence information and hence, suitability to nonmodel as well as model organisms, 2) high level of resolution in determining the precise location of transcription boundaries, 3) extremely low background signal, 4) the ability to detect a wide range of expression levels (both extremely low and high), 5) high reproducibility across technical and biological replicates, and 6) relatively low cost (reviewed by Wang et al. 2009).
Spontaneous MA experiments provide a powerful framework to investigate divergence in global transcription profiles due to accumulated genetic changes without interference from the effects of purifying selection. Expression profiles of MA lines relative to the ancestral control in themselves offer key insights into the divergence of expression profiles due to the input of new genetic variants. However, if all ensuing genetic changes in MA lines have been characterized via genome sequencing a priori, it further enables the dissection of gene expression alteration as a function of the particular characteristics of the mutation in question, both with respect to its genomic location and mutation class (coding vs. regulatory, single nucleotide polymorphisms vs. CNVs vs. small indels, etc.). To date, only six studies have examined long-term MA lines of three eukaryotic species to offer the first glimpses into the influence of spontaneous MA on gene expression divergence with the majority (all but two) using hybridization-based, microarray technology. It remains to be seen if the initial conclusions of the microarray studies can be recapitulated with the application of the more modern approach of RNA-Seq.
Denver et al. (2005) applied a microarray approach to four Caenorhabditis elegans MA lines propagated across 280 consecutive MA generations, their ancestral N2 control, and five natural isolates in order to examine and contrast global expression patterns under conditions of genetic drift (MA lines) versus strong natural selection (natural isolates). Rifkin et al. (2005) conducted a similar transcriptome analysis of 12 D. melanogaster lines following their passage through 200 MA generations using microarray technology. Gene expression levels were measured at two developmental stages, namely the third larval instar and at puparium formation. Landry et al. (2007) extended these investigations to a unicellular eukaryote by examining four MA lines of S. cerevisiae propagated for 4,000 generation at Ne = 10. Huang et al. (2016) assessed transcriptional divergence of 25 D. melanogaster lines maintained by full-sib mating at N = 20 following 60 MA generations. Most recently, Zalts and Yanai (2017) conducted the first RNA-Seq analysis of gene expression during the embryonic development of 19 Caenorhabditis elegans MA lines following 250 generations followed by Konrad et al. (2018) who investigated the transcriptional consequences of copy-number changes in Caenorhabditis elegans MA lines subjected to varying intensity of selection.
Relative Roles of Selection versus Drift in Shaping the Evolution of Expression Divergence
Phenotypic variation within a population (including gene expression) can be partitioned into genetic (Vg) and/or environmental (Ve) components (Falconer and Mackay 1996; Lynch and Walsh 1998). In the case of MA lines, between-line genetic variation can be attributed to the input of novel spontaneous mutations (Vm), and the within-line phenotypic variation due to environmental or technical noise (Ve, or its proxy, the residual variance Vr). The relative roles of neutral evolution versus selection in shaping expression divergence can be investigated by comparing the gene-specific ratios of transcriptional genetic variance (Vg) in the natural isolates with the transcriptional mutational variance (Vm) in the MA lines. Specifically, Vm is defined as the per-generation increase in trait variance across a population that is due to mutation alone whereas Vg represents the among-line or standing genetic variance. If gene expression divergence is neutral, the expected Vg/Vm ratio is equal to 4Ne in a self-fertilizing diploid species, such as Caenorhabditis elegans (Lynch and Hill 1986). An increasing role for purifying selection in constraining transcript abundance will manifest as smaller observed Vg/Vm ratios. Denver et al. (2005) found all the observed Vg/Vm ratios to be well below the neutral expectation, which suggests that strong stabilizing selection constrains gene expression in the wild. Patterns of expression divergence in two independent sets of Drosophila MA lines (Rifkin et al. 2005; Huang et al. 2016) recapitulate the conclusion from the Caenorhabditis elegans study that strong stabilizing selection has far greater influence than drift in shaping the evolution of gene expression. The observed expression divergence between species (D. melanogaster, Drosophilasimulans, and Drosophilayakuba) was much lower than expected given the Vm estimates for transcription in the MA lines and a neutral model for comparison (Rifkin et al. 2005).
Gene Functionality and the Potential for Transcriptional Evolution
Are genes equally mutable in their ability to diverge at the transcriptional level? Patterns of observed nucleotide divergence among orthologous genes in diverse organisms would suggest otherwise, given that some genes can remain virtually unchanged in sequence over lengthy evolutionary periods whereas others exhibit accelerated sequence evolution. These divergent patterns in the rates of sequence evolution of different genes have long been taken to imply that selective constraints can vary considerably among genes involved in different biological processes. An examination of gene expression profiles offers a more direct approach to investigate the differential capacity of genes to evolve at the transcriptional level and determine whether gene-specific patterns are shared across diverse species.
In Caenorhabditis elegans, genes involved in carbohydrate, amino acid, and lipid metabolism as a class appeared to be under the least influence of stabilizing selection. In contrast, genes implicated in the signal transduction pathway exhibited a strong signature of stabilizing selection (Denver et al. 2005). A similar pattern was recapitulated in D. melanogaster. Genes involved in essential cellular functions relating to transcription, translation, cell cycle, and energy metabolism displayed significantly lower variability in expression suggesting stringent selective constraints, whereas those encoding enzymes and structural proteins involved in chitin metabolism, iron binding, and sensory perception of chemical stimuli displayed a significant capacity for gene expression evolution (Rifkin et al. 2005; Huang et al. 2016). Zalts and Yanai (2017) used an RNA-Seq platform to explore gene expression variation during embryonic development in Caenorhabditis elegans spanning seven stages, from a four-cell embryo to a newly hatched L1 larva. Gene expression divergence was found to be significantly depleted in mid-embryogenesis which marks a highly constrained developmental stage across diverse species, with homeodomain transcription factors and genes responsible for the integration of germ layers during morphogenesis evolving under stringent selection.
Relative Roles of Cis- versus Trans-acting Changes in the Evolution of Gene Expression
MA experiments are especially amenable to understanding the rate of evolution of expression divergence, given that the evolutionary time since divergence from the ancestral control is precisely known. A determination of the rate of expression divergence relative to the rate of genic changes further enables the disentangling of the relative roles of cis- versus trans-mutations in effecting the evolution of gene expression. Approximately two-thirds of the differentially expressed genes in the Caenorhabditis elegans MA lines were restricted to seven sets of coregulated genes, which suggests that most of the observed global change in transcription patterns was due to mutations at relatively few trans-acting loci with pleiotropic effects (Denver et al. 2005). Mutations with multiple trans-acting effects are likely to be deleterious and would be weeded out by purifying selection in natural populations. Furthermore, genes in close proximity to one another were also overrepresented among the set of differentially expressed genes, which suggests an influence of cis-acting regulatory mutations, changes in chromatin organization or novel CNVs. Indeed, Gibson (2005) examined Denver et al.’s (2005)Caenorhabditis elegans data and estimated that the rate of gene expression divergence is approximately an order of magnitude higher than the rate of genic change per line per generation, implicating the contribution of both cis- and trans-acting mutations toward changes in expression.
Are Gene Expression Patterns Associated with Particular Features of the Genetic and Genomic Architecture?
Given the considerable variation in the genome organization of different groups of organisms, how might a species’ prevailing genomic and genetic architecture impinge on the evolution of its transcriptome? The genomes of eukaryotic species are highly variable in size and can comprise large expanses of repetitive, gene-poor regions of low complexity as well as a high incidence of selfish genetic elements. Furthermore, there exists genomic variation in recombination frequency which in conjunction with selection further influences the patterns of nucleotide variation. In Caenorhabditis elegans, gene organization is nonrandom within and between chromosomes (Cutter et al. 2009) comprising gene-poor autosomal arms with high rates of recombination versus gene-rich, centrally located autosomal clusters/cores exhibiting limited recombination (Barnes et al. 1995; Rockman and Kruglyak 2009). Caenorhabditiselegans MA lines with differential gene expression were not significantly biased toward autosomal arms versus core regions. In contrast, differentially expressed genes in the natural isolate lines exhibited a significant distributional bias toward autosomal arms (Denver et al. 2005) which was taken to represent stronger purifying selection against expression divergence of core-residing genes. Additionally, Huang et al. (2016) used Vm/Vg as an indicator of the strength of the apparent stabilizing selection to observe stronger constraints on the expression of X-linked genes in D. melanogaster, with a more pronounced effect in males relative to females.
Transcriptional Consequences of Copy-Number Changes
The three previously mentioned studies investigated genome-wide changes in transcription following MA, but did not analyze the transcriptional consequences of any particular class of mutation. Gene duplications, a class of copy-number changes, have the potential to alter transcript abundance of any gene contained within the duplication tract as well as other genes whose transcription is under the direct or indirect control of the duplicated genes. A handful of recent studies aiming to investigate the role of segmental gene duplications in shaping gene expression patterns have arrived at contrasting conclusions. Some studies of gene duplications in natural or laboratory populations of yeast, Drosophila, and mammals have concluded a minimal or no change in gene expression associated with an increase in gene copy-number (Qian et al. 2010; Guschanski et al. 2017; Rogers et al. 2017). In stark contrast, an engineered duplication inserted into different locations in the Drosophila genome often resulted in a >2-fold increase in transcript abundance (Loehlin and Carroll 2016). Konrad et al. (2018) specifically investigated the transcriptional consequences of gene copy-number changes in Caenorhabditis elegans MA lines under minimal selection (N = 1) and observed that the average increase in transcript abundance following gene duplication significantly exceeded 2-fold. This suggests that the lack of significant increase in transcript abundance of gene duplicates in wild or laboratory populations is either the result of selection against duplications that lead to increased transcription, or secondary mutations that downregulate the transcription of duplicated genes. Konrad et al.’s (2018) study in Caenorhabditis elegans also implemented a modified MA approach with different population bottlenecks of N = 1, 10, and 100 individuals per generation, thereby modulating the intensity of selection during experimental evolution. Bottlenecks of single individuals allow genetic drift to operate to the maximum degree possible, and larger MA populations are expected to experience greater selection intensity against deleterious mutations, inversely proportional to the Ne. MA lines with larger population bottlenecks (N = 10 and 100 individuals) had a significantly lower increase in average transcript abundance of duplicated genes relative to standard MA lines with single individual bottlenecks in every generation (N = 1). Furthermore, the genes duplicated in MA lines maintained at larger population sizes had significantly lower ancestral transcript abundance than the genes duplicated in the N = 1 lines. Together, these results show that 1) duplications of highly expressed genes are more detrimental than duplications of genes with low transcript abundance, and 2) the deleterious fitness consequences of duplications are associated with the increase in transcript abundance they engender.
Evolution of Canalization in Response to Genetic versus Environmental Perturbations
Phenotypic variability in organisms can display remarkable robustness despite exposure to persistent genetic and environmental perturbations, often referred to as canalization (Waddington 1942). While genetic and environmental perturbations appear to be distinct processes, the mechanism of buffering, itself, may be an evolutionarily shared, generic response to constrain the effects of any class of perturbations (Meiklejohn and Hartl 2002). Under this scenario, traits that are buffered against the effects of environmental perturbations may also be buffered to a similar degree against the effects of genetic mutations. In other words, does genetic variation accumulate faster (or slower) in genes exhibiting greater (or lowered) plasticity in response to environmental perturbations? In technical terms, this would be manifested as a significant positive correlation between the mutational variance (Vm) and environmental (residual) variance (Ve or Vr) which has been observed in three studies studying gene expression divergence in D. melanogaster (Rifkin et al. 2005; Huang et al. 2016) and S. cerevisiae (Landry et al. 2007). These results would imply that perturbations, irrespective of source (genetic or environmental), affect gene expression in similar ways and the evolved genetic mechanism(s) for promoting or buffering the transcriptional response may be the same.
Epigenetic Changes during MA
Cytosine methylation is a widespread form of DNA modification in eukaryotes and is associated with epigenetic silencing of genes and transposons. The rate at which epigenetic modifications to the DNA are gained and lost (epimutations) is essential for understanding the population dynamics of epigenetic variation and its contribution to adaptation or the genetic load (Slatkin 2009; Furrow and Feldman 2014; van der Graaf et al. 2015). The introduction of a sodium bisulfate treatment to genomic DNA, which converts unmethylated cytosines to uracil, allows for the genome-wide analysis of cytosine methylation. Several studies have applied these methods to MA lines of Arabidopsis to measure the rate and spectrum of epigenetic mutations (Becker et al. 2011; Schmitz et al. 2011; Jiang et al. 2014; van der Graaf et al. 2015). The estimated epigenetic mutation rate of CpG dinucleotides in Arabidopsis ranges from 2.56 × 10−4 to 6.30 × 10−4 per nucleotide per generation, with methylation losses close to 3-fold more common than methylation gains (Schmitz et al. 2011; van der Graaf et al. 2015). The excess of gains over losses is consistent with the proportion of CpG sites that are methylated in the genome. However, plant transposable elements, which are heavily methylated at CpG sites, have a methylation loss rate that is much lower, at ∼1/30 of the gain rate. It appears that the methylation patterns of transposable elements can be explained by a low ratio of gains to loss of CpG methylation. The environment can influence both the rate of mutations as well as the rate of epimutations. One set of experiments with Arabidopsis measured the mutation rate and the rate of changes in methylated cytosines in plants reared in a standard soil versus highly saline soil (Jiang et al. 2014). The mutation rate was 2-fold higher for plants grown in a high-salinity soil, with the rate of transversions exceeding that of transitions. Furthermore, differentially methylated CpG sites were increased by 40% in plants from the high-saline soil.
A common objection to the long-term evolutionary potential of epimutations is that they are too unstable (Slatkin 2009; Furrow 2014). The high rate of epimutations is certainly borne out with the analyses of these MA lines as the per-nucleotide epimutation rate is 5 orders of magnitude higher than the DNA-based mutation rate. Nonetheless, epimutations may be stable enough to respond to selection (van der Graaf et al. 2015).
Conclusions and Future Directions
The mutation rate is a fundamental parameter for understanding a multitude of biological phenomena. Attempts to estimate mutation rates have a long history in evolutionary biology and have utilized a wide variety of methods, including direct observations of mutant phenotypes under laboratory conditions, estimates from polymorphisms in natural populations, and analysis of silent site divergence between taxa (reviewed by Kondrashov FA and Kondrashov AS 2010). The wide availability of cost-effective next-generation sequencing methods and computing power has provided unprecedented opportunities for direct measurements of mutation rates in a wide variety of taxa. In some cases, the measurements can be made by parent–offspring genotype comparisons (parent–offspring trios) and counting the number of mutations across a single generation. This is a reasonable approach for taxa that have a relatively high number of mutations per generation. Humans, for example, have a base substitution rate ranging from 1.1 to 1.7 × 10−8/site/generation yielding ∼100 new mutations in an offspring (Kondrashov 2002; Lynch 2016, and references therein). However, many taxa have much lower mutation rates and no new mutations in the majority of their offspring. For example, model organisms, such as D. melanogaster, Caenorhabditis elegans and A. thaliana, have base substitution rates on the order of 10−9/nucleotide site/generation whereas bacteria and protists have even lower mutation rates on the order of 10−10 and 10−11–10−12/nucleotide site/generation (table 1). Multigenerational MA experiments partnered with high-throughput genomic technologies have proved indispensable in enabling robust measures of mutation rates and their properties for these organisms.
MA experiments can be labor- and time-intensive, and it can take a substantial time investment to reap rewards in the form of new and exciting data. However, many processes that contribute to heritable variation and evolutionary change are rare, and if we are to investigate them experimentally rather than being content with retrospective analysis of extant organisms, MA experiments are still an unparalleled experimental approach. MA experiments continue to provide us with important information about mutational processes and their consequences. The broad variation in mutation rates across the tree of life, most of which have been measured in MA lines, has resulted in an original theory of the evolution of mutation rates, the drift-barrier hypothesis (Sung, Ackerman, et al. 2012). MA-WGS studies have been crucial in revealing a significant contribution of copy-number changes to standing genetic variation across diverse genomes, by enabling direct estimation of the spontaneous rates of gene duplication and deletion, on the order of 10−5–10−7/gene/generation (table 3). This discovery has engendered a recognition of a significant role of CNVs in generating intraspecific genetic variation, the full functional and phenotypic consequences of which remain obscure. Future investigations should focus on further elucidating the transcriptional, phenotypic, and fitness consequences of this form of genetic variation that until now has been largely ignored. Indeed, Konrad et al.’s study (2018) on the transcriptional consequences of copy-number changes in Caenorhabditis elegans MA lines has taken a first step in this direction to demonstrate that while gene duplications play a unique role in adaptation and the origin of evolutionary novelties, their immediate transcriptional consequences are deleterious with respect to fitness. The application of WGS to novel mutations in organelles is giving insights into the population dynamics of mutations at a different level altogether, within the cytoplasm. These examples come from only a few species and it is of great importance to expand this sample to include more taxa beyond the traditional model organisms to elucidate general patterns and, perhaps, important and illustrative exceptions. Next-generation sequencing technology has also aided in the detection of low-frequency heteroplasmic variants and demonstrated their pervasiveness within mitochondrial genomes (Haag-Liautard et al. 2008; Konrad et al. 2017). This in turn suggests that the mitochondrial effective population size may be greater than previously recognized.
MA as an experimental system was originally conceived as a method to measure the rate of deleterious mutations, but it is now emerging as a powerful framework to analyze the molecular spectrum of mutations and their transcriptional consequences. The MA model should also be extended beyond standard DNA-based genotyping of base substitutions, indels, and structural variants. In this respect, we have already seen a handful of MA experiments that have investigated the transcriptional consequences of mutations. It is possible that changes in gene regulation are of greater importance in evolution than changes in protein structure. The first few experiments analyzing transcriptional changes in MA lines highlight that regulation of gene expression is under strong selection. This is an area that has a lot of untapped potential and should be extended. Another important topic that can be addressed with MA experiments is the rate and stability of epimutations.
Perhaps, one of the most promising future directions that MA experiments can take is the use of a modified MA design with differing population size treatments. Thus far, the vast majority of MA studies have maintained the focal organism at a constant minimal Ne for the purpose of drastically reducing the efficacy of selection and enabling the accumulation of the vast majority of mutations (all but the most deleterious mutation that confer complete sterility or mortality). A recent spontaneous MA study in Caenorhabditis elegans (Konrad et al. 2017, 2018) maintained multiple replicate lines at the minimal population size (N = 1) but additionally encompassed replicate populations maintained at incrementally increasing population sizes of N = 10 and N = 100 individuals per generation. The varying Ne treatment offers a powerful framework to assess how spontaneous mutational input in conjunction with varying strengths of natural selection shapes genomes. Indeed, Halligan and Keightley (2009) highlighted a sore need for future studies exploring MA in populations of different sizes in order to reveal the distribution of fitness effects of new mutations. MA experiments of varying population size would provide an unprecedented resource to further delineate the evolutionary role of natural selection versus genetic drift 1) at multiple phenotypic scales (including but not limited to behavior, immunity, morphology, and physiology), 2) at the DNA level with implications for genome evolution, 3) at the level of transcriptome to investigate the evolution of gene expression and smRNAs, and 4) in the evolution of protein function and protein interactions (fig. 4).
Fig. 4.

—Mutation accumulation with varying population sizes (Ne) as a valuable biological resource. The differential intensity of genetic drift and natural selection among different population size treatments facilitates investigations into the joint influence of spontaneous mutation and selection on the evolution of phenotypic traits, DNA sequences, transcription, and protein function.
As sequencing technologies become more cost-effective and analytical methods for WGS data become more refined, genome sequencing of parent–offspring trios or three-generation pedigrees has the potential to generate reliable estimates of the genomic mutation rate in a wide range of taxa that are not amenable to MA experiments and hence remain under- or unrepresented in the set of organisms with known mutation rates (Venn et al. 2014; Keightley et al. 2015; Yang et al. 2015; Smeds et al. 2016; Jónsson et al. 2017; Pfeifer 2017; Tatsumoto et al. 2017; Smith et al. 2018). This particularly pertains to species with longer generation times such as vertebrate species (most, if not all, mammals, birds, amphibians, and reptiles) as well as plants. Plants as a large and diverse clade have been traditionally underrepresented in MA experiments with minimal information available on their rates and spectra of mutations in both the nuclear and organellar genomes. To date, there has been no effort to determine the genome-wide spontaneous mutation rates in plant mitochondria and chloroplasts, despite their intriguing evolutionary history and divergent patterns and rates of mutation. Greater species and taxa representation will serve to further refine our understanding of basic mutational parameters and their shared versus discernible features across diverse taxa, as well as advance our understanding of the fitness consequences of mutations and their role in shaping genomes, one of the cornerstones of modern biology. MA-WGS approaches bear immense potential to provide a unified account of evolution at the genetic and phenotypic levels, while yielding significant insights into the evolutionary process at multiple fundamental scales—the genetic basis of variation, the evolutionary dynamics of mutations under the forces of natural selection and genetic drift, and their range of fitness effects.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
V.K. was supported by a National Science Foundation (Grant MCB-1330245). U.B. and V.K. were additionally supported by start-up funds from the Department of Veterinary Integrative Biosciences, College of Veterinary Medicine and Biomedical Sciences at Texas A&M University. The authors wish to acknowledge Associate Editor Dr. Kateryna Makova for the invitation to write this review article, her steadfast patience during the extended preparation phase, and her assistance with revisions. The authors are grateful to two anonymous referees for valuable suggestions.
Literature Cited
- Abzhanov A, Protas M, Grant BR, Grant PR, Tabin CJ.. 2004. Bmp4 and morphological variation of beaks in Darwin’s finches. Science 305(5689):1462–1465. [DOI] [PubMed] [Google Scholar]
- Alexander MP, Begins KJ, Crall WC, Holmes MP, Lippert MJ.. 2013. High levels of transcription stimulate transversions at GC base pairs in yeast. Environ Mol Mutagen. 54(1):44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI, Hughes D.. 1996. Muller’s ratchet decreases fitness of a DNA-based microbe. Proc Natl Acad Sci U S A. 93(2):906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeithuber B, Betancourt AJ, Ebner T, Tiemann-Boege I.. 2015. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc Natl Acad Sci U S A. 112(7):2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaf ZJ, Tilk S, Park J, Siegal ML, Petrov DA.. 2017. Deep sequencing of natural and experimental populations of Drosophila melanogaster reveals biases in the spectrum of new mutations. Genome Res. 27(12):1988–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ávila A, Garcia-Dorado A.. 2002. The effects of spontaneous mutation on competitive fitness in Drosophila melanogaster. J Evol Biol. 15(4):561–566. [Google Scholar]
- Avise JC. 2000. Phylogeography: the history and formation of species. Cambridge: Harvard University Press. [Google Scholar]
- Azevedo RBR, et al. 2002. Spontaneous mutational variation for body size in Caenorhabditis elegans. Genetics 162:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer CF, et al. 2005. Comparative evolutionary genetics of spontaneous mutations affecting fitness in rhabditid nematodes. Proc Natl Acad Sci U S A. 102(16):5785–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer CF, Miyamoto MM, Denver DR.. 2007. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 8(8):619–631. [DOI] [PubMed] [Google Scholar]
- Ballard JWO, Whitlock MC.. 2004. The incomplete natural history of mitochondria. Mol Ecol. 13(4):729–744. [DOI] [PubMed] [Google Scholar]
- Barnes TM, Kohara Y, Coulson A, Hekimi S.. 1995. Meiotic recombination, noncoding DNA and genomic organization in Caenorhabditis elegans. Genetics 141(1):159–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CM, Neiman M, Taylor DR.. 2005. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. l68:39–50. [DOI] [PubMed] [Google Scholar]
- Becker C, et al. 2011. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480(7376):245–249. [DOI] [PubMed] [Google Scholar]
- Begun DJ, Aquadro CF.. 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356(6369):519–520. [DOI] [PubMed] [Google Scholar]
- Behringer MG, Hall DW.. 2016. Genome-wide estimates of mutation rates and spectrum in Schizosaccharomyces pombe indicate CpG sites are highly mutagenic despite the absence of DNA methylation. G3 12:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beldade P, Brakefield PM, Long AD.. 2002. Contribution of distal-less to quantitative variation in butterfly eyespots. Nature 415(6869):315–318. [DOI] [PubMed] [Google Scholar]
- Benzer S. 1961. On the topography of genetic fine structure. Proc Natl Acad Sci U S A. 47(3):403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom CT, Pritchard J.. 1998. Germline bottlenecks and the evolutionary maintenance of mitochondrial genomes. Genetics 149(4):2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergthorsson U, Adams KL, Thomason B, Palmer JD.. 2003. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 424(6945):197–201. [DOI] [PubMed] [Google Scholar]
- Bergthorsson U, Katju V.. 2016. Gene Copy-Number Changes in Evolution. In eLS, John Wiley & Sons, Ltd (Ed.). doi:10.1002/9780470015902.a0026319 [Google Scholar]
- Birky CW. 2001. The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms and models. Annu Rev Genet. 35(1):125–148. [DOI] [PubMed] [Google Scholar]
- Bowe LM, Coat G, dePamphilis CW.. 2000. Phylogeny of seed plants based on all three genomic compartments: extant gymnosperms are monophyletic and Gnetales’ closest relatives are conifers. Proc Natl Acad Sci U S A. 97(8):4092–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Beaupré HC, Stewart DT, Hoeh WR, Blier PU.. 2007. The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 23(9):465–474. [DOI] [PubMed] [Google Scholar]
- Brown WM, Prager EM, Wan A, Wilson AC.. 1982. Mitochondrial DNA sequences in primates: tempo and mode of evolution. J Mol Evol. 18(4):225–239. [DOI] [PubMed] [Google Scholar]
- Caballero A, Keightley PD.. 1994. A pleiotropic nonadditive model of variation in quantitative traits. Genetics 138(3):883–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellana S, Vicario S, Saccone C.. 2011. Evolutionary patterns of the mitochondrial genome in Metazoa: exploring the role of mutation and selection in mitochondrial protein coding genes. Genome Biol Evol. 3:1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 1990. Mutation-selection balance and the evolutionary advantage of sex and recombination. Genet Res. 55(3):199–221. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 10(3):195–205. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Borthwick H, Bartolomé C, Pignatelli P.. 2004. Estimates of the genomic mutation rate for detrimental alleles in Drosophila melanogaster. Genetics. 167(2):815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D, Morgan MT.. 1990. Genetic loads and estimates of mutation rates in highly inbred plant populations. Nature 347:308–382. [Google Scholar]
- Charlesworth B, Hughes KA.. 1996. Age-specific inbreeding depression and components of genetic variance in relation to the evolution of senescence. Proc Natl Acad Sci U S A. 93(12):6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Hughes KA.. 1999. The maintenance of genetic variation in life history traits In Singh RS, Krimbas CB, editors.. Evolutionary genetics from molecules to morphology. Vol. 1.Cambridge: Cambridge University Press; p. 369–392. [Google Scholar]
- Charlesworth D, Charlesworth B.. 1987. Inbreeding depression and is evolutionary consequences. Annu Rev Ecol Syst. 18(1):237–368. [Google Scholar]
- Charlesworth D, Morgan MT, Charlesworth B.. 1993. Mutation accumulation in finite outbreeding and inbreeding populations. Genet Res. 61(01):39–56. [Google Scholar]
- Chen X, Zhang J.. 2013. No gene-specific optimization of mutation rate in Escherichia coli. Mol Biol Evol. 30(7):1559–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Choi JY.. 2010. Natural selection shapes nucleotide polymorphism across the genome of the nematode Caenorhabditis briggsae. Genome Res. 20(8):1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Dey A, Murray RL.. 2009. Evolution of the Caenorhabditis elegans genome. Mol Biol Evol. 26(6):1199–1234. [DOI] [PubMed] [Google Scholar]
- Davies EK, Peters AD, Keightley PD.. 1999. High frequency of cryptic deleterious mutations in Caenorhabditis elegans. Science 285(5434):1748–1751. [DOI] [PubMed] [Google Scholar]
- Deng W-H, Lynch M.. 1996. Estimation of deleterious-mutation parameters in natural populations. Genetics 144:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, et al. 2005. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat Genet. 37(5):544–548. [DOI] [PubMed] [Google Scholar]
- Denver DR, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci U S A. 106(38):16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, et al. 2012. Variation in base-substitution mutation in experimental and natural lineages of Caenorhabditis nematodes. Genome Biol Evol. 4(4):513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Vassilieva LL, Thomas WK.. 2000. High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science 289(5488):2342–2344. [DOI] [PubMed] [Google Scholar]
- Dettman JR, Sztepanacz JL, Kassen R.. 2016. The properties of spontaneous mutations in the opportunistic pathogen Pseudomonas aeruginosa. BMC Genomics 17:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MM, Cooper VS.. 2016. The fitness effects of spontaneous mutations nearly unseen by selection in a bacterium with multiple chromosomes. Genetics 204(3):1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MM, Sung W, Lynch M, Cooper VS.. 2015. The rate and molecular spectrum of spontaneous mutations in the GC-Rich multichromosome genome of Burkholderia cenocepacia. Genetics 200(3):935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MM, Sung W, Sebra R, Lynch M, Cooper VS.. 2017. Genome-wide biases in the rate and molecular spectrum of spontaneous mutations in Vibrio cholerae and Vibrio fischeri. Mol Biol Evol. 34(1):93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci U S A. 88(16):7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. 2006. Chaos and order in spontaneous mutation. Genetics 173(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan BK, Miller JH.. 1980. Mutagenic deamination of cytosine residues in DNA. Nature 287(5782):560–561. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N.. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 10:285–311. [DOI] [PubMed] [Google Scholar]
- Ellegren H. 2007. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc R Soc B. 274(1606):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS, Mackay TCF.. 1996. Introduction to quantitative genetics. London: Longman. [Google Scholar]
- Farlow A, et al. 2015. The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 201(2):737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wittkopp PJ.. 2008. Evaluating the role of natural selection in the evolution of gene regulation. Heredity 100(2):191–199. [DOI] [PubMed] [Google Scholar]
- Fisher RA. 1930. The genetical theory of natural selection. Oxford: Clarendon Press. [Google Scholar]
- Flynn JM, Chain FJ, Schoen DJ, Cristescu ME.. 2017. Spontaneous mutation accumulation in Daphnia pulex in selection-free vs. competitive environments. Mol Biol Evol. 34(1):160–173. [DOI] [PubMed] [Google Scholar]
- Force A, et al. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Hanson AJ, Lee H, Popodi EM, Tang H.. 2013. On the mutational topology of the bacterial genome. G3 3(3):399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Lee H, Popodi EM, Townes JP, Tang H.. 2015. Determinants of spontaneous mutation in the bacterium Escherichia coli as revealed by whole-genome sequencing. Proc Natl Acad Sci U S A. 112(44):E5990–E5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francioli LC, et al. 2015. Genome-wide patterns and properties of de novo mutations in humans. Nat Genet. 47(7):822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese E. 1962. On the evolution of the base composition of DNA. J Theor Biol. 3(1):82–101. [Google Scholar]
- Fry JD, Keightley PD, Heinsohn SL, Nuzhdin SV.. 1999. New estimates of the rates and effects of mildly deleterious mutation in Drosophila melanogaster. Proc Natl Acad Sci U S A. 96(2):574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrow RE. 2014. Epigenetic inheritance, epimutation, and the response to selection. PLoS One 9(7):e101559.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrow RE, Feldman MW.. 2014. Genetic variation and the evolution of epigenetic regulation. Evolution 68(3):673–683. [DOI] [PubMed] [Google Scholar]
- Gabriel W, Lynch M, Bürger R.. 1993. Muller’s ratchet and mutational meltdowns. Evolution 47(6):1744–1757. [DOI] [PubMed] [Google Scholar]
- García-Dorado A, López-Fanjul C, Caballero A.. 1999. Properties of spontaneous mutations affecting quantitative traits. Genet Res. 74(3):341–350. [DOI] [PubMed] [Google Scholar]
- García-Dorado A, Monedero JL, López-Fanjul C.. 1998. The mutation rate and distribution of mutational effects of viability and fitness in Drosophila melanogaster. Genetica 103:255–265. [PubMed] [Google Scholar]
- Gibson G. 2005. Mutation accumulation of the transcriptome. Nat Genet. 37(5):458–460. [DOI] [PubMed] [Google Scholar]
- Gong Y, Woodruff RC, Thompson JN.. 2005. Deleterious genomic mutation rate viability in Drosophila melanogaster. Biol Lett. 1(4):492–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grollman AP, Moriya M.. 1993. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 9(7):246–249. [DOI] [PubMed] [Google Scholar]
- Gu X, Li WH.. 1994. A model for the correlation of mutation rate with GC content and the origin of GC-rich isochores. J Mol Evol. 38(5):468–475. [DOI] [PubMed] [Google Scholar]
- Guschanski K, Warnefors M, Kaessmann H.. 2017. The evolution of duplicate gene expression in mammalian organs. Genome Res. 27(9):1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyllensten U, Wharton D, Josefsson A, Wilson AC.. 1991. Paternal inheritance of mitochondrial DNA in mice. Nature 352(6332):255–257. [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C, et al. 2008. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 6(8):e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagström E, Freyer C, Battersby BJ, Stewart JB, Larsson N-G.. 2014. No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res. 42(2):1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. 1935. The rate of spontaneous mutation of a human gene. J Genet. 31:317–326. [DOI] [PubMed] [Google Scholar]
- Hall DW, Fox S, Kuzdzal-Fick JJ, Strassmann JE, Queller DC.. 2013. The rate and effects of spontaneous mutation on fitness traits in the social amoeba, Dictyostelium discoideum. G3 (Bethesda) 3(7):1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD.. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst. 40(1):151–172. [Google Scholar]
- Hamilton WD. 1966. The moulding of senescence by natural selection. J Theor Biol. 12(1):12–45. [DOI] [PubMed] [Google Scholar]
- Hasan MS, Wu X, Zhang L.. 2015. Performance evaluation of indel calling tools using real short-read data. Hum Genomics. 9:20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havey MJ. 1997. Predominant paternal inheritance of the mitochondrial genome in cucumber. J Hered. 88(3):232–235. [Google Scholar]
- Houle D, Hoffmaster DK, Assimacopoulos S, Charlesworth B.. 1992. The genomic mutation rate for fitness in Drosophila. Nature 359(6390):58–60. [DOI] [PubMed] [Google Scholar]
- Howe DK, Baer CF, Denver DR.. 2010. High rate of large deletions in Caenorhabditis briggsae mitochondrial genome mutation processes. Genome Biol Evol. 2:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, et al. 2016. Spontaneous mutations and the origin and maintenance of quantitative genetic variation. eLife 5:e14625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RE, Bergthorsson U, Ochman H.. 2003. Transcription increases multiple spontaneous point mutations in Salmonella enterica. Nucleic Acids Res. 31(15):4517–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, et al. 2014. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res. 24(11):1821–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson H, et al. 2017. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549(7673):519–522. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Hall DW.. 2004. Spontaneous mutations in diploid Saccharomyces cerevisiae: more beneficial than expected. Genetics 168(4):1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katju V. 2012. In with the old, in with the new: the promiscuity of the duplication process engenders diverse pathways for novel gene creation. Int J Evol Biol. 2012:341932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katju V, Bergthorsson U.. 2013. Copy-number changes in evolution: rates, fitness effects and adaptive significance. Front Genet. 4:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katju V, Packard LB, Keightley PD.. 2018. Fitness decline under osmotic stress in Caenorhabditis elegans populations subjected to spontaneous mutation accumulation at varying population sizes. Evolution 72(4):1000–1008. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Caballero A.. 1997. Genomic mutation rates for lifetime reproductive output and lifespan in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 94(8):3823–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, et al. 2009. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 19(7):1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, et al. 2015. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol. 32(1):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Eyre-Walker A.. 1999. Terumi Mukai and the riddle of deleterious mutation rates. Genetics 153:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith N, et al. 2016. High mutational rates of large-scale duplication and deletion in Daphnia pulex. Genome Res. 26(1):60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibota TT, Lynch M.. 1996. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature 381(6584):694–696. [DOI] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S.. 2012. Transcription as a source of genome instability. Nat Rev Genet. 13(3):204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1962. On the probability of fixation of mutant genes in a population. Genetics 47:713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1983. The neutral theory of molecular evolution. Cambridge: Cambridge University Press. [Google Scholar]
- Kimura M, Ohta T.. 1969. Average number of generations until fixation of a mutant gene in a finite population. Genetics 61:763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapacz J, Bhagwat AS.. 2002. Transcription-dependent increase in multiple classes of base substitution mutations in Escherichia coli. J Bacteriol. 184(24):6866–6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo R, et al. 1990. Incomplete maternal transmission of mitochondrial DNA in Drosophila. Genetics 126:657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS. 1988. Deleterious mutations and the evolution of sexual reproduction. Nature 336(6198):435–440. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS. 2002. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian disease. Hum Mutat. 21(1):12–27. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS, Crow JF.. 1991. Haploid or diploid: which is better? Nature 351(6324):314–315. [DOI] [PubMed] [Google Scholar]
- Kondrashov FA, Kondrashov AS.. 2010. Measurements of spontaneous rates of mutations in the recent past and in the near future. Philos Trans R Soc B. 365(1544):1169–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, et al. 2012. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488(7412):471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A, et al. 2017. Mitochondrial mutation rate, spectrum and heteroplasmy in Caenorhabditis elegans spontaneous mutation accumulation lines of differing population size. Mol Biol Evol. 34(6):1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A, et al. 2018. Mutational and transcriptional landscape of spontaneous gene duplications and deletions in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 115(28):7386–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, et al. 2016. Fitness effects of spontaneous mutations in picoeukaryotic marine green algae. G3 (Bethesda) 6(7):2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, Eyre-Walker A, Sanchez-Ferandin S, Piganeau G.. 2017. Spontaneous mutation rate in the smallest photosynthetic eukaryotes. Mol Biol Evol. 34(7):1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraytsberg Y, et al. 2004. Recombination of human mitochondrial DNA. Science 304(5673):981. [DOI] [PubMed] [Google Scholar]
- Kucukyildirim S, et al. 2016. The rate and spectrum of spontaneous mutations in Mycobacterium smegmatis, a bacterium naturally devoid of the postreplicative mismatch repair pathway. G3 6(7):2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvist L, Martens J, Nazarenko AA, Orell M.. 2003. Paternal leakage of mitochondrial DNA in the great tit (Parus major). Mol Biol Evol. 20(2):243–247. [DOI] [PubMed] [Google Scholar]
- Ladoukakis ED, Eyre-Walker A.. 2004. Evolutionary genetics: direct evidence of recombination in human mitochondrial DNA. Heredity 93(4):321.. [DOI] [PubMed] [Google Scholar]
- Lande R. 1994. The risk of population extinction from new deleterious mutations. Evolution 48(5):1460–1469. [DOI] [PubMed] [Google Scholar]
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL.. 2007. Genetic properties influencing the evolvability of gene expression. Science 317(5834):118–121. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H.. 1997. Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 44(4):383–397. [DOI] [PubMed] [Google Scholar]
- Lee H, Popodi E, Tang H, Foster PL.. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A. 109(41):E2774–E2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, et al. 2010. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am J Hum Genet. 87(2):237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W-H. 1980. Rate of gene silencing at duplicate loci: a theoretical study and interpretation of data from tetraploid fishes. Genetics 95(1):237–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Andersson DI.. 2008. Whole-genome mutational biases in bacteria. Proc Natl Acad Sci U S A. 105(46):17878–17883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link V, Aguilar-Gómez D, Ramírez-Suástegui C, Hurst LD, Cortez D.. 2017. Male mutation bias is the main force shaping chromosomal substitution rates in monotreme mammals. Genome Biol Evol. 9(9):2198–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski KJ, et al. 2011. High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr Biol. 21(4):306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry JR. 1996. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 13(5):660–665. [DOI] [PubMed] [Google Scholar]
- Loehlin DW, Carroll SB.. 2016. Expression of tandem gene duplicates is often greater than twofold. Proc Natl Acad Sci U S A. 113(21):5988–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, et al. 2015. Background mutational features of the radiation-resistant bacterium Deinococcus radiodurans. Mol Biol Evol. 32(9):2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, et al. 2016. Low base-substitution mutation rate in the germline genome of the ciliate Tetrahymena thermophila. Genome Biol Evol. 8:3629–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale DM, Brears T, Hodge TP, Melville SE, Rottman WH.. 1988. The plant mitochondrial genome: homologous recombination as a mechanism for generating heterogeneity. Philos Trans R Soc Lond B. 319(1193):149–163. [Google Scholar]
- Lynch M. 2010a. Evolution of the mutation rate. Trends Genet. 26(8):345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010b. Rate, molecular spectrum and consequences of human mutation. Proc Natl Acad Sci U S A. 107(3):961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2016. Mutation and human exceptionalism: our future genetic load. Genetics 202(3):869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery J, Bürger R.. 1995a. Mutational accumulation and the extinction of small populations. Am Nat. 146(4):489–518. [Google Scholar]
- Lynch M, Conery J, Bürger R.. 1995b. Mutational meltdowns in sexual populations. Evolution 49(6):1067–1080. [DOI] [PubMed] [Google Scholar]
- Lynch M, et al. 1999. Perspective: spontaneous deleterious mutation. Evolution 53(3):645–663. [DOI] [PubMed] [Google Scholar]
- Lynch M, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 105(27):9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, et al. 2016. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. 17(11):704–714. [DOI] [PubMed] [Google Scholar]
- Lynch M, Gabriel W.. 1990. Mutation load and the survival of small populations. Evolution 44(7):1725–1737. [DOI] [PubMed] [Google Scholar]
- Lynch M, Hill WG.. 1986. Phenotypic evolution by neutral mutation. Evolution 40(5):915–935. [DOI] [PubMed] [Google Scholar]
- Lynch M, Walsh B.. 1998. Genetics and analysis of quantitative traits. Sunderland (MA: ): Sinauer Associates. [Google Scholar]
- MacAlpine DM, Perlman PS, Butow RA.. 1998. The high mobility group protein Abf2p influences the level of yeast mitochondrial DNA recombination intermediates in vivo. Proc Natl Acad Sci U S A. 95(12):6739–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Seshasayee AS, Luscombe NM.. 2012. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 485(7396):95–98. [DOI] [PubMed] [Google Scholar]
- McCauley DE, Bailey MF, Sherman NA, Darnell MZ.. 2005. Evidence for paternal transmission and heteroplasmy in the mitochondrial genome of Silene vulgaris, a gynodioecious plant. Heredity 95(1):50–58. [DOI] [PubMed] [Google Scholar]
- McGaugh SE, et al. 2012. Recombination modulates how selection affects linked sites in Drosophila. PLoS Biol. 10(11):e1001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Hartl DL.. 2002. A single mode of canalization. Trends Ecol Evol. 17(10):468–473. [Google Scholar]
- Mira A, Ochman H, Moran NA.. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17(10):589–596. [DOI] [PubMed] [Google Scholar]
- Miyata T, Hayashida H, Kuma K, Mitsuyasu K, Yasunaga T.. 1987. Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harb Symp Quant Biol. 52:863–867. [DOI] [PubMed] [Google Scholar]
- Molnar RI, Bartelmes G, Dinkelacker I, Witte H, Sommer RJ.. 2011. Mutation rates and intraspecific divergence of the mitochondrial genome of Pristionchus pacificus. Mol Biol Evol. 28(8):2317–2326. [DOI] [PubMed] [Google Scholar]
- Montooth KL, Rand DM.. 2008. The spectrum of mitochondrial mutation differs across species. PLoS Biol. 6(8):e213.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T. 1964. The genetic structure of natural populations of Drosophila melanogaster. I. Spontaneous mutation rate of polygenes controlling viability. Genetics 50:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T, Chigusa SI, Mettler LE, Crow JF.. 1972. Mutation rate and dominance of genes affecting viability in Drosophila melanogaster. I. Genetics 72:333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1928. The measurement of gene mutation rate in Drosophila, its high variability, and its dependence on temperature. Genetics 13:279–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1950. Our load of mutations. Am J Hum Genet. 2(2):111–176. [PMC free article] [PubMed] [Google Scholar]
- Nakabachi A, et al. 2006. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 314(5797):267.. [DOI] [PubMed] [Google Scholar]
- Neale DB, Marshall KA, Sederoff RR.. 1989. Chloroplast and mitochondrial DNA are paternally inherited in Sequoia sempervirens D. Don Endl. Proc Natl Acad Sci U S A. 86(23):9347–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman M, Hehman G, Miller JT, Logsdon JM Jr, Taylor DR.. 2010. Accelerated mutation accumulation in asexual lineages of a freshwater snail. Mol Biol Evol. 27(4):954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman M, Taylor DR.. 2009. The causes of mutation accumulation in mitochondrial genomes. Proc Biol Sci. 276(1660):1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Colegrave N, Keightley PD.. 2012. Estimate of the spontaneous mutation rate in Chlamydomonas reinhardtii. Genetics 192(4):1447–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD.. 2015. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res. 25(11):1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson AI, et al. 2005. Bacterial genome size reduction by experimental evolution. Proc Natl Acad Sci U S A. 102(34):12112–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant KT, et al. 2010. The baker’s yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genet. 6(9):e1001109.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi O. 1977a. Spontaneous and ethyl methanesuflate-induced mutations controlling viability in Drosophila melanogaster. I. Recessive lethal mutations. Genetics 87:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi O. 1977b. Spontaneous and ethyl methanesuflate-induced mutations controlling viability in Drosophila melanogaster. II. Homozygous effect of polygenic mutations. Genetics 87:529–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi O. 1977c. Spontaneous and ethyl methanesuflate-induced mutations controlling viability in Drosophila melanogaster. III. Heterozygous effect of polygenic mutations. Genetics 87:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. New York: Springer. [Google Scholar]
- Ohta T. 1992. The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst. 23(1):263–286. [Google Scholar]
- Okoniewski MJ, Miller CJ.. 2006. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinformatics 7:276.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rawe J, et al. 2013. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 5(3):28.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, et al. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327(5961):92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP, Michalakis Y.. 1998. The evolution of recombination in changing environments. Trends Ecol Evol. 13(4):145–151. [DOI] [PubMed] [Google Scholar]
- Pamilo P, Nei M, Li W-H.. 1987. Accumulation of mutations in sexual and asexual populations. Genet Res. 49(2):135–146. [DOI] [PubMed] [Google Scholar]
- Partridge L, Barton NH.. 1993. Optimality, mutation and the evolution of aging. Nature 362(6418):305–311. [DOI] [PubMed] [Google Scholar]
- Passamonti M, Boore JL, Scali V.. 2003. Molecular evolution and recombination in gender-associated mitochondrial DNAs of the Manila clam Tapes philippinarum. Genetics 164(2):603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck JR, Barreau G, Heath SC.. 1997. Imperfect genes, Fisherian mutation and the evolution of sex. Genetics 145(4):1171–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot VS, Richerd S, Valero M.. 1991. Transition from haploidy to diploidy. Nature 351(6324):315–317. [DOI] [PubMed] [Google Scholar]
- Pfeifer SP. 2017. Direct estimate of the spontaneous germ line mutation rate in African green monkeys. Evolution 71(12):2858–2870. [DOI] [PubMed] [Google Scholar]
- Piganeau G, Gardner M, Eyre-Walker A.. 2004. A broad survey of recombination in animal mitochondria. Mol Biol Evol. 21(12):2319–2325. [DOI] [PubMed] [Google Scholar]
- Qian W, Liao B-Y, Chang AY-F, Zhang J.. 2010. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 26(10):425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM. 2001. The units of selection on mitochondrial DNA. Annu Rev Ecol Syst. 32(1):415–448. [Google Scholar]
- Raynes Y, Gazzara MR, Sniegowski PD.. 2011. Mutator dynamics in sexual and asexual experimental populations of yeast. BMC Evol Biol. 11(158):158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle C, Colin M, Matagne RF.. 1995. Genetic mapping of mitochondrial markers by recombinational analysis in Chlamydomonas reinhardtii. Mol Gen Genet. 249(2):185–190. [DOI] [PubMed] [Google Scholar]
- Rifkin SA, Houle D, Kim J, White KP.. 2005. A mutation accumulation assay reveals a broad capacity for rapid evolution of gene expression. Nature 438(7065):220–223. [DOI] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L.. 2009. Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet. 5(3):e1000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Shao L, Thornton KR.. 2017. Tandem duplications lead to novel expression patterns through exon shuffling in Drosophila yakuba. PLoS Genet. 13(5):e1006795.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y.. 2012. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet. 13(7):505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford RA, Cole JR, Tiedje JM.. 2002. Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl Environ Microbiol. 68(2):893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxer G, et al. 2012. Whole genome sequencing of mutation accumulation lines reveals a low mutation rate in the social amoeba Dictyostelium discoideum. PLoS One 7(10):e46759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, et al. 2011. Transgenerational epigenetic instability is a source of novel methylation variants. Science 334(6054):369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen DJ. 2005. Deleterious mutation in related species of the plant genus Amsinckia with contrasting mating systems. Evolution 59(11):2370–2377. [PubMed] [Google Scholar]
- Schrider DR, Houle D, Lynch M, Hahn MW.. 2013. Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics 194(4):937–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz ST, Lynch M, Willis JH.. 1999. Spontaneous deleterious mutation in Arabidopsis. Proc Natl Acad Sci U S A. 96(20):11393–11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serero A, Jubin C, Loeillet S, Legoix-Né P, Nicolas AG.. 2014. Mutational landscape of yeast mutator strains. Proc Natl Acad Sci U S A. 111(5):1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MD, et al. 2004. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 428(6984):717–723. [DOI] [PubMed] [Google Scholar]
- Sharp NP, Agrawal AF.. 2016. Low genetic quality alters key dimension of the mutational spectrum. PLoS Biol. 14(3):e1002419.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RG, Byers DL, Darmo E.. 2000. Spontaneous mutational effects on reproductive traits of Arabidopsis thaliana. Genetics 155(1):369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewaramani S, et al. 2017. Anaerobically grown Escherichia coli has an enhanced mutation rate and distinct mutational spectra. PLoS Genet. 13(1):e1006570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski DOF, Gallagher C, Beynon CM.. 1994. Sex-limited mitochondrial DNA transmission in the marine mussel Mytilus edulis. Genetics 138:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. 2009. Epigenetic inheritance and the missing heritability problem. Genetics 182(3):845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeds L, Qvarnström A, Ellegren H.. 2016. Direct estimate of the rate of germline mutation in a bird. Genome Res. 26(9):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TCA, Arndt PF, Eyre-Walker A.. 2018. Large scale variation in the rate of germ-line de novo mutation, base composition, divergence and diversity in humans. PLoS Genet. 14(3):e1007254.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Lenski RE.. 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387(6634):703–705. [DOI] [PubMed] [Google Scholar]
- Städler T, Delph LF.. 2002. Ancient mitochondrial haplotypes and evidence for intragenic recombination in a gynodioecious plant. Proc Natl Acad Sci U S A. 99:11730–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueoka N. 1962. On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci U S A. 48:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueoka N. 1988. Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci U S A. 85(8):2653–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueoka N. 1995. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J Mol Evol. 40(3):318–325. [DOI] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, et al. 2012. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci U S A. 109(45):18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Tucker AE, et al. 2012. Extraordinary genome stability in the ciliate Paramecium tetraurelia. Proc Natl Acad Sci U S A. 109(47):19339–19344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, et al. 2015. Asymmetric context-dependent mutation patterns revealed through mutation-accumulation experiments. Mol Biol Evol. 32(7):1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei F, et al. 1997. Role of mutator alleles in adaptive evolution. Nature 387(6634):700–702. [DOI] [PubMed] [Google Scholar]
- Tatsumoto S, et al. 2017. Direct estimation of de novo mutation rates in a chimpanzee parent-offspring trio by ultra-deep whole genome sequencing. Sci Rep. 7:13561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JW. 1986. Topical review: fungal evolutionary biology and mitochondrial DNA. Exp Mycol. 10(4):259–269. [Google Scholar]
- Uchimura A, et al. 2015. Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Res. 25(8):1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Graaf A, et al. 2015. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc Natl Acad Sci U S A. 112(21):6676–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilieva LL, Hook AM, Lynch M.. 2000. The fitness effects of spontaneous mutations in Caenorhabditis elegans. Evolution 151:119–129. [DOI] [PubMed] [Google Scholar]
- Venn O, et al. 2014. Strong male bias drives germline mutation in chimpanzees. Science 344(6189):1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. 1942. Canalization of development and the inheritance of acquired characters. Nature 150(3811):563–565. [Google Scholar]
- Wallace DC. 2015. Mitochondrial DNA variation in human radiation and disease. Cell 163(1):33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Chalkia D.. 2013. Mitochondrial DNA genetics and the heteroplasmy conundrum I evolution and disease. Cold Spring Harb Perspect Biol. 5(11):a021220.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JB. 1995. How often do duplicated genes evolve new functions? Genetics 110:345–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M.. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller AM, Rödelsperger C, Eberhardt G, Molnar RI, Sommer RJ.. 2014. Opposing forces of A/T-biased mutations and G/C-biased gene conversions shape the genome of the nematode Pristionchus pacificus. Genetics 196(4):1145–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DJ, Wolff JB, Pierson M, Gemmell NJ.. 2008. Revealing the hidden complexities of mtDNA inheritance. Mol Ecol. 17(23):4925–4942. [DOI] [PubMed] [Google Scholar]
- Wilson Sayres MA, Makova KD.. 2011. Genome analyses substantiate male mutation bias in many species. Biosessays 33(12):938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Williams BL, Selegue JE, Carroll SB.. 2003. Drosophila pigmentation evolution: divergent genotypes underlying convergent phenotypes. Proc Natl Acad Sci U S A. 100(4):1808–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Li W-H, Sharp PM.. 1987. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast and nuclear DNAs. Proc Natl Acad Sci U S A. 84(24):9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Sharp PM, Li WH.. 1989. Mutation rates differ among regions of the mammalian genome. Nature 337(6204):283–285. [DOI] [PubMed] [Google Scholar]
- Wong WSW, et al. 2016. New observations on maternal age effect on germline de novo mutations. Nat Commun. 7:10486.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA, et al. 2003. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 20(9):1377–1419. [DOI] [PubMed] [Google Scholar]
- Wu C-I. 1991. DNA strand asymmetry. Nature 352(6331):114.. [DOI] [PubMed] [Google Scholar]
- Wu C-I, Maeda N.. 1987. Inequality of mutation rates of the two strands of DNA. Nature 327(6118):169–170. [DOI] [PubMed] [Google Scholar]
- Xu S, et al. 2012. High mutation rates in the mitochondrial genomes of Daphnia pulex. Mol Biol Evol. 29(2):763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yampolsky LY, Stoltzfus A.. 2001. Bias in the introduction of variation as an orienting factor in evolution. Evol Dev. 3(2):73–83. [DOI] [PubMed] [Google Scholar]
- Yang S, et al. 2015. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523(7561):463–467. [DOI] [PubMed] [Google Scholar]
- Zalts H, Yanai I.. 2017. Developmental constraints shape the evolution of the nematode mid-developmental transition. Nat Ecol Evol. 1:0113. [DOI] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA.. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 111(22):E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E, Ball AO, Saavedra C, Freeman KR.. 1994. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci U S A. 91(16):7463–7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.