

Abstract
The NAD+-dependent protein lysine deacylases of the Sirtuin family regulate various physiological functions, from energy metabolism to stress responses. The human Sirtuin isoforms, SIRT1-7, are considered attractive therapeutic targets for aging-related diseases, such as type 2 diabetes, inflammatory diseases and neurodegenerative disorders. We review the status of Sirtuin-targeted drug discovery and development. Potent and selective pharmacological Sirt1 activators and inhibitors are available, and initial clinical trials have been carried out. Several promising inhibitors and activators have also been described for other isoforms. Progress in understanding the mechanisms of Sirtuin modulation by such compounds provides a rational basis for further drug development.
Keywords: Deacetylase, deacylase, inhibition, activation, drug discovery and development
The Sirtuin family of NAD+-dependent protein deacylases
Physiological and pharmacological relevance of Sirtuins and their modulators
The Sirtuins are named after the prototypical silent information regulator 2 gene (SIR2) that is essential for the formation of silent heterochromatin in budding yeast (Rine and Herskowitz, 1987). SIR2 was considered an unremarkable gene until 1995 when it was identified as a key regulator of yeast replicative lifespan (Kennedy et al., 1995). Inserting an extra copy of the SIR2 gene (but not SIR3 or SIR4) stabilized the yeast genome and extended lifespan by 30% (Kaeberlein et al., 1999; Sinclair and Guarente, 1997). This importance of SIR2 in aging was recently corroborated by an unbiased genome-wide associated study (GWAS) that identified the SIR2 locus as the most significant regulator of replicative lifespan (Stumpferl et al., 2012). Yeast Sir2 is believed to have evolved to boost energy production and cellular defenses in response to conditions of adversity such as DNA damage and a lack of nutrients (Mills et al., 1999; Sinclair, 2002).
Sir2 homologs in other species have since been identified by virtue of their homology to the conserved central catalytic core (Michan and Sinclair, 2007). Mammals have seven sirtuins, SIRT1-7. SIRT1, 6, and 7 are primarily nuclear, SIRT3, 4, and 5 are mitochondrial, and SIRT2 is primarily cytosolic (Houtkooper et al., 2012). In 2000, Sir2 was reported to have histone deacetylase activity that required nicotinamide adenine dinucleotide (NAD+) as a cosubstrate (Imai et al., 2000). There is a growing list of chemical reactions that the mammalian sirtuins catalyze, including demalonylation, desuccinylation, decrotonylation, depropyonylation, delipoamidation, other long-chain fatty acid deacylations, which are generally referred to as “deacylation reactions”, and mono-ADP-ribosylation, which is discussed as being a main or just a side activity of Sirtuins (Du et al., 2009; Feldman et al., 2012; Mathias et al., 2014; Roessler et al., 2014).
Yeast sirtuins (Sir2, Hst1-4) regulate gene expression by directly deacetylating histones H3, H4 and H1 (Toiber et al., 2011). Similar to the yeast sirtuins, the mammalian sirtuins SIRT1, SIRT6 and SIRT7 also deacetylate histones but they also serve many additional roles, including the control of energy metabolism, cell survival, DNA repair, tissue regeneration, inflammation, neuronal signaling, and even circadian rhythms (Bonkowski and Sinclair, 2016). For example, SIRT1, a predominately nuclear protein, deacetylates histones H3, H4 and H1 (Toiber et al., 2011) but also modifies more than 50 non-histone targets, including transcription factors (e.g., p53, NF-κB, p65 and PGC-1α) and DNA repair proteins (e.g., Ku70, PARP1) (Bonkowski and Sinclair, 2016). Other mammalian sirtuins localize to other subcellular compartments where they target non-histone proteins (Gertz and Steegborn, 2016; Haigis and Sinclair, 2010). For example, SIRT3 modifies numerous mitochondrial metabolic enzymes to up-regulate β-oxidation of fatty acids, the TCA cycle, and the urea cycle (Gertz and Steegborn, 2016; Yang et al., 2016). For a comprehensive review see (Bonkowski and Sinclair, 2016).
Sirtuins are regulated at the level of transcription, translation, protein stability, oxidation, and by protein-protein interactions, natural inhibitory molecules such as nicotinamide, microRNAs, localization within the cell and within organelles, but their NAD+-dependence is the main way they monitor an organism’s external and internal conditions (Cohen et al., 2004; Lin et al., 2000). In fact, upregulation of NAD+ biosynthesis dramatically extends yeast lifespan (Anderson et al., 2003).
Sirtuins are believed to be the major reason why calorie restriction (CR) and exercise improve health (Bordone et al., 2007; Guarente, 2005; Rogina and Helfand, 2004). The best evidence comes from lower organisms where individuals lacking sirtuins or NAD recovery via the salvage pathway fail to live longer when calorie restricted (Anderson et al., 2003; Lin et al., 2000; Moroz et al., 2014) and live longer when sirtuins are overexpressed (Banerjee et al., 2012; Rizki et al., 2011; Rogina and Helfand, 2004; Tissenbaum and Guarente, 2001), although the magnitudes of the effects were challenged. Upregulation of the NAD+ salvage pathway extends lifespan in yeast and flies via a pathway that overlaps with CR (Anderson et al., 2003; Balan et al., 2008; Moroz et al., 2014).
Mammalian SIRT1, SIRT3 and SIRT6 are induced by calorie restriction and exercise, and their genetic ablation prevents many of the health benefits and longevity provided by these interventions (Boily et al., 2008; Chen et al., 2005; Cohen et al., 2004; Hebert et al., 2013; Qiu et al., 2010; Someya et al., 2010). Conversely, over-expression of SIRT1 or SIRT6 in mice mimics many of the physiological effects of CR and exercise, and extends healthspan and lifespan (Kanfi et al., 2012; Satoh et al., 2010). It is further protective in many murine disease models, including cancer, type 2 diabetes, and cardiovascular disease (Banks et al., 2008; Bordone et al., 2007; Firestein et al., 2008; Hsu et al., 2008; Oberdoerffer et al., 2008; Pfluger et al., 2008; Qin et al., 2006; Ramadori et al., 2011; Satoh et al., 2010). The key functions of the mammalian sirtuins with regard to diet are inducing the mitochondrial biogenesis and oxidative metabolism during CR (Lagouge et al., 2006; Shi et al., 2005; Someya et al., 2010), as well as reducing reactive oxygen species and inflammation (Chalkiadaki and Guarente, 2012; Hirschey et al., 2011; Pfluger et al., 2008; Yoshizaki et al., 2009). These findings indicate that molecules that activate sirtuins in humans may not only provide broad health benefits with potent anti-inflammatory, cardio-protective, neuroprotective, and anti-tumor activities (Kugel et al., 2016). The SIRT1 activators SRT1720 and 2104 were indeed shown to extend mouse healthspan and lifespan (Mercken et al., 2014; Minor et al., 2011), and STACs are now in clinical trials for a variety of indications (see below). With regards to cancer and metabolic disorders, some studies indicate that inhibitors of SIRT1 and other Sirtuin isoforms might also be useful (Chalkiadaki and Guarente, 2015; Jeong and Haigis, 2015). Examples for Sirtuin activators and inhibitors and their potential therapeutic applications are shown in Figure 1. Sirtuin modulating compounds and their mechanisms as well as first clinical trials with STACs will be discussed in detail below, and comprehensive discussions of the (patho)physiological roles of Sirtuins and their potential therapeutic use can be found, e.g., in (Haigis and Sinclair, 2010; Chalkiadaki and Guarente, 2015; Gertz and Steegborn, 2016; Hubbard and Sinclair, 2014).
Figure 1. Potential therapeutic applications for Sirtuin activating compounds (STACs) and Sirtuin inhibiting compounds (STICs).
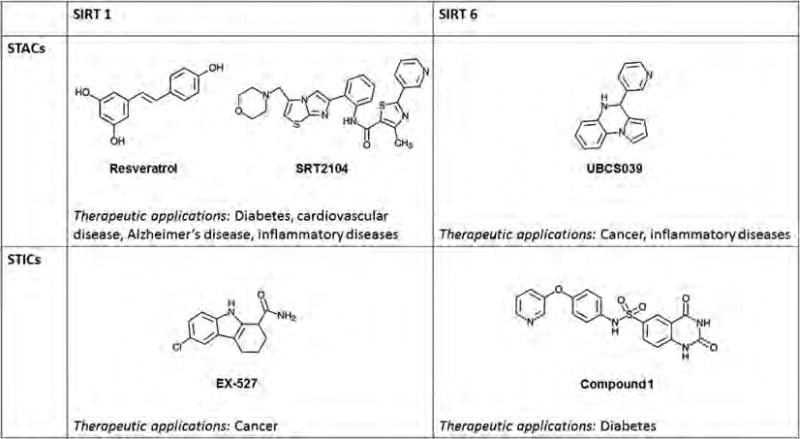
Chemical structures for examples of activators and inhibitors for Sirt1 and Sirt6 as isoforms with examples for prospective therapeutic applications.
In light of the profound health benefits conferred on mice when sirtuins are overexpressed, discovering “Sirtuin activating compounds” (STACs) has been a major goal of the field (Howitz et al., 2003; Hubbard and Sinclair, 2014). Drugs that target enzymes are typically inhibitors. In rarer cases, drugs that enhance the activity of their target enzymes have been developed. Examples include glucokinase (GK), alpha amylase, phosphoinositide-dependent protein kinase 1 (PDK1), AMPK-activated kinase (AMPK), and protein phosphatase 1 (PP1) (Kashani-Amin et al., 2013; Zorn and Wells, 2010). The reason for their rarity might be the fact that an allosteric binding site is required, distinct from the substrate sites often exploited by inhibitors, and that it is generally easier to disturb catalytically needed conformational changes rather than to improve these evolutionary optimized systems. Although activators are more challenging to develop, they have distinct advantages. They generally do not have to be as potent as inhibitors to induce cellular and physiological effects, and they typically have greater target specificity and illicit fewer side effects (Zorn and Wells, 2010). Although finding potent activators began without knowledge of sirtuin structures (Howitz et al., 2003), in recent years, drug development has been aided by a detailed molecular understanding of the sirtuin structure and catalysis (Dai et al., 2015; Gertz et al., 2012; You et al., 2017) and promises to yield helpful chemical tools and attractive new drugs for therapy. In addition to direct modulators of sirtuins, there has been increasing interest in small molecules that raise NAD+ levels, to combat the decline in NAD+ during aging (Bonkowski and Sinclair, 2016).
Sirtuin architecture and catalysis
Sirtuins share an evolutionarily conserved, ~270 residue catalytic core (Schutkowski et al., 2014). N- and C-terminal extensions of differing lengths and sequences contribute to isoform-specific localization and regulation. SIRT1 features the largest extensions among mammalian isoforms, including an N-terminal STAC binding domain (SBD) (Dai et al., 2015; Hubbard et al., 2013) and intrinsically disordered regions (Lakshminarasimhan et al., 2013a; Pan et al., 2012). The mitochondrial Sirtuins SIRT3, 4, 5 have only short extensions, in particular N-terminal mitochondrial localization sequences (MLS) (Gertz and Steegborn, 2016). For SIRT3’s MLS, an autoinhibitory function was reported (Schwer et al., 2002), and the N- and C-termini of SIRT6 and SIRT7 were reported to contribute to DNA and chromatin binding (Tennen et al., 2010; Tong et al., 2016).
The generic Sirtuin catalytic core comprises an NAD+-binding Rossmann-fold subdomain and a small Zn2+-binding module, and the active site is located in the cleft between these subdomains (Figure 2a) (Sanders et al., 2010; Schutkowski et al., 2014). Human SIRT1-7 show only weak, isoform-specific substrate sequence preferences due to subtle differences in their polypeptide binding grooves (Rauh et al., 2013). The active site pockets accommodating the substrate acyls show larger variations among isoforms, however, resulting in isoform-specific selectivities for differing protein Lys acylations. While SIRT1-SIRT3 show robust deacetylation activities, a SIRT5-specific Arg at the bottom of the acyl binding channel mediates a preference for the dicarboxylate modifications malonylation, succinylation, and glutarylation (Du et al., 2011; Feldman et al., 2013; Roessler et al., 2014; Tan et al., 2014). These modifications belong to a growing list of more recently identified physiological protein Lys acylations, several of which are efficiently hydrolyzed by a Sirtuin (Lin et al., 2012; Jiang et al., 2013; Tan et al., 2014; Anderson et al., 2017). SIRT6 prefers long chain fatty acylations over acetylations, at least in vitro, due to a hydrophobic, wider acyl binding region than in other isoforms (Pan et al., 2011; Jiang et al., 2013). Interestingly, free fatty acids can also bind and activate SIRT6’s deacetylation activity, possibly explaining the isoform’s robust in vivo deacetylation activity (Feldman et al., 2013). Recent insights in SIRT4 structure and function also revealed novel, preferred acyl substrates, in particular hydroxymethylglutarylation and structurally related acylations, and an isoform-specific acyl binding site (Pannek et al., 2017; Anderson et al., 2017). The isoform-specific acyl site features are exploited by pharmacological SIRT6 activators, peptide-based Sirt5 inhibitors, and likely by the SIRT4 inhibitor lipoic acid, and they appear attractive for further development of isoform-specific Sirtuin modulators (You et al., 2017; Pannek et al., 2017; Rajabi et al., 2017) (see below).
Figure 2. Sirtuin structure, catalytic mechanism, and activation.
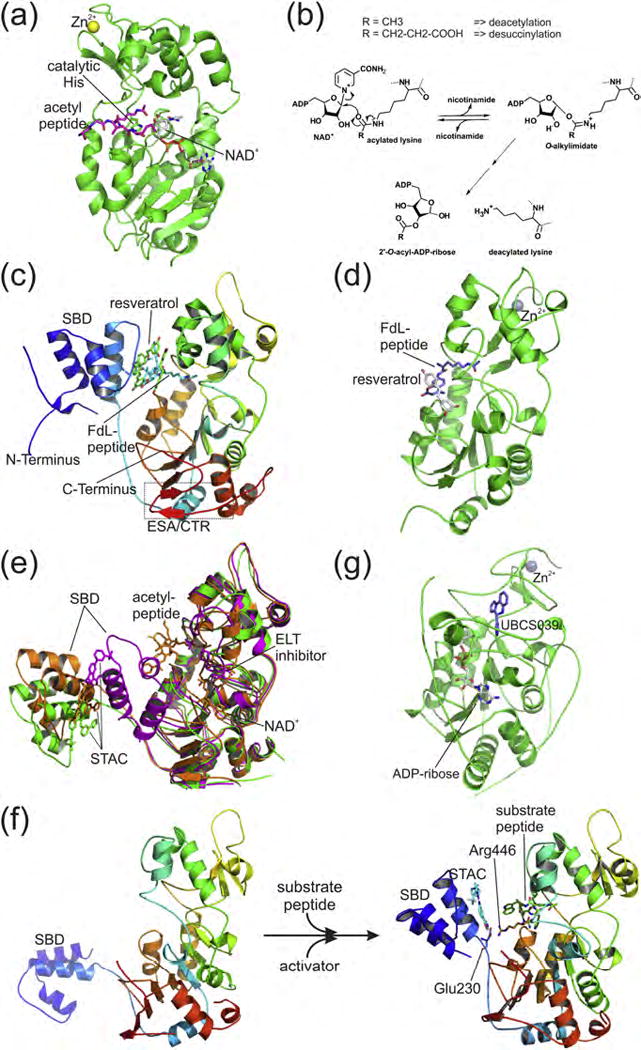
(a) Sirtuin catalytic core structure. The crystal structure of human SIRT3 in complex with substrate peptide (magenta) and non-hydrolysable NAD+ analog (gray; PDB ID 4FVT) illustrates the substrate binding sites. (b) Mechanism of Sirtuin-catalyzed NAD+-dependent protein Lys deacylation. (c) Crystal structure of human SIRT1 in complex with FdL substrate peptide (cyan) and three molecules of the STAC resveratrol (green; PDB ID 5BTR). The SIRT1-specific N-terminal STAC binding domain (SBD) and C-terminal regulatory (CTR) segment are indicated. (d) Crystal structure of human SIRT5 in complex with FdL substrate peptide (blue) and the STAC resveratrol (gray; PDB ID 4HDA). (e) Crystal structures of SIRT1 complexes with three different synthetic STACS (green [PDB ID 4ZZH], magenta [PDB ID 4ZZI], orange [PDB ID 4ZZJ]), illustrating that the linker to the SBD allows varying relative orientations of the two domains. Acetyl-peptide and non-hydrolysable NAD+ analog (orange) respective a competitive ELT inhibitor (magenta) are also bound and indicate substrate binding sites. (f) Model for transition of the apo SIRT1 open conformation (left) to the “closed” conformation of SIRT1 (right), with the STAC-bound SBD placed on top of the substrate-bound catalytic core. The important electrostatic interaction between Arg446 and Glu230 is indicated. (g) Crystal structure of human SIRT6 in complex with the NAD+ fragment ADP-ribose (gray) and the STAC UBCS039 (blue; PDB ID 5MF6).
Despite their different acyl selectivities, the homologous catalytic cores of the seven Sirtuin isoforms catalyze via the same deacylation mechanism (Schutkowski et al., 2014). In a first step, the nicotinamide group of NAD+ is replaced by the substrate acyl oxygen to form a 1′-O-alkylamidate intermediate (Figure 2b). Rearrangement to a bicyclic 1′–2′-acetal and subsequent hydrolysis yield the deacylated protein and 2′-O-acyl-ADP-ribose. This mechanism deviates strongly from the Zn2+-dependent water activation employed by all other deacylase families, and the unique NAD+ co-substrate renders Sirtuins metabolic sensors (Sauve et al., 2006). Binding of NAD+ and acylated protein substrate induce a relative reorientation of the Zn2+-binding and Rossmann-fold subdomains, and a so-called “cofactor binding loop” assumes a more rigid conformation, followed by a closed state upon acyl transfer, to support catalysis (Moniot et al., 2013; Sanders et al., 2010; Schutkowski et al., 2014).
Pharmacological Sirtuin modulation
As Sirtuins are attractive therapeutic targets, considerable effort has been directed towards developing specific Sirtuin activators and inhibitors, as tools for studying Sirtuin function and potentially as treatments for age-related conditions. Advances in Sirtuin biochemistry, assays and crystal structures of Sirtuin/modulator complexes revealed an intricate interplay of compounds with the enzymes’ structure and mechanism, and these insights are now beginning to support the development of pharmacological Sirtuin modulators.
SIRT1 activation
Sirtuin activating compounds (STACs)
The therapeutic potential of increased mammalian SIRT1 activity, demonstrated in animal models and initial clinical trials (see below), makes this enzyme an attractive drug target (Bonkowski and Sinclair, 2016; Hubbard and Sinclair, 2014). Seminal efforts by David Sinclair’s lab in 2003 led to the discovery of the first generation of SIRT1 activators in a high throughput screen using a so-called “Fluor de Lys” (FdL) peptide substrate, Ac-Arg-His-Lys-(acetyl-Lys)-AMC (Howitz et al., 2003). Several classes of plant polyphenols, such as butein, piceatannol, isoliquiritigenin, were shown to activate recombinant SIRT1 and to extend the lifespan of Saccharomyces cerevisiae (Table 1). The most effective of these STACs, activating SIRT1 by more than 10 fold, is resveratrol (3, 5, 4′-trihydroxystilbene; Table 1), a natural product existing in grapes and red wines. A variety of synthetic resveratrol derivatives with modifications at the B ring 4′ position resulted in lower toxicity toward human cells and higher potency with respect to SIRT1 activation and lifespan extension in budding yeast, showing that it is possible to improve upon naturally occurring STACs (Yang et al., 2007).
Table 1.
Selected Sirtuin activators and inhibitors.
| Compound name | Compound structure | Sirtuin effect(s), other information | Reference(s) |
|---|---|---|---|
| Sirtuin activators | |||
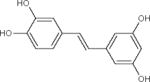
| Piceatannol |
|
Natural STAC. Activates SIRT1, but also effects on SIRT3 and SIRT5 as well as other, unrelated targets. | (Gertz et al., 2012; Howitz et al., 2003) |
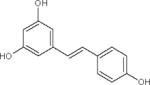
| Resveratrol |
|
Natural STAC. Activates SIRT1, but also effects on SIRT3 and SIRT5 as well as, other, unrelated targets. | (Gertz et al., 2012; Howitz et al., 2003) |
| SRT1720 |
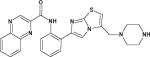
|
Synthetic SIRT1 activator, but has off-target effects. | (Milne et al., 2007) (Dai et al., 2010) |
| SRT2104 |
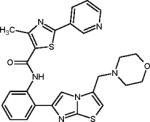
|
Highly specific synthetic SIRT1 activator | (Hoffmann et al., 2013) (Krueger et al., 2015) |
| 1,4-DHP derivative |
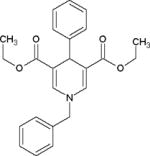
|
SIRT1 activator in vitro and in vivo, activating effect on SIRT2 and SIRT3 in the FdL assay | (Mai et al., 2009; Valente et al., 2016) |
| UBCS039 |
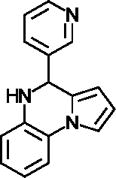
|
Synthetic deacetylation activator for SIRT6; also activates SIRT5 desuccinylase activity | (You et al., 2017) |
| Sirtuin inhibitors | |||
| Cambinol |
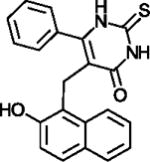
|
One of the first, weak Sirtuin inhibitors | (Grozinger et al., 2001) |
| Ex-527 |
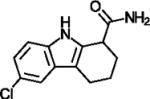
|
Potent SIRT1 inhibitor selective over SIRT2 and SIRT3 | (Gertz et al., 2013; Napper et al., 2005) |
| AGK2 |
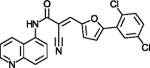
|
Widely used SIRT2 inhibitor | (Outeiro et al., 2007) |
| 3′-(3-fluoro-phenethyloxy)-2-anilinobenzamide |
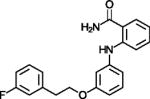
|
Potent SIRT2 inhibitor with high selectivity over SIRT1 | (Suzuki et al., 2012) |
| SirReal2 |
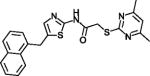
|
Potent SIRT2 inhibitor with high selectivity over SIRT1,3,4,5,6 | (Rumpf et al., 2015) |
| Compound 15e |
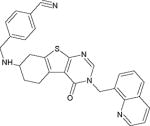
|
Potent SIRT2 inhibitor with high selectivity over SIRT1,3 | (Sundriyal et al., 2017) |
| UBCS0137 (Compound 39) |
|
Potent SIRT2 inhibitor selective over SIRT1,3,5 | (Moniot et al., 2017) |
| ELT-11c |
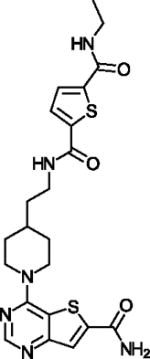
|
Very potent pan SIRT1,2,3 inhibitor | (Disch et al., 2013) |
| Compound 28e |
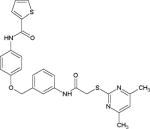
|
Highly potent SIRT2 inhibitor selective over SIRT1 and SIRT3 | (Yang et al., 2017) |
| Compound 8 |
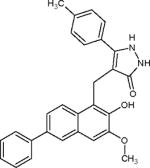
|
Potent SIRT3 inhibitor with selectivity over SIRT1 and SIRT2 | (Mahajan et al., 2014) |
The implication of Sirtuins in aging and disease and the discovery and shortcomings of natural STACs, such as limited bioavailability and specificity (Pirola and Frojdo, 2008), prompted pharmaceutical companies to screen for synthetic activators. These campaigns were supported by evolution of Sirtuin assays to more robust and flexible formats, e.g. to sensitive fluorescence polarization and robust mass spectrometry setups (Milne et al., 2007; Schutkowski et al., 2014). Milne and colleagues reported synthetic STACs with an imidazo[1,2-b]thiazole core, such as SRT1720 (Table 1), that are structurally unrelated to resveratrol and activate SIRT1 much more potently (Milne et al., 2007). These activators have been frequently used in research studies and also generated preclinical and clinical proof-of-concept efficacy data, with the most advanced compound in this series, SRT2104 (Table 1), appearing to hold promise for the treatment of Psoriasis (Krueger et al., 2015) (see below). STACs of a variety of chemotypes, including oxazolo[4,5-b]pyridine, thiazolopyridine, benzimidazole, imidazo[4,5-c]pyridine, and bridged urea have been developed to improve the activation potency, physicochemical properties and developability, and they appear to hold even greater therapeutic potential (Bemis et al., 2009; Dai et al., 2015; Dai et al., 2010; Hubbard et al., 2013; Vu et al., 2009). Another, unrelated class of 1,4-dihydropyridine (DHP) based compounds bearing a benzyl group at the N1 position (Table 1) was reported to activate SIRT1, SIRT2 and SIRT3 in FdL assays and SIRT1 also in coupled enzymatic assays (Mai et al., 2009; Valente et al., 2016).
Sirtuin/STAC complex structures
Structural insights in mammalian Sirtuins started with apo SIRT2 (Finnin et al., 2001; Moniot et al., 2013), and insights in other isoforms and Sirtuin/modulator complexes followed only slowly (Gertz and Steegborn, 2016; Sanders et al., 2010). In particular for SIRT1, the most studied isoform, structural biology of the enzyme and its modulators has been lagging behind the significant advances in SIRT1 biology and pharmacology. The first human SIRT1 structure comprised only the catalytic domain bound to NAD+ and an Ex-527-related inhibitor (see below), confirming the generic catalytic core structure (Zhao et al., 2013). Subsequently, a SIRT1 catalytic domain complexed with a peptide representing its ~20 residue “C-terminal regulatory” (CTR) region was characterized, rationalizing how it potentiates SIRT1 activity (Davenport et al., 2014; Kang et al., 2011; Lakshminarasimhan et al., 2013a; Pan et al., 2012). The CTR forms a β-hairpin and extends the β-sheet of the catalytic domain (Figure 2c). The most recent advance in SIRT1 structural biology revealed the mode of SIRT1 modulation by either the natural STAC resveratrol or a synthetic STAC of the bridged urea chemotype (Cao et al., 2015; Dai et al., 2015). Key to these studies was the use of SIRT1 protein constructs that include the N-terminal region abutting the catalytic domain, which had been suggested to form a folded domain (Hubbard et al., 2013).
Cao et al. reported a ternary complex of SIRT1/FdL peptide/resveratrol using a construct SIRT1(143–512) directly linked with SIRT1(641–665) and cysteine-to-serine mutations (SIRT1-143CS: C235S, C268S, C501S and C502S) (Cao et al., 2015). N-terminal to the catalytic domain, it features a three-helix bundle STAC binding domain (SBD), and C-terminally the CTR β-hairpin (Figure 2c) The catalytic domain forms few direct interactions with the SBD, except for the connecting polypeptide chain and a hydrogen bond/electrostatic interaction between Glu230 and Arg446. The SIRT1/FdL/resveratrol co-crystal structure revealed the binding of three resveratrol molecules to SIRT1, two of which form hydrogen bonds with both SBD and substrate peptide, thereby bridging allosteric SBD site and active site. The third resveratrol molecule interacts only with catalytic domain and substrate peptide and appears less likely to be crucial for SIRT1 activation (Cao et al., 2015), consistent with the observation that efficient SIRT1 activation against other substrates requires the SBD (Hubbard et al., 2013). However, in the SIRT1/FdL/resveratrol complex the substrate fluorophore, which occupies the channel normally accommodating residues +1 and +2 of regular substrates, forms pronounced hydrophobic interactions with all resveratrol molecules, likely rationalizing the influence of the substrate sequence on compound effects (Hubbard et al., 2013; Lakshminarasimhan et al., 2013b). Interestingly, a previous SIRT5/FdL/resveratrol structure had also revealed a direct substrate/activator interaction dominated by hydrophobic contacts via the fluorophore, and a position roughly related to the third resveratrol molecule in SIRT1/FdL/resveratrol, bound between loops from small and Rossmann-fold lobe of the catalytic core, respectively (Figure 2d) (Gertz et al., 2012). Similarly, in a SIRT3/FdL complex with the SIRT3-inhibitory resveratrol derivative piceatannol the compound pose resembles that of the second resveratrol molecule in the SIRT1/FdL/resveratrol complex (Cao et al., 2015; Gertz et al., 2012), suggesting some convergence in terms of the resveratrol binding mode among different sirtuin isoforms as well as divergence due to the lack of SBD in isoforms other than SIRT1.
Around the same time as the SIRT1/FdL/resveratrol complex, Dai et al. reported structures of SIRT1 complexed with a synthetic STAC of the bridged urea chemotype (Dai et al., 2015). In this study, a “mini-SIRT1” construct was employed, which encompasses SBD, catalytic domain, and the CTR peptide. The overall structure is similar to that of SIRT1-143CS, except for the orientation of the N-terminal SBD relative to the catalytic domain (Figure 2e). In the three mini-SIRT1/STAC-11 complexes, the N-terminal SBD is pointing further away from the catalytic domain. Comparison of the relative orientations in the three mini-SIRT1/STAC-11 structures indicates that they are likely influenced by crystal packing. Unlike resveratrol, STAC-11 binds to SIRT1 with a 1:1 stoichiometry, to a defined binding site in the SBD (Figure 2e). The binding site is a shallow hydrophobic surface depression with a single polar interaction partner, Asn226, and an off-center, deeper hydrophobic pocket accommodating the CF3 group of STAC-11. This binding mode is consistent with the structure-activity relationships observed across multiple STAC chemotypes, indicating the requirement of overall flatness of the core scaffold maintained by an intramolecular hydrogen bond (Dai et al., 2015; Dai et al., 2010; Hubbard et al., 2013). Mutagenesis supports the compound binding mode for full-length SIRT1 and for STACs of several other chemotypes. Structures and biochemical results suggest an activated “closed” conformation where the SBD packs on top of the active site/substrate complex (Figure 2f), allowing SBD-bound activator to interact with the substrate as observed in the FdL complexes of SIRT1 and SIRT5 (Figure 2c,d) (Cao et al., 2015; Dai et al., 2015; Gertz et al., 2012). A hydrogen-deuterium exchange study indeed showed a coupling between SBD site and active site (Hubbard et al., 2013). This coupling is impaired by a Glu230Lys mutation, which abolishes SIRT1 activation by resveratrol and a wide range of chemically diverse STACs (Hubbard et al., 2013). An electrostatic interaction between this Glu230 and Arg446 was proposed to stabilize the activated “closed” conformation, based on a modeling with the SBD rotated as a rigid body around the flexible linker, bringing Glu230 close to Arg446 and the bound STAC-11 close to active site and bound substrate (Figure 2f), and supported by an activation rescue of the double charge reversal mutant (Glu230Lys,Arg446Glu). A Glu230-Arg446 interaction was indeed observed in the SIRT1-143CS/FdL peptide/resveratrol complex, corroborating the importance of this “closed” conformation of SBD and catalytic domain. Interestingly, a related arrangement of catalytic domain and N-terminal extension (albeit with no significant sequence homology to SIRT1) was observed for yeast Sir2 in its complex with a fragment of its activator protein Sir4, which is positioned on top of the active site via binding to the N-terminal domain (Hsu et al., 2013). An experimental structure of the activated SIRT1 conformation with a natural substrate remains to be elucidated, however.
Small molecule activation with physiological substrates has more recently also been established and structurally characterized for SIRT6. SIRT6 deacetylation activity was first found to be activated by free fatty acids, but several hundred micromolar concentrations are required for this effect, which competition experiments indicated to involve the acyl binding channel (Feldman et al., 2013). More recently, first synthetic and more potent SIRT6 activators (IC50 ~38 μM for the strongest activator, UBCS039; Table 1) were described (You et al., 2017). They are based on a pyrrolo[1,2-a]quinoxaline scaffold, and crystal structures of SIRT6 complexes with ADP-ribose and activators or an inactive derivative revealed binding site and interaction details (Figure 2g). The compounds occupy with their pyrrolo[1,2-a]quinoxaline scaffold the rather hydrophobic cone at the exit region of the enzyme’s acyl binding channel. The hydrophobic, non-targeted interactions positions the bottom of the pyrrolo[1,2-a]quinoxaline toward solvent but allow variations in the plane orientation of several related compounds within the hydrophobic exit cone. Orientation details appear determined by dominant interaction contributions from substituents at N5 of the pyrrolo[1,2-a]quinoxaline (N-group) and, in particular, at C4 (C-group). The different plane tilts enable three slightly different derivatives to position their identical 3-pyridyl C-groups into a hydrophobic “C-group pocket” within the acyl channel. Comparisons of these compound complexes suggest that the C-group serves as a major anchoring moiety, while pyrrolo[1,2-a]quinoxaline and N-group contribute mostly weaker and less specific binding interactions (You et al., 2017). These analyses suggest that modifying in particular the N-group for improvements in solubility and potency will be required for wider pharmacological applications. Interestingly, comparing the SIRT6/activator complexes with SIRT6/substrate complexes further revealed that the compounds occupy an acyl channel region that accommodates the distal end of longer acyl substrates. Consistently, myristoyl peptide and compound bind competitively, illustrating that these compounds and their binding site enable to differentially modulate Sirt6’s deacetylation (allosteric activation) and demyristoylation (competitive inhibition) activities.
Mechanisms of Sirtuin activation
Enzymatic characterization of SIRT1 activation by resveratrol suggested a lowering of the Km for the acetylated peptide substrate (Howitz et al., 2003; Milne et al., 2007), consistent with the proposed active site closure upon activator binding. Initially, the observation that no activation is obtained for resveratrol or early generation synthetic STACs when the fluorophore of the FdL substrate peptide is removed (Borra et al., 2005; Kaeberlein et al., 2005) had elicited controversies on Sirtuin activation. An opposing model to direct SIRT1/activator binding suggested that STACs produced complexes with the substrate fluorophore in solution, leading the authors of that study to conclude that STAC effects on cells and animals may be indirect (Pacholec et al., 2010). However, extensive mechanistic and structural studies of the past five years have conclusively established that STACs directly interact with SIRT1 and other isoforms and that the FdL fluorophore actually mimics natural amino acids that occur on native Sirtuin substrates (Cao et al., 2015; Dai et al., 2015; Dai et al., 2010; Gertz et al., 2012; Hubbard et al., 2013; Lakshminarasimhan et al., 2013b). Mostly hydrophobic substrate amino acids apparently form contacts that are similar to those of the FdL fluorophore (Figure 2c,d), mediating the activated closed conformation and rationalizing the substrate-specific activation effect (Gertz et al., 2012; Hubbard et al., 2013; Lakshminarasimhan et al., 2013b). In the case of SIRT1, the additional STAC interaction with the SBD results in a composite binding site (Figure 2c,f) and enables the potent and pronounced activation of this isoform (Dai et al., 2015; Hubbard et al., 2013). Resveratrol-responsive sirtuin isoforms lacking the SBD, such as SIRT3 and SIRT5, have small loop/compound interactions instead (Gertz et al., 2012) and thus appear to rely more heavily on the substrate/STAC interaction, which is particularly strong with the FdL substrate, rendering these isoforms less amenable to this activation mechanism. Cumulatively, these data suggest that SIRT1-activating STACs bind to its SBD and bring the STAC-bound SBD close to active site and substrate, resulting in a composite binding site to enhance substrate binding and thereby to yield an activating effect. Which physiological ligand(s) or regulation mechanisms exploit the SBD and induce the activated SIRT1 conformation remains an open question.
1,4-dihydropyridine (DHP)-derived compounds were also described as SIRT1 activators (Table 1), in vitro as well as in cellular systems (Mai et al., 2009; Valente et al., 2016), but no mechanistic information is yet available for them. Some of the DHPs also increased SIRT2 and SIRT3 activity in the FdL assay (Mai et al., 2009). Despite a lack of the SIRT1 SBD, these isoforms might be amenable to a SIRT1-like activation mechanism, at least with the FdL substrate and its strongly hydrophobic fluorophore that enables pronounced interactions with resveratrol-like STACs even without the SBD (see above) (Gertz et al., 2012; Lakshminarasimhan et al., 2013a). However, to substantiate the relevance of the activating DHP effects on SIRT2 and SIRT3 it will be essential to confirm them with non-modified, physiological substrates. Interestingly, the DHP-related lignan honokiol was reported to activate SIRT3-dependent deacetylation of superoxide dismutase in vivo, but the compound also increased SIRT3 expression and a detailed characterization of its potential SIRT3 activating effect remains to be awaited.
The binding site for the SIRT6-activating pyrrolo[1,2-a]quinoxalines is clearly distinct from that characterized for SIRT1 STACs, yet both activator types appear to exploit alternative active site openings and might share mechanistic features. The SIRT6 activators bind, independent from substrate, to the acyl channel and might induce an enzyme conformation with increased substrate peptide affinity as suggested by such kinetic effects of free fatty acids, which are assumed to also bind to the acyl channel based on competition experiments (Feldman et al., 2013; You et al., 2017). Comparison of SIRT6 conformational states with a SIRT6/activator complex revealed no significant conformational changes, however, and the exact kinetic and structural mechanism of activation remains to be determined (You et al., 2017). Nevertheless, activation is mediated by a unique SIRT6 site, which is created by a reduced cofactor binding loop and neighboring helix bundle as compared to SIRT1 and SIRT5 (Figure 2c,d,g), rendering it highly attractive for the development of SIRT6-specific modulators. In contrast, the activator site in the SIRT5/resveratrol complex, which appears to also contribute to the SIRT1 STAC site (see above) (Cao et al., 2015; Dai et al., 2015; Gertz et al., 2012), is a SIRT1/5 active site opening occluded in SIRT6 by its N-terminus. Interestingly, Ser10 within this N-terminus was shown to serve as a phosphorylation site mediating activation of the related SIRT6 ADP-ribosylation activity (Van Meter et al., 2016), and it is tempting to speculate that there might be common mechanistic features for this effect and SIRT1/5 activation due to the similar regulator sites on the catalytic cores.
Indirect approaches for increasing Sirtuin activity
In addition to direct Sirtuin activation, indirect strategies have also been attempted. In principle, increasing expression levels of Sirtuins could be an approach for boosting Sirtuin activity, but the regulation of their gene expression is complex and coupled to other proteins (Buler et al., 2016), rendering this approach unsuitable for specific activation. Iso-nicotinamide (iso-NAM), a competitor to the endogenous Sirtuin inhibitor nicotinamide, prevents the base-exchange reaction resulting from inhibitory rebinding of nicotinamide to a Sirtuin/intermediate complex (see below) and reformation of the substrates (Sauve et al., 2005). Iso-NAM thus suppresses Sirtuin inhibition, resulting in an apparent activation. However, iso-NAM concentrations need to reach the millimolar range for effective sirtuin activation, and the stimulatory effect is limited to the level of initial nicotinamide inhibition. Also, Iso-NAM activates all Sirtuin isoforms similarly. For a more specific “activation by derepression” strategy, identification of molecules that disrupt the interaction between SIRT1 and its inhibitory partner protein, Dbc1 (Deleted in Breast Cancer 1) (Kim et al., 2008), has been attempted but no promising compounds are yet available. Similarly, one could imagine to identify compounds that strengthen the interaction of SIRT1 with the protein AROS (Active Regulator of Sirt1) (Kim et al., 2007), although the exact effect of this protein on SIRT1 is controversial (Knight et al., 2013; Kokkola et al., 2014; Lakshminarasimhan et al., 2013a). These strategies could also be used for other Sirtuin isoforms, but few regulator proteins have yet been identified. A related strategy for Sirtuin activation is to increase intracellular NAD+ concentrations, either by inhibiting NAD+-consuming enzymes such as CD38 or by stimulating NAD+ biosynthesis (Bonkowski and Sinclair, 2016). The NAD+ precursors nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) have successfully been used for Sirtuin stimulation via increasing NAD+ levels (Bonkowski and Sinclair, 2016; Yoshino et al., 2011), but such an effect is of course non-specific, stimulating all Sirtuin isoforms and affecting any other NAD+-dependent enzymes and processes such as PARP-1 regulation (Li et al., 2017).
Sirtuin inhibition
For some pathophysiologies and Sirtuin isoforms, inhibition rather than activation is desired, e.g. for SIRT2 in neurodegenerative diseases (Donmez and Outeiro, 2013). Advances in assays and an increasing number of Sirtuin/inhibitor complex structures support the ongoing efforts to develop highly potent and specific Sirtuin-targeting inhibitors (Schutkowski et al., 2014).
Substrate mimics and other acyl-peptide competitive inhibitors
Many Sirtuin inhibitors target the polypeptide binding cleft and/or the NAD+ pocket for competitive inhibition (Cen, 2010; Chen, 2011). Isoform-specific features, reflected by differing preferences concerning substrate sequence and acyl modification (Du et al., 2011; Jiang et al., 2013; Roessler et al., 2014), indicate that the acyl-peptide site might be promising for selective inhibition. A straightforward approach exploiting it are thioacyl-peptides, which form a stable thioalkylimidate intermediate and thereby trap the enzyme (Gertz et al., 2013; Hawse et al., 2008; Smith and Denu, 2007). Highly potent thioacetyl inhibitors were developed, e.g., for SIRT3 and SIRT1 (Fatkins and Zheng, 2008; Kiviranta et al., 2009), but their selectivity is poor due to the limited sequence specificity of sirtuins. In addition exploiting their differing acyl selectivities (Feldman et al., 2013; Jiang et al., 2013; Roessler et al., 2014; Rajabi et al., 2017) yielded potent and/or isoform-specific inhibitors. An ethoxymalonyl modification enables selective SIRT1 inhibition (Asaba et al., 2009), and a 3-methyl-3-phenylsuccinyl group potent and specific SIRT5 inhibition by exploiting its unique binding pocket for a distal carboxylate (Figure 3a) (Roessler et al., 2014). A thiosuccinyl penta-peptide was described for moderately potent SIRT5 inhibition in a cellular environment (He et al., 2012). However, most peptides are unable to cross membranes and unstable in vivo, stimulating efforts to reduce such ligands to their elements essential for binding. Cell active compounds were obtained, e.g., by modifying the α-amino and α-carboxy groups of acyl-Lys with a benzyloxycarbonyl and an anilino group, respectively (Asaba et al., 2009), and a protected Lys with benzyloxycarbonyl-5-aminoglutaryl modification yielded Sirt5 selectivity and cellular activity (Polletta et al., 2015). Such larger acyl groups allow exploiting the SIRT5-specific carboxylate pocket and simultaneously the C-site as a preferred inhibitor binding pocket (see below) (Roessler et al., 2014), and further refinement of acyl-Lys derived compounds thus appears attractive for Sirtuin inhibitor development.
Figure 3. Sirtuin inhibition.
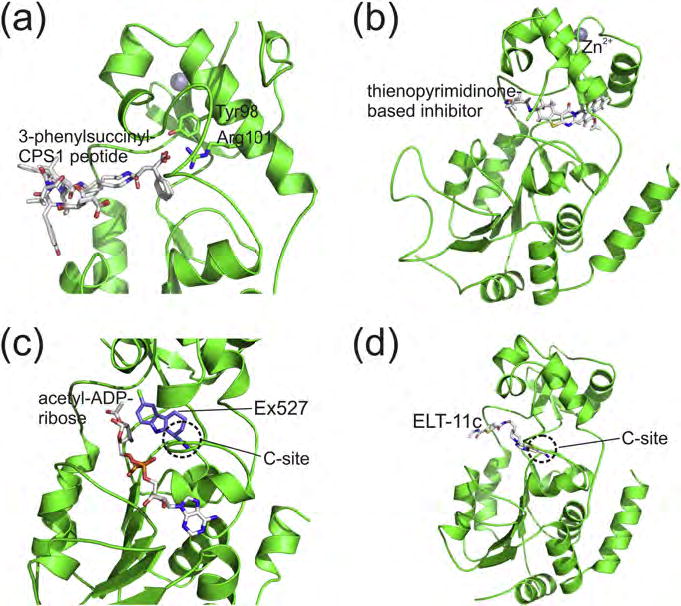
(a) Crystal structure of a SIRT5 complex with inhibitory 3-phenyl-succinyl-modified CPS1 peptide (gray; PDB ID 4UTV), which is closely related to the highly potent and SIRT5 selective 3-phenyl-3-methyl-succinyl-CPS1. Two ligands indicate the two enantiomers. The two key residues recognizing the distal carboxylate are indicated. (b) Crystal structure of SIRT2 in complex with a potent and selective thienopyrimidinone-based inhibitor (PDB ID 5MAT), which exploits the extended acyl channel. (c) Crystal structure of SIRT3 in complex with the product 2′-O-acetyl-ADP-ribose (gray) and the ECS inhibitor Ex-527 (blue; PDB ID 4BVH). The NAM accommodating C-site is indicated by a dotted circle. (d) Crystal structure of SIRT3 in complex with the highly potent pan Sirtuin inhibitor ELT-11c (PDB ID 4JSR), which blocks C site (center; dotted circle) and the channel accommodating the substrate Lys side chain (left part of the inhibitor).
N-alkylated thiobabiturates are assumed to exploit, with a certain specificity, SIRT5’s acyl-Lys pocket (Maurer et al., 2012). Acyl-peptide competition was demonstrated for cambinol (Table 1), a hydroxynaphthyl distantly related to Sirtinol, one of the first, weak Sirtuin inhibitors reported (Grozinger et al., 2001). Cambinol inhibits with moderate specificity and potency (Heltweg et al., 2006), but a more potent and moderately SIRT3-specific cambinol derivative (Mahajan et al., 2014) indicates potential for this compound class. The huge diarylurea suramin blocks both the acyl-peptide and the NAD+ binding site potently, but its size and non-specific effects on Sirtuins and many other targets render it less promising for further development (McGeary et al., 2008; Schuetz et al., 2007; Suenkel et al., 2013; Trapp et al., 2007). The sulfobenzol-based brain-permeable SIRT2 specific inhibitor Ak7 also appears to block NAD+ and peptide site and seems also more promising due to its more compact size and neuroprotective effects in a Parkinson’s Disease model (Chen et al., 2015). Docking studies yielded pseudopeptidic inhibitors with a certain SIRT2/SIRT3 selectivity but poor potency (Salo et al., 2013) and SIRT2 selective inhibitors with chemical features not yet suitable for pharmacological applications (Schlicker et al., 2011). One of the latter compounds could be developed into the potent, SIRT2 selective yet compact oxadiazole inhibitor UBCS0137 (Table 1), and a SIRT2/oxadiazole crystal structure confirmed that these compounds exploit an isoform-specific extension of the substrate acyl pocket (Moniot et al., 2017). The acyl binding extension was also proposed to accommodate SIRT1 and SIRT2 inhibiting 2-anilinobenzamides (Suzuki et al., 2012), with the potent 3′-(3-fluoro-phenethyloxy)-2-anilinobenzamide (Table 1) showing selective SIRT2 inhibition, and the highly potent (IC50 42 nM) and SIRT2 over SIRT1/3 selective N-(4-((3-(2-((4,6-dimethylpyrimidin-2-yl)thio)acetamido)benzyl)oxy)phenyl)thiophene-2-carboxamide (Table 1). The SIRT2 acyl channel and extension can undergo conformational changes that contribute to potent and SIRT2 selective inhibition by SirReal2 (Table 1) and thienopyrimidinones (Table 1; Figure 3b) (Rumpf et al., 2015; Sundriyal et al., 2017), and the SIRT2 specific pocket and its dynamics might explain why this isoform appears particular amenable to specific inhibition by drug-like compounds.
A potent and partly selective SIRT3 inhibitor acting competitively with the acetyl-Lys substrate and un-competitively with NAD+ is SRT1720 (Table 1) (Jin et al., 2009; Nguyen et al., 2013b). A SIRT3/SRT1720/NAD+-analog structure shows that the inhibitor occupies part of the acetyl-Lys binding channel and interacts tightly with NAD+ and a unique cofactor binding loop conformation (Nguyen et al., 2013b). Interestingly, the compound was initially described as a SIRT1 activator (Milne et al., 2007), illustrating the significant isoform differences.
C-site and extended C-site inhibitors
Productive NAD+ binding places its nicotinamide moiety in the Sirtuin C-pocket (Gertz et al., 2013; Sanders et al., 2010). After alkylimidate formation and nicotinamide release, free nicotinamide can rebind to the C-pocket and reverse this step (so-called base-exchange), resulting in inhibition of the deacylation reaction (Sauve, 2010). Nicotinamide thus is a pan-Sirtuin inhibitor, possibly with physiological relevance (Bitterman et al., 2002), but potencies differ among isoforms and even among deacylation activities (Fischer et al., 2012; Pannek et al., 2017). It inhibits SIRT5’s desuccinylation activity with a potency typical for Sirtuin effects (IC50 21 μM) yet hardly affects its deacetylation activity likely due to steric hindrance by the SIRT5-specific arginine recognizing distal substrates carboxylates (Fischer et al., 2012). The kinase and Sirtuin inhibitor GW5074 also showed an acyl dependent effect, by inhibiting SIRT5’s desuccinylase activity but not its deacetylase activity (Suenkel et al., 2013). Thus, isoform-specific features next to the C-site exist and should enable the development of selective inhibitors. A potent Sirtuin inhibitor that exploits the C-site and which shows some isoform selectivity is Ex-527 (Table 1). It exploits the unique Sirtuin deacylation mechanism to inhibit SIRT1 strongly, SIRT2/3/6 more moderately, and not SIRT5 (Gertz et al., 2013; Napper et al., 2005; Schuster et al., 2016), and it entered a clinical trial for Huntington’s disease treatment (see below) (Sussmuth et al., 2015). The compound binds to the C-site and a hydrophobic patch next to it (“extended C-site”, ECS; Figure 3c) (Gertz et al., 2013; Zhao et al., 2013). Its complex with apo Sirtuin or in presence of NAD+ is easily dissociated by acyl substrate, and a stable, inhibitory complex forms only after generation of the co-product 2′-O-acetyl-ADP-ribose, which provides part of the inhibitor binding pocket (Figure 3c) (Gertz et al., 2013; Napper et al., 2005). Interestingly, all residues directly interacting with Ex-527 are identical in the differently inhibited isoforms, and the kinetics of product formation and/or dissociation appears to determine their Ex-527 sensitivities (Gertz et al., 2013). The Ex-527 binding site thus enables potent Sirtuin inhibition, but for further improvement of Ex-527 modifications that reach more distant, isoform-specific regions will be required.
AGK2 (Table 1) is a widely used, potent SIRT2 inhibitor with >10-fold isoform-specificity and protective effects in Parkinson’s Disease models (Outeiro et al., 2007), and the thiazole MIND4 is a potent SIRT2 inhibitor with neuroprotective effects in Huntington’s Disease models (Quinti et al., 2016). Both compounds are assumed to exploit the C-site, and MIND4 in addition the SIRT2 acyl extension (see above), but structural validations remain to be awaited. C-site binding is established for the most potent Sirtuin inhibitors reported so far, thieno[3,2-d]pyrimidine-6-carboxamides obtained through screening of a combinatorial “Encoded Library Technology” (ELT) compound library (Disch et al., 2013). The most potent compound, ELT-11c (Table 1), inhibits SIRT3 with an IC50 of 4 nM, and SIRT3/ELT complex structures show that the inhibitors occupy C-pocket plus acetyl-Lys channel (Figure 3d). The thieno[3,2-d]pyrimidine forms π-stacking interactions with a conserved Phe of the cofactor binding loop, and the carboxamide H-bonds to C-pocket residues comparable to nicotinamide and Ex-527 (Disch et al., 2013; Gertz et al., 2013). However, consistent with the conservation of these Sirtuin features, ELT compounds inhibit SIRT1, 2, and 3 with comparable potencies. The ELT compounds nevertheless appear interesting leads for Sirtuin inhibition since they show no strong off-target effects (Disch et al., 2013).
In summary, the Sirtuin C-site appears to be a prominent ligand binding site and neighboring variations should – and likely do for several specific compounds - enable isoform-specific inhibition.
Resveratrol-related Sirtuin inhibitors
Resveratrol and related polyphenols (Table 1) were first identified as Sirtuin activators (Baur and Sinclair, 2006; Howitz et al., 2003), but resveratrol can also inhibit SIRT1, depending on the substrate used (see above) (Dai et al., 2010; Hubbard et al., 2013; Lakshminarasimhan et al., 2013b). It also inhibits SIRT3 and SIRT5, again depending on the substrate (Fischer et al., 2012; Gertz et al., 2012). Resveratrol-related compounds were speculated to act on Sirtuins via two different binding sites (Nguyen et al., 2013a), since SIRT1 activation with regular substrates depends on the isoform’s specific SBD (Hubbard et al., 2013), but structural insights in Sirtuin activation instead suggest a bipartite binding site consisting of active site elements (including bound substrate) and catalytic core loops or the SBD in SIRT1 (see above) (Cao et al., 2015; Dai et al., 2015; Gertz et al., 2012). The direct activator/substrate interactions apparently influence substrate binding details and thereby catalysis (Gertz et al., 2012). However, the substrate contacts appear artificially enhanced in FdL complexes through the hydrophobic FdL fluorophore, making these substrates more sensitive to modulators and SBD contacts less relevant, and further studies will be required for fully understanding the mechanisms of Sirtuin modulation and the complex pattern of isoform- and substrate-dependent effects. In addition to Sirtuins, resveratrol affects a variety of other targets (e.g., kinases, ATP synthase) (Pirola and Frojdo, 2008), and molecular conclusion from physiological studies with resveratrol should thus be drawn with extreme caution. The pleiotropic effects together with its moderate potency against Sirtuins and limited bioavailability render resveratrol less likely to be suited as a therapeutic drug. Therefore, derivatives such as the more soluble piceatannol (Table 1) and polydatin and other polyphenols have been investigated as Sirtuin inhibitors but yielded mostly weak and/or non-specific effects (Gertz et al., 2012; Howitz et al., 2003; Kahyo et al., 2008; Nguyen et al., 2013a). However, 4′-bromo-resveratrol showed promising potency against SIRT1 and SIRT3, with excellent discrimination (i.e., no effect) against Sirt5 (Nguyen et al., 2013a). The compound inhibits via C-site and a neighboring “ECS III” pocket (Gertz and Steegborn, 2016; Nguyen et al., 2013a), i.e. exploits a different binding site than the parent compound. The ECS III pocket accommodating the bromophenyl moiety varies among Sirtuin isoforms and might allow the development of isoforms-selective compounds that exploit the C-site (Gertz and Steegborn, 2016; Nguyen et al., 2013a), which appears to be a preferred binding pocket but lacks the isoform differences required for selectivity (see above). Interestingly, the more distantly resveratrol-related compound SDX-437 was reported to inhibit SIRT3 potently, with little effect on SIRT1 (Patel et al., 2015), but a contribution of the 4′-bromo-resveratrol pocket remains to be analyzed.
Other Sirtuin inhibitors
Many additional compounds, from different compound families, have been described to have effects on Sirtuins or in biological systems via Sirtuin modulation. Examples are the acylthioureas tenovin-1 and tenovin-6 (Lain et al., 2008) and the benzothiazol AC-93253 (Zhang et al., 2009); or natural compounds such as phloroglucinols (Gey et al., 2007) and tanikolides (Gutierrez et al., 2009). Such compounds, while lacking either the combination of potency, selectivity or pharmacological properties required for a wider use or even a rigorous characterization of their selectivity, have added to the insights in chemical structures modulating Sirtuins. These chemical compound classes with reported Sirtuin effects are comprehensively listed in previous reviews (Chen, 2011; Yoon and Oon, 2016).
Sirtuin modulators as therapeutic drugs – state of the art
Several Sirtuin isoforms appear to bear potential for being used as therapeutic targets, but to date only modulators of SIRT1 have entered into the clinic. The natural STAC resveratrol suffered from low bioavailability and potency, and - like early synthetic STACs such as SRT1720 – from limited target specificity (Pirola and Frojdo, 2008; Huber et al., 2010; Nguyen et al., 2013b; Park et al., 2017). This review will concentrate on a SIRT1 inhibitor and novel synthetic SIRT1 activators with superior properties that have advanced into the clinic. Clinical experience with the promiscuous compound resveratrol can be found in the review by (Borriello et al., 2013).
SIRT1 inhibitors
The only selective SIRT1 inhibitor to enter the clinic is selisistat, also known as Ex-527 or SEN0014196, which has been taken into a study in normal healthy volunteers (Westerberg et al., 2015) and also into a study in patients with Huntington’s Disease (HD) (Sussmuth et al., 2015) by Siena Biotech. In the trial in healthy volunteers selisistat was given at single doses up to 600 mg and in repeat dosing up to 300 mg. Selisistat was considered safe and well tolerated at all doses and showed dose proportional exposure up to 300 mg in this study (Westerberg et al., 2015). In the study in HD patients (Sussmuth et al., 2015) selisistat was given once daily at either 10 or 100 mg for 14 days in a double-blind, placebo-controlled study. Selisistat was found to be well tolerated with no adverse effects and circulating levels of soluble huntingtin were not affected by selisistat in this short study. A 12-week study with selisistat at doses of 50 and 200 mg has also been carried out in HD patients (ClinicalTrials.gov) but no results have been published from this study. Siena Biotech filed for bankruptcy in April 2015 and it appears that all clinical work with selisistat has stopped.
SIRT1 activators
Three small molecule SIRT1 activators (SRT2104, SRT2379, SRT3025) have been taken into the clinic by GlaxoSmithKline. SRT2104 is the most studied of the small molecule activators having been in fourteen clinical trials (www.clinicaltrials.gov). SRT2379 was shown to be well tolerated in a clinical study in healthy male volunteers but it showed no effect on the cytokine response to an LPS challenge at doses up to 1,000 mg and further development was stopped (Wiewel et al., 2013). SRT3025 was tested in healthy male volunteers at doses up to 3,000 mg and was well tolerated but showed a dose-dependent prolongation effect on QTc and although none of the telemetry findings were considered clinically significant further development of SRT3025 was stopped (GSK Clinical Study Register).
In the first-time-in-human (FTIH) study with SRT2104, the compound was administered orally once or in a seven day repeat study at doses up to 3,000 mg. SRT2104 was well tolerated in these studies and while systemic exposure was limited in the single administration study exposure was increased by the addition of food and by repeated administration (Hoffmann et al., 2013). In a study in elderly volunteers (Libri et al., 2012), SRT2104 was well tolerated at doses up to 2,000 mg and produced a decrease in serum cholesterol, LDL levels and triglycerides but had no significant effect in an oral glucose tolerance test. Similar reductions in serum cholesterol, LDL levels and triglycerides was seen in a study in otherwise healthy cigarette smokers with 2,000 mg of SRT2104 administered once daily for 28 days (Venkatasubramanian et al., 2013). In a separate study in Type 2 diabetics (Baksi et al., 2014) SRT2104 administered once daily for 28 days at doses of 250, 500, 1,000 and 2,000 mg also had a beneficial effect on lipids but did not improve glucose or insulin control. This study was characterized by highly variable pharmacokinetics and resulting low exposure of SRT2104.
SRT2104 was also examined in healthy volunteers for effects on cytokine release and coagulation induced by lipopolysaccharide (LPS) (van der Meer et al., 2015). 2000 mg of SRT2104 reduced LPS-induced release of IL-6 and IL-8 but not TNFα or IL-10 and also inhibited LPS activation of coagulation.
SRT2104 was tested in a clinical trial to assess its safety and anti-inflammatory effects in patients with mild-to-moderate ulcerative colitis (Sands et al., 2016). The compound showed modest but not statistically significant efficacy in these patients. Limitations in the clinical study include a less than optimal design including the limited size of the treatment groups, the lack of a placebo group, and variable oral pharmacokinetics of the molecule, and therefore the study did not allow for definitive conclusions. Despite this, levels of fecal calprotectin, a biomarker of colitis disease activity, were reduced by ~75% upon treatment with SRT2104. The reduction in fecal calprotectin was similar to that obtained with the JAK inhibitor tofacitinib (Xeljanz®) in its successful ulcerative colitis trial (Sands et al., 2016). Fecal calprotectin levels have been shown to correlate well with histological and endoscopic findings in IBD and can be used to detect disease activity, to predict relapse and to monitor the response to therapy. There is also emerging data in its utility to assess mucosal healing (Ikhtaire et al., 2016).
The longest duration study with SRT2104 was carried out in moderate-to-severe psoriasis patients where SRT2104 was dosed once daily for 12 weeks at 250, 500 and 1000 mg (Krueger et al., 2015). Despite sub-optimal PK, SRT2104 showed efficacy based on histological examination and physician assessment scores. Moreover, skin levels of ST2104 were found to be predictive of improvement as measured by Psoriasis Area and Severity Index (PASI) scores. In addition, patients who responded to SRT2104 showed significant changes in expression levels of genes known to be involved in psoriasis (Tian et al., 2012).
The clinical data with the first generation SIRT1 activator SRT2104 is instructive. Firstly, the drug shows poor and variable pharmacokinetics upon oral administration. Attempts were made to reduce the pharmacokinetic variability and to increase the systemic exposure by modifying the release characteristics of the SRT2104 formulations but these attempts were unsuccessful (McCallum et al., 2014). Secondly SRT2104 in multiple studies showed an effect on lipid parameters including cholesterol and triglycerides, and thirdly there was also a broad anti-inflammatory effect including in patients with ulcerative colitis and psoriasis.
Overall the data with SRT2104 argues that if newer small molecule SIRT1 activators can be developed with a good pharmacokinetic and tolerability profile then they could provide important new treatment paradigms particularly in inflammatory disease.
Outlook
The intense efforts in the past ~15 years to develop isoform specific small molecule modulators of the Sirtuins have yielded key insights into the pathophysiological roles of Sirtuins and seen great strides in understanding the enzymatic reaction mechanisms. Initial drug development efforts focused on SIRT1 and SIRT2 have yielded a number of potent SIRT2 inhibitors and SIRT1 activators that have now been through the first clinical trials, with evidence of safety and efficacy. The promising early clinical data with SRT2104 in psoriasis and in metabolic syndrome strongly advocate for continued research and development into novel SIRT1 activators which either have better pharmacokinetic properties allowing for oral administration or have physicochemical properties amenable for topical administration e.g in psoriasis. Mechanistic and structural insights obtained in recent years will support the development of STACs for other Sirtuin isoforms. There is particular excitement around finding SIRT6 activators suitable for the clinic, given the health benefits of SIRT6 in mice. Functional insights, such as SIRT1-regulating proteins or the interactions of SIRT6 and SIRT7 domains with nucleosomes and chromatin, open additional opportunities for modulating Sirtuins with small molecules. The field of Sirtuin modulators has clearly matured into an exciting path of drug development that holds the promise of treating common and rare diseases that have considerable unmet needs. Looking further into the future, if the remarkable effects of Sirtuins on rodents hold up in humans, STACs could one day revolutionize medicine and increase the average human healthspan, if not even lifespan.
Acknowledgments
We are grateful to Alice Kane for help with figure preparation and to our colleagues in the field for helpful discussions. Work on Sirtuins in the authors’ labs was supported by Deutsche Forschungsgemeinschaft grants STE1701/5 and STE1701/15 (to CS) and NIH grants DK100263 and AG028730 (to DS).
Nonstandard abbreviations used
- CR
calorie restriction
- DHP
1,4-dihydropyridine
- ECS
extended C-site
- ELT
Encoded Library Technology
- FdL
Fluor de Lys
- HD
Huntington’s Disease
- LPS
lipopolysaccharide
- LDL
low density lipoprotein
- MLS
mitochondrial localization sequences
- NAM
nicotinamide
- SBD
STAC binding domain
- SIR
silent information regulator
- SIRT
Sirtuin
- STAC
Sirtuin activating compound
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
HD and JLE are employees of GlaxoSmithKline, which has filed patent applications relating to Sirtuin modulators. DAS is a consultant to and/or inventor on patents licensed to GlaxoSmithKline, Metrobiotech, Jumpstart Fertility and Liberty Biosecurity. All other authors declare that they have no conflict of interest.
References
- Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, Wagner GR, Thompson JW, Madsen AS, Green MF, Sivley RM, Ilkayeva OR, Stevens RD, Backos DS, Capra JA, Olsen CA, Campbell JE, Muoio DM, Grimsrud PA, Hirschey MD. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 2017;25:838–855 e815. doi: 10.1016/j.cmet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaba T, Suzuki T, Ueda R, Tsumoto H, Nakagawa H, Miyata N. Inhibition of human sirtuins by in situ generation of an acetylated lysine-ADP-ribose conjugate. J Am Chem Soc. 2009;131:6989–6996. doi: 10.1021/ja807083y. [DOI] [PubMed] [Google Scholar]
- Baksi A, Kraydashenko O, Zalevkaya A, Stets R, Elliott P, Haddad J, Hoffmann E, Vlasuk GP, Jacobson EW. A phase II, randomized, placebo-controlled, double-blind, multi-dose study of SRT2104, a SIRT1 activator, in subjects with type 2 diabetes. Br J Clin Pharmacol. 2014;78:69–77. doi: 10.1111/bcp.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balan V, Miller GS, Kaplun L, Balan K, Chong ZZ, Li F, Kaplun A, VanBerkum MF, Arking R, Freeman DC, Maiese K, Tzivion G. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J Biol Chem. 2008;283:27810–27819. doi: 10.1074/jbc.M804681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee KK, Ayyub C, Ali SZ, Mandot V, Prasad NG, Kolthur-Seetharam U. dSir2 in the adult fat body, but not in muscles, regulates life span in a diet-dependent manner. Cell Rep. 2012;2:1485–1491. doi: 10.1016/j.celrep.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- Bemis JE, Vu CB, Xie R, Nunes JJ, Ng PY, Disch JS, Milne JC, Carney DP, Lynch AV, Jin L, Smith JJ, Lavu S, Iffland A, Jirousek MR, Perni RB. Discovery of oxazolo[4,5-b]pyridines and related heterocyclic analogs as novel SIRT1 activators. Bioorg Med Chem Lett. 2009;19:2350–2353. doi: 10.1016/j.bmcl.2008.11.106. [DOI] [PubMed] [Google Scholar]
- Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, Xuan J, Evans M, Harper ME, McBurney MW. SirT1 regulates energy metabolism and response to caloric restriction in mice. PloS One. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016;17:679–690. doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- Borriello A, Bencivenga D, Caldarelli I, Tramontano A, Borgia A, Pirozzi AV, Oliva A, Della Ragione F. Resveratrol and cancer treatment: is hormesis a yet unsolved matter? Current pharmaceutical design. 2013;19:5384–5393. doi: 10.2174/1381612811319300007. [DOI] [PubMed] [Google Scholar]
- Buler M, Andersson U, Hakkola J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. 2016;30:3942–3960. doi: 10.1096/fj.201600410RR. [DOI] [PubMed] [Google Scholar]
- Cao D, Wang M, Qiu X, Liu D, Jiang H, Yang N, Xu RM. Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 2015;29:1316–1325. doi: 10.1101/gad.265462.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen Y. Sirtuins inhibitors: The approach to affinity and selectivity. Biochim Biophys Acta. 2010;1804:1635–1644. doi: 10.1016/j.bbapap.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 2012;16:180–188. doi: 10.1016/j.cmet.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015;15:608–624. doi: 10.1038/nrc3985. [DOI] [PubMed] [Google Scholar]
- Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- Chen L. Medicinal chemistry of sirtuin inhibitors. Curr Med Chem. 2011;18:1936–1946. doi: 10.2174/092986711795590057. [DOI] [PubMed] [Google Scholar]
- Chen X, Wales P, Quinti L, Zuo F, Moniot S, Herisson F, Rauf NA, Wang H, Silverman RB, Ayata C, Maxwell MM, Steegborn C, Schwarzschild MA, Outeiro TF, Kazantsev AG. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS One. 2015;10:e0116919. doi: 10.1371/journal.pone.0116919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, Zhao H, Jiang Y, Sweitzer SM, Pietrak B, Schwartz B, Blum CA, Disch JS, Caldwell R, Szczepankiewicz B, Oalmann C, Yee Ng P, White BH, Casaubon R, Narayan R, Koppetsch K, Bourbonais F, Wu B, Wang J, Qian D, Jiang F, Mao C, Wang M, Hu E, Wu JC, Perni RB, Vlasuk GP, Ellis JL. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat Commun. 2015;6:7645. doi: 10.1038/ncomms8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Kustigian L, Carney D, Case A, Considine T, Hubbard BP, Perni RB, Riera TV, Szczepankiewicz B, Vlasuk GP, Stein RL. SIRT1 activation by small molecules: kinetic and biophysical evidence for direct interaction of enzyme and activator. J Biol Chem. 2010;285:32695–32703. doi: 10.1074/jbc.M110.133892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AM, Huber FM, Hoelz A. Structural and functional analysis of human SIRT1. J Mol Biol. 2014;426:526–541. doi: 10.1016/j.jmb.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disch JS, Evindar G, Chiu CH, Blum CA, Dai H, Jin L, Schuman E, Lind KE, Belyanskaya SL, Deng J, Coppo F, Aquilani L, Graybill TL, Cuozzo JW, Lavu S, Mao C, Vlasuk GP, Perni RB. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem. 2013;56:3666–3679. doi: 10.1021/jm400204k. [DOI] [PubMed] [Google Scholar]
- Donmez G, Outeiro TF. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med. 2013;5:344–352. doi: 10.1002/emmm.201302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Jiang H, Lin H. Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry. 2009;48:2878–2890. doi: 10.1021/bi802093g. [DOI] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatkins DG, Zheng W. Substituting N(epsilon)-thioacetyl-lysine for N(epsilon)-acetyl-lysine in peptide substrates as a general approach to inhibiting human NAD(+)-dependent protein deacetylases. Int J Mol Sci. 2008;9:1–11. doi: 10.3390/ijms9010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem. 2013;288:31350–31356. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Dittenhafer-Reed KE, Denu JM. Sirtuin catalysis and regulation. J Biol Chem. 2012;287:42419–42427. doi: 10.1074/jbc.R112.378877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Pavletich NP. Structure of the histone deacetylase SIRT2. Nat Struct Biol. 2001;8:621–625. doi: 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Gertz M, Suenkel B, Lakshminarasimhan M, Schutkowski M, Steegborn C. Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One. 2012;7:e45098. doi: 10.1371/journal.pone.0045098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Natl Acad Sci USA. 2013;110:E2772–2781. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Nguyen GT, Fischer F, Suenkel B, Schlicker C, Franzel B, Tomaschewski J, Aladini F, Becker C, Wolters D, Steegborn C. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS One. 2012;7:e49761. doi: 10.1371/journal.pone.0049761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Steegborn C. Using mitochondrial sirtuins as drug targets: disease implications and available compounds. Cell Mol Life Sci. 2016;73:2871–2896. doi: 10.1007/s00018-016-2180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gey C, Kyrylenko S, Hennig L, Nguyen LH, Buttner A, Pham HD, Giannis A. Phloroglucinol derivatives guttiferone G, aristoforin, and hyperforin: inhibitors of human sirtuins SIRT1 and SIRT2. Angew Chem Int Ed Engl. 2007;46:5219–5222. doi: 10.1002/anie.200605207. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- Guarente L. Calorie restriction and SIR2 genes-towards a mechanism. Mech Ageing Dev. 2005;126:923–928. doi: 10.1016/j.mad.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Gutierrez M, Andrianasolo EH, Shin WK, Goeger DE, Yokochi A, Schemies J, Jung M, France D, Cornell-Kennon S, Lee E, Gerwick WH. Structural and synthetic investigations of tanikolide dimer, a SIRT2 selective inhibitor, and tanikolide seco-acid from the Madagascar marine cyanobacterium Lyngbya majuscula. J Org Chem. 2009;74:5267–5275. doi: 10.1021/jo900578j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse WF, Hoff KG, Fatkins DG, Daines A, Zubkova OV, Schramm VL, Zheng W, Wolberger C. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure. 2008;16:1368–1377. doi: 10.1016/j.str.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Du J, Lin H. Thiosuccinyl peptides as Sirt5-specific inhibitors. J Am Chem Soc. 2012;134:1922–1925. doi: 10.1021/ja2090417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, Westphall MS, Pagliarini DJ, Prolla TA, Assadi-Porter F, Roy S, Denu JM, Coon JJ. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA, Bedalov A. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368–4377. doi: 10.1158/0008-5472.CAN-05-3617. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV, Jr, Kahn CR, Verdin E. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E, Wald J, Lavu S, Roberts J, Beaumont C, Haddad J, Elliott P, Westphal C, Jacobson E. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br J Clin Pharmacol. 2013;75:186–196. doi: 10.1111/j.1365-2125.2012.04340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Hsu CP, Odewale I, Alcendor RR, Sadoshima J. Sirt1 protects the heart from aging and stress. Biol Chem. 2008;389:221–231. doi: 10.1515/BC.2008.032. [DOI] [PubMed] [Google Scholar]
- Hsu HC, Wang CL, Wang M, Yang N, Chen Z, Sternglanz R, Xu RM. Structural basis for allosteric stimulation of Sir2 activity by Sir4 binding. Genes Dev. 2013;27:64–73. doi: 10.1101/gad.208140.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, Riera TV, Lee JE, E SY, Lamming DW, Pentelute BL, Schuman ER, Stevens LA, Ling AJ, Armour SM, Michan S, Zhao H, Jiang Y, Sweitzer SM, Blum CA, Disch JS, Ng PY, Howitz KT, Rolo AP, Hamuro Y, Moss J, Perni RB, Ellis JL, Vlasuk GP, Sinclair DA. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013;339:1216–1219. doi: 10.1126/science.1231097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–154. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JL, McBurney MW, Distefano PS, McDonagh T. SIRT1-independent mechanisms of the putative sirtuin enzyme activators SRT1720 and SRT2183. Future Med Chem. 2010;2:1751–1759. doi: 10.4155/fmc.10.257. [DOI] [PubMed] [Google Scholar]
- Ikhtaire S, Shajib MS, Reinisch W, Khan WI. Fecal calprotectin: its scope and utility in the management of inflammatory bowel disease. J Gastroenterology. 2016;51:434–446. doi: 10.1007/s00535-016-1182-4. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jeong SM, Haigis MC. Sirtuins in Cancer: a Balancing Act between Genome Stability and Metabolism. Molecules and cells. 2015;38:750–758. doi: 10.14348/molcells.2015.0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Galonek H, Israelian K, Choy W, Morrison M, Xia Y, Wang X, Xu Y, Yang Y, Smith JJ, Hoffmann E, Carney DP, Perni RB, Jirousek MR, Bemis JE, Milne JC, Sinclair DA, Westphal CH. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 2009;18:514–525. doi: 10.1002/pro.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, Bedalov A, Kennedy BK. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahyo T, Ichikawa S, Hatanaka T, Yamada MK, Setou M. A novel chalcone polyphenol inhibits the deacetylase activity of SIRT1 and cell growth in HEK293T cells. J Pharmacol Sci. 2008;108:364–371. doi: 10.1254/jphs.08203fp. [DOI] [PubMed] [Google Scholar]
- Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- Kang H, Suh JY, Jung YS, Jung JW, Kim MK, Chung JH. Peptide switch is essential for Sirt1 deacetylase activity. Mol Cell. 2011;44:203–213. doi: 10.1016/j.molcel.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashani-Amin E, Larijani B, Ebrahim-Habibi A. Neohesperidin dihydrochalcone: presentation of a small molecule activator of mammalian alpha-amylase as an allosteric effector. FEBS Lett. 2013;587:652–658. doi: 10.1016/j.febslet.2013.01.022. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Austriaco NR, Jr, Zhang J, Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80:485–496. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Kiviranta PH, Suuronen T, Wallen EA, Leppanen J, Tervonen J, Kyrylenko S, Salminen A, Poso A, Jarho EM. N(epsilon)-thioacetyl-lysine-containing tri-, tetra-, and pentapeptides as SIRT1 and SIRT2 inhibitors. J Med Chem. 2009;52:2153–2156. doi: 10.1021/jm801401k. [DOI] [PubMed] [Google Scholar]
- Knight JR, Allison SJ, Milner J. Active regulator of SIRT1 is required for cancer cell survival but not for SIRT1 activity. Open Biol. 2013;3:130130. doi: 10.1098/rsob.130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola T, Suuronen T, Molnar F, Maatta J, Salminen A, Jarho EM, Lahtela-Kakkonen M. AROS has a context-dependent effect on SIRT1. FEBS Lett. 2014;588:1523–1528. doi: 10.1016/j.febslet.2014.03.020. [DOI] [PubMed] [Google Scholar]
- Krueger JG, Suarez-Farinas M, Cueto I, Khacherian A, Matheson R, Parish LC, Leonardi C, Shortino D, Gupta A, Haddad J, Vlasuk GP, Jacobson EW. A Randomized, Placebo-Controlled Study of SRT2104, a SIRT1 Activator, in Patients with Moderate to Severe Psoriasis. PLoS One. 2015;10:e0142081. doi: 10.1371/journal.pone.0142081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S, Sebastian C, Fitamant J, Ross KN, Saha SK, Jain E, Gladden A, Arora KS, Kato Y, Rivera MN, Ramaswamy S, Sadreyev RI, Goren A, Deshpande V, Bardeesy N, Mostoslavsky R. SIRT6 Suppresses Pancreatic Cancer through Control of Lin28b. Cell. 2016;165:1401–1415. doi: 10.1016/j.cell.2016.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJ, Pass G, Woods J, Lane DP, Westwood NJ. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Curth U, Moniot S, Mosalaganti S, Raunser S, Steegborn C. Molecular architecture of the human protein deacetylase Sirt1 and its regulation by AROS and resveratrol. Biosci Rep. 2013a;33 doi: 10.1042/BSR20120121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Rauth D, Schutkowski M, Steegborn C. Sirt1 activation by resveratrol is substrate sequence-selective. Aging (Albany NY) 2013b;5:151–154. doi: 10.18632/aging.100542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJ, Rajman LA, Qin B, Lou Z, Gorbunova V, Aravind L, Steegborn C, Sinclair DA. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science. 2017;355:1312–1317. doi: 10.1126/science.aad8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri V, Brown AP, Gambarota G, Haddad J, Shields GS, Dawes H, Pinato DJ, Hoffman E, Elliot PJ, Vlasuk GP, Jacobson E, Wilkins MR, Matthews PM. A pilot randomized, placebo controlled, double blind phase I trial of the novel SIRT1 activator SRT2104 in elderly volunteers. PLoS One. 2012;7:e51395. doi: 10.1371/journal.pone.0051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol. 2012;7:947–960. doi: 10.1021/cb3001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Mahajan SS, Scian M, Sripathy S, Posakony J, Lao U, Loe TK, Leko V, Thalhofer A, Schuler AD, Bedalov A, Simon JA. Development of pyrazolone and isoxazol-5-one cambinol analogues as sirtuin inhibitors. J Med Chem. 2014;57:3283–3294. doi: 10.1021/jm4018064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A, Valente S, Meade S, Carafa V, Tardugno M, Nebbioso A, Galmozzi A, Mitro N, De Fabiani E, Altucci L, Kazantsev A. Study of 1,4-dihydropyridine structural scaffold: discovery of novel sirtuin activators and inhibitors. J Med Chem. 2009;52:5496–5504. doi: 10.1021/jm9008289. [DOI] [PubMed] [Google Scholar]
- Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, Kang Y, Shenk T, Cristea IM. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell. 2014;159:1615–1625. doi: 10.1016/j.cell.2014.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer B, Rumpf T, Scharfe M, Stolfa DA, Schmitt ML, He W, Verdin E, Sippl W, Jung M. Inhibitors of the NAD dependent protein desuccinylase and demalonylase Sirt5. ACS Med Chem Lett. 2012;3:1050–1053. doi: 10.1021/ml3002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum SW, Wald JA, Hoffmann E, Jayaramachandran S, Bhatt K, Englehart S, Hernandez C, Shukla R, Sueda K, Hussaini A, Ellis JL. Modified Release Formulations Do Not Enhance the PK of the Novel SIRT1 Activator SRT2104 (GSK2245840B). American Association of Pharmaceutical Scientists - 2014 Annual Meeting & Exposition; San Diego, CA, USA. 2014. [Google Scholar]
- McGeary RP, Bennett AJ, Tran QB, Cosgrove KL, Ross BP. Suramin: clinical uses and structure-activity relationships. Mini Rev Med Chem. 2008;8:1384–1394. doi: 10.2174/138955708786369573. [DOI] [PubMed] [Google Scholar]
- Mercken EM, Mitchell SJ, Martin-Montalvo A, Minor RK, Almeida M, Gomes AP, Scheibye-Knudsen M, Palacios HH, Licata JJ, Zhang Y, Becker KG, Khraiwesh H, Gonzalez-Reyes JA, Villalba JM, Baur JA, Elliott P, Westphal C, Vlasuk GP, Ellis JL, Sinclair DA, Bernier M, de Cabo R. SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell. 2014;13:787–796. doi: 10.1111/acel.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin YK, Canto C, Scheibye-Knudsen M, Krawczyk M, Irusta PM, Martin-Montalvo A, Hubbard BP, Zhang Y, Lehrmann E, White AA, Price NL, Swindell WR, Pearson KJ, Becker KG, Bohr VA, Gorospe M, Egan JM, Talan MI, Auwerx J, Westphal CH, Ellis JL, Ungvari Z, Vlasuk GP, Elliott PJ, Sinclair DA, de Cabo R. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniot S, Forgione M, Lucidi A, Hailu GS, Nebbioso A, Carafa V, Baratta F, Altucci L, Giacche N, Passeri D, Pellicciari R, Mai A, Steegborn C, Rotili D. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure-Activity Relationship, X-ray Crystal Structure, and Anticancer Activity. J Med Chem. 2017;60:2344–2360. doi: 10.1021/acs.jmedchem.6b01609. [DOI] [PubMed] [Google Scholar]
- Moniot S, Schutkowski M, Steegborn C. Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J Struct Biol. 2013;182:136–143. doi: 10.1016/j.jsb.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA, Blackwell TK. Dietary restriction involves NAD(+)-dependent mechanisms and a shift toward oxidative metabolism. Aging Cell. 2014;13:1075–1085. doi: 10.1111/acel.12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, Thomas RJ, Kates M, Jones S, Navia MA, Saunders JO, DiStefano PS, Curtis R. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- Nguyen GT, Gertz M, Steegborn C. Crystal structures of sirt3 complexes with 4′-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem Biol. 2013a;20:1375–1385. doi: 10.1016/j.chembiol.2013.09.019. [DOI] [PubMed] [Google Scholar]
- Nguyen GT, Schaefer S, Gertz M, Weyand M, Steegborn C. Structures of human sirtuin 3 complexes with ADP-ribose and with carba-NAD(+) and SRT1720: binding details and inhibition mechanism. Acta Crystallogr D Biol Crystallogr. 2013b;69:1423–1432. doi: 10.1107/S0907444913015448. [DOI] [PubMed] [Google Scholar]
- Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, Wright SM, Mills KD, Bonni A, Yankner BA, Scully R, Prolla TA, Alt FW, Sinclair DA. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Yuan H, Brent M, Ding EC, Marmorstein R. SIRT1 contains N- and C-terminal regions that potentiate deacetylase activity. J Biol Chem. 2012;287:2468–2476. doi: 10.1074/jbc.M111.285031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. J Biol Chem. 2011;286:14575–14587. doi: 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannek M, Simic Z, Fuszard M, Meleshin M, Rotili D, Mai A, Schutkowski M, Steegborn C. Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat Commun. 2017;8:1513. doi: 10.1038/s41467-017-01701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Ahmad F, Um JH, Brown AL, Xu X, Kang H, Ke H, Feng X, Ryall J, Philp A, Schenk S, Kim MK, Sartorelli V, Chung JH. Specific Sirt1 Activator-mediated Improvement in Glucose Homeostasis Requires Sirt1-Independent Activation of AMPK. EBioMedicine. 2017;18:128–138. doi: 10.1016/j.ebiom.2017.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K, Sherrill J, Mrksich M, Scholle MD. Discovery of SIRT3 Inhibitors Using SAMDI Mass Spectrometry. J Biomol Screen. 2015;20:842–848. doi: 10.1177/1087057115588512. [DOI] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola L, Frojdo S. Resveratrol: one molecule, many targets. IUBMB Life. 2008;60:323–332. doi: 10.1002/iub.47. [DOI] [PubMed] [Google Scholar]
- Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, Steegborn C, Nowak T, Schutkowski M, Pellegrini L, Sansone L, Villanova L, Runci A, Pucci B, Morgante E, Fini M, Mai A, Russo MA, Tafani M. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. 2015;11:253–270. doi: 10.1080/15548627.2015.1009778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Quinti L, Casale M, Moniot S, Pais TF, Van Kanegan MJ, Kaltenbach LS, Pallos J, Lim RG, Naidu SD, Runne H, Meisel L, Rauf NA, Leyfer D, Maxwell MM, Saiah E, Landers JE, Luthi-Carter R, Abagyan R, Dinkova-Kostova AT, Steegborn C, Marsh JL, Lo DC, Thompson LM, Kazantsev AG. SIRT2- and NRF2-Targeting Thiazole-Containing Compound with Therapeutic Activity in Huntington’s Disease Models. Cell Chem Biol. 2016;23:849–861. doi: 10.1016/j.chembiol.2016.05.015. [DOI] [PubMed] [Google Scholar]
- Rajabi N, Auth M, Troelsen KR, Pannek M, Bhatt DP, Fontenas M, Hirschey MD, Steegborn C, Madsen AS, Olsen CA. Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure-Activity Relationship, Biostructural, and Kinetic Insight. Angew Chem Int Ed Engl. 2017;56:14836–14841. doi: 10.1002/anie.201709050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadori G, Fujikawa T, Anderson J, Berglund ED, Frazao R, Michan S, Vianna CR, Sinclair DA, Elias CF, Coppari R. SIRT1 deacetylase in SF1 neurons protects against metabolic imbalance. Cell Metab. 2011;14:301–312. doi: 10.1016/j.cmet.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh D, Fischer F, Gertz M, Lakshminarasimhan M, Bergbrede T, Aladini F, Kambach C, Becker CFW, Zerweck J, Schutkowski M, Steegborn C. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat Commun. 2013;4:2327. doi: 10.1038/ncomms3327. [DOI] [PubMed] [Google Scholar]
- Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki G, Iwata TN, Li J, Riedel CG, Picard CL, Jan M, Murphy CT, Lee SS. The evolutionarily conserved longevity determinants HCF-1 and SIR-2.1/SIRT1 collaborate to regulate DAF-16/FOXO. PLoS Genetics. 2011;7:e1002235. doi: 10.1371/journal.pgen.1002235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler C, Nowak T, Pannek M, Gertz M, Nguyen GT, Scharfe M, Born I, Sippl W, Steegborn C, Schutkowski M. Chemical probing of the human sirtuin 5 active site reveals its substrate acyl specificity and peptide-based inhibitors. Angew Chem Int Ed Engl. 2014;53:10728–10732. doi: 10.1002/anie.201402679. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf T, Schiedel M, Karaman B, Roessler C, North BJ, Lehotzky A, Olah J, Ladwein KI, Schmidtkunz K, Gajer M, Pannek M, Steegborn C, Sinclair DA, Gerhardt S, Ovadi J, Schutkowski M, Sippl W, Einsle O, Jung M. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat Commun. 2015;6:6263. doi: 10.1038/ncomms7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo HS, Laitinen T, Poso A, Jarho E, Lahtela-Kakkonen M. Identification of novel SIRT3 inhibitor scaffolds by virtual screening. Bioorg Med Chem Lett. 2013;23:2990–2995. doi: 10.1016/j.bmcl.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Sanders BD, Jackson B, Marmorstein R. Structural basis for sirtuin function: What we know and what we don’t. Biochim Biophys Acta. 2010;1804:1604–1616. doi: 10.1016/j.bbapap.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands BE, Joshi S, Haddad J, Freudenberg JM, Oommen DE, Hoffmann E, McCallum SW, Jacobson E. Assessing Colonic Exposure, Safety, and Clinical Activity of SRT2104, a Novel Oral SIRT1 Activator, in Patients with Mild to Moderate Ulcerative Colitis. Inflammatory bowel diseases. 2016;22:607–614. doi: 10.1097/MIB.0000000000000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Brace CS, Ben-Josef G, West T, Wozniak DF, Holtzman DM, Herzog ED, Imai S. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci. 2010;30:10220–10232. doi: 10.1523/JNEUROSCI.1385-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA. Sirtuin chemical mechanisms. Biochim Biophys Acta. 2010;1804:1591–1603. doi: 10.1016/j.bbapap.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Moir RD, Schramm VL, Willis IM. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol Cell. 2005;17:595–601. doi: 10.1016/j.molcel.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- Schlicker C, Boanca G, Lakshminarasimhan M, Steegborn C. Structure-based Development of Novel Sirtuin Inhibitors. Aging (Albany NY) 2011;3:852–857. doi: 10.18632/aging.100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A, Min J, Antoshenko T, Wang CL, Allali-Hassani A, Dong A, Loppnau P, Vedadi M, Bochkarev A, Sternglanz R, Plotnikov AN. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure. 2007;15:377–389. doi: 10.1016/j.str.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Schuster S, Roessler C, Meleshin M, Zimmermann P, Simic Z, Kambach C, Schiene-Fischer C, Steegborn C, Hottiger MO, Schutkowski M. A continuous sirtuin activity assay without any coupling to enzymatic or chemical reactions. Sci Rep. 2016;6:22643. doi: 10.1038/srep22643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutkowski M, Fischer F, Roessler C, Steegborn C. New assays and approaches for discovery and design of Sirtuin modulators. Exp Opin Drug Disc. 2014;9:183–99. doi: 10.1517/17460441.2014.875526. [DOI] [PubMed] [Google Scholar]
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- Sinclair DA. Paradigms and pitfalls of yeast longevity research. Mech Ageing Dev. 2002;123:857–867. doi: 10.1016/s0047-6374(02)00023-4. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles–a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Smith BC, Denu JM. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry. 2007;46:14478–14486. doi: 10.1021/bi7013294. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpferl SW, Brand SE, Jiang JC, Korona B, Tiwari A, Dai J, Seo JG, Jazwinski SM. Natural genetic variation in yeast longevity. Genome research. 2012;22:1963–1973. doi: 10.1101/gr.136549.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenkel B, Fischer F, Steegborn C. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg Med Chem Lett. 2013;23:143–146. doi: 10.1016/j.bmcl.2012.10.136. [DOI] [PubMed] [Google Scholar]
- Sundriyal S, Moniot S, Mahmud Z, Yao S, Di Fruscia P, Reynolds CR, Dexter DT, Sternberg MJ, Lam EW, Steegborn C, Fuchter MJ. Thienopyrimidinone Based Sirtuin-2 (SIRT2)-Selective Inhibitors Bind in the Ligand Induced Selectivity Pocket. J Med Chem. 2017;60:1928–1945. doi: 10.1021/acs.jmedchem.6b01690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussmuth SD, Haider S, Landwehrmeyer GB, Farmer R, Frost C, Tripepi G, Andersen CA, Di Bacco M, Lamanna C, Diodato E, Massai L, Diamanti D, Mori E, Magnoni L, Dreyhaupt J, Schiefele K, Craufurd D, Saft C, Rudzinska M, Ryglewicz D, Orth M, Brzozy S, Baran A, Pollio G, Andre R, Tabrizi SJ, Darpo B, Westerberg G. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br J Clin Pharmacol. 2015;79:465–476. doi: 10.1111/bcp.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Khan MN, Sawada H, Imai E, Itoh Y, Yamatsuta K, Tokuda N, Takeuchi J, Seko T, Nakagawa H, Miyata N. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J Med Chem. 2012;55:5760–5773. doi: 10.1021/jm3002108. [DOI] [PubMed] [Google Scholar]
- Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, Ro J, Wagner GR, Green MF, Madsen AS, Schmiesing J, Peterson BS, Xu G, Ilkayeva OR, Muehlbauer MJ, Braulke T, Muhlhausen C, Backos DS, Olsen CA, McGuire PJ, Pletcher SD, Lombard DB, Hirschey MD, Zhao Y. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennen RI, Berber E, Chua KF. Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech Ageing Dev. 2010;131:185–192. doi: 10.1016/j.mad.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Krueger JG, Li K, Jabbari A, Brodmerkel C, Lowes MA, Suarez-Farinas M. Meta-analysis derived (MAD) transcriptome of psoriasis defines the “core” pathogenesis of disease. PLoS One. 2012;7:e44274. doi: 10.1371/journal.pone.0044274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Toiber D, Sebastian C, Mostoslavsky R. Characterization of nuclear sirtuins: molecular mechanisms and physiological relevance. Handb Exp Pharmacol. 2011;206:189–224. doi: 10.1007/978-3-642-21631-2_9. [DOI] [PubMed] [Google Scholar]
- Tong Z, Wang Y, Zhang X, Kim DD, Sadhukhan S, Hao Q, Lin H. SIRT7 Is Activated by DNA and Deacetylates Histone H3 in the Chromatin Context. ACS Chem Biol. 2016;11:742–747. doi: 10.1021/acschembio.5b01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M. Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins) ChemMedChem. 2007;2:1419–1431. doi: 10.1002/cmdc.200700003. [DOI] [PubMed] [Google Scholar]
- Valente S, Mellini P, Spallotta F, Carafa V, Nebbioso A, Polletta L, Carnevale I, Saladini S, Trisciuoglio D, Gabellini C, Tardugno M, Zwergel C, Cencioni C, Atlante S, Moniot S, Steegborn C, Budriesi R, Tafani M, Del Bufalo D, Altucci L, Gaetano C, Mai A. 1,4-Dihydropyridines Active on the SIRT1/AMPK Pathway Ameliorate Skin Repair and Mitochondrial Function and Exhibit Inhibition of Proliferation in Cancer Cells. J Med Chem. 2016;59:1471–1491. doi: 10.1021/acs.jmedchem.5b01117. [DOI] [PubMed] [Google Scholar]
- van der Meer AJ, Scicluna BP, Moerland PD, Lin J, Jacobson EW, Vlasuk GP, van der Poll T. The Selective Sirtuin 1 Activator SRT2104 Reduces Endotoxin-Induced Cytokine Release and Coagulation Activation in Humans. Critical care medicine. 2015;43:e199–202. doi: 10.1097/CCM.0000000000000949. [DOI] [PubMed] [Google Scholar]
- Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, Bredbenner K, Park R, Sinclair DA, Bohr VA, Gorbunova V, Seluanov A. JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Rep. 2016;16:2641–2650. doi: 10.1016/j.celrep.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubramanian S, Noh RM, Daga S, Langrish JP, Joshi NV, Mills NL, Hoffmann E, Jacobson EW, Vlasuk GP, Waterhouse BR, Lang NN, Newby DE. Cardiovascular effects of a novel SIRT1 activator, SRT2104, in otherwise healthy cigarette smokers. J Am Heart Assoc. 2013;2:e000042. doi: 10.1161/JAHA.113.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu CB, Bemis JE, Disch JS, Ng PY, Nunes JJ, Milne JC, Carney DP, Lynch AV, Smith JJ, Lavu S, Lambert PD, Gagne DJ, Jirousek MR, Schenk S, Olefsky JM, Perni RB. Discovery of imidazo[1,2-b]thiazole derivatives as novel SIRT1 activators. J Med Chem. 2009;52:1275–1283. doi: 10.1021/jm8012954. [DOI] [PubMed] [Google Scholar]
- Westerberg G, Chiesa JA, Andersen CA, Diamanti D, Magnoni L, Pollio G, Darpo B, Zhou M. Safety, pharmacokinetics, pharmacogenomics and QT concentration-effect modelling of the SirT1 inhibitor selisistat in healthy volunteers. Br J Clin Pharmacol. 2015;79:477–491. doi: 10.1111/bcp.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiewel MA, van der Meer AJ, Haddad J, Jacobson EW, Vlasuk GP, van der Poll T. SRT2379, a small-molecule SIRT1 activator, fails to reduce cytokine release in a human endotoxemia model. Critical Care. 2013;17:P8–P8. [Google Scholar]
- Yang H, Baur JA, Chen A, Miller C, Adams JK, Kisielewski A, Howitz KT, Zipkin RE, Sinclair DA. Design and synthesis of compounds that extend yeast replicative lifespan. Aging Cell. 2007;6:35–43. doi: 10.1111/j.1474-9726.2006.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Ma X, Yuan C, He Y, Li L, Fang S, Xia W, He T, Qian S, Xu Z, Li G, Wang Z. Discovery of 2-((4,6-dimethylpyrimidin-2-yl)thio)-N-phenylacetamide derivatives as new potent and selective human sirtuin 2 inhibitors. European J Med Chem. 2017;134:230–241. doi: 10.1016/j.ejmech.2017.04.010. [DOI] [PubMed] [Google Scholar]
- Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, Hayes SD, Jedrychowski MP, Vyas FS, Zaganjor E, Guarani V, Ringel AE, Gygi SP, Harper JW, Haigis MC. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell. 2016;167:985–1000 e1021. doi: 10.1016/j.cell.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YK, Oon CE. Sirtuin inhibitors: An overview from medicinal chemistry perspective. Anticancer Agents Med Chem. 2016;16:1003–1016. doi: 10.2174/1871520616666160310141622. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, Babendure JL, Lu JC, Smith JJ, Jirousek MR, Olefsky JM. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Molecular and cellular biology. 2009;29:1363–1374. doi: 10.1128/MCB.00705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You W, Rotili D, Li TM, Kambach C, Meleshin M, Schutkowski M, Chua KF, Mai A, Steegborn C. Structural Basis of Sirtuin 6 Activation by Synthetic Small Molecules. Angew Chem Int Ed Engl. 2017;56:1007–1011. doi: 10.1002/anie.201610082. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Au Q, Zhang M, Barber JR, Ng SC, Zhang B. Identification of a small molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem Biophys Res Commun. 2009;386:729–733. doi: 10.1016/j.bbrc.2009.06.113. [DOI] [PubMed] [Google Scholar]
- Zhao X, Allison D, Condon B, Zhang F, Gheyi T, Zhang A, Ashok S, Russell M, Macewan I, Qian Y, Jamison JA, Luz JG. The 2.5 A crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analog) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem. 2013;56:963–9. doi: 10.1021/jm301431y. [DOI] [PubMed] [Google Scholar]
- Zorn JA, Wells JA. Turning enzymes ON with small molecules. Nature Chem Biol. 2010;6:179–188. doi: 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]