

SUMMARY
DNA damage provokes mutations and cancer, and results from external carcinogens or endogenous cellular processes. Yet, the intrinsic instigators of endogenous DNA damage are poorly understood. Here we identify proteins that promote endogenous DNA damage when overproduced: the DNA “damage-up” proteins (DDPs). We discover a large network of DDPs in Escherichia coli and deconvolute them into six function clusters, demonstrating DDP mechanisms in three: reactive-oxygen increase by transmembrane transporters, chromosome loss by replisome binding, and replication stalling by transcription factors. Their 284 human homologs are over-represented among known cancer drivers, and their RNAs in tumors predict heavy mutagenesis and poor prognosis. Half of tested human homologs promote DNA damage and mutation when overproduced in human cells, with DNA-damage-elevating mechanisms like those in E. coli. Together, our work identifies networks of DDPs that provoke endogenous DNA damage and may reveal DNA-damage-associated functions of many human known and newly implicated cancer-promoting proteins.
Keywords: cancer, DNA-damage response, DNA damage-up proteins, DNA double-strand breaks, endogenous DNA damage, Escherichia coli, evolvability, evolution, genome instability, human cells, microbial cancer models, mutagenesis, replication-fork reversal, replication stress, SOS response
In Brief
A large network of proteins in bacteria promotes endogenous DNA damage when upregulated and these are shown to have human homologs that form a cancer-predictive network.
INTRODUCTION
DNA damage often underlies “spontaneous” mutations (Tubbs and Nussenzweig, 2017) which drive cancer, genetic diseases, and evolution. DNA damage can result from exogenous agents such as radiation or tobacco smoke; indeed most known carcinogens are DNA-damaging agents and mutagens. Most DNA damage is generated endogenously in cells (Tubbs and Nussenzweig, 2017) by intrinsic cellular processes that involve macromolecules including proteins. Presumably, misregulateion of functional proteins may disturb cellular homeostasis in ways that provoke endogenous DNA damage. The identities and functions of endogenous DNA damage-promoting proteins in any organism are poorly understood (Figure 1A).
Figure 1. Comprehensive Discovery of E. coli DNA “Damage-up” Protein Network.
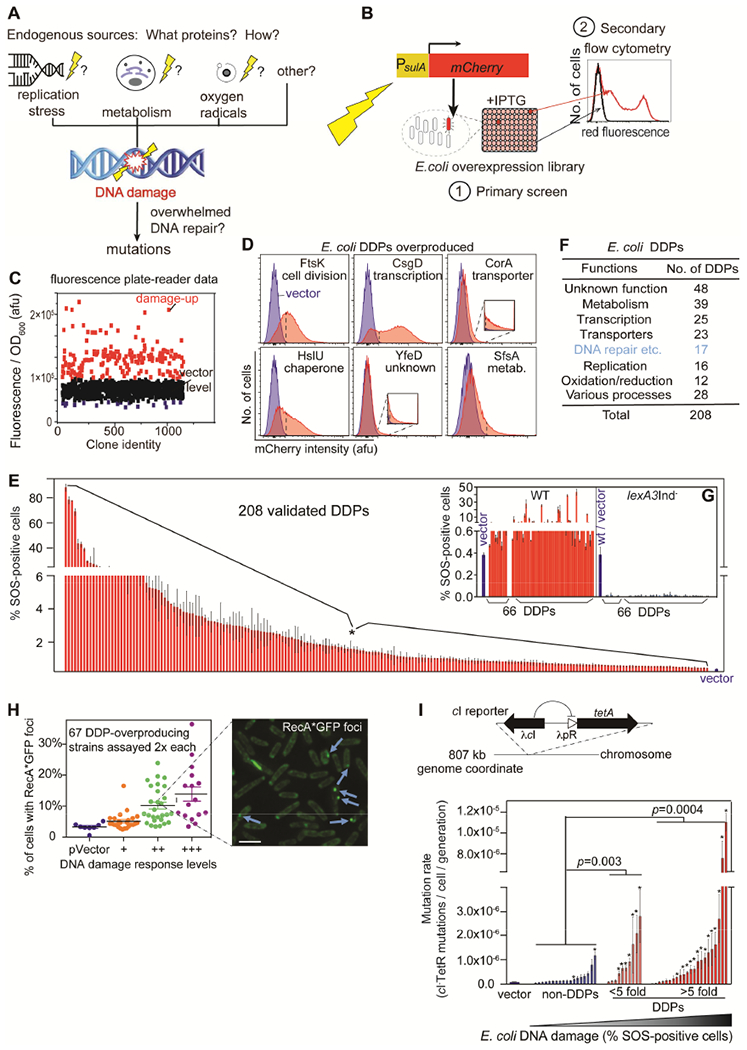
(A) Endogenous DNA damage may promote mutations and occurs by unknown means.
(B) Screen for overproduction DNA damage-up proteins (DDPs). (1) Fluorescence plate-reader screen of E. coli Mobile overexpression library for fluorescence from SOS-DNA-damage-response reporter (Nehring et al., 2016). (2) Elimination of false-positives by flow-cytometry–single-cell assay.
(C) Plate-reader representative results: afu, arbitrary fluorescence units, per OD600, (biomass). Red, potential DDPs with fold change >30%.
(D) Representative flow-cytometry validation of SOS-positive DDPs. Dashed line, “gate” for SOS-positives (significance, STAR Methods). Blue, vector control; red, DDP producers.
(E) % SOS-positive cells for the 208 validated E. coli DDPs (Table S1).
(F) DDP network summary; proteins of many different functions are DDPs.
(G) LexA-dependence of fluorescence from DDPs shows SOS-response activation/DNA damage.
(H) SOS-positive phenotype correlated with RecA*GFP foci, indicating persistent single-stranded DNA. 67 representative DDPs show 32 (48%) with elevated RecA*GFP foci (p < 0.05, unpaired two-tail t-test), r = 0.7, p = 1.3×10−10, Pearson’s correlation, (data, Table S1). Scale bar: 2μm.
(I) Mutation-rate increase with DNA-damage levels in representative DDP-producing clones. Above, assay (STAR Methods). Each bar, the mean mutation rate (± SEM) of each strain, N=3 (STAR Methods; Table S1). P-values, fraction of clones with mutation rate significantly higher than vector-only control, one-way Fisher’s exact test.
One way to discover proteins that promote endogenous DNA damage is by overproduction—a natural event that occurs frequently by stochastic cell-to-cell variation (Elowitz et al., 2002) or copy-number alteration—and a major driver of cancers (Zack et al., 2013). Given DNA biology conservation across life (Makarova and Koonin, 2013), proteins that promote spontaneous DNA damage may be conserved and their identification could potentially inform strategies for prevention, diagnosis, and treatment of disease, including cancer, aging, and pathogen evolution (Fitzgerald et al., 2017).
Some proteins that prevent or reduce endogenous DNA damage have been identified by loss-of-function mutations/knock-downs that increase DNA damage (Alvaro et al., 2007; Lovejoy et al., 2009; Paulsen et al., 2009). DNA-repair proteins are in this category. By contrast, no unbiased screen has been reported for proteins that actively promote endogenous DNA damage in cells. Though a nucleus-specific screen identified some (Lovejoy et al., 2009), the numbers and range of functions of proteins and mechanisms that cause spontaneous endogenous DNA damage remain elusive.
Here we report comprehensive discovery in Escherichia coli of a large, diverse network of proteins that, when overproduced, lead to increased endogenous DNA damage: the DNA “damage-up” proteins (DDPs). Their human homologs behave similarly in human cells, and predict cancer mortality and mutation loads. Bacterial and human DDPs increase mutation rates, and share common mechanisms of DNA-damage instigation, three of which we identify. The identities and functions of the bacterial and human DDPs provide important general models for illuminating mechanisms of genesis of spontaneous endogenous DNA damage, and may inform discovery of cancer-promoting functions of many known and newly discovered cancer-driving proteins.
RESULTS
Diverse Protein Network Promotes DNA Damage
We screened an inducible overexpression library of all E. coli genes in two steps to identify clones with increased endogenous DNA damage (Figure 1B). For both, we measured fluorescence of cells with a fluorescence-reporter gene driven by an SOS DNA-damage-response-activated promoter (Nehring et al., 2016) (Figure 1B), which reports DNA-damage-response induction (Pennington and Rosenberg, 2007) (Figure S1A-B). The primary screen used a high-throughput but low-resolution fluorescence plate-reader to identify potential clones with increased DNA damage (Figure 1B-C, STAR Methods), then false positives were eliminated using high-resolution flow-cytometry (Figure 1B, D, STAR Methods), reporting DNA damage at the single-cell level (Pennington and Rosenberg, 2007). Flow cytometry validated 208 proteins as genuine DDPs that increase DNA-damage when overproduced (Figure 1E,F Table S1). Additional confirmations of their DNA-damage promotion are shown in Figures 1G and S1C; Table S1. Three additional, independent strategies demonstrated that all 208 are genuine DDPs (STAR Methods), including an assay for persistent single-stranded (damaged) DNA detected as foci of a fluorescent partial-function DNA-damage-sensor protein RecA*GFP (Renzette et al., 2005) (Figure 1H).
DDPs span diverse cellular roles (Figure 1F; Table S1), with only 8% known DNA-repair proteins. Normally, DNA-repair proteins reduce DNA damage; their overproduction, here, might perturb undamaged DNA, titrate repair partner proteins away from DNA damage, and/or inhibit DNA repair—direct or indirect means. We call all of these proteins DDPs because they increase cellular levels of endogenous DNA damage when overproduced.
The 208 DDPs display properties of protein networks. Functionally, all cause increased DNA damage on overproduction and show increased protein-protein associations compared with random sets of E. coli proteins, and slight but significant co-expression (Figure S2A legend), all indicating a network.
We estimate the E. coli DDP network to exceed the 208 proteins identified with a predicted ≥331: about 8% of E. coli protein-coding genes (STAR Methods). That perturbation of diverse aspects of cell biology can indirectly or directly promote DNA damage (Figure 1F; Table S1) may explain why such a large fraction of genes promote endogenous DNA damage when dysregulated.
DDPs Promote Mutagenesis
We tested the hypothesis that triggering endogenous DNA damage would increase mutation rates (Figure 1A) with 32 representative E. coli DDPs (STAR Methods, Table S1). The data (Figure 1I) show that increased endogenous DNA-damage corresponds with elevated mutation rates, confirmed by sequencing (Figure S1F), which also revealed various kinds of mutations including base substitutions, indels, transpositions, and gross chromosomal rearrangements (GCRs, Figure S1F). These mimic increased small mutations and GCRs in various cancers (Stratton, 2011). Although the SOS response induces mutations by upregulating error-prone DNA polymerases (Pols) V and IV, many of the mutations differ from common Pol V/IV errors, suggesting that types of DNA damage, not merely SOS-response induction, may influence the kinds and rates of mutations made (Figure S1F, Table S1). Thus, overproduction of diverse E. coli proteins causes DNA-damage and mutations of essentially all kinds (Figures 1I and S1F; Table S1).
Human Homolog Network Associated with Cancers
We identified 284 human proteins with amino-acid similarity to 58 E. coli DDPs (homologs, STAR Methods, Figure 2A; Table S2). These are candidate human (h)DDPs. The remaining E. coli DDPs are mostly analogs of human proteins (function similarly but are not homologous, Table S1). 163 E. coli DDPs have identifiable human analogs, homologs, or both.
Figure 2. Human Homologs of E. coli DDPs a Network Associated with Cancers.
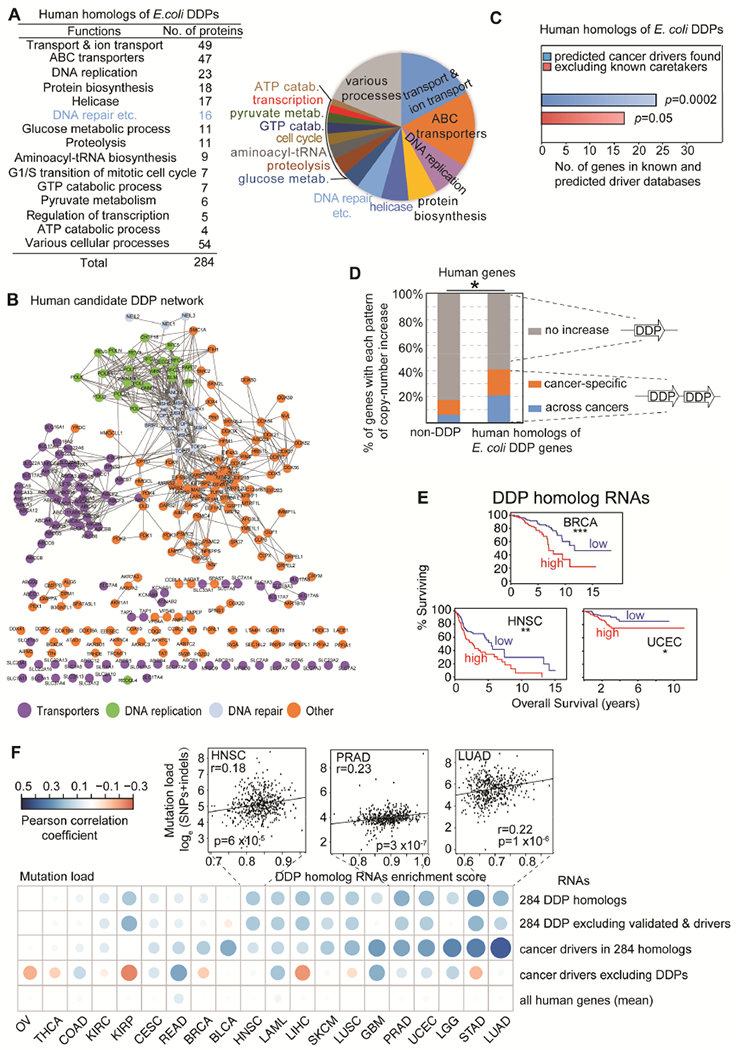
(A) Summary of 284 human homologs of E. coli DDPs (Table S2).
(B) Protein-protein associations of human DDP homologs (Figure S2; Table S2).
(C) Human homologs are overrepresented among known and predicted cancer drivers (blue bar), even without known DNA-repair “caretakers”.
(D) hDDP candidates are enriched among genes with cancer-associated copy-number increases, indicating overexpression in cancers (Figure S3A-C, Table S4).
(E) Decreased cancer survival with high DDP-homolog RNA levels in cancers: our analyses of TCGA data (STAR Methods). Cancer types, STAR Methods. *, **, ***, high versus low levels of 284 RNAs p ⩽ 0.05; ⩽ 0.01, and ⩽ 0.001, log-rank test.
(F) High hDDP candidate RNA levels predict tumor mutation loads (TCGA data). Each dot, Pearson correlation coefficient 284 homolog RNAs/total RNAs versus tumor mutation load. The average correlation strength was in the top 0.5% of correlations for randomly selected groups of genes. X-axis, cancer types.
The human homologs constitute a protein-protein association network, more than random human homologs of E. coli proteins (Figures 2B, S2B). Only 5.6% are known DNA-repair proteins (Figure 2A).
The human homologs are overrepresented among known (Forbes et al., 2015) and predicted (D’Antonio and Ciccarelli, 2013) gain- and loss-of-function cancer-driving genes, with or without inclusion of known DNA-repair proteins (Figure 2C; Table S3). Human homologs of random E. coli proteins are not overrepresented, showing that DDP homologs, not conserved proteins generally are cancer associated (STAR Methods). Our analysis of data from an overexpression screen in human cells (Lovejoy et al., 2009) also shows cancer association (STAR Methods). Only one protein, FIGNL1, overlaps with our 284 hDDP candidates, indicating many new genes revealed by the E. coli screen.
hDDP candidate genes show increased copy numbers in human cancers in patients in The Cancer Genome Atlas (TCGA) (Gao et al., 2013). About 40% have increased copy numbers (GISTIC threshold copy-number gain ⩾1), compared with fewer than 20% of non-DDP genes increased in those cancers (Figure 2D, p = 0.04, one-way Fisher’s exact test), suggesting that their overexpression is associated with cancers. Table S4 shows homologs with copy-number increases (Figure S3A-C).
In at least three cancer types, overexpression of the 284 homolog RNAs, relative to total RNAs, is associated with decreased overall survival (Figure 2E), even with known/predicted drivers excluded (Figure S3D-H), indicating the cancer relevance of network genes not known previously to drive cancers. Moreover, increased levels of the 284 RNAs is strongly associated with total tumor mutation loads in ≥12 cancer types (Figure 2F, STAR Methods), including with known/predicted drivers excluded (Figure 2F). The data imply that previously unsuspected cancer-gene candidates also promote mutagenesis. These data highlight the network properties of the 284 DDP-homologs, their frequent overexpression in cancers, and predictive power for poor survival and high tumor mutation loads.
Human Homologs Promote DNA Damage and Mutation
We validated a sample of candidate hDDPs as genuine DNA-damage instigators in human cells (Figure 3), cloning several de novo (STAR Methods) to create 70 full-length sequence-verified overexpression GFP-fusion genes, 3 human homologs of E. coli damage-down proteins, as possible negative controls, and 20 negative-control random non-DDPs (Table S5, ~half random human homologs of E. coli proteins). About half the homologs are amplified in cancers in TCGA (Gao et al., 2013). We performed three flow-cytometric assays (Figure 3A) for increased DNA damage (STAR Methods). The assays measure—increased (i) γH2AX levels, a DNA double-strand-break (DSB) marker (Kinner et al., 2008); (ii) γH2AX in cells treated with a nonhomologous-break-repair (DNA-PK) inhibitor (sensitized screen); and (iii) phospho-p53, a DNA-damage marker (Sakaguchi et al., 1998). 45% of the human homologs (33 of 73) showed increased DNA damage (Figure 3B), a highly significant enrichment compared with 20 random human proteins (p < 0.0001 one-way Fisher’s exact test; Figure S3I-K; Table S6). The 45% validation rate is higher than a candidate DNA/nucleus-associated-protein screen for increased DNA damage, in which 1.7% of candidate proteins were validated (Lovejoy et al., 2009) (p=1 × 10−36, Fisher’s exact test), demonstrating the predictive power of the E. coli screen.
Figure 3. Human Homologs Promote DNA Damage in Human Cells.
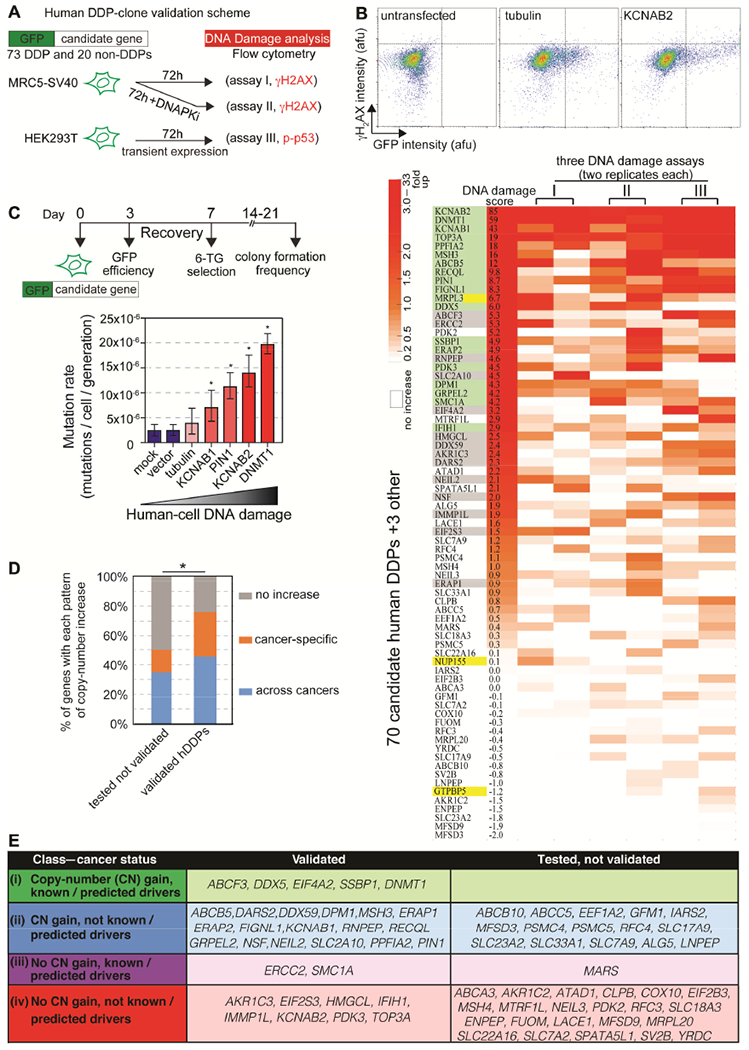
(A) hDDP-candidate-GFP N-terminal fusions (and 3 damage-down-, plus 20 non-DDP controls) were transiently overproduced and green cells screened for DNA damage by flow cytometry.
(B) 33 validated hDDPs. Upper: representative flow cytometric assay (STAR Methods, Figure S3I-K; Table S6). Lower: heatmap, flow-cytometric data normalized to GFP-tubulin. Data ranked by cumulative DNA-damage score. Green, damage-up in ⩾ 2 assays; gray, one assay; yellow, damage-down homologs; white, not damage-up. 45% validated; more than among 20 random human genes (p <0.0001, two-tailed unpaired t-test with FDR correction, Figure S3I-K).
(C) Increased mutation with overproduced validated hDDPs in human-cell HPRT forward-mutation assays. Lower: mutation rates of selected hDDP overproducers; error bars, 95% CIs.
(D) Validated hDDP genes enriched among cancer-associated copy-number increases (p = 0.02, one-way Fisher’s exact test).
(E) New and known potential cancer-promoters predicted among 33 validated hDDPs, suggesting potential overexpression cancer-promoting roles for all of these genes. Classes (i) 16%; (ii) 53%; (iii) 6%; and (iv) 25%.
Our validation rate is probably underestimated because the human assays are biased towards DSB detection and only 41% of E. coli DDPs promote DSBs (Table S1)—a number similar to the 45% hDDPs validated in our assays. The γH2AX assays (Figure 3AI and II) are DSB-specific (Shee et al., 2013); and only 4 of 33 validated hDDPs show phospho-p53 signal without γH2AX (Figure 3B), implying DSB-bias of phospho-p53. Thus, many additional hDDP candidates may be genuine hDDPs that cause non-DSB DNA damage not detected by these assays.
Overproduction of 4 of 4 validated hDDPs increased mutation rates compared with controls in human cells in forward-mutation fluctuation-test assays for hypoxanthine-guanine phosphoribosyl transferase (HPRT) deficiency (Figure 3C). Thus, hDDPs may drive cancers by increasing genome instability—a cancer hallmark (Hanahan and Weinberg, 2011).
The validated hDDPs encompass four classes (Figure 3E), none predicted previously to promote DNA damage, and some with no hypothesized link to cancer (e.g., classes iii and iv). The rates of validation in each class allow us to estimate that many additional hDDPs populate the 284-protein candidate hDDP network, that would test positive in our assays (STAR Methods). −75% of the validated hDDP genes show cancer-associated copy-number increases in TCGA cancers (Gao et al., 2013) (GISTIC threshold copy-number gain ⩾ 1, Figure 3D), exceeding the candidates not validated (Figure 3D). The data imply that validated hDDPs may drive cancer via DNA damage when overproduced, and that our DNA-damage assays relate to human cancer biology.
Functional Systems Biology
We used the tractable E. coli model to discover DDP functions, to bin the 208 E. coli DDPs into function clusters that reflect the molecular kinds, causes, and consequences of DNA damage provoked by their overproduction. The phenotypes are from seven quantitative functional assays, many at the single-cell level (Figure 4). We use the data to predict and demonstrate mechanisms by which diverse conserved proteins increase endogenous DNA damage.
Figure 4. Kinds, Causes and Consequences of DNA Damage from E. coli DDPs.
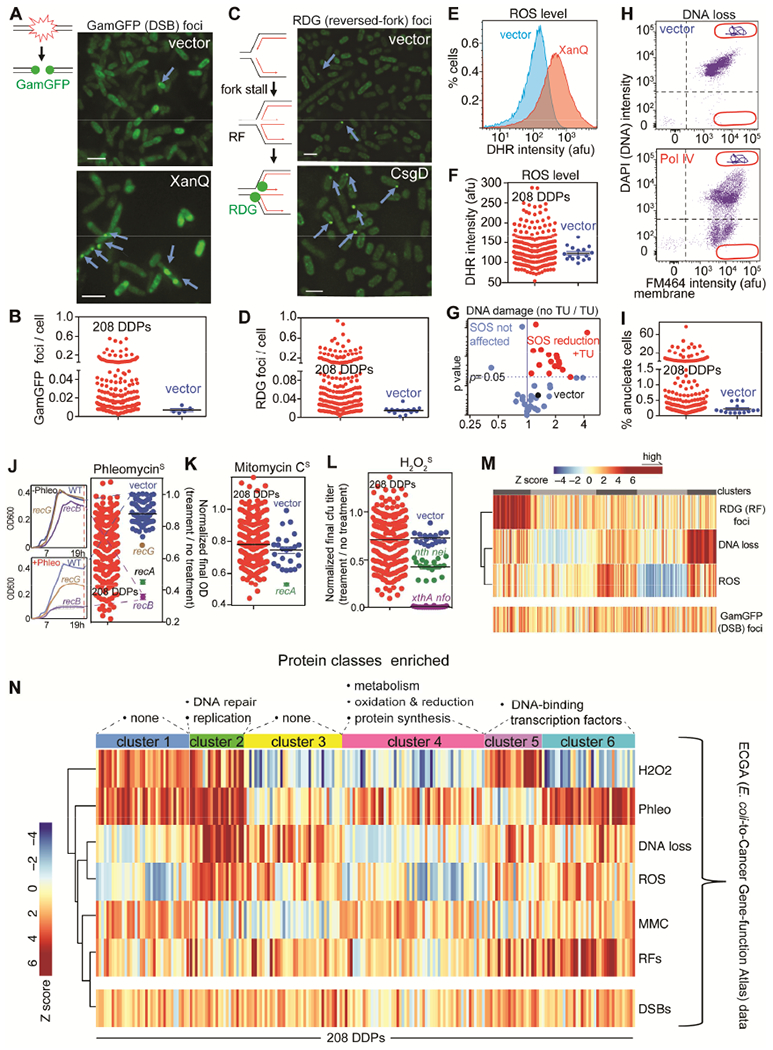
Clone by clone data Table S1.
(A) DDPs that increase DNA double-strand breaks (DSBs), detected as GamGFP foci. Scale bar: 2μm. Lines, DNA strands; green balls, GamGFP.
(B) 87 of the 208 (45% of) E. coli DDPs promote DSBs.
(C) Stalled, reversed replication forks (RFs) detected as RDG foci in recA− cells, per (Xia et al., 2016). Scale bar: 2μm. Lines, DNA strands; red lines, new strands; green balls, RDG.
(D) 106 of the 208 (51% of) E. coli DDPs promote fork stalling and reversal.
(E-I) Flow-cytometric assays for—
(E) elevated ROS, DHR fluorescence, example.
(F) 56 (27% of) DDPs induce high ROS.
(G) DNA damage (% SOS-on cells) from 17 of 43 high-ROS DDPs tested is reduced by ROS-quenching agent thiourea (TU).
(H) DNA loss: “anucleate” cells with no DNA, example. Events below the horizontal line, anucleate.
(I) 67 of 208 (32% of) DDPs induce DNA loss.
(J-L) Sensitivity to DNA-damaging agents implies DNA-repair-pathway reduction (potential saturation), possibly from elevated DNA damage. Relevant DNA-repair-defective controls shown. Assays for slowed growth per J left (STAR Methods).
(J) 106 of 208 DDP clones (51%) show phleomycin (DSB) sensitivity.
(K) 10 of 208 DDP clones (5%) sensitive to DNA cross-linker mitomycin C (MMC) (reduced NER and/or HDR).
(L) H2O2 sensitivity (reduced BER) from 75 of 208 DDPs (36%).
(M) Stalled replication (RFs) clusters with particular DDPs; DNA breakage does not (STAR Methods).
(N) Clustering Z scores reveal DNA-damage signatures. H2O2, hydrogen-peroxide sensitivity; Phleo, phleomycin sensitivity; DNA loss (anucleate cells); ROS levels; MMC, (MMC sensitivity); RFs (reversed forks); DSBs. Vertical bars: phenotype scores of each DDP clone. The 6 clusters/DNA-damage signatures suggest at least 6 mechanisms of DNA-damage generation. Protein category enrichment (above, one-way Fisher’s exact test): clusters 2 p = 0.01; 4; p = 0.01, 5 and 6 p = 0.03.
DNA Double-Strand Breaks from 40% of E. coli DDPs
We quantified DSBs in single cells as fluorescent foci of engineered DSB-end-specific binding protein GamGFP (Shee et al., 2013). GamGFP “traps” DSBs, prevents their repair in E. coli and mammalian cells, and labels DSB ends as foci at an estimated 70% efficiency (Shee et al., 2013). 87 of the 208 E. coli DDP-overproducing clones showed more GamGFP foci than vector-only controls, and 25 random SOS-negative (non-DDP) clones (Figures 4A,B and S4A, Table S1, STAR Methods). Our finding that 121 (59% of) DDP-overproducing clones do not increase GamGFP foci suggests that single-stranded (ss)DNA, the SOS-response inducing signal, frequently accumulates at sites other than DSBs. The 41% of E. coli DDPs that elevate GamGFP/DSB foci are not enriched in any gene-function category (Table S1), implying that DSBs result from diverse cellular processes.
Half of DDPs Promote Replication Stalling
Stalling of DNA replication leads to DNA damage and can create four-way DNA junctions when stalled replication forks “reverse” (illustrated Figure 4C). Reversed forks (RFs) block resumption of DNA replication, lead to replication-fork breakage (Seigneur et al., 1998), and reflect a cause of DNA damage—stalled replication. We quantified RFs as foci of RuvCDefGFP (RDG) engineered 4-way-junction-specific DNA-binding protein in cells that lack homology-directed-repair (HDR) protein RecA, in which most/all RDG foci represent RFs (Xia et al., 2016). We found that 106 of 208 DDP clones showed more RDG foci than vector-only and 30 other control clones (Figure 4D, Figure S4B; Table S1) and 49 also showed increased DSBs (Table S1), a significant correlation (p = 0.03, r=0.15 Spearman’s correlation). Thus, ≥51% of DDPs promote replication stalling, indicating the importance of DNA replication to DNA-damage generation by many, but not all, of the proteins.
DNA Damage by Reactive Oxygen: Transporters and Metabolism
We measured intracellular levels of reactive oxygen species (ROS) using the peroxide-indicator dye, dihydrorhodamine (DHR) (Gutierrez et al., 2013) and flow cytometry, and found that 56 DDPs increased ROS levels (Figures 4F and S4C, D). In at least 17 of the 56, DNA damage was reduced by ROS-quenching agent thiourea (Figure 4G, Table S1). Thus, high endogenous ROS underlie DNA damage in a subset of the DDP-producing clones. These comprise five membrane-spanning transporters (investigated below), an excess compared with transporters among E. coli proteins (p=0.002, hypergeometric test). 40% of the other 12 proteins relate to metabolism (Table S1), implying that perturbation of metabolic pathways can cause DNA damage by increasing ROS.
DDPs Promote DNA Loss
Cells can lose DNA via various problems including chromosome-segregation failure, for example by incomplete DNA replication or HDR between chromosomes, which can leave two chromosomes attached at cell division. We identified 67 DDPs that increase DNA-depleted (“anucleate”) cells, using flow cytometry with DNA and cell membranes stained separately (Figures 4H, I and S4E, F; Table S1). Overproduced DNA-repair and replication proteins are enriched among these clones (p = 0.04, one-way Fisher’s exact test, Table S1), implying that excessive DNA-repair and replication proteins promote DNA damage resulting in DNA erosion or chromosome-segregation failure.
DDPs Reduce DNA-repair Capacity
We assayed DNA-repair capacity indirectly, as sensitivity to DNA-damaging agents that induce damage repaired by specific DNA-repair mechanisms: DSB-, ssDNA-break-, and ROS-instigator phleomycin; base-oxidizing agent hydrogen peroxide (H2O2); and DNA cross-linking agent mitomycin-C (MMC). These cause damage repaired by HDR (phleomycin), base-excision repair (“BER”, H2O2), and both nucleotide-excision repair and HDR (MMC). Of the 208 DDPs, 106, 75, and 10 were sensitive to phleomycin, H2O2, and MMC, respectively (Figures 4J-L and S5A-E). Collectively, 140 DDP-overproducers were sensitive to at least one DNA-damaging agent, and 45 to multiple drugs (Table S1, Figure S5G). The DDPs are enriched for phleomycin and H2O2, (but not MMC) sensitivity, compared with control non-DDP clones (Figure S5). We excluded the possibilities that most DNA-damage sensitivities resulted from DDP-induced heritable mutations, or transcriptional downregulation of DNA-repair genes (Figure S5F,H). Specific DNA-repair pathways could be either inhibited directly or saturated by DNA damage caused by specific DDPs implying that dysregulating diverse proteins can mimic DNA-repair deficiency without mutations in DNA-repair genes.
E. coli Function-data Clusters Implicate Mechanisms
We grouped quantitative data from the functional assays using stability-based clustering (Figures 4M,N and S5G). The data on RDG (RF) foci analyzed with three other single-cell quantitative parameters—ROS levels, DNA loss, and DSBs—showed high RF loads in a specific cluster (Figure 4M) enriched for DNA-binding transcription factors (TFs), with 29%, compared with 12% among the network as a whole (p = 0.002, one-way Fisher’s exact test, Figure 4M; Table S1). The data imply that distinct protein functions preferentially stall replication. The distinct groups shown by clustering the bulk-culture data are shown in Figure S5G. Grouping all quantitative data sets revealed six discreet function clusters (Figure 4N; Table S1), which may indicate at least six different potential mechanisms and/or consequences of DNA-damage via DDPs (Figures S2A legend and S5G).
Transcription Factor Binding Promotes Replication Stalls
The reversed fork (RF)-dense cluster of Figure 4M (clusters 5 and 6, Figure 4N) is most enriched for DNA-binding TFs (Table S1): transcriptional activators and repressors. Persistent binding of a protein to DNA might create a replication “roadblock”, stall and reverse forks. Supporting this hypothesis, mutational ablation of the DNA-binding-domains (DBDs) of TFs CsgD, HcaR and MhpR abolished their promotion of SOS-inducing DNA damage, and RDG (RF) foci (Figures 5A-E and S6A). Thus, these TFs must bind DNA to provoke DNA damage and RFs. Also, TFs CsgD- or HcaR-mCherry fusions form foci DBD-dependently (Figures 5F and S6B), suggesting that foci reflect the DNA-bound TF. Most CsgD-mCherry and HcaR-mCherry foci co-localized with RDG (RF) foci (Figure 5F-H). Foci are distinguishable at ~50kb apart on DNA (Shee et al., 2013); thus, RDG/RF foci accumulate in the vicinity of TF-bound DNA. High-resolution genomic mapping of RDG by ChIP-seq in CsgD-overproducing cells showed RDG (RFs) enriched near the TF’s DNA-binding sites CsgD-DBD-dependently (Figure 5I). CsgD has 10 experimentally determined binding sites, and we found CsgD-dependent RDG/RF ChIP-seq peaks to be very significantly enriched in 10kb regions around these sites (Figures 5I and S7 legend). The RDG peaks occurred both upstream and downstream of the binding sites in the replication paths, CsgD- and DBD-dependently (Figure S7 legend). Our data support a model (Figure 5J) in which overproduced DNA-bound TFs create replication roadblocks, causing fork stalling and reversal near TF-bound sites, generating DNA damage.
Figure 5. E. coli Transcription Factors Promote Replication-fork Stalling via DNA Binding.
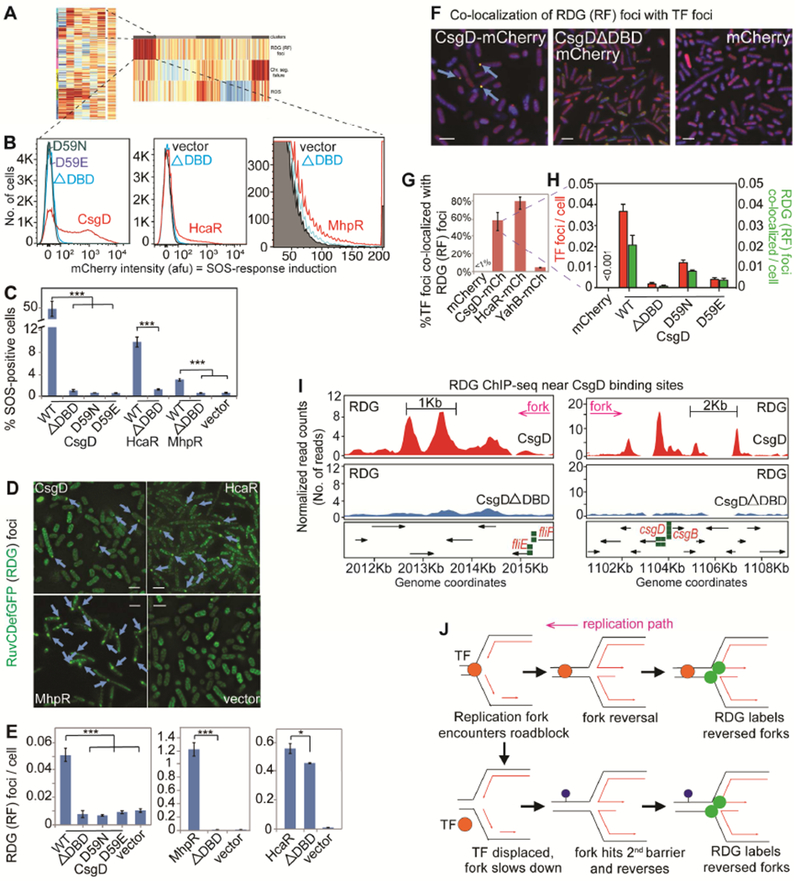
(A) DNA-binding transcription factors (TFs) enriched in DDP clones with high reversed forks (RFs, p =0.002, one-way Fisher’s exact test).
(B) TF DNA-binding required for DNA-damage promotion. Representative data, TFs and corresponding mutants: ΔDBD, DNA-binding domain deletion; and single amino-acid changes that reduce DNA-binding.
(C) Mean ± SEM of ≥3 experiments.
(D) DNA-binding required for RDG (RF) foci (blue arrows). Representative images, scale bar: 2μm. Figure S6A, all genotypes.
(E) Mean ± SEM of ≥3 experiments.
(F) DNA-binding TF-mCherry foci co-localize with RDG RF foci. Representative data. Blue arrows, co-localized foci, scale bar: 2μm.
(G) Mean ± SEM of ≥3 experiments. Figure S6B, images.
(H) Co-localization of TF with RDG foci requires TF DNA-binding.
(I) RDG ChIP-Seq RF peaks (in ΔrecA) enriched near CsgD-binding sites (green squares; p =0.01, two-tailed z-test versus simulated data, Figure S7 legend). Figure S7A-C, complete set RF peaks.
(J) Model: overproduced TFs (orange circles) bound to DNA (parallel lines) induce replication roadblock RFs. Lower model, how RFs might appear downstream of a DNA-bound TF: first, the bound TF slows/impairs the fork; second, a downstream replication-slowing site/occurrence that otherwise would not have stalled replication.
E. coli and Human Transporters Elevate ROS
Membrane-spanning transporters dominate human homologs of the E. coli DDPs (Figure 2A, Table S2), and several overrepresented among known cancer drivers also provoke DNA damage on overproduction (Figure 3B, Tables S2 and S3). The E. coli transporters are overrepresented at 26% in the high-ROS cluster (Figure 4M) compared with 11% over the whole network (p = 0.004, one-way Fisher’s exact test, Table S1), and 17 DDP clones with high ROS caused DNA damage ROS-dependently (Figure 4G). These include five transporters (Figure 6A-D). Three are H+ symporters—significantly enriched compared with H+ symporters in the genome (p = 2.7×10−5, hypergeometric test); one transports polypeptides, and the remaining one metal ions.
Figure 6. E. coli and Human Transporters Promote DNA Damage via ROS.
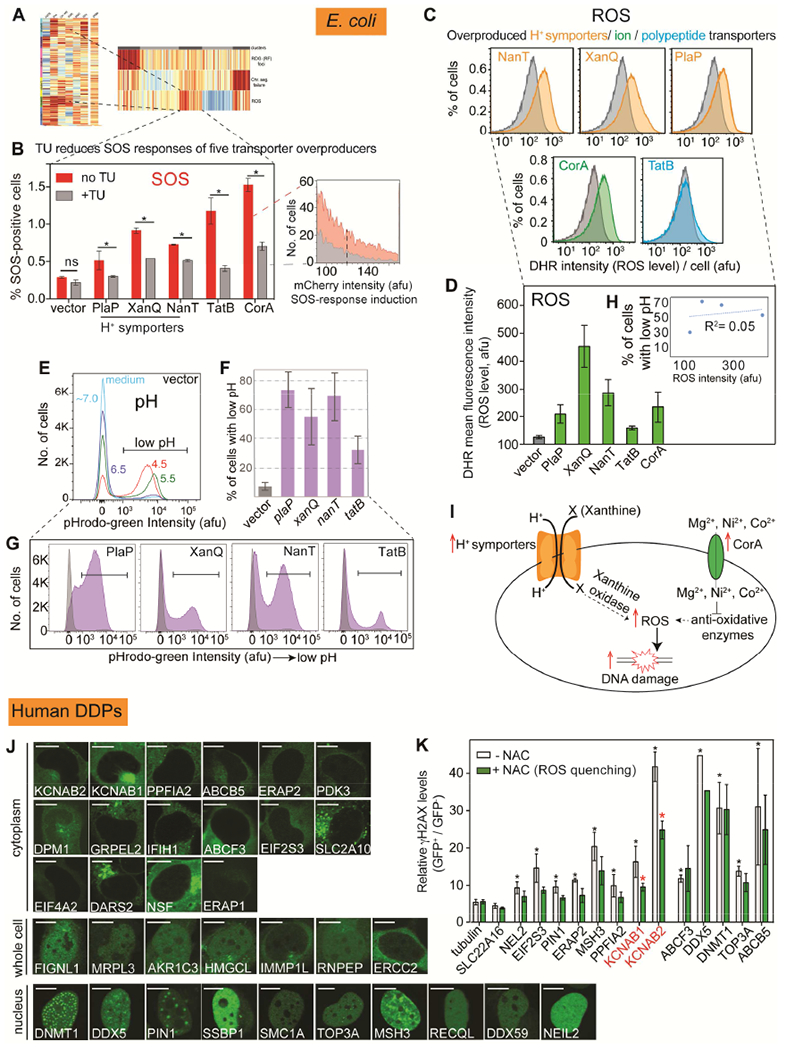
(A) E. coli high-ROS clusters enriched for membrane-spanning transporters (p = 0.004 one-way Fisher’s exact test).
(B) DNA damage from 5 E. coli transporters reduced by ROS-scavenger thiourea (TU): ROS-dependent. Mean ± range, N=2; representative data.
(C) Transporter overproduction elevates ROS levels. (Table S1). Gray, vector.
(D) Mean ± range, N=2.
(E) pHrodo-green pH stain and flow cytometry in buffers with varied pH show cell subpopulations with decreased pH.
(F) Overproducing E. coli H+ symporters increases activity/reduces pH: gain-of-function. Grey, vector only. Mean ± range of two experiments.
(G) Representative flow-cytometry data.
(H) Reduced pH is not correlated quantitatively with increased ROS (R2=0.05, Pearson’s correction), suggesting that the specific cargoes may promote DNA damage.
(I) Models. Discussed Figure S4D legend.
(J) Overproduced GFP-tagged hDDPs cellular localization. Bar: 5 μm.
(K) ROS underlie DNA damage caused by human KCNAB1/2 transporter overproduction. NAC: N-acetyl-cysteine. * p < 0.05 relative to untreated GFP-tubulin control; * p < 0.05 relative to the corresponding NAC-untreated control, unpaired two-tailed t-test.
Proton (H+) symporters import molecules concurrently with H+. Overproduction of H+ symporters reduced intracellular pH (increased H+, Figure 6E-G), implying that overproduction increased their symporter activities. Their induction of ROS was not well correlated with their reduction of pH (Figure 6H), suggesting either that other cargos that they import, or that compromising membrane integrity may provoke ROS/DNA damage. Specific models for XanQ, the strongest ROS-promoter among them, and CorA are suggested (Figure 6I, Figure S4D legend). Overall, the data reveal that increased transporter activity can provoke high ROS levels that can damage DNA (Figure 6A-G), and suggest that disturbing cellular boundaries may cause DNA damage via ROS.
In human cells, the 33 validated hDDPs probably promote multiple DNA-damage mechanisms, because of their apparent localization to different cellular compartments: 16 cytoplasmic; 10 nuclear; and 7 throughout the cell (Figure 6J), suggesting that direct contact with DNA is not required to instigate DNA damage. Among 13 validated hDDP-producers KCNAB1 or KCNAB2 were suppressed by ROS quencher N-acetyl cysteine (NAC, Figure 6K). KCNAB1/2 are subunits of intracellular voltage-gated K+ channels that function in redox transformations of xenobiotics (Hlavac et al., 2014). Increased KCNAB2 mRNA appears in breast cancer (Hlavac et al., 2014), but how KCNAB1/2 overproduction might promote cancer is unknown. DNA-damage production could be a potential mechanism.
E. coli Pol IV, Human DNMT1 Damage DNA via Replisome-Clamp Interaction
DNA Pol IV, encoded by dinB, is a very high DNA-damage instigators in E. coli (Figure 7A-E; Table S1), generating DSBs (GamGFP foci), chromosome loss, and ssDNA gaps. SOS induction by overproduced Pol IV was partially RecB- and partially RecF-dependent (Figure 7D-E), implicating DSB and ssDNA-gap promotion (Pennington and Rosenberg, 2007).
Figure 7. E. coli DNA Pol IV, Human DNMT1 Promote DNA Damage via Binding the Replisome Clamp.
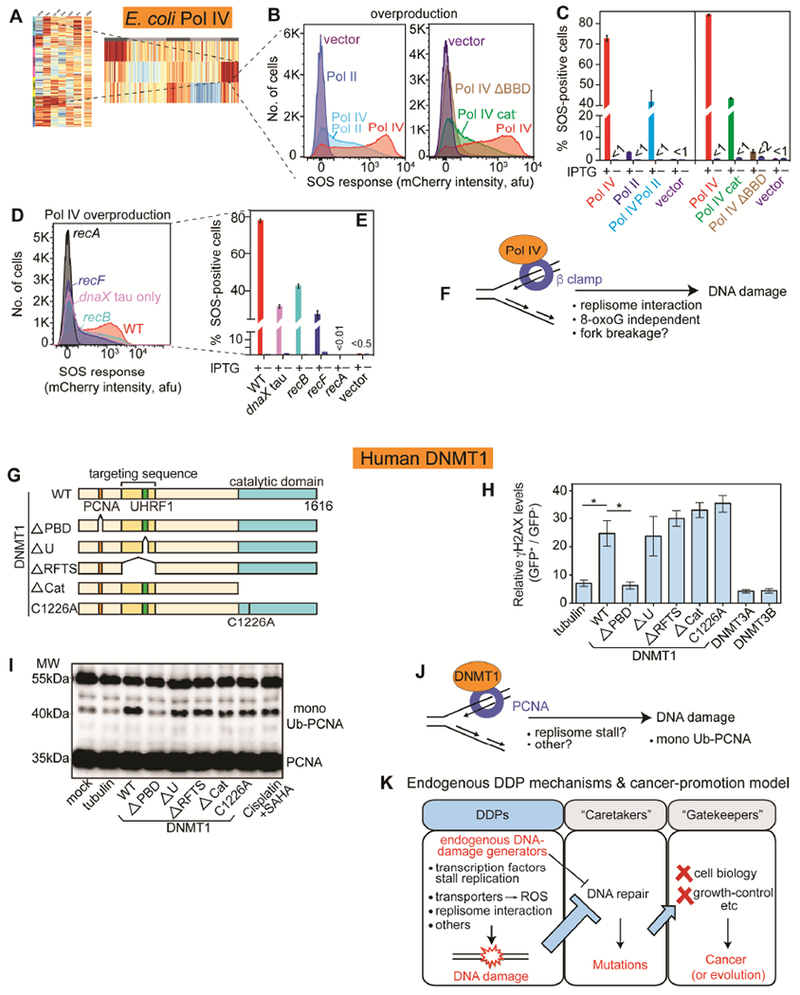
(A) E. coli Pol IV in cluster with high DNA loss.
(B) Left: Pol IV promotion of DNA damage is reduced by overproducing its competitor Pol II. Right: Pol IV promotion of DNA damage requires interaction with beta (β) replisome sliding clamp—ΔBBD, deleted β-binding domain—and is partly independent of Pol IV catalytic activity (cat− mutant).
(C) Mean ± SEM of ≥3 experiments.
(D) DNA damage reduced by reduction of Pol IV-β interaction with tau-only mutant, which favors Pol III. RecB- and RecF-dependence of Pol IV-induced DNA damage implicate DSBs and single-strand gaps.
(E) Mean ± SEM of ≥3 experiments. Pol IV is induced by IPTG.
(F) Model: Pol IV induces DNA damage by excess binding the β clamp. Excess β interaction might slow the replisome causing fork breakage/collapse, or displace β-binding DNA-repair proteins, among other possibilities. 8-oxo-dG-independence, Figure S7F,G.
(G) Mutant derivatives of human DNA methyltransferase DNMT1 (WT, wild-type). PBD, PCNA-binding domain; U, UHRF1 (ubiquitin-like PHD and RING-finger 1 interacting domain; RFTS, (recruits DNMT1 to DNA-methylation sites); Cat and C1226A, catalytically inactivate mutants, all N-terminally GFP tagged.
(H) Human DNMT1 overproduction in human cells promotes γH2AX accumulation methylase-independently and replisome-clamp-interaction dependently.
(I) Elevated DNMT1 promotes PCNA monoubiquitination (replication-stress) replisome-interaction dependently. Western blot with anti-PCNA antibody.
(J) Model/hypotheses for how excess DNMT1 promotes DNA damage.
(K) Hypothesis: DDPs, a cancer-protein function class upstream of DNA repair. Excessive endogenous DNA damage could titrate (thick blue -|) or inhibit (thin black -|) DNA repair causing DNA-repair-protein deficiency without a DNA-repair-gene mutation. Repair deficiency increases mutation rate, and cancer- (or evolution-) driving mutations in cell-biology-altering “gatekeeper” genes that cause the cancer phenotypes.
Pol IV is a poorly processive, low-fidelity DNA polymerase that traverses otherwise replication-blocking damaged bases. Pol IV competes with more processive DNA polymerases (Frisch et al., 2010; Hastings et al., 2010), by competition for binding the replisome sliding clamp protein, beta (β) (Dohrmann et al., 2016; Heltzel et al., 2012). Overproducing Pol IV simultaneously with its competitor DNA Pol II, caused ~50% reduction of Pol IV-dependent DNA damage (Figure 7B-C). A mutant replisome clamp-loader protein that reduces Pol IV loading in favor of replicative DNA polymerase, Pol III (Dohrmann et al., 2016), also reduced DNA damage from Pol IV (Figure 7D-E, dnaX tau-only), but did not inhibit SOS on its own (Figure S7D). A C-terminal Pol IV deletion that abolishes interaction with the replisome β clamp (Uchida et al., 2008) also abolished Pol IV-promoted DNA damage (ΔBBD, Figure 7B-C). Thus, Pol IV interaction with the replisome clamp is required for its promotion of DNA damage.
Perhaps surprisingly, Pol IV catalytic activity (DNA synthesis) was not required for all of its DNA-damage induction; the catalytically-inactive Pol IV R49F mutant (Wagner et al., 1999) reduced only half the DNA damage (Figure 7B-C), without reducing Pol IV protein levels (Figure S7E). Thus, binding of Pol IV to the replisome, not just its catalytic activity, appears sufficient to cause DNA damage. The synthesis-dependent component of DNA damage might have resulted from the ability of Pol IV to incorporate oxidized guanine (8-oxo-dG) into DNA, leading to strand-breaking BER that begins with base removal by MutM and MutY DNA glycosylases (Foti et al., 2012). However, loss of neither glycosylase diminished the DNA damage (Figure S7F-G), implying that BER at 8-oxo-dG is not how Pol IV causes most DNA damage. Overall, Pol IV promotes DNA damage via replisome-clamp interaction, and only partly dependently on catalysis (model, Figure 7F).
Like E. coli Pol IV, human DNMT1 induced DNA damage in human cells via binding the replisome clamp, independently of its catalytic activity: a non-canonical potential cancerdriving role. DNMT1 is the major human DNA methyltransferase that methylates DNA upon replication. Hypomorphic mutations in DNMT1 promote microsatellite instability (Jin and Robertson, 2013). Increased DNMT1 occurs in several cancer types, and causes hypermethylation, proposed to downregulate tumor-suppressor genes (Biniszkiewicz et al., 2002). Surprisingly, DNMT1 promoted DNA damage independently of its DNA-methylation activity (Figure 7G-I). Two other DNA methylases did not increase DNA-damage levels (Figure 7H). DNMT1 promotion of DNA-damage required its PCNA-binding domain (PBD), which binds the replisome sliding clamp: PCNA (Figures 7G,H, S3O and S3P). Rad18-mediated monoubiquitination of PCNA, a DNA-damage response (Mortusewicz et al., 2005), also resulted from DNMT1 overproduction, also methylase-independently and PBD-dependently (Figures 7I and S3P). Thus, mere binding of overproduced DNMT1 to PCNA promotes DNA damage independently of methylation. The finding that both E. coli DNA Pol IV and human DNMT1 promote DNA damage via replisome-clamp binding and independently of their catalytic activities (Figure 7A-J) indicates generality of this mechanism. Promotion of DNA damage by DNMT1-PCNA complexes (Figure 7G-J), and resulting mutagenesis (Figure 3C), may promote cancers other than or in addition to via the known gene-regulatory function of DNMT1 in DNA methylation, and many clamp-binding proteins may act similarly when overproduced.
DISCUSSION
Endogenous DNA Damage in Cancer
The 284 human DDP homologs are overrepresented among known and predicted cancer drivers (Figure 2C, Table S3), overrepresented in cancers as amplified genes (Figures 2D and S3A-C, Table S2), and their increased RNAs in human cancers accompany poor outcomes and heavy mutation loads (Figures 2E,F and S3D-H). The correlations of the 284 human-homolog RNAs with tumor mutation loads and poor survival remain strong even when both known/predicted cancer drivers and human proteins validated as DNA-damage instigating here are removed (Figures 2F and S3H). Thus, many new and previously difficult-to-predict cancer-relevant functions are likely to populate the network (Figure 3E), and implicating those proteins as probable candidates for overproduction oncoproteins.
The DDPs appear to represent a new broad function class of cancer-promoting proteins, that may propel tumor evolution. Cancer-gene functions have been grouped into multiple specific categories (Hanahan and Weinberg, 2011) that fit into two broad classes (Kinzler and Vogelstein, 1997). Mutations in the cancer-cell-biology-altering “gatekeepers” make cell biology more cancer-like, and genomic “caretaker”-(DNA-repair)-gene mutations elevate mutation rate, driving cancer by promoting gatekeeper mutations (Figure 7K). The DDPs are expected to act before/upstream of DNA-repair functions promoting the endogenous DNA damage that necessitates repair (Figure 7K), and so promote cancer development. hDDP upregulation may decrease DNA-repair capacity by saturating repair pathways with excessive DNA damage, or inhibit repair directly, either way—increasing mutation rate without a DNA-repair-gene mutation (Figures 4J-L and S5). Thus, “mutator” mutations will be difficult to predict in cancer genomes, with most being previously hypothesized “gatekeepers”, or genes not thought to affect cancer.
Many hDDPs span diverse protein functions, the cancer-driving roles of which may be obscure or mis-assigned. Some of the mechanisms of hDDP action may necessitate reevaluation of their cancer-driving mechanisms, and also of the drugs designed to inhibit them. For example, human DNA methyltransferase DNMT1 caused DNA damage independently of its methylation activity, via its interaction with the replisome sliding clamp (Figure 7G-J). Current cancer drugs target the DNMT1 methylase activity (Jones et al., 2016), and not replisome binding. Our finding may inform the development/use of DNMT1-targeting strategies, taking into account its multifunctionality.
E. coli-to-Cancer Gene-function Atlas
Our data from seven quantitative phenotype assays for kinds, causes, and consequences of DNA damage promoted by E. coli DDPs provide a resource for within- and cross-species discovery of conserved DNA-damage-generating mechanisms. These data revealed six main function/phenotype clusters (Figure 4N), which drive discovery of three DDP mechanisms, two found also in human cells (Figures 5–7). The E. coli phenotype data may aid prediction of conserved protein functions in human. We created a minable web-based resource for searching the complete E. coli function/quantitative-phenotype data, and the functional data from the validated hDDPs: the E. coli-to-Cancer Gene-function Atlas (ECGA, STAR Methods). The ECGA data can be searched via human-homolog or bacterial-protein names or by function/phenotype key words. ECGA can be used to query/generate hypotheses for potential conserved human-protein functions, and for other organisms.
Mechanisms that Cause Endogenous DNA Damage
E. coli DNA-binding TFs caused replication-fork stalling and reversal by apparent blocking of replication forks by the bound TFs (Figure 5). Though replication-transcription conflicts have been engineered by altering chromosomes (Tehranchi et al., 2010), or knock-out of RNA-removal proteins (Wahba et al., 2016), the resulting DNA damage types were unidentified, and the mechanisms not known to occur in natural genomes with mere upregulation of an endogenous protein, a frequent occurrence. Transcription-generated RNA/DNA hybrid molecules (R-loops) promote DNA damage (Wahba et al., 2011) including DSBs (Hamperl et al., 2017; Wimberly et al., 2013), and form often in regions transcribed “head-on” to DNA replication (Hamperl et al., 2017; Lang et al., 2017), a bias not seen for reversed forks from TF binding here (Figure S7 legend).
Our data indicate that fork reversal is common and protein-function specific—promoted by TFs on DNA. Nearly 10% of human genes encode TFs (Levine and Tjian, 2003), including many cancer-driving overproduction (onco-)proteins, some known to promote DNA damage when overproduced, e.g., c-Myc (Vafa et al., 2002). Based on our observation of TF binding and DNA-damage in E. coli, many onco-protein TFs might promote cancer by instigating mutagenic DNA damage, similarly to E. coli TFs, possibly by replication blocking and fork reversal (Figure 5J).
We discovered two additional mechanisms in both E. coli and human cells. First, increased transmembrane transporter activities, including human KCNAB1/2 transporter, elevated ROS causing DNA damage (Figures 4G and 6A-I, K). This mechanism might explain the KCNAB2 association with cancers (Hlavac et al., 2014). Second, overproduced E. coli DNA Pol IV and human DNMT1 provoked DNA damage via binding their respective replisome sliding clamps, independently of their catalytic activities (Figures 7A-J, and S3O,P). Disruption of the replisome leading to replication-fork collapse (or other means Figure 7F,J) may promote much DNA damage (Kuzminov, 2011), and, our data imply, is likely to result from dysregulation of many replisome-binding proteins.
DNA Damage as Potential Cancer Biomarker
The existence of diverse mechanisms that increase endogenous DNA damage, and the predicted large sizes and diversity of both bacterial and human DDP networks, indicate that mis-regulation of many proteins is likely to be mutagenic via DNA damage (Figures 1I, 2F, 3C and S1F). Because many different proteins/mechanisms instigate DNA damage, DNA damage itself might predict cancer and genetic-disease susceptibility. The ability to detect high DNA-damage loads in cells could potentially make DNA-damage screening attractive for early identification of at-risk individuals, useable before genome-sequencing would identify disease-associated mutations. Additionally, cancer immune therapy “checkpoint inhibitor” use is limited to high-mutagenesis cancers, apparently because diverse tumor antigens can be attacked by the stimulated immune system (Germano et al., 2017). Our data suggest that DNA damage or upregulation of DDPs may predict tumorigenic processes and susceptibilities in various cancers.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resource sharing should be directed to and will be fulfilled by the Lead Contact S. M. Rosenberg (smr@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli K-12 (strains MG1655 and W3110) and isogenic derivatives were used for all bacterial experiments. Human MRC5-SV40 (male) and HEK293T (female) cells were used for all human cell line experiments.
METHOD DETAILS
Escherichia coli strains and media
E. coli K12 strains and plasmids used in this work are shown in Table S7. Strains were grown in Luria Bertani Herskowitz (LBH) rich medium or M9 minimal medium supplemented with thiamine (10μg/ml) and 0.1% glucose or glycerol as a carbon source (Xia et al., 2016). Other additives were used at the following concentrations: ampicillin (100μg/ml), carbenecillin (20μg/ml), chloramphenicol (25μg/ml), kanamycin (30μg/ml), and sodium citrate (20 mM). P1 transductions were performed as by (Xia et al., 2016). Genotypes were verified by antibiotic resistance, polymerase chain reaction (PCR) followed by sequencing, and, when relevant, UV sensitivity.
Synthesis and generation of E. coli mutant and fusion genes
Mutant or truncated genes were synthesized to introduce site-specific mutations or small deletions in (GenScript) pUC57 backbone plasmids, and subsequently cloned into plasmid pNT3-SD to allow E. coli conjugation. Genes that encode wild-type and mutant DNA-binding transcription factors were fused with mCherry of (Shee et al., 2013) with a 4-6 alanine linker as described. The plasmids mentioned above are shown in Table S7.
E. coli Mobile plasmid overexpression library
The Mobile-plasmid collection is an ordered library of all 4229 E. coli protein-coding genes in a conjugation-transferrable plasmid (Saka et al., 2005). Of these genes, 1017 (or 24%) encode the native E. coli protein, whereas 3212 (or 76%) encode the E. coli protein with an additional three N-terminal amino acids (Met-Arg-Ala) and an additional two C-terminal amino acids (Gly-Leu), with genes randomly distributed to one or the other kind. We found that native proteins were over-represented significantly as positive for DNA damage in our screens (Estimate of additional E. coli DDPs not discovered, below), implying that the 5 additional amino acids in some of the clones are more likely to confer false-negative results for the proteins that carry them than false-positive results. Table S1 shows the 208 validated DDPs and indicates the clones that produce native proteins with * next to the clone-ID number, and those with the five additional amino acids with an unmarked clone-ID number.
Whole-genome primary DDP screen of ordered E. coli overexpression library
The ordered Mobile-plasmid collection of 4229 E. coli genes in a conjugation transferrable plasmid (Saka et al., 2005) was mobilized into SOS-response-reporter strain SMR17962 (Nehring et al., 2016) to generate a DNA-damage screenable E. coli overproduction library. Protein overproduction is controlled by the IPTG-inducible Ptac promoter in cells that fluoresce red when they experience SOS-inducing DNA damage (single-stranded DNA) (Nehring et al., 2016). We adapted a high-throughput 96-well plate reader and robotics to screen for potential DDP-positive strains with increased mCherry fluorescence. Fluorescence intensity per unit of OD600 was compared in each well. Primary screens were performed on cells grown in M9 glucose or M9 glycerol medium (to survey two different conditions), each in duplicate. Ordered E. coli overproduction strains were grown to saturation overnight with shaking at 37°C in clear 96-well plates containing 150μl medium per well, then each well diluted 1:100 into 150μl IPTG-containing medium in 96-well plates (μ-clear, black, Greiner Bio-One, Monroe, NC, USA). The plates were shaken at 37°C for another 24h and analyzed in a Synergy 2 fluorescence plate reader (BioTek, Winooski, VT). We set thresholds of 20% (for glucose with IPTG induction) or 30% (for glycerol with IPTG induction) compared with the median fluorescence intensity per unit of OD600 of each individual 96-well plate to identify primary hits. Primary hits were called when two replicates done in the same medium were both above the threshold. Altogether, 414 candidate proteins were identified in this high-throughput plate-reader screen, then tested by flow cytometry for increased endogenous DNA-damage levels to eliminate false positives from the lower resolution/noisier plate-reader assay.
E coli flow-cytometry secondary screen for increased endogenous DNA damage
We screened candidate-protein hits from the primary (plate-reader) screen with our more sensitive flow-cytometric assays for SOS-induction/DNA damage in single cells (Pennington and Rosenberg, 2007). Each strain identified as positive from the primary plate-reader screen was grown at 37°C to saturation overnight in M9 glucose medium, diluted 1:100 into M9 glycerol medium, and grown for 9 hrs to early exponential phase at which time IPTG was added to 100μM to induce plasmid-protein overproduction. After 8 hours of induction, the cultures were diluted 1:100 in filtered M9 glycerol medium. Samples were analyzed in a LSR Fortessa flow cytometer (BD Biosciences) and analyzed with BD FACSDivaTM and FlowJo software. For these analyses, 105 events were collected per strain, per experiment, with each strain assayed in three independent repeats. Student’s t-test (p value ⩽ 0.05) and False Discovery Rate (FDR) q < 0.1 were calculated and applied based on Benjamini multiple comparison to determine whether overproduction strains had significantly increased levels of endogenous DNA damage. Two-hundred and eight of the original 414 E. coli candidate proteins were validated as genuine DNA-damage-inducing DDPs when overproduced (shown Table S1).
Confirmation of DNA damage in all 208 E. coli DDP network clones
The following three assays were used in addition to the fluorescence flow-cytometry secondary screen to confirm that all 208 validated DDPs promote DNA damage on overproduction. First, 95% of the 208 validated E. coli DDPs displayed an independent DNA-damage-related phenotype in at least one of nine assays that are either less sensitive or more DNA-damage-type specific than the SOS-flow cytometric assay in the secondary screen, results of all of which are shown for each clone in Table S1. The assay for persistent single-stranded (damaged) DNA, detected as microscopically visible foci of a fluorescent partial-function DNA-damage-sensor protein RecA*GFP (Figure 1H), is less sensitive because it uses a partially functional RecA protein (Renzette et al., 2005). With it, a representative sample of 67 of the DDP-overproducing clones showed significant association of RecA*GFP foci with SOS-positive (DDP) clones; 32 of the 67 showed increased foci (r = 0.7, p = 1.3×10−10, Pearson’s correlation; Figure 1H; Table S1). In work in Figure 4, we explored the kinds and causes of endogenous DNA damage promoted by the 208 E. coli DDPs, using seven assays for specific kinds of DNA damage—all more DNA-damage-type-specific than the SOS-response assay, and we also assayed DDPs for increased mutagenesis (Figure 1I, rates Table S1). All but 12 (95%) of the 208 clones were positive in the RecA*GFP-focus assay, and/or at least one of the seven more specific assays (Figure 4, Table S1) and/or mutagenesis (rates Table S1). We then demonstrated that the twelve remaining DDPs that were not further validated in a non-SOS-based assay (Table S1) show RecA-FexA-dependence of their increase fluorescence, confirming a genuine SOS-response (Figures 1G and S1C; Table S1), and not general fluorescence increase (Figure S1D; Table S1).
E. coli assay for RecA*GFP foci indicating single-stranded DNA
E. coli containing the chromosomal recA4155gfp allele, encoding RecA*GFP (Renzette et al., 2005), and the flow-cytometrically validated Mobile-plasmid carriers were grown to saturation in M9 glucose medium at 37°C, then diluted 1:100 into M9 0.1% glycerol and grown for 9h to early log phase. IPTG was added to 100μM to induce protein overproduction for 8h as described above, then images taken and analyzed.
E. coli assays for GamGFP and RDG foci
Saturated cultures of E. coli strains [SMR14334 for GamGFP; and SMR19406 for RDG (RuvCDefGFP)] containing each of the 208 validated DDP-encoding Mobile plasmids were grown and induced as described in flow-cytometric assays for DNA damage. lOOng/ml of doxycycline were added to induce GamGFP for 3h and RDG for 2h prior to harvesting. Cells were fixed with 1% paraformaldehyde for 15 min. and washed with PBS buffer three times before being concentrated for microscopy.
E. coli microscopy and image analysis for RecA*GFP, GamGFP and RDG foci
Images were acquired using a 100× /NA = 1.4 immersion oil objective (Olympus) on a DeltaVision Elite deconvolution microscope (Applied Precision, GE). A z-series was acquired sampling every 0.2 microns for a total of 15-25 sections. The z-series was then deconvolved, and a maximum projection image rendered using Softworx (GE). Image analysis was performed using the Advanced Imaging collection in Pipeline Pilot 8.5 or 9.2 (Biovia-Dassault Systemes, San Diego). Projected images from the DeltaVision were read into Pipeline Pilot and metadata data parsed from the file name and path. A rolling ball background subtraction was applied to improve the signal-to-background ratio, and to facilitate further segmentation. Individual bacterial cells were then identified and segmented by applying a global threshold on images of the fluorescently labeled protein. Morphological manipulations (smoothing, opening and closing) were applied to refine the segmentation edges and a watershed was then performed to separate neighboring objects. Filtering was then applied to remove bacteria that fell outside a certain area threshold and that did not contain DNA. Foci were then identified using a more aggressive per-object background subtraction and peak identification method. Objects tentatively identified by this method were subsequently filtered by circularity, signal-to-background ratio, and size. Focus-positive bacteria were then determined using the co-localized objects component in the Advanced-imaging library in Pipeline Pilot. A binary metric, whether the cells were focuspositive or not, was calculated in addition to recording the total area and count of foci for those bacteria that were positive.
STRING/network analyses
Known protein-protein interactions were displayed using CytoScape V3.4.0 software. Protein-protein interaction linkage scores were taken from the STRING 10.0 database (Szklarczyk et al., 2015) to identity interaction pairs. We used STRING, all parameters, with an interaction score cut-off of ⩾0.6 (medium-to-high confidence). Random controls were produced by examining equal-size groups of random E. coli genes. P values were calculated with a hypergeometric test. The E. coli DDP network has network properties that are defined as scale-free and “small-world”, and it has significantly more edges (connectivity) compared with a random network (Figure S2A). The human candidate-DDP network was generated similarly, and also has more connectivity than a random human-gene network or random human genes with E. coli homologs (Figures 2B, S2B).
Estimate of additional E. coli DDPs not discovered
The 208 demonstrated E. coli DDPs (Figure 1E,F, Table S1) are likely to represent just over half of overproduction DDPs encoded in the E. coli genome. Per Figure S1E, 1 of 99 random proteins not identified in the primary screen tested positive in the sensitive flow-cytometry secondary assay, predicting an additional undiscovered 38 DDPs in the overproduction library used (Figure S1E). Further, although it is the most complete and least adulterated E. coli overexpression library, the Mobile plasmid library (Saka et al., 2005) contains some genes that encode five additional amino acids, which our data indicate were biased against in our screens. Twenty-four percent of clones in the library (STAR Methods) produce native E. coli proteins, and the rest produce proteins with three extra N-terminal (Met-Arg-Ala) and two extra C-terminal (Gly-Leu) amino acids, with the composition of genes in each class being random (Saka et al., 2005) (STAR Methods). We found that both the initial DDP candidates identified in the plate-reader primary screen and the 208 flow-cytometry-validated DDPs carried significantly higher fractions of native proteins than the library; there were 158 native proteins in the initial 414 candidates identified in the primary plate-reader screen (38%, differs from the library at p = 1.7 × 10−11, Fisher’s exact test), and 85 native proteins in the 208 validated DDPs, or 41% (shown in Table S1, differs from the library at p = 4.1 × 10−8, Fisher’s exact test). The data imply that some of the non-native proteins may have lost full function, and, because of that, gave false-negative readings in the screens. The native genes in the library were “hit” in the primary screen at 15.6% (158 discovered out of 1017 native genes in the library), whereas the non-native genes were identified at 8% efficiency (256 discovered out of 3212 non-native genes in the library). If there are an additional 7.6% of the non-native proteins that would score as DNA-damage-promoting in our primary screen, if they did not carry the extra amino acids (15.6% predicted - 8% found), then among the 3212 non-native-protein-encoding genes in the library, we predict that there would be an additional 244 overproduction DDP candidates found in the primary screen (7.6% of 3212). We found that candidates from the primary screen were validated in the secondary screen at 208 validated out of 414 candidates (Figure 1F; Tables S1), or just over 50%, which predicts 123 additional genuine DDPs among the predicted additional candidates.
E. coli forward-mutation assay
We used the forward-mutation assay of Matic and colleagues (Gutierrez et al., 2013) in which E. coli wild-type strain MG1655 harbors a chromosomal phage lambda cI transcriptional repressor gene, and a CI-repressible tetA gene, such that mutations that inactivate cI are scored as tetracycline-resistant (TetR) mutant cfu. Into this strain, we conjugated 32 validated E. coli DDP genes in their Mobile-plasmid-library vector (genes tested Table S1; below and Table S1 for their Mobile-library clone names). We developed a modified higher-throughput fluctuation-test assay for determining numbers of cultures with TetR mutants from which to calculate TetR mutation rates. Each DDP overproducer was grown overnight to saturation in M9 glucose with 20μg/ml carbenicillin at 37°C shaking, then diluted 1:10,000 into M9 glycerol carbenicillin and each culture split into 24 or 32 wells in 96-well-plates at 100μl per well. The plates were shaken at 37°C for 15h (early log phase), and IPTG added to attain 100μM in each well to induce protein overproduction for 8h, as described in flow-cytometric validation. From the end cultures, 5-10 μl were moved into LBH medium containing 10μg/ml tetracycline to determine the fraction of cultures that contained no TetR cells after incubation and scoring of the wells in the plate reader for OD (TetR cells) versus failure to grow (no TetR cells). The viable cell counts were estimated by sampling three wells chosen randomly. The P0 method was used to estimate mutation rates for each genotype as described with correction for the fraction sampled. The data reported (Figure 1I; Table S1) are the mean mutation rates (± SEM) of three experiments of at least 24 cultures per strain for each of the 32 strains assayed.
E. coli clones assayed for mutation rate
The respresentative E. coli DDPs assayed for mutation rate follow. DDPs that cause < 5-fold increase in DNA damage: DsbG, YijF, CadA, FolD, YddG, LeuO, UvrB, YajR, YbgQ, ORF 6106.1. DDPs that cause ≥ 5-fold increase in DNA damage: HypF, ZipA, YedA, CueO, YefU, MacB, HcaR, MdtB, SetB, DinD, RusA, YdcR, CsgD, HslU, SfsA, TopB, CorA, YegI, GrpE, PgrR, Mrr, MhpR. Non-DDPs: AceF, HprT, AceE, YaeG, YadF, PdhR, HrpB, MrcB, FhuD, YadG, Dgt, FhuA, HtrE, EcpD, FhuC, YacH, YadK.
E. coli TetR mutation verification by sequencing
We selected strains that overproduce the following 10 different DDPs with strong DNA-damage-up phenotypes: CsgD, TopB, CheA, YegL, MdtA, GrpE, HslU, YicR, UvrA, and Mrr. We selected 3-10 independent TetR mutant colonies, each from a separate culture from each strain, from which to sequence cI mutations. For the vector-only negative control, 19 independent TetR colonies were isolated. We amplified and sequenced a 1122nt region encompassing the cI gene as described (Gutierrez et al., 2013) to identify the mutations. For those TetR mutants that failed to yield PCR products, implying deletion of the cI gene, further outside primers (forward: ACCGCGGCGTGGGTAGTAAAGT, and reverse: GCCAATCCCCATGGCATCGAGTAAC) were used for PCR, and the products sequenced. In two cases (both TopB overproducers), whole-genome sequencing (WGS) was performed to determine the end-points for deletions that could not be determined via PCR and sequencing.
E. coli whole-genome sequencing and analysis
TetR mutants were grown at 37°C to saturation overnight in LBH with 10μg/ml tetracycline, and genomic DNA was extracted and purified using DNeasy Blood & Tissue kits (Qiagen). Libraries were prepared using Nextera XT kits (Illumina); sequencing was performed on an Illumina Mi-Seq, and sequencing data analyzed as described (Xia et al., 2016). Sequencing reads were mapped to the MG1655 genome (NCBI RefSeq Accession: NC_000913.3). Low-quality reads and duplicates were removed. Whole-genome sequence files were visualized and deletion endpoints were analyzed using IGV software (Broad Institute, MA).
Additional controls for E. coli DDP-function assays
While analyzing RNA-Seq data, we identified a 2177bp deletion including the lacIq region in the Mobile-plasmid library pNT3 empty vector. We determined that this deletion does not alter results in any of our assays or any of our conclusions. The phenotypes of the truncated empty vector were compared with 10 non-DDP overproducers, and then with the full-length empty vector, in all 7 functional assays in Figure 4, and, by one-way ANOVA analysis, there were no significant differences between the means of all 11 strains in any of the 7 assays (p=0.19 GamGFP foci; p=0.28 RDG (reversed-fork) foci; p=0.99 ROS; p=0.99 anucleate cells/DNA loss; p=0.26 phleomycin sensitivity; p=0.08 H2O2 sensitivity; p=0.21 mitomycin C sensitivity), and no difference between it and the full-length vector (p=0.31; p=0.44; p=0.62; p=0.32; p=0.62; p=0.28; and p=0.78, respectively, two-tailed unpaired t-test).
Flow-cytometric assays for DNA loss
Saturated cultures of E. coli strains derived from SMR21384 containing each of the 208 validated DDP-producing mobile plasmids were grown and induced as described in flow-cytometric assays for DNA damage. Cells were resuspended in 100 μl PBS, and stained with membrane dye FM® 4-64FX (Thermo Fisher) with a final concentration of 10 μg/ml. The mix was kept on ice for 10 min. and then washed three times with PBS. A final concentration of 70% ethanol (pre-chilled) was used to fix the cells at −20°C for lh, after which cells were washed with twice with PBS and resuspended in 100 μl PBS. 100 μl DAPI (5μg/μl) were used to stain DNA at room temperature (RT) for another 10 min. Samples were filtered and analyzed as described above.
Flow-cytometric assay for intracellular ROS levels
Saturated cultures of E. coli strains derived from SMR21384 containing each of the 208 validated DDP-producing mobile plasmids were grown and induced as described in flow-cytometric assays for DNA damage. The ROS measurement protocol was modified from Gutierrez et al. (Gutierrez et al., 2013). In brief, cells were incubated with ROS-staining dye DHR123 (Invitrogen), which measures H2O2, for 30 min. at 4°C in M9 buffer. After washing twice with M9 buffer, flow cytometry analyses were performed immediately as described above.
Flow-cytometric assay for intracellular pH
pHrodo® Green AM Intracellular pH Indicator (Thermo Fisher) was used to measure intracellular pH in live E. coli. Cells were first washed with live-cell imaging solution (LCIS) and then 10 μl of pHrodo™ Green AM with 100 μl of PowerLoad™ concentrate were added to 10 ml of LCIS. The pHrodo™ AM/PowerLoad™/LCIS was mixed with cells and incubated at 37°C for 30 minutes. Cells were then washed twice with PBS to remove excess dye before flow-cytometric analysis. Intracellular pH calibration buffers (Thermo Fisher) were used as standards.
Assays for sensitivity to DNA-damaging agents
Cultures of E. coli strain (SMR21384) containing each of the 208 validated DDP-producing mobile plasmids were grown as described in flow-cytometric assays for DNA damage with the following modifications: For hydrogen-peroxide (H2O2) treatment, 100 μM IPTG was used to induce overproduction of each DDP. Each culture was split into two tubes, prior to addition of 5mM H2O2 into one of the tubes for 15min. The cells with and without H2O2 were immediately diluted and plated onto LBH plates for assay of viable cells as cfu after incubation for a day at 37°C. For phleomycin or mitomycin C (MMC) treatment, saturated M9 glucose cultures were diluted into M9 glycerol medium with 100 μM IPTG to induce overproduction in 96-well plates. The plates were grown with shaking for 8 hours at 37°C to early log phase prior to addition of 1 μg/ml phleomycin or 0.05μg/ml MMC to each well. After 20 hours of continuous shaking, the OD600 was read using a BioTek microplate reader Synergy 2 (BioTek). DNA-damaging-agent sensitivities of the DDP-producing clones are normalized to sensitivity of vector-only controls: (treated/untreated DDP overproducer) / (treated/untreated vector-only) so that values < 1 indicate sensitivity. For all three assays for sensitivity to DNA-damaging agents, Student’s t-test (p value ⩽ 0.05) with FDR adjustment (q ⩽ 0.1) was used to determine whether DDP-overproducing strains were significantly more sensitive to DNA-damaging agents than the vector-only control.
Clustering methods
For each DDP and DNA-damage outcome measure, raw data for each functional assay (overproduction versus vector) were converted into z scores and were used to delineate groupings of proteins with similar properties and patterns of response. Unsupervised discovery methods K-means in combination with Progeny Clustering (Hu et al., 2015) were performed using the R package ProgenyClust to determine the optimal number of protein clusters for the 208 DDPs. Seven functional tests were clustered by hierarchical clustering to assess the association of kinds, causes, and consequences of DNA damage.
RNA-seq library preparation and sequencing
E. coli cultures were grown as described for flow-cytometric assays for DNA damage, and RNA was isolated from 1 ml of culture (~108 cells) for each of two biological replicates. Total RNA was isolated using the RNeasy Mini Kit (Qiagen), according to the manufacturer’s protocol. RNAprotect Bacterial Reagent (Qiagen) was used to stabilize RNA during harvest and enzymatic cell lysis. After elution, total RNA was treated with RNase-free DNase I (NEB), according to the manufacturer’s protocol. RNA was recovered by phenol-chloroform extraction and ethanol precipitation. Ribosomal RNA was depleted using RiboZero (Epicentre/Illumina), according to the manufacturer’s protocol. Remaining RNA was concentrated by ethanol precipitation and approximately 100 ng of rRNA-depleted RNA was used to construct libraries using the TruSeq Stranded mRNA Library Preparation Kit (Illumina). Libraries were prepared according to the manufacturer’s protocol, using recommended modifications for previously isolated mRNA (McClure et al., 2013) (poly-A RNA enrichment steps excluded). Final RNA-seq libraries were run on a BioAnalyzer (Agilent) to estimate the average fragment size (~800 bp) and the concentration of adapter-ligated library fragments was determined using the qPCR-based Illumina Library Quantification Kit (KAPA Biosystems). Libraries were pooled and sequenced on an Illumina NextSeq 500 using a High Output v2 Kit (2 × 75 bp paired-end reads).
Analysis and deposition of RNA-seq data
Read mapping, transcript assembly, and differential expression analysis were performed using Rockhopper (McClure et al., 2013), a bacteria-specific RNA-seq analysis pipeline, using MG1655 (NC_000913.3) as the reference genome. Genes were considered as differentially expressed if the fold change was greater than or equal to 2 and q-value was less than 0.01. Sequencing data are available in the European Nucleotide Archive (ENA) under study accession no. E-MTAB-7361.
RDG ChIP-seq library preparation, sequencing, and data analysis
Cells were grown as for focus quantification, then crosslinked, lysed and sonicated as described (Xia et al., 2016). Immunoprecipitation and library preparation methods are also based on those of (Xia et al., 2016) with small modifications as follows. RuvC antibody (Santa Cruz) was first pre-incubated with Dynabead protein A, then the RuvC-antibody-coated Dynabeads were incubated with cell lysates at 4°C overnight. Library preparation was performed while DNA fragments were still on Dynabeads. Samples were barcoded using NEBNext Multiplex Oligos for Illumina. Size selection of adaptor ligated DNA was performed on AMPure XP Beads as described in NEBNext ChIP-Seq Library Prep guidelines. Because the concentrations of eluted ChIP DNA are low, samples were amplified briefly prior to size selection, and a second amplification was performed after size selection. Sequencing was performed on an Illumina MiSeq. The pipeline for data analysis consists of the following steps: (i) reads were trimmed by Trimmomatic removing sequencing adaptors and low quality bases; (ii) reads were aligned by BWA-MEM (Li, 2013) to the W3110 genome [National Center for Biotechnology Information (NCBI) Reference Sequence (RefSeq) Database accession: NC_007779.1] and the plasmid pNT3 (Saka et al., 2005); (iii) Secondary alignment and multiple-mapped reads were discarded, this results in zero coverage in repetitive regions and regions present in both the genome and the plasmid, including the csgD gene; (iv) potential PCR duplicates were removed by Picard Tools MarkDuplicates; (v) bedGraph files were generated with deepTools and imported to R for plotting; and (vi) peak calling was performed with MOSAiCS. Sequencing data are available in the European Nucleotide Archive (ENA) under study accession no. PRJEB21035.
No bias in transcription direction of ChIP-seq reversed-fork RDG peaks
Transcription-generated RNA/DNA hybrids, or R-loops, form preferentially in regions of DNA transcribed “head-on” to DNA replication in bacterial and human cells (Hamperl et al., 2017; Lang et al., 2017), and provoke DNA damage in multiple species (Hamperl et al., 2017; Wahba et al., 2011; Wimberly et al., 2013). We found no similar bias in the locations of reversed forks induced by DNA-binding transcription factors (Figures 5I and S7A-C), detected by ChIP-seq of RDG 4-way-junction-trapping protein in ΔrecA cells, which lack HR-generated junctions (Xia et al., 2016), as follows. The 155 RDG peaks detected with CsgD transcription-factor production cover a total of 42,938bp of DNA, among which 27.6% is in an open reading frame (ORF) that is transcribed “head-on” (HO) to the DNA replication path, 33.2% is in an ORF that is transcribed co-directionally (CO), and the remaining 39.2% occupies regions outside any known ORF (referred to as “intergenic regions”). In the E. coli genome, the DNA is 41.0% HO-ORF, 49.7% CO-ORF, and 9.3% intergenic. The ChIP-seq signal of reversed-fork 4-way junctions is enriched in intergenic regions (Fisher exact test, p<2.2×10−16), and the signal in known transcription units shows no bias for “head-on” or co-directional with DNA replication. The ratio of RDG signal is HO:CO=0.83 compared with the whole genome’s ratio of HO: CO=0.82 (p = 0.5, Fisher exact test). The obvious bias of RDG peaks to intergenic region, and the lack of any detectable bias in the direction of transcription for RDG signal in transcribed DNA implies that R-loops from replication/transcription head-on collisions are not the main cause of DNA damage/reversed forks that results from CsgD binding to DNA.
Western analyses of Pol IV protein levels
M9 glycerol cultures inducing wild-type, catalytically inactive, and β-binding-defective Pol IV were normalized to OD600 of 1.0, and 1 ml of each was pelleted, resuspended and boiled as described (Kim et al., 2001). Proteins were separated by 10% SDS-PAGE and transferred to PVDF membrane according to the manufacturer’s instructions (Amersham, GE Healthcare). The membranes were blocked with ECL Prime blocking agent (GE Healthcare) and probed with primary anti-Pol IV polyclonal antibody (Kim et al., 2001) (1:2000). The membrane was further probed with secondary polyclonal goat anti-rabbit IgG-Cy5 antibody (Bethyl Laboratories) and visualized by scanning in multicolor imager Typhoon detection system (GE Healthcare).
Identification of human homologs using BLASTp and delta BLAST
“Homologs” are defined here as proteins with amino-acid similarity that could result from possible evolutionary relatedness. We used two basic local alignment search tools: the BLASTp and Delta-BLAST algorithms, searching protein sequences obtained from GenBank and other NCBI database resources. For both we used e-value < 0.01 (⩽1 gene is identified by random chance in 100 queries) and sequence identity of ⩾20%. Note that ⩾20% sequence identity between E. coli and human is considerable. Given a protein query, BLASTp returns the most similar protein sequences from the protein database with e-value < 0.01 and identity ⩾ 20%. Delta-BLAST uses multiple sequence alignment with conserved domains found in the CDD (Conserved Domains database from NCBI) and computes a Position Specific Score Matrix (PSSM) with e-value < 0.01. Both methods were compared against the human protein database of NCBI. Proteins identified from either algorithm were identified as human homologs of the E. coli DDPs.
Cancer association of DDP homologs and human proteins from a select DNA-damage screen
The 284 human homologs of the E. coli DDPs are significantly overrepresented among known (Forbes et al., 2015) and predicted (D’Antonio and Ciccarelli, 2013) cancer driving genes in a curated consensus in the Sanger Institute’s Catalogue of Somatic Mutations In Cancer (COSMIC) (Forbes et al., 2015) and the database of D’Antonio and Ciccarelli (2013) (p = 0.0002, Fisher’s exact test; Figure 2C; Table S3), which contain gain- and loss-of-function drivers. Human homologs of random E. coli proteins are not overrepresented (p = 0.48, Fisher’s exact test), showing that cancer association is specific to DDP homologs, not conserved proteins generally. The human homologs remain overrepresented among known and predicted drivers when homologs of E. coli DNA-repair proteins and other known human DNA-repair proteins are excluded (Figure 2C).
We also analyzed published data from the limited human overexpression DNA-damage-up screen of (Lovejoy et al., 2009) by Chi-square test against known (Forbes et al., 2015) and predicted (D’Antonio and Ciccarelli, 2013) cancer-driving genes. This overexpression screen of a set of nucleus/DNA-associated proteins discovered 96 human proteins (Lovejoy et al., 2009), which we found are overrepresented among known and predicted cancer drivers at p = 0.0001 and p = 0.0002, with DNA-repair proteins excluded (Chi-square test, identities, Table S3). Only one protein was identified in common between the E. coli DDP homologs and the human overproduction screen (FIGNL1), indicating that the E. coli screen identified many new hDDP candidates, then validated hDDPs. We note that an unbiased screen of all human proteins for DNA damage on overproduction is challenging because the best human overexpression libraries contain a fraction of all human protein-coding genes, and many clones that are not full length (following section). Additionally, the DSB bias of current assays for DNA damage in human cells (RESULTS, Human Homologs Promote DNA Damage and Mutation), is likely to cause many non-DSB-promoting hDDPs to be missed when screened for in human cells. This contrasts with the E. coli SOS-response-based screen, which recognizes single-stranded DNA, including single strands not at DSBs [references in (Pennington and Rosenberg, 2007)]. Our data showing that only 41% of E. coli DDPs show elevated DSBs (Figure 4A, Table S1), the rest having non-DSB DNA damage, may explain the apparent greater sensitivity of the E. coli screen for finding human DDPs (45% of those tested were validated, compared with 1.7% of human nucleus specific genes tested, Lovejoy et al., 2009).
Analyses of cancer survival and mutation loads
High RNA levels of the 284 hDDP candidates are associated with tumor mutation loads in data from TCGA (Gao et al., 2013), and these relationships are still robust when—(1) validated DDPs (Figure 3B, Table S2) are removed; (2) known and predicted drivers are removed; (3) both drivers and validated DDPs are removed (Figure 2F). The average correlation strength of the above RNA sets with mutation load was in the top 3% of correlations for randomly selected groups of genes across all human genes. The average correlation strength of the validated DDPs (Figure 3B, Table S2) was among about the top 10% of correlations for randomly selected groups of genes across all human genes—still correlated but the weaker correlation probably reflecting the much smaller gene set: 32 validated and 284 homologs total. These data show that the correlation with cancer mutation loads holds not only for RNAs of validated hDDPs, shown to promote DNA damage in human cells (Figure 3), in assays that are double-strand-break (DSB) biased (discussed, RESULTS, Human Homologs Promote DNA Damage and Mutation), but also in the many candidate hDDPs of the whole network. Many of these may promote non-DSB DNA damage and may not be validated by our assays, or have not yet been tested (Table S2).
RNA-sequencing data from The Cancer Genome Atlas (TCGA) (Gao et al., 2013) were processed in the form of transcripts per million (TPM) and obtained via Gene Expression Omnibus (accession number GSE62944). Only the TCGA cancer types that had over 100 patients with RNA- and DNA-sequencing data were analyzed. Upon defining our gene sets of interest, RNA data were subjected to single sample Gene-Set Enrichment Analysis (ssGSEA) using GSVA package in R. The resulting gene-set enrichment score for each sample was used as a representation of gene-set RNA level in each sample. Somatic mutation data for TCGA cancers were obtained in the form of mutation annotation files (raw or final) from the Broad Institute Genome Data Analysis Center (GDAC). For each sample, the sum of base-substitution and indel mutations was taken as the total mutation count, and loge of this value was referred to as “mutation load.” Correlation analysis for “all human genes” was performed via bootstrapping. Briefly, we computed the mean correlation coefficient of mutation load with gene-set enrichment scores for 1000 randomly sampled gene sets, each consisting of a random number, between 10 to 1000, of genes out of over all human genes for which expression data were available. Kaplan Meier survival analysis was performed using “survival” package in R comparing the top and bottom tertiles of samples based on their gene-set enrichment score. Correlation analyses with mutation loads was performed in base R and correlation coefficients were plotted using the “corrplot” package in R.
Cancer type designations (Figure 2F): OV ovarian serous cystadenocarcinoma; THCA thyroid carcinoma; COAD colon adenocarcinoma; KIRC kidney renal clear cell carcinoma; KIRP kidney renal papillary cell carcinoma; CESC cervical squamous cell carcinoma and endocervical adenocarcinoma; READ rectum adenocarcinoma; BLCA bladder urothelial carcinoma; LAML acute myeloid leukemia; LIHC liver hepatocellular carcinoma; SKCM skin cutaneous melanoma; LUSC lung squamous cell carcinoma; PRAD prostate adenocarcinoma; STAD stomach adenocarcinoma; LUAD lung adenocarcinoma.
Cloning of human genes for DNA-damage analyses in human cells
Of the 284 human homologs, we identified 121 candidates of particular interest according to the following criteria: (i) Many are encoded by genes amplified at high frequencies in cancer genomes from TCGA (Gao et al., 2013) (Table S4). (ii) For a minority, the genes are mutated or deleted at impressive, high frequencies in TCGA (Gao et al., 2013). (iii) Full-length clones that encode 90 of these appeared to be available in the Orfeome V8.1 or CCsBroad cDNA-clone collections. We determined by restriction mapping that many of the human genes in those libraries are not full length (Table S5), and cloned 18 genes including 15 candidate hDDP genes and three controls de novo as full-length cDNA clones that we sequence-verified (below), ultimately creating 70 full-length overexpression GFP-fusion clones of human homologs of E. coli DDP genes, and overexpression GFP-fusion clones of 3 human homologs of E. coli damage-down genes, as possible negative controls (STAR Methods, Table S5), 9 random human genes, and 11 random human homologs of E. coli non-DDP genes (Tables S2 and S5).
Fifty-eight human DDP and 19 non-DDP cDNA clones (Table S5) in the Gateway entry vectors pDONR221 and pDONR223 (Invitrogen) were subcloned from an augmented library of ~32,000 Orfeome V8.1 stated to contain sequenced human full-length cDNA clones, and additional full-length and commonest splice-variant length clones obtained from others including CCsBroad gene libraries. The size of cDNA from each gene was confirmed by restriction enzyme digestions. We also cloned, de novo, 15 candidate hDDP genes, one non-hDDP gene, tubulin and two de novo methylase genes (Table S5) that were not present as full-length clones in the Orfeome V8.1 or CCsBroad gene libraries. These candidate genes were amplified from cDNAs generated from mRNAs extracted from the human cancer-cell lines U2OS or MRC5-SV40. PCR products of the correct size were cloned into the Gateway entry vector pENTR11 at restriction enzyme cut sites or into pDONR201 using attB site-specific recombination sites. Five DNMT1 truncated constructs were modified by using site-directed mutagenesis (Table S5). Clones were sequenced and verified as the correct gene sequence based on the Reference Sequence (RefSeq) database from NCBI. We subcloned each gene into a mammalian expression vector containing a GFP epitope tag (pcDNA6.2/N-EmGFP-DEST, Invitrogen), which allows us to analyze transfection efficiency and visualize protein localization in transfected cells. All human-cell overexpression plasmids used in this study are listed in Table S5.
Human cell lines, plasmids, and reagents
MRC5-SV40 and HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 μg/mL penicillin, 100 μg/mL and streptomycin. Transient transfections into human cells were performed using GenJet (SignaGen Laboratories) for MRC5-SV40 and PEI (polyethylenimine, Sigma) for HEK293T. Transfections for siRNA were carried out with lipofectamine RNAiMax (Invitrogen) following the manufacturer’s instructions. The siRNAs were siNT: non-targeting pool (Dharmacon) and siRAD18: ACUCAGUGUCCAACUUGCU (Sigma). DNA-PK inhibitor (NU7441, Tocris Bioscience) was used at 2.5 μM 6 h prior to harvesting cells for flow cytometry. NAC (N-acetyl-cysteine, Sigma) treatment was performed twice, with a final concentration of 5 mM, post-24hr and −48hr transfection. To create inducible stable clones to verify DNMT1 and PCNA interaction, GFP-tubulin, GFP-DNMT1 and GFP-DNMT1-ΔPBD cDNAs were cloned into pcDNA5/FRT/TO/Intron vector (Invitrogen, CA). Inducible HEK293T FlpIn Trex GFP-tubulin, GFP-DNMT1 and GFP-DNMT1-ΔBD cells were generated followed by manufacturer’s protocol and were cultured in the same normal medium with 15μg/ml Blasticidin and 80μg/ml hygromycin. Doxycycline (Sigma) was added to medium to trigger the production of GFP fusions.
Human-cell DNA-damage screens by flow cytometry
We screened for increased DNA damage by flow-cytometric quantification of γH2AX- and phospho-P53-antibody signals among GFP-positive transfectants. Immunostaining was performed according to a standard procedure with minor modifications. Seventy-two hours post-transfection, cells were collected and approximately 1×106 cells taken for staining. For staining, cells were fixed with 2% (v/v) formalin for 15 min on ice, washed twice in cold-PBS and permeabilized with 0.05% (v/v) Triton-X for 15 min on ice followed by two washes with PBS. The fixed cells were then blocked with 5% BSA-PBS for 1 hr, and stained with either γH2AX (Millipore) or phosphorylated p53 primary antibodies (Cell Signaling) overnight at 4°C. Cells were washed three times in 1% BSA-PBS followed by an incubation of Alexa Fluor 647 goat anti-mouse IgG in 5% BSA-PBS (Invitrogen) for 1 hr at room temperature in the dark, then washed three times with 1% BSA-PBS. Stained samples were measured by a BD LSRFortessa flow cytometer and analyzed using FlowJo software. Cells without transfection were used to set the threshold gating to determine the percentage of GFP- and γH2AX- or phosphorylated p53-positive cells, with 0.5% of control cells gated as the damage threshold. The DNA-damage ratio caused by protein overproduction is defined by (Q2/Q3)/(Q1/Q4), where Q2 is the number of transfected damage-positive cells; Q3 is the number of transfected damage-negative cells; Q1 is the number of untransfected damage-positive cells; and Q4 is the number of untransfected damage-negative cells. Results were obtained from at least two independent experiments. Statistical significance (p value) was determined using two-tailed unpaired Student’s t-test followed by false discovery rate (q value) correction. Both the γH2AX and phosphorylated-p53 assays show linear responses to exogenous DNA damage caused by ionizing radiation (Figure S3L and M), indicating their quantitative validity.
Superiority of transient transfection to stable integration of genes encoding hDDP candidates
We found transient transfection to be superior to creation of stable clones because of apparent selection for mutations in the inducible hDDP candidate genes upon integration. Mutations in the hDDP candidates or other DNA damage-response pathways are selected probably because the gene products are toxic when overproduced and the genes are difficult to keep tightly “off.” The GFP-hDDP-gene fusions allow transient transfection assays to identify immediate effects of the DNA damage and to analyze only the minority population of cells that have been transfected successfully and produce the protein of interest. This cell subpopulation is GFP-positive, and easily identified in the flow-cytometric assays (e.g., Figure 3B).
HPRT mutagenesis assay
MRC5-SV40 cells were transfected with the plasmids indicated, and harvested 72 hours post-transfection. The percentage of GFP-positive cells of each transfectant was scored as transfection efficiency using a BD Accuri flow cytometer. The remaining cells were re-grown in 15 cm dishes for an additional 4 days. After a week of transfection, 3×106 cells were plated in 15 cm dishes containing medium with 20 mM 6-thioguanine (Sigma), with five 15 cm dishes for each gene. In addition, 600 cells were plated in triplicate, per well, in a 6-well plate without 6-TG to determine plating efficiency. The plates were incubated at 37°C in a humidified incubator until colonies formed. The colonies were stained with 0.005% crystal violet. These colonies were counted, and mutation rates determined using the MSS-maximum likelihood estimator method with correction for transfection efficiency. We verified that 6-TG resistant clones result from HPRT mutations by sequencing the cloned HPRT cDNAs from four independent mutants. The mutations are: a single-basepair insertion between the 206-207nt of HPRT gene, and three identical deletions (from 403nt to 485nt). Two of the sequenced clones were independent DNMT1-overproducing transfectants, and two were from independent vector-only control transfected cells. The HPRT cDNA is 657bp long, whereas HPRT including introns is 42kb, making sequencing the cDNAs more practical.
Estimation of additional human DDPs demonstrable in assays used here
We evaluated the validation efficiencies of four classes of human homologs of E. coli DDP genes (shown Figure 3E): genes that are—(i) both known (Forbes et al., 2015) or predicted (D’Antonio and Ciccarelli, 2013) cancer drivers and amplified in TCGA cancers (Table S4); (ii) amplified in cancers and not known or predicted drivers; (iii) known/predicted cancer drivers that are not known to be amplified in cancers; and (iv) neither amplified in cancers nor previously known/predicted cancer drivers. Based on the number of candidates that we tested in each class among the 70 DDP homologs tested, these data correspond to the following validation rates as DNA-damage-promoting for each class: (i) 100%; (ii) 53%; (iii) 67%; and (iv) 27%. Based on the numbers of homologs not yet tested in each of these classes, our data predict that the following numbers of proteins would be likely to be validated among the remaining 214 as-yet untested human homologs: (i) 6; (ii) 38; (iii) 34; and (iv) 7, for a total of 85 more demonstrable hDDPs predicted among the 284-protein candidate hDDP network. We note, however, that the human-cell DNA-damage assays used favor detection of DNA double-strand breaks, not all DNA-damage types comprehensively (RESULTS, Human Homologs Promote DNA Damage and Mutation). Thus, many more of the human homologs may be DNA-damage promoting for other kinds of DNA damage than is estimated here. Only 41% of E. coli DDPs promote DSBs (Table S1, Figure 4B); if the hDDP candidates contained as many non-DSB instigating DDPs, nearly all of tested-but-not-validated candidates would be genuine DDPs that cause non-DSB DNA damage, which is not detected in the human-cell assays used.
Human-cell immunoprecipitation and western blot analysis
After induction of protein production using doxycycline in FlpIn-inducible HEK293T cells producing GFP-tubulin, GFP-DNMT1-WT or GFP-DNMT1^PBD, cells were lysed with NETN buffer (150 mM NaCl, 1mM EDTA, 10 mM Tris-HCl, pH 8.0, and 0.5% NP-40) containing TurboNuclease (Accelagen) and 1 mM MgCl2 for 1 h at 4°C. Cell lysates were then centrifuged for 30 min at 4°C. GFP-tagged proteins were immunoprecipitated with 20 μl of GFP-Trap_A (Chromotek) for 1 h at 4°C. Beads were then washed three times with NETN buffer. Protein mixtures were eluted by boiling at 95°C with Laemmli buffer (4% (v/v) SDS, 20% (v/v) glycerol and 120 mM Tris-HCl, pH 6.8). For whole cell extracts, cells were collected with Laemmli buffer, and heated for 5 min at 95°C before loading. Samples were resolved by SDS-PAGE followed by western blot analysis. Primary antibodies were used as follows: anti-GFP (Invitrogen), anti-PCNA (Santa Cruz), anti-beta tubulin (Abcam), anti-RAD18 (Cell Signaling). Blots were analyzed by standard chemiluminescence (GE Healthcare, Amersham ECL Prime system) using a Bio-Rad molecular imager ChemiDoc XRS+ system.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details can be found in the main text, figure legends, or here. All E. coli wet-bench experiments were performed at least three times independently, and a two-tailed unpaired t-test was used to determine significant differences, unless otherwise specified. Error bars represent 1 SEM except where otherwise indicated. Pearson’s correlation coefficient was computed to assess the relationship between two parameters. STRING enrichment analysis was performed using hypergeometric tests with the correction for multiple comparisons. False discovery rate (FDR) adjustments are used to limit the overall type I errors in both E. coli and human DNA-damage flow-cytometry assays. The FDR (Benjamini Hochberg) method is the default p-value adjustment method in this paper. Fisher exact or Chi-square tests are used to determine whether two proportions are different. Wilcoxon rank-sum test was used to determine whether each gene has cancer-associated copy-number increases.
DATA AND SOFTWARE AVAILABILITY
RNA-Seq data and RDG ChIP-seq generated from this study are deposited in the European Nucleotide Archive (ENA) under accession numbers E-MTAB-7361 and PRJEB21035.
Supplementary Material
Figure S1. Genuine SOS Response, Detection of DNA Damage by the SOS-reporter Gene, and Evaluation of False Negatives in the Primary Screen, Related to Figure 1
The E. coli DNA-damage assay detects fluorescence caused by upregulation of the SOS DNA-damage-response-activated promoter PsulA fused to mCherry in a non-genic chromosomal site (Nehring et al., 2016; Pennington and Rosenberg, 2007). This assay was shown to report on DNA damage, not spurious promoter firing with the demonstration that fluorescence induction requires the DNA-damage-sensing protein RecA, and is inhibited by a mutant SOS-response transcriptional repressor, LexAInd−, which does not de-repress the SOS genes during DNA damage (Pennington and Rosenberg, 2007), both also shown here (A). Further, of the spontaneous SOS-inducing DNA damage, previously 60% was shown to reflect DNA doublestrand breaks (DSBs) and 40% single stranded DNA not at DSBs (Pennington and Rosenberg, 2007), and the spontaneous DSB frequency and rates per cell division and chromosome replication were confirmed in a direct, independent assay for DSB ends (Shee et al., 2013). Thus, this reporter reports on DNA damage.
(A) Fluorescence requires the ability to activate the SOS response, and so is absent in SOS-induction-deficient ΔrecA or lexA3(Ind−) mutant cells, demonstrating that DNA damage, not spurious promoter firing, underlies fluorescence increases, per (Pennington and Rosenberg, 2007). Using these negative controls, a flow-cytometry gate (vertical dashed line) is set in each experiment, per (Pennington and Rosenberg, 2007), at fluorescence at which 10−4 of the lexA3(Ind−) negative- control cells are positive. The SOS-positive cells in the wild-type (WT) strain represent spontaneous DNA damage, per (Pennington and Rosenberg, 2007). Representative flow-cytometry histograms generated under the growth conditions used in the screens reported here.
(B) Positive control, mCherry+ lexA51(Def) cells with constitutively activated SOS response, and negative control, lexA3(Ind−) SOS-off cells.
(C) For a representative sample of 66 of the validated DDPs (Table S1), we confirmed that their increased fluorescence results from increased SOS response by showing its dependence on SOS-response-activator RecA, and a functioning (inducible) LexA SOS repressor (Pennington and Rosenberg, 2007). All 66 showed RecA- (here) and LexA-dependent high fluorescence (Figure 1G), demonstrating the presence of DNA damage and a genuine SOS response (Table S1). We also ruled out the possibility that DDP overproduction might increase mCherry protein fluorescence itself (Figure S1D; Table S1).
(D) Representative DDPs do not enhance mCherry fluorescence generally, when mCherry is controlled by non-SOS promoter PBAD.
(E) Screen of 99 random E. coli proteins not identified in the primary plate-reader screen finds one SOS-positive clone. Because of its inherent noise level, the plate-reader primary screen is likely to generate some false-negative results; i.e., miss clones with subtle but real DNA damage-up phenotypes. If the 3815 E. coli proteins that were not identified in the primary screen (4229 protein-coding genes in the library - 414 proteins found in the primary screen) also harbor 1% real but undiscovered DDPs, then an additional 38 overproduction DDPs are predicted to reside in the E. coli overproduction library, for an estimated total network size of 246 (38 + 208) DDP genes in the overexpression library used. In STAR Methods, we estimate an additional 123 E. coli overexpression DDP genes that are likely to be in the E. coli genome, but would not be discoverable using the Mobile plasmid overexpression library.
(F) E. coli TetR clones from cI mutation assay harbor genuine mutations. Sequences of cI mutations from 10 different DDP-overproducing strains with strong damage-up phenotypes: CsgD, TopB, YegL, GrpE, HslU, Mrr, CheA, MdtA, YicR, and UvrA. The compiled sequences from the 10 DDP clones are shown in red and blue (n = 3-10 independent TetR mutants per clone). The 69 cI mutations sequenced include 3 IS (mobile)-element insertions, and 2 clones without a PCR product, which whole-genome sequencing revealed to result from large deletions (STAR Methods). Mutations in the vector-only control strain are shown in black (19 independent mutants). Red font, mutations attributable to common errors of the SOS-upregulated error-prone DNA polymerases V and IV (Kobayashi et al., 2002; Maor-Shoshani et al., 2000; Wagner and Nohmi, 2000); blue font, mutations not attributable to common Pol V or Pol IV errors. Capital letters, basepairs that were changed or deleted; carrots, insertion points of bases indicated; Δs, deletions of the bases indicated. Upper right, diagram of gross chromosomal rearrangements (GCRs) found in TopB-overproducing cells, among which, 18% (7/40) of the TetR mutations are GCRs, including large deletions (from 200bp-6200bp), an inversion, and a transposon insertion.
Figure S5. Sensitivities to DNA-Damaging Agents, Related to Figure 4
(A) Phleomycin sensitivity detected by growth inhibition in known mutants with reduced homologous recombinational (HR) repair. recF: defective in single-strand gap HR-repair; recG: reduced ability to branch migrate Holliday junctions; recB: defective in HR DSB repair (Kuzminov, 2011).
(B) 106 E. coli DDP overproducers are sensitive to phleomycin. Data normalized to vector-only controls: (treated/untreated DDP overproducer) / (treated/untreated vector-only) so that values < 1 indicate sensitivity. Sensitive clones include 11 more sensitive than a recB mutant, 72 within the range of recB and recF mutants, and 23 more sensitive than the vector-only control but less than recF mutant. One of the 16 non-DDP-overproducers was phleomycin sensitive, which differs from the DDP-overproducers at p = 0.0003 (one-way Fisher’s exact test).
(C) Sensitivity of 75 E. coli DDP clones to oxidative-damage-inducing agent H2O2 detected as colony forming units in known mutants with reduced base excision repair (BER) efficiency: xthA (inset) encodes Exonuclease III; nfo encodes Endonuclease IV. xthA nfo (green) double mutants are more sensitive than each single mutant (Galhardo et al., 2000). nth encodes Endonuclease III; nei encodes Endonuclease VIII. The nth nei (purple) double mutant has almost no AP lyase activity, and has extreme sensitivity (Saito et al., 1997).. Data of the DDP clones are normalized: (treated/untreated DDP overproducer) / (treated/untreated vector-only) so that values < 1 indicate sensitivity. The H2O2-sensitive DDP clones include 36 more sensitive than xthA nfo cells; 39 more sensitive than the vector-only control but less sensitive than xthA nfo cells. None of the 15 nonDDP-overproducers had H2O2 sensitivity, which differs from the DDP clones at p = 0.002 (one-way Fisher’s exact test).
(D) Sensitivity to interstrand-crosslinking agent Mitomycin C (MMC) detected by growth inhibition in known mutants with defective HR repair or nucleotide excision repair (NER): recA, HR- and SOS response-defective; and uvrC, NER-defective (shown in E, cyan).
(E) Ten DDP clones were MMC sensitive. DDP clone data are normalized: (treated/untreated DDP overproducer) / (treated/untreated vector-only) so that values < 1 indicate sensitivity. None of the 16 non-DDP overproducers was MMC sensitive, which does not differ from the DDP clones, p = 0.47 (one-way Fisher’s exact test).
(F) Representative DDPs do not downregulate DNA-repair-gene mRNAs upon overproduction. Seven DDPs representative of the six DDP function clusters (Figure 4N; Table S1) were assayed for RNA levels of 32 DNA-repair genes by RNA-seq with and without overproduction of each of 7 DDPs, which confer sensitivity to the following agents: TopA and BamC (H2O2); CusR, HemX, AroP, YicR, and YqjD (phleomycin). The repair pathways represented were nucleotide excision repair (uvrA, uvrB, uvrC, uvrD), base-excision repair (BER: mutT, mutM, xthA, nfo, ung, mug, nth, tag, alkA, nei), mismatch repair (mutS, mutL, uvrD, mutH), homology-directed repair (recA, radA, ruvA, ruvB, ruvC, recT, recF, recO, recR, recG, recN), and homology-directed DSB repair (as for homology-directed repair with the addition of recB, recC, recD, and without recFOR). Log2 ratio (DDP overproducer / vector) for each DNA-repair gene in each of the DDP clones was calculated and clustered by hierarchy clustering. Most of the DNA-repair genes are up-regulated during DDP overproduction. Upregulation presumably results from SOS or other DNA-damage or oxidative-stress responses. Only mutM RNA was is downregulated (in the TopA-overproducing strain). mutM downregulation is unlikely to account for the H2O2 sensitivity of the TopA-overproducing strain because even mutM null mutants are not sensitive to H2O2 (Asad et al., 2004). The data exclude the hypothesis that most or all of the DNA-damage sensitivities caused by overproduced DDPs (Figures 4J-L, A-E here; Table S1) result from transcriptional downregulation of DNA-repair genes. Potential post-transcriptional regulation mechanisms are not excluded. The data support the hypothesis that the DNA-damage sensitivities result from excess DNA damage overwhelming DNA-repair capacity, potentially titrating DNA-repair enzymes, or direct inhibition of DNA repair by the overproduced protein. Reduction of DNA-repair capacity could produce phenotypes like those of DNA-repair mutants, which would drive genome instability, in many DDP-gene dysregulated cells that do not possess DNA-repair-gene mutations or downregulated mRNAs.
(G) Cluster analysis of quantitative data on DNA-damaging-agent sensitivities in the E. coli DDP network. Each bar, a quantitative phenotype of each strain producing each of the 208 E. coli DDPs. Assays indicated above the heatmap: H2O2, hydrogen-peroxide sensitivity (reduced base excision repair); PHLEO, phleomycin sensitivity (reduced DSB-repair); MMC, mitomycin-C sensitivity (reduced NER and/or HR repair). Z score are compared with the vector-only negative control. Increased resistance (blue) to H2O2, MMC, and phleomycin are apparent in clusters in each, as is decreased ROS levels in a cluster Figure 4M, and reduced anucleate cells (DNA loss) in that figure. Similarly, clusters 3, 4, and 6 show reduced sensitivity (greater resistance than wild-type cells) to H2O2, and 2 and 3 show greater resistance to mitomycin C (Figure 4N). A probable explanation for these “better-than-wild-type” phenotypes is that some imbalance in these cells may induce a stress response, one of the consequences of which may be improvement of the fidelity of, e.g., chromosome segregation (preventing anucleate cells), reduction of ROS levels, or resistance to DNA-damaging agents, as is documented, for example, for DNA-damage resistance induced by mild induction of the SOS response, among other stress responses (reviewed, Fitzgerald et al., 2017).
(H) Heritable mutations do not account for DNA-damage sensitivities of seven highly DNA-damage-sensitive DDP clones. The DDP overproducers were tested for sensitivity with overproduction of the DDP (red, “before”), then again after the drug treatment, with the DDP-gene repressed (blue, “after”) to determine whether their sensitivity resulted from induction of mutations in genes needed for resistance. After drug treatment, three colonies were recovered on plates with no DDP-gene-inducing IPTG, and then tested for DNA-damaging-agent sensitivity again. In all cases, the recovered colonies not induced for DDP overproduction showed no sensitivity to DNA-damaging agents (blue, after). Thus, their DNA-damage sensitivities did not result from heritable mutations.
Figure S6. DNA-binding Transcription-Factor and Replication-Stall (RDG) Foci, Examples, Related to Figure 5
(A) Representative data show induction of RDG reversed-fork foci with production of DNA-binding transcription factors (TFs), DNA-binding-domain dependently. ΔDBD, in-frame deletion of the DNA-binding domain; CsgDD59N, D59E: single amino-acid changes that reduce CsgD DNA binding (Ogasawara et al., 2011). Three wild-type TFs and the corresponding mutants with reduced DNA-binding ability overproduced in ΔrecA cells, in which RDG foci represent reversed-forks (RFs), and quantified (Figure 5E; Table S1). Representative images shown, scale bar: 2μm.
(B) Representative data show co-localization of RDG reversed-fork foci with TF-mCherry foci, which form DNA-binding-domain dependently. Representative examples, scale bar: 2μm. Most of the CsgD-mCherry and HcaR-mCherry foci were co-localized with RDG/RF foci (Figure 5G, H; Table S1). Showing weak co-localization, about 5% of the YahB-mCherry TF foci were colocalized with RDG/RF foci (Figure 5G). Negative-control mCherry alone rarely forms foci (Figure 5G).
Figure S7. RDG ChIP-seq Detects Stalled-Fork Enrichment Near CsgD-Binding Sites and Controls for Pol IV, Related to Figures 5 and 7
Nine of the 10 known, validated CsgD-binding sites (Keseler et al., 2017; Ogasawara et al., 2011) showed an RDG peak nearby induced by CsgD overproduction: within 10kb (median 2.8kb), which differs from random simulations of the genomic distribution of the total number of RDG peaks (p = 0.01 two-tailed z test,. Isogenic cells overproducing—CsgD, red; CsgD ΔDBD (deletion of the DNA-binding domain), blue; vector-only, green. Green boxes, CsgD binding sites.
The RDG ChIP-seq experiments (Figures 5I and here A-C) were performed twice, with peaks called against the matched control DNA-binding-domain mutant, CsgDΔDBD. We identified 155 total reproducible RDG ChIP-seq peaks with at least 2.5-fold increase in both repeats compared with CsgDADBD done in parallel. The following control simulations show that RDG ChIP-seq signals are enriched near known CsgD binding sites. We simulated CsgD-overproduction ChIP-Seq data by randomly distributing the called RDG peaks across the genome and analyzed, first, the number of binding sites with at least one RDG peak within 10kb, and, second, the distance between each known CsgD-binding site and the nearest RDG peak (median), for each of the 10 experimentally validated CsgD-binding sites (Keseler et al., 2017; Ogasawara et al., 2011). Strikingly, there is at least one observed (actual) RDG peak within 10kb of 9 out of the 10 known CsgD-binding sites (A-C), whereas there are only 5 known binding sites with at least one simulated RDG peak within 10kb of the 10 known binding sites in our simulations, which is significantly less than the observed (p = 0.01, two-tailed z-test). Also, the median distance between a given known CsgD-binding site and the nearest RDG ChIP-seq peak is 2.8kb for the real peaks, which is significantly closer than the 10.3kb median distance in our simulations (p = 0.009).
RDG peaks not at known CsgD-binding sites. For the 142 RDG peaks that are not significantly near a known CsgD binding site, these could result from—(i) binding of CsgD to sites not yet identified in the literature, in which no comprehensive high-resolution (ChIP-seq) genome-wide binding study has been done; (ii) relaxation of the binding specificity of CsgD when overproduced such that new sites are hit; and (iii) downstream (indirect) effects of regulation of CsgD-regulated genes by CsgD binding to its sites. For example, CsgD upregulation of other DNA-binding transcription factors could cause peaks at their binding sites, among other indirect but biologically real (CsgD-DBD-dependent) possibilities. The 142 of 155 RDG ChIP-seq peaks that are not near known CsgD binding sites do not overlap significantly with palindromic REP sequences, which are prone to form DNA cruciform structures (four RDG peaks overlap with REPs, p =0.43, two-tailed z-test compared with median random distribution), or terA-terC regions, which have higher frequencies of fork convergence (11 RDG peaks overlap with ter sequences, p =0.47, two-tailed z-test, compared with compared with median random distribution).
Upstream bias of RDG stalled-fork peaks at CsgD binding sites. The 9 known CsgD-binding sites with one or more observed RDG peaks within 10kb are of two types: six show RDG peaks at or upstream of the binding sites in the replication path and five show RDG peaks downstream. The observed number of CsgD binding sites with upstream or co-localized RDG peaks is significantly higher than the median in the simulation (p = 0.04, two-tailed z-test), whereas the number of CsgD binding sites with RDG peaks downstream is not quite significant, at p = 0.13, two-tailed z-test. The analysis above suggests that direct binding of CsgD to its known binding sites underlies the RDG upstream and co-localized peaks, whereas the downstream peaks may reflect a component of direct CsgD-DNA-induced fork reversal, and a component of indirect effects, or binding to as yet unknown CsgD binding sites. One way that an RDG stalled-fork peak might result downstream in the replichore, but still via direct interaction of replication with bound CsgD, could be if some of the downstream stalled forks are caused by two events (illustrated Figure 5J, lower): first, slowing of replication forks by CsgD binding at its site might make the replisome susceptible to a second, otherwise surmountable barrier of any kind downstream in the replication path.
(A) RDG ChIP-seq peaks overlap with three known CsgD-binding sites DBD dependently.
(B) RDG ChIP-seq peaks near three known CsgD-binding sites, upstream in the replication path. Increased negative supercoiling between the oncoming fork and the CsgD-bound site may stall replication and causes fork reversal, illustrated Figure 5J.
(C) RDG ChIP-seq peaks located near three known CsgD binding sites, downstream in the replication path. Because the upstream peaks (B) are more significantly associated with the known CsgD-binding sites relative to simulation of random genomic distributions (above), it is possible that the downstream RDG peaks could result from either CsgD binding to sites not yet known, or from indirect consequences of CsgD DNA binding, such as effects of CsgD-regulated gene products interacting with other sites in DNA (above). Alternatively, some of the downstream RDG peaks could result from CsgD binding its known site, slowing replication, then encountering an otherwise surmountable obstacle downstream, per Figure 5J.
(D) The clamp-loader dnaX tau-only mutant is not generally deficient in SOS-response induction, but merely reduces the DNA damage (SOS activity) caused by Pol IV upregulation (Figure 7D,E).
(E) The Pol IV R49F catalytic-mutant (Pol IV cat−) and ΔBBD-mutant proteins do not display reduced protein levels in western blots, in agreement with previous studies (Uchida et al., 2008; Wagner et al., 1999).
(F) Neither the E. coli MutM 8-oxo-dG glycosylase nor the MutY adenine glycosylase is required for DNA damage instigated by Pol IV, indicating DNA damage produced independently of incorporation of 8-oxo-dG into DNA. Representative data, quantified (G).
(G) Mean ± SEM ≥ 3 experiments. Pol IV is induced by IPTG.
Table S1. Summary of E. coli DNA-damage, mutation, function and phenotype data from overproduced DDPs, Related to Figures 1, 2, 3, 4, 6
Table S3. Human DDPs-/candidate-DDPs overrepresented among known and predicted cancer-driving genes, Related to Figure 2
Table S4. Human-homolog candidate-DDP genes are overrepresented in cancer genomes as copy-number gains, Related to Figure 2
Table S5. Sources of full-length and truncated clones of candidate hDDPs and 20 non-DDPs/human overexpression plasmids constructed and used in this study, Related to STAR Methods and Figure 3B
Table S6. DNA-damage levels in human cells overproducing candidate hDDPs and random human proteins, Related to Figure 3
Table S7. Escherichia coli K12 strains and plasmids used in this study, Related to STAR Methods.
Figure S2. Protein-Protein Associations in E. coli and Human DDP Networks via STRING, Related to Figures 1 and 2
(A) Protein-protein associations in the E. coli DDP network detected with STRING and co-expression analysis. CytoScape software generated from STRING 10.0 database. Other, defined Table S1. STRING measures protein-protein associations of many kinds including indirect associations (e.g., co-occurrence in different organisms’ genomes, or in papers in the literature) and direct protein-protein interactions, among others (STAR Methods). Using STRING with an interaction score cut-off of ⩾0.6 (medium-to-high confidence, STAR Methods), we found that the 208 DDPs form a significant network via protein-protein interaction data with more interactions than random sets of 208 E. coli proteins (p = 4.4 × 10−31, hypergeometric test). When known DNA-repair proteins are excluded, the STRING network is still significant compared with random sets of the same number of E. coli proteins (p = 8 × 10−6, hypergeometric test), indicating that the associations are not solely via DNA-repair-protein associations. When both DNA-repair and DNA-replication proteins are excluded—both known to interact directly with DNA—the STRING network is no longer significant compared with random sets of the same number of E. coli proteins (p = 0.47, hypergeometric test). These data suggest that, as might be expected from the highly diverse protein functions (Figure 1F), these proteins work in many different cellular processes that may share only their various effects on DNA damage, seen by significant association as a network only when DNA-repair and replication proteins are included, which we identify as the hubs of the DDP STRING interaction network (blue and green circles shown). Comparison of the six phenotype/function clusters of the E. coli DDPs (Supplement Figure 3) with the STRING protein-protein-associations reveals that cluster 2 shows even more protein-protein interactions than the DDP network as a whole (p = 0.0007, one-was Fisher’s exact test), supporting the function clusters (Figures 4M,N and S5G) as biological groups.
Co-expression of E. coli DDP RNAs. Normalized RNA abundance (gene expression) data available at Many Microbial Microarrays Database (http://m3d.mssm.edu/norm/) show RNA levels under a few hundred growth conditions. We calculated pairwise co-expression Pearson correlation coefficients for the 208 E. coli DDP genes, and for 1000 iterations of 208 random E.coli genes. We find that the 208 DDP-gene RNAs have a median correlation coefficient of 0.035, which is slightly but significantly higher than 1000 iterations of random E. coli genes (0.02 ± 0.007, mean ± SD, p<0.01), implying a network property of these genes. We tested whether, because of their DNA-damage provoking properties, the RNAs of the 208 DDPs might be produced in smaller ranges of RNA levels than random genes, and found no such difference (p=0.09, two-tailed unpaired t-test). Thus, DDP-encoding RNAs are not subject to special narrower-range accumulation relative to other E. coli genes. A complexity of comparison with random genes is that we predict both many more overproduction DDPs among them (RESULTS, Human Homologs Promote DNA Damage and Mutation), and also greater numbers of loss-of-function drivers of DNA damage.
(B) Specific protein-protein interactions of human candidate DDP network. Diagram details are as in A. Image from Figure 2B enlarged. Although, the human homolog (candidate hDDP) network has DNA-repair and -replication genes at its center, removal of these proteins leaves the remainder with still significant connectivity: p = 4.1 × 10−215 (hypergeometric test), though less than the whole network (p = 1.2 × 10−327, hyper geometric test). Random human proteins do not form robust protein-protein association networks. Although not associated nearly as strongly as the human homologs of DDPs (1.2 × 10−327), the significant association of random human homologs of E. coli proteins (1.8 × 10−49) compared with random human proteins (p = 0.80) could potentially result if highly conserved proteins generally have more interactions with each other than random proteins. This might be because the most highly conserved, fundamental aspects of biology, and proteins that participate in them, are enriched for conserved protein machines (ribosomes, replisomes, transcription complexes, etc.), and/or fundamental pathways the actors in which have remained associated. Alternatively, it might be that proteins that function as part of interacting protein groups evolve more slowly, and so are overrepresented among conserved proteins.
Figure S3. Association of Human Homolog Network with Cancers: Copy-number Gain, Survival, and Mutation Load, and Controls for Human DNA-Damage Assays and DNMT1, Related to Figures 2, 3 and 7
(A to C) Twenty-six cancer types in TCGA data (Gao et al., 2013) are displayed along the x axes. Blue, median % of patient cancers with increased copy number of any gene in their genome; red, % of patient cancers with increased copy number of the gene indicated. Copy-number-increases in human homologs of E. coli DDP genes are higher than those of human homologs of random E. coli genes (p < 0.05, FDR < 0.10, Wilcoxon test). Examples of the Pan-Cancer copy-number-increase analysis (GISTIC threshold copy-number gain ⩾ 1) of the 284 human homologs of E coli DDP genes in 26 cancer types are shown here (complete analysis Table S4). The human genes fell into three categories:
(A) genes with increased DDP-gene copy numbers across cancers (fold change > 1.5, p < 0.05, FDR < 0.10, Wilcoxon test);
(B) genes with cancer-specific copy-number-increases (p < 0.05); and
(C) not particularly cancer associated.
(D) Decreased cancer survival is associated with high DDP-homolog RNA levels in cancers—our analyses of data from TCGA (Gao et al., 2013), STAR Methods. BRCA, breast invasive carcinoma; HNSC, head and neck squamous cell carcinoma; UCEC, uterine corpus endometrial carcinoma. *, **, *** indicate that survival of the cancers with high and low levels of the 284 RNAs differ at p ⩽ 0.05; ⩽ 0.01, and ⩽ 0.001 respectively, log-rank test.
(E) Decreased cancer survival is not confined to the previously known cancer drivers in the network. RNAs of the known (Forbes et al., 2015) cancer-driving genes among the homologs show less association with poor survival than the whole homolog network.
(F) RNAs of the DDP homologs excluding known (Forbes et al., 2015) drivers are still associated with decreased patient survival, indicating that genes in the homolog network other than and in addition to the previously known drivers are also associated with decreased survival.
(G-H) RNAs of the human DDP homologs are still robustly associated with decreased patient survival, when (G) validated DDPs or (H) validated DDPs and known drivers are removed.
(I-K) Human MRC5-SV40 or HEK293T were transfected with human candidate DDP and random human genes and random human non-DDP homologs of E. coli genes, and DNA-damage markers were analyzed by flow cytometry.
(I) Assay I: γH2AX in MRC5-SV40 cells.
(J) Assay II: γH2AX in MRC5-SV40 cells treated with DNA-PK inhibitor, which inhibits DNA break repair by non-homologous end joining and so is a sensitizing DNA-damage screen.
(K) Assay III: phosphorylated p53 in HEK293T cells. Data represent mean ± range, n ⩾ 2. The candidate hDDP genes differ from the 20 random human genes, p < 0.0001, Fisher exact test.
(L and M) Finear response of both human-cell DNA-damage-detection assays with exogenous ionizing radiation treatment indicates quantitative validity of these assays. Percent of MRC5-SV40 cells that are positive for (F) γH2AX, and (M) phosphorylated p53 (p53-S15p), in flow-cytometric assays. MRC5-SV40 cells were treated with IR with the indicated dose and analyzed by flow cytometry.
(N) Validated hDDP genes are enriched among genes with cancer-associated copy-number increases (bar 4 versus bar 1, one-way Fisher’s exact test, p = 0.0001). The level of enrichment is higher (one-way Fisher’s exact test) than total human homologs of E. coli DDP genes (bar 4 versus bar 2, p = 0.0002), and higher than the candidates that were tested but not validated (bar 4 versus bar 3, p = 0.02).
(O) DNMT1 wild-type (WT) but not ΔPBD interacts with replisome sliding clamp, PCNA. The tubulin negative control also does not interact with PCNA. GFP-trap immunoprecipitation was performed in flipln stable inducible HEK293T cells expressing GFP-tubulin, GFP-DNMT1-WT or GFP-DNMT1-ΔPBD. Interactions were determined in immunoprecipitation samples by western blotting with anti-GFP and -PCNA antibodies.
(P) Mono-ubiquitylation of the replisome clamp, PCNA, by overproduced DNMT1 is RAD18-mediated. Knockdown of RAD18 reduced the level of ubiquitylated PCNA in DNMT-WT overexpressing cells. Western analyses of PCNA ubiquitylation in MRC5-SV40 cells transfected with the plasmids indicated in combination with non-targeting (NT) or RAD18 siRNA.
Figure S4. DSBs, Reversed Forks, Reactive Oxygen, and DNA Loss in DDP Clones, Related to Figure 4
(A) Increased GamGFP foci indicate DSBs caused by overproduction of 87 of the 208 E. coli DDPs. 87 of the 208 clones, were significantly different from the vector-only control atp < 0.05, q <0.10 (unpaired two-tailed t-test with FDR adjustment). Each bar, a DDP clone (mean ± range, n=2 experiments of >1000 cells per strain, STAR Methods). DDP-overproducing clones are grouped by the fold change of GamGFP focus levels compared with the vector-only strain. Grey, < 2-fold increase; brown, 2-5-fold increase; green, 5-10-fold increase; magenta, > 10-fold increase, per Table S1 (data summary). Representative images shown, scale bar: 2μm. None of 25 non-DDP overproducers had increased GamGFP foci; thus, clones with high levels of GamGFP foci are enriched in the DDP-overproducing clones (p = 3.6×10−6, one-way Fisher’s exact test).
(B) Stalled reversed replication forks in most DDP-overproducing strains. Significantly increased reversed forks (RFs), visualized as RDG foci in ΔrecA cells (Xia et al., 2016) occur in 106 of the 208 (51% of) E. coli DDP-overproducing clones (significantly different from vector-only control, p < 0.05, q <0.10 unpaired two-tailed t-test with FDR adjustment). Each bar, a DDP clone (mean ± range, n=2 experiments of >1000 cells per strain). Labeling per (A), data Table S1. Representative images shown, scale bar: 2μm. No non-DDP overproducers had increased RDG foci; high RDG-focus/RF loads are enriched in the DDP-overproducing strains (p =4.0×10−9, one-way Fisher’s exact test).
(C-D) Increased intracellular ROS levels in 56 E. coli DDP-overproducing clones identified by di-hydrorhodamine (DHR) staining and flow cytometry. Fifty-six of the 208 (27%) showed mean fluorescence intensities significantly different from the vector-only control, p < 0.05, q <0.10 unpaired two-tail t-test with FDR adjustment. Each bar, a DDP clone (mean ± range, of two experiments). Labeling per (A), data Table S1. Representative flow-cytometry histograms shown. None of 17 non-DDP overproducers had increased ROS, and high-ROS clones are enriched in the DDP-overproducing clones at p = 0.006 (one-way Fisher’s exact test).
Models for DNA-damage promotion by overproduced XanQ xanthine-proton symporter and CorA ion transporter (shown in Figure 6I). Membrane-spanning transporters including proton (H+) symporters are overrepresented among DDPs that cause increased ROS when overproduced (Figure 6A-D), and cause DNA damage ROS-dependently (Figure 6B). The strongest generator of ROS-dependent DNA damage and ROS is the XanQ xanthine symporter. Although overproduction of each of three proton symporters decreases pH (increases H+, Figure 6F,G), their ROS promotion is not well correlated with decreased pH (Figure 6H), suggesting that the molecules they transport with H+ might underlie generation of ROS and DNA damage. For XanQ, overproduction of which causes high ROS but a moderate drop in pH (Figure 6F,G), one possibility is that the excess xanthine imported may be oxidized by ROS-generator xanthine oxidase (Kelley et al., 2010), causing the increased ROS, which damages DNA (Figure 6I). Other explanations are possible. CorA, an inner membrane Mg2+ transporter, which transports Co2+ and Ni2+ less efficiently (Kehres and Maguire, 2002), elevates ROS (Figure 6C-D) and DNA damage (Figure 6B) when overproduced. Both might occur by increasing the usually minimal import of Co2+ and Ni2+ (Figure 6I). Ni2+ is toxic and induces DNA damage via oxidative stress (Cameron et al., 2011) because Ni2+ binds sulfhydryl groups commonly found in anti-oxidative enzymes (Schmidt et al., 2009), which causes DNA damage ROS-dependently (Cameron et al., 2011). Regardless of specific hypotheses, overproduction of membrane transporters generally may promote DNA damage, and for several, increased ROS, by any of many mechanisms that result from compromising compartmentalization of the cell from its environment.
(E) Flow cytometry assay for DNA loss (anucleate cells). Increased anucleate cells illustrated in a parCTS mutant, which has a severe chromosome-segregation defect at non-permissive temperature. Cells with positive FM464 (membrane) but negative DAPI (DNA) staining are anucleate cells (lower right quadrant). Controls for both wild-type (DB81) and parCTS (DB193) strains: cell-only, cells not stained; DAPI, cells stained only with DAPI; FM464, cells stained only with FM464 membrane dye; double, cells stained with both FM464 and DAPI.
(F) DNA loss in 67 E. coli DDP-overproducing clones. Quantification of assays per part E. The 67 (32% of DDPs) differ significantly from the vector-only control, p < 0.05, q <0.10 unpaired two-tail t-test with FDR adjustment. Each bar, a DDP clone (mean ± range, n=2). DDP overproducers are grouped by the fold change of anucleate cells levels compared with the vector-only strain. Labeling per (A), data Table S1. Representative flow-cytometry results shown. Only one of 16 non-DDP-overproducing clones had increased anucleate cells, and clones with high-levels of anucleate cells are enriched in the DDP clones at p = 0.02 (one-way Fisher’s exact test).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-PCNA | Santa Cruz | sc-56 |
| anti-γH2AX | Millipore | 05-636 |
| anti-phospho-P53 | Cell Signaling | 9286 |
| anti-RAD18 | Cell Signaling | 9040S |
| anti-Tubulin | Abeam | ab6046 |
| anti-GFP | Thermo Fisher | A11122 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | 7074 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | 7076 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | A21236 |
| anti-Pol IV polyclonal | Kim et al., 2001 | N/A |
| anti-RuvC (5G9/3) monoclonal | Santa Cruz | sc-53437 |
| anti-Mouse IgG | Bethyl Laboratories | A90-116D5 |
| anti-Goat IgG | Bethyl Laboratories | A50-100D5 |
| Bacterial and Virus Strains | ||
| E. coli mobile plasmid overexpression library | (Saka et al., 2005) | N/A |
| See Table S7 for all bacterial strains used | ||
| Human cell lines | ||
| MRC5-SV40 | Stephen P. Jackson Lab | N/A |
| HEK293T | ATCC | CRL3216 |
| Plasmid | ||
| See Tables S5 and S7 for all plasmids used | ||
| Chemicals | ||
| GenJet™ In Vitro DNA Transfection Reagent | SignaGen Laboratories | SL100489 |
| Polyethylenimine | Polysciences | 23966 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | 13778030 |
| N-acetyl cysteine | Sigma | A7250 |
| DNA-PK inhibitor | Tocris Bioscience | NU7441 |
| 6-thioguanine | Sigma | A4882 |
| Critical Commercial assays | ||
| FM® 4-64FX | Thermo Fisher | F34653 |
| DHR123 | Thermo Fisher | D23806 |
| pHrodo® Green AM Intracellular pH indicator | Thermo Fisher | P35373 |
| RNeasy Mini Kit | Qiagen | 74104 |
| RiboZero | Illumina | MRZB12424 |
| TruSeq Stranded mRNA Library Preparation Kit | Illumina | RS-122-2001 |
| qPCR-based Illumina Library Quantification Kit | KAPA Biosystems | KK4828 |
| Dneasy Blood & Tissue kits | Qiagen | 69506 |
| QIAprep Spin Miniprep Kit | Qiagen | 27106 |
| Gateway™ LR Clonase™ II Enzyme mix | Invitrogen | 11791100 |
| SuperScript™ III Reverse Transcriptase | Invitrogen | 18080-093 |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs | M0491S |
| Oligonucleotides | ||
| ON-TARGETplus Non-targeting Pool | Dharmacon | D-001810-10-05 |
| siRAD18 ACUCAGUGUCCAACUUGCU | Sigma | N/A |
| cI forward primer ACCGCGGCGTGGGTAGTAAAGT | (Gutierrez et al., 2013) | N/A |
| cI reverse primer GCCAATCCCCATGGCATCGAGTAAC | (Gutierrez et al., 2013) | N/A |
| Deposited Data | ||
| RNA-Seq data | This paper | ENA: E-MTAB-7361 |
| ChIP-Seq data | This paper | ENA: PRJEB21035 |
| Software and Algorithms | ||
| cBioportal | Gao et al., 2013 | http://www.cbioportal.org/ |
| R programming language | R Development Core Team, 2015. | https://www.R-project.org/ |
| E. coli-to-Cancer Gene-function Atlas (ECGA) | This paper | https://microbialphenotypes.org/wiki/index.php/Special:ECGA |
| R package for progeny clustering: ProgenyClust | CRAN | https://cran.r-project.org/ |
| Trimmomatic | The Usadel lab | http://www.usadellab.org/cms/?page=trimmomatic |
| BWA-MEM | Li, 2013 | N/A |
| deepTools | Bioinformatics Facility at the Max Planck Institute for Immunobiology and Epigenetics, Freiburg | https://deeptools.readthedocs.io/en/develop/ |
| MOSAICS | CRAN | https://cran.r-project.org/ |
| Rockhopper | McClure et al., 2013 | N/A |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R programming language | R Development Core Team, 2015. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. | https://www.R-project.org/ |
| FlowJo 10.2 | FLOWJO | https://www.flowjo.com/ |
| STRING 10.0 | (Szklarczyk et al., 2015) | https://string-db.org/ |
| FACSDivaTM | BD Biosciences | http://www.bdbiosciences.com |
| Advanced Imaging collection In Pipeline pilot 8.5 or 9.2 | Biovia-Dassault Systems | N/A |
| Softworx | GE | N/A |
| BLASTp and delta BLAST | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgI |
| RNA-sequencing data | The Cancer Genome Atlas | N/A |
| Gene-Set Enrichment Analysis (ssGSEA) using GSVA package | CRAN | https://cran.r-project.org/ |
Highlights.
E. coli carcinogen-like proteins cause DNA damage and mutation when upregulated
Human homologs form a cancer-predictive network, promote DNA damage and mutation
Conserved endogenous DNA-damage-promoting mechanisms identified
DNA damage-up proteins (DDPs) a broad class of cancer-protein functions
ACKNOWLEDGMENTS
This paper is dedicated to the memory of Ken Scott whose intensity and generosity inspired us. We thank M Ellis, G Ira, H Dierick, V Lundblad, RS Harris, FW Stahl, M Wang, H Zoghbi, and C Zong for comments on the manuscript, M Yamada, T Nohmi, I Matic, SJ Sandler and JD Wang for antisera or strains. Supported by National Institutes of Health (NIH) Director’s Pioneer Award DP1-CA174424 (SMR); the WM Keck Foundation (SMR, KMM); NIH grants DP20-OD008371 (CQ); R01 grants GM102679 (DB); CA198279 and CA201268 (KMM); GM088653 (CH); GM089636 (JCH, DAS); GM106373 (PJH); and CA175486 and U24 CA209851 (HL); T32-GM008231 (JPP); U01-CA168394 (KLS); and R35-GM122598 (SMR); CPRIT grants R1116 (KMM); RP170295 and RP170005 (CC); RP150578 (MAM); RP140462 (HL); RP140553 (SMR, KMM); RP160283 (DMF); American Cancer Society Postdoctoral Fellowship 132206-PF-18-035-01-DMC (DMF); the BCM Cytometry and Cell Sorting Core NIH P30-AI036211, P30-CA125123, and S10-RR024574; and Integrated Microscopy Core NIH HD007495, DK56338, and CA125123; the Dan L Duncan Comprehensive Cancer Center, and John S. Dunn Gulf Coast Consortium for Chemical Genomics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures and seven tables.
REFERENCES
- Alvaro D, Lisby M, and Rothstein R (2007). Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet 3, e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asad NR, Asad LMBO, Almeida C.E.B.d., Felzenszwalb I, Cabral-Neto JB, and Leitão AC (2004). Several pathways of hydrogen peroxide action that damage the E. coli genome. Genetics and Molecular Biology 27, 291–303. [Google Scholar]
- Biniszkiewicz D, Gribnau J, Ramsahoye B, Gaudet F, Eggan K, Humpherys D, Mastrangelo MA, Jun Z, Walter J, and Jaenisch R (2002). Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol Cell Biol 22, 2124–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron KS, Buchner V, and Tchounwou PB (2011). Exploring the molecular mechanisms of nickel-induced genotoxicity and carcinogenicity: a literature review. Rev Environ Health 26, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antonio M, and Ciccarelli FD (2013). Integrated analysis of recurrent properties of cancer genes to identify novel drivers. Genome Biol 14, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrmann PR, Correa R, Frisch RL, Rosenberg SM, and McHenry CS (2016). The DNA polymerase III holoenzyme contains gamma and is not a trimeric polymerase. Nucleic Acids Res 44, 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, and Swain PS (2002). Stochastic gene expression in a single cell. Science 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DM, Hastings P, and Rosenberg SM (2017). Stress-induced mutagenesis: implications in cancer and drug resistance. Annu Rev Cancer Biol 1, 119–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. (2015). COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 43, D805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti JJ, Devadoss B, Winkler JA, Collins JJ, and Walker GC (2012). Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch RL, Su Y, Thornton PC, Gibson JL, Rosenberg SM, and Hastings PJ (2010). Separate DNA Pol II- and Pol IV-dependent pathways of stress-induced mutation during doublestrand-break repair in Escherichia coli are controlled by RpoS. J Bacteriol 192, 4694–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galhardo RS, Almeida CE, Leitao AC, and Cabral-Neto JB (2000). Repair of DNA lesions induced by hydrogen peroxide in the presence of iron chelators in Escherichia coli: participation of endonuclease IV and Fpg. J Bacteriol 182, 1964–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et al. (2017). Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120. [DOI] [PubMed] [Google Scholar]
- Gutierrez A, Laureti L, Crussard S, Abida H, Rodriguez-Rojas A, Blazquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, et al. (2013). beta-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun 4, 1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786 e719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Hersh MN, Thornton PC, Fonville NC, Slack A, Frisch RL, Ray MP, Harris RS, Leal SM, and Rosenberg SM (2010). Competition of Escherichia coli DNA polymerases I, II and III with DNA Pol IV in stressed cells. PLoS One 5, e10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltzel JM, Maul RW, Wolff DW, and Sutton MD (2012). Escherichia coli DNA polymerase IV (Pol IV), but not Pol II, dynamically switches with a stalled Pol III* replicase. J Bacteriol 194, 3589–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavac V, Brynychova V, Vaclavikova R, Ehrlichova M, Vrana D, Pecha V, Trnkova M, Kodet R, Mrhalova M, Kubackova K, et al. (2014). The role of cytochromes p450 and aldo-keto reductases in prognosis of breast carcinoma patients. Medicine (Baltimore) 93, e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CW, Kornblau SM, Slater JH, and Qutub AA (2015). Progeny Clustering: A Method to Identify Biological Phenotypes. Sci Rep 5, 12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, and Robertson KD (2013). DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol 754, 3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Issa JP, and Baylin S (2016). Targeting the cancer epigenome for therapy. Nat Rev Genet 17, 630–641. [DOI] [PubMed] [Google Scholar]
- Kehres DG, and Maguire ME (2002). Structure, properties and regulation of magnesium transport proteins. Biometals 15, 261–270. [DOI] [PubMed] [Google Scholar]
- Kelley EE, Khoo NK, Hundley NJ, Malik UZ, Freeman BA, and Tarpey MM (2010). Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic Biol Med 48, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseler IM, Mackie A, Santos-Zavaleta A, Billington R, Bonavides-Martinez C, Caspi R, Fulcher C, Gama-Castro S, Kothari A, Krummenacker M, et al. (2017). The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res 45, D543–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Matsui K, Yamada M, Gruz P, and Nohmi T (2001). Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics 266, 207–215. [DOI] [PubMed] [Google Scholar]
- Kinner A, Wu W, Staudt C, and Iliakis G (2008). Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 36, 5678–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, and Vogelstein B (1997). Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 386, 761, 763. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Valentine MR, Pham P, O’Donnell M, and Goodman MF (2002). Fidelity of Escherichia coli DNA polymerase IV. Preferential generation of small deletion mutations by dNTP-stabilized misalignment. J Biol Chem 277, 34198–34207. [DOI] [PubMed] [Google Scholar]
- Kuzminov A (2011). Homologous Recombination-Experimental Systems, Analysis, and Significance. EcoSal Plus 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, Pollock AJ, Woodward JJ, Dreifus JE, and Merrikh H (2017). Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 170, 787–799 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, and Tjian R (2003). Transcription regulation and animal diversity. Nature 424, 147–151. [DOI] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997.
- Lovejoy CA, Xu X, Bansbach CE, Glick GG, Zhao R, Ye F, Sirbu BM, Titus LC, Shyr Y, and Cortez D (2009). Functional genomic screens identify CINP as a genome maintenance protein. Proc Natl Acad Sci U S A 106, 19304–19309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, and Koonin EV (2013). Archaeology of eukaryotic DNA replication. Cold Spring Harb Perspect Biol 5, a012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor-Shoshani A, Reuven NB, Tomer G, and Livneh Z (2000). Highly mutagenic replication by DNA polymerase V (UmuC) provides a mechanistic basis for SOS untargeted mutagenesis. Proc Natl Acad Sci U S A 97, 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, and Tjaden B (2013). Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41, e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, and Leonhardt H (2005). Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci U S A 102, 8905–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehring RB, Gu F, Lin HY, Gibson JL, Blythe MJ, Wilson R, Bravo Nunez MA, Hastings PJ, Louis EJ, Frisch RL, et al. (2016). An ultra-dense library resource for rapid deconvolution of mutations that cause phenotypes in Escherichia coli. Nucleic Acids Res 44, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara H, Yamamoto K, and Ishihama A (2011). Role of the biofilm master regulator CsgD in cross-regulation between biofilm formation and flagellar synthesis. J Bacteriol 193, 2587–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. (2009). A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington JM, and Rosenberg SM (2007). Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39, 797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N, Gumlaw N, Nordman JT, Krieger M, Yeh SP, Long E, Centore R, Boonsombat R, and Sandler SJ (2005). Localization of RecA in Escherichia coli K-12 using RecA-GFP. Mol Microbiol 57, 1074–1085. [DOI] [PubMed] [Google Scholar]
- Saito Y, Uraki F, Nakajima S, Asaeda A, Ono K, Kubo K, and Yamamoto K (1997). Characterization of endonuclease III (nth) and endonuclease VIII (nei) mutants of Escherichia coli K-12. J Bacteriol 179, 3783–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka K, Tadenuma M, Nakade S, Tanaka N, Sugawara H, Nishikawa K, Ichiyoshi N, Kitagawa M, Mori H, Ogasawara N, et al. (2005). A complete set of Escherichia coli open reading frames in mobile plasmids facilitating genetic studies. DNA Res 12, 63–68. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, and Appella E (1998). DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Wolfe DM, Stiller B, and Pearce DA (2009). Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem Biophys Res Commun 383, 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD, and Michel B (1998). RuvAB acts at arrested replication forks. Cell 95, 419–430. [DOI] [PubMed] [Google Scholar]
- Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu LY, Magnan D, Halliday JA, Frisch RL, Gibson JL, et al. (2013). Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife 2, e01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR (2011). Exploring the genomes of cancer cells: progress and promise. Science 331, 1553–1558. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, Peng J, Herman C, and Wang JD (2010). The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell 141, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, and Nussenzweig A (2017). Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Furukohri A, Shinozaki Y, Mori T, Ogawara D, Kanaya S, Nohmi T, Maki H, and Akiyama M (2008). Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol Microbiol 70, 608–622. [DOI] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, and Wahl GM (2002). c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 9, 1031–1044. [DOI] [PubMed] [Google Scholar]
- Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RP, and Nohmi T (1999). The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol Cell 4, 281–286. [DOI] [PubMed] [Google Scholar]
- Wagner J, and Nohmi T (2000). Escherichia coli DNA polymerase IV mutator activity: genetic requirements and mutational specificity. J Bacteriol 182, 4587–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, and Vuica-Ross M (2011). RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Costantino L, Tan FJ, Zimmer A, and Koshland D (2016). S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev 30, 1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly H, Shee C, Thornton PC, Sivaramakrishnan P, Rosenberg SM, and Hastings PJ (2013). R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat Commun 4, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Chen LT, Mei Q, Ma CH, Halliday JA, Lin HY, Magnan D, Pribis JP, Fitzgerald DM, Hamilton HM, et al. (2016). Holliday junction trap shows how cells use recombination and a junction-guardian role of RecQ helicase. Sci Adv 2, e1601605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genuine SOS Response, Detection of DNA Damage by the SOS-reporter Gene, and Evaluation of False Negatives in the Primary Screen, Related to Figure 1
The E. coli DNA-damage assay detects fluorescence caused by upregulation of the SOS DNA-damage-response-activated promoter PsulA fused to mCherry in a non-genic chromosomal site (Nehring et al., 2016; Pennington and Rosenberg, 2007). This assay was shown to report on DNA damage, not spurious promoter firing with the demonstration that fluorescence induction requires the DNA-damage-sensing protein RecA, and is inhibited by a mutant SOS-response transcriptional repressor, LexAInd−, which does not de-repress the SOS genes during DNA damage (Pennington and Rosenberg, 2007), both also shown here (A). Further, of the spontaneous SOS-inducing DNA damage, previously 60% was shown to reflect DNA doublestrand breaks (DSBs) and 40% single stranded DNA not at DSBs (Pennington and Rosenberg, 2007), and the spontaneous DSB frequency and rates per cell division and chromosome replication were confirmed in a direct, independent assay for DSB ends (Shee et al., 2013). Thus, this reporter reports on DNA damage.
(A) Fluorescence requires the ability to activate the SOS response, and so is absent in SOS-induction-deficient ΔrecA or lexA3(Ind−) mutant cells, demonstrating that DNA damage, not spurious promoter firing, underlies fluorescence increases, per (Pennington and Rosenberg, 2007). Using these negative controls, a flow-cytometry gate (vertical dashed line) is set in each experiment, per (Pennington and Rosenberg, 2007), at fluorescence at which 10−4 of the lexA3(Ind−) negative- control cells are positive. The SOS-positive cells in the wild-type (WT) strain represent spontaneous DNA damage, per (Pennington and Rosenberg, 2007). Representative flow-cytometry histograms generated under the growth conditions used in the screens reported here.
(B) Positive control, mCherry+ lexA51(Def) cells with constitutively activated SOS response, and negative control, lexA3(Ind−) SOS-off cells.
(C) For a representative sample of 66 of the validated DDPs (Table S1), we confirmed that their increased fluorescence results from increased SOS response by showing its dependence on SOS-response-activator RecA, and a functioning (inducible) LexA SOS repressor (Pennington and Rosenberg, 2007). All 66 showed RecA- (here) and LexA-dependent high fluorescence (Figure 1G), demonstrating the presence of DNA damage and a genuine SOS response (Table S1). We also ruled out the possibility that DDP overproduction might increase mCherry protein fluorescence itself (Figure S1D; Table S1).
(D) Representative DDPs do not enhance mCherry fluorescence generally, when mCherry is controlled by non-SOS promoter PBAD.
(E) Screen of 99 random E. coli proteins not identified in the primary plate-reader screen finds one SOS-positive clone. Because of its inherent noise level, the plate-reader primary screen is likely to generate some false-negative results; i.e., miss clones with subtle but real DNA damage-up phenotypes. If the 3815 E. coli proteins that were not identified in the primary screen (4229 protein-coding genes in the library - 414 proteins found in the primary screen) also harbor 1% real but undiscovered DDPs, then an additional 38 overproduction DDPs are predicted to reside in the E. coli overproduction library, for an estimated total network size of 246 (38 + 208) DDP genes in the overexpression library used. In STAR Methods, we estimate an additional 123 E. coli overexpression DDP genes that are likely to be in the E. coli genome, but would not be discoverable using the Mobile plasmid overexpression library.
(F) E. coli TetR clones from cI mutation assay harbor genuine mutations. Sequences of cI mutations from 10 different DDP-overproducing strains with strong damage-up phenotypes: CsgD, TopB, YegL, GrpE, HslU, Mrr, CheA, MdtA, YicR, and UvrA. The compiled sequences from the 10 DDP clones are shown in red and blue (n = 3-10 independent TetR mutants per clone). The 69 cI mutations sequenced include 3 IS (mobile)-element insertions, and 2 clones without a PCR product, which whole-genome sequencing revealed to result from large deletions (STAR Methods). Mutations in the vector-only control strain are shown in black (19 independent mutants). Red font, mutations attributable to common errors of the SOS-upregulated error-prone DNA polymerases V and IV (Kobayashi et al., 2002; Maor-Shoshani et al., 2000; Wagner and Nohmi, 2000); blue font, mutations not attributable to common Pol V or Pol IV errors. Capital letters, basepairs that were changed or deleted; carrots, insertion points of bases indicated; Δs, deletions of the bases indicated. Upper right, diagram of gross chromosomal rearrangements (GCRs) found in TopB-overproducing cells, among which, 18% (7/40) of the TetR mutations are GCRs, including large deletions (from 200bp-6200bp), an inversion, and a transposon insertion.
Figure S5. Sensitivities to DNA-Damaging Agents, Related to Figure 4
(A) Phleomycin sensitivity detected by growth inhibition in known mutants with reduced homologous recombinational (HR) repair. recF: defective in single-strand gap HR-repair; recG: reduced ability to branch migrate Holliday junctions; recB: defective in HR DSB repair (Kuzminov, 2011).
(B) 106 E. coli DDP overproducers are sensitive to phleomycin. Data normalized to vector-only controls: (treated/untreated DDP overproducer) / (treated/untreated vector-only) so that values < 1 indicate sensitivity. Sensitive clones include 11 more sensitive than a recB mutant, 72 within the range of recB and recF mutants, and 23 more sensitive than the vector-only control but less than recF mutant. One of the 16 non-DDP-overproducers was phleomycin sensitive, which differs from the DDP-overproducers at p = 0.0003 (one-way Fisher’s exact test).
(C) Sensitivity of 75 E. coli DDP clones to oxidative-damage-inducing agent H2O2 detected as colony forming units in known mutants with reduced base excision repair (BER) efficiency: xthA (inset) encodes Exonuclease III; nfo encodes Endonuclease IV. xthA nfo (green) double mutants are more sensitive than each single mutant (Galhardo et al., 2000). nth encodes Endonuclease III; nei encodes Endonuclease VIII. The nth nei (purple) double mutant has almost no AP lyase activity, and has extreme sensitivity (Saito et al., 1997).. Data of the DDP clones are normalized: (treated/untreated DDP overproducer) / (treated/untreated vector-only) so that values < 1 indicate sensitivity. The H2O2-sensitive DDP clones include 36 more sensitive than xthA nfo cells; 39 more sensitive than the vector-only control but less sensitive than xthA nfo cells. None of the 15 nonDDP-overproducers had H2O2 sensitivity, which differs from the DDP clones at p = 0.002 (one-way Fisher’s exact test).
(D) Sensitivity to interstrand-crosslinking agent Mitomycin C (MMC) detected by growth inhibition in known mutants with defective HR repair or nucleotide excision repair (NER): recA, HR- and SOS response-defective; and uvrC, NER-defective (shown in E, cyan).
(E) Ten DDP clones were MMC sensitive. DDP clone data are normalized: (treated/untreated DDP overproducer) / (treated/untreated vector-only) so that values < 1 indicate sensitivity. None of the 16 non-DDP overproducers was MMC sensitive, which does not differ from the DDP clones, p = 0.47 (one-way Fisher’s exact test).
(F) Representative DDPs do not downregulate DNA-repair-gene mRNAs upon overproduction. Seven DDPs representative of the six DDP function clusters (Figure 4N; Table S1) were assayed for RNA levels of 32 DNA-repair genes by RNA-seq with and without overproduction of each of 7 DDPs, which confer sensitivity to the following agents: TopA and BamC (H2O2); CusR, HemX, AroP, YicR, and YqjD (phleomycin). The repair pathways represented were nucleotide excision repair (uvrA, uvrB, uvrC, uvrD), base-excision repair (BER: mutT, mutM, xthA, nfo, ung, mug, nth, tag, alkA, nei), mismatch repair (mutS, mutL, uvrD, mutH), homology-directed repair (recA, radA, ruvA, ruvB, ruvC, recT, recF, recO, recR, recG, recN), and homology-directed DSB repair (as for homology-directed repair with the addition of recB, recC, recD, and without recFOR). Log2 ratio (DDP overproducer / vector) for each DNA-repair gene in each of the DDP clones was calculated and clustered by hierarchy clustering. Most of the DNA-repair genes are up-regulated during DDP overproduction. Upregulation presumably results from SOS or other DNA-damage or oxidative-stress responses. Only mutM RNA was is downregulated (in the TopA-overproducing strain). mutM downregulation is unlikely to account for the H2O2 sensitivity of the TopA-overproducing strain because even mutM null mutants are not sensitive to H2O2 (Asad et al., 2004). The data exclude the hypothesis that most or all of the DNA-damage sensitivities caused by overproduced DDPs (Figures 4J-L, A-E here; Table S1) result from transcriptional downregulation of DNA-repair genes. Potential post-transcriptional regulation mechanisms are not excluded. The data support the hypothesis that the DNA-damage sensitivities result from excess DNA damage overwhelming DNA-repair capacity, potentially titrating DNA-repair enzymes, or direct inhibition of DNA repair by the overproduced protein. Reduction of DNA-repair capacity could produce phenotypes like those of DNA-repair mutants, which would drive genome instability, in many DDP-gene dysregulated cells that do not possess DNA-repair-gene mutations or downregulated mRNAs.
(G) Cluster analysis of quantitative data on DNA-damaging-agent sensitivities in the E. coli DDP network. Each bar, a quantitative phenotype of each strain producing each of the 208 E. coli DDPs. Assays indicated above the heatmap: H2O2, hydrogen-peroxide sensitivity (reduced base excision repair); PHLEO, phleomycin sensitivity (reduced DSB-repair); MMC, mitomycin-C sensitivity (reduced NER and/or HR repair). Z score are compared with the vector-only negative control. Increased resistance (blue) to H2O2, MMC, and phleomycin are apparent in clusters in each, as is decreased ROS levels in a cluster Figure 4M, and reduced anucleate cells (DNA loss) in that figure. Similarly, clusters 3, 4, and 6 show reduced sensitivity (greater resistance than wild-type cells) to H2O2, and 2 and 3 show greater resistance to mitomycin C (Figure 4N). A probable explanation for these “better-than-wild-type” phenotypes is that some imbalance in these cells may induce a stress response, one of the consequences of which may be improvement of the fidelity of, e.g., chromosome segregation (preventing anucleate cells), reduction of ROS levels, or resistance to DNA-damaging agents, as is documented, for example, for DNA-damage resistance induced by mild induction of the SOS response, among other stress responses (reviewed, Fitzgerald et al., 2017).
(H) Heritable mutations do not account for DNA-damage sensitivities of seven highly DNA-damage-sensitive DDP clones. The DDP overproducers were tested for sensitivity with overproduction of the DDP (red, “before”), then again after the drug treatment, with the DDP-gene repressed (blue, “after”) to determine whether their sensitivity resulted from induction of mutations in genes needed for resistance. After drug treatment, three colonies were recovered on plates with no DDP-gene-inducing IPTG, and then tested for DNA-damaging-agent sensitivity again. In all cases, the recovered colonies not induced for DDP overproduction showed no sensitivity to DNA-damaging agents (blue, after). Thus, their DNA-damage sensitivities did not result from heritable mutations.
Figure S6. DNA-binding Transcription-Factor and Replication-Stall (RDG) Foci, Examples, Related to Figure 5
(A) Representative data show induction of RDG reversed-fork foci with production of DNA-binding transcription factors (TFs), DNA-binding-domain dependently. ΔDBD, in-frame deletion of the DNA-binding domain; CsgDD59N, D59E: single amino-acid changes that reduce CsgD DNA binding (Ogasawara et al., 2011). Three wild-type TFs and the corresponding mutants with reduced DNA-binding ability overproduced in ΔrecA cells, in which RDG foci represent reversed-forks (RFs), and quantified (Figure 5E; Table S1). Representative images shown, scale bar: 2μm.
(B) Representative data show co-localization of RDG reversed-fork foci with TF-mCherry foci, which form DNA-binding-domain dependently. Representative examples, scale bar: 2μm. Most of the CsgD-mCherry and HcaR-mCherry foci were co-localized with RDG/RF foci (Figure 5G, H; Table S1). Showing weak co-localization, about 5% of the YahB-mCherry TF foci were colocalized with RDG/RF foci (Figure 5G). Negative-control mCherry alone rarely forms foci (Figure 5G).
Figure S7. RDG ChIP-seq Detects Stalled-Fork Enrichment Near CsgD-Binding Sites and Controls for Pol IV, Related to Figures 5 and 7
Nine of the 10 known, validated CsgD-binding sites (Keseler et al., 2017; Ogasawara et al., 2011) showed an RDG peak nearby induced by CsgD overproduction: within 10kb (median 2.8kb), which differs from random simulations of the genomic distribution of the total number of RDG peaks (p = 0.01 two-tailed z test,. Isogenic cells overproducing—CsgD, red; CsgD ΔDBD (deletion of the DNA-binding domain), blue; vector-only, green. Green boxes, CsgD binding sites.
The RDG ChIP-seq experiments (Figures 5I and here A-C) were performed twice, with peaks called against the matched control DNA-binding-domain mutant, CsgDΔDBD. We identified 155 total reproducible RDG ChIP-seq peaks with at least 2.5-fold increase in both repeats compared with CsgDADBD done in parallel. The following control simulations show that RDG ChIP-seq signals are enriched near known CsgD binding sites. We simulated CsgD-overproduction ChIP-Seq data by randomly distributing the called RDG peaks across the genome and analyzed, first, the number of binding sites with at least one RDG peak within 10kb, and, second, the distance between each known CsgD-binding site and the nearest RDG peak (median), for each of the 10 experimentally validated CsgD-binding sites (Keseler et al., 2017; Ogasawara et al., 2011). Strikingly, there is at least one observed (actual) RDG peak within 10kb of 9 out of the 10 known CsgD-binding sites (A-C), whereas there are only 5 known binding sites with at least one simulated RDG peak within 10kb of the 10 known binding sites in our simulations, which is significantly less than the observed (p = 0.01, two-tailed z-test). Also, the median distance between a given known CsgD-binding site and the nearest RDG ChIP-seq peak is 2.8kb for the real peaks, which is significantly closer than the 10.3kb median distance in our simulations (p = 0.009).
RDG peaks not at known CsgD-binding sites. For the 142 RDG peaks that are not significantly near a known CsgD binding site, these could result from—(i) binding of CsgD to sites not yet identified in the literature, in which no comprehensive high-resolution (ChIP-seq) genome-wide binding study has been done; (ii) relaxation of the binding specificity of CsgD when overproduced such that new sites are hit; and (iii) downstream (indirect) effects of regulation of CsgD-regulated genes by CsgD binding to its sites. For example, CsgD upregulation of other DNA-binding transcription factors could cause peaks at their binding sites, among other indirect but biologically real (CsgD-DBD-dependent) possibilities. The 142 of 155 RDG ChIP-seq peaks that are not near known CsgD binding sites do not overlap significantly with palindromic REP sequences, which are prone to form DNA cruciform structures (four RDG peaks overlap with REPs, p =0.43, two-tailed z-test compared with median random distribution), or terA-terC regions, which have higher frequencies of fork convergence (11 RDG peaks overlap with ter sequences, p =0.47, two-tailed z-test, compared with compared with median random distribution).
Upstream bias of RDG stalled-fork peaks at CsgD binding sites. The 9 known CsgD-binding sites with one or more observed RDG peaks within 10kb are of two types: six show RDG peaks at or upstream of the binding sites in the replication path and five show RDG peaks downstream. The observed number of CsgD binding sites with upstream or co-localized RDG peaks is significantly higher than the median in the simulation (p = 0.04, two-tailed z-test), whereas the number of CsgD binding sites with RDG peaks downstream is not quite significant, at p = 0.13, two-tailed z-test. The analysis above suggests that direct binding of CsgD to its known binding sites underlies the RDG upstream and co-localized peaks, whereas the downstream peaks may reflect a component of direct CsgD-DNA-induced fork reversal, and a component of indirect effects, or binding to as yet unknown CsgD binding sites. One way that an RDG stalled-fork peak might result downstream in the replichore, but still via direct interaction of replication with bound CsgD, could be if some of the downstream stalled forks are caused by two events (illustrated Figure 5J, lower): first, slowing of replication forks by CsgD binding at its site might make the replisome susceptible to a second, otherwise surmountable barrier of any kind downstream in the replication path.
(A) RDG ChIP-seq peaks overlap with three known CsgD-binding sites DBD dependently.
(B) RDG ChIP-seq peaks near three known CsgD-binding sites, upstream in the replication path. Increased negative supercoiling between the oncoming fork and the CsgD-bound site may stall replication and causes fork reversal, illustrated Figure 5J.
(C) RDG ChIP-seq peaks located near three known CsgD binding sites, downstream in the replication path. Because the upstream peaks (B) are more significantly associated with the known CsgD-binding sites relative to simulation of random genomic distributions (above), it is possible that the downstream RDG peaks could result from either CsgD binding to sites not yet known, or from indirect consequences of CsgD DNA binding, such as effects of CsgD-regulated gene products interacting with other sites in DNA (above). Alternatively, some of the downstream RDG peaks could result from CsgD binding its known site, slowing replication, then encountering an otherwise surmountable obstacle downstream, per Figure 5J.
(D) The clamp-loader dnaX tau-only mutant is not generally deficient in SOS-response induction, but merely reduces the DNA damage (SOS activity) caused by Pol IV upregulation (Figure 7D,E).
(E) The Pol IV R49F catalytic-mutant (Pol IV cat−) and ΔBBD-mutant proteins do not display reduced protein levels in western blots, in agreement with previous studies (Uchida et al., 2008; Wagner et al., 1999).
(F) Neither the E. coli MutM 8-oxo-dG glycosylase nor the MutY adenine glycosylase is required for DNA damage instigated by Pol IV, indicating DNA damage produced independently of incorporation of 8-oxo-dG into DNA. Representative data, quantified (G).
(G) Mean ± SEM ≥ 3 experiments. Pol IV is induced by IPTG.
Table S1. Summary of E. coli DNA-damage, mutation, function and phenotype data from overproduced DDPs, Related to Figures 1, 2, 3, 4, 6
Table S3. Human DDPs-/candidate-DDPs overrepresented among known and predicted cancer-driving genes, Related to Figure 2
Table S4. Human-homolog candidate-DDP genes are overrepresented in cancer genomes as copy-number gains, Related to Figure 2
Table S5. Sources of full-length and truncated clones of candidate hDDPs and 20 non-DDPs/human overexpression plasmids constructed and used in this study, Related to STAR Methods and Figure 3B
Table S6. DNA-damage levels in human cells overproducing candidate hDDPs and random human proteins, Related to Figure 3
Table S7. Escherichia coli K12 strains and plasmids used in this study, Related to STAR Methods.
Figure S2. Protein-Protein Associations in E. coli and Human DDP Networks via STRING, Related to Figures 1 and 2
(A) Protein-protein associations in the E. coli DDP network detected with STRING and co-expression analysis. CytoScape software generated from STRING 10.0 database. Other, defined Table S1. STRING measures protein-protein associations of many kinds including indirect associations (e.g., co-occurrence in different organisms’ genomes, or in papers in the literature) and direct protein-protein interactions, among others (STAR Methods). Using STRING with an interaction score cut-off of ⩾0.6 (medium-to-high confidence, STAR Methods), we found that the 208 DDPs form a significant network via protein-protein interaction data with more interactions than random sets of 208 E. coli proteins (p = 4.4 × 10−31, hypergeometric test). When known DNA-repair proteins are excluded, the STRING network is still significant compared with random sets of the same number of E. coli proteins (p = 8 × 10−6, hypergeometric test), indicating that the associations are not solely via DNA-repair-protein associations. When both DNA-repair and DNA-replication proteins are excluded—both known to interact directly with DNA—the STRING network is no longer significant compared with random sets of the same number of E. coli proteins (p = 0.47, hypergeometric test). These data suggest that, as might be expected from the highly diverse protein functions (Figure 1F), these proteins work in many different cellular processes that may share only their various effects on DNA damage, seen by significant association as a network only when DNA-repair and replication proteins are included, which we identify as the hubs of the DDP STRING interaction network (blue and green circles shown). Comparison of the six phenotype/function clusters of the E. coli DDPs (Supplement Figure 3) with the STRING protein-protein-associations reveals that cluster 2 shows even more protein-protein interactions than the DDP network as a whole (p = 0.0007, one-was Fisher’s exact test), supporting the function clusters (Figures 4M,N and S5G) as biological groups.
Co-expression of E. coli DDP RNAs. Normalized RNA abundance (gene expression) data available at Many Microbial Microarrays Database (http://m3d.mssm.edu/norm/) show RNA levels under a few hundred growth conditions. We calculated pairwise co-expression Pearson correlation coefficients for the 208 E. coli DDP genes, and for 1000 iterations of 208 random E.coli genes. We find that the 208 DDP-gene RNAs have a median correlation coefficient of 0.035, which is slightly but significantly higher than 1000 iterations of random E. coli genes (0.02 ± 0.007, mean ± SD, p<0.01), implying a network property of these genes. We tested whether, because of their DNA-damage provoking properties, the RNAs of the 208 DDPs might be produced in smaller ranges of RNA levels than random genes, and found no such difference (p=0.09, two-tailed unpaired t-test). Thus, DDP-encoding RNAs are not subject to special narrower-range accumulation relative to other E. coli genes. A complexity of comparison with random genes is that we predict both many more overproduction DDPs among them (RESULTS, Human Homologs Promote DNA Damage and Mutation), and also greater numbers of loss-of-function drivers of DNA damage.
(B) Specific protein-protein interactions of human candidate DDP network. Diagram details are as in A. Image from Figure 2B enlarged. Although, the human homolog (candidate hDDP) network has DNA-repair and -replication genes at its center, removal of these proteins leaves the remainder with still significant connectivity: p = 4.1 × 10−215 (hypergeometric test), though less than the whole network (p = 1.2 × 10−327, hyper geometric test). Random human proteins do not form robust protein-protein association networks. Although not associated nearly as strongly as the human homologs of DDPs (1.2 × 10−327), the significant association of random human homologs of E. coli proteins (1.8 × 10−49) compared with random human proteins (p = 0.80) could potentially result if highly conserved proteins generally have more interactions with each other than random proteins. This might be because the most highly conserved, fundamental aspects of biology, and proteins that participate in them, are enriched for conserved protein machines (ribosomes, replisomes, transcription complexes, etc.), and/or fundamental pathways the actors in which have remained associated. Alternatively, it might be that proteins that function as part of interacting protein groups evolve more slowly, and so are overrepresented among conserved proteins.
Figure S3. Association of Human Homolog Network with Cancers: Copy-number Gain, Survival, and Mutation Load, and Controls for Human DNA-Damage Assays and DNMT1, Related to Figures 2, 3 and 7
(A to C) Twenty-six cancer types in TCGA data (Gao et al., 2013) are displayed along the x axes. Blue, median % of patient cancers with increased copy number of any gene in their genome; red, % of patient cancers with increased copy number of the gene indicated. Copy-number-increases in human homologs of E. coli DDP genes are higher than those of human homologs of random E. coli genes (p < 0.05, FDR < 0.10, Wilcoxon test). Examples of the Pan-Cancer copy-number-increase analysis (GISTIC threshold copy-number gain ⩾ 1) of the 284 human homologs of E coli DDP genes in 26 cancer types are shown here (complete analysis Table S4). The human genes fell into three categories:
(A) genes with increased DDP-gene copy numbers across cancers (fold change > 1.5, p < 0.05, FDR < 0.10, Wilcoxon test);
(B) genes with cancer-specific copy-number-increases (p < 0.05); and
(C) not particularly cancer associated.
(D) Decreased cancer survival is associated with high DDP-homolog RNA levels in cancers—our analyses of data from TCGA (Gao et al., 2013), STAR Methods. BRCA, breast invasive carcinoma; HNSC, head and neck squamous cell carcinoma; UCEC, uterine corpus endometrial carcinoma. *, **, *** indicate that survival of the cancers with high and low levels of the 284 RNAs differ at p ⩽ 0.05; ⩽ 0.01, and ⩽ 0.001 respectively, log-rank test.
(E) Decreased cancer survival is not confined to the previously known cancer drivers in the network. RNAs of the known (Forbes et al., 2015) cancer-driving genes among the homologs show less association with poor survival than the whole homolog network.
(F) RNAs of the DDP homologs excluding known (Forbes et al., 2015) drivers are still associated with decreased patient survival, indicating that genes in the homolog network other than and in addition to the previously known drivers are also associated with decreased survival.
(G-H) RNAs of the human DDP homologs are still robustly associated with decreased patient survival, when (G) validated DDPs or (H) validated DDPs and known drivers are removed.
(I-K) Human MRC5-SV40 or HEK293T were transfected with human candidate DDP and random human genes and random human non-DDP homologs of E. coli genes, and DNA-damage markers were analyzed by flow cytometry.
(I) Assay I: γH2AX in MRC5-SV40 cells.
(J) Assay II: γH2AX in MRC5-SV40 cells treated with DNA-PK inhibitor, which inhibits DNA break repair by non-homologous end joining and so is a sensitizing DNA-damage screen.
(K) Assay III: phosphorylated p53 in HEK293T cells. Data represent mean ± range, n ⩾ 2. The candidate hDDP genes differ from the 20 random human genes, p < 0.0001, Fisher exact test.
(L and M) Finear response of both human-cell DNA-damage-detection assays with exogenous ionizing radiation treatment indicates quantitative validity of these assays. Percent of MRC5-SV40 cells that are positive for (F) γH2AX, and (M) phosphorylated p53 (p53-S15p), in flow-cytometric assays. MRC5-SV40 cells were treated with IR with the indicated dose and analyzed by flow cytometry.
(N) Validated hDDP genes are enriched among genes with cancer-associated copy-number increases (bar 4 versus bar 1, one-way Fisher’s exact test, p = 0.0001). The level of enrichment is higher (one-way Fisher’s exact test) than total human homologs of E. coli DDP genes (bar 4 versus bar 2, p = 0.0002), and higher than the candidates that were tested but not validated (bar 4 versus bar 3, p = 0.02).
(O) DNMT1 wild-type (WT) but not ΔPBD interacts with replisome sliding clamp, PCNA. The tubulin negative control also does not interact with PCNA. GFP-trap immunoprecipitation was performed in flipln stable inducible HEK293T cells expressing GFP-tubulin, GFP-DNMT1-WT or GFP-DNMT1-ΔPBD. Interactions were determined in immunoprecipitation samples by western blotting with anti-GFP and -PCNA antibodies.
(P) Mono-ubiquitylation of the replisome clamp, PCNA, by overproduced DNMT1 is RAD18-mediated. Knockdown of RAD18 reduced the level of ubiquitylated PCNA in DNMT-WT overexpressing cells. Western analyses of PCNA ubiquitylation in MRC5-SV40 cells transfected with the plasmids indicated in combination with non-targeting (NT) or RAD18 siRNA.
Figure S4. DSBs, Reversed Forks, Reactive Oxygen, and DNA Loss in DDP Clones, Related to Figure 4
(A) Increased GamGFP foci indicate DSBs caused by overproduction of 87 of the 208 E. coli DDPs. 87 of the 208 clones, were significantly different from the vector-only control atp < 0.05, q <0.10 (unpaired two-tailed t-test with FDR adjustment). Each bar, a DDP clone (mean ± range, n=2 experiments of >1000 cells per strain, STAR Methods). DDP-overproducing clones are grouped by the fold change of GamGFP focus levels compared with the vector-only strain. Grey, < 2-fold increase; brown, 2-5-fold increase; green, 5-10-fold increase; magenta, > 10-fold increase, per Table S1 (data summary). Representative images shown, scale bar: 2μm. None of 25 non-DDP overproducers had increased GamGFP foci; thus, clones with high levels of GamGFP foci are enriched in the DDP-overproducing clones (p = 3.6×10−6, one-way Fisher’s exact test).
(B) Stalled reversed replication forks in most DDP-overproducing strains. Significantly increased reversed forks (RFs), visualized as RDG foci in ΔrecA cells (Xia et al., 2016) occur in 106 of the 208 (51% of) E. coli DDP-overproducing clones (significantly different from vector-only control, p < 0.05, q <0.10 unpaired two-tailed t-test with FDR adjustment). Each bar, a DDP clone (mean ± range, n=2 experiments of >1000 cells per strain). Labeling per (A), data Table S1. Representative images shown, scale bar: 2μm. No non-DDP overproducers had increased RDG foci; high RDG-focus/RF loads are enriched in the DDP-overproducing strains (p =4.0×10−9, one-way Fisher’s exact test).
(C-D) Increased intracellular ROS levels in 56 E. coli DDP-overproducing clones identified by di-hydrorhodamine (DHR) staining and flow cytometry. Fifty-six of the 208 (27%) showed mean fluorescence intensities significantly different from the vector-only control, p < 0.05, q <0.10 unpaired two-tail t-test with FDR adjustment. Each bar, a DDP clone (mean ± range, of two experiments). Labeling per (A), data Table S1. Representative flow-cytometry histograms shown. None of 17 non-DDP overproducers had increased ROS, and high-ROS clones are enriched in the DDP-overproducing clones at p = 0.006 (one-way Fisher’s exact test).
Models for DNA-damage promotion by overproduced XanQ xanthine-proton symporter and CorA ion transporter (shown in Figure 6I). Membrane-spanning transporters including proton (H+) symporters are overrepresented among DDPs that cause increased ROS when overproduced (Figure 6A-D), and cause DNA damage ROS-dependently (Figure 6B). The strongest generator of ROS-dependent DNA damage and ROS is the XanQ xanthine symporter. Although overproduction of each of three proton symporters decreases pH (increases H+, Figure 6F,G), their ROS promotion is not well correlated with decreased pH (Figure 6H), suggesting that the molecules they transport with H+ might underlie generation of ROS and DNA damage. For XanQ, overproduction of which causes high ROS but a moderate drop in pH (Figure 6F,G), one possibility is that the excess xanthine imported may be oxidized by ROS-generator xanthine oxidase (Kelley et al., 2010), causing the increased ROS, which damages DNA (Figure 6I). Other explanations are possible. CorA, an inner membrane Mg2+ transporter, which transports Co2+ and Ni2+ less efficiently (Kehres and Maguire, 2002), elevates ROS (Figure 6C-D) and DNA damage (Figure 6B) when overproduced. Both might occur by increasing the usually minimal import of Co2+ and Ni2+ (Figure 6I). Ni2+ is toxic and induces DNA damage via oxidative stress (Cameron et al., 2011) because Ni2+ binds sulfhydryl groups commonly found in anti-oxidative enzymes (Schmidt et al., 2009), which causes DNA damage ROS-dependently (Cameron et al., 2011). Regardless of specific hypotheses, overproduction of membrane transporters generally may promote DNA damage, and for several, increased ROS, by any of many mechanisms that result from compromising compartmentalization of the cell from its environment.
(E) Flow cytometry assay for DNA loss (anucleate cells). Increased anucleate cells illustrated in a parCTS mutant, which has a severe chromosome-segregation defect at non-permissive temperature. Cells with positive FM464 (membrane) but negative DAPI (DNA) staining are anucleate cells (lower right quadrant). Controls for both wild-type (DB81) and parCTS (DB193) strains: cell-only, cells not stained; DAPI, cells stained only with DAPI; FM464, cells stained only with FM464 membrane dye; double, cells stained with both FM464 and DAPI.
(F) DNA loss in 67 E. coli DDP-overproducing clones. Quantification of assays per part E. The 67 (32% of DDPs) differ significantly from the vector-only control, p < 0.05, q <0.10 unpaired two-tail t-test with FDR adjustment. Each bar, a DDP clone (mean ± range, n=2). DDP overproducers are grouped by the fold change of anucleate cells levels compared with the vector-only strain. Labeling per (A), data Table S1. Representative flow-cytometry results shown. Only one of 16 non-DDP-overproducing clones had increased anucleate cells, and clones with high-levels of anucleate cells are enriched in the DDP clones at p = 0.02 (one-way Fisher’s exact test).
Data Availability Statement
RNA-Seq data and RDG ChIP-seq generated from this study are deposited in the European Nucleotide Archive (ENA) under accession numbers E-MTAB-7361 and PRJEB21035.