

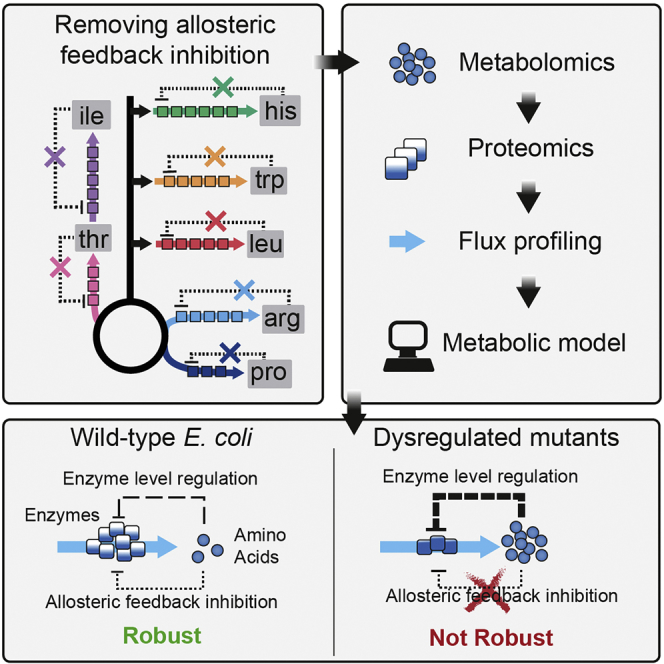
Summary
Microbes must ensure robust amino acid metabolism in the face of external and internal perturbations. This robustness is thought to emerge from regulatory interactions in metabolic and genetic networks. Here, we explored the consequences of removing allosteric feedback inhibition in seven amino acid biosynthesis pathways in Escherichia coli (arginine, histidine, tryptophan, leucine, isoleucine, threonine, and proline). Proteome data revealed that enzyme levels decreased in five of the seven dysregulated pathways. Despite that, flux through the dysregulated pathways was not limited, indicating that enzyme levels are higher than absolutely needed in wild-type cells. We showed that such enzyme overabundance renders the arginine, histidine, and tryptophan pathways robust against perturbations of gene expression, using a metabolic model and CRISPR interference experiments. The results suggested a sensitive interaction between allosteric feedback inhibition and enzyme-level regulation that ensures robust yet efficient biosynthesis of histidine, arginine, and tryptophan in E. coli.
Keywords: allosteric feedback inhibition, metabolic robustness, amino acid biosynthesis, multi-omics, CRISPR interference, kinetic modeling
Graphical Abstract
Highlights
-
•
Amino acid biosynthesis enzymes do not normally operate at maximum capacity
-
•
Allosteric feedback inhibition ensures that enzymes are overabundant
-
•
Enzyme overabundance provides robustness against decreases in gene expression
Sander et al. explore the consequences of removing allosteric feedback inhibition in seven amino acid biosynthesis pathways in Escherichia coli. Their work suggests that allosteric feedback inhibition and transcriptional regulation of enzyme abundance work together to ensure robust yet efficient biosynthesis of histidine, arginine, and tryptophan.
Introduction
Regulation of microbial metabolism involves a wide range of mechanisms that act on different cellular layers and together control the abundance and activity of enzymes (Chubukov et al., 2014). An example is end-product inhibition of amino acid biosynthesis in Escherichia coli, which can act on enzyme abundance through transcriptional regulatory cues and enzyme activities through allosteric feedback inhibition. However, since metabolic reaction rates are determined by both enzyme abundance and enzyme activity, it has been difficult to disentangle the specific roles of the two regulatory layers and to understand how they interact to control metabolism (Chubukov et al., 2013, Daran-Lapujade et al., 2007, ter Kuile and Westerhoff, 2001).
Allosteric feedback inhibition of the committed step in biosynthetic pathways is thought to maintain homeostasis of end-products (Umbarger, 1956), and 16 out of 20 amino acids in E. coli feedback inhibit enzymes of their own biosynthesis pathway (Reznik et al., 2017). The consequences of dysregulating these enzymes were mainly studied in vitro (Schomburg et al., 2013) or in the context of biotechnological overproduction strains (Hirasawa and Shimizu, 2016). For the case of nucleotide biosynthesis in E. coli, a detailed in vivo study showed that removing allosteric feedback inhibition did not perturb nucleotide homeostasis (Reaves et al., 2013). In the absence of allosteric feedback inhibition, additional regulatory mechanisms accomplished proper control of the pathway by channeling the excess of nucleotides into degradation pathways (so-called directed overflow). Theoretical analyses, in contrast, suggest a key role of allosteric feedback inhibition in achieving end-product homeostasis (Hofmeyr and Cornish-Bowden, 2000), metabolic robustness (Grimbs et al., 2007), flux control (Kacser and Burns, 1973, Schuster and Heinrich, 1987), and optimal growth (Goyal et al., 2010).
The abundance of enzymes in E. coli amino acid metabolism is mainly regulated at the level of transcription, either by transcriptional attenuation (Yanofsky, 1981) or transcription factors (Cho et al., 2008, Cho et al., 2012). For example, a set of four transcription factors (ArgR, TrpR, TyrR, and Lrp) control expression of 19 out of 20 amino acid pathways by sensing the availability of amino acids via allosteric binding (Cho et al., 2012). This regulation ensures that enzymes in amino acid pathways are only made when they are needed (Schmidt et al., 2016, Zaslaver et al., 2004). As a consequence of such need-based enzyme level regulation, one would expect that enzyme levels are not higher than absolutely needed for amino acid biosynthesis. However, recent data suggest that cells express the majority of enzymes at higher levels than necessary to fulfill biosynthetic demands, and that such enzyme overabundance provides a benefit in changing environments (Davidi and Milo, 2017, O’Brien et al., 2016). For example, enzyme overabundance enables a quick activation of the pentose phosphate pathway upon stresses (Christodoulou et al., 2018), and similar benefits were attributed to overabundant ribosomes (Mori et al., 2017) and coenzymes (Hartl et al., 2017).
Here, we constructed seven E. coli mutants, each with a different feedback-dysregulated amino acid biosynthesis pathway (arginine, histidine, tryptophan, leucine, isoleucine, threonine, and proline), and measured their proteins, metabolites, fluxes, and growth. In all seven feedback-dysregulated pathways, the concentration of amino acid end products increased, and in five pathways, we measured lower enzyme levels. Despite the lower enzyme levels, biosynthetic flux was not limited, indicating that these enzymes are not operating at maximal capacity in wild-type cells. By combining theoretical and experimental analysis, we showed that this enzyme overabundance provides a robustness benefit against genetic perturbations in the arginine, tryptophan, and histidine pathways.
Results
Dysregulating Allosteric Enzymes Changes Levels of Specific Amino Acids in E. coli
To explore the function of allosteric feedback inhibition in the arginine, histidine, tryptophan, leucine, isoleucine, threonine, and proline biosynthesis pathways, we first created a panel of seven allosterically dysregulated E. coli mutants (Figure 1A; Table S1). Using a scarless CRISPR method (Reisch and Prather, 2015), we introduced point mutations into genes encoding the allosteric enzyme that catalyzes the committed reaction in each pathway (argA, hisG, trpE, leuA, ilvA, thrA, and proB). These mutations have been shown previously to abolish the allosteric interaction while not affecting enzyme activity, thereby allowing us to study regulation of the pathway in the absence of allosteric feedback (Caligiuri and Bauerle, 1991, Csonka et al., 1988, Doroshenko et al., 2013, Gusyatiner et al., 2005, LaRossa et al., 1987, Lee et al., 2003, Rajagopal et al., 1998). For N-acetylglutamate synthase (ArgA), we confirmed with in vitro assays that the mutation does not affect enzymatic activity and abolishes inhibition by arginine (Figure S1). To analyze the metabolism of the mutants we quantified intracellular metabolites during exponential growth on glucose by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Guder et al., 2017). Stronger metabolic changes were restricted to amino acid biosynthesis, with specific increases between 2- and 16-fold of only the amino acid products of the dysregulated pathways (Figure 1B). Despite these changes within the dysregulated pathways, the remaining amino acid concentrations as well as the global metabolite profile remained relatively stable (Figures 1B and S2). Thus, dysregulating allosteric enzymes in E. coli amino acid biosynthesis elevated the intracellular concentration of the corresponding amino acid product.
Figure 1.

Amino Acid Profile of Feedback-Dysregulated E. coli Mutants
(A) Seven amino acid pathways were dysregulated by genomic point mutations in the indicated genes. See also Table S1. Negative allosteric feedbacks of amino acids on enzymes in the biosynthetic pathways are shown as dotted lines. Negative transcriptional feedbacks of amino acids are shown as dashed lines. Boxes indicate enzymes in the biosynthesis pathways.
(B) Relative concentrations of intracellular amino acids in wild-type E. coli and the seven dysregulated mutants. Bar plots show absolute concentrations of the amino acid in the dysregulated pathways. See also Figure S2. Data are represented as mean, and error bars are ± SD (n = 3).
Lower Expression of Enzymes in Feedback-Dysregulated Pathways
With the exception of proline biosynthesis, all of the dysregulated pathways are additionally controlled at the layer of enzyme abundance via either transcription factors or transcriptional attenuation. To probe if elevated amino acid concentrations in our mutants affected enzyme levels in the corresponding pathways we measured their proteomes (Figure 2A). The data covered relative abundances of 173 out of the 204 enzymes annotated to amino acid metabolism in the latest E. coli metabolic model (Monk et al., 2017). Enzyme expression was indeed lower in five of the seven dysregulated pathways (argA∗, trpE∗, hisG∗, leuA∗, and thrA∗), indicating that the elevated amino acid concentrations caused a compensatory downregulation of their associated pathway (Figure 2A). Enzyme levels did not change in the proB∗ and ilvA∗ mutants, which is expected because proline biosynthesis lacks enzyme level regulation and isoleucine biosynthesis is subject to a second allosteric feedback that was not removed (Figures 1A and 2A). The leuA∗ mutant showed more global changes in enzyme levels compared to the other mutants. The high leucine concentration in this strain likely activates the leucine responsive transcription factor Lrp, which acts on many genes in amino acid metabolism (Cho et al., 2008). In the argA∗ mutant we observed an expected accompanying decrease in histidine biosynthesis enzymes, which are additional targets of the transcription factor ArgR (Gama-Castro et al., 2016). Apart from the compensatory downregulation of biosynthetic enzymes, enzymes in dedicated amino acid degradation pathways were upregulated in three mutants (AstC in the arginine mutant, TnaA in the tryptophan mutant, and PutA in the proline mutant, Figure 2A). This likely constitutes an additional compensatory mechanism similar to the directed overflow reported for nucleotides (Reaves et al., 2013).
Figure 2.

Expression of Enzymes in Feedback-Dysregulated Pathways
(A) Abundance of 173 enzymes in amino acid metabolism (out of 204 enzymes in total), relative to the level in the wild-type. Data are represented as mean (n = 3). For each strain the enzymes in the dysregulated pathway are shown as colored dots. Enzymes in degradation pathways of arginine, tryptophan, and proline are indicated by their names.
(B) GFP-fluorescence measured by flow cytometry. GFP-promoter fusions were transformed in wild-type cells and the indicated mutant. Upper panel: pPargA-gfp; middle panel: pPtrpL-gfp; lower panel: pPthrL-gfp. Histograms represent fluorescence of 10,000 single cells. Mean fluorescence was calculated from 10,000 single cells of n = 3 independent cultures. See also Figure S3.
To obtain additional evidence for lower enzyme levels in the dysregulated pathways, we used GFP-promoter fusions and measured fluorescence in single cells (Figure 2B). GFP expression from an ArgR-regulated promoter was indeed ∼3-fold lower in the argA∗ mutant compared to the wild-type. Similarly, a TrpR-regulated promoter was ∼3-fold stronger repressed in the trpE∗ mutant. The cell-to-cell variation in GFP content was similar in wild-type cells and the mutants, thus indicating that all cells in the population of allosteric feedback mutants have lower enzyme levels in the dysregulated pathway. A GFP reporter with the thrL leader peptide was only 17% repressed in the thrA∗ mutant compared to the wild-type, which is consistent with the small decrease of enzyme levels in the dysregulated threonine pathway (Figures 2A and 2B). We also fused GFP to the hisL and leuL leader peptides, but they did not report repression by amino acids even when they were added to the medium (Figure S3). Probably, transcriptional attenuation by hisL and leuL requires the genomic context and cannot function on plasmids. In summary, proteome data revealed a lower expression of enzymes for five of the seven dysregulated pathways (argA∗, trpE∗, hisG∗, leuA∗, and thrA∗). GFP-promoter fusions confirm this enzyme level regulation at the single-cell level and indicate that downregulation of enzymes in the argA∗, trpE∗, and thrA∗ mutants occurs at the transcriptional layer.
Allosteric Feedback Inhibition Enforces Enzyme Overabundance
Next, we wondered if lower expression of enzymes limits the biosynthetic capacity of the mutants. First, we tested steady-state growth on glucose minimal medium and seven other carbon sources (Figure S4). All mutants showed wild-type like growth, except the leuA∗ mutant, which grew on average 10% slower than the wild-type. To test if lower enzyme levels affect biosynthetic capacity in dynamics shifts, we starved cells for carbon and measured growth resumption on glucose minimal medium (Figure 3A). During the initial phase of growth resumption all mutants had the same growth rate as the wild-type. Only the leuA∗, ilvA∗, and thrA∗ mutants reached lower growth rates than the wild-type during the subsequent 4 hr. The three strains had also lower ODs after 20 hr starvation. Similarly, nutritional up- and downshifts between glucose and galactose had only a tangible effect on the growth of the leuA∗, ilvA∗, and thrA∗ mutants during the downshift (Figure S5). The three strains with the highest reduction in enzyme levels (argA∗, trpE∗, and hisG∗) grew like the wild-type in all tested conditions, indicating that biosynthetic capacity is not limited by lower enzyme levels. The advantage of lower protein costs in these pathways was either too subtle to be detected by growth assays or counterbalanced by negative effects of feedback dysregulation.
Figure 3.

Growth and Biosynthetic Flux of Feedback-Dysregulated E. coli Mutants
(A) Growth resumption after 20 hr carbon starvation of wild-type E. coli and the seven dysregulated mutants. Cells were starved in minimal medium and glucose was added at t = 0 hr. OD was measured in 5 min intervals in a plate reader. Shown are means of n = 3 cultures. Inserts show the specific growth rate in h−1 during the same time period. Growth rates were estimated by linear regression over a moving 30 min window. The same wild-type growth curve and growth rate is shown in each graph in black as a reference. See also Figures S4 and S5.
(B) Decay of unlabeled amino acids in the wild-type E. coli (black) and the seven dysregulated mutants (color). The measured amino acid is indicated above each graph. Cells were loaded from shake flasks onto filters and perfused with 15N-medium for different lengths of time (0, 30, 60, 120, and 180 seconds). Dots are means of n = 2 samples for each time point. Lines are means of 1,000 fits of decay rates based on equations for kinetic flux profiling. Box plots show fluxes based on the 1,000 fits, relative to the median flux estimate in the wild-type. Boxes contain 50% and whiskers 99% of the flux estimates.
To directly probe biosynthetic capacity, we traced intracellular fluxes of amino acids with 15N labeling experiments (Figure 3B). Labeling of arginine, tryptophan, and proline was similar in the respective mutant and the wild-type, whereas histidine, (iso)-leucine, and threonine labeled slower in the mutants. However, it is important to consider that labeling rates depend on fluxes and absolute pool sizes of amino acids. Because amino acid pools were higher in the mutants, we used a method for quantitative analysis of the labeling profiles to estimate fluxes (Yuan et al., 2008). To account for unknown labeling profiles of upstream nitrogen precursors, we calculated fluxes for a wide range of precursor labeling rates in the literature (Yuan et al., 2006). The flux estimates show that none of the mutants had lower flux through the dysregulated pathways than the wild-type (Figure 3B). In most cases, biosynthetic flux was even higher, indicating that downregulation of enzyme levels could not fully compensate the loss of allosteric feedback inhibition in some of the mutants. This might be the reason for the growth phenotype of the leuA∗, ilvA∗, and thrA∗ mutants in dynamic growth experiments (Figure 3A).
In conclusion, the feedback-dysregulated mutants showed the same or higher flux through the dysregulated amino acid pathways than wild-type cells, although in five mutants (argA∗, trpE∗, hisG∗, leuA∗, and thrA∗) enzyme levels in the dysregulated pathway were lower. Especially, the argA∗, trpE∗, and hisG∗ mutants, which had ∼2-fold lower enzyme levels in the dysregulated pathways compared to the wild-type, showed 1- to 2-fold higher fluxes and normal growth. This indicates that these enzymes are not operating at maximal capacity in wild-type E. coli during growth on glucose. We then hypothesized that this enzyme overabundance emerges from allosteric feedback inhibition by maintaining a low concentration of end-products, which in turn increases production of enzymes (e.g., by de-repression of transcription). Next, we explored this interplay between control of enzyme activity and enzyme abundance and its relevance for cellular metabolism.
Interdependence of Allosteric Feedback Inhibition and Enzyme-Level Regulation
To obtain a better mechanistic understanding of the interplay between allosteric feedback inhibition and enzyme level regulation, we developed a kinetic model of metabolism and enzyme expression (Figure 4A). Briefly, the model includes two enzymes, e1 and e2, and two metabolites, m1 and m2, in a two-step pathway. The end-product m2 represents an amino acid, which is consumed in the last reaction for protein synthesis and growth. The end-product m2 feedback inhibits the expression of both enzymes as well as the activity of the first enzyme. The first reaction and the expression of both enzymes follow simple inhibition kinetics, whereas the second reaction follows Michaelis-Menten kinetics (Figure 4A). As such, this model is a simplified representation of an amino acid biosynthesis pathway that is controlled at two layers (Figure 1A).
Figure 4.

A Kinetic Model Predicts a Robustness-Efficiency Tradeoff
(A) Stoichiometry and structure of the kinetic model. m1 and m2 are metabolites, e1 and e2 are enzymes. Kinetics of the enzyme catalyzed reactions r1 and r2, as well as kinetics of enzyme expression rates β1 and β2, are sampled in the indicated intervals.
(B) Steady-state concentrations of e1, e2, m1, and m2 calculated with 5,000 random parameter sets for the complete model (grey), and the model with only enzyme level regulation (blue). Boxes contain 50% and whiskers 99% of the simulated concentrations. All concentrations are normalized to the median concentrations of the complete model. See also Figures S6 and S7.
(C) Enzyme levels (sum of e1 and e2) and robustness against perturbations of β2,max for 5,000 simulations of the complete model (dots). The color of each dot shows the ratio of inhibition constants for allosteric feedback inhibition (K1) and enzyme level regulation (K2) in the respective model. Robustness corresponds to the percentage downregulation of β2,max that was tolerated by each model. 100% enzyme abundance corresponds to the maximum theoretical enzyme concentration in the model.
(D) Abundance of enzymes in amino acid metabolism in the ΔargR, ΔtrpR, and ΔhisL mutants, relative to the wild-type. Data are represented as mean (n = 3). For each strain the enzymes in the dysregulated pathway are shown as orange dots.
As a starting point for the model analysis, we fixed the flux in the pathway to the amino acid requirement given by the growth rate of E. coli on glucose. We randomly sampled seven model parameters (maximal rates and binding constants) 5,000 times from physiologically meaningful ranges based on literature values (Davidi and Milo, 2017, Li et al., 2014, Milo et al., 2010). For each of the thus derived 5,000 parameter sets, we calculated concentrations of e1, e2, m1, and m2, for a model including feedback on enzyme activity and enzyme abundance (complete model, gray in Figure 4B), and also for a model including only feedback on enzyme abundance (single feedback model, blue in Figure 4B). The simulated concentrations of e1, e2, m1, and m2 matched qualitatively the measured protein and metabolite data: the two enzymes decreased in the single feedback model (Figure 2A), whereas the end-product m2 increased (Figure 1B). Also, the simulated concentration of the intermediate m1 matched the measured increase of intermediates in amino acid pathways (Figure S6). Thus, a simple model confirms our hypothesis that allosteric feedback inhibition enforces enzyme overabundance. In theory, other types of enzyme inhibition could cause a similar increase in enzyme expression. To test this, we replaced the allosteric feedback in the model with competitive product inhibition of the second reaction (Figure S7). However, removing competitive product inhibition was compensated by lower substrate concentrations (m1) and not by lower enzyme levels. This model result indicates that enzyme overabundance does not emerge from all types of enzyme inhibition.
The Interplay of Two Feedbacks Solves a Robustness-Efficiency Tradeoff
Next, we set out to investigate the function that emerges from the interplay between feedback on enzyme activity and enzyme abundance. While low enzyme levels are obviously advantageous due to lowering protein cost, high enzyme levels could provide a cellular benefit by improving robustness against perturbations in enzyme expression. To test this with the model, we made use of a numerical parameter continuation method to quantify robustness (Lee et al., 2014). This method iteratively decreases a model parameter until instabilities occur in the model. Robustness can then be defined as the percentage change of this parameter that is tolerated. Using this method, we calculated robustness against perturbations of the maximal expression rate of the second enzyme (β2,max) in the complete model with 5,000 randomly sampled parameter sets (Figure 4C). Changing β2,max reflects genetic or environmental perturbations of gene expression that can lead to a bottleneck in the pathway. Consistent with our expectations, models with high enzyme levels showed increased robustness, while models with lower enzyme levels were more sensitive to perturbations of enzyme expression (Figure 4C). However, robustness was not proportional to the enzyme level: a relatively small increase of enzyme levels already conferred a large robustness benefit. Very high enzyme levels, in comparison, did not increase robustness substantially over more subtle changes in enzyme abundance. Our model thus reveals a tradeoff between protein costs and robustness, which can be solved by sensitively balancing enzyme levels.
The optimal balance of enzyme levels occurs in models occupying the middle of the tradeoff frontier: those models with equally strong feedback on enzyme activity and enzyme abundance (indicated by similar inhibition constants Ki, black dots in Figure 4C). We then wondered if amino acid biosynthesis in E. coli operates in the middle of the tradeoff frontier, meaning that both feedbacks are simultaneously active. In particular, enzyme levels in the argA∗, trpE∗, and hisG∗ mutants demonstrated that wild-type E. coli does not operate with minimal enzyme levels in these pathways (blue dots in Figure 4C). To test if enzymes in these pathways are maximally expressed (orange dots in Figure 4C), we removed their transcriptional regulation, which functions by different mechanisms: a transcription factor (arginine), transcriptional attenuation (histidine), or both (tryptophan). In the arginine and tryptophan pathway, we deleted the respective transcription factor (ΔargR and ΔtrpR), and in histidine biosynthesis we removed the leader peptide hisL. Removing transcriptional regulation of all three pathways resulted in higher expression of enzymes in the respective pathway (Figure 4D): arginine enzymes increased between 5- and 60-fold; histidine enzymes, about 6-fold; and tryptophan enzymes, about 8-fold. This shows that E. coli does not operate at maximal expression of arginine, tryptophan, and histidine enzymes but rather in the middle of the tradeoff frontier. Previous studies that support this observation showed that ArgR binds to promoters of arginine genes more than 80% of the time when E. coli grows on glucose (Gerosa et al., 2014). Deletion of ArgR caused more global changes of amino acid enzymes than removing TrpR or HisL. This reflects the potential of ArgR to control metabolism of almost all amino acid pathways (Cho et al., 2012).
Taken together, both model and dysregulated mutants indicate a regulatory interplay in the arginine, tryptophan, and histidine pathways: removing transcriptional regulation increased enzyme levels (Figure 4D), whereas removing allosteric regulation decreased enzyme levels (Figure 2A). The model shows that if feedback on enzyme activity and enzyme abundance are simultaneously active, inhibition constants of the two feedbacks must have similar values (black dots in Figure 4C). Inhibition constants and binding affinities in the literature show that feedbacks on enzyme activity and enzyme abundance are indeed equally strong for many amino acids (Table S5), corroborating the existence of a two-pronged regulation strategy.
Enzyme Overabundance Provides Robustness against Genetic Perturbations
To test if arginine, tryptophan, and histidine biosynthesis are more robust against perturbations of gene expression in wild-type cells compared to the feedback-dysregulated mutants, we used CRISPR interference (CRISPRi) (Larson et al., 2013). We designed single guide RNAs (sgRNAs) targeting the genes argE in arginine biosynthesis, hisB in histidine biosynthesis, and trpA in tryptophan biosynthesis. The sgRNAs were cloned on a plasmid that harbors an inducible dCas9 and the constitutively expressed sgRNA. The three CRISPRi plasmids and a control without target sequence were transformed into the wild-type as well as into the argA∗, trpE∗, and hisG∗ mutants. This resulted in 16 strains with all combinations of genetic perturbations and dysregulation of the three pathways (Figure 5A). All strains expressing the control sgRNA without target sequence grew almost identically and induction of dCas9 did not affect growth (Figure 5B).
Figure 5.

Enzyme Overabundance Achieves Robustness against Perturbations of Gene Expression by CRISPR Interference
(A) CRISPR interference in wild-type cells and the allosteric feedback mutants argA∗, hisG∗, and trpE∗. Strains were transformed with single guide RNAs targeting genes of the arginine (argE), histidine (hisB), and tryptophan (trpA) pathways, as well as an empty sgRNA without target.
(B) Growth of wild-type, argA∗, hisG∗, and trpE∗ with the empty control sgRNA. Upper panels show uninduced cultures and lower panel induced cultures (100 μM IPTG). Growth curves show means from n=3 cultures cultivated in minimal glucose medium in a plate reader. Numbers are specific growth rates (in h−1) and were estimated by linear regression between OD 0.2 and 0.6.
(C) Growth of wild-type, argA∗, hisG∗, and trpE∗ with sgRNAs targeting argE, hisB, and trpA. dCas9 expression was induced with 100 μM IPTG. Growth curves are means of n=3 cultures; two curves per graph show experiments that were performed at different days. Numbers and colors indicate specific growth rates (in h−1), which were estimated by linear regression between 5 and 15 hr. All axes have ranges shown in the lower left graph.
(D) Same as (C) but without induction of dCas9. Growth rates were estimated by linear regression between OD 0.2 and 0.6. All axes have ranges shown in the lower left graph.
Induction of dCas9 in strains with sgRNAs targeting argE, hisB, and trpA reduced growth of all strains by more than 50% (Figure 5C). However, we observed the strongest growth defect when perturbing a gene in a dysregulated pathway. For example, CRISPRi of argE reduced growth of the argA∗ mutant more than twice as much as the other strains. Similarly, the hisG∗ and trpE∗ mutants were most sensitive to perturbations of expression of hisB and trpA, respectively. The argA∗mutant was also sensitive to a perturbation of hisB, which matches the lower expression of histidine enzymes in this mutant (Figure 2A). These data confirm that feedback-dysregulated mutants are indeed more sensitive to a perturbation of gene expression. Notably, the mutants were only more sensitive to a perturbation within pathways that had lower enzyme levels, and they did not lack a general robustness.
While these data support the hypothesis that high enzyme levels render arginine, histidine, and tryptophan biosynthesis more robust against perturbations of gene expression, bacteria would hardly face such strong perturbations in nature. Therefore, we designed the sgRNAs in such a way that the wild-type showed only a small growth defect without induction of dCas9 (Figure 5D). The mild perturbations in uninduced cultures still affected the respective mutants stronger than the other strains, causing unstable growth and lower growth rates (Figure 5D). Thus, feedback dysregulation renders the arginine, tryptophan, and histidine pathways more sensitive against perturbations of gene expression, which may arise in nature due to the stochasticity of gene expression.
Discussion
In this study, we explored the consequences of missing allosteric feedback inhibition in seven E. coli mutants with dysregulated amino acid biosynthesis pathways: arginine (argA∗), histidine (hisG∗), tryptophan (trpE∗), leucine (leuA∗), threonine (thrA∗), isoleucine (ilvA∗), and proline (proB∗). In all mutants, the amino acid product of the feedback-dysregulated pathway increased, showing that allosteric feedback inhibition is relevant to maintain end-products at a desired level. In five mutants (argA∗, trpE∗, hisG∗, thrA∗, and leuA∗), we observed a downregulation of enzymes in the dysregulated pathways, presumably because high end-products caused stronger inhibition of enzyme expression. However, these low enzyme levels did not limit biosynthetic flux, thus indicating that wild-type cells maintain higher enzyme levels than would be necessary to ensure sufficient supply of amino acids (enzyme overabundance). These results are consistent with enzyme overabundance in other pathways (Davidi and Milo, 2017, O’Brien et al., 2016) and the observation that enzymes are rarely operating at maximal capacity (Fendt et al., 2010, Hackett et al., 2016).
Both model analysis and dysregulated mutants indicate that enzyme overabundance is enforced by allosteric feedback inhibition, which maintains low end-product levels and thereby increases production of enzymes. In case of amino acid biosynthesis, it is likely that low end-products de-repress transcription because amino acid levels are known signals for transcription factors and transcriptional attenuation (Cho et al., 2012). Additionally, GFP-promoter fusions indicated regulation at the transcriptional layer in the argA∗, trpE∗, and thrA∗ mutants. It will be important to clarify if enzyme overabundance also emerges from other inhibitory interactions, which are abundant in metabolic networks (Alam et al., 2017). Besides inhibition of enzymes by metabolites, other sources for enzyme overabundance might be post-translational modifications. For example, it was recently shown that deleting kinases in yeast has a strong effect on enzyme levels (Zelezniak et al., 2018), pointing toward a similar interplay between post-translational modifications of enzymes and enzyme-level regulation.
The strongest and most localized decrease of enzyme levels occurred when we removed allosteric feedback inhibition in the arginine, tryptophan, and histidine pathways. Removing transcriptional regulation in the same pathways caused higher expression of enzymes, which is in agreement with previous reports of a role for transcriptional regulation in minimizing protein costs in metabolic pathways (Chubukov et al., 2012, You et al., 2013). This antagonistic regulation by allosteric feedback inhibition and transcriptional regulation enables an optimal balance of enzyme levels in amino acid metabolism of wild-type cells. Optimization of enzyme levels has been shown for the global E. coli proteome (Scott et al., 2010, You et al., 2013), for the lac system (Dekel and Alon, 2005), and for a single enzyme in the methionine pathway (Li et al., 2014). Here we provided first indication that enzyme abundance is optimized in the arginine, histidine, and tryptophan pathways, to meet multiple, conflicting objectives—robustness and efficiency. Using a simplified model of amino acid metabolism, we showed that cells can solve this tradeoff between protein costs and robustness through the interplay of allosteric feedback inhibition and enzyme level regulation. CRISPRi of metabolic enzymes in the dysregulated arginine, tryptophan, and histidine pathways showed that allosteric feedback inhibition provides a substantial robustness benefit against perturbations of gene expression. While such robustness effects were attributed to allosteric feedback by previous modeling approaches (Chandra et al., 2011, Grimbs et al., 2007), we quantified it in vivo by studying mutants lacking allosteric control. During the lifetime of a cell, perturbations of gene expression could result from stochastic effects at the level of transcription or in response to fluctuating environments.
In conclusion, our case study of E. coli amino acid metabolism demonstrated that regulation of enzyme activity and enzyme abundance are not isolated from each other but interact to control metabolism. Allosteric feedback inhibition sets amino acid concentrations, which are signals for enzyme level regulation. Considering the high precision of metabolite concentrations (Fuhrer et al., 2017, Mülleder et al., 2016), it seems possible that the proposed regulatory principle goes beyond E. coli amino acid metabolism.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli TOP10: F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(araleu)7697 galU galK rpsL (StrR) endA1 nupG | Invitrogen, Thermo Fischer Scientific | Cat#C404003 |
| Escherichia coli MG1655: wild-type: F-, lambda-, rph-1 | DZMS-German Collection of Microorganisms and Cell Cultures | DSM-No.: 18039 |
| MG1655: argA∗:F-, lambda-, rph-1, argA(H15Y) | This study | N/A |
| MG1655: ilvA∗:F-, lambda-, rph-1, ilvA(L447F) | This study | N/A |
| MG1655: hisG∗:F-, lambda-, rph-1, hisG(E271K) | This study | N/A |
| MG1655: leuA∗:F-, lambda-, rph-1, leuA(G462D) | This study | N/A |
| MG1655: proB∗:F-, lambda-, rph-1, proB(D107N) | This study | N/A |
| MG1655: thrA∗:F-, lambda-, rph-1, thrA(S345F) | This study | N/A |
| MG1655: trpE∗:F-, lambda-, rph-1, trpE(S40F) | This study | N/A |
| MG1655: ΔargR: F-, lambda-, rph-1, ΔargR | This study | N/A |
| MG1655: ΔtrpR: F-, lambda-, rph-1, ΔtrpR | This study | N/A |
| MG1655: ΔhisL: F-, lambda-, rph-1, ΔhisL | This study | N/A |
| Escherichia coli BW25113: JW2000-1: F-, Δ (araD-araB)567, ΔlacZ4787(::rrnB-3), λ-,ΔhisL787::kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 | CGSC | CGSC#9646 |
| MG1655: wild-type CRISPRi-ctrl: F-, lambda-, rph-1, pNUT1533-ctrl | This study | N/A |
| MG1655: wild-type CRISPRi-argE: F-, lambda-, rph-1, pNUT1533-argE | This study | N/A |
| MG1655: wild-type CRISPRi-hisB: F-, lambda-, rph-1, pNUT1533-hisB | This study | N/A |
| MG1655: wild-type CRISPRi-trpA: F-, lambda-, rph-1, pNUT1533-trpA | This study | N/A |
| MG1655: argA∗ CRISPRi-ctrl: F-, lambda-, rph-1, argA (H15Y) pNUT1533-ctrl | This study | N/A |
| MG1655: argA∗ CRISPRi-argE: F-, lambda-, rph-1, argA (H15Y) pNUT1533-argE | This study | N/A |
| MG1655: argA∗ CRISPRi-hisB: F-, lambda-, rph-1, argA (H15Y) pNUT1533-hisB | This study | N/A |
| MG1655: argA∗ CRISPRi-trpA: F-, lambda-, rph-1, argA (H15Y) pNUT1533-trpA | This study | N/A |
| MG1655: hisG∗ CRISPRi-ctrl: F-, lambda-, rph-1, hisG (E271K) pNUT1533-ctrl | This study | N/A |
| MG1655: hisG∗ CRISPRi-hisB: F-, lambda-, rph-1, hisG (E271K) pNUT1533-hisB | This study | N/A |
| MG1655: hisG∗ CRISPRi-argE: F-, lambda-, rph-1, hisG (E271K) pNUT1533-argE | This study | N/A |
| MG1655: hisG∗ CRISPRi-trpA: F-, lambda-, rph-1, hisG (E271K) pNUT1533-trpA | This study | N/A |
| MG1655: trpE∗ CRISPRi-ctrl: F-, lambda-, rph-1, trpE (S40F) pNUT1533-ctrl | This study | N/A |
| MG1655: trpE∗ CRISPRi-trpA: F-, lambda-, rph-1, trpE (S40F) pNUT1533-trpA | This study | N/A |
| MG1655: trpE∗ CRISPRi-argE: F-, lambda-, rph-1, trpE (S40F) pNUT1533-argE | This study | N/A |
| MG1655: trpE∗ CRISPRi-hisB: F-, lambda-, rph-1, trpE (S40F) pNUT1533-hisB | This study | N/A |
| MG1655: pPargA-gfp: F-, lambda-, rph-1, | This study | N/A |
| MG1655: argA∗ pPargA-gfp: F-, lambda-, rph-1, argA (H15Y) | This study | N/A |
| MG1655: pPtrpL-gfp: F-, lambda-, rph-1, | This study | N/A |
| MG1655: trpE∗ pPtrpL-gfp: F-, lambda-, rph-1, trpE (S40F) | This study | N/A |
| MG1655: pPthrL-gfp: F-, lambda-, rph-1, | This study | N/A |
| MG1655: thrA∗ pPthrL-gfp: F-, lambda-, rph-1, thrA (S345F) | This study | N/A |
| MG1655: pPhisL-gfp: F-, lambda-, rph-1, | This study | N/A |
| MG1655: hisG∗ pPhisL-gfp: F-, lambda-, rph-1, hisG (E271K) | This study | N/A |
| MG1655: pPleuL-gfp: F-, lambda-, rph-1, | This study | N/A |
| MG1655: leuA∗ pPleuL-gfp: F-, lambda-, rph-1, leuA (G462D) | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetonitrile | Honeywell Riedel-de Haën | Cat#14261-2L |
| Methanol | VWR | Cat#83638.320 |
| Anhydrotetracycline | Sigma-Aldrich | Cat#1035708-25MG |
| IPTG | Roth | Cat#CN08.2 |
| Ampicillin | Roth | Cat#K029.2 |
| Kanamycin | Roth | Cat#T832.3 |
| Gentamycin | Roth | Cat#0233.3 |
| Spectinomycin | Roth | Cat#HP66.2 |
| Critical Commercial Assays | ||
| PierceTM Quantitative Colometric Peptide Assay | Thermo Fisher Scientific | Cat#23275 |
| His GraviTrapTM | Merck | 11-0033-99 |
| Deposited Data | ||
| Metabolome, proteome and labeling data | This study | Table S7 |
| kcat-values for enzymes in amino-acid biosynthesis (Table S3) | (Schomburg et al., 2013) (Davidi and Milo, 2017) |
BRENDA [doi:https://doi.org/10.1016/j.copbio.2017.02.007] |
| Amino acid requirement of Escherichia coli (Table S4) | (Monk et al., 2017) | [doi:https://doi.org/10.1038/nbt.3956] |
| Inhibition Constants (Table S5) | (Keseler et al., 2017) (Schomburg et al., 2013); (Gama-Castro et al., 2016) |
EcoCyc BRENDA; RegulonDB |
| Oligonucleotides | ||
| Oligonucleotides are listed in Table S6 | ||
| Recombinant DNA | ||
| pKDsgRNA-ack | Reisch and Prather (2015) | Addgene plasmid # 62654 |
| pCas9-CR4 | Reisch and Prather (2015) | Addgene plasmid # 62655 |
| pKDsgRNA-p15 | Reisch and Prather (2015) | Addgene plasmid # 62656 |
| pdCas9 | Larson et al. (2013) | Addgene plasmid # 44249 |
| pgRNA | Larson et al. (2013) | Addgene plasmid # 44251 |
| pKDsgRNA-argA(H15Y) | This study | N/A |
| pKDsgRNA-ilvA(L447F) | This study | N/A |
| pKDsgRNA-hisG(E271K) | This study | N/A |
| pKDsgRNA-leuA(G462D) | This study | N/A |
| pKDsgRNA-proB(D107N) | This study | N/A |
| pKDsgRNA-thrA(S345F) | This study | N/A |
| pKDsgRNA-trpE(S40F) | This study | N/A |
| pKDsgRNA-ΔargR | This study | N/A |
| pKDsgRNA-ΔtrpR | This study | N/A |
| pNUT542 | Singh et al. (2017) | |
| pNUT1533-ctrl | This study | N/A |
| pNUT1533-argE | This study | N/A |
| pNUT1533-trpA | This study | N/A |
| pNUT1533-hisA | This study | N/A |
| pUA66-PargA-gfp: pPargA-gfp | Zaslaver et al. (2006) | N/A |
| pUA66-PtrpL-gfp: pPtrpL-gfp | Zaslaver et al. (2006) | N/A |
| pUA66 based plasmid with PhisL | This study | pPhisL-gfp |
| pUA66 based plasmid with PleuL | This study | pPleuL-gfp |
| pUA66 based plasmid with PthrA | This study | pPthrA-gfp |
| Software and Algorithms | ||
| MATLAB codes for model analysis | This study | https://github.com/nfarke/Sander_et_al |
| Matlab Version 9.3.0.713579 (R2017b) for the modelling section and analysis of experimental data | mathworks.com | N/A |
| BD FACSDiva software version 8.0 | BD Biosciences, NJ, USA | N/A |
| FlowJo v10.4.1 | FlowJo LLC, Ashland, OR, USA | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hannes Link (hannes.link@synmikro.mpi-marburg.mpg.de).
Experimental Model and Subject Details
Strains and Culture
E. coli MG1655 (DSMZ No. 18039) was the wild-type strain. Chemically competent E. coli TOP10 (One Shot™ TOP10, Invitrogen) were used for cloning. All mutants created in this study derive from the MG1655 strain and are listed in Key Resources Table. Genomic point mutations were created by scarless Cas9 Assisted recombineering (Reisch and Prather, 2015). Therefore we constructed 7 specific sgRNA-plasmids, derived from the backbone plasmid pKDsgRNA-ack (Addgene #62654). The sgRNAs consist of a gene specific 20 base pair region (argA: ggtcgagggattccgccatt; trpE: acacaactggtgaaaaagcg; hisG: tggaaaaactgaaagcgctg; thrA: tggtgctgattacgcaatca; leuA: cggtaaagatgcgctgggtc; ilvA: caacacgctgggtacgtact; proB: cgacaccctgcgagcgttgc), which pairs adjacent to a NGG PAM site. Each sgRNA-plasmid was transformed together with pCas9-CR4 (Addgene #62655) into MG1655 wild-type cells. The resulting strains were grown at 30°C (pKDsgRNA-ack is temperature sensitive at 37°C) and supplemented with arabinose (final concentration 1.2 %) to induce the λ-Red recombinase genes which are located on the sgRNA-plasmid. The induced strains were then transformed with the 70-80 bp homologous oligonucleotides (Table S2), which contain the desired base pair exchanges of PAM site and the point mutation disrupting allosteric feedback (argAH15Y, trpES40F, hisGE271K, thrAS345F, leuAG462D, ilvAL447F, proBD107N). Cells were plated on LB agar containing 100 ng ml-1 anhydrotetracycline (aTc) to induce Cas9 expression, which recognizes the sgRNA adjacent to the PAM sequence and cleaves the chromosomal DNA. Only cells that successfully integrated the homologous oligonucleotides will survive due to the modified PAM sequence which prevents Cas9 recognition. Thereby we counter selected for clones harboring the desired amino acid exchanges, which were verified by sequencing. The transcriptional knockout mutants ΔargR and ΔtrpR were constructed with the same cloning procedure according to the noSCAR protocol, while ΔhisL was constructed by P1 Phage transduction with the donor strain JW2000-1 (ΔhisL) from the Keio collection (Baba et al., 2006).
All cultivations were performed using M9 minimal medium with 5 g L-1 glucose (or the respective carbon source in Figure S4). The M9 medium consisted of the following components (per liter): 7.52 g Na2HPO4 2 H2O, 5 g KH2PO4, 1.5 g (NH4)2SO4, 0.5 g NaCl. The following components were sterilized separately and then added (per liter of final medium): 1 ml 0.1 M CaCl2, 1 ml 1 M MgSO4, 0.6 ml 0.1 M FeCl3, 2 ml 1.4 mM thiamine-HCL and 10 ml trace salts solution. The trace salts solution contained (per liter): 180 mg ZnSO4 7 H2O, 120 mg CuCl2 2 H2O, 120 mg MnSO4 H2O, 180 mg CoCl2 6 H2O. Where appropriate, 50 μg mL-1 kanamycin, 34 μg mL-1 chloramphenicol, 15 μg mL-1 gentamycin, 50 μg mL-1 spectinomycin or 100 μg mL-1 ampicillin was added. For cultivations in microtiter plates, LB pre-culture in 96-deep-well format plates were inoculated from glycerol stocks and grown to an exponential stage. From this first pre-culture a second M9 pre-culture in 96-deep-well plates was inoculated 1:100 and incubated overnight at 37°C under shaking. Finally, 96-well flat transparent plates (Greiner Bio-One International) containing 150 μl M9 minimal medium were inoculated 1:150 from the overnight culture. Online measurements of optical density at 600 nm (OD600) were performed at 37°C with shaking in a plate reader (Epoch, BioTek Instruments Inc, USA; Spark 10M, Tecan Trading AG, Switzerland). For induction of CRISPRi, IPTG was added to the main culture to a final concentration of 100 μM. Growth rates were calculated as dln(OD)/dt by linear regression over the indicated time windows. For cultivations in shake flask, 5 ml LB pre-culture in cultivation tubes were inoculated from glycerol stocks and grown to an exponential stage. From this first pre-culture, 5 ml of a second M9 glucose pre-culture in cultivation tubes was inoculated 1:100 and incubated overnight at 37°C in a rotary shaker. For the main culture, a 500 ml shake flask containing 35 ml M9 minimal medium (5 g L-1 glucose) was inoculated 1:150 from the overnight culture, and incubated at 37°C under shaking at 220 rpm.
Method Details
CRISPR Interference
CRISPR interference experiments were performed with a single plasmid (pNUT1533) expressing the sgRNA from a constitutive and the dCas9 protein from an IPTG inducible Ptac promotor. For construction of this plasmid, the sgRNA and its constitutive promotor were amplified from the pgRNA plasmid (Addgene #44251) and the dCas9 gene was amplified from the pdCas9 plasmid (Addgene #44249). The promotor of dCas9 was replaced by an IPTG inducible Ptac promotor. To assure tight regulation of dCas9 expression, the gene coding for the lacIQ1 repressor (Glascock and Weickert, 1998) was added to the vector. The two single fragments were joined together by PCR and the resulting fragment was inserted into pNUT542 with PacI and NotI restriction enzymes (New England Biolabs, USA). This plasmid was used as a backbone for cloning of the specific plasmids targeting the arginine (pNUT1533-argE), histidine (pNUT1533-hisB) and tryptophan pathway (pNUT1533-trpA). Therefore sgRNAs guide sequences were customized by site-directed mutagenesis using the primer listed in Table S6.
Metabolite Measurements
Shake flask cultivations on M9 glucose were performed as described above. Cells were grown to an optical density (OD600) of 0.5 and 2 mL culture aliquots were vacuum-filtered on a 0.45 μm pore size filter (HVLP02500, Merck Millipore). Filters were immediately transferred into 40:40:20 (v-%) acetonitrile/methanol/water at -20°C for extraction. Extracts were centrifuged for 15 minutes at 13,000 rpm at -9°C. Centrifuged extracts were mixed with 13C-labeled internal standard and analyzed by LC-MS/MS, with an Agilent 6495 triple quadrupole mass spectrometer (Agilent Technologies) as described previously (Guder et al., 2017). An Agilent 1290 Infinity II UHPLC system (Agilent Technologies) was used for liquid chromatography. Temperature of the column oven was 30°C, and the injection volume was 3 μL. LC solvents A were water with 10 mM ammonium formate and 0.1% formic acid (v/v) (for acidic conditions); and water with 10 mM ammonium carbonate and 0.2% ammonium hydroxide (for basic conditions). LC solvents B were acetonitrile with 0.1% formic acid (v/v) for acidic conditions and acetonitrile without additive for basic conditions. LC columns were an Acquity BEH Amide (30 x 2.1 mm, 1.7 μm) for acidic conditions, and an iHILIC-Fusion(P) (50 x 2.1 mm, 5 μm) for basic conditions. The gradient for basic and acidic conditions was: 0 min 90% B; 1.3 min 40 % B; 1.5 min 40 % B; 1.7 min 90 % B; 2 min 90 % B. Absolute concentrations of amino acids in the 13C-labeled internal standard were determined with authentic standards. Quantification of intracellular metabolite concentrations was based on the ratio of 12C and 13C peak heights, and a specific cell volume of 2 μL mg-1 was used to calculate the cell volume.
Proteomics
Shake flask cultivations on M9 glucose were performed as described above. Cells were grown to an optical density (OD600) of 0.5 and 2 mL culture aliquots were transferred into 2 ml reaction tubes and washed two times with PBS buffer (0.14 mM NaCl, 2.7 mM KCL, 1.5 KH2PO4, 8.1 Na2HPO4). Cell pellets were resuspended in 300 μl lysis buffer containing 100 mM ammonium bicarbonate, 0.5 % sodium laroyl sarcosinate (SLS) and 5 mM Tris(2-carboxyethyl)phosphine (TCEP). Cells were lysed by 5 minutes incubation at 95°C and ultra-sonication for 10 seconds (Vial Tweeter, Hielscher). Cells were again incubated for 30 minutes at 90°C followed by alkylation with 10 mM iodoacetamide for 30 minutes at 25°C. To clear the cell lysate, samples were centrifuged for 10 minutes at 15,000 rpm and the supernatant transferred into a new tube. Proteins in the cell lysates were digested with 1 μg trypsin (Promega) overnight at 30°C. To remove the SLS by precipitation, trifluoroacetic acid (TFA) was added to a final concentration of 1.5 % and rested at room temperature for 10 minutes. Samples were centrifuged for 10 minutes at 10,000 rpm and the supernatant used for C18 purification. The peptide purification was performed using the C18 microspin columns according to the manufactors instructions (Harvard Apparatus). Eluted peptide solutions were dried and resuspended in 0.1 % TFA. The concentration of peptides in the samples was measured with a colorimetric peptide assay (Pierce™ Quantitative Colorimetric Peptide Assay, Thermo Fischer Scientific). Analysis of peptides was performed by liquid chromatography-mass spectrometry. Analysis of peptides was performed by liquid chromatography-mass spectrometry, carried out on a Q-Exactive Plus instrument connected to an Ultimate 3000 RSLC nano with a Prowflow upgrade and a nanospray flex ion source (Thermo Scientific). Peptide separation was performed on a reverse-phase HPLC column (75 μm x 42 cm) packed in-house with C18 resin (2.4 μm, Dr. Maisch GmbH, Germany). The following separating gradient was used: 98 % solvent A (0.15% formic acid) and 2 % solvent B (99.85 acetonitrile, 0.15 % formic acid) to 25 % solvent B over 105 minutes and to 35 % solvent B for additional 35 minutes at a flow rate of 300 nl/min. The data acquisition mode was set to obtain one high resolution MS scan at a resolution of 70,000 full width at half maximum (at m/z 200) followed by MS/MS scans of the 10 most intense ions. To increase the efficiency of MS/MS attempts, the charged state screening modus was enabled to exclude unassigned and singly charged ions. The dynamic exclusion duration was set to 30 seconds. The ion accumulation time was set to 50 ms for MS and 50 ms at 17,500 resolution for MS/MS. The automatic gain control was set to 3x106 for MS survey scans and 1x105 for MS/MS scans. Label-free quantification (LFQ) of the data was performed using Progenesis QIP (Waters), and for MS/MS searches of aligned peptide features MASCOT (v2.5, Matrix Science) was used. The following search parameters were used: full tryptic search with two missed cleavage sites, 10ppm MS1 and 0.02 Da fragment ion tolerance. Carbamidomethylation (C) as fixed, oxidation (M) and deamidation (N,Q) as variable modification. Progenesis outputs were further processed with SafeQuant.
Kinetic Flux Profiling
Incorporation of 15N label into amino acids was measured with a filter cultivation method (Link et al., 2013). Briefly, cells were cultured on M9 glucose medium, which contains unlabeled ammonium sulfate as sole nitrogen source. At mid-exponential phase when cells reached ODs between 0.4 and 0.6, 2 mL of the culture was vacuum-filtered, and cell-loaded filters were continuously perfused with M9 glucose medium containing labeled ammonium-15N sulfate. Filters were repeatedly loaded and perfused with 15N-medium for different lengths of time: 0, 30, 60, 120 and 180 seconds. Subsequently, filters were immediately transferred into 40:40:20 (v-%) acetonitrile/methanol/water kept at -20°C. Extracts were centrifuged for 15 minutes at 13,000 r.p.m. at -9°C and the supernatant was directly used for LC-MS/MS. For LC separation of tryptophan, proline, threonine and (iso)leucine a ZIC-pHILIC column (150 x 2.1 mm, 5 μm, Merck) was used, and an Acquity BEH Amide (100 x 2.1 mm, 1.7 μm, Waters) for LC separation of histidine and arginine. Buffers were as described for metabolite measurements and gradients were for Acquity BEH Amide: 0 min 90% B; 2.6 min 40 % B; 3 min 40 % B; 3.4 min 90 % B; 5 min 90 % B. For ZIC-pHILIC: 0 min 90% B; 4.5 min 40 % B; 5 min 40 % B; 6 min 90 % B; 8 min 90 % B. Transitions for all isotopologues per amino acid were measured by LC-MS/MS and the amount of each isotopologue was used to calculate the fraction of unlabeled amino acid FU as:
Where M0 is the amount of the unlabeled amino acid, M+1 is the amount of all isotopologues with one 15N atom, etc. N is the number of 15N atoms in the amino acid: arginine (N = 4 from 2x glutamate, 1x glutamine, 1x aspartate), tryptophan (N = 2 from 1x glutamine, 1x serine), histidine (N = 3 from ATP, 1x glutamate), threonine (N = 1 from glutamate), proline (N = 1 from glutamate), iso-/leucine (N = 1 from glutamate). Fluxes were estimated based on equations for kinetic flux profiling (Yuan et al., 2008), which considers the decay of the unlabeled fraction FU:
The rate constant kaa is the flux into the amino acid (fluxaa) divided by their absolute concentration: kaa = fluxaa/caa. The rate constant kaa was obtained by fitting the equation to the measured unlabeled fraction FU. The rate constant kpc describes labeling of upstream nitrogen precursor. Because amino acids like arginine receive 15N label from several sources, the rate constant of precursor labeling kpc was unknown. To account for this uncertainty the parameter kpc was randomly sampled between boundaries of 0.8 min-1 and 14.2 min-1, which are the highest and lowest first order rate constants measured for nitrogen assimilation in E. coli (Yuan et al., 2006). a and b consider amino acid production from degradation of protein and other macromolecules and they were estimable parameters within bounds of 0 and 0.2.
GFP-promoter Fusions
GFP reporter plasmids for detection of promotor activity of argA, trpL, hisL and leuL were obtained from a library of fluorescent transcriptional reporters for E. coli (Zaslaver et al., 2006). Since the original plasmids pUA66-PhisL-gfp and pUA66-PleuL-gfp lacked parts of the attenuator region, we modified the respective promotor resulting in the plasmids pPhisL-gfp and pPleuL-gfp. Therefore we amplified leader sequence including the rho-independent terminator of hisL and leuL from chromosomal DNA of E. coli MG1655 (PhisL: hisL_fwd_gfp ccgctcgaggctttcatcattgttgccg, hisL_rev_gfp ccgggatcccgcagaatatcaatcggc; PleuL: leuL_fwd_gfp ccgctcgagttgtcccctttttcctcg, leuL_rev_gfp ccgggatccgatggtttgcaccgattc). The resulting two single fragments were introduced into an empty pUA66 backbone with the restriction enzymes XhoI and BamHI. The threonine reporter plasmid which was not available in the library was constructed with the same strategy. The attenuator region of thrL was amplified with the primer pair thrA_fwd_gfp (ccgctcgagactgcaacgggcaatatg) and thrA_rev_gfp (ccgggatcctcggcatcgctgatattg) and the single fragment was introduced into pUA66 (XhoI and BamHI) resulting in pPthrL-gfp.
Flow Cytometry
Activity of the argA, trpL and thrL promoter was assayed using plasmid-based GFP reporters that were described in the previous section. Strains for flow cytometry were cultivated in three independent shake flasks (100 ml) containing 10 ml M9 minimal medium (5 g L-1 glucose; 50 μg mL-1 kanamycin) as described in Strains and Culture. After reaching an OD between 0.5 and 0.8 cells were diluted 1:2000 in tethering buffer (10 mM KH2PO4, 100 μM EDTA, 1 μM L-methionine and 10 mM lactic acid, pH=7.0) and fluorescence was measured with BD LSRFortessa SORP cell analyser (BD Biosciences, Germany). 488-nm lasers, 600 long pass and a 520/30 band pass filters were used for detection of green fluorescence. Per sample, fluorescence of 10,000 single cells was measured. Before the measurements, cell aggregates were dispersed by vigorous mixing. BD FACSDiva software version 8.0 (BD Biosciences, NJ, USA) and FlowJo v10.4.1 (FlowJo LLC, Ashland, OR, USA) were used for analysis of the acquired data.
Purification and In Vitro Activity Assays of N-Acteylglutamate Synthase
E. coli BL21 cells harboring the overexpression vector pET28a(+)-argA respectively pET28a(+)-argA(H15Y) were cultivated at 37°C (220 rpm) in 500 ml of LB medium (5 L shake flasks) containing 30 μg ml-1 kanamycine. When cells reached OD600 0.6, the culture was shifted to 16°C to cool down the cell broth. To induce protein expression, 10 μl of IPTG stock solution (final concentration is 10 μM) were added. The culture was incubated overnight at 16°C (220 rpm). The cells were harvested by centrifugation at 6000 x g for 10 minutes at 4°C. The supernatant was completely removed. The cell pellet was resuspended in Lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM Imidazol) (2-5 ml per gram wet weight). 50 μl protease inhibitor cocktail and 5 mg of DNAse I powder were added. Lysis of cells was performed by french press (1100 bar). The lysate was centrifuged at 4,000 x g for 45 minutes at 4°C to pellet the cellular debris. The supernatant was filtered using a 0.2-μm-pore-size syringe filter and transferred into a new collection tube. Purification was performed with columns purchased from GE Healthcare Life Science (His GraviTrap; 11-0033-99). 10 ml of equilibration buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM Imidazol) was added to the column. As soon as equilibration buffer flowed through, up to 35 ml of filtered supernatant were added to the column. The column was washed twice with 10 ml washing buffer (same as equilibration buffer). Elution of the protein was performed 3 times with 3 ml elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM Imidazol). Protein concentration of all fractions was determined (660 nm protein assay, life technologies PIERCE™ #22660). Activity of purified N-acetylglutamate-synthase (ArgA) as well as for the feedback-resistant version ArgA (H15Y) was assayed in 30 mM TRIS buffer (with 40 mM L-glutamate, 0.65 mM N-acetyl-CoA and 10 mM MgCl2). To start the enzymatic reaction 10 μL of enzyme stock solution (0.15 mg/ml) was transferred to 90 μL assay buffer and mixed by pipetting up and down. To stop the reaction, 10 μL were transferred into 40 μl of 50:50 (v-%) acetonitrile/methanol at -20°C. Samples were taken every minute in a total time interval of 8 minutes. The reaction product N-acetylglutamate was measured by LC-MS and calibrated with authentic standards.
Kinetic Model
The stoichiometry of the model is shown in Figure 4A. Mass balancing results in the system of ordinary differential equations (ODEs), F, that is a temporal function of the state variables x and the kinetic parameters p:
| (Equation 1) |
The five reactions (r1, r2, β1, β2, μ) are described by the following kinetic equations:
Reaction 1 is feedback inhibited by m2 according to normal inhibition kinetics:
| (Equation 2) |
In the model without allosteric regulation the equation reduces to:
| (Equation 3) |
Reaction 2 follows Michaelis-Menten kinetics:
| (Equation 4) |
Expression rates of enzyme 1 and enzyme 2 follow inhibition kinetics
| (Equation 5) |
| (Equation 6) |
The growth rate depends on availability of the end-product m2:
| (Equation 7) |
Dilution of metabolites by growth was not considered, due to large difference in time scales between growth dilution and metabolic flux. Dilution of enzymes by growth is included in Equation 1, because the time scales of enzyme synthesis and growth dilution are closer.
Together, the kinetic equations include eight kinetic parameters kcat1, kcat2, β1,max, β2,max, K1, K2, Km and α. The physiological ranges for these parameters were derived from literature values. The boundaries of enzyme turnover number (kcat,1 and kcat,2) are based on in vitro measured kcat values of enzymes in amino acid biosynthesis (Table S3) and have values between 930 min-1 and 4140 min-1. The maximal enzyme expression rates (β1,max and β2,max) are defined by the translation rate of ribosomes according to Equation 8. The equation considers the following parameters that were derived from the Bionumbers Database (Milo et al., 2010): average translation rate (rT = 8.4 amino acids s-1), the median and abundance weighted protein length (L = 209 amino acids), the fraction of active ribosomes (fR = 0.8), the cellular volume (Vc,0.6 = 3 x 10-15 L) at a growth rate of μ = 0.6 h-1, the Avogadro number (NA = 6.02 x 1023 mol-1), the amount of ribosomes per cell at that growth rate (R0.6 = 8000 ribosomes cell-1) and the fraction of ribosomes (p) that synthesize the enzyme:
| (Equation 8) |
The limits of βk,max are then derived by varying the fraction of ribosomes (p) that synthesize the enzymes in the pathway. According to the literature the maximal number for a single amino acid biosynthesis enzyme in E. coli is 7% (Li et al., 2014), therefore we set the boundaries to 1% and 10% (p = 0.01 - 0.1). The parameter limits for the Ki and Km values were set to 0.01 mM and 1 mM. The amino acid requirement (α = 86.6 mM) was a fixed parameter based on the average amino acid requirement of an E. coli cell (Table S4). We assumed that the amino acid limits the growth rate reaction only at very low concentrations. This reflects the low Km values of tRNA ligases. Therefore we fixed Kμ at a low value of 10-5 mM and set μmax to the measured growth rate on glucose of 0.6 h-1.
Steady State and Robustness Analysis
For steady state analysis a parameter set was randomly sampled from the intervals given above. With a specific parameter set the steady state concentrations of e1, e2, m1 and m2 were calculated numerically for each of the two models (complete model and single feedback model). Starting values of the numerical solver were 0.01 mM for m1 and m2, and 10-5 mM for e1 and e2. The convergence criterion was defined as <10-8 change in all variables. To test stability of the steady state we calculated eigenvalues of the Jacobian matrix, and tested if all eigenvalues are negative (λ < -10-5). This procedure was repeated until 5,000 steady states (with different parameter sets) were achieved. Note that both models share the same parameter sets and reach the same steady state flux. In order to estimate robustness of the model against perturbations of the maximal enzyme expression rate , we used a numerical parameter continuation method (Lee et al., 2014). The method is based on finding a connected path of steady state concentrations (xss: steady state concentration vector containing e1,ss, e2,ss, m1,ss, m2,ss), as a parameter, p, is varied. As the system is in steady state it follows that:
| (Equation 9) |
The derivative of with respect to the parameters is also zero:
| (Equation 10) |
After rearranging Equation 10, Equation 11 is obtained:
| (Equation 11) |
which describes the changes in the steady-state concentrations as a kinetic parameter is varied iteratively. The iteration stops when one of the following three stability criteria is no longer fulfilled. 1st criterion: all real parts of the eigenvalues of the system’s Jacobian need to be negative. This implies stability of a steady state. Furthermore, in Equation 11 the inverse of the Jacobian Matrix is required. The inversion is only possible as long as the matrix is regular. Once an eigenvalue reaches zero, the Jacobian becomes singular and matrix inversion is no longer possible. This bifurcation point defines the boundary between the stable and unstable parameter space. In other words: after this point is passed, the system no longer returns to a stable steady state. By checking the eigenvalues of the Jacobian at each step, we make sure that the iteration is terminated when one eigenvalue becomes bigger than λ = -10-5. 2nd criterion: all variables are required to be positive. 3rd criterion: a model is considered unstable when a certain time limit (t > 1 s) is exceeded, which can be the case when numerical errors occur during the numerical integration process. The maximum theoretical enzyme amount in the model was calculated as:
| (Equation 12) |
After rearranging Equation 12 and substituting the upper parameter bound of the maximum protein translation rate (), the maximum theoretical enzyme amount of each enzyme is:
| (Equation 13) |
Considering that the model includes two enzymes, the maximum amount of total enzyme is 0.17 mM, which was defined as the maximal enzyme level (100%).
Quantification and Statistical Analysis
Statistical analysis was done with MATLAB. The statistical details of each experiment can be found in the respective figure caption. For proteomics and metabolomics n represents the number of independent shake flask cultures. In growth assays, n represents the number of independent microtiter plate cultures. For in vitro assays, n represents the number of independent reaction vessels.
Data and Software Availability
Software
All codes for model analysis are available in the Github repository: https://github.com/nfarke/Sander_et_al.
Data Resources
Table S7 contains metabolome data as relative concentrations of 110 intracellular metabolites (related to Figure 1 and S1), proteome data as relative abundance of 1870 proteins (related to Figure 2), 15N labelling and absolute concentrations of amino acids (related to Figures 1 and 3).
Acknowledgments
We thank E. Bremer, K. Drescher, T.J. Erb, J. Guder, E. Noor, K. Kochanowski, F.D. Pascual, J. Plumbridge, R. Thauer, and S. Vonesch for discussions. This work was supported by Deutsche Forschungsgemeinschaft grant LI 1993/2-1, and ERC starting grant 715650. M.K. acknowledges founding of the IMPRS graduate school for environmental, cellular, and molecular microbiology from the Max Planck Society.
Author Contributions
T.S., C.D., and M.K. performed experiments. N.F. and H.L. performed kinetic modeling. T.S., C.D., and H.L performed data analysis. T.G. performed proteome measurements. T.S., N.F., and H.L. co-wrote the manuscript. H.L. directed the project.
Declaration of Interests
The authors declare no competing interests.
Published: January 9, 2019
Footnotes
Supplemental Information includes seven figures and seven tables and can be found with this article online at https://doi.org/10.1016/j.cels.2018.12.005.
Supplemental Information
Metabolome data as relative concentrations of 110 intracellular metabolites (related to Figures 1 and S1), proteome data as relative abundance of 1870 proteins (related to Figure 2), 15N labelling and absolute concentrations of amino acids (related to Figures 1 and 3).
References
- Alam M.T., Olin-Sandoval V., Stincone A., Keller M.A., Zelezniak A., Luisi B.F., Ralser M. The self-inhibitory nature of metabolic networks and its alleviation through compartmentalization. Nat. Commun. 2017;8:16018. doi: 10.1038/ncomms16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri M.G., Bauerle R. Subunit communication in the anthranilate synthase complex from Salmonella typhimurium. Science. 1991;252:1845–1848. doi: 10.1126/science.2063197. [DOI] [PubMed] [Google Scholar]
- Chandra F.A., Buzi G., Doyle J.C. Glycolytic oscillations and limits on robust efficiency. Science. 2011;333:187. doi: 10.1126/science.1200705. [DOI] [PubMed] [Google Scholar]
- Cho B.-K., Barrett C.L., Knight E.M., Park Y.S., Palsson B.O. Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc. Natl. Acad. Sci. USA. 2008;105:19462–19467. doi: 10.1073/pnas.0807227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B.-K., Federowicz S., Park Y.-S., Zengler K., Palsson B.Ø. Deciphering the transcriptional regulatory logic of amino acid metabolism. Nat. Chem. Biol. 2012;8:65. doi: 10.1038/nchembio.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou D., Link H., Fuhrer T., Kochanowski K., Gerosa L., Sauer U. Reserve flux capacity in the pentose phosphate pathway enables Escherichia coli’s rapid response to oxidative stress. Cell Syst. 2018;6:569–578.e7. doi: 10.1016/j.cels.2018.04.009. [DOI] [PubMed] [Google Scholar]
- Chubukov V., Zuleta I.A., Li H. Regulatory architecture determines optimal regulation of gene expression in metabolic pathways. Proc. Natl. Acad. Sci. USA. 2012;109:5127–5132. doi: 10.1073/pnas.1114235109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubukov V., Uhr M., Le Chat L., Kleijn R.J., Jules M., Link H., Aymerich S., Stelling J., Sauer U. Transcriptional regulation is insufficient to explain substrate-induced flux changes in Bacillus subtilis. Mol. Syst. Biol. 2013;9:709. doi: 10.1038/msb.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubukov V., Gerosa L., Kochanowski K., Sauer U. Coordination of microbial metabolism. Nat. Rev. Microbiol. 2014;12:327–340. doi: 10.1038/nrmicro3238. [DOI] [PubMed] [Google Scholar]
- Csonka L.N., Gelvin S.B., Goodner B.W., Orser C.S., Siemieniak D., Slightom J.L. Nucleotide sequence of a mutation in the proB gene of Escherichia coli that confers proline overproduction and enhanced tolerance to osmotic stress. Gene. 1988;64:199–205. doi: 10.1016/0378-1119(88)90335-6. [DOI] [PubMed] [Google Scholar]
- Daran-Lapujade P., Rossell S., van Gulik W.M., Luttik M.A.H., de Groot M.J.L., Slijper M., Heck A.J.R., Daran J.-M., de Winde J.H., Westerhoff H.V. The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc. Natl. Acad. Sci. USA. 2007;104:15753–15758. doi: 10.1073/pnas.0707476104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidi D., Milo R. Lessons on enzyme kinetics from quantitative proteomics. Curr. Opin. Biotechnol. 2017;46:81–89. doi: 10.1016/j.copbio.2017.02.007. [DOI] [PubMed] [Google Scholar]
- Dekel E., Alon U. Optimality and evolutionary tuning of the expression level of a protein. Nature. 2005;436:588–592. doi: 10.1038/nature03842. [DOI] [PubMed] [Google Scholar]
- Doroshenko V.G., Lobanov A.O., Fedorina E.A. The directed modification of Escherichia coli MG1655 to obtain histidine-producing mutants. Appl. Biochem. Microbiol. 2013;49:130–135. doi: 10.7868/s0555109913020037. [DOI] [PubMed] [Google Scholar]
- Fendt S.-M., Buescher J.M., Rudroff F., Picotti P., Zamboni N., Sauer U. Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol. Syst. Biol. 2010;6:356. doi: 10.1038/msb.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer T., Zampieri M., Sévin D.C., Sauer U., Zamboni N. Genomewide landscape of gene–metabolome associations in Escherichia coli. Mol. Syst. Biol. 2017;13:907. doi: 10.15252/msb.20167150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Castro S., Salgado H., Santos-Zavaleta A., Ledezma-Tejeida D., Muñiz-Rascado L., García-Sotelo J.S., Alquicira-Hernández K., Martínez-Flores I., Pannier L., Castro-Mondragón J.A. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016;44:D133–D143. doi: 10.1093/nar/gkv1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerosa L., Kochanowski K., Heinemann M., Sauer U. Dissecting specific and global transcriptional regulation of bacterial gene expression. Mol. Syst. Biol. 2014;9:658. doi: 10.1038/msb.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glascock C.B., Weickert M.J. Using chromosomal lacIQ1 to control expression of genes on high-copy-number plasmids in Escherichia coli. Gene. 1998;223:221–231. doi: 10.1016/s0378-1119(98)00240-6. [DOI] [PubMed] [Google Scholar]
- Goyal S., Yuan J., Chen T., Rabinowitz J.D., Wingreen N.S. Achieving optimal growth through product feedback inhibition in metabolism. PLoS Comput. Biol. 2010;6:e1000802. doi: 10.1371/journal.pcbi.1000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimbs S., Selbig J., Bulik S., Holzhütter H.-G., Steuer R. The stability and robustness of metabolic states: identifying stabilizing sites in metabolic networks. Mol. Syst. Biol. 2007;3:146. doi: 10.1038/msb4100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guder J.C., Schramm T., Sander T., Link H. Time-optimized isotope ratio LC-MS/MS for high-throughput quantification of primary metabolites. Anal. Chem. 2017;89:1624–1631. doi: 10.1021/acs.analchem.6b03731. [DOI] [PubMed] [Google Scholar]
- Gusyatiner, M.M., Ivanovskaya, L.V., Kozlov, Y.I., Lunts, M.G., and Voroshilova, E.B. (2005). DNA coding for mutant isopropylmalate synthase, l-leucine-producing microorganism and method for producing l-leucine. US patent application publication 6,403,342 B1, filed July 10, 2000, and published June 11, 2002.
- Hackett S.R., Zanotelli V.R.T., Xu W., Goya J., Park J.O., Perlman D.H., Gibney P.A., Botstein D., Storey J.D., Rabinowitz J.D. Systems-level analysis of mechanisms regulating yeast metabolic flux. Science. 2016;354 doi: 10.1126/science.aaf2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl J., Kiefer P., Meyer F., Vorholt J.A. Longevity of major coenzymes allows minimal de novo synthesis in microorganisms. Nat. Microbiol. 2017;2:17073. doi: 10.1038/nmicrobiol.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa T., Shimizu H. Recent advances in amino acid production by microbial cells. Curr. Opin. Biotechnol. 2016;42:133–146. doi: 10.1016/j.copbio.2016.04.017. [DOI] [PubMed] [Google Scholar]
- Hofmeyr J.-H.S., Cornish-Bowden A. Regulating the cellular economy of supply and demand. FEBS Lett. 2000;476:47–51. doi: 10.1016/s0014-5793(00)01668-9. [DOI] [PubMed] [Google Scholar]
- Kacser H., Burns J.A. The control of flux. Symp. Soc. Exp. Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- Keseler I.M., Mackie A., Santos-Zavaleta A., Billington R., Bonavides-Martínez C., Caspi R., Fulcher C., Gama-Castro S., Kothari A., Krummenacker M. The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res. 2017;45:D543–D550. doi: 10.1093/nar/gkw1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Kuile B.H., Westerhoff H.V. Transcriptome meets metabolome: hierarchical and metabolic regulation of the glycolytic pathway. FEBS Lett. 2001;500:169–171. doi: 10.1016/s0014-5793(01)02613-8. [DOI] [PubMed] [Google Scholar]
- LaRossa R.A., Van Dyk T.K.V., Smulski D.R. Toxic accumulation of alpha-ketobutyrate caused by inhibition of the branched-chain amino acid biosynthetic enzyme acetolactate synthase in Salmonella typhimurium. J. Bacteriol. 1987;169:1372–1378. doi: 10.1128/jb.169.4.1372-1378.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M.H., Gilbert L.A., Wang X., Lim W.A., Weissman J.S., Qi L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013;8:2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Lee D.E., Lee B.U., Kim H.S. Global analyses of transcriptomes and proteomes of a parent strain and an L-threonine-overproducing mutant strain. J. Bacteriol. 2003;185:5442–5451. doi: 10.1128/JB.185.18.5442-5451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Lafontaine Rivera J.G., Liao J.C. Ensemble modeling for robustness analysis in engineering non-native metabolic pathways. Metab. Eng. 2014;25:63–71. doi: 10.1016/j.ymben.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Li G.-W., Burkhardt D., Gross C., Weissman J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link H., Kochanowski K., Sauer U. Systematic identification of allosteric protein-metabolite interactions that control enzyme activity in vivo. Nat. Biotechnol. 2013;31:357–361. doi: 10.1038/nbt.2489. [DOI] [PubMed] [Google Scholar]
- Milo R., Jorgensen P., Moran U., Weber G., Springer M. BioNumbers—the database of key numbers in molecular and cell biology. Nucleic Acids Res. 2010;38:D750–D753. doi: 10.1093/nar/gkp889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk J.M., Lloyd C.J., Brunk E., Mih N., Sastry A., King Z., Takeuchi R., Nomura W., Zhang Z., Mori H. iML1515, a KnowledgeBase that computes Escherichia coli traits. Nat. Biotechnol. 2017;35:904–908. doi: 10.1038/nbt.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M., Schink S., Erickson D.W., Gerland U., Hwa T. Quantifying the benefit of a proteome reserve in fluctuating environments. Nat. Commun. 2017;8:1225. doi: 10.1038/s41467-017-01242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mülleder M., Calvani E., Alam M.T., Wang R.K., Eckerstorfer F., Zelezniak A., Ralser M. Functional metabolomics describes the yeast biosynthetic regulome. Cell. 2016;167:553–565.e12. doi: 10.1016/j.cell.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien E.J., Utrilla J., Palsson B.O. Quantification and classification of E. coli proteome utilization and unused protein costs across environments. PLoS Comput. Biol. 2016;12:e1004998. doi: 10.1371/journal.pcbi.1004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal B.S., DePonte J., Tuchman M., Malamy M.H. Use of inducible feedback-resistant N-acetylglutamate synthetase (argA) genes for enhanced arginine biosynthesis by genetically engineered Escherichia coli K-12 strains. Appl. Environ. Microbiol. 1998;64:1805–1811. doi: 10.1128/aem.64.5.1805-1811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves M.L., Young B.D., Hosios A.M., Xu Y.-F., Rabinowitz J.D. Pyrimidine homeostasis is accomplished by directed overflow metabolism. Nature. 2013;500:237–241. doi: 10.1038/nature12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisch C.R., Prather K.L.J. The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Sci. Rep. 2015;5:15096. doi: 10.1038/srep15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznik E., Christodoulou D., Goldford J.E., Briars E., Sauer U., Segrè D., Noor E. Genome-scale architecture of small molecule regulatory networks and the fundamental trade-off between regulation and enzymatic activity. Cell Rep. 2017;20:2666–2677. doi: 10.1016/j.celrep.2017.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A., Kochanowski K., Vedelaar S., Ahrné E., Volkmer B., Callipo L., Knoops K., Bauer M., Aebersold R., Heinemann M. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016;34:104–110. doi: 10.1038/nbt.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomburg I., Chang A., Placzek S., Söhngen C., Rother M., Lang M., Munaretto C., Ulas S., Stelzer M., Grote A. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res. 2013;41:D764–D772. doi: 10.1093/nar/gks1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S., Heinrich R. Time hierarchy in enzymatic reaction chains resulting from optimality principles. J. Theor. Biol. 1987;129:189–209. doi: 10.1016/s0022-5193(87)80012-7. [DOI] [PubMed] [Google Scholar]
- Scott M., Gunderson C.W., Mateescu E.M., Zhang Z., Hwa T. Interdependence of cell growth and gene expression: origins and consequences. Science. 2010;330:1099–1102. doi: 10.1126/science.1192588. [DOI] [PubMed] [Google Scholar]
- Singh P.K., Bartalomej S., Hartmann R., Jeckel H., Vidakovic L., Nadell C.D., Drescher K. Vibrio cholerae Combines Individual and Collective Sensing to Trigger Biofilm Dispersal. Curr. Biol. 2017;27 doi: 10.1016/j.cub.2017.09.041. 3359–3366.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbarger H.E. Evidence for a negative-feedback mechanism in the biosynthesis of isoleucine. Science. 1956;123:848. doi: 10.1126/science.123.3202.848. [DOI] [PubMed] [Google Scholar]
- Yanofsky C. Attenuation in the control of expression of bacterial operons. Nature. 1981;289:751–758. doi: 10.1038/289751a0. [DOI] [PubMed] [Google Scholar]
- You C., Okano H., Hui S., Zhang Z., Kim M., Gunderson C.W., Wang Y.-P., Lenz P., Yan D., Hwa T. Coordination of bacterial proteome with metabolism by cyclic AMP signalling. Nature. 2013;500:301–306. doi: 10.1038/nature12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Fowler W.U., Kimball E., Lu W., Rabinowitz J.D. Kinetic flux profiling of nitrogen assimilation in Escherichia coli. Nat. Chem. Biol. 2006;2:529–530. doi: 10.1038/nchembio816. [DOI] [PubMed] [Google Scholar]
- Yuan J., Bennett B.D., Rabinowitz J.D. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat. Protoc. 2008;3:1328–1340. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslaver A., Mayo A.E., Rosenberg R., Bashkin P., Sberro H., Tsalyuk M., Surette M.G., Alon U. Just-in-time transcription program in metabolic pathways. Nat. Genet. 2004;36:486–491. doi: 10.1038/ng1348. [DOI] [PubMed] [Google Scholar]
- Zaslaver A., Bren A., Ronen M., Itzkovitz S., Kikoin I., Shavit S., Liebermeister W., Surette M.G., Alon U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods. 2006;3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- Zelezniak A., Vowinckel J., Capuano F., Messner C.B., Demichev V., Polowsky N., Mülleder M., Kamrad S., Klaus B., Keller M.A. Machine learning predicts the yeast metabolome from the quantitative proteome of kinase knockouts. Cell Syst. 2018;7:269–283.e6. doi: 10.1016/j.cels.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Metabolome data as relative concentrations of 110 intracellular metabolites (related to Figures 1 and S1), proteome data as relative abundance of 1870 proteins (related to Figure 2), 15N labelling and absolute concentrations of amino acids (related to Figures 1 and 3).
Data Availability Statement
Software
All codes for model analysis are available in the Github repository: https://github.com/nfarke/Sander_et_al.
Data Resources
Table S7 contains metabolome data as relative concentrations of 110 intracellular metabolites (related to Figure 1 and S1), proteome data as relative abundance of 1870 proteins (related to Figure 2), 15N labelling and absolute concentrations of amino acids (related to Figures 1 and 3).