

Abstract
The neurotransmitter acetylcholine (ACh) acts as an autocrine growth factor for human lung cancer. Several lines of evidence show that lung cancer cells express all of the proteins required for the uptake of choline (choline transporter 1, choline transporter-like proteins) synthesis of ACh (choline acetyltransferase, carnitine acetyltransferase), transport of ACh (vesicular acetylcholine transport, OCTs, OCTNs) and degradation of ACh (acetylcholinesterase, butyrylcholinesterase). The released ACh binds back to nicotinic (nAChRs) and muscarinic receptors on lung cancer cells to accelerate their proliferation, migration and invasion. Out of all components of the cholinergic pathway, the nAChR-signaling has been studied the most intensely. The reason for this trend is due to genome-wide data studies showing that nicotinic receptor subtypes are involved in lung cancer risk, the relationship between cigarette smoke and lung cancer risk as well as the rising popularity of electronic cigarettes considered by many as a “safe” alternative to smoking. There are a small number of review articles which review the contribution of the other cholinergic proteins in the pathophysiology of lung cancer. The primary objective of this review article is to discuss the function of the acetylcholine-signaling proteins in the progression of lung cancer. The investigation of the role of cholinergic network in lung cancer will pave the way to novel molecular targets and drugs in this lethal malignancy.
Keywords: Lung cancer, acetylcholine, cholinergic, proliferation, invasion, anti-cancer drugs
1. Introduction
Lung cancer is comprised of a spectrum of malignancies. Small cell lung cancer (SCLC; formerly known as oat cell carcinoma) is a neuroendocrine carcinoma and accounts for about 15–20% of all lung cancer cases (Gazdar, Bunn, Minna, 2017). All other forms of lung cancers are included in a heterogeneous group called non-small cell lung cancer (NSCLC). NSCLC includes lung adenocarcinoma (LAC), squamous cell carcinoma (SCC-L), large cell carcinoma (LCC) and neuroendocrine lung carcinoid tumors (Fig. 1; Doroshow & Herbst, 2018; Herbst, Morgensztern, & Boshoff, 2018). LAC originates from the mucus secreting glands in the lungs (Meza, Meernik, Jeon, & Cote, 2015). A substantial number of early published reports involved a type of lung cancer called bronchioalveolar carcinoma (BAC). According to the new WHO classification, BAC is now included in the category of LAC. SCC-Ls usually develop in the tissues comprising the air passages of the lung. Due to cigarette smoking, SCC-L is often preceded by a columnar-to-squamous metaplasia, which lasts for years before developing into an in situ carcinoma (Soldera & Leighl, 2017). Traditionally SCC-L has also been called as epidermoid carcinoma, arising in central large bronchi which join the trachea to the lung.
Figure 1.
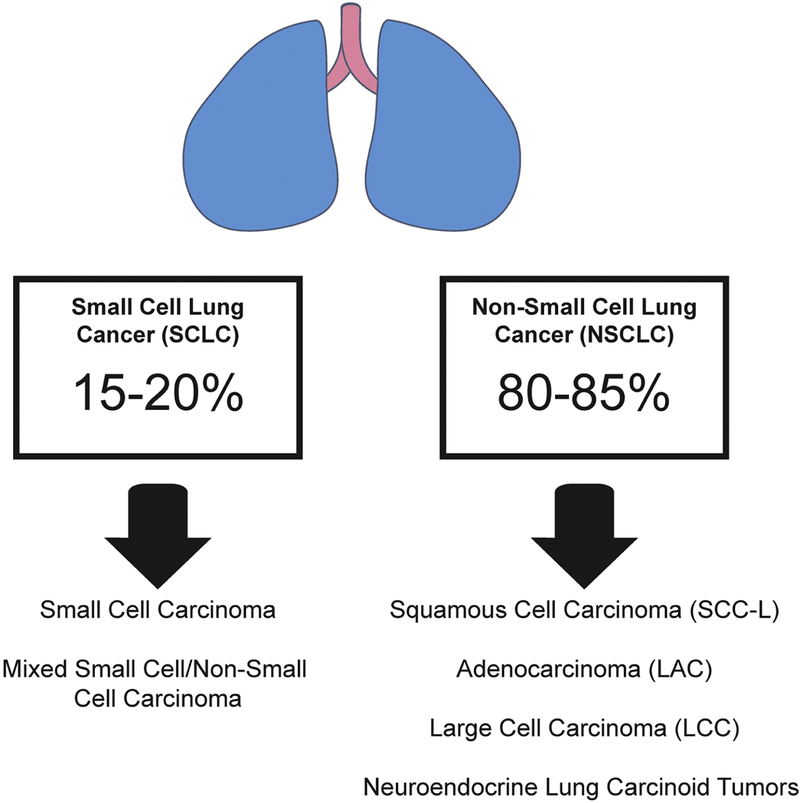
The spectrum of malignancies which comprise lung cancers. Small cell lung cancer (SCLC; also called oat cell carcinoma) comprises the morphologically of tiny cells. All other lung malignancies are put into a heterogenous group termed non-small cell lung cancer (NSCLC). Out of NSCLCs lung adenocarcinoma (LAC) accounts for majority of cases followed by squamous cell carcinoma of the lung (SCC-L). Large cell carcinoma (LCC) and neuroendocrine carcinoid tumors of the lung are relatively less common than LAC and SCC-Ls.
Epidemiological data indicates that cigarette smoking bears a strong etiological association with the development of all histological types of lung cancer (Furrukh, 2013). The association between smoking and lung cancer is stronger with SCLC and SCC-L than with other forms of lung cancer (Khuder, 2001; Khuder & Mutgi, 2001). Nicotine is the addictive component of cigarette smoke. Several lines of evidence show that nicotine accelerates the growth, angiogenesis and metastasis of lung cancers (Dasgupta, Rastogi, et al., 2006; Dasgupta, et al., 2011; Dasgupta, et al., 2009; Davis, et al., 2009; C Heeschen, et al., 2001; C. Heeschen, Weis, Aicher, Dimmler, & Cooke, 2002; Singh, Pillai, & Chellappan, 2011; Spindel, 2016; Zoli, Pucci, Vilella, & Gotti, 2018). Furthermore, nicotine protects lung cancers from cell death induced by chemotherapeutic drugs, oxidative stress and ionizing radiation (Dasgupta, Kinkade, et al., 2006; Egleton, Brown, & Dasgupta, 2008; Jin, Gao, Flagg, & Deng, 2004; Mai, May, Gao, Jin, & Deng, 2003; Maneckjee & Minna, 1994; West, Linnoila, Belinsky, Harris, & Dennis, 2004; Zeidler, Albermann, & Lang, 2007). The growth-stimulatory effects of nicotine are mediated via nicotinic acetylcholine receptors (nAChRs) on lung tumors and the surrounding stroma (S. Wang & Hu, 2018; Zhao, 2016; Zoli, et al., 2018). The endogenous ligand for nAChRs is the neurotransmitter acetylcholine (ACh; Kirkpatrick, et al., 2001; Kummer & Krasteva-Christ, 2014; Mucchietto, Crespi, Fasoli, Clementi, & Gotti, 2016; Niu & Lu, 2014; Saracino, Zorzetto, Inghilleri, Pozzi, & Stella, 2013). Genome-wide association studies (GWAS) identified a genetic component of the association between tobacco components and the development of lung cancer. Data collected from European populations have discovered a locus in the long arm of chromosome 15 (15q24/15q25.1) as the ‘top hit’ for genomic association with lung cancer. The region includes three genes that encode nicotinic acetylcholine receptor subunits α5, α3, and β4-nAChR (CHRNA5, CHRNA3 and CHRNB4; Amos, et al., 2008; Hung, et al., 2008; Improgo, Scofield, Tapper, & Gardner, 2010; P. Liu, et al., 2008; Thorgeirsson, et al., 2008a). Such observations underscore a role for the cholinergic pathway in the development and progression of lung cancer (Gao, Zhang, Breitling, & Brenner, 2016; Tournier & Birembaut, 2011; Wen, Jiang, Yuan, Cui, & Li, 2016; I. A. Yang, Holloway, & Fong, 2013).
Traditionally, ACh is a neurotransmitter and mediates synaptic transmission (Arias, et al., 2009; Barman, Barrett, Boitano, & Brooks, 2016; Kopelman, 1986; Lindstrom, 1996; Phillips, et al., 2010; Picciotto, Higley, & Mineur, 2012). ACh and cholinergic proteins have been detected in non-neuronal tissues like lung, colon, pancreas, skin, gall bladder, and small/large intestine tissues (Beckmann & Lips, 2013; S. A. Grando, 2008; S.A. Grando, Kist, Qi, & Dahl, 1993; Lindstrom, 1997; Wessler, Kirkpatrick, & Racke, 1998). The bronchial epithelium has been shown to synthesize, transport and degrade ACh (Kistemaker & Gosens, 2015; Kummer & Krasteva-Christ, 2014; Proskocil, et al., 2004; Saracino, et al., 2013; Wessler, et al., 1998). These observations suggest that ACh plays a vital role in the lung homeostasis (Pieper, 2012). Published data demonstrate that ACh functions as an autocrine and paracrine growth factor for lung epithelial cells (Proskocil, et al., 2004). It is also a regulator of airway remodeling, airway muscle contraction, mucus secretion and immune functions of the lungs (Fujii, et al., 2017a, 2017b; Koarai & Ichinose, 2018; Kummer & Krasteva-Christ, 2014; Pieper, Chaudhary, & Park, 2007; Proskocil, et al., 2004; Wessler, et al., 1998). ACh is synthesized in the cytoplasm by the enzyme choline acetyltransferase (ChAT) from choline and acetyl-coenzyme A (acetyl-coA; Kummer & Krasteva-Christ, 2014). An alternative route for ACh synthesis is provided by carnitine acetyltransferase (CarAT), which has been detected in the respiratory tract (Fig. 2; Kummer & Krasteva-Christ, 2014; Kummer, Lips, & Pfeil, 2008; Lips, Wunsch, et al., 2007). Subsequently, the ACh is packaged in vesicles by the vesicular acetylcholine transporter (VAChT) and transported to the plasma membrane, where it is released into the extracellular space by exocytosis (Barman, et al., 2016; de Castro, et al., 2009). In addition, a V-ATPase containing proteolipid complex called “mediatophore” also releases ACh from the cytoplasm to the extracellular space (Birman, et al., 1990; Brochier, Israel, & Lesbats, 1993; Brochier & Morel, 1993; Fujii, Takada-Takatori, Horiguchi, & Kawashima, 2012). The released ACh binds to its cognate receptors, namely the nAChRs and muscarinic receptors, on the target cells. The excess ACh is rapidly degraded by acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) to generate choline (Patocka, Kuca, & Jun, 2004; Silman & Sussman, 2005; Xi, Wu, Liu, Zhang, & Li, 2015). The choline is then transported back to the cytoplasm by choline transporter1 (ChT1) for another round of ACh synthesis (Barman, et al., 2016). Apart from ChT1, choline transporter-like proteins 1–5 (CTL1–5) have been shown to have a role in choline uptake and its transport to the cytoplasm in non-neuronal cells (Inazu, 2014; Song, Sekhon, Duan, Mark, & Spindel, 2007; Traiffort, O’Regan, & Ruat, 2013). Similarly, polyspecific organic cationic transporters (OCT1–2 and OCTN1–2) facilitate the bidirectional transport of choline and ACh in lung cells (Lips, et al., 2005; Pochini, Scalise, Galluccio, & Indiveri, 2012, 2013; Pochini, Scalise, Galluccio, Pani, et al., 2012; Tamai, 2013; Volk, 2014).
Figure 2.
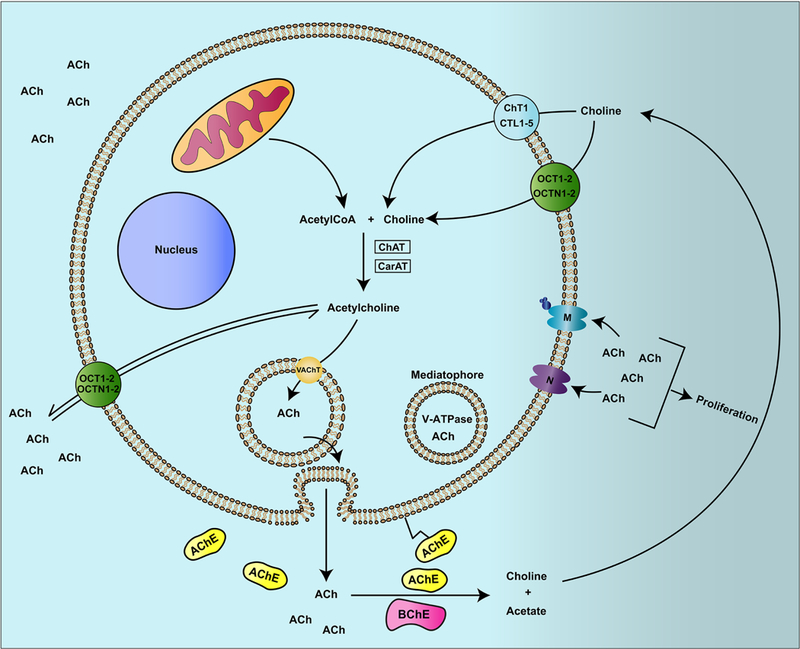
A simplified diagram of the acetylcholine (ACh)-signaling pathway in human lung cells. ACh is synthesized in the cytoplasm by the enzyme choline acetyltransferase (ChAT). In the absence of ChAT, an enzyme carnitine acetyltransferase (CarAT) synthesizes ACh from Choline by adding an acetyl group to it (from acetyl-CoA). The ACh is packaged into vesicles by the vesicular acetylcholine transporter (VAChT) and exocytosed into the extracellular milieu. The ACh so released can bind back to its cognate receptors namely the nicotinic acetylcholine receptor (nAChR) and the muscarinic acetylcholine receptor in an autocrine (or paracrine) manner to recruit downstream cellular signaling pathways. The excess ACh is quickly hydrolyzed by the enzymes acetylcholinesterase (AChE) and butrylcholinesterase (BuCHE) to generate choline. This choline is transported back in the cytoplasm by choline transporter 1 (ChT1). In the absence of ChT1 choline transporter like proteins (CTLP) 1–5 facilitate the uptake of choline back to the cytoplasm for another round of ACh synthesis. Polyspecific organic cations OCTs and OCTNs have the ability to transport ACh and choline bidirectionally in and out of the cell.
A survey of literature shows that several components of the cholinergic pathway are altered in human lung cancers (Dang, Meng, & Song, 2016; S. A. Grando, 2008; Sergei A. Grando, 2014; Improgo, Soll, Tapper, & Gardner, 2013; Spindel, 2016; S. Wang & Hu, 2018). Pioneering studies by Song et al., (2003, 2007, 2008) showed that ACh acts as a growth factor for human SCLC and NSCLC (Song, et al., 2008; Song, Sekhon, Jia, et al., 2003; Song, Sekhon, Lu, et al., 2007). ACh also promotes migration and invasion of human lung cancers (Niu & Lu, 2014; Spindel, 2016; Wessler, et al., 1998). Data from several studies suggest that the ACh signaling pathway in lung cancers is modified to elevate the production of the growth factor ACh. This may include upregulation of ACh, ChAT, VAChT, CTLs and OCTs or a decrease in the function/expression of AChE (Lau, et al., 2013; Niu & Lu, 2014; Spindel, 2016; Wessler, et al., 1998; Zoli, et al., 2018). The present review describes the functional role of the ACh signaling pathway in human lung cancers. We will discuss the feasibility of the cholinergic network as a molecular target for detection and treatment of lung cancer. Out of all components of the cholinergic signaling axis, nAChRs have been most extensively studied in the context of lung cancer (Sergei A. Grando, 2014; Improgo, et al., 2013; Schaal & Chellappan, 2016; Schuller, 2012; Spindel, 2016). The primary reason for this trend may be due to the fact that cigarette smoking is closely correlated with lung cancer (Pesch, et al., 2012; Proctor, 2012). Tobacco components like nicotine, nicotine-derived nitrosamine ketone (NNK), N-Nitrosonornicotine (NNN) and N-Nitrosodiethylamine (DEN) are high-affinity ligands for nAChRs (Schuller, 1992, 2007; Schuller, Jull, Sheppard, & Plummer, 2000; Schuller & Orloff, 1998; Schuller, Plummer, & Jull, 2003). The rising popularity of electronic cigarettes has led to further research in the field of nAChR signaling in the lungs. Many people view electronic cigarettes as a “cessation device” or a “safe alternative” to cigarettes (Dinakar & O’Connor, 2016; M. Hua & Talbot, 2016; Springer, 2014). Furthermore, single nucleotide polymorphisms (SNPs) involving the nAChRs locus chromosome 15q25 region (CHRNA5, CHRNA3, CHRNB4) have been associated with an increased risk of lung cancer in European populations comprised of heavy smokers (Amos, et al., 2008; Hung, et al., 2008; Improgo, et al., 2010; P. Liu, et al., 2008; Thorgeirsson, et al., 2008a). All these factors have led to intense research involving the role of nAChRs in progression of lung cancer. Several state-of-the-art reviews are already published on this subject (Sergei A. Grando, 2014; Improgo, et al., 2013; Schaal & Chellappan, 2016; Schuller, 2012; Spindel, 2016; S. Wang & Hu, 2018; Zoli, et al., 2018). On the other hand, there is a paucity of reviews which contain in-depth knowledge involving the role of other cholinergic proteins in lung cancer. The primary emphasis of this review is to discuss the role of the acetylcholine-signaling pathway in lung cancer. In the light of this rationale, we will only discuss the most recent (past three years) findings involving nicotine-NNK-nAChRs signaling pathway. The potential applications of cholinergic modulators in the detection and treatment of human lung cancer will be summarized. This review will include recently identified nAChR modulators which have potential applications in lung cancer therapy. Finally, we will discuss the signaling pathways underlying the anti-neoplastic activity of cholinergic modulators in lung cancer and normal lung cells.
2. Acetylcholine (ACh)
The presence of ACh in non-neuronal tissues has raised intriguing questions about its role in non-neuronal systems (for review articles, Spindel, 2016; Niu & Lu, 2014; Fujii, et al., 2017a, 2017b; Koarai & Ichinose, 2018; Kummer & Krasteva-Christ, 2014; Pieper, et al., 2007; Proskocil, et al., 2004; Wessler, et al., 1998). ACh has important immunomodulatory functions and triggers both initiation and termination of cytokine synthesis (Fujii, et al., 2017a, 2017b). The synthesis of ACh in immune cells is sensitive to phytohemagglutinin (PHA), lipopolysaccharide and toll-like receptors (TLR), which emphasize its role in immune functions (for reviews see Fujii, et al., 2017a, 2017b; Fujii, Takada-Takatori, & Kawashima, 2012; Kawashima, Fujii, Moriwaki, & Misawa, 2012; Koarai & Ichinose, 2018; Yoo & Mazmanian, 2017).
ACh acts as an autocrine and paracrine growth factor for bronchial epithelial cells (BECs). ACh has been detected in human bronchi, mouth, trachea and pulmonary pleura (Kummer & Krasteva-Christ, 2014; Kummer, et al., 2008; Wessler, et al., 1998). High-performance liquid chromatography (HPLC) analysis revealed that ACh was secreted in cultured BECs isolated from one-year-old monkeys and from humans (Proskocil, et al., 2004). When SV-40 immortalized human BECs were stimulated with cigarette smoke extract elevated production of ACh was observed both in lysates and supernatant (Albano, et al., 2018; Montalbano, et al., 2014; Profita, et al., 2009). This phenomenon is believed to play a role in the context of pro-inflammatory lung diseases like chronic obstructive pulmonary disease (COPD; Profita, et al., 2009). ACh is also generated by pulmonary arteries, human umbilical cord endothelial cells (HUVEC) and human angiosarcoma endothelial cells (HAEND; Haberberger, Bodenbenner, & Kummer, 2000; Kirkpatrick, Bittinger, Nozadze, & Wessler, 2003). ACh released by the endothelium plays a vital role in endothelial calcium signaling, vasodilation/relaxation of arteries and maintenance of vascular homeostasis (Chataigneau, et al., 1999; Wilson, Lee, & McCarron, 2016; M. Zhao, et al., 2015).
Schuller al., (1995) induced lung carcinogenesis in hamsters via subcutaneous injection of nicotine and simultaneous exposure to 60% hyperoxia for 12 weeks (Schuller, McGavin, Orloff, Riechert, & Porter, 1995). Subsequently, they isolated neuroendocrine lung tumor epithelial cells from these tumors. They observed that ACh stimulated the proliferation of these neuroendocrine lung cancer cell lines via nAChR receptors (Schuller, et al., 1995). Song et al., (2003) demonstrated (for the first time) that ACh is produced by a panel of human SCLC cell lines, namely H345, NCI-H69 (H69), NCI-H82 (H82), H1694 and H592 (Song, Sekhon, Jia, et al., 2003; Song, Sekhon, Lu, et al., 2007; Song, Sekhon, Proskocil, et al., 2003; Song & Spindel, 2008). Furthermore, they went on to show that ACh acts as an autocrine growth factor for H82 human SCLC cells (Song, Sekhon, Jia, et al., 2003; Song, Sekhon, Proskocil, et al., 2003). The magnitude of ACh secreted by H82 human SCLC cells was upregulated by neostigmine (Fig. 3A, an antagonist of acetylcholinesterase; see section 7; Song, Sekhon, Jia, et al., 2003) and choline (Song, et al., 2013). In contrast, ACh production in H82 cells was inhibited by vesamicol (Fig. 3B, an antagonist of vesicular acetylcholine transporter; section 3.1; Song, Sekhon, Jia, et al., 2003) and hemicholinium-3 (HC-3; Fig. 3C, an antagonist of choline transporters, see section 3.2; Song, Sekhon, Jia, et al., 2003). The treatment of quiescent SBC3 human SCLC cells with 100 μM-1 mM ACh increased the viability of these cells at 48 and 72 hours (S. Zhang, et al., 2010). ACh activated mitogenic pathways, namely the mitogen-activated protein kinase (MAPK) pathway, intracellular calcium pathway and Akt pathway in H82 human SCLC cells (Song, Sekhon, Lu, et al., 2007). Subsequent studies from their research group showed that homogenates of human SCC-L (isolated from patients) produced an increased amount of ACh relative to adjacent normal lung tissue (Song, et al., 2008). The role of ACh as a growth factor for human lung cancer is further re-enforced by the co-expression of ChAT (the enzyme synthesizing ACh) and muscarinic receptor type 3 (M3R) in human lung cancers (Song, Sekhon, Lu, et al., 2007; Spindel, 2012). Tobacco components like nicotine elevate the levels of ACh in human lung cancer cells (Lau, et al., 2013; Song, et al., 2008). Data from our laboratory show that the streatment of A549, H358 and H650 human LAC cells with 10 nM-10 μM nicotine caused a concentration-dependent increase in the levels of ACh over 24 hours (Lau, et al., 2013; Song, et al., 2008). Subsequently, we analyzed the mitogenic effects of ACh (at levels produced in nicotine-treated cells) in A549 and H358 human LAC cells ACh, using the bromodeoxyuridine (BrdU) assay. BrdU is a thymidine analog which gets incorporated into the DNA of cells entering S-phase (Lau, et al., 2013). We found that ACh (at levels present in the supernatant of nicotine treated LAC cells) induced a 4–4.5 fold increase in the proliferation of A549 and H838 human LAC cells. A relevant aspect of the above-mentioned studies was that they were performed using nicotine concentrations found in the plasma of moderate-heavy smokers (Lau, et al., 2013). Xu et al., (2015) studied the mitogenic effects of ACh in A549 and H1299 human NSCLC cells using the cell counting kit-8 (CCK-8) assay (R. Xu, et al., 2015). They observed that ACh stimulated the proliferation of the above-mentioned cell lines in a concentration-dependent manner from 50–300 μM in 24 hours, with the maximal cell proliferation being observed at 200 μM (N. Hua, et al., 2012; R. Xu, et al., 2015). In contrast, Hua et al., (2007) reported that exogeneous ACh caused no change in cell viability in H1299 cells within the concentration range 0–100 μM at 72 hours. These differences may be attributed to the time points used in these studies. It is probable that ACh produces a rapid proliferative response in human lung cancer cells at 24 hours, which is ablated by 72 hours. The basal concentration of ACh secreted by the human lung cancer cells ranges from 5–50 nM. When the AChE inhibitor neostigmine is added the levels of ACh range between 125–175 nM (Lau, et al., 2013; Song, et al., 2013; Song, Sekhon, Duan, et al., 2007; Song, et al., 2008; Song, Sekhon, Jia, et al., 2003; Song, Sekhon, Lu, et al., 2007; Song, Sekhon, Proskocil, et al., 2003; Spindel, 2012, 2016). Such elevation in ACh levels are observed due to neostigmine-induced blockage of ACh degradation by AChE. Data from Song et al., (2007) estimates the basal ACh content of SCLC tumors (xenografted on athymic mice) as approximately 400 nM (Song, Sekhon, Lu, et al., 2007). Therefore, it is unclear why several of the above-mentioned studies have used unusually high concentrations of ACh for their experiments (N. Hua, et al., 2012; R. Xu, et al., 2015). The reason may have been that they did not use neostigmine for their experiments. Furthermore, ACh is rapidly degraded and a high initial concentration may be required for physiological steady-state levels of ACh in the extracellular milieu.
Figure 3.
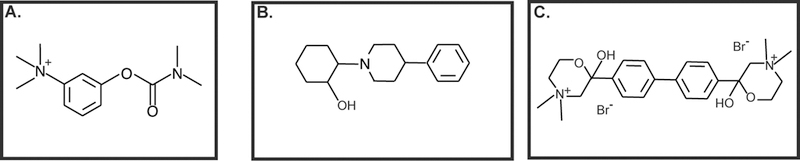
ACh production is sensitive to cholinergic pathway ligands. A. Neostigmine, an antagonist to AChE B. Vesamicol, an inhibitor of VAChT C. Hemicholinum-3, an antagonist of choline transporters.
Apart from being an autocrine growth factor, ACh potently stimulates the adhesion, migration and invasion of human lung cancer cells (Fig. 4). The treatment of SBC3 human SCLC cells with 100 μM ACh caused 2–3-fold increase in adhesion to fibronectin and migration through fibronectin-coated filters (S. Zhang, et al., 2010). The pro-adhesive and pro-migratory effect of ACh involved functional regulation of αvβ1 and α5β1 integrins (S. Zhang, et al., 2010). Xu et al. (2015) found that the ACh displayed robust pro-invasive and pro-migratory activity in human NSCLC cell lines within the concentration range of 100–300 μM (R. Xu, et al., 2015). The highest magnitude of invasion and migration was observed at 200 μM ACh in A549 and H1299 human NSCLC cells (R. Xu, et al., 2015). This data agrees with the observations of Lin et al., (2014) that 100 μM ACh stimulated the invasion (and migration) of two NSCLC cell lines, A549 and L78 (Lin, Sun, Wang, Guo, & Xie, 2014). Real-time PCR analysis showed that 200 μM ACh induced the expression of cytokines IL-1, IL-6, IL-8 from A549 human NSCLC cells. Out of these genes, ACh-induced upregulation of IL-8 was confirmed by ELISA (R. Xu, et al., 2015). The cytokines IL-1, IL-6 and IL-8 induce growth, angiogenesis and metastasis of human NSCLCs (Neufeld & Kessler, 2006; Nishida, Yano, Nishida, Kamura, & Kojiro, 2006; Z. Wang, et al., 2015). Lin et al., (2014) observed that 100 μM ACh increased the expression (and functional activity) of MMP-9, as well as downregulated E-cadherin expression in A549 and L78 human NSCLC cells (Lin, et al., 2014). Both of these signaling events required the phosphoinositol-3 kinase (PI-3 kinase)/Akt signaling pathway in A549 and L78 cells (Lin, et al., 2014; R. Xu, et al., 2015). MMPs play a vital role in the invasion and metastasis of human lung cancers (Gong, et al., 2016; Merchant, et al., 2017). The downregulation of E-cadherin is a marker for epithelial-to-mesenchymal transition (EMT), which confers a migratory phenotype on tumor cells, allowing them to invade into the surrounding stroma, blood vessels and lymph (Nieto, Huang, Jackson, & Thiery, 2016; Tsoukalas, et al., 2017; Xiao & He, 2010). The fact that ACh is upregulating the levels of these proliferative, angiogenic and pro-invasive pathways suggests that it plays an essential role in the progression and metastasis of human NSCLC. A drawback of these experiments is that the authors have used very high concentrations of ACh in their studies (100 μM and 200 μM), which makes it difficult to extrapolate their results to the pathophysiology of NSCLC (Lin, et al., 2014; Song, Sekhon, Lu, et al., 2007; R. Xu, et al., 2015). Once again, a plausible explanation may be the lack of neostigmine (AChE antagonist) in their experiments. ACh-induced proliferation, migration and invasion of A549 and H1299 human NSCLC cells were found to require the M3R (see section 4) which transactivated the epidermal growth factor receptor (EGFR) followed by downstream activation of PI-3 kinase/Akt pathway.
Figure 4.
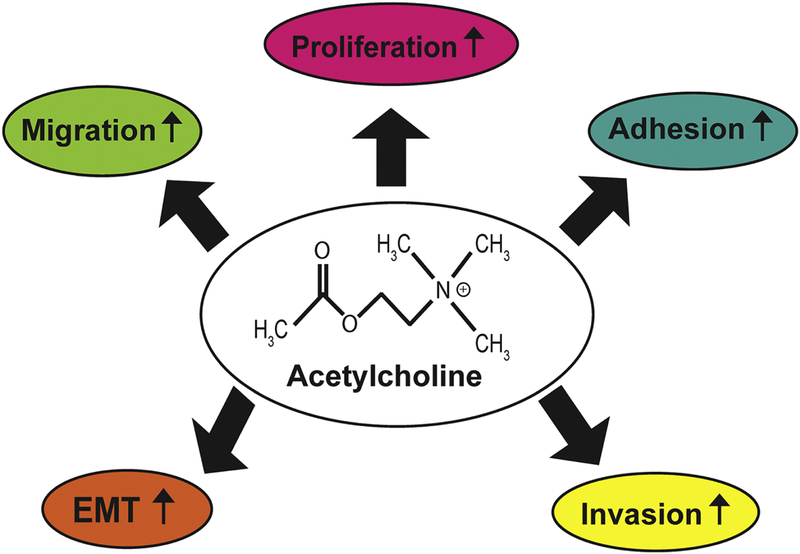
A simplified schematic of the multiple functions of acetylcholine (ACh) in human lung cancer.
The cholinergic signaling axis has been found to play a role in TGF-β1-induced EMT in A549 human NSCLC and immortalized human BECs (K. Yang, et al., 2014). The treatment of A549 human NSCLC cells with 5 ng/ml TGF-β1 caused a 1.5-fold increase in ACh secretion from A549 human NSCLC cells (K. Yang, et al., 2014). TGF-β1-induced EMT was primarily mediated by the muscarinic receptor subtype 1 (M1R) and M3R in human NSCLCs.
2.1. Choline Acetyltransferase (ChAT)
The enzyme ChAT catalyzes the synthesis of ACh from choline. The gene encoding ChAT is comprised of multiple exons, which undergo alternate splicing to generate six transcripts of the gene (Barman, et al., 2016; Oda, 1999). A unique feature of the ChAT gene locus is that the open reading frame of the VAChT gene is located within the first intron of the ChAT gene (Oda, 1999). Out of all the ChAT transcripts, four (denoted as H, R, N1 and N2) translate to a 69 kDa protein which is the predominant form of ChAT. The remaining two transcripts (called M and S) yield two isoforms of ChAT of molecular weights 74 kDa and 82 kDa, respectively (Oda, 1999). In addition, Tooyama and Kimura (2000) have identified a new form of ChAT called pChAT (molecular weight of 50 kDa) generated by alternate splicing and exon skipping of the regions between exon 6 and 9 (Bellier & Kimura, 2011; Nakajima, Tooyama, Yasuhara, Aimi, & Kimura, 2000; Tooyama & Kimura, 2000). A smaller ChAT protein (molecular weight of 27 kDa) has also been characterized. This protein lacks catalytic activity but is believed have a regulatory role on the activity of full-length ChAT (Grosman, Lorenzi, Trinidad, & Strauss, 1995).
Recombinant ChAT (69 kDa) and its isoform (82 kDa) are modified by phosphorylation via protein kinase-C (PK-C), protein kinase CK2, and α-Ca2+/calmodulin-dependent protein kinase II (CaM-kinase; Dobransky, Davis, & Rylett, 2001; Dobransky, Davis, Xiao, & Rylett, 2000; Dobransky, et al., 2004; Dobransky & Rylett, 2003, 2005; Pahud, Bontron, & Eder-Colli, 2001; Schmidt, 1993). SNPs in the ChAT gene have been correlated with nicotine dependence and prospective smoking cessation (R. Ray, et al., 2010).
Immunoreactive ChAT and ChAT activity has been detected in multiple tissues of the human lung epithelium (Krasteva, et al., 2011; Kummer & Krasteva-Christ, 2014; Kummer, et al., 2008; Proskocil, et al., 2004; Song & Spindel, 2008). This includes immortalized normal lung epithelial cells, primary normal human alveolar epithelial cells, normal BECs and SAECs (Table 1). Electron microscopy experiments show that ChAT in airway epithelial cells is localized to the cytosol, nucleus and extracellular fluids like plasma (Kummer & Krasteva-Christ, 2014; Kummer, et al., 2008; Matsuo, et al., 2011). The expression of ChAT on normal airway epithelium is regulated by inflammatory stimuli, cigarette smoke and nicotine (Albano, et al., 2018; Lau, et al., 2013; Montalbano, et al., 2014; Profita, et al., 2009). ChAT is vigorously expressed in immortalized human BECs, SCLC and NSCLC (Akers, et al., 2018; Dasgupta, et al., 2018; Dasgupta, et al., 2016; N. Hua, et al., 2012; Song, Sekhon, Jia, et al., 2003; Song, Sekhon, Proskocil, et al., 2003; Song & Spindel, 2008). Song et al., (2003) demonstrated for the first time, the existence of a functional cholinergic loop in human SCLC. They performed Southern blotting to demonstrate the presence of N, R and S ChAT transcript in a panel of six human SCLC cell lines (Song, Sekhon, Jia, et al., 2003; Song, Sekhon, Proskocil, et al., 2003; Song & Spindel, 2008). Apart from SCLCs, ChAT has been detected in many human LAC and SCC-Ls cell lines (Table 1). The expression of ChAT in human lung cancer cells is sensitive to mitogenic factors like TGF-β1 and nicotine (K. Yang, et al., 2014; Lau, et al., 2013). The treatment of human LAC cell lines with 100 nM nicotine (which is within the range of nicotine concentrations found in the plasma of average smoker) increased ChAT levels, ACh production and cell proliferation (Lau, et al., 2013). Similarly, the multifunctional cytokine TGF-β1 increased ChAT expression and ACh secretion in A549 human NSCLC cells, which in turn correlated with the induction of EMT in these cells (K. Yang, et al., 2014). The aforesaid findings confirm the mitogenic, pro-migratory and pro-invasive activity of ACh in human lung cancer cells. Hence, we surmised that human LAC cells should express higher amounts of ChAT (which in turn would produce increased amounts of ACh) relative to normal bronchial epithelial cells. We performed ELISA and immunoblotting experiments to analyze the expression of ChAT in a panel of human LAC cell lines and in primary normal bronchial epithelial cells (NHBEs). We observed elevated amounts of ChAT in human LAC cells, relative to NHBEs (Dasgupta, et al., 2018; Dasgupta, et al., 2016). We repeated the experiments using two other types of normal lung epithelial cells namely small airway epithelial cells (SAEC) and human pulmonary alveolar epithelial cells (HPAEpiCs) and obtained similar results.
Table 1.
ChAT Expression in normal human bronchial epithelial cells and lung cancer cell lines
| Cell Line | Nature of lung cells |
Expression of ChAT |
References |
|---|---|---|---|
| NHBE | Normal human bronchial epithelium |
+ | 1, 2, 53, 54 |
| SAEC | Small airway epithelial cells |
+ | 1, 2, 53, 54 |
| HLF-1 | Human lung fibroblasts |
+ | 254 |
| 16-HBE | Immortalized human bronchial epithelial cells |
+ | 5, 213, 254 |
| BEP2D | Immortalized human bronchial epithelial cells |
+ | 128 |
| HPAEpics | Human pulmonary alveolar epithelial cells |
+ | 171 |
| H82, H69, H345, H417, H378, H592 |
Small cell lung cancer |
+ | 198, 300, 302 |
| H520, H226, SK- MES, H157 |
Squamous cell carcinoma of the lung |
+ | 53, 54, 128, 198 |
| H460, H1299, A549, H358, H650, H2228, H1975, H1563, H1650, H23, H1944, H838, H1355, PC-9 |
Lung adenocarcinoma |
+ |
1, 2, 53, 54, 128, 171, 198, 359, 376, 377 |
Numerous research studies have demonstrated the presence of ChAT in human SCLC and NSCLC tumors, isolated from patients (Table 2; Dasgupta, et al., 2018; Dasgupta, et al., 2016; Song, et al., 2008; Song, Sekhon, Proskocil, et al., 2003; Song & Spindel, 2008; Spindel, 2012). A noteworthy observation is that the muscarinic receptor M3R is co-expressed with ChAT in a large fraction of SCLC, SCC-Ls and LAC tumors isolated from patients (Song, Sekhon, Lu, et al., 2007; Spindel, 2012). Such co-expression may have important ramifications for the progression of lung cancers. Song et al., (2008) compared the levels of ChAT between human SCC-L tumors and adjacent normal tissue isolated from patients using real-time PCR techniques. They found that ChAT mRNA was virtually undetectable in normal tissue whereas it was highly expressed in the SCC-L tissue (Song, et al., 2008; Spindel, 2012, 2016). They also measured the ChAT levels in a panel of well differentiated to poorly differentiated human SCC-L tumors from patients. They did not find any statistically significant differences in ChAT expression between human well differentiated SCC-Ls and poorly differentiated SCC-L tumors (Song, et al., 2008). Studies in our laboratory examined relative ChAT expression patterns in human LACs tumor tissues (isolated from patients) and adjacent matched normal lung tissue using ELISA, immunoblotting and immunohistochemistry techniques (Lau, et al., 2013). The expression of ChAT in all human LAC tumor tissue was higher than adjacent normal lung tissue. Song et al., (2008) measured the abundance of ChAT in human SCC-L tumors isolated from patients (Dasgupta, et al., 2018; Dasgupta, et al., 2016). They found that about 60% of all the SCC-L tumors expressed ChAT, which underscores the vital function of this protein in the progression of human lung cancers (Song, et al., 2008).
Table 2.
Expression of ChAT in lung cancer tissue adjacent normal tissue (isolated from patients)
| Type of lung tissue | Expression of ChAT | References |
|---|---|---|
| Normal lung tissue | + | 171, 299, 302 |
| Small cell lung cancer | + | 300, 302, 305 |
| Squamous cell lung cancer tissue |
+ | 299, 300, 301, 302 |
| Lung adenocarcinoma tissue |
+ | 171, 301, 302, 305 |
The initial quest for pharmacological ligands of ChAT was aimed at using these compounds for the diagnosis and treatment of neurological diseases like Alzheimer’s disease, related dementias, Down’s syndrome and Lewy body disorders (Barman, et al., 2016; Oda, 1999). An early research study describing small molecule ChAT inhibitors was that of Mehta and Musso (1985) who synthesized water soluble styryloxazine compounds that displayed potent ChAT-inhibitory activity in isolated brain tissue (Mehta, Musso, & White, 1985). Out of these compounds BW813U (Fig. 5A ) is an irreversible non-competitive inhibitor of ChAT, which has been studied extensively in neuronal systems (Mehta, et al., 1985). BW813U does not show any effect on AChE activity. The possible side-effects of ChAT disrupters on the brain and nervous system may be of concern for clinical applications of ChAT-ligands. While early studies aiming to disrupt ChAT activity by inducing morphological lesions in a murine model resulted in a worsened performance in the radial maze test, recent studies using small molecule inhibitors of ChAT activity demonstrated no such disruption in spatial brain functions (Russell, 1988). The administration of BW813U (50 mg/kg bodyweight intraperitoneally) in rats did not impair their performance in the radial maze test (Meck, 2006; Wenk, Sweeney, Hughey, Carson, & Olton, 1986). The authors showed that BW813U did indeed reduce ChAT activity by 66–80%. Such experiments confirm that BW813U does not cause detrimental side-effects on spatial memory and cognition. This may be explained by the fact that the functional activity of ChAT is not the rate-limiting step for ACh synthesis. The inhibition of ChAT activity (close to 90%) had only minimal effect on ACh production. Another plausible explanation may be the compensatory effects by non-cholinergic neural systems.
Figure 5.
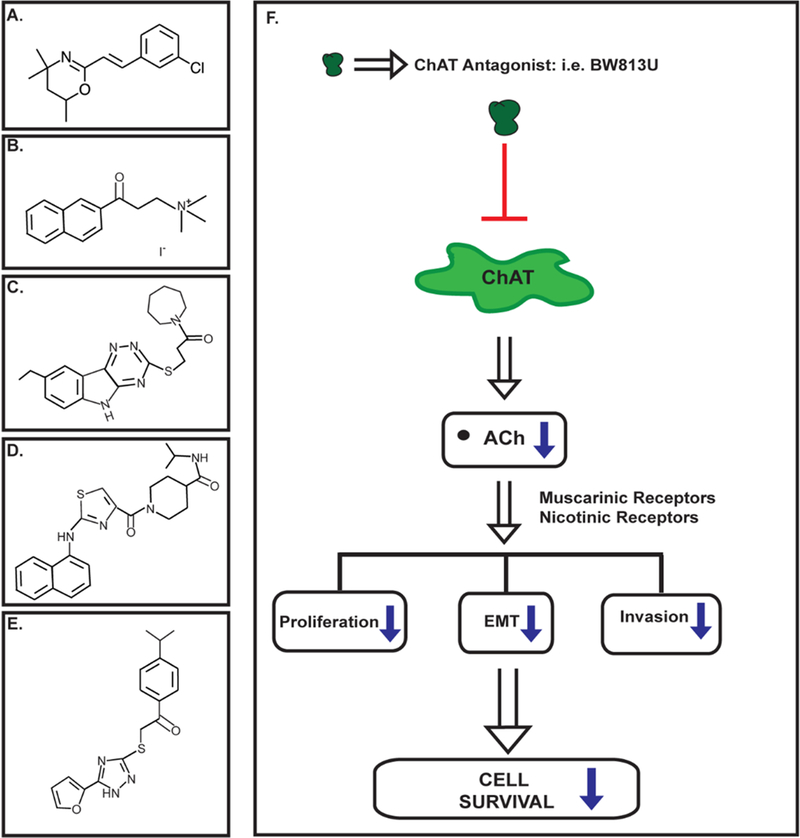
Structure of synthetic ChAT inhibitors. A. BW813U B. alpha-NETA C. ASN07441713 D. BAS11101702 E. BAS03014741 F. Pharmacological disruptors of ChAT may be useful for suppressing the growth of human lung cancers. ChAT antagonists like BW813U (represented by dot) bind and inhibit ChAT enzymatic activity, which diminishes the downstream production of acetylcholine (ACh). The decreased ACh levels translates to reduced growth and progression of human lung cancer cells.
The elucidation of the crystal structure of ChAT has enabled the rational design of potent ChAT inhibitors. Sastry et al., (1988) examined the ability of two compounds namely 2-(α-naphthoyl)ethyltrimethylammonium (α-NETA; Fig. 5B) and its beta isomer to suppress the enzymatic activity of ChAT in isolated organ systems. They observed that both α-NETA and β-NETA functioned as specific ChAT inhibitors (Sastry, Jaiswal, Janson, Day, & Naukam, 1988; Sastry, Jaiswal, Owens, Janson, & Moore, 1988). However, the binding affinity of α-NETA for ChAT (IC50=9 μM) is higher relative to the beta isomer (IC50=76 μM). Both of these compounds displayed no cross-reactivity at carnitine acetyltransferase, cholinesterases, muscarinic and nicotinic receptors. Recent experiments have used high-throughput virtual screening of commercial compound libraries (comprising of about 300,000 compounds) to identify potential ChAT modulators. The hits obtained after the virtual screen were tested for their ability to suppress ChAT activity in vitro (Kumar, Kumar, Langstrom, & Darreh-Shori, 2017) and subjected to molecular docking studies. The authors found that the three compounds namely ASN07441713 (Fig. 5C), BAS11101702 (Fig. 5D) and BAS03014741 (Fig. 5E) to be the most effective inhibitors of ChAT activity. The authors intend to use these compounds as a starting platform for developing a second generation of ChAT ligands which would be used as imaging probes for early diagnosis of neurodegenerative diseases (Kumar, Kumar, Langstrom, & Darreh-Shori, 2017).
The decrease in ChAT expression or disruption in its enzyme activity has been investigated as a possible drug target in the treatment of human lung cancers. The depletion of ChAT by small interfering RNA (siRNA) methodology decreased the viability of H1299 human LAC cells at 48 and 72 hours (N. Hua, et al., 2012). Studies in our laboratory have attempted to assess the anti-cancer activity of the small molecule water-soluble ChAT antagonist BW813U. We observed that BW813U decreased the viability of human SCC-L and LAC cell lines in vitro in a concentration dependent manner (Akers, et al., 2017; Dasgupta, et al., 2018). Subsequently we analyzed the anti-tumor activity of BW813U in athymic mouse models of human LAC. The administration of BW813U (at a dose of 2.5 mg/kg bodyweight, thrice a week by intraperitoneal injection) robustly decreased the growth rate of H838 human LAC tumors xenografted into athymic mice. Most importantly, the treatment of tumor-bearing athymic mice with BW813U did not cause in any gross toxicity or behavioral discomfort to mice; weights and food/water consumption of the BW813U-treated athymic mice were similar to vehicle-treated athymic mice (Akers, et al., 2018). The small molecule ChAT enzyme inhibitors namely ASN07441713, BAS11101702 and BAS03014741 decreased the viability of HEK293 human embryonic kidney fibroblasts at 10 and 50 μM (Kumar, et al., 2017). The growth-inhibitory activity of α-NETA has not been tested in human cell lines. Our ongoing studies are aimed at dissecting out the molecular mechanisms underlying the anti-neoplastic activity of ChAT antagonists like BW813U. We believe that ChAT antagonists (like BW813U) block ChAT enzyme activity, which in turn induces a decline in the secretion of ACh by human lung cancer cells. Traditionally, ACh acts via nAChR and muscarinic receptors to stimulate the proliferation, induction of EMT, migration and invasion of human lung cancer cells. The fall in ACh levels will suppress the abovementioned signaling pathways and abrogate the growth and survival of human lung cancers (Fig. 5F).
Therefore, ChAT disruptors may represent a new generation of drugs relevant for lung cancer therapy. However, these compounds were not tested for their growth-inhibitory activity in human cancer cells.
3. Choline Transporters (ChTs)
3.1. Vesicular Acetylcholine Transporter (VAChT)
The primary function of VAChT is to package ACh (synthesized in the cytoplasm) into vesicles, which store ACh at much higher concentrations than that available in the cytoplasm (Prado, Roy, Kolisnyk, Gros, & Prado, 2013; Usdin, Eiden, Bonner, & Erickson, 1995). These vesicles transport ACh to the cellular membrane where it is released into the extracellular space by exocytosis (Barman, et al., 2016). Several lines of evidence show that VAChT is localized in the vesicle membrane (Y. Liu & Edwards, 1997; Weihe, Tao-Cheng, Schafer, Erickson, & Eiden, 1996). Each molecule of ACh transported by VAChT is exchanged for two vesicular protons, which leads to loading of synaptic vesicles with ACh (Barman, et al., 2016). Molecular cloning and hydrophobic analysis studies have revealed that the structure of VAChT is comprised of twelve transmembrane domains. The carboxy terminus of VAChT contains structural motifs such as di-leucine motif, which are vital for its cellular trafficking and localization (Eiden, Schafer, Weihe, & Schutz, 2004; Erickson, et al., 1996; Prado, et al., 2013; Usdin, et al., 1995).
The detection of VAChT in multiple types of cells in normal lung tissue and lung cancer tissue has led to intense about its possible role in lung maintenance and homeostasis (Song, Sekhon, Proskocil, et al., 2003; Song & Spindel, 2008; Spindel, 2016; Wessler, et al., 1998). Studies in VAChT-mutant mice have indicated role for VAChT in pulmonary inflammation (Lips, Luhrmann, et al., 2007; Pinheiro, et al., 2015). VAChT has been robustly expressed in a diverse array of human lung cancer cell lines (Table 3). VAChT also has been detected in human LAC and SCC-L tissues (isolated from patients) and in matched normal tissue (Table 4). Immunohistochemistry experiments reveal that HUVEC human microvascular endothelial cells express VAChT (Kirkpatrick, Bittinger, Nozadze, Wessler, 2003; Kirkpatrick, et al., 2001). Electron microscopy experiments demonstrate that endothelial VAChT is localized to endocytotic vesicles (Kirkpatrick, et al., 2001). This observation supports the possibility that VAChT is responsible for packaging ACh and transporting it to extracellular space, in a manner analogous to neuronal cells. Shao et al., (2016) explored the effect of autonomic nervous infiltration on the risk and prognosis of patients diagnosed with LAC (Shao, et al., 2016). VAChT was used as a biomarker for cholinergic nerve infiltration (Prado, et al., 2013). They observed that the upregulation of VAChT was correlated with increased risk and increased recurrence in surviving LAC patients (Shao, et al., 2016).
Table 3.
VAChT Expression in normal human bronchial epithelial cells and lung cancer cell lines
| Cell Line | Nature of lung cells |
Expression of VAChT |
References |
|---|---|---|---|
| NHBE | Normal human bronchial epithelium |
+ | 53, 54 |
| SAEC | Small airway epithelial cells |
+ |
53, 54 |
| HPAEpics | Human pulmonary alveolar epithelial cells |
+ |
171 |
| H82, H69, H345, H417, H592, H378 |
Small cell lung cancer |
+ |
300, 302 |
| H520, H226, SK- MES |
Squamous cell carcinoma of the lung |
+ |
53, 54 |
| A549, H358, H650, H2228, H1975, H1563, H1650, H23, H1944, H1355 |
Lung adenocarcinoma |
+ |
53, 54, 171 |
Table 4.
VAChT Expression in lung cancer tissue and adjacent normal tissue (isolated from patients)
| Type of lung tissue | Expression of VAChT |
References |
|---|---|---|
| Normal lung tissue | + | 171, 299, 302 |
| Squamous cell lung cancer tissue |
+ | 53, 54, 299 |
| Lung adenocarcinoma tissue |
+ | 171 |
Studies in our laboratory have analyzed the effect of nicotine on VAChT levels in human LAC cell lines. The treatment of A549 and H358 human LAC cell lines with 100 nM nicotine (which is within the range of nicotine concentrations found in the plasma of an average smoker) elevated the magnitude of VAChT. We observed that VAChT was robustly expressed in human LAC tumors (isolated from patients) and adjacent normal lung tissue (Lau, et al., 2013). Furthermore, Song et al., (2008) detected the presence of VAChT mRNA in human SCC-L tumor tissue (Song, et al., 2008). They performed VAChT immunohistochemistry on 31 SCC-L tumors and observed that VAChT is robustly expressed by about 65% of the tumors (Song, et al., 2008).
The vesicular transporter activity of VAChT is blocked by the non-competitive antagonist, vesamicol. The growth-inhibitory activity of vesamicol has been studied in both SCLCs and NSCLCs (Table 5). Song et al., (2003) demonstrated that vesamicol suppressed the viability of asynchronous H82 human SCLC cells (in a concentration-dependent manner) at nine days and twelve days post treatment (Song, Sekhon, Jia, et al., 2003). Our laboratory examined the growth-suppressive activity vesamicol in a panel of human LAC cells (Table 5). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assays revealed that vesamicol potently decreased the viability of nicotine-treated human LAC cell lines (Dasgupta, et al., 2018; Dasgupta, et al., 2016; Lau, et al., 2013). Caspase-3 activity and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) apoptosis assays revealed that vesamicol induced 2.5–3.0-fold apoptosis in nicotine-treated A549 and H358 human LAC cells. The administration of vesamicol (at a dose of 50 mg vesamicol/kg food) decreased the growth rate of nicotine-treated A549 human LAC tumors xenografted in athymic mice (Lau, et al., 2013). Although, Song et al. (2003) showed the growth-inhibitory effects of vesamicol in H82 human SCLC cells in cell culture, they did not examine the anti-neoplastic activity of vesamicol in vivo (Song, Sekhon, Jia, et al., 2003). SCLCs have a robust cholinergic signaling axis and occur exclusively in smokers. Therefore, it may be probable that vesamicol will display anti-neoplastic activity in athymic mouse models of SCLC.
Table 5.
Growth-suppressive activity of VAChT inhibitor vesamicol in human lung cancer cell lines and athymic mouse models
| Cell Line | Nature of lung cells |
Growth- inhibitory Activity of Vesamicol |
Model used | References |
|---|---|---|---|---|
| H82 | Small cell lung cancer |
+ | Cell culture | 300, 306 |
| A549 | Lung adenocarcinoma |
+ | Cell culture, athymic mice |
171 |
| H1975, H838 H358 |
Lung adenocarcinoma |
+ | Cell culture | 53, 54, 171 |
3.2. ChT1, CTLs, OCTs and OCTNs in lung cancer
Choline plays a vital role in cellular homeostasis and survival. Mammalian cells utilize choline for the synthesis of membrane phospholipids namely phosphatidylcholine, sphingomyelin and betaine (Farine, Niemann, Schneider, & Butikofer, 2015; Lagace & Ridgway, 2013; Ridgway, 2013). Additionally, choline is the precursor for the synthesis of ACh, which acts as an autocrine and paracrine growth factor for bronchial epithelium, SCLCs, SCC-Ls, and LACs. (S. A. Grando, 2008; Kummer & Krasteva-Christ, 2014; Kummer, et al., 2008; Proskocil, et al., 2004; Song & Spindel, 2008; Wessler, et al., 1998; Lau, et al., 2013; Song, et al., 2008; Song, Sekhon, Proskocil, et al., 2003; Song & Spindel, 2008; Spindel, 2012, 2016). The choline transport system in the lung is mediated by three major families of proteins: i) High affinity choline transporter 1 (ChT1/SLC5A7; Okuda and Haga, 2003), ii) Choline transporter like proteins (CTL1–5; Inazu, 2014; Traiffort, et al., 2013) with moderate affinity towards choline, and iii) Polyspecific organic transporters (OCT1–3/SLC22A1–2) and carnitine/cation transporters (OCTN1 and OCTN2; Pochini, et al., 2013; Tamai, 2013; Volk, 2014). OCTN3 has been only detected in mouse tissues (Tamai, et al., 2000). OCT1 and OCT2 also transport ACh in BECs (Kummer, et al., 2006; Lips, et al., 2005). Pochini et al., (2012) investigated the ability of human OCTN1 to transport ACh using proteoliposomal model systems. They developed human OCTN1 reconstituted proteoliposomes and analyzed their ability to mediate the uptake, transport and efflux of ACh. The results from their experiments revealed that OCTN1 (in proteoliposomal preparation) efficiently catalyzed the bidirectional transport of ACh, and this process was asymmetrically regulated by sodium ions (Pochini, Scalise, Galluccio, & Indiveri, 2012; Pochini, Scalise, Galluccio, Pani, et al., 2012). However, these studies have not been extended to normal lung or lung cancer cells.
Five types of CTL like proteins have been characterized in humans (Inazu, 2014; Traiffort, et al., 2013). Studies in rat models indicate that the major form of CTL1 originates from a 3.5 kb transcript and is present in the diverse regions of the brain and spinal cord. A minor form of CTL1 (arising from a 5 kb transcript) is detected in the colon, lung and spinal cord (O’Regan, et al., 2000; Traiffort, et al., 2013). The CTL1 protein has lower affinity for choline than ChT1. However, both CTL1 and ChT1 are inhibited by HC-3 (Inazu, Takeda, & Matsumiya, 2005; Kouji, et al., 2009; Uchida, et al., 2009). The lung expresses two isoforms of CTL2, namely CTL2-P1 and CTL2-P2. Human CTL-P1 does not participate in choline transport. However, CTL2-P2 is a functional choline transporter (Kommareddi, et al., 2010). CTL2 and CTL4 have been shown to transport choline in human lung cells (Nakamura, et al., 2010; Song, et al., 2013). No studies have addressed the choline transport properties of CTL3 and 5. The affinity of OCT1 and OCT2 for choline is lower than CTL1 and CTL2 (Inazu, 2014). OCT3 does not have the ability to transport choline (Inazu, 2014). Both CTL1 and CTL2 play a vital role in physiological functions of the lung like surfactant production, cell growth and cell repair (Traiffort, et al., 2013). Choline transporters play a vital role in multiple lung diseases, like infant respiratory distress syndrome, drug-induced interstitial lung diseases, transfusion-related lung injury and lung cancer (Curtis, et al., 2010; Greinacher, et al., 2010; Inazu, 2014; Nakamura, et al., 2010; Traiffort, et al., 2013).
Data from several research laboratories have shown that cancer cells have enhanced choline uptake and transport in comparison to normal cells (Inazu, 2014; Ingoglia, et al., 2015; Salomon, et al., 2014; Tamai, 2013; Volk, 2014). The reason for this may be due to the vital role of choline transport in cell growth, membrane integrity and cell repair process (Glunde, Bhujwalla, & Ronen, 2011; Glunde, Penet, Jiang, Jacobs, & Bhujwalla, 2015; Mori, Wildes, Takagi, Glunde, & Bhujwalla, 2016). The enhanced uptake of choline has formed the basis of imaging of tumors by magnetic resonance imaging (MRI) and positron emission tomography (PET; Glunde, et al., 2015; Hara, Bansal, & DeGrado, 2006). Radiolabeled choline transporter ligands like tritiated HC-3, 18F-Choline, 18F-FA-4 and 11C-pipzA-4 (Fig. 6) have been investigated as imaging agents for a variety of human tumors including lung cancer (Challapalli & Aboagye, 2016; Gilissen, et al., 2003; M. Li, et al., 2013; Ramirez de Molina, et al., 2007). CTLs, OCTs and OCTNs are robustly expressed by multiple human lung cancer cell lines (Table 6). CTL1 and CTL2 mediate choline transport in human SCLCs and LACs (Inazu, Yamada, Kubota, & Yamanaka, 2013; Nakamura, et al., 2010). The choline-transport activity of CTL4 has been only investigated in SCLC cells (Song, et al., 2013). Song et al., (2013) studied expression of CTLs in two SCLC tumors isolated from patients. Both the tumors expressed CTL1–5 (Song, et al., 2013). OCT1–2 and OCTN1–2 were detected in lung tumors (isolated from patients) and matched normal lung tissue samples (Table 7; More, et al., 2010; T. Wang, et al., 2007). However, no clear-cut trends were obtained in the expression pattern of OCTs or OCTNs between normal and lung tumor tissue.
Figure 6.
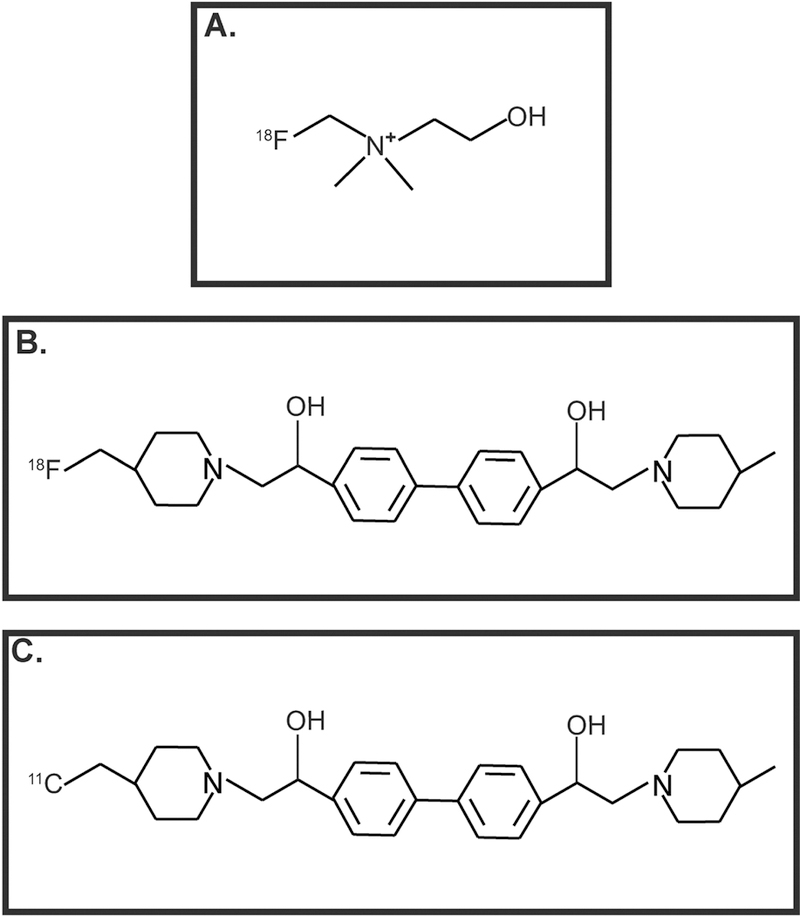
Choline-transporter-based imaging agents used in PET scans. A. 18F-choline B. 18FA-4 C. 11C-pipzA-4
Table 6.
The expression of choline transporters in normal human bronchial epithelial cells and lung cancer cell lines
| Cell Line | Nature of lung cells |
ChT1 | CTL1- 5 |
OCT1-2 | OCTN1 | References |
|---|---|---|---|---|---|---|
| NHBE | Normal human bronchial epithelium |
+ | 53, 54 | |||
| SAEC | Small airway epithelial cells |
+ | 53, 54 | |||
| HPAEpics | Human pulmonary alveolar epithelial cells |
+ | 171 | |||
| H146, H417, H82 |
Small cell lung cancer |
+ | 297 | |||
| H69, H345 |
Small cell lung cancer |
+ | + | 136, 297 | ||
| H592 | Small cell lung cancer |
CTL1- 3, CTL5 |
297 | |||
| H1694 | Small cell lung cancer |
+ | 297 | |||
| H520, H226 |
Squamous cell carcinoma of the lung |
+ | 53, 54, 171 | |||
| H1703 | Squamous cell carcinoma of the lung |
OCT1, OCT2 |
214 | |||
| H1975, H1563, H1734, H1650, H23, H650, H358 |
Lung adenocarcinoma |
+ | 53, 54, 171 | |||
| H838 | Lung adenocarcinoma |
+ | OCT1, OCT2 |
53, 54 | ||
| A549 | Lung adenocarcinoma |
+ | CTL1 | OCT1 | OCTN1, OCTN2 |
171, 335 |
| H460 | Lung adenocarcinoma |
OCT1, OCT2 |
214 | |||
| SPC-A-1, H1299 |
Lung adenocarcinoma |
CTL1 | OCTN1, OCTN2 |
335 | ||
| H441 | Lung adenocarcinoma |
OCT1, OCT2 |
OCTN1, OCTN2 |
273 |
Blanks indicate Not Determined (ND)
Table 7.
Expression of choline transporters in lung cancer tissue and adjacent normal tissue (isolated from patients)
| Type of lung tissue |
ChT1 | CTL1- 5 |
OCT1- 2 |
OCTN1- 3 |
References |
|---|---|---|---|---|---|
| Normal lung tissue |
+ |
CLT-1 | OCT1 | OCTN1 | 241, 299, 300, 335 |
| Small cell lung carcinoma tissue |
+ | + | + | 302, 297 | |
| Squamous cell lung cancer tissue |
+ | OCT1, OCT2 |
299, 302 | ||
| Lung adenocarcinoma tissue |
+ | CTL1 | OCTN1, OCTN2 |
302, 335 |
Blanks indicate Not Determined (ND)
The observation that neoplastic cells have higher choline metabolism and uptake (relative to normal cells) has been exploited to develop innovative therapeutic approaches in lung cancer. The depletion of CTL4 by siRNA methodology suppressed the proliferation of SCLC cells (Song, et al., 2013). Inazu et al., (2013) found that the transfection of CTL1-siRNA decreased the viability and enhanced apoptosis of H69 human SCLC cells (Inazu, et al., 2013). Similarly, CTL1-siRNA displayed small yet significant anti-proliferative activity in H82 human SCLC cells (Song, Mark, & Spindel, 2010). H82 is a variant human SCLC cell line, whereas H69 is a classical human SCLC cell line. The variant SCLC cells are associated with accelerated doubling time, greater invasive phenotype and lower sensitivity to growth-inhibitory agents than classical SCLC cell lines like H69 (Broers, et al., 1988; Gazdar, Carney, Nau, & Minna, 1985). This may explain the lower growth-suppressive activity of CTL1-siRNA in H82 relative to H69 human SCLC cells.
The CLT1 inhibitor HC-3, shows growth-inhibitory activity in human SCLC and LAC cell lines (Table 8). Data from our laboratory indicates that 50 μM HC-3 induces robust apoptosis in nicotine-treated H1975 and H838 human LAC cells over 24 hours (Dasgupta, et al., 2018; Dasgupta, et al., 2016). We did not observe any apoptotic activity of HC-3 in untreated human LAC cells. Our data is divergent relative to the results of Inazu et al., (2013) who treated H69 human SCLC cells with 1 mM HC-3 for 2 days (Inazu, et al., 2013). Subsequently, they measured cell viability by a luminescence ATP detection assay. They found that 1 mM HC-3 decreased the viability of H69 cells at 48 hours (Inazu, et al., 2013). Such differences in results may be due to the fact the two studies involved two different histological types of lung cancer (LAC versus SCLC). Inazu et al., (2013) explored the apoptotic activity of HC-3 using caspase-3/7 activity assay and immunofluorescence. They observed that HC-3 induced 2–2.5-fold increase in apoptosis in H69 cells. DAPI staining of HC-3 treated H69 cells showed morphology typical of apoptotic cells including condensed nuclei and apoptotic bodies (Inazu, et al., 2013). Such data indicate that multiple dosing with high concentrations of the drug may be needed for the pro-apoptotic activity of HC-3 in asynchronous human SCLC cells. This is confirmed by the data of Wang et al., (2007) who observed that HC-3 induced cell cycle arrest in A549 and SPC-A-1 human LAC cells. They added 200 μM HC-3 daily for eight days, changing the drug daily and measured S-phase entry by BrdU assay (T. Wang, et al., 2007). They found that HC-3 decreased S-phase entry of A549 and SPC-A-1 cells by approximately 64% relative to the vehicle-treated cells. Choline uptake blockers like phenoxybenzamine (PbA), tetraethylammonium (TEA) and norepinephrine (NEP) block choline uptake and proliferation of A549 and SPC-A-1 LAC cells (T. Wang, et al., 2007). Likewise, organic cationic drugs like quinine, quinidine, desipramine, imipramine, clomipramine, fluvoxamine, diphenhydramine, paroxetine, reboxetine, citalopram and fluoxetine inhibit choline uptake and the viability of H69 human SCLC cells (T. Wang, et al., 2007). Such results seem to emphasize the importance of choline metabolism regulating the viability of lung cancer cells. This is confirmed by the data of More et al., (2010) showing a role for OCT1 and OCT2 in enhancing the cytotoxicity of picoplatin in a panel of human LAC cell lines (More, et al., 2010). The overexpression of OCT1 and OCT2 (but not OCT3) augmented the cytotoxicity of picoplatin in both cell culture and mice models of human LAC. Cimetidine, a small molecule inhibitor of OCT1 and OCT2, reduced the growth-inhibitory activity of picoplatin in human LAC cell lines (More, et al., 2010). This appears to be contradictory to the results obtained with ChT1 and CTLs antagonists. However, the effect of OCT1 and OCT2 on the anti-cancer activity of picoplatin is mediated by their effects on the uptake of the drug and increasing the formation of intracellular DNA-picoplatin adducts (More, et al., 2010). Therefore, cationic transporters like OCT1 and OCT2 also regulate drug trafficking and biodistribution in lung cancer cells, apart from their role in choline uptake and transport.
Table 8.
The choline transporter antagonist hemicholinium-3 blocks the growth of lung cancer cell lines in vitro
| Cell Line | Nature of lung cells |
Growth- inhibitory Activity of Hemicholinium- 3 |
Model used | References |
|---|---|---|---|---|
|
H69, H82 |
Small cell lung cancer |
+ |
Cell culture | 136, 300, 306 |
| A549, SPC- A-1 |
Lung adenocarcinoma |
+ | Cell culture | 335 |
| H1975, H838 | Lung adenocarcinoma |
+ | Cell culture | 53, 54 |
Blanks indicate Not Determined (ND)
4. Muscarinic Receptors
The biological activity of ACh is mediated by its binding to the nicotinic acetylcholine receptors and the muscarinic receptors on target cells (Barman, et al., 2016). Muscarinic receptors belong to the superfamily of G-protein-coupled receptors. These receptors are comprised of seven hydrophobic domains and an intracytoplasmic loop between hydrophobic domains 5 and 6 (Wess, 1996). This loop is considered to be responsible for the G-protein coupling functions of the muscarinic activity. Five subtypes of the muscarinic receptor (M1R to M5R) have been identified in mammalian cells (Barman, et al., 2016; Wess, 1996). These receptors may be broadly divided in two categories, based on their G-protein coupling activity (D. A. Brown, 2018; Kruse, et al., 2014; Zenko & Hislop, 2018). The M1R, M3R and M5R are coupled to Gq-type proteins and activate phospholipase C to recruit phosphoinositol triphosphate-signaling cascade. The M2R and M4R receptors are coupled to pertussis toxin-sensitive Gi/0 proteins which inhibit adenylyl cyclase activity (D. A. Brown, 2018; Kruse, et al., 2014; Zenko & Hislop, 2018). The muscarinic receptor system regulates several lung functions including airway remodeling, airway smooth muscle contraction, inflammation and wound healing (Kistemaker & Gosens, 2015; Roth, 2015).
Immunoblotting and RT-PCR reveal that muscarinic receptor subtypes are expressed in human lung cancer cell lines (Table 9). However, research studies aimed at analyzing the role of muscarinic receptors in lung cancer have yielded divergent results, which may be attributed to the nature of the muscarinic ligand (Figueroa, Griffin, & Ehlert, 2009) used to activate the muscarinic receptors on human lung cancer cells. Early studies revealed that the activation of muscarinic receptors (by carbachol) inhibited cell cycle progression and voltage-dependent calcium influx in SCC-9 human SCLC cells (Williams, 2003; Williams & Lennon, 1990, 1991). In contrast, Song et al., (2003) observed that carbachol at concentrations of 1 μM and 10 μM increased the viability of H82 human SCLC cells at nine and twelve days post-treatment (Song, Sekhon, Jia, et al., 2003). Such variations in the results could be due to the fact that carbachol activates nicotinic and muscarinic receptors. Therefore, the biological effects of carbachol is dependent on the cell membrane specific expression (and abundance) of nAChRs and muscarinic receptors on SCLC cells (Spindel, 2012, 2016). The expression pattern of nAChRs and muscarinic receptors varies across diverse human SCLC cell lines and such differences may be playing a central role in mediating the observed differences in response to carbachol treatment.
Table 9.
Expression of muscarinic receptors in normal human bronchial epithelial cells and lung cancer cell lines
| Cell line | Nature of cells | Nature of muscarinic receptor | Referenc e |
||||
|---|---|---|---|---|---|---|---|
| M1R | M2R | M3R | M4R | M5 R |
|||
| BEP2D | Immortalized normal human bronchial epithelium |
+ | 128 | ||||
| 16-HBE | Immortalized normal human bronchial epithelium |
+ |
5, 213 |
||||
| HLF | Human lung fibroblasts |
+ | 177, 254 | ||||
| SCC-15, SCC-9 |
Small cell lung cancer |
+ | 257, 346- 348 |
||||
| SBC3 | Small cell lung cancer |
+ | 31, 369 | ||||
| NCI-H146 | Small cell lung cancer |
+ | 346 | ||||
| NCI-H209 | Small cell lung cancer |
+ | 346 | ||||
| H69, H82, H345, H592, H417, H1694 |
Small cell lung cancer |
+ | + | + | + | + | 300, 301 |
| H398 | Small cell lung cancer |
+ | + | 300 | |||
| H157 | Squamous cell carcinoma |
+ | + | + | + | + | 128 |
| L78, SPCA-1 | Squamous cell carcinoma |
+ | 177 | ||||
| A549, PC9 | Lung adenocarcinoma |
+ | + | + | + | + | 128, 177, 376, 377 |
| GLC82 | Lung adenocarcinoma |
+ | 177 | ||||
| H1299, H460 | Lung adenocarcinoma |
+ | + | + | + | + | 128 |
(Blanks indicate Not Determined (ND)
Carbachol upregulated the invasive phenotype of A549 human NSCLC cells by inducing EMT. The treatment of A549 cells with 1 μM carbachol produced a dose-dependent and time-dependent decrease of the epithelial junction protein E-cadherin with concomitant increase in the levels of mesenchymal proteins namely vimentin and α-smooth muscle actin. These results were confirmed in immortalized BECs (K. Yang, et al., 2014). Apart from inducing EMT, carbachol upregulated the expression of MMP-9 in BECs (K. Yang, et al., 2014). MMPs degrade the basement membrane to enable the invasion of tumor cells in the blood and lymph thereby facilitating distant metastasis of tumors (Gong, et al., 2016; Merchant, et al., 2017).
Yang et al., (2014) examined the role of the muscarinic receptor pathway in carbachol-induced EMT in A549 human NSCLC and immortalized bronchial epithelial cells. Carbachol stimulated the production of TGF-β1 and MMP-9 from A549 human NSCLC cells (K. Yang, et al., 2014). Carbachol-induced EMT and MMP-9 expression was reversed by the M1R antagonist pirenzepine (Fig. 7A) and M3R antagonist 4-diphenyl-acetoxy-N-methyl-piperidine (4-DAMP; Fig. 7B) indicating that these biological activities of carbachol were mediated by M1R and M3R. The authors further showed that carbachol induced EMT required downstream activation of the Smad and ERK pathways (K. Yang, et al., 2014). Conflicting reports exist about the biological role of M2R in lung cancer. The activation of M2R inhibits proliferation of H1694 human SCLC cells (Song, Sekhon, Duan, et al., 2007). These findings are in alignment with the fact that M2R staining in poorly differentiated human SCC-L tissue (isolated from patients) is decreased relative to adjacent normal tissue (Song, et al., 2008). Data by Zhao et al., (2015a and b) reveals that the activation of muscarinic receptors by pilocarpine induces proliferation, EMT, migration and invasion of human NSCLCs via the MAP kinase and the Akt pathway (Fig. 8A; Q. Zhao, X. Gu, et al., 2015; Q. Zhao, J. Yue, et al., 2015). These biological effects of pilocarpine are antagonized by M2R-short-hairpin RNA (shRNA; Q. Zhao, J. Yue, et al., 2015). An innovative aspect of this study was that the authors demonstrated the growth-inhibitory effects of M2R-shRNA in athymic mouse model (Q. Zhao, X. Gu, et al., 2015). A549 cells transfected with control (non-targeting)-shRNA or transfected with M2R-shRNA were subcutaneously injected in the flank of athymic mice. After four weeks, the authors observed that the growth rate of the A549-M2R-shRNA tumors were significantly lower (P≤0.05) than A549-control-shRNA tumors (Q. Zhao, X. Gu, et al., 2015). The results obtained from M2R-shRNA were verified by using the synthetic M2R antagonist namely methoctramine (Fig. 9A). Methoctramine suppressed the viability of A549 and PC9 cells in a concentration dependent manner over 72 hours. Furthermore, methoctramine displayed anti-neoplastic activity in athymic mouse models of human NSCLCs (Q. Zhao, X. Gu, et al., 2015; Q. Zhao, J. Yue, et al., 2015).
Figure 7.
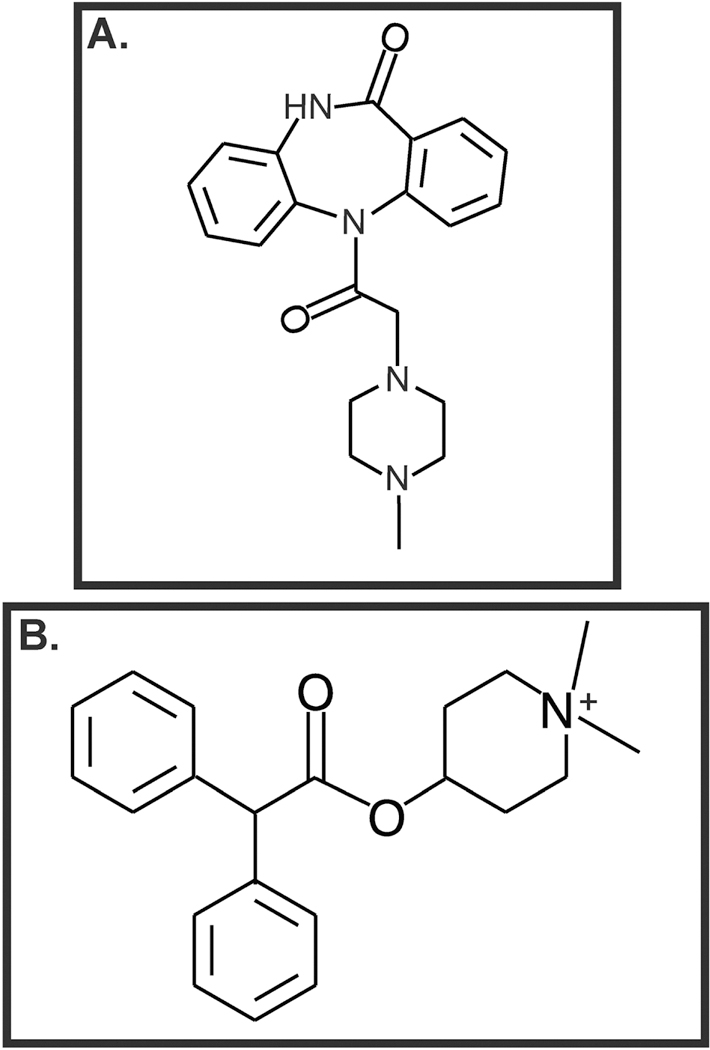
Muscarinic receptor antagonists display growth suppressive activity in human lung cancers. A. Pirenzepine B. 4-DAMP
Figure 8.
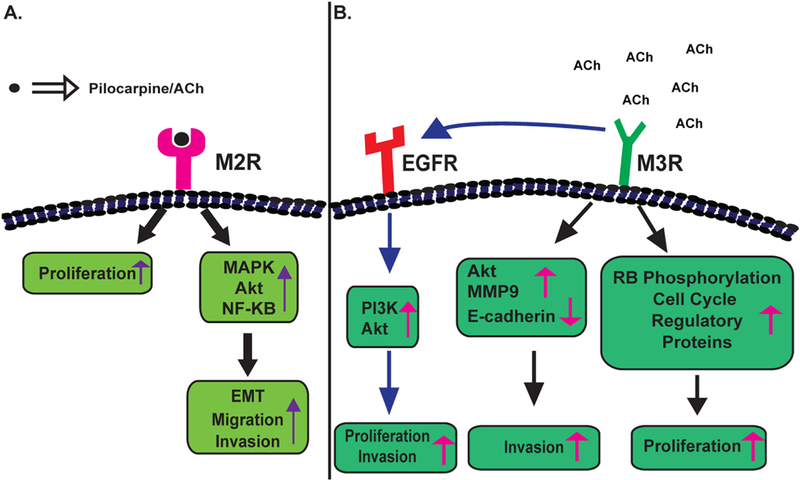
A flow chart summarizing muscarinic receptor-induced signaling pathways. A. Signal-transduction pathways downstream of M2R in human lung cancers. B. Cellular signaling pathways underlying the proliferative activity of M3R. EMT: Epithelial to mesenchymal transition
Figure 9.
Structure of muscarinic receptor antagonists analyzed for their growth-inhibitory activity in human lung cancer A. Methoctramine B. AFDX-116 C. Darifenacin D. Tiotropium E. R2HBJJ F. R2-PHC G. J-115311
A majority of research papers have explored the role of M3R in the proliferative and pro-invasive effects of ACh in lung cancer (Fig. 8B). M3R is robustly expressed on several SCLC and NSCLC cell lines (Table 9). The depletion of M3R by siRNA abolished ACh-induced cell growth and elevation of intracellular calcium in H82 and H1694 human SCLC cells (Song, Sekhon, Lu, et al., 2007). ACh-induced calcium influx in H82 cells was unaffected by the M1R antagonist pirenzepine, M2R/M4R antagonist {11-[[2-[(Diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one; (AFDX-116, Fig. 9B), M1R-siRNA and M5R-siRNA. Immunoblotting experiments show that ACh induces the activation of MAP kinase via M3R in H82 human SCLC cells (Song, Sekhon, Lu, et al., 2007). Furthermore, M3R is involved in the adhesion and migration of lung cancers. Boyden chamber assays confirmed that ACh accelerated the migration of SBC3 human SCLC cells towards fibronectin (as a chemoattractant). Similarly, ACh increased the adhesion of SBC3 cells on fibronectin-coated dishes (S. Zhang, et al., 2010). The proliferative effects of ACh on SBC3 cells required both nicotinic and muscarinic receptors. However, ACh-induced adhesion and migration of SBC3 were exclusively mediated by the M3R (S. Zhang, et al., 2010). In contrast, Quigley et al., (1998) found that carbachol increased the adhesion of SCLC cells on laminin and collagen, but not on fibronectin (Quigley, Shafer, & Williams, 1998). It must be recognized that ACh and carbachol activate both nAChRs and muscarinic receptors. The muscarinic receptor antagonist atropine blocked carbachol-induced adhesion of SCC-9 cells on collagen IV, suggesting that the adhesion-stimulatory activity of carbachol was primarily mediated by muscarinic receptors on SCC-9 cells. Both the studies by Zhang et al., (2010) and Quigley et al., (1998) found that muscarinic receptors alter the functional activity of α1-containing integrins to elevate the adhesion of SCLC to extracellular matrix proteins (Quigley, et al., 1998; S. Zhang, et al., 2010).
The treatment of SCC-9 human SCLC cells with 100 μM carbachol increased cell-cell adhesion and compaction by about 2–3 fold relative to untreated SCC-9 cells. Carbachol-induced cell-cell adhesion was reversed by the muscarinic receptor antagonist atropine, indicating that the cell-compaction activity of carbachol was mediated by the muscarinic receptor family. Furthermore, carbachol-induced cell-cell adhesion of SCC-9 (human SCLC cells) was ablated by the M3R receptor antagonist 4-DAMP suggesting that the cell-cell adhesion activity of carbachol required the activation of M3Rs on SCC-9 human SCLC cells (Williams, 2003). Subsequently, the authors overexpressed GFP-tagged constitutively active Rac (Rac1val-12) or GFP-tagged dominant negative Rac1 (Rac1Asn-17) in SCC-9 cells and activated muscarinic receptors using carbachol. Phase contrast microscopy revealed that the adhesion protein GFP-Rac1val-12 was localized to cell-cell junctions in carbachol-treated cells, probably indicating a role for the Rac pathway in this process (Williams, 2003). Taken together, these studies emphasize the vital role of M3R in the growth and progression of human SCLCs.
Liu et al., (2014) compared the expression of M3R in a panel of human NSCLC cell lines with normal human lung fibroblasts. Out of these NSCLC cell lines, four were human LACs and one was SCC-L (Lin, et al., 2014). All NSCLC cell lines showed elevated levels of M3R, relative to normal human lung fibroblasts. However, no difference in M3R expression was observed between LAC and SCC-L cell lines. Subsequently, the authors explored the role of M3R in the progression of NSCLC using siRNA methodology. The transfection of M3R-siRNA caused a small but significant reduction of cell viability in A549 human LAC and L78 human SCC-L cells (Lin, et al., 2014). However, the depletion of M3R (by siRNA techniques) had dramatic effects on the migration and invasion in both A549 and L78 human NSCLC cells (Lin, et al., 2014). The knockdown of M3R in these two human NSCLC cell lines attenuated cell invasion and migration by 50–70% relative to control scrambled-siRNA-transfected cells. M3R-siRNA-transfected A549 human LAC cells and L78 human SCC-L cells showed decreased expression of MMP-9 expression and activity relative to control-siRNA-transfected cells. M3R-siRNA also decreased the activation of Akt and increased levels of E-cadherin in both A549 and L78 cells. An intriguing observation was that the changes in expression/activity of the above-mentioned genes was more pronounced in L78 human SCC-L cells than A549 human LAC cells (Lin, et al., 2014). Both E-cadherin and MMP-9 play a vital role in the invasion and distant metastasis of human NSCLC cells (Gong, et al., 2016; Merchant, et al., 2017; Nieto, et al., 2016; Tsoukalas, et al., 2017; Xiao & He, 2010). A noteworthy observation is that the authors performed these experiments on only one LAC cell line (A549) and one SCC-L cell line (L78). It would be interesting to determine if this pattern is maintained in a panel of human LAC and SCC-L cell lines.
A role for cross-talk between M3R and EGFR has been indicated in the proliferative, pro-migratory and pro-invasive activity of ACh (Fig. 8B). The treatment of A549 cells with 200 μM ACh led to phosphorylation of EGFR, PI-3 kinase and Akt (R. Xu, et al., 2015). The knockdown of M3R using siRNA methodology abrogated ACh-induced proliferation, migration and invasion. M3R-siRNA also suppressed ACh-induced activation of EGFR, PI-3 kinase and Akt (R. Xu, et al., 2015). Such interaction between cell surface receptors and EGFR has been reported for other muscarinic receptor subtypes and nicotinic receptors in human cancer cells (Di Bari, et al., 2018; H. Li, et al., 2015).
Immunohistochemistry experiments demonstrate that M3R is expressed by 70% of SCLC, 85% of LAC and 70% of SCC-L tissues (Song, et al., 2008; Song, Sekhon, Lu, et al., 2007; Spindel, 2012). Apart from M3R, SCC-Ls also express M2R and M4R (Table 10). The M3R co-localizes with ChAT in about 70% of SCLC and LACs, which further confirms the growth-stimulatory role of ACh in human lung cancers (Song, Sekhon, Lu, et al., 2007; Spindel, 2012). Lin et al., (2014) observed that M3R levels in NSCLC tumors was considerably higher than adjacent matched normal tissue. They further confirmed their results using immunohistochemistry in 148 cases of archived paraffin-embedded sections of NSCLC tumors (Lin, et al., 2014). M3R was robustly expressed in approximately 57% samples. In agreement with their cell culture data, no trends were observed within the histological types of NSCLC (LAC vs. SCC-L vs. LCC; Lin, et al., 2014). Statistical analysis revealed that the levels of M3R were inversely correlated with five-year survival rate of patients. There are no significant trends of M3R expression for age and sex of patients. A similar study was performed by Wu et al., (2013) which analyzed the levels of M3R in 192 NSCLC tumors isolated from patients. They observed that M3R levels were elevated in metaplasia/dysplastic tissue relative to matched adjacent normal tissue (J. Wu, et al., 2013). The expression of M3R showed a strong association with stage, Ki67 (biomarker for proliferation) expression, tumor size, lymphatic vessel size and lymph node metastasis. M3R staining was higher in LAC relative to other types of NSCLC. M3R expression was elevated in Stage II and III NSCLC relative to Stage I of the disease (J. Wu, et al., 2013). Furthermore, NSCLC patients whose tumors expressed high levels of M3R displayed lower disease free survival and overall survival relative to NSCLC patients with low M3R levels.
Table 10.
Expression of muscarinic receptor subtypes in lung cancer tissue and adjacent normal tissue (isolated from patients)
| Nature of cells | Nature of muscarinic receptor | Reference | ||||
|---|---|---|---|---|---|---|
| M1R | M2R | M3R | M4R | M5R | ||
| Normal lung tissue | + | + | + | 177, 299, 352 |
||
| Lung tissue (metaplasia/dysplasia ) |
+ |
352 |
||||
| Small cell lung carcinoma tissue |
+ | 302, 305 | ||||
| Squamous cell lung cancer tissue |
+ | + | + | 177, 299, 305 |
||
| Lung adenocarcinoma tissue |
+ | 177, 301, 305, 352 |
||||
Blanks indicate Not Determined (ND)
COPD is an independent risk factor for NSCLC (Durham & Adcock, 2015; Takiguchi, Sekine, Iwasawa, Kurimoto, & Tatsumi, 2014). Muscarinic receptors play a crucial role in the pathophysiology and airway remodeling associated with COPD (Gosens, Zaagsma, Meurs, & Halayko, 2006; Mastrodicasa, et al., 2017). This led to extensive research on the status of muscarinic receptors in NSCLC (Song & Spindel, 2008; Spindel, 2012, 2016). Lin et al., (2014) examined the status of the M3R in NSCLC patients suffering from COPD (hereby referred to as NSCLC-COPD patients). They observed that high levels of M3R expression in NSCLC-COPD patients correlated with poorer survival rates than NSCLC-COPD patients displaying low M3R expression (Lin, et al., 2014). Elevated levels of M3R in NSCLC-COPD patients correlated with high smoking history and poor lung function. Furthermore, tumors isolated from NSCLC-COPD patients showed substantially higher M3R expression than NSCLC patients without COPD (Lin, et al., 2014). Both univariate and multivariate Cox’s regression analyses showed that M3R expression was an independent predictor of prognosis in NSCLC patients.
The muscarinic acetylcholine signaling system has been extensively used for cancer drug discovery (Table 11). Cell culture experiments reveal that generalized muscarinic antagonists like atropine decrease the viability of human SCLC cell lines (Song, Sekhon, Proskocil, et al., 2003; Spindel, 2016). In addition, atropine abrogates TGF-β1-induced EMT in A549 human NSCLC cells at concentrations ranging from 0.1–10 μM over 72 hours (K. Yang, et al., 2014).
Table 11.
Anti-cancer drugs targeting muscarinic receptor subtypes in lung cancer
| Name of drug | Specificit y |
Cell line | Activity | Model | Reference s |
|---|---|---|---|---|---|
| Pirenzepine | M1R | A549 | Inhibition of TGF-β1 and carbachol- induced EMT |
Cell culture | 128, 359 |
| Methoctramine | M2R | A549, PC-9 | Inhibition of EMT, invasion, migration |
Cell culture athymic mice |
376, 377 |
| Darifenacin | M3R | H520, H82 | Inhibition of cell growth |
Cell culture athymic mice |
7, 299, 301, 305 |
| Darifenacin | M3R | H1299 | Inhibition of cell growth |
Cell culture | 128 |
| 4-DAMP | M3R | H82, H1694, SBC3 |
Inhibition of proliferation, viability, angiogenesis and carbachol- induced EMT |
Cell culture, athymic mice |
31, 301, 306, 359, 369 |
| Tiotropium | M3R | H520 | Inhibition of cell growth |
Cell culture, athymic mice |
295, 296 |
| R2BHJJ and R2PHC |
M3R | A549, H1299, H157, H460 |
Inhibition of viability, cell cycle progression |
Cell culture | 128 |
| AFDX-116 | M2R/M4 R |
H1299 | Inhibition of viability |
Cell culture |
128 |
| P-F-HHSiD | M3R | H82 | Inhibition of viability |
Cell culture |
301 |
| J-115311 | M3R | H82 | Inhibition of viability, cell growth |
Cell culture, orthotopic mouse model |
7 |
The dual nicotinic/muscarinic receptor agonist carbachol induced EMT in A549 human NSCLC and immortalized BECs in a concentration-dependent manner at 72 hours post treatment. ELISA experiments revealed that carbachol-induced EMT in A549 human NSCLC cells correlated to increased secretion of TGF-β1 from A549 cells (K. Yang, et al., 2014). The pro-EMT effects of carbachol were abolished by the M1R receptor antagonist pirenzepine and unaffected by the M2R antagonist methoctramine (Fig. 9A). However, Zhao et al. (2015) observed that the M2R antagonist methoctramine inhibited EMT, invasion and migration of A549 and PC-9 human NSCLC cells by multiple pathways (Q. Zhao, X. Gu, et al., 2015; Q. Zhao, J. Yue, et al., 2015). The anti-invasive, anti-migratory and EMT-inhibitory activity of methoctramine was triggered by inhibition of the PI-3kinase/Akt, MAPK, ERK and NF-κB pathway (Q. Zhao, J. Yue, et al., 2015). Methoctramine showed robust anti-tumor activity in A549 human NSCLC tumors xenografted in athymic mice. Two doses of methoctramine (2 mg/kg bodyweight/day and 5 mg/kg bodyweight/day) were administered intraperitoneally to these tumor-bearing athymic mice for three weeks (Q. Zhao, X. Gu, et al., 2015). Methoctramine (5 mg/kg bodyweight/day) decreased the growth of A549 tumors (in athymic mice) by about five-fold relative to vehicle-treated mice. Saline was used as the vehicle in the study. The methoctramine dose 2 mg/kg bodyweight/day did not show significant anti-tumor activity (P≤0.05) relative to saline-treated mice. The studies by Yang et al. (2014) used carbachol to activate the muscarinic receptor whereas Zhao et al., (2015) used pilocarpine as the muscarinic agonist in A549 human NSCLC cells. Such differences in data may be due to the fact that carbachol activates to both nicotinic receptors and muscarinic receptors, whereas pilocarpine is thought to be a generalized muscarinic receptor agonist with minimal nAChR-activating ability (Figueroa, Griffin, & Ehlert, 2009).
The M3R antagonist 4-DAMP, abrogated ACh-induced cell proliferation and calcium influx in human SCLCs (Song, Sekhon, Lu, et al., 2007). The treatment of H82 human SCLC cells with 100 pM-100 nM 4-DAMP robustly decreased ACh-induced activation of Akt and MAP kinase. Furthermore, 4-DAMP also decreased the viability of asynchronous H82, H1694 and SBC3 human SCLC cells in a concentration-dependent manner. The growth-inhibitory activity of 4-DAMP on asynchronous H82 human SCLC cells correlated with downregulation of phospho-Akt and phospho-MAPK (Song, Sekhon, Lu, et al., 2007; Spindel, 2012; S. Zhang, et al., 2010). Data from Yang et al., (2014) demonstrated that 4-DAMP decreases carbachol-induced EMT in A549 human NSCLC cells (K. Yang, et al., 2014). The growth-inhibitory activity of 4-DAMP was investigated in athymic mice models xenografted with SBC3 human SCLC cells (Caihong & Shuxiang, 2017). Real time PCR and immunoblotting experiments were used to monitor M3R expression, vascular endothelial growth factor (VEGF) levels and microvessel density (MVD) in these tumor-bearing athymic mice. Three doses of 4-DAMP (0.5, 1, and 2 mg/kg body weight) were administered intraperitoneally to these mice. They observed that all three doses of 4-DAMP decreased the growth rate and weights of SBC3 tumors implanted in athymic mouse (Caihong & Shuxiang, 2017). Furthermore, 4-DAMP decreased M3R expression, VEGF levels and MVD in these SBC3 tumors. This is the first study which demonstrates the anti-angiogenic activity of M3R antagonists (Caihong & Shuxiang, 2017).
Apart from 4-DAMP, multiple M3R antagonists like darifenacin (Fig. 9C), tiotropium (Fig. 9D) and para-fluoro-hexahydrosila-difenidol (P-F-HHSiD) suppressed the growth of human SCLC and SCC-L cell lines in vitro (Song, Olivas, & Spindel, 2009; Song, et al., 2008; Song, Sekhon, Lu, et al., 2007; Spindel, 2012, 2016). The M2R/M4R antagonist AFDX-116 had no impact on the viability of asynchronous H82 and H1694 human SCLC cells (Song, Sekhon, Lu, et al., 2007; Spindel, 2012).
Song et al., (2008) observed that the M3R-antagonist darifenacin suppressed nicotine-induced proliferation of H520 human SCC-L cells (Song, et al., 2008). Subsequently, they investigated the anti-tumor activity of darifenacin in athymic mouse model of human SCC-Ls using osmotic pumps. They injected H520 human SCC-L cells subcutaneously in the right flank of male athymic mice (Song, et al., 2008). The tumors were administered darifenacin at a dose of 6 mg/kg bodyweight/day using osmotic pumps. The mice in the control group were administered the vehicle (50% DMSO in PBS) via osmotic pumps. After four weeks of treatment, the authors observed that darifenacin decreased the tumor weight and volume of SCC-L tumors xenotransplanted in athymic mice (Song, et al., 2008). A later study by the same research group demonstrated the anti-tumor activity of tiotropium in athymic mice bearing SCC-L tumors. An interesting aspect of this experiment was that tiotropium was administered via inhalation, which would minimize any possible pleotropic effects of this compound on other tissues (Song, Olivas, & Spindel, 2010).
Song et al., (2008) continued their studies to investigate the anti-tumor activity of darifenacin in H82 human SCLC tumors xenografted on athymic mice (Song, et al., 2008; Spindel, 2012) The methodology of the experiment was similar to the previously discussed study involving the anti-cancer activity of H520 SCC-L tumor-bearing athymic mice. Three doses of darifenacin (0.3, 1, 3 mg/kg/day) were administered (using osmotic pumps) to the athymic mice xenografted with H82 human SCLC cells. Two of the doses of darifenacin, namely 1 mg/kg/day and 3 mg/kg/day, significantly decreased the growth rate of H82 tumors, whereas the dose of 3 mg/kg/day darifenacin decreased both tumor volume and tumor weight of H82 human SCLC tumor-bearing mice (Song, Sekhon, Lu, et al., 2007). The authors detected darifenacin in the plasma in all the three doses of drug administered to mice. The anti-tumor activity of darifenacin correlated with decreased levels of phospho-Akt and phospho-MAPK (Song, Sekhon, Lu, et al., 2007). A noteworthy aspect of these studies was that the authors did not report any gross toxicity of darifenacin on mice in either of the studies.
Hua et al., (2012) examined the anti-neoplastic activity of a novel synthetic muscarinic receptor antagonist R2HBJJ (Fig. 9E) in human NSCLC. The design of R2HBJJ was based on structure-activity relationship (SAR) studies on the potent anti-cholinergic compound R2-PHC (Fig. 9F), developed in their laboratory (N. Hua, et al., 2012). Receptor binding assays showed that the R2HBJJ showed maximal affinity for M3R followed by M1R, M4R, M5R and M2R. The authors investigated the growth-inhibitory effect of R2BHJJ on a panel of four NSCLC cell lines (A549, H1299, H157, and H460) and an immortalized normal lung epithelial cell line BEP2D. The growth-suppressive activity of R2BHJJ in H460, H157 and H1299 cells was greater than in BEP2D normal lung cells (N. Hua, et al., 2012). R2BHJJ minimally affected the viability of A549 human NSCLC cells. This may be due to the fact that A549 expressed the lowest amount of M3R out of all the cell lines tested. The authors also compared the growth-inhibitory activity of R2BJJ with atropine, pirenzepine, AFDX-116, darifenacin and their parent compound R2PHC in H1299 cells (N. Hua, et al., 2012). The cells were treated with varying concentrations of the drugs (1–100 μM) for 72 hours. Out of all of the compounds tested, atropine and pirenzepine had the least effect on the viability of H1299 cells. AFDX-116 displayed growth inhibitory activity at high concentrations, namely 30 μM and 100 μM. The growth-inhibitory activity of R2BHJJ was similar to the parent compound R2-BHC. However, R2BHJJ displayed a better side effect profile than R2-PHC. The growth-suppressive effect of darifenacin was intermediate between AFDX-116 and R2BHJJ. The transfection of M3R-siRNA ablated the growth-inhibitory activity of R2BHJJ in human NSCLC cells, suggesting that R2BHJJ predominantly targeted the M3R for its growth-suppressive activity. The binding of R2BHJJ to M3R induced cell cycle arrest (G0/G1 phase) in H1299 cells (N. Hua, et al., 2012). R2BHJJ was shown to have the ability to inhibit Rb phosphorylation and downregulate the expression of cell cycle-regulatory proteins.
The muscarinic receptor antagonist J-115311 (Fig. 9G) has a high binding affinity for M3R and is about 50- to 400-fold more selective for M3R than other muscarinic receptor subtypes. J-115311 had a greater affinity and selectivity for M3R than darifenacin (Sagara, et al., 2002; Song, et al., 2008; Song, Sekhon, Lu, et al., 2007). Ami et al., (2011) explored the anti-cancer activity of J-115311 in human SCLCs. The authors compared the ability of J-115311 and darifenacin to decrease the viability of H82 human SCLC cells. WST-8 assays reveal that the growth-inhibitory activity of J-115311 was about three-times greater than darifenacin in H82 cells (Ami, et al., 2011). J-115311 suppressed the growth of H82 cells by inducing apoptosis. This report by Ami et al., (2011) is the first to examine the anti-tumor activity of an M3R antagonist using the orthotopic mouse model of SCLC (Ami, et al., 2011). Several congruent studies show that the orthotopic mouse model reproduces the morphology, tumor microenvironment and metastatic pattern of human lung cancers (Justilien & Fields, 2013; Kellar, Egan, & Morris, 2015). H82 cells were injected into the upper left lung of athymic mice. The tumors were allowed to grow for a week after which the mice were daily administered two doses of J-115311 of 25 mg/kg bodyweight and 50 mg/kg bodyweight subcutaneously. After 10 days, the tumors were visualized by microCT scans (Ami, et al., 2011). The administration of 50 mg/kg bodyweight J-115311 caused a reduction in tumor weight and tumor volumes by about 40%. An interesting observation was that the anti-tumor activity of darifenacin was observed at lower doses (6 mg/kg bodyweight/day) than J-115311 (Ami, et al., 2011; Song, et al., 2008). This may be due to differences in mouse models and duration of experiments used in both studies. Data from Sagara et al., (2002) also indicates that darifenacin has a higher cross-reactivity towards other muscarinic receptor subtypes relative to J-115311 (Sagara, et al., 2002). The M2R has been shown to play a role in promoting invasion and EMT in human lung cancers (Q. Zhao, X. Gu, et al., 2015; Q. Zhao, J. Yue, et al., 2015). It is possible that the greater anti-tumor activity of darifenacin may be due to its ability to target multiple muscarinic receptor subtypes. The design and discovery of synthetic compounds with improved specificity for M2R, M3R or both may have potential applications in the management and treatment of lung cancer.
5. Nicotinic Receptors (nAChRs)
The role of nAChRs has been widely studied in the growth, angiogenesis and metastasis of lung cancer (S. A. Grando, 2008; Sergei A. Grando, 2014; Pillai Chellappan, 2012; Schuller, 2012; Spindel, 2016; S. Wang & Hu, 2018; Zoli, et al., 2018). The proliferative effects of ACh in SBC3 human SCLC cells are partially mediated by nAChRs (S. Zhang, et al., 2010). In addition, nAChRs protect lung cancer cells against cell death induced by chemotherapeutic drugs, ionizing radiation, EGFR-tyrosine kinase inhibitors (EGFR-TKI) and other extracellular stress signals (Campoy, et al., 2016; Jin, et al., 2004; Mai, et al., 2003; Maneckjee & Minna, 1994; Mucchietto, et al., 2016; Togashi, et al., 2015; West, et al., 2003; West, et al., 2004; Wright, Zhong, Zheng, & Larrick, 1993; J. Xu, et al., 2007; Zeidler, et al., 2007; T. Zhang, et al., 2006). The reviews (mentioned above) comprehensively discuss the role of nAChRs in lung cancer progression and therapy. We will chronicle the most recent developments in the field of nAChR-biology (over the past three years) in this review.
The nAChRs are ligand-gated ion channel receptors, with five subunit proteins tightly wrapped around a central ion pore (Barman, et al., 2016; Gotti & Clementi, 2004). These pentameric proteins are comprised of α and β subunits (Egleton, et al., 2008; Egleton, Brown, & Dasgupta, 2009; Gotti & Clementi, 2004). The α-subunit contains the binding site for ACh (Lindstrom, 1996, 1997). Non-neuronal nAChRs are broadly classified into two categories, heteromeric and homomeric nAChRs. Heteromeric nAChRs contain a combination of α and β subunits (Dang, et al., 2016; Improgo, et al., 2013; Improgo, Tapper, & Gardner, 2011; Mucchietto, et al., 2016; Zhao, 2016; Zoli, et al., 2018). Homomeric nAChRs are composed of five α subunits (Gotti & Clementi, 2004; Lindstrom, 1996, 1997). There are many isoforms of α (α1–10) and β (β1–4) subunits. Examples of homomeric nAChRs are α7-nAChR, α6-nAChR and α9-nAChR (Gotti & Clementi, 2004). Apart from ACh, physiological modulators of nAChRs include lynx1, lynx2, SLURP-1 and SLURP-2 (section 6; Chernyavsky, Arredondo, Galitovskiy, Qian, & Grando, 2010; Durek, et al., 2017; Fu, Rekow, & Spindel, 2012; Ibanez-Tallon, et al., 2002; Lyukmanova, et al., 2018; Lyukmanova, Shulepko, Kudryavtsev, et al., 2016; Miwa, et al., 1999; O’Neill, et al., 2012; Sekhon, Song, Jia, Lindstrom, & Spindel, 2005; Tekinay, et al., 2009). The nAChRs are ubiquitously expressed in all tissues of the lung, even in fetal lung tissue (Sekhon, et al., 2005). Such observations underscore the importance of nAChRs in lung development and homeostasis (Mucchietto, et al., 2016; Schuller, 2012; Song & Spindel, 2008; Spindel, 2016; Zoli, et al., 2018).
GWAS studies have indicated that SNPs in the α5-α3-β4 nAChR cluster (CHRNA5-CHRNA3-CHRNB4) located on chromosome 15q25 region confers risk for lung cancer in European populations who are heavy smokers (Amos, et al., 2008; Hung, et al., 2008; Spitz, Amos, Dong, Lin, & Wu, 2008; Thorgeirsson, et al., 2008a). Genetic variations in the 15q25 chromosome (in smokers) are associated with increased risk of death from lung cancer, COPD and tobacco-related cancers (Hallden, et al., 2016; Nedeljkovic, et al., 2018; Qu, et al., 2016; Saccone, et al., 2010). Polymorphisms in CHRNA3 and CHRNA5 are also associated with familial lung cancer (Byun, et al., 2018). Other genomic variances near CHRNA2 gene were correlated to increased overall risk for developing lung cancer (McKay, et al., 2017). It must be remembered that the occurrence of these SNPs is dependent of race and ethnicity of the population selected for the study (Luo, et al., 2008). Studies in Chinese and African-American populations have implicated different SNPs of CHRNA3 and duplicated CHRNB4 as risk factors for lung cancers (Amos, et al., 2010; Hansen, et al., 2010; Y. Zhang, et al., 2016; Zhou, et al., 2015). Studies in European populations of heavy smokers have shown that the rs1051370 variant of CHRNA3 correlates with larger tumor size in SCC-L patients (X. Chen, et al., 2011; Ware, van den Bree, & Munafo, 2012). The rs16969968 genotype of CHRNA5 may comprise of a G to A (G-A), G to G (G-G) or A to A (A-A) missense variant in the CHRNA5 gene. The G-A variant translates to a substitution of aspartic acid 398 to asparagine (Lassi, et al., 2016; Pandey, et al., 2017; Russo, et al., 2011; Wen, et al., 2016). This is also known as the D398N form of CHRNA5, which displays a reduced response to the nAChR agonist epibatidine (Bierut, et al., 2008; Hung, et al., 2008; Kuryatov, Berrettini, & Lindstrom, 2011). The rs16969968 genotype of CHRNA5 containing the A-A variant is considered to be a high-risk variant for the development of lung cancer. In contrast, the CHRNA5 rs16969968 genotype containing G-G variant is a low-risk variant for lung cancer (Bierut, et al., 2008; Kuryatov, et al., 2011). Epidemiological studies in European populations of active heavy smokers show that the high risk variant (A-A; rs1696996 CHRNA5) displayed a strong association with lung cancer risk, developing lung cancer four years earlier than individuals having the low risk variant (G-G; rs16969968 CHRNA5; L. S. Chen, et al., 2016; L. S. Chen, et al., 2015; Hall, 2016).
Recent studies have focused on the role of α5-nAChR in the development and progression of lung cancer. Conventionally, the α7-nAChR is thought to mediate the proliferative, pro-angiogenic and pro-metastatic activity of nicotine in lung cancer (C Heeschen, et al., 2001; C. Heeschen, et al., 2002; Pillai & Chellappan, 2012; Schuller, 2012; S. Wang & Hu, 2018; C. Zhang, et al., 2016). The role of α7-nAChR is further reinforced by the anti-tumor activity of α7-nAChR antagonists in multiple experimental models of lung cancer (K. C. Brown, et al., 2012; Mucchietto, et al., 2018; S. Wang & Hu, 2018). Zhang et al., (2017) have shown that tobacco compounds like nicotine increase the levels of α5-nAChR in A549 human NSCLC cells (Y. Zhang, et al., 2017). Nicotine-induced elevation of α5-nAChR coincided with increased levels of phopho-STAT3 and recruitment of the JAK-STAT pathway (Y. Zhang, et al., 2017). Chromatin immunoprecipitation experiments confirmed the presence of STAT binding sites and STAT-regulatory elements on the α5-nAChR promoter (Y. Zhang, et al., 2017). The α5-nAChR receptor potently influences addiction, cigarette consumption and nicotine dependence (Berrettini & Doyle, 2012; Bierut, et al., 2008; Kuryatov, et al., 2011; Lassi, et al., 2016; Spitz, et al., 2008; Thorgeirsson, et al., 2008b). These factors have to be taken into consideration when translating basic research findings to patient-oriented studies involving the role of α5-nAChR in lung cancer.
A recent study mapped the transcriptome of A549 cells that were transfected with or without α5-nAChR-siRNA (H. J. Sun, Jia, & Ma, 2017). The gene expression profile revealed that α5-nAChR modulates cell cycle genes, DNA replication genes, oncogenes and the p53 signaling pathway. Out of all of the signaling pathways, genes involved in cell cycle progression (cyclin D1, E2 and D3) were most significantly downregulated by α5-nAChR-siRNA (H. J. Sun, et al., 2017). The treatment of α5-nAChR-siRNA transfected A549 human NSCLC cells with nicotine caused a robust decrease in S-phase entry. Notably, the depletion of α5-nAChR also suppressed basal cell cycle progression in A549 cells. Flow cytometry experiments revealed that α5-nAChR-siRNA triggered apoptosis in both untreated and nicotine-treated A549 cells. Such observations argue for a role for α5-nAChR in cell cycle progression and apoptosis (H. J. Sun, et al., 2017). Sun et al., (2015) showed that nicotine-induced migration and invasion of A549 human NSCLC cells was mediated by α5-nAChR (H. Sun & Ma, 2015). The ablation of α5-nAChR by siRNA techniques abrogated nicotine-induced invasion and migration of A549 cells. The transfection of α5-nAChR-siRNA led to an increase in the cell adhesion protein E-cadherin (H. Sun & Ma, 2015). The decrease of E-cadherin is a vital event in the EMT of lung cancer cells, which endows the cells with a migratory phenotype, allowing them to invade surrounding blood vessels and eventually metastasize to distant sites (Gheldof & Berx, 2013; Wong, Fang, Chuah, Leong, & Ngai, 2018). This is in alignment with studies of athymic mouse models demonstrating a role for α5-nAChR in the progression of NSCLCs. A549 human NSCLC cells transfected with α5-nAChR-siRNA were subcutaneously implanted in the flank of athymic mice. The presence of α5-nAChR-siRNA decreased the tumor growth rates of both nicotine-treated and untreated A549 tumors (H. J. Sun, et al., 2017).
An analysis of α5-nAChR expression in NSCLC tumors isolated from patients reveal that α5-nAChR expression in tumor tissue is greater than adjacent matched normal tissue. Zhang et al., (2017) analyzed the levels of α5-nAChR in 130 LAC specimens. They observed that 60% of the tumors showed elevated levels of α5-nAChR and about 67% contained phosphorylated STAT3 (Y. Zhang, et al., 2017). Statistical analysis showed an association between α5-nAChR and phopho-STAT3 expression. The expression of α5-nAChR in LAC tumors isolated from patients showed a positive correlation with smoking status, STAT3 expression and decreased survival times in LAC patients (Y. Zhang, et al., 2017). Emerging data show that α5-nAChR accelerates the development of lung cancer by indirect mechanisms. The indirect mechanisms by which α5-nAChR facilitates the development of lung cancers are thought to be behavioral in nature. The α5-nAChR mediates tobacco addiction, deeper inhalation of cigarettes and increased intake of nicotine (Berrettini & Doyle, 2012; Brunzell, Stafford, & Dixon, 2015; L. S. Chen, et al., 2015; Kuryatov, et al., 2011; Lassi, et al., 2016). All of these factors translate to the development of aggressive lung tumors, reduced response to chemotherapy and poorer survival rates in lung cancer patients who are smokers.
There are no known synthetic antagonists to α5-nAChR. Polymorphisms in the α5-α3-β4 nAChR cluster are associated with both nicotine dependence and lung cancer risk (Amos, et al., 2010; Hung, et al., 2008; Thorgeirsson, et al., 2008b). However, it is not known how these three subunits regulate each other in the brain or in the lung. The discovery of synthetic molecules specific for α5-nAChR may shed light on how this receptor regulates other nAChR subunits. An interaction between the α5-nAChR and α7-nAChR-signaling pathways has already been reported in normal and malignant lung cells. With this background, Ray et al. (2017) used a directed compound library of 275 nAChR antagonists to identify compounds which would display selectivity for α5-nAChR over α3-nAChR and β2-nAChRs. In addition, they screened for compounds which would differentially bind to α5- and α5-D398N-nAChR (C. Ray, et al., 2017). These landmark studies are the first to report of small molecule antagonists which selectively bind to α5-nAChR. They identified three compounds AK-968/12117231, AN-038/15563010 and AE-641/30177001 (as named by the authors; Fig. 10A-C) display differential binding to α5- and α5-D398N-nAChRs. Furthermore, the compounds AE-s641/30177001 and AK-968/40218701 (Fig. 10C-D) distinguish between α3β4-and α3β4α5-D398N-nAChRs (C. Ray, et al., 2017). It would be interesting to determine if these compounds display any anti-cancer activity in human lung cancers.
Figure 10.
Structures of nicotinic receptor antagonists selective for α5-nAChR and α5-D398N-nAChR. A. AK-968/12117231: 1-(5-bromo-2-furoyl)-4-(4-fluorophenyl)piperazine B. AN-038/15563010: allyl 2-[(5-bromo-2-furoyl)amino]-3-phenylacrylate C. AE-641/30177001: N,N,4-trimethyl-5,6-diphenyl-1-hexanamine D. AK-968/40218701: 1-decyl-1-(2-undecynyl)piperidium
The role of α7-nAChR in non-neuronal systems has been predominantly studied using tobacco components like nicotine and NNK (Schuller, 2007; Schuller, et al., 2000; Schuller & Orloff, 1998; Schuller, et al., 2003; Zoli, et al., 2018). Studies with genetically modified mice reveal that α7nAChRs play a vital role in susceptibility and onset of lung diseases (like cancer and fibrosis) upon chronic exposure to cigarette smoke (Gahring, Myers, Dunn, Weiss, & Rogers, 2017). The binding of nicotine to α7-nAChR leads to the recruitment of the adapter protein β-arrestin, which in turn induces the activation of Src kinase. The activation of Src kinase triggers a mitogenic signaling cascade comprising of the Rb-Raf pathway, PI-3 kinase/Akt and the MAP kinase pathways which ultimately causes cell proliferation (Dasgupta & Chellappan, 2006; Dasgupta, Rastogi, et al., 2006; C Heeschen, et al., 2001; C. Heeschen, et al., 2002; Mucchietto, et al., 2018; Schuller, et al., 2000; West, et al., 2003; C. Zhang, et al., 2017; Zheng, Ritzenthaler, Roman, & Han, 2007). Apart from cytoplasmic mitogenic signaling pathways, chronic nicotine and NNK exposure elevates the levels of α7-nAChR (Al-Wadei, Al-Wadei, Masi, & Schuller, 2010; Schaal & Chellappan, 2016). A similar upregulation of α7-nAChR is observed when electronic cigarette extracts are used instead of nicotine (Alasmari, et al., 2017; Schaal & Chellappan, 2016). Unlike neuronal cells, nicotine-induced upregulation of α7-nAChR is a transcriptional event. The α7-nAChR promoter is controlled by many regulatory elements and transcription factors. Recent studies show that the transcription factor E2F1 activates α7-nAChR transcription, whereas STAT1 represses the promoter (Schaal & Chellappan, 2016). The activation of α7-nAChR transcription by E2F1 recruits a complex signaling network, such as the Src, MEK, PI-3 kinase/Akt and CDK4/6 which in turn form an autoregulatory feed-forward loop to further increase the expression of α7-nAChRs (Schaal & Chellappan, 2016).
The α7-nAChR-signaling network intermingles with other growth factor and nicotinic receptor-signaling pathways (Krais, et al., 2011; Mucchietto, et al., 2018). Chernyavsky et al. (2015) showed that nAChRs synergize with growth factors like EGF and IGF-1 to elevate the proliferation of BEP2D BECs. The synergistic effect of nicotine (used as a nAChR agonist) and EGF was reversed by α-bungarotoxin indicating that the combinatorial mitogenic effects of nicotine and EGF required α7-nAChR function. These experiments were repeated in NNK-transformed BEP2D BECs and similar results were obtained (Chernyavsky, Shchepotin, & Grando, 2015). Fan et al., (2017) have shown an interaction between the nicotinic receptor and the prostanoid receptor signaling pathway. The prostanoid receptor EP4 is overexpressed in human lung cancer cells relative to normal lung cells (Fan Wang, 2017). The α7-nAChR was found to play a vital role in nicotine-induced upregulation of EP4 expression and proliferation of human LAC cells. Nicotine-induced proliferation of A549 and H1838 human LAC cells was mediated by α7-nAChR-induced activation of PI-3 kinase, JNK and protein kinase C pathways. Nicotine-induced EP4 expression was found to be a transcriptional event and correlates with the decreased binding of AP2α on the EP4 promoter (Fan & Wang, 2017).
Bordas et al., (2017) examined the α7-nAChR expression of 40 SCC-L tissues, 38 LAC tissues and adjacent normal tissues isolated from patients. All tumors showed dysregulation of the 15q25 chromosomal locus, which contains the CHRNA3, CHRNA5 and CHRNAB4 genes. Both SCC-Ls and LACs showed decreased CHRFAM7A (dupa α7-nAChR subunit) expression relative to normal tissue (Bordas, et al., 2017; Gault, et al., 1998). The dupa α7-nAChR subunit negatively regulates α7-nAChR activity in Xenopus oocytes (de Lucas-Cerrillo, et al., 2011). However, SCC-Ls expressed higher levels of α3-, α5-, α7-, α9-, β2- and β4-nAChR mRNA relative to adjacent normal tissue. The levels of dupa α7-nAChR mRNA was decreased in SCC-L tumors relative to adjacent normal tissue. LAC tumors showed elevated expression of α3-, α5-, α7-, and β4-nAChR mRNA, compared to adjacent normal tissue. Expression analysis revealed α4-nAChR and dupa α7-nAChR mRNA was decreased in LAC tumors when compared to normal lung tissue. The ratio of α7-nAChR/dupa α7-nAChR mRNA was higher for SCC-L than LACs tissue (Bordas, et al., 2017).
Bordas et al., (2017) also compared the expression of nAChR mRNA in 35 SCC-Ls tumors originating from never smokers and non-smokers exposed to second hand smoke versus SCC-L patients who were active smokers. They found that increased expression of α5- and α7-nAChRs correlated with a poor prognosis and decreased survival in SCC-L patients. No trends were found in the human LAC tumor samples (Bordas, et al., 2017). Clinical studies show that SCC-L shows a stronger correlation with smoking than LACs (Gandara, Hammerman, Sos, Lara, & Hirsch, 2015; Heist, Sequist, & Engelman, 2012). Such genetic studies may provide a novel insight into the differential correlation of smoking with different kinds of lung cancers.
Recent studies have revealed that functional nAChRs are not only localized on the outer cell plasma membrane but are also expressed on outer mitochondrial membranes of lung tissues (Gergalova, et al., 2012; Gergalova,; S. A. Grando, Kawashima, Kirkpatrick, Kummer, & Wessler, 2015; Kalashnyk, Gergalova, Komisarenko, & Skok, 2012; Skok, et al., 2016). Experiments involving C57BL/6J mice show that α3-, α4-, α7-, β2- and β4-nAChRs are robustly expressed on the outer mitochondrial membranes of the lungs of these mice (Lykhmus, Komisarenko, & Skok, 2014). Chernyavsky et al., (2015) compared the tumor promoting activities of cell membrane-associated nAChRs (cm-nAChRs) and mitochondrial-nAChRs (mt-nAChRs) in the human SCC-L cell line SW900. They observed that the treatment of SW900 cells with a combination of nicotine and growth factors, namely VEGF and EGF, resulted in a synergistic increase in cell proliferation relative to SW900 cells treated with either agent alone. The combinatorial proliferative effects of nicotine and VEGF or EGF were correlated with concomitant increase in the functional activity of cyclin D1 and ERK1/2. Subsequently, the authors analyzed the molecular mechanisms underlying the synergistic activity of nicotine and the abovementioned growth factors in SW900 human SCC-L cells. Co-immunoprecipitation western blotting experiments showed that the activation of (cm)-nAChRs (by ACh or nicotine) induces their direct binding with VEGFR and EGFR via the α7- and β2-(cm)-nAChRs on SW900 human lung cancer cells (Chernyavsky, Shchepotin, & Grando, 2015). The (mt)-nAChRs were primarily found to be responsible for the ability of nicotine to protect SW900 human SCC-Ls against apoptotic agents like hydrogen peroxide and staurosporine. The mt-nAChRs mediated the anti-apoptotic effects of nicotine via regulation of the of the mitochondria permeability transition pore (mPTP), which required the functional activity of both α7-(mt) and non-α7-(mt)-nAChRs. Most interestingly, the ACh-induced activation of (mt)-nAChRs induced the association of (mt)-nAChRs with Src and PI-3 intramitochondrial kinases (Chernyavsky, Shchepotin, & Grando, 2015).
The same research group performed a parallel study where they showed that nicotine displayed synergistic growth-stimulatory activity with EGF in normal BECs, BEP2D immortalized BECs, NNK-transformed BEP2D cells and SW900 human SCC-L cells via the activation of α7- and α9-(cm)-nAChRs. Similarly, the synergistic proliferative effect of nicotine and insulin growth factor (IGF) was observed in BEP2D cells, NNK-transformed BEP2D cells and SW900 human SCC-L cells via α4- and α9-(cm)-nAChRs. The authors noted that nicotine and VEGF only displayed synergistic growth-promoting activity in SW900 human SCC-L cells and this required the α4- and α9-(cm)-nAChRs (Chernyavsky, Shchepotin, Galitovkiy, & Grando, 2015). An interesting data obtained in this research paper was that the treatment of BEP2D BECs with the tobacco carcinogen NNK altered the gamut of (mt)-nAChRs on these cells. Furthermore, NNK-induced malignant transformation (of BEP2D cells) increased the magnitude of (mt)-nAChRs coupled to inhibition of mPTP activity, indicating that NNK treatment could elevate the anti-apoptotic effects of (mt)-nAChRs. Therefore, agents which can lower (mt)-nAChR-induced inhibition of mPTP activity could revive mitochondrial apoptotic pathways and arrest the progression of human lung cancers (Chernyavsky, Shchepotin, Galitovkiy, & Grando, 2015).
Innovative studies by Schaal and Chellappan (2016) show that the overexpression of α5- and α3-nAChR by all histological types of NSCLC in men (who were smokers) correlated with increased survival probability. On the other hand, the overexpression of α7-nAChR in NSCLC correlated with decreased survival probability (Schaal & Chellappan, 2016). Kaplan-Meir plots from their study demonstrate that the survival probability of NSCLC patients whose tumors contained high levels of α7-nAChR mRNA was greater than NSCLC patients who expressed low levels of α7-nAChR mRNA in their tumors. This is indeed a surprising observation because a plethora of research papers have indicated that the α7-nAChR plays a pivotal role in accelerating the growth, angiogenesis and distant metastasis of human NSCLCs. Although, further research is required to fully explain the findings of Schaal and Chellappan (2016), it is probable that there are multiple cellular events underlying their observations. A somewhat analogous result was also obtained by Medjeber et al., (2015), who studied the differential subcellular localization of nAChRs in primary cultures established from NSCLC tumors isolated from patients. They observed that α5-, α7-, β2- and β4-nAChR subunits were expressed by all LAC tumors and SCC-L tumors. These nAChR subunits were localized in the glandular structures of human LAC cells. Most interestingly α5-, β2- and β4-nAChR subunit were expressed on the invasive front of human SCC-L tumors, (Medjber, et al., 2015). The unexpected result was that α7-nAChR was not expressed at the invasive front of these tumors.
As discussed in their elegant study, Schaal and Chellappan (2016) suggest that it may be possible that human NSCLC tumors express high magnitude of the α7-nicotinic receptors at early phases of tumorigenesis (enabling the tumor to grow rapidly, acquire angiogenic phenotype and propensity for metastasis) and that the expression of α7-nAChRs decline as the tumor progresses to an advanced stage, where other signaling pathways take over and control tumor growth (Schaal & Chellappan, 2016). This idea is supported (at least, in part) by the studies of Medjeber et al., (2015) who observed that well-differentiated human SCC-L tumors express higher levels of α7-nAChR, relative to poorly differentiated SCC-L (Medjber, et al., 2015). Conventionally, well-differentiated SCC-Ls tend to be slow-growing and associated with a relatively good prognosis. On the other hand, poorly-differentiated SCC-Ls are the very aggressive tumors, associated with dismal prognosis (Doroshow & Herbst, 2018; Herbst, Morgensztern, & Boshoff, 2018). These results lend support to the hypothesis of Schaal and Chellappan (2016) that the expression/functional activity of α7-nAChR may decline with tumor progresses to advanced stages. A reason for the decline in the functional activity of α7-nAChR in NSCLCs could be due to receptor desensitization which would attenuate its tumor-promoting ability (Schaal & Chellappan, 2016). Support for their hypothesis comes from the experiments of Medjeber et al., (2015) who observed an inverse correlation between the levels of the cell proliferation marker Ki-67 and α7-nAChR expression in the poorly differentiated NSLC tumors, (isolated from patients) used in their experiments (Medjber, et al., 2015).
Apart from α7-nAChR desensitization, variations in the subcellular localization of α7- and other nAChR subtypes (cell membrane versus mitochondrial membrane) could possibly contribute to increase the survival of human NSCLCs patients. The mitochondrial (mt)-nAChRs mediate the pro-survival functions of nAChRs in human lung cancer cells. Several congruent studies reveal that the pro-survival activity of (mt)-nAChRs are mediated by a combination α7-(mt) and non α7- (α3-, β2- and β4-)-nAChRs (mt). The upregulation of α7-nAChR may be accompanied by concomitant decrease in mitochondrial membrane bound α3-, β2-and β4-nAChRs, which would enable the NSCLC tumors to respond better to chemotherapy and radiation.
Clinical studies show that the acquisition of chemoresistance is primarily responsible for the dismal survival rates observed in NSCLC patients. NSCLC patients initially respond well to chemotherapy; however, they inevitably relapse and subsequently the NSCLC tumors become unresponsive to chemotherapeutic drugs and radiation (Doroshow & Herbst, 2018; Herbst, Morgensztern, & Boshoff, 2018). The α3/β2-nAChR subunits (not α7-nAChR) confer resistance on human NSCLC cells against the apoptotic activity of chemotherapeutic drugs (Dasgupta & Chellappan, 2006). The data of Schaal and Chellappan (2016) show that high expression of α3-nAChR significantly correlates with decreased survival probability in all histological subtypes and variants of NSCLC. The seminal findings of Schaal and Chellappan (2016) emphasize the need for patient-oriented studies to precisely identify the role of α7-nAChR and other nAChR subtypes in the progression of human NSCLCs.
Small molecule antagonists of α7-nAChR have been extensively investigated for their anti-cancer activity in human lung cancer. The suppression of α7-nAChR expression by short-hairpin RNA (shRNA) suppresses nicotine-induced growth of H1299 human LACs in both cell culture and athymic mouse models (C. Zhang, et al., 2016; C. Zhang, et al., 2017). Nicotine elevated the levels of the extracellular matrix (ECM) protein vimentin in both in vitro and in vivo. Vimentin is a vital regulator of EMT, a process which facilitates the invasion and metastasis of neoplastic cells (Kidd, Shumaker, & Ridge, 2014; C. Y. Liu, Lin, Tang, & Wang, 2015). The α7-nAChR-shRNA inhibited nicotine-induced vimentin expression via the MEK pathway. The α7-nAChR is also required for nicotine-induced invasion and EMT of human NSCLC cells (Dasgupta, et al., 2009; C. Zhang, et al., 2016). Apart from α7- and α5-nAChR, the α9-nAChR has emerged as a novel molecular target in lung cancer therapy. Depletion of α7-nAChR or α9-nAChR by siRNA methodology ablated nicotine-induced proliferation on A549 and H1975 human LAC cells. The α9-nAChR antagonist RGIA4 (Fig. 11A) and α7-nAChR antagonist ArIB (V11L:V16D) peptide (Fig. 11B) abolished nicotine-induced proliferation of nicotine-treated A549 cells (Mucchietto, 2016; Mucchietto, et al., 2018); The design and synthesis of these compounds is detailed in Romero, et al., 2017 and Whiteaker, et al., 2007 (Romero, et al., 2017; Whiteaker, et al., 2007). The growth-inhibitory effects of RGIA4 or ArIB (V11L:V16D) peptide involved the Akt pathway. Dual α7-nAChR and α9-nAChR antagonists like α-bungarotoxin (α-BT; Fig. 12A), methyllycaconitine (MLA; Fig. 12B) and MG624 (Fig. 12C) displayed greater growth-suppressive activity than RGIA4 or ArIB (V11L:V16D) peptides (Mucchietto, et al., 2016; Mucchietto, et al., 2018). These observations seem to suggest that both of these receptors interact in a synergistic manner to mediate nicotine-induced proliferation of human LAC cells. MLA, α-BT and MG624 inhibited the activation of both ERK and Akt in A549 human LAC cells. Moreover, MG624 also induced oxidative stress in A549 cells (Mucchietto, et al., 2018). Natural compounds like β-Cryptoxanthin (BCX; Fig. 12D) suppress α7-nAChR expression at both mRNA and protein levels in A549 human LAC and BEAS-2B immortalized human lung epithelial cells. BCX inhibited α7-nAChR-induced proliferative and pro-survival pathways like PI-3 kinase/Akt pathway, ERK pathway and its downstream effectors (Iskandar, et al., 2016). Furthermore, BCX also suppressed cell migration, actin remodeling and lamellipodia formation in immortalized human lung epithelial cells. The anti-migratory effects of BCX were mediated by the MMP-2 pathway (Iskandar, et al., 2016).
Figure 11.
Structure of nAChR subunit antagonists. A. RGIA4 B. ArIB (V11L;V16D). The disulfide bridges are formed between the cysteine residues of the peptides depicted in A and B.
Figure 12.
Nicotinic Receptor Antagonists targeting both α7-nAChR and α9-nAChR or α7-nAChR alone. A. α-bungarotoxin B. Methyllycaconitine C. MG624 β-cryptoxanthine
Radiolabeled nicotinic receptor ligands have been extensively used to visualize multiple regions of the brain (Lotfipour, Mandelkern, & Brody, 2011; Rotering, et al., 2014). The potential of nAChR ligands for imaging lung tumors was examined using the radiolabeled α4-nAChR ligand 18F-Nifene (Fig. 13A) in an A/J mice NNK lung carcinogenesis model (Galitovskiy, et al., 2013). Western blot analysis and immunofluorescence experiments of the excised tumors revealed that the tumors nodules of NNK-treated A/J mice displayed higher expression of α4-nAChRs than the lungs of tumor-free A/J mice. Subsequently, the authors performed positron emission tomography (PET)/CT scans using 18F-Nifene over a period of eight months after NNK administration. The uptake of 18F-Nifene in the lungs of NNK-treated A/J mice (which developed lung tumors) was higher than the uptake of 18F-Nifene in lungs of untreated A/J mice (Galitovskiy, et al., 2013). Furthermore, 18F-Nifene demonstrated a higher lung tumor to non-tumor uptake ratio than radiolabeled fluorodeoxyglucose (18F-FDG), which is the conventional ligand for all PET/CT scans in the detection of lung cancers in patients. These results may pave the way for a novel application of nAChR ligands involving the detection and imaging of human lung cancers (Galitovskiy, et al., 2013).
Figure 13.
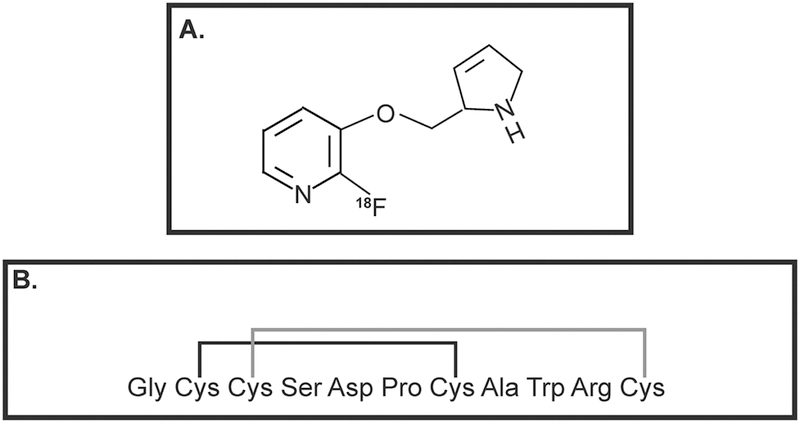
Novel applications of nicotinic receptor ligands in human lung cancer. A. 18F-Nifene B. α-conotoxin Iml. The disulfide bridges are formed between the cysteine residues of the peptides depicted in B.
Apart from nicotine, the tobacco nitrosamine NNK is a high-affinity ligand of nAChRs. However, a subset of the tumorigenic activities of NNK is also mediated by the β-adrenergic receptor on lung cancer cells (Schuller, 2002; Schuller, Tithof, Williams, & Plummer, 1999). NNK-induced activation of insulin growth factor-1 receptor (IGF-1R) is mediated by β-adrenergic receptor pathway (Boo, et al., 2016). On the other hand, NNK-induced exocytosis of insulin growth factor-2 (IGF-2) was mediated by the nAChRs via elevation of intracellular calcium in human lung epithelial cells. Calcium channel blockers like amlodipine (0.5 mg/kg bodyweight/day) and nifedipine (10 mg/kg bodyweight/day) ablated NNK-induced lung carcinogenesis in FVB mice (Boo, et al., 2016).
An innovative study by Mei et al., (2018) has used the α7-nAChR ligand α-conotoxin Iml (α-CT; Fig. 13B) as a molecular target for delivery of the chemotherapeutic drug docetaxel to α7-nAChR overexpressing human lung cancers (Mei, et al., 2018). As a “proof-of-concept” the authors used the human LAC cell line A549 which robustly expresses α7-nAChR. They synthesized α-CT-poly(ethylene glycol)-(distearoyl-sn-glycero-3-phosphoethanolaminen) (PEG-DSPE) based micelles and loaded these micelles with docetaxel. The micelles were characterized and their growth inhibitory effect was measured in A549 human LAC cells by the Sulforhodamine B assay (Mei, et al., 2018). α-CT-PEG-DSPE-docetaxel displayed significantly improved growth-suppressive activity than docetaxel alone in A549 cells (Mei, et al., 2018). Such data indicate that α7-nAChR is not only a drug target in human lung cancers but can also be used to improve the intracellular delivery of chemotherapeutic drugs in α7-nAChR-positive lung cancers.
6. Lymphocyte Antigen 6 (Ly-6) Proteins as allosteric modulators of Nicotinic Acetylcholine Receptor (nAChR): Lynx and Secreted ly6/urokinase-type plasminogen activator receptor related peptide (SLURP)
The three-dimensional structure of the Ly-6 family of proteins resembles three finger snake venom toxins, like α-bungarotoxin (V. Tsetlin, 1999; V. I. Tsetlin, 2015). The Ly-6 protein family is comprised of six members; lynx1, lynx2, SLURP-1, SLURP-2, PSCA and Pate-B (Fu, Song, & Spindel, 2015). All of these proteins have been found to be allosteric and orthosteric modulators of nAChRs (Arredondo, Chernyavsky, Jolkovsky, Webber, & Grando, 2006; Horiguchi, et al., 2009; Hruska, et al., 2009; Levitin, et al., 2008; Miwa, et al., 1999; Tekinay, et al., 2009). Studies in pre- and post-natal monkey lungs has indicated a role for lynx1 (ly from ly-6 and nx for neurotoxin) in fetal lung development. Sekhon et al., (2004) have shown that prenatal nicotine exposure alters the development of pulmonary structures and functions in the lungs of fetal monkeys by regulation of lynx1 expression. (Sekhon, et al., 2005). Furthermore, experiments in human BECs have revealed a role for lynx1 in airway physiology, mucus production, development of asthma and COPD (Fu, et al., 2012; Fu, et al., 2015).
The proteins lynx1 and lynx2 are negative allosteric regulators of multiple nAChR subunits (Ibanez-Tallon, et al., 2002; Miwa, et al., 1999; Tekinay, et al., 2009). As described in Section 4, the α7-nAChR is primarily responsible for the proliferative, pro-angiogenic and pro-metastatic activity of nicotine in lung cancer (S. A. Grando, 2008; Sergei A. Grando, 2014; Pillai & Chellappan, 2012; Schuller, 2012; Spindel, 2016; S. Wang & Hu, 2018; Zoli, et al., 2018). Immunofluorescence experiments showed that lynx1 and lynx2 were expressed in the airways and co-localized with α7-nAChR (Fu, et al., 2012). Several congruent studies indicate that lynx1 and lynx2 are negative allosteric regulators of α7-nAChR (Fu, et al., 2012; Ibanez-Tallon, et al., 2002; V. Tsetlin, Utkin, & Kasheverov, 2009). Therefore, lynx1 expression should be decreased in lung cancer relative to normal adjacent tissue. This is in alignment with data showing that human SCC-L tissue (isolated from patients) contained lower levels of lynx1 relative to adjacent normal lung tissue. The levels of lynx1 correlated with the degree of differentiation of the SCC-Ls; poorly differentiated SCC-Ls showed lower expression of lynx1 than moderately or well differentiated tumors (Fu, et al., 2015; Song, et al., 2008). Electrophysiology experiments suggest that the binding pocket for α7-nAChR lies in the extracellular domain of lynx1 (Lyukmanova, et al., 2013). Lynx2 is also a negative modulator of α7-nAChR (Fu, et al., 2015). However, no studies have addressed the potential role of lynx2 in the cell biology of lung cancer.
The effect of lynx1 on cell proliferation was studied using siRNA techniques (Fu et al., 2015). Lynx1-siRNA stimulated the proliferation of A549 human lung cancer cells. Conversely, the overexpression of lynx1 (using lentiviral vectors) suppressed the growth of A549 cells. The precise molecular mechanisms underlying the growth-inhibitory activity of lynx1 has yet to be identified. It is tempting to speculate that the growth-suppressive actions of lynx1 are mediated via inhibition of α7-nAChR (Fu, et al., 2012; Fu, et al., 2015). However, lynx1 also interacts with α4β2-nAChR and α6-nAChR (O’Neill, et al., 2012). Studies have shown that NSCLC cells and tumors overexpress α6-nAChR (Lam, et al., 2007). Similarly, CHRNA4 is one of the genes located on the 15q25 chromosomal region of the human genome, which has been shown to confer susceptibility to lung cancer in European populations (Amos, et al., 2008; Hung, et al., 2008; P. Liu, et al., 2008; Thorgeirsson, et al., 2008a). Such observations reveal that lynx1 modulates multiple nAChR subunits involved in the progression of lung cancer.
SLURP-1 is a member of Ly-6 family of proteins and functions as a positive allosteric regulator of α7-nAChR (Arredondo, Chernyavsky, Webber, & Grando, 2005; Favre, et al., 2007; Lyukmanova, et al., 2018). SLURP-1 is robustly expressed in the ciliated bronchial epithelium of the lungs (Horiguchi, et al., 2009). SLURP-1 displays potent anti-inflammatory activity and regulates ciliary beat frequency in the airways (Narumoto, et al., 2013). SLURP-1 also is profoundly downregulated in inflammatory lung diseases like asthma (Narumoto, et al., 2010; Narumoto, et al., 2013).
There are very few research papers which have examined the role of SLURP-1 in lung cancer. SLURP-1 suppresses NNK-induced transformation of human BECs. Kalantari-Dehagi et al., (2012) treated BEP2D immortalized human BECs with 1 μM of the tobacco carcinogen NNK in the presence or absence of 1 μg/ml recombinant SLURP-1 for 24 hours (Kalantari-Dehaghi, Parnell, Armand, Bernard, & Grando, 2015). Subsequently, they performed a PCR-array analysis for oncogenes and tumor suppressor genes. NNK upregulated the expression of the EGF, the pro-survival gene RB1, CTNBB1 gene (encoding for the EMT gene β-catenin), and oncogenes MYB, PIK3A, AKT and KIT in BEP2D cells. The expression of pro-apoptotic genes, namely Bax and caspase-8, were downregulated by NNK. The presence of recombinant SLURP-1 (along with NNK) completely or partially abrogated of the expression pattern of the above-mentioned genes (Kalantari-Dehaghi, et al., 2015). The gene expression signature obtained with NNK facilitated the malignant transformation of BECs, whereas SLURP-1 had a reciprocal effect on NNK-induced gene expression (Kalantari-Dehaghi, et al., 2015). Several convergent studies show that the α7-nAChR is responsible for the proliferative effects of nicotine and NNK (Sergei A. Grando, 2014; Schuller, 2012; Spindel, 2016; S. Wang & Hu, 2018; Zoli, et al., 2018). Therefore, SLURP-1 (a positive allosteric modulator of α7-nAChR) should amplify NNK-induced changes in gene expression, not reverse them (Kalantari-Dehaghi, et al., 2015). The authors speculate that perhaps there is a common binding site for NNK and SLURP-1 on α7-nAChR. The presence of SLURP-1 knocks out NNK from its cognate binding site on α7-nAChR, thereby abolishing the NNK-induced gene expression profile (Kalantari-Dehaghi, et al., 2015; Lyukmanova, Shulepko, Kudryavtsev, et al., 2016). Durek et al., (2010) generated synthetic SLURP-1 using solid phase peptide synthesis methodology. Synthetic SLURP-1 was found to potently inhibit the functional activity of rat and human α3β4-nAChR (and to a lesser extent α3β2-α4β4-nAChRs) at high concentrations of ACh (Durek, et al., 2017). Similarly, SLURP-1 suppressed ACh-induced current traces in rat and human α9-nAChRs and α10-nAChRs (Lyukmanova, Shulepko, Kudryavtsev, et al., 2016). However, these experiments were performed by transfecting the indicated nAChRs in Xenopus oocytes, not in human lung cells (Durek, et al., 2017). Published data show that the S442 naturally occurring variant of α9-nAChR promotes the growth of BEP2D BECs. Most importantly, the S442 variant stimulated spontaneous as well as NNK-induced transformation of BEP2D cells (Chikova & Grando, 2011). It may be probable that the reciprocal effects of SLURP-1 on NNK-induced gene expression is mediated by its interaction with α9-nAChRs in BECs. Lyukmanova et al., (2018) observed that SLURP-1 suppresses the viability of A549 human NSCLC cells over 24 hours. WST cell viability assays reveal that SLURP-1 had a low IC50 (1.8±0.7 nM) in A549 cells (Lyukmanova, et al., 2018). In contrast, SLURP-2 stimulated the growth of A549 cells (1.5-fold increase compared to the untreated cells) over 24 hours (Lyukmanova, et al., 2018). The role of SLURP-2 in cell proliferation is not very well understood. SLURP-2-transfected Het-1A human oral keratinocytes suppressed NNK-induced tumorigenesis (Arredondo, Chernyavsky, & Grando, 2007), whereas exogenous SLURP-2 induced proliferation of Het-1A cells (Lyukmanova, Shulepko, Shenkarev, et al., 2016). Based on such observations, further studies are required to clarify the role of SLURP-2 in the cholinergic signaling pathway in human lung cancer. Studies involving transfected Chinese hamster ovary (CHO) cells show that SLURP-2 interacts with M1R and M3R (Lyukmanova, Shulepko, Shenkarev, et al., 2016). Itmay be probable that some of the biological effects of SLURP-2 are mediated its interaction with muscarinic receptors on target cells. SLURP-2 interacts with several nAChRs and muscarinic receptor subtypes, so it is probable that such oligomeric complexes are responsible for the spectrum of downstream signaling pathways in neoplastic cells.
Swamynathan et al. (2017) observed that the treatment of HUVECs with exogenous histidine-tagged SLURP-1 suppressed TNF-α-induced angiogenic tubule formation in Matrigel models (Swamynathan, et al., 2015). Similarly, SLURP-1 decreased the adhesion of HUVECs to a panel of extracellular matrix proteins namely collagen1, collagen4, vitronectin and fibronectin. Although, these studies were performed in the context of corneal angiogenesis, they may have potential applications in the angiogenesis of lung cancers (Swamynathan, et al., 2015).
7. Cholinesterases (ChEs)
A survey of literature shows that vertebrates express two types of ChEs (Barman, et al., 2016; Pohanka, 2011). The first is AChE (also called acetylcholineacetylhydrolase) and the second is BChE (also called acylcholine acylhydrolase; Barman, et al., 2016; Colovic, Krstic, Lazarevic-Pasti, Bondzic, & Vasic, 2013; Soreq & Seidman, 2001). Classically, the central function of AChE is the hydrolysis of ACh, terminating cholinergic signaling (Barman, et al., 2016). The substrate for AChE is ACh. The identification of ChEs in non-neuronal tissue, blood and body fluids indicates that these proteins have cellular functions which are independent of its ACh-hydrolytic activity (Jiang & Zhang, 2008; Pickett, Dush, & Nascone-Yoder, 2017; Pohanka, 2011; Soreq & Seidman, 2001; Xi, et al., 2015). HUVECs express enzymatically active AChE (Carvalho, Graca, Martins-Silva, & Saldanha, 2005). The expression of AChE in HUVECs is higher than other cholinesterases. The functions of endothelial AChE are yet to be fully understood. Yeast two-hybrid experiments have revealed that AChE interacts with extracellular matrix protein like laminin and heparin, suggesting a role for this protein in cell-cell recognition and transmembrane-receptor-mediated cytoplasmic signaling pathways (Paraoanu & Layer, 2008).
The presence of multiple AChE-isoforms supports the notion that these proteins have functions independent of their role in the cholinergic signaling pathway (Zimmermann, 2013). These isoforms are generated by alternate mRNA splicing, post-translational modifications, assembly of catalytic domains and the binding of non-catalytic structural domains (Lockridge, Norgren, Johnson, & Blake, 2016; Nazim, et al., 2017; Soreq & Seidman, 2001). We will be discussing the isoforms AChE-T (tailed), AChE-R (readthrough) and AChE-S (synaptic) in this review because they have been studied in human lung cancers. Three alternative exons 4, 5, 6 (E4, E5 and E6) constitute the C-terminal motif of AChE subunits. AChE-S is a tetrameric protein containing a C-terminal collagen tail or a hydrophobic domain which enables it to anchor to the membrane (Meshorer & Soreq, 2006; Meshorer, et al., 2004; Taylor & Radic, 1994). AChE-T is generated by alternate splicing of E4 to E6. The AChE-E (erythrocyte) is a dimeric protein that is produced by joining E4 to E5. It is also called AChE-H (hydrophobic) and contains a glycosylphosphoinositol (GPI)-anchor (Paraoanu & Layer, 2008). Additional alternative splicing leads to the generation of the monomeric AChE-R protein. The effect of AChE in cellular survival is varied in an isoform-dependent manner (Meshorer & Soreq, 2006; Meshorer, et al., 2004). AChE-S is highly expressed in apoptotic cells and tissues (Jiang & Zhang, 2008). Overexpression of the N-terminally extended form of AChE-S (N-AChE-S) induces cell death in a diverse array of cells and tissues (Toiber, et al., 2008; Toiber, Greenberg, & Soreq, 2009). In contrast, AChE-R promotes cell proliferation and confers protection against injurious stimuli (Deutsch, et al., 2002; Dori & Soreq, 2006; Grisaru, Keidar, Schreiber, Lessing, & Deutsch, 2007; Jiang & Zhang, 2008; Perry, Sklan, & Soreq, 2004).
A survey of literature shows that BChE is encoded by a single gene (Allderdice, et al., 1991; Arpagaus, et al., 1990). It is a glycoprotein found in both the central and peripheral nervous system. The structure of BChE bears a 65% homology to AChE (Nicolet, Lockridge, Masson, Fontecilla-Camps, & Nachon, 2003). Both of these proteins contain a catalytic active site, a deep gorge and a peripheral anionic site (PAS; Brus, et al., 2014; Shafferman, et al., 1992). It is a pseudocholinesterase or serum cholinesterase, hydrolyzing choline, butryl- and succinyl-choline and aliphatic esters (Q. Li, Yang, Chen, & Sun, 2017). Studies have indicated that BChE can replace AChE in degrading ACh, when AChE is absent or inhibited (Chatonnet & Lockridge, 1989).
The role of AChE in human lung cancers is not very well defined. Human SCLC, SCC-L and LAC cell lines express AChE (Table 12). Our laboratory has compared the AChE expression and activity in a panel of human SCC-L cell lines and normal lung cells. We results revealed that the expression (and activity) of AChE in normal lung cells is higher than in SCC-L cell lines (Dasgupta, et al., 2018; Dasgupta, et al., 2016). We also observed that treatment with nicotine decreased the levels of AChE in human LACs relative to untreated cells (Lau, et al., 2013). However, Zhang et al., (2014) performed immunoblotting and found that the levels of AChE in H520 human SCC-Ls cells and H460 human LAC cells was higher than immortalized lung cell lines (B. Zhang, et al., 2014). Depletion of AChE by siRNA did not affect the viability of H520 human SCC-L cells. However, the transfection of AChE-siRNA abrogated the apoptotic activity of cisplatin in human SCC-L and LAC cells (B. Zhang, et al., 2014). Such observations may be explained by the fact that several anti-cancer drugs increase AChE to trigger apoptosis (Steinritz, et al., 2007; Ye, Zhang, Chen, & Zhou, 2015; X. J. Zhang & Greenberg, 2012). Another fact to note was that Zhang et al., (2014) cultured cells in fetal bovine serum-containing media (B. Zhang, et al., 2014). Fetal bovine serum contains substantial amounts of AChE (Ralston, Rush, Doctor, & Wolfe, 1985). Our laboratory conducted studies involving measurement of AChE expression and activity in serum-free medium containing bovine serum albumin (BSA) and insulin-transferrin-selenium supplement (as described by Song et al., (2003)), and measured the levels of intracellular AChE (Song, Sekhon, Jia, et al., 2003). Additionally, we used primary SAEC and NHBE for our experiments. Zhang et al., (2014) used the immortalized BEAS-2B cells for their studies (B. Zhang, et al., 2014). Such differences in the experimental design may at least in part, explain the divergent results obtained by different research groups.
Table 12.
Expression of AChE in normal human bronchial epithelial cells and lung cancer cell lines
| Cell Line | Nature of lung cells | Expression of AChE |
References |
|---|---|---|---|
| BEAS-2B | Immortalized human bronchial epithelial cells |
+ | 189, 366 |
| NHBE | Normal human bronchial epithelium |
+ | 53 |
| SAEC | Small airway epithelial cells |
+ | 53 |
|
HPAEpics |
Human pulmonary alveolar epithelial cells |
+ |
171 |
| H82, H69, H187, N417 |
Small cell lung cancer | + | 198 |
| H157, H520, H226, SK-MES-1 |
Squamous cell carcinoma of the lung |
+ | 53, 54,198 |
| A549, H358, H650, H2228, H1975, H1563, H1650, H23, H460, H1944, H1355 |
Lung adenocarcinoma | + | 53, 54, 171 198 |
Clinical studies in NSCLC patients show that the biological activity of AChE in human lung cancers varies according to the histological type of lung tumors (Table 13). Martinez-Moreno et al., (2005) compared the levels of AChE expression and functional activity in a spectrum of NSCLC and adjacent normal lung tissue isolated from patients. They found that total AChE enzyme activity in SCC-L tissue was significantly lower than adjacent normal tissue (Martinez-Moreno, et al., 2005; Martinez-Moreno, et al., 2006). The AChE activity in human SCC-L tissue is represented by the symbol “+”, whereas the AChE activity of normal lung tissue is represented by the symbol “++” to indicate that the enzymatic activity of AChE is higher in normal lung tissue relative to human SCC-Ls (Table 13; rows 1–5). The AChE expression (and activity) of human SCLCs, LACs and LCCs was comparable to normal lung tissue. A similar result was obtained from bronchial aspirates of human SCC-L patients. The AChE activity of bronchial aspirates of SCC-L patients was found to be about half of that in non-cancerous patients (Martinez-Lopez de Castro, et al., 2008). A similar representation schema has been used to describe the data involving bronchial aspirates of SCC-L patients versus those of normal individuals (using the symbol * in Table 13; rows 6–9). The data from Song et al., (2008) show that the levels of AChE in human SCC-L tissues is dependent on the extent of differentiation of the tumor (Song, et al., 2008). The well differentiated SCC-Ls did not display a significant difference of AChE levels compared to adjacent normal tissue. In contrast, AChE levels in moderately to poorly differentiated SCC-Ls showed substantially lower AChE expression relative to normal lung tissue (Song, et al., 2008). The role of BChE in lung cancer has been not been studied extensively. Martinez-Moreno et al., (2005) detected BChE activity in normal lung tissue, SCLC and NSCLC tissues (Martinez-Moreno, et al., 2005). The activity of BChE is significantly decreased in human SCC-L, LAC and LCC (isolated from patients) relative adjacent normal non-cancerous lung tissue (Martinez-Moreno, et al., 2005; Martinez-Moreno, et al., 2006; Song, et al., 2008). The decrease in functional activity of AChE and BChE in human lung cancer tissue in may be a mechanism to maximize the levels of the growth factor ACh, thereby promoting the proliferation of human lung cancer cells.
Table 13.
Expression of AChE, BuChE, total ChE activity in lung cancer tissue, bronchial aspirates, serum and adjacent normal tissue (isolated from normal individuals and patients)
| Type of lung tissue | Expression of AChE |
AChE Activity |
BuChE Expression/ Activity |
Total ChE Activity |
References | |
|---|---|---|---|---|---|---|
| 1. | Normal lung tissue | ++ | ++ | ++ | 199, 200, 299 |
|
| 2. | Small cell lung cancer tissue |
++ | ++ | ++ | 199, 200 | |
| 3. | Squamous cell lung cancer tissue |
+ | + | + | 199, 200, 299 |
|
| 4. | Lung adenocarcinoma tissue |
++ | ++ | ++ | 199, 200 | |
| 5. | Large cell lung carcinoma |
++ | ++ | ++ | 199, 200 | |
| 6 | Bronchial aspirates associated with normal lung tissue |
** | ** | 198 | ||
| 7 | Bronchial aspirates from SCLC patients |
** | ** | 198 | ||
| 8 | Bronchial aspirates from SCC-L patients |
* | * | 198 | ||
| 9 | Bronchial aspirates from LAC patients |
** | ** | 198 | ||
| 10 | Serum from normal individuals |
x | x | x | x | 362, 363 |
| 11 | Serum from lung cancer (all histological subtypes) |
x | x | x | xx | 362, 363 |
Blanks indicate Not Determined (ND)
Zakut et al., (1988) compared the total serum cholinesterase activity of 88 patients with 21 healthy volunteers who served as controls. Out of the 88 patients enrolled in the study, seven patients suffered from lung cancer. They found that total serum cholinesterase (sChE) activity of human lung patients was significantly (P≤0.05) higher than normal controls (Table 13; Zakut, et al., 1988). The total serum cholinesterase activity of lung cancer patients (denoted by xx) was higher than the cholinesterase activity of normal individuals (shown by the symbol “x” in Table 13, rows 10–11). An analogous study by Zanini et al., (2013) measured AChE activity in whole blood of NSCLC patients receiving gemcitabine or cisplatin-based chemotherapy (Zanini, et al., 2013). For normal controls, they isolated the whole blood from healthy individuals and treated them with a range of cisplatin or gemcitabine concentrations in vitro. The concentration ranges of cisplatin and gemcitabine were based on pharmacokinetic data and reflected the amount of these drugs found in the serum of patients (Zanini, et al., 2013). The enzymatic activity AChE in the whole blood of NSCLC patients showed a slight but significant (P≤0.05) elevation (231±12.81 μmol ACh/mg protein), relative to normal controls (191.4±10.89 μmol ACh/mg protein). The levels of BChE in serum remained constant across all whole blood samples (Zanini, et al., 2013).
ACh acts as a growth factor for human SCLC and NSCLCs. The regulation of AChE activity by small molecules has been investigated as a strategy for lung cancer therapy (Table 14). It may be envisaged that drugs which elevate the levels of AChE in human lung cancer cells, will cause increased degradation of the ACh and thereby decrease the concentration of ACh in the cellular milieu (Fig. 14). The downregulation of ACh production will suppress its proliferative, pro-invasive and EMT-promoting activity and block the growth and survival of human lung cancers. Lu et al., (2013) identified the synaptic microRNA-212 (miR-212) as a negative regulator of AChE expression. They also observed that miR-212 acts as a tumor suppressor in human NSCLC by targeting AChE. Furthermore, they generated AChE-S-overexpressing H520 human SCC-L cells (hereby referred to as H520-AChE cells). Experiments in athymic mouse models revealed that the growth rate of H520-AChE cells was decreased relative to mock transfected H520 tumors (L. Lu, Zhang, Zhang, Wu, & Zhang, 2013). This is in alignment with data from our laboratory and those of other laboratories showing that AChE expression (or activity) in normal lung tissue is greater than that of NSCLCs (Dasgupta, et al., 2018; Dasgupta, et al., 2016; Martinez-Lopez de Castro, et al., 2008; Martinez-Moreno, et al., 2005; Song, et al., 2008). Athymic mice studies by Lu et al., (2013) suggest that elevation of AChE expression may suppresses the growth of human SCC-Ls (L. Lu, et al., 2013). The inhibition of AChE (by eserine) stimulated the growth of rat mammary tumors (Calaf, Parra, & Garrido, 2007). Although this study was not performed in human lung cancer cells, it seems to provide the important proof-of-concept that elevation of AChE activity could attenuate cell growth and trigger apoptosis. Studies in neuronal systems have shown that the amyloid-beta peptide 1–40 fragment inhibits the release of ACh via stimulation of AChE activity (W. Hu, Gray, & Brimijoin, 2003; Kar, et al., 1998). We tested the growth-inhibitory activity of amyloid beta (Aβ) peptide fragment 1–40 and Aβ peptide fragment 1–28 in a panel of human SCC-L cell lines (Dasgupta, et al., 2018). We observed that both Aβ peptide fragment 1–40 and 1–28 elevated AChE activity in H520, H226 and SK-MES human SCC-L cells (Dasgupta, et al., 2018). Furthermore, MTT assays revealed that these peptides suppressed the viability of aforementioned SCC-L cell lines.
Table 14.
AChE-based anti-cancer drugs tested in human lung cancer cells in cell culture model.
| Name of drug | Cell line | Nature of cells | References |
|---|---|---|---|
| Poly APS polymers from Halicona (reniera) sarai |
A549, Primary NSCLC cell lines (isolated from patients) |
NSCLC | 383 |
| Aβ1–40, Aβ1–28 | H520, H226, SK- MES |
Squamous cell lung cancer |
53 |
| miRNA-132 | H520 | Squamous cell lung carcinoma |
366 |
| miRNA-212 | H520 | Squamous cell lung carcinoma |
189 |
| Methoxy azomethine- dihydroquinazolinone |
H157 | Squamous cell lung carcinoma |
138 |
| Rhodolactouchal | NCI-H460 | Lung adenocarcinoma |
225 |
| Methanolic extracts of Anonaceae |
NCI-H460, NCI-H460/ADR |
Lung adenocarcinoma |
82 |
| Dehydrocordaline | H1299 | Lung adenocarcinoma |
172 |
The term ADR denotes for Adriamycin-resistant cell line
Figure 14.
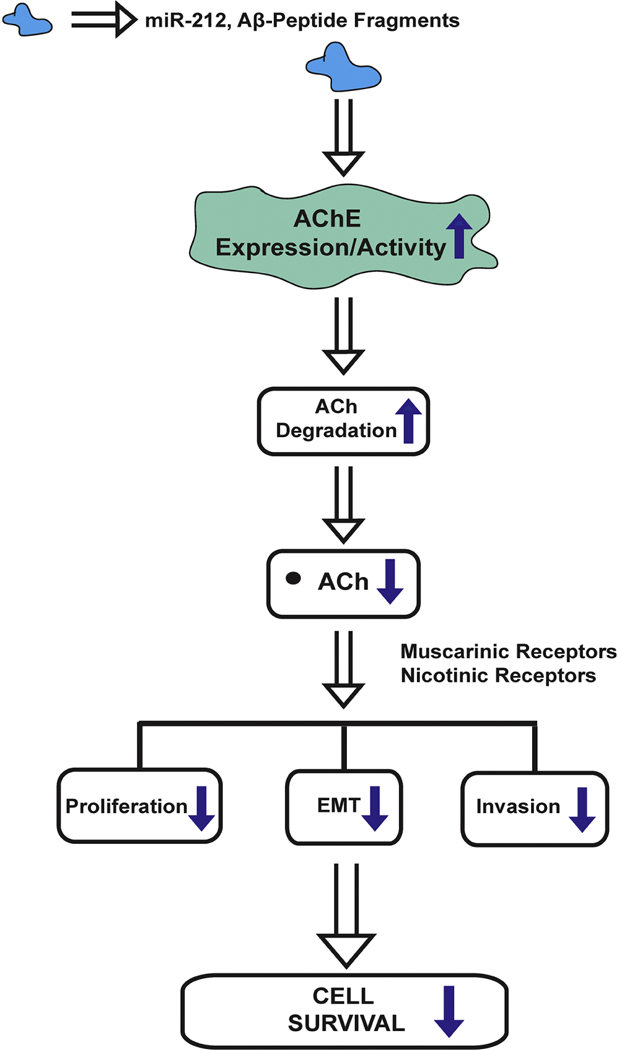
Pharmacological manipulation of AChE expression/activity may have potential applications in lung cancer therapy. AChE modulators like miR-212 and Aβ-peptide fragments increase the expression (and activity) of AChE, which in turn accelerates the degradation of acetylcholine (ACh) in human lung cancer cells. Such a decline in ACh levels decreases its tumor promoting activities like proliferation, induction of EMT and invasion potentially suppressing the progression of human lung cancers.
An interesting observation is that AChE inhibitors have also been investigated for their growth-inhibitory activity in human lung cancer cell lines. Several natural products potently inhibit AChE activity (Patel, Raghuwanshi, Masood, Acharya, & Jain, 2018). Methanolic extracts of the medicinal plant belonging Annonaceae genus were tested for their ability to inhibit AChE and cell growth in five human cancer cells (Formagio, et al., 2015). Eleven Annonaceae extracts were tested for their AChE-inhibitory/growth-suppressive activity in parent NCI-H460 and Adriamycin-resistant NCI-H460 cells. The extracts isolated from Annonaceae coriacea and Annonaceae crassiflora seeds displayed the maximal cytostatic activity in both NCI-H60 and Adriamycin-resistant NCI-H460 cell lines (Formagio, et al., 2015). A panel of compounds isolated from the plant Sonneratia ovata Backer were tested for their ability to inhibit the enzyme activity of AChE. One of the compounds, namely Rhodolatouchol (Fig. 15A) suppressed AChE activity and the growth of NCI-H460 cells in cell culture models (Nguyen, et al., 2015). 3-alkyl pyridium polymers (poly-APS), a class of compounds isolated from marine sponge extracts displayed potent AChE-inhibitory activity (Zovko, Specici, & Turk, 2009). Polymers comprising of 3-octylpyridinium units isolated from the marine sponge Haliclona (reniera) sarai displayed the greatest inhibition of AChE activity. These poly-APS compounds displayed decreased viability of A549 human NSCLC cells, as well as primary human NSCLC cell lines (isolated from NSCLC patients). These results were confirmed in colony formation assays and similar results were obtained. Poly-APS compounds induced robust apoptosis in lung cancer cells, as measured by TUNEL assays, via the activation of caspases and loss of mitochondrial membrane potential (Zovko, et al., 2009). These compounds also decreased the growth rate of A549 tumors xenografted in athymic mice. These poly-APS compounds were selective towards tumor cells and displayed minimal growth-inhibitory activity in healthy lymphocytes. In addition, these compounds displayed no toxicity (or inflammation) in the liver, heart or kidney of C57BL/6N mice (Zovko, et al., 2009).
Figure 15.
Structure of some AChE inhibitors investigated for their growth inhibitory activity. A. Rhodolatouchol B. Dehydrocorydaline
Synthetic phenylcinnamide based compounds are potent inhibitors of both AChE and BChE (Saeed, et al., 2014). These compounds displayed robust growth-inhibitory activity in H157 human SCC-L cells in vitro. Enzyme activity and molecular docking experiments demonstrate AChE-inhibitory and BChE-inhibitory activity of azomethine-dihydroquinazolinone conjugates. These compounds also displayed cytotoxic activity in H157 human lung cancer cells, as determined by Sulfarhodamine B assay (Iqbal, Saeed, Shah, al-Rashida, & Shams-ul, 2016). The 2-O-methoxy and 4-bromo derivatives of these azomethine-dihydroquinazolinone conjugates displayed the greatest selectivity for AChE over BChE. However, the growth-inhibitory activity of 2-O-methoxy-derivative was greatest (out of all the compounds tested) in H157 human lung cancer cells (Iqbal, et al., 2016). The natural alkaloid dehydrocorydaline is a potent inhibitor of AChE activity (Fig. 15B). Dehydrocorydaline abrogated migration and invasion of H1299 human LAC cells, as measured by wound-healing assays (Lee, et al., 2017). However, the caveat of these studies is that none of them have used overexpression or siRNA-based experiments to definitively prove that these compounds are mediating their growth-inhibitory activity via the AChE pathway. Natural compounds have many pleotropic biological effects apart from suppressing AChE activity. This is clearly exemplified by the data of Zhang et al., (2014) who identified AChE as a potential target of microRNA-132 (hsa-miR-132). Subsequently, they observed that hsa-miR-132 showed potent apoptotic activity in a panel of human NSCLC cell lines. However, the apoptotic activity of hsa-miR-132 was independent of its inhibition of AChE (B. Zhang, et al., 2014).
8. Conclusions and future directions
Lung cancer is the leading cause of cancer-related deaths in the United States. The mortality due to lung cancer exceeds the combined deaths from breast, prostate and colon cancers (Dela Cruz, Tanoue, & Matthay, 2011). Recent studies have shed light on molecular events contributing to LAC, leading to the development of targeted therapies (Chan & Hughes, 2015; Rolfo, et al., 2015). A major challenge with these targeted therapies is that they are minimally effective in lung cancer patients exposed to tobacco smoke. For example, targeted treatments based on EGFR and anaplastic lymphoma kinase are most effective in LAC patients who are never-smokers (Han, et al., 2012; Waller, Miller, & Petty, 2010; Y. Zhang, et al., 2015). Apart from LAC, the quest for molecular targets, suitable for lung cancer therapy has proved to be elusive. Anti-angiogenic therapies have proved problematic in NSCLC (especially SCC-L) due to severe pulmonary hemorrhage in patients and discontinuation of treatment (Johnson, et al., 2004; Piperdi, Merla, & Perez-Soler, 2014). Similarly, clinical trials involving tyrosine kinase inhibitors like sunitinib have been associated with severe toxicity in SCLC patients (H. Lu & Jiang, 2017; Ready, et al., 2015). Such sobering statistics define the arena where novel molecular targets and anti-cancer drugs are urgently required to combat this lethal malignancy.
The cholinergic pathway is functional in both SCLC and NSCLCs. Different components of the cholinergic pathway have altered functional activity between normal lung cells and lung cancer cells. Such differences have proved to the basis of investigating cholinergic ligands for treatment of lung cancers (S. A. Grando, 2008; Sergei A. Grando, 2014; Mucchietto, et al., 2016; Song & Spindel, 2008; Spindel, 2012, 2016). An important concern may be that these cholinergic ligands may display unwanted adverse side effects on the brain, muscles and the peripheral nervous system. The aim of cholinergic anti-cancer therapies is not to perturb the elements of the acetylcholine-signaling pathway but to restore them to normal levels. For example, SCC-Ls display decreased AChE activity, so AChE modulators would suppress the growth of SCC-Ls by bringing the AChE levels to those observed in normal lung tissue (Martinez-Moreno, et al., 2005; Martinez-Moreno, et al., 2006). Moreover, studies in athymic mice models and orthotopic mice models have demonstrated that the administration of these cholinergic compounds was not associated with any adverse cognitive or behavioral side effects (Akers, et al., 2017; Ami, et al., 2011; Song, Olivas, et al., 2010; Song, et al., 2008). An advantage with a few of these compounds like BW813U (ChAT antagonist) is that they have been extensively studied in neuronal models. Therefore, their effects on the brain and peripheral nervous system are well established. BW813U does not induce any adverse cognitive or behavioral effects in rats (Meck, 2006; Wenk, et al., 1986). Similarly, muscarinic receptor antagonists are being used in the clinic to treat asthma and COPD in patients. The therapeutic potential of these drugs in lung cancer is has shown promising results in preclinical model systems (Song, Sekhon, Lu, et al., 2007; Song & Spindel, 2008; Spindel, 2012, 2016). The re-purposing of these anti-asthma (or anti-COPD) drugs for lung cancer will accelerate their eventual clinical development in the treatment of human lung cancer.
An exciting new development in the field of ACh signaling pathway in lung cancer has been the design and synthesis if selective ligands suitable for imaging tumors. Selective ligands of ChAT, ChT1, nAChRs and nAChR subunits may prove useful for early detection and diagnosis of lung cancers in which these cholinergic proteins are overexpressed relative to normal tissue (Challapalli & Aboagye, 2016; Galitovskiy, et al., 2013; Gilissen, et al., 2003; Kumar, et al., 2017; M. Li, et al., 2013; Ramirez de Molina, et al., 2007; C. Ray, et al., 2017). In addition, α7-nAChR ligands have also been used as a strategy to improve the targeting and delivery of chemotherapeutic drugs to lung tumors (Mei, et al., 2018). It is hoped that selective ligands to other nAChR subunits (like the α5-nAChR and α5-nAChR-D398N) will improve the targeting efficacy of conventional anti-cancer drugs and minimize unwanted side effects (C. Ray, et al., 2017).
The nAChR-signaling pathway has transpired as an important drug target in the management and treatment of human lung cancer. Emerging studies have shown that competitive α9-nAChR and α7-nAChR antagonists may be useful for lung cancer therapy (Mucchietto, et al., 2016; Mucchietto, et al., 2018). An alternate promising drug-design strategy is to design allosteric modulators of nAChRs, which do not suppress the physiological function of nAChRs. Endogenous allosteric modulators of nAChRs namely to the Ly-6 family of proteins have been detected in the bronchial epithelium. The mechanisms by which these Ly-6 proteins regulate nAChRs are poorly understood (Fu, et al., 2015; Lyukmanova, Shulepko, Shenkarev, et al., 2016; Song, et al., 2008; Spindel, 2016). The identification of molecular targets downstream of the lynx and SLURP proteins will foster the design of allosteric nAChR ligands with an improved biological activity and side effect profile (Arredondo, et al., 2007; Chernyavsky, et al., 2010; Durek, et al., 2017; Horiguchi, et al., 2009; Kalantari-Dehaghi, Bernard, & Grando, 2012; Lyukmanova, et al., 2018; Lyukmanova, Shulepko, Kudryavtsev, et al., 2016; Lyukmanova, Shulepko, Shenkarev, et al., 2016).
The signaling proteins like CTL1–5, OCTs and OCTNs are promising drug targets for lung cancer (Inazu, 2014; Pochini, Scalise, Galluccio, & Indiveri, 2012; Pochini, et al., 2013; Pochini, Scalise, Galluccio, Pani, et al., 2012; Song, Mark, et al., 2010; Song, et al., 2013; Spindel, 2016; Tamai, 2013; Traiffort, et al., 2013; Volk, 2014). Choline is also a vital component of phospholipid synthesis (Inazu, 2014; Traiffort, et al., 2013). Therefore, drugs targeting choline uptake will suppress the growth of lung cancer cells by two mechanisms; first, by blocking the production of the growth factor ACh and second by suppressing cell membrane synthesis in lung cancer cells.
Future research will clarify the signaling pathways regulating the production of ACh in human lung cancer cells. An interesting observation is that human microvascular endothelial cells secrete, transport and degrade ACh (Carvalho, et al., 2005; Haberberger, Bodenbenner, & Kummer, 2000; Kirkpatrick, et al., 2003; Kirkpatrick, et al., 2001). Endothelial ACh has been implicated in the transport of intracellular calcium, relaxation of arteries and protecting the endothelium from hypoxia/reoxygenation injury (Chataigneau, et al., 1999; Wilson, Lee, & McCarron, 2016; M. Zhao, et al., 2015). The pro-angiogenic activity of nAChRs in human lung cancers has been extensively studied (Cooke, 2007; Cooke & Ghebremariam, 2008; J. C. Wu, et al., 2009). Similarly, the M3R receptor antagonist 4-DAMP displayed robust anti-angiogenic activity in athymic mouse models xenografted with human SCLC (C. Hu & Zhang, 2017). However, the potential role of other cholinergic proteins namely ChAT, CTLs, VAChT and AChE in neovascularization of lung tumors is yet to be understood. A few cholinergic proteins like ChAT, CTLs and AChE exist in multiple isoforms. AChE and CTL isoforms have been detected in human lung cancer tissues, as well as bronchial aspirates of lung cancer patients (Martinez-Moreno, et al., 2005; Martinez-Moreno, et al., 2006). The function of these cholinergic protein isoforms in the pathophysiology of lung cancer is unknown. Studies in human lung, oral cancers and colon cancers have revealed the existence of two types of nAChRs, one localized on the cell membranes and the other on mitochondrial membranes (Chernyavsky, Shchepotin, Galitovkiy, et al., 2015; Chernyavsky, Shchepotin, & Grando, 2015; S. A. Grando, et al., 2015). The proliferative activity of nAChRs are controlled by membrane-bound nAChRs, whereas the pro-survival effects of nAChRs are mediated via the mitochondrial nAChRs. Although experiments in mouse lungs have detected mitochondrial nAChRs, their functions are yet to be understood in normal lung epithelial cells and in lung cancer. The development of selective high affinity cholinergic modulators will provide valuable insights into non-neuronal ACh-mediated-signaling pathways and will foster the hope of novel acetylcholine pathway-based therapies in lung cancer.
Acknowledgements
We acknowledge Dr. S. Chellappan and his laboratory for their continuous support. We would like to thank Ashley Duff for proofreading the manuscript. SDR and JCM are recipients of NSF-SURE and WV-NASA Space Consortium undergraduate fellowships respectively. PD is supported by a National Institutes of Health R15 Academic Research Enhancement Award (Grants 1R15CA161491–01A1 and 2R15CA161491–02).
List of abbreviations used 3 or more times
- α-NETA
2-(alpha-naphthoyl)ethyltrimethylammonium
- ACh
Acetylcholine
- AChE
Acetylcholinesterase or acetylcholineacetylhydrolase
- AFDX-116
{11-[[2-[(Diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one}
- BAC
Bronchioalveolar carcinoma
- BChE
Butyrylcholinesterase or acylcholine acylhydrolase
- BCX
β-Cryptoxanthin
- BECs
Bronchial epithelial cells
- BrdU
Bromodeoxyuridine
- CarAT
Carnitine acetyltransferase
- ChAT
Choline acetyltransferase
- ChE
Cholinesterase
- ChT
Choline transporter
- ChT1/SLC5A7
High affinity choline transporter 1
- COPD
Chronic obstructive pulmonary disease
- CTL1–5
Choline transporter-like proteins 1–5
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- EGFR
Epithelial-to-mesenchymal transition
- ERK
Extracellular related kinase
- H69
NCI-H69
- H82
NCI-H82
- HC-3
Hemicholinium-3
- HMEC-L
Human microvascular endothelial cells of the lung
- hsa-miR-132
microRNA-132
- HUVECs
Human umbilical cord microvascular endothelial cells
- IGF-2
Insulin growth factor-2
- LAC
Lung adenocarcinoma
- LCC
Large cell carcinoma
- Ly-6
Lymphocyte antigen 6
- M1R to M5R
Muscarinic Receptor 1–5
- MAPK
Mitogen activated protein kinase
- MMP
Matrix metalloproteinases
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
- nAChR
Nicotinic acetylcholine receptors
- NEP
Norepinephrine
- NETs
Lung neuroendocrine tumors
- NHBE
Normal bronchial epithelial cells
- NNK
Nicotine-derived nitrosamine ketone, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NSCLC
Non-small cell lung cancer
- OAT
Organic anion transporter
- OCT
Organic cation transporter
- OCT1–3/SLC22A1–2
Polyspecific organic transporters
- OCTN1 and OCTN2
Carnitine/cation transporters
- PI-3 kinase
Phosphoinositol-3 kinase
- poly-APS
3-alkyl pyridium polymers
- SAEC
Primary small airway epithelial cells
- SCC-L
Squamous cell carcinoma of the lung
- SCLC
Small cell lung cancer
- shRNA
Short-hairpin RNA
- siRNA
Small interfering RNA
- SLURP
Secreted ly6/urokinase-type plasminogen activator receptor related peptide
- SNPs
Single nucleotide polymorphisms
- STAT3
Signal transducer and activator of transcription 3
- TGF-β1
Transforming growth factor β1
- VAChT
Vesicular acetylcholine transporter
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
- 1.Akers AT, Brown KC, Colclough KW, Nolan NA, Friedman JR, Miles SL, Bow EW, Rimoldi JM, Hardman EW, & Dasgupta P (2018). Inhibition of choline acetyltransferase activity abrogates the growth of lung adenocarcinoma patients who are exposed to tobacco smoke. In Experimental Biology April 21–25, 2018 San Diego, CA: Abstract #677.18. [Google Scholar]
- 2.Akers AT, Colclough KW, Friedman JR, Bow EW, Rimoldi JM, Cutler SJ, Hardman EW, & Dasgupta P (2017). The choline acetyltransferaseinhibitor BW813U surpresses the growth of lung adenocarcinoma from smokers. In Experimental Biology April 22–26, 2017 Chicago, IL: Abstract 996.11. [Google Scholar]
- 3.Al-Wadei HA, Al-Wadei MH, Masi T, & Schuller HM (2010). Chronic exposure to estrogen and the tobacco carcinogen NNK cooperatively modulates nicotinic receptors in small airway epithelial cells. Lung Cancer, 69, 33–39. [DOI] [PubMed] [Google Scholar]
- 4.Alasmari F, Crotty Alexander L. E., Nelson JA, Schiefer IT, Breen E, Drummond CA, & Sari Y (2017). Effects of chronic inhalation of electronic cigarettes containing nicotine on glial glutamate transporters and alpha-7 nicotinic acetylcholine receptor in female CD-1 mice. Prog Neuropsychopharmacol Biol Psychiatry, 77, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albano GD, Bonanno A, Moscato M, Anzalone G, Di Sano C, Riccobono L, Wenzel SE, & Profita M (2018). Crosstalk between mAChRM3 and beta2AR, via acetylcholine PI3/PKC/PBEP1/Raf-1 MEK1/2/ERK1/2 pathway activation, in human bronchial epithelial cells after long-term cigarette smoke exposure. Life Sci, 192, 99–109. [DOI] [PubMed] [Google Scholar]
- 6.Allderdice PW, Gardner HA, Galutira D, Lockridge O, LaDu BN, & McAlpine PJ (1991). The cloned butyrylcholinesterase (BCHE) gene maps to a single chromosome site, 3q26. Genomics, 11, 452–454. [DOI] [PubMed] [Google Scholar]
- 7.Ami N, Koga K, Fushiki H, Ueno Y, Ogino Y, & Ohta H (2011). Selective M3 muscarinic receptor antagonist inhibits small-cell lung carcinoma growth in a mouse orthotopic xenograft model. J Pharmacol Sci, 116, 81–88. [DOI] [PubMed] [Google Scholar]
- 8.Amos CI, Gorlov IP, Dong Q, Wu X, Zhang H, Lu EY, Scheet P, Greisinger AJ, Mills GB, & Spitz MR (2010). Nicotinic acetylcholine receptor region on chromosome 15q25 and lung cancer risk among African Americans: a case-control study. J Natl Cancer Inst, 102, 1199–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, Wang Y, Mills G, Doheny K, Tsai YY, Chen WV, Shete S, Spitz MR, & Houlston RS (2008). Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet, 40, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arias HR, Richards VE, Ng D, Ghafoori ME, Le V, & Mousa SA (2009). Role of non-neuronal nicotinic acetylcholine receptors in angiogenesis. Int J Biochem Cell Biol, 41, 1441–1451. [DOI] [PubMed] [Google Scholar]
- 11.Arpagaus M, Kott M, Vatsis KP, Bartels CF, La Du BN, & Lockridge O (1990). Structure of the gene for human butyrylcholinesterase. Evidence for a single copy. Biochemistry, 29, 124–131. [DOI] [PubMed] [Google Scholar]
- 12.Arredondo J, Chernyavsky AI, & Grando SA (2007). Overexpression of SLURP-1 and −2 alleviates the tumorigenic action of tobacco-derived nitrosamine on immortalized oral epithelial cells. Biochem Pharmacol, 74, 1315–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arredondo J, Chernyavsky AI, Jolkovsky DL, Webber RJ, & Grando SA (2006). SLURP-2: A novel cholinergic signaling peptide in human mucocutaneous epithelium. J Cell Physiol, 208, 238–245. [DOI] [PubMed] [Google Scholar]
- 14.Arredondo J, Chernyavsky AI, Webber RJ, & Grando SA (2005). Biological effects of SLURP-1 on human keratinocytes. J Invest Dermatol, 125, 1236–1241. [DOI] [PubMed] [Google Scholar]
- 15.Barman SM, Barrett KE, Boitano S, & Brooks HL (2016). Neurotransmitters and Neuromodulators. In Weitz M & Kearns B (Eds.), Ganong’s Review of Medical Physiology (25 ed., pp. 137–156): McGraw-Hill Education. [Google Scholar]
- 16.Beckmann J, & Lips KS (2013). The non-neuronal cholinergic system in health and disease. Pharmacology, 92, 286–302. [DOI] [PubMed] [Google Scholar]
- 17.Bellier JP, & Kimura H (2011). Peripheral type of choline acetyltransferase: biological and evolutionary implications for novel mechanisms in cholinergic system. J Chem Neuroanat, 42, 225–235. [DOI] [PubMed] [Google Scholar]
- 18.Berrettini WH, & Doyle GA (2012). The CHRNA5-A3-B4 gene cluster in nicotine addiction. Mol Psychiatry, 17, 856–866. [DOI] [PubMed] [Google Scholar]
- 19.Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, Kuperman S, Madden PA, Mayo K, Nurnberger J Jr., Pomerleau O, Porjesz B, Reyes O, Schuckit M, Swan G, Tischfield JA, Edenberg HJ, Rice J sP., & Goate AM (2008). Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry, 165, 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birman S, Meunier FM, Lesbats B, Le Caer JP, Rossier J, & Israel M (1990). A 15 kDa proteolipid found in mediatophore preparations from Torpedo electric organ presents high sequence homology with the bovine chromaffin granule protonophore. FEBS Lett, 261, 303–306. [DOI] [PubMed] [Google Scholar]
- 21.Boo HJ, Min HY, Jang HJ, Yun HJ, Smith JK, Jin Q, Lee HJ, Liu D, Kweon HS, Behrens C, Lee JJ, Wistuba II, Lee E, Hong K, & Lee HY (2016). The tobacco-specific carcinogen-operated calcium channel promotes lung tumorigenesis via IGF2 exocytosis in lung epithelial cells. Nat Commun, 7, 12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bordas A, Cedillo JL, Arnalich F, Esteban-Rodriguez I, Guerra-Pastrian L, de Castro J, Martin-Sanchez C, Atienza G, Fernandez-Capitan C, Rios JJ, & Montiel C (2017). Expression patterns for nicotinic acetylcholine receptor subunit genes in smoking-related lung cancers. Oncotarget, 8, 67878–67890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brochier G, Israel M, & Lesbats B (1993). Immunolabelling of the presynaptic membrane of Torpedo electric organ nerve terminals with an antiserum towards the acetylcholine releasing protein mediatophore. Biol Cell, 78, 145–154. [DOI] [PubMed] [Google Scholar]
- 24.Brochier G, & Morel N (1993). The same 15 kDa proteolipid subunit is a constituent of two different proteins in Torpedo, the acetylcholine releasing protein mediatophore and the vacuolar H+ ATPase. Neurochem Int, 23, 525–539. [DOI] [PubMed] [Google Scholar]
- 25.Broers JL, Pahlplatz MM, Katzko MW, Oud PS, Ramaekers FC, Carney DN, & Vooijs GP (1988). Quantitative description of classic and variant small cell lung cancer cell lines by nuclear image cytometry. Cytometry, 9, 426–431. [DOI] [PubMed] [Google Scholar]
- 26.Brown DA (2018). Regulation of neural ion channels by muscarinic receptors. Neuropharmacology, 136, 383–400. [DOI] [PubMed] [Google Scholar]
- 27.Brown KC, Lau JK, Dom AM, Witte TR, Luo H, Crabtree CM, Shah YH, Shiflett BS, Marcelo AJ, Proper NA, Hardman WE, Egleton RD, Chen YC, Mangiarua EI, & Dasgupta P (2012). MG624, an alpha7-nAChR antagonist, inhibits angiogenesis via the Egr-1/FGF2 pathway. Angiogenesis, 15, 99–114. [DOI] [PubMed] [Google Scholar]
- 28.Brunzell DH, Stafford AM, & Dixon CI (2015). Nicotinic receptor contributions to smoking: insights from human studies and animal models. Curr Addict Rep, 2, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brus B, Kosak U, Turk S, Pislar A, Coquelle N, Kos J, Stojan J, Colletier JP, & Gobec S (2014). Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J Med Chem, 57, 8167–8179. [DOI] [PubMed] [Google Scholar]
- 30.Byun J, Schwartz AG, Lusk C, Wenzlaff AS, de Andrade M, Mandal D, Gaba C, Yang P, You M, Kupert EY, Anderson MW, Han Y, Li Y, Qian D, Stilp A, Laurie C, Nelson S, Zheng W, Hung RJ, Gaborieau V, McKay J, Brennan P, Caporaso NE, Landi MT, Wu X, McLaughlin JR, Brhane Y, Bosse Y, Pinney SM, Bailey-Wilson JE, & Amos CI (2018). Genome-Wide Association Study of Familial Lung Cancer. Carcinogenesis [DOI] [PMC free article] [PubMed]
- 31.Caihong H, & Shuxiang Z (2017). Muscarinic cholinergic receptor antagonist inhibits the growth and angiogenesis of small cell lung cancer in vivo. TUMOR, 37, 324–333. [Google Scholar]
- 32.Calaf GM, Parra E, & Garrido F (2007). Cell proliferation and tumor formation induced by eserine, an acetylcholinesterase inhibitor, in rat mammary gland. Oncol Rep, 17, 25–33. [DOI] [PubMed] [Google Scholar]
- 33.Campoy FJ, Vidal CJ, Munoz-Delgado E, Montenegro MF, Cabezas-Herrera J, & Nieto-Ceron S (2016). Cholinergic system and cell proliferation. Chem Biol Interact, 259, 257–265. [DOI] [PubMed] [Google Scholar]
- 34.Carvalho FA, Graca LM, Martins-Silva J, & Saldanha C (2005). Biochemical characterization of human umbilical vein endothelial cell membrane bound acetylcholinesterase. FEBS J, 272, 5584–5594. [DOI] [PubMed] [Google Scholar]
- 35.Challapalli A, & Aboagye EO (2016). Positron Emission Tomography Imaging of Tumor Cell Metabolism and Application to Therapy Response Monitoring. Front Oncol, 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan BA, & Hughes BG (2015). Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res, 4, 36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chataigneau T, Feletou M, Huang PL, Fishman MC, Duhault J, & Vanhoutte PM (1999). Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br J Pharmacol, 126, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatonnet A, & Lockridge O (1989). Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem J, 260, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen LS, Baker T, Hung RJ, Horton A, Culverhouse R, Hartz S, Saccone N, Cheng I, Deng B, Han Y, Hansen HM, Horsman J, Kim C, Rosenberger A, Aben KK, Andrew AS, Chang SC, Saum KU, Dienemann H, Hatsukami DK, Johnson EO, Pande M, Wrensch MR, McLaughlin J, Skaug V, van der Heijden EH, Wampfler J, Wenzlaff A, Woll P, Zienolddiny S, Bickeboller H, Brenner H, Duell EJ, Haugen A, Bruske I, Kiemeney LA, Lazarus P, Le Marchand L, Liu G, Mayordomo J, Risch A, Schwartz AG, Teare MD, Wu X, Wiencke JK, Yang P, Zhang ZF, Spitz MR, Amos CI, & Bierut LJ (2016). Genetic Risk Can Be Decreased: Quitting Smoking Decreases and Delays Lung Cancer for Smokers With High and Low CHRNA5 Risk Genotypes - A Meta-Analysis. EBioMedicine, 11, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen LS, Hung RJ, Baker T, Horton A, Culverhouse R, Saccone N, Cheng I, Deng B, Han Y, Hansen HM, Horsman J, Kim C, Lutz S, Rosenberger A, Aben KK, Andrew AS, Breslau N, Chang SC, Dieffenbach AK, Dienemann H, Frederiksen B, Han J, Hatsukami DK, Johnson EO, Pande M, Wrensch MR, McLaughlin J, Skaug V, van der Heijden HF, Wampfler J, Wenzlaff A, Woll P, Zienolddiny S, Bickeboller H, Brenner H, Duell EJ, Haugen A, Heinrich J, Hokanson JE, Hunter DJ, Kiemeney LA, Lazarus P, Le Marchand L, Liu G, Mayordomo J, Risch A, Schwartz AG, Teare D, Wu X, Wiencke JK, Yang P, Zhang ZF, Spitz MR, Kraft P, Amos CI, & Bierut LJ (2015). CHRNA5 risk variant predicts delayed smoking cessation and earlier lung cancer diagnosis--a meta-analysis. J Natl Cancer Inst, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, Gorlov IP, Merriman KW, Weng SF, Foy M, Keener G, Amos CI, Spitz MR, Kimmel M, & Gorlova OY (2011). Association of smoking with tumor size at diagnosis in non-small cell lung cancer. Lung Cancer, 74, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chernyavsky AI, Arredondo J, Galitovskiy V, Qian J, & Grando SA (2010). Upregulation of nuclear factor-kappaB expression by SLURP-1 is mediated by alpha7-nicotinic acetylcholine receptor and involves both ionic events and activation of protein kinases. Am J Physiol Cell Physiol, 299, C903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chernyavsky AI, Shchepotin IB, Galitovkiy V, & Grando SA (2015). Mechanisms of tumor-promoting activities of nicotine in lung cancer: synergistic effects of cell membrane and mitochondrial nicotinic acetylcholine receptors. BMC Cancer, 15, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chernyavsky AI, Shchepotin IB, & Grando SA (2015). Mechanisms of growth-promoting and tumor-protecting effects of epithelial nicotinic acetylcholine receptors. Int Immunopharmacol, 29, 36–44. [DOI] [PubMed] [Google Scholar]
- 45.Chikova A, & Grando SA (2011). Naturally occurring variants of human Alpha9 nicotinic receptor differentially affect bronchial cell proliferation and transformation. PLoS One, 6, e27978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colovic MB, Krstic DZ, Lazarevic-Pasti TD, Bondzic AM, & Vasic VM (2013). Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol, 11, 315–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooke JP (2007). Angiogenesis and the role of the endothelial nicotinic acetylcholine receptor. Life Sci, 80, 2347–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooke JP, & Ghebremariam YT (2008). Endothelial nicotinic acetylcholine receptors and angiogenesis. Trends Cardiovasc Med, 18, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curtis BR, Cox NJ, Sullivan MJ, Konkashbaev A, Bowens K, Hansen K, & Aster RH (2010). The neutrophil alloantigen HNA-3a (5b) is located on choline transporter-like protein 2 and appears to be encoded by an R>Q154 amino acid substitution. Blood, 115, 2073–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dang N, Meng X, & Song H (2016). Nicotinic acetylcholine receptors and cancer. Biomed Rep, 4, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dasgupta P, & Chellappan SP (2006). Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle, 5, 2324–2328. [DOI] [PubMed] [Google Scholar]
- 52.Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, & Chellappan S (2006). Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A, 103, 6332–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dasgupta P, Lau JK, Brown KC, Bow EW, Robateau ZR, Rollyson WD, Stover CA, Rimoldi JM, Cutler S, Hardman EW, Carpenter AB, & Chen YC (2018). Acetylcholine signaling pathway: A novel target for lung cancer in smokers. In Experimental Biology April 21–25 2018 San Diego, CA: Abstract #677.19. [Google Scholar]
- 54.Dasgupta P, Lau JK, Brown KC, Rollyson WD, Stover CA, Rimoldi JM, Cutler S, Hardman EW, Carpenter AB, & Chen YC (2016). Acetylcholine-signaling inhibitors for lung cancer therapy. In Experimental Biology April 2–6, 2016 San Diego, CA: Abstract #699.6. [Google Scholar]
- 55.Dasgupta P, Rastogi S, Pillai S, Ordonez-Ercan D, Morris M, Haura E, & Chellappan S (2006). Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest, 116, 2208–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dasgupta P, Rizwani W, Pillai S, Davis R, Banerjee S, Hug K, Lloyd M, Coppola D, Haura E, & Chellappan SP (2011). ARRB1-mediated regulation of E2F target genes in nicotine-induced growth of lung tumors. J Natl Cancer Inst, 103, 317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, & Chellappan S (2009). Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer, 124, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis R, Rizwani W, Banerjee S, Kovacs M, Haura E, Coppola D, & Chellappan S (2009). Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS One, 4, e7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Castro BM, De Jaeger X, Martins-Silva C, Lima RD, Amaral E, Menezes C, Lima P, Neves CM, Pires RG, Gould TW, Welch I, Kushmerick C, Guatimosim C, Izquierdo I, Cammarota M, Rylett RJ, Gomez MV, Caron MG, Oppenheim RW, Prado MA, & Prado VF (2009). The vesicular acetylcholine transporter is required for neuromuscular development and function. Mol Cell Biol, 29, 5238–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Lucas-Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R, Cruces J, Sanchez-Pacheco A, Andres-Mateos E, & Montiel C (2011). Function of partially duplicated human alpha77 nicotinic receptor subunit CHRFAM7A gene: potential implications for the cholinergic anti-inflammatory response. J Biol Chem, 286, 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dela Cruz C. S., Tanoue LT, & Matthay RA (2011). Lung cancer: epidemiology, etiology, and prevention. Clin Chest Med, 32, 605–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deutsch VR, Pick M, Perry C, Grisaru D, Hemo Y, Golan-Hadari D, Grant A, Eldor A, & Soreq H (2002). The stress-associated acetylcholinesterase variant AChE-R is expressed in human CD34(+) hematopoietic progenitors and its C-terminal peptide ARP promotes their proliferation. Exp Hematol, 30, 1153–1161. [DOI] [PubMed] [Google Scholar]
- 63.Di Bari M, Bevilacqua V, De Jaco A, Laneve P, Piovesana R, Trobiani L, Talora C, Caffarelli E, & Tata AM (2018). Mir-34a-5p Mediates Cross-Talk between M2 Muscarinic Receptors and Notch-1/EGFR Pathways in U87MG Glioblastoma Cells: Implication in Cell Proliferation. Int J Mol Sci, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dinakar C, & O’Connor GT (2016). The Health Effects of Electronic Cigarettes. N Engl J Med, 375, 1372–1381. [DOI] [PubMed] [Google Scholar]
- 65.Dobransky T, Davis WL, & Rylett RJ (2001). Functional characterization of phosphorylation of 69-kDa human choline acetyltransferase at serine 440 by protein kinase C. J Biol Chem, 276, 22244–22250. [DOI] [PubMed] [Google Scholar]
- 66.Dobransky T, Davis WL, Xiao GH, & Rylett RJ (2000). Expression, purification and characterization of recombinant human choline acetyltransferase: phosphorylation of the enzyme regulates catalytic activity. Biochem J, 349, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobransky T, Doherty-Kirby A, Kim AR, Brewer D, Lajoie G, & Rylett RJ (2004). Protein kinase C isoforms differentially phosphorylate human choline acetyltransferase regulating its catalytic activity. J Biol Chem, 279, 52059–52068. [DOI] [PubMed] [Google Scholar]
- 68.Dobransky T, & Rylett RJ (2003). Functional regulation of choline acetyltransferase by phosphorylation. Neurochem Res, 28, 537–542. [DOI] [PubMed] [Google Scholar]
- 69.Dobransky T, & Rylett RJ (2005). A model for dynamic regulation of choline acetyltransferase by phosphorylation. J Neurochem, 95, 305–313. [DOI] [PubMed] [Google Scholar]
- 70.Dori A, & Soreq H (2006). ARP, the cleavable C-terminal peptide of “readthrough” acetylcholinesterase, promotes neuronal development and plasticity. J Mol Neurosci, 28, 247–255. [DOI] [PubMed] [Google Scholar]
- 71.Doroshow DB, & Herbst RS (2018). Treatment of Advanced Non-Small Cell Lung Cancer in 2018. JAMA Oncol, 4, 569–570. [DOI] [PubMed] [Google Scholar]
- 72.Durek T, Shelukhina IV, Tae HS, Thongyoo P, Spirova EN, Kudryavtsev DS, Kasheverov IE, Faure G, Corringer PJ, Craik DJ, Adams DJ, & Tsetlin VI (2017). Interaction of Synthetic Human SLURP-1 with the Nicotinic Acetylcholine Receptors. Sci Rep, 7, 16606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Durham AL, & Adcock IM (2015). The relationship between COPD and lung cancer. Lung Cancer, 90, 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Egleton RD, Brown KC, & Dasgupta P (2008). Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci, 29, 151–158. [DOI] [PubMed] [Google Scholar]
- 75.Egleton RD, Brown KC, & Dasgupta P (2009). Angiogenic activity of nicotinic acetylcholine receptors: implications in tobacco-related vascular diseases. Pharmacol Ther, 121, 205–223. [DOI] [PubMed] [Google Scholar]
- 76.Eiden LE, Schafer MK, Weihe E, & Schutz B (2004). The vesicular amine transporter family (SLC18): amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine. Pflugers Arch, 447, 636–640. [DOI] [PubMed] [Google Scholar]
- 77.Erickson JD, Weihe E, Schafer MK, Neale E, Williamson L, Bonner TI, Tao-Cheng JH, & Eiden LE (1996). The VAChT/ChAT “cholinergic gene locus”: new aspects of genetic and vesicular regulation of cholinergic function. Prog Brain Res, 109, 69–82. [DOI] [PubMed] [Google Scholar]
- 78.Fan Y, & Wang K (2017). Nicotine induces EP4 receptor expression in lung carcinoma cells by acting on AP-2alpha: The intersection between cholinergic and prostanoid signaling. Oncotarget, 8, 75854–75863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Farine L, Niemann M, Schneider A, & Butikofer P (2015). Phosphatidylethanolamine and phosphatidylcholine biosynthesis by the Kennedy pathway occurs at different sites in Trypanosoma brucei. Sci Rep, 5, 16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Favre B, Plantard L, Aeschbach L, Brakch N, Christen-Zaech S, de Viragh PA, Sergeant A, Huber M, & Hohl D (2007). SLURP1 is a late marker of epidermal differentiation and is absent in Mal de Meleda. J Invest Dermatol, 127, 301–308. [DOI] [PubMed] [Google Scholar]
- 81.Figueroa KW, Griffin MT, & Ehlert FJ (2009). Selectivity of agonists for the active state of M1 to M4 muscarinic receptor subtypes. J Pharmacol Exp Ther, 328, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Formagio AS, Vieira MC, Volobuff CR, Silva MS, Matos AI, Cardoso CA, Foglio MA, & Carvalho JE (2015). In vitro biological screening of the anticholinesterase and antiproliferative activities of medicinal plants belonging to Annonaceae. Braz J Med Biol Res, 48, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fu XW, Rekow SS, & Spindel ER (2012). The ly-6 protein, lynx1, is an endogenous inhibitor of nicotinic signaling in airway epithelium. Am J Physiol Lung Cell Mol Physiol, 303, L661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fu XW, Song PF, & Spindel ER (2015). Role of Lynx1 and related Ly6 proteins as modulators of cholinergic signaling in normal and neoplastic bronchial epithelium. Int Immunopharmacol, 29, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, Kawashima K (2017a). Expression and Function of the Cholinergic System in Immune Cells. Front Immunol, 8, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, & Kawashima K (2017b). Physiological functions of the cholinergic system in immune cells. J Pharmacol Sci, 134, 1–21. [DOI] [PubMed] [Google Scholar]
- 87.Fujii T, Takada-Takatori Y, Horiguchi K, & Kawashima K (2012). Mediatophore regulates acetylcholine release from T cells. J Neuroimmunol, 244, 16–22. [DOI] [PubMed] [Google Scholar]
- 88.Fujii T, Takada-Takatori Y, & Kawashima K (2012). Regulatory mechanisms of acetylcholine synthesis and release by T cells. Life Sci, 91, 981–985. [DOI] [PubMed] [Google Scholar]
- 89.Furrukh M (2013). Tobacco Smoking and Lung Cancer: Perception-changing facts. Sultan Qaboos Univ Med J, 13, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gahring LC, Myers EJ, Dunn DM, Weiss RB, & Rogers SW (2017). Lung epithelial response to cigarette smoke and modulation by the nicotinic alpha 7 receptor. PLoS One, 12, e0187773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galitovskiy V, Kuruvilla SA, Sevriokov E, Corches A, Pan ML, Kalantari-Dehaghi M, Chernyavsky AI, Mukherjee J, & Grando SA (2013). Development of novel approach to diagnostic imaging of lung cancer with (18)F-Nifene PET/CT using A/J mice treated with NNK. J Cancer Res Ther (Manch), 1, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gandara DR, Hammerman PS, Sos ML, Lara PN Jr., & Hirsch FR (2015). Squamous cell lung cancer: from tumor genomics to cancer therapeutics. Clin Cancer Res, 21, 2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao X, Zhang Y, Breitling LP, & Brenner H (2016). Tobacco smoking and methylation of genes related to lung cancer development. Oncotarget, 7, 59017–59028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, Moore T, Jacobs S, Meriwether J, Choi MJ, Kim EJ, Walton K, Buiting K, Davis A, Breese C, Freedman R, & Leonard S (1998). Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics, 52, 173–185. [DOI] [PubMed] [Google Scholar]
- 95.Gazdar AF, Bunn PA, & Minna JD (2017). Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer, 17, 765. [DOI] [PubMed] [Google Scholar]
- 96.Gazdar AF, Carney DN, Nau MM, & Minna JD (1985). Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties. Cancer Res, 45, 2924–2930. [PubMed] [Google Scholar]
- 97.Gergalova G, Lykhmus O, Kalashnyk O, Koval L, Chernyshov V, Kryukova E, Tsetlin V, Komisarenko S, & Skok M (2012). Mitochondria express alpha7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: study on isolated mitochondria. PLoS One, 7, e31361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gergalova G, Lykhmus O, Komisarenko S, & Skok M (2014). alpha7 nicotinic acetylcholine receptors control cytochrome c release from isolated mitochondria through kinase-mediated pathways. Int J Biochem Cell Biol, 49, 26–31. [DOI] [PubMed] [Google Scholar]
- 99.Gheldof A, & Berx G (2013). Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci, 116, 317–336. [DOI] [PubMed] [Google Scholar]
- 100.Gilissen C, de Groot TJ, Bronfman F, van Leuven F, Verbruggen AM, & Bormans GM (2003). Evaluation of 18F-FA-4 and 11C-pipzA-4 as radioligands for the in vivo evaluation of the high-affinity choline uptake system. J Nucl Med, 44, 269–275. [PubMed] [Google Scholar]
- 101.Glunde K, Bhujwalla ZM, & Ronen SM (2011). Choline metabolism in malignant transformation. Nat Rev Cancer, 11, 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Glunde K, Penet MF, Jiang L, Jacobs MA, & Bhujwalla ZM (2015). Choline metabolism-based molecular diagnosis of cancer: an update. Expert Rev Mol Diagn, 15, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gong L, Wu D, Zou J, Chen J, Chen L, Chen Y, Ni C, & Yuan H (2016). Prognostic impact of serum and tissue MMP-9 in non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget, 7, 18458–18468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gosens R, Zaagsma J, Meurs H, & Halayko AJ (2006). Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res, 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gotti C, & Clementi F (2004). Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol, 74, 363–396. [DOI] [PubMed] [Google Scholar]
- 106.Grando SA (2008). Basic and clinical aspects of non-neuronal acetylcholine: biological and clinical significance of non-canonical ligands of epithelial nicotinic acetylcholine receptors. J Pharmacol Sci, 106, 174–179. [DOI] [PubMed] [Google Scholar]
- 107.Grando SA (2014). Connections of nicotine to cancer. Nature Reviews Cancer, 14, 419. [DOI] [PubMed] [Google Scholar]
- 108.Grando SA, Kawashima K, Kirkpatrick CJ, Kummer W, & Wessler I (2015). Recent progress in revealing the biological and medical significance of the non-neuronal cholinergic system. Int Immunopharmacol, 29, 1–7. [DOI] [PubMed] [Google Scholar]
- 109.Grando SA, Kist DA, Qi M, & Dahl MV (1993). Human keratinocytes synthesize, secrete, and degrade acetylcholine. J. Invest. Dermatol, 101, 32–36. [DOI] [PubMed] [Google Scholar]
- 110.Greinacher A, Wesche J, Hammer E, Furll B, Volker U, Reil A, & Bux J (2010). Characterization of the human neutrophil alloantigen-3a. Nat Med, 16, 45–48. [DOI] [PubMed] [Google Scholar]
- 111.Grisaru D, Keidar R, Schreiber L, Lessing JB, & Deutsch V (2007). The effect of the readthrough acetylcholinesterase variant (AChE-R) on uterine muscle and leiomyomas. Mol Hum Reprod, 13, 351–354. [DOI] [PubMed] [Google Scholar]
- 112.Grosman DD, Lorenzi MV, Trinidad AC, & Strauss WL (1995). The human choline acetyltransferase gene encodes two proteins. J Neurochem, 65, 484–491. [DOI] [PubMed] [Google Scholar]
- 113.Haberberger RV, Bodenbenner M, & Kummer W (2000). Expression of the cholinergic gene locus in pulmonary arterial endothelial cells. Histochem Cell Biol, 113, 379–387. [DOI] [PubMed] [Google Scholar]
- 114.Hall FS (2016). Genetic Risk for Lung Cancer and the Benefits of Quitting Smoking. EBioMedicine, 11, 19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hallden S, Sjogren M, Hedblad B, Engstrom G, Hamrefors V, Manjer J, & Melander O (2016). Gene variance in the nicotinic receptor cluster (CHRNA5-CHRNA3-CHRNB4) predicts death from cardiopulmonary disease and cancer in smokers. J Intern Med, 279, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Han JY, Park K, Kim SW, Lee DH, Kim HY, Kim HT, Ahn MJ, Yun T, Ahn JS, Suh C, Lee JS, Yoon SJ, Han JH, Lee JW, Jo SJ, & Lee JS (2012). First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol, 30, 1122–1128. [DOI] [PubMed] [Google Scholar]
- 117.Hansen HM, Xiao Y, Rice T, Bracci PM, Wrensch MR, Sison JD, Chang JS, Smirnov IV, Patoka J, Seldin MF, Quesenberry CP, Kelsey KT, & Wiencke JK (2010). Fine mapping of chromosome 15q25.1 lung cancer susceptibility in African-Americans. Hum Mol Genet, 19, 3652–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hara T, Bansal A, & DeGrado TR (2006). Choline transporter as a novel target for molecular imaging of cancer. Mol Imaging, 5, 498–509. [PubMed] [Google Scholar]
- 119.Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, & Cooke JP (2001). Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nature Medicine, 7, 833–839. [DOI] [PubMed] [Google Scholar]
- 120.Heeschen C, Weis M, Aicher A, Dimmler S, & Cooke JP (2002). A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Invest, 110, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heist RS, Sequist LV, & Engelman JA (2012). Genetic changes in squamous cell lung cancer: a review. J Thorac Oncol, 7, 924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Herbst RS, Morgensztern D, & Boshoff C (2018). The biology and management of non-small cell lung cancer. Nature, 553, 446–454. [DOI] [PubMed] [Google Scholar]
- 123.Horiguchi K, Horiguchi S, Yamashita N, Irie K, Masuda J, Takano-Ohmuro H, Himi T, Miyazawa M, Moriwaki Y, Okuda T, Misawa H, Ozaki H, & Kawashima K (2009). Expression of SLURP-1, an endogenous alpha7 nicotinic acetylcholine receptor allosteric ligand, in murine bronchial epithelial cells. J Neurosci Res, 87, 2740–2747. [DOI] [PubMed] [Google Scholar]
- 124.Hruska M, Keefe J, Wert D, Tekinay AB, Hulce JJ, Ibanez-Tallon I, & Nishi R (2009). Prostate stem cell antigen is an endogenous lynx1-like prototoxin that antagonizes alpha7-containing nicotinic receptors and prevents programmed cell death of parasympathetic neurons. J Neurosci, 29, 14847–14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hu C, & Zhang S (2017). Muscarinic cholinergic receptor antagonist inhibits the growth and angiogenesis of small cell lung cancer in vivo.
- 126.Hu W, Gray NW, & Brimijoin S (2003). Amyloid-beta increases acetylcholinesterase expression in neuroblastoma cells by reducing enzyme degradation. J Neurochem, 86, 470–478. [DOI] [PubMed] [Google Scholar]
- 127.Hua M, & Talbot P (2016). Potential health effects of electronic cigarettes: A systematic review of case reports. Prev Med Rep, 4, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hua N, Wei X, Liu X, Ma X, He X, Zhuo R, Zhao Z, Wang L, Yan H, Zhong B, & Zheng J (2012). A novel muscarinic antagonist R2HBJJ inhibits non-small cell lung cancer cell growth and arrests the cell cycle in G0/G1. PLoS One, 7, e53170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Chen C, Goodman G, Field JK, Liloglou T, Xinarianos G, Cassidy A, McLaughlin J, Liu G, Narod S, Krokan HE, Skorpen F, Elvestad MB, Hveem K, Vatten L, Linseisen J, Clavel-Chapelon F, Vineis P, Bueno-de-Mesquita HB, Lund E, Martinez C, Bingham S, Rasmuson T, Hainaut P, Riboli E, Ahrens W, Benhamou S, Lagiou P, Trichopoulos D, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Macfarlane G, Talamini R, Simonato L, Lowry R, Conway DI, Znaor A, Healy C, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, & Brennan P (2008). A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature, 452, 633–637. [DOI] [PubMed] [Google Scholar]
- 130.Ibanez-Tallon I, Miwa JM, Wang HL, Adams NC, Crabtree GW, Sine SM, & Heintz N (2002). Novel modulation of neuronal nicotinic acetylcholine receptors by association with the endogenous prototoxin lynx1. Neuron, 33, 893–903. [DOI] [PubMed] [Google Scholar]
- 131.Improgo MR, Scofield MD, Tapper AR, & Gardner PD (2010). From smoking to lung cancer: the CHRNA5/A3/B4 connection. Oncogene, 29, 4874–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Improgo MR, Soll LG, Tapper AR, & Gardner PD (2013). Nicotinic acetylcholine receptors mediate lung cancer growth. Front Physiol, 4, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Improgo MR, Tapper AR, & Gardner PD (2011). Nicotinic acetylcholine receptor-mediated mechanisms in lung cancer. Biochem Pharmacol, 82, 1015–1021. [DOI] [PubMed] [Google Scholar]
- 134.Inazu M (2014). Choline transporter-like proteins CTLs/SLC44 family as a novel molecular target for cancer therapy. Biopharm Drug Dispos, 35, 431–449. [DOI] [PubMed] [Google Scholar]
- 135.Inazu M, Takeda H, & Matsumiya T (2005). Molecular and functional characterization of an Na+-independent choline transporter in rat astrocytes. J Neurochem, 94, 1427–1437. [DOI] [PubMed] [Google Scholar]
- 136.Inazu M, Yamada T, Kubota N, & Yamanaka T (2013). Functional expression of choline transporter-like protein 1 (CTL1) in small cell lung carcinoma cells: a target molecule for lung cancer therapy. Pharmacol Res, 76, 119–131. [DOI] [PubMed] [Google Scholar]
- 137.Ingoglia F, Visigalli R, Rotoli BM, Barilli A, Riccardi B, Puccini P, & Dall’Asta V (2015). Functional characterization of the organic cation transporters (OCTs) in human airway pulmonary epithelial cells. Biochim Biophys Acta, 1848, 1563–1572. [DOI] [PubMed] [Google Scholar]
- 138.Iqbal J, Saeed A, Shah SJ, al-Rashida M, & Shams-ul M (2016). Biological Evaluation of Azomethine-dihydroquinazolinone Conjugates as Cancer and Cholinesterase Inhibitors. Med Chem, 12, 74–82. [DOI] [PubMed] [Google Scholar]
- 139.Iskandar AR, Miao B, Li X, Hu KQ, Liu C, & Wang XD (2016). beta-Cryptoxanthin Reduced Lung Tumor Multiplicity and Inhibited Lung Cancer Cell Motility by Downregulating Nicotinic Acetylcholine Receptor alpha7 Signaling. Cancer Prev Res (Phila), 9, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jiang H, & Zhang XJ (2008). Acetylcholinesterase and apoptosis. A novel perspective for an old enzyme. FEBS J, 275, 612–617. [DOI] [PubMed] [Google Scholar]
- 141.Jin Z, Gao F, Flagg T, & Deng X (2004). Nicotine induces multi-site phosphorylation of Bad in association with suppression of apoptosis. J Biol Chem, 279, 23837–23844. [DOI] [PubMed] [Google Scholar]
- 142.Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, Langer CJ, DeVore RF 3rd, Gaudreault J, Damico LA, Holmgren E, & Kabbinavar F (2004). Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol, 22, 2184–2191. [DOI] [PubMed] [Google Scholar]
- 143.Justilien V, & Fields AP (2013). Utility and applications of orthotopic models of human non-small cell lung cancer (NSCLC) for the evaluation of novel and emerging cancer therapeutics. Curr Protoc Pharmacol, 62, Unit 14 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kalantari-Dehaghi M, Bernard HU, & Grando SA (2012). Reciprocal effects of NNK and SLURP-1 on oncogene expression in target epithelial cells. Life Sci, 91, 1122–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kalantari-Dehaghi M, Parnell EA, Armand T, Bernard HU, & Grando SA (2015). The nicotinic acetylcholine receptor-mediated reciprocal effects of the tobacco nitrosamine NNK and SLURP-1 on human mammary epithelial cells. Int Immunopharmacol, 29, 99–104. [DOI] [PubMed] [Google Scholar]
- 146.Kalashnyk OM, Gergalova GL, Komisarenko SV, & Skok MV (2012). Intracellular localization of nicotinic acetylcholine receptors in human cell lines. Life Sci, 91, 1033–1037. [DOI] [PubMed] [Google Scholar]
- 147.Kar S, Issa AM, Seto D, Auld DS, Collier B, & Quirion R (1998). Amyloid beta-peptide inhibits high-affinity choline uptake and acetylcholine release in rat hippocampal slices. J Neurochem, 70, 2179–2187. [DOI] [PubMed] [Google Scholar]
- 148.Kawashima K, Fujii T, Moriwaki Y, & Misawa H (2012). Critical roles of acetylcholine and the muscarinic and nicotinic acetylcholine receptors in the regulation of immune function. Life Sci, 91, 1027–1032. [DOI] [PubMed] [Google Scholar]
- 149.Kellar A, Egan C, & Morris D (2015). Preclinical Murine Models for Lung Cancer: Clinical Trial Applications. Biomed Res Int, 2015, 621324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Khuder SA (2001). Effect of cigarette smoking on major histological types of lung cancer: a meta-analysis. Lung Cancer, 31, 139–148. [DOI] [PubMed] [Google Scholar]
- 151.Khuder SA, & Mutgi AB (2001). Effect of smoking cessation on major histologic types of lung cancer. Chest, 120, 1577–1583. [DOI] [PubMed] [Google Scholar]
- 152.Kidd ME, Shumaker DK, & Ridge KM (2014). The role of vimentin intermediate filaments in the progression of lung cancer. Am J Respir Cell Mol Biol, 50, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kirkpatrick CJ, Bittinger F, Nozadze K, & Wessler I (2003). Expression and function of the non-neuronal cholinergic system in endothelial cells. Life Sci, 72, 2111–2116. [DOI] [PubMed] [Google Scholar]
- 154.Kirkpatrick CJ, Bittinger F, Unger RE, Kriegsmann J, Kilbinger H, & Wessler I (2001). The non-neuronal cholinergic system in the endothelium: evidence and possible pathobiological significance. Jpn J Pharmacol, 85, 24–28. [DOI] [PubMed] [Google Scholar]
- 155.Kistemaker LE, & Gosens R (2015). Acetylcholine beyond bronchoconstriction: roles in inflammation and remodeling. Trends Pharmacol Sci, 36, 164–171. [DOI] [PubMed] [Google Scholar]
- 156.Koarai A, & Ichinose M (2018). Possible involvement of acetylcholine-mediated inflammation in airway diseases. Allergol Int [DOI] [PubMed]
- 157.Kommareddi PK, Nair TS, Thang LV, Galano MM, Babu E, Ganapathy V, Kanazawa T, McHugh JB, & Carey TE (2010). Isoforms, expression, glycosylation, and tissue distribution of CTL2/SLC44A2. Protein J, 29, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kopelman MD (1986). The cholinergic neurotransmitter system in human memory and dementia: a review. Q J Exp Psychol A, 38, 535–573. [DOI] [PubMed] [Google Scholar]
- 159.Kouji H, Inazu M, Yamada T, Tajima H, Aoki T, & Matsumiya T (2009). Molecular and functional characterization of choline transporter in human colon carcinoma HT-29 cells. Arch Biochem Biophys, 483, 90–98. [DOI] [PubMed] [Google Scholar]
- 160.Krais AM, Hautefeuille AH, Cros MP, Krutovskikh V, Tournier JM, Birembaut P, Thepot A, Paliwal A, Herceg Z, Boffetta P, Brennan P, & Hainaut PL (2011). CHRNA5 as negative regulator of nicotine signaling in normal and cancer bronchial cells: effects on motility, migration and p63 expression. Carcinogenesis, 32, 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Krasteva G, Canning BJ, Hartmann P, Veres TZ, Papadakis T, Muhlfeld C, Schliecker K, Tallini YN, Braun A, Hackstein H, Baal N, Weihe E, Schutz B, Kotlikoff M, Ibanez-Tallon I, & Kummer W (2011). Cholinergic chemosensory cells in the trachea regulate breathing. Proc Natl Acad Sci U S A, 108, 9478–9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, & Wess J (2014). Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat Rev Drug Discov, 13, 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kumar R, Kumar A, Langstrom B, & Darreh-Shori T (2017). Discovery of novel choline acetyltransferase inhibitors using structure-based virtual screening. Sci Rep, 7, 16287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kummer W, & Krasteva-Christ G (2014). Non-neuronal cholinergic airway epithelium biology. Curr Opin Pharmacol, 16, 43–49. [DOI] [PubMed] [Google Scholar]
- 165.Kummer W, Lips KS, & Pfeil U (2008). The epithelial cholinergic system of the airways. Histochem Cell Biol, 130, 219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kummer W, Wiegand S, Akinci S, Wessler I, Schinkel AH, Wess J, Koepsell H, Haberberger RV, & Lips KS (2006). Role of acetylcholine and polyspecific cation transporters in serotonin-induced bronchoconstriction in the mouse. Respir Res, 7, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kuryatov A, Berrettini W, & Lindstrom J (2011). Acetylcholine receptor (AChR) alpha5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (alpha4beta2)(2)alpha5 AChR function. Mol Pharmacol, 79, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lagace TA, & Ridgway ND (2013). The role of phospholipids in the biological activity and structure of the endoplasmic reticulum. Biochim Biophys Acta, 1833, 2499–2510. [DOI] [PubMed] [Google Scholar]
- 169.Lam DC, Girard L, Ramirez R, Chau WS, Suen WS, Sheridan S, Tin VP, Chung LP, Wong MP, Shay JW, Gazdar AF, Lam WK, & Minna JD (2007). Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res, 67, 4638–4647. [DOI] [PubMed] [Google Scholar]
- 170.Lassi G, Taylor AE, Timpson NJ, Kenny PJ, Mather RJ, Eisen T, & Munafo MR (2016). The CHRNA5-A3-B4 Gene Cluster and Smoking: From Discovery to Therapeutics. Trends Neurosci, 39, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lau JK, Brown KC, Thornhill BA, Crabtree CM, Dom AM, Witte TR, Hardman WE, McNees CA, Stover CA, Carpenter AB, Luo H, Chen YC, Shiflett BS, & Dasgupta P (2013). Inhibition of cholinergic signaling causes apoptosis in human bronchioalveolar carcinoma. Cancer Res, 73, 1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lee J, Sohn EJ, Yoon SW, Kim CG, Lee S, Kim JY, Baek N, & Kim SH (2017). Anti-Metastatic Effect of Dehydrocorydaline on H1299 Non-Small Cell Lung Carcinoma Cells via Inhibition of Matrix Metalloproteinases and B Cell Lymphoma 2. Phytother Res, 31, 441–448. [DOI] [PubMed] [Google Scholar]
- 173.Levitin F, Weiss M, Hahn Y, Stern O, Papke RL, Matusik R, Nandana SR, Ziv R, Pichinuk E, Salame S, Bera T, Vincent J, Lee B, Pastan I, & Wreschner DH (2008). PATE gene clusters code for multiple, secreted TFP/Ly-6/uPAR proteins that are expressed in sreproductive and neuron-rich tissues and possess neuromodulatory activity. J Biol Chem, 283, 16928–16939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li H, Wang S, Takayama K, Harada T, Okamoto I, Iwama E, Fujii A, Ota K, Hidaka N, Kawano Y, & Nakanishi Y (2015). Nicotine induces resistance to erlotinib via cross-talk between alpha 1 nAChR and EGFR in the non-small cell lung cancer xenograft model. Lung Cancer, 88, 1–8. [DOI] [PubMed] [Google Scholar]
- 175.Li M, Peng Z, Liu Q, Sun J, Yao S, & Liu Q (2013). Value of 11C-choline PET/CT for lung cancer diagnosis and the relation between choline metabolism and proliferation of cancer cells. Oncol Rep, 29, 205–211. [DOI] [PubMed] [Google Scholar]
- 176.Li Q, Yang H, Chen Y, & Sun H (2017). Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur J Med Chem, 132, 294–309. [DOI] [PubMed] [Google Scholar]
- 177.Lin G, Sun L, Wang R, Guo Y, & Xie C (2014). Overexpression of muscarinic receptor 3 promotes metastasis and predicts poor prognosis in non-small-cell lung cancer. J Thorac Oncol, 9, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lindstrom J (1996). Neuronal nicotinic acetylcholine receptors. Ion Channels, 4, 377–450. [DOI] [PubMed] [Google Scholar]
- 179.Lindstrom J (1997). Nicotinic acetylcholine receptors in health and disease. Mol Neurobiol, 15, 193–222. [DOI] [PubMed] [Google Scholar]
- 180.Lips KS, Luhrmann A, Tschernig T, Stoeger T, Alessandrini F, Grau V, Haberberger RV, Koepsell H, Pabst R, & Kummer W (2007). Down-regulation of the non-neuronal acetylcholine synthesis and release machinery in acute allergic airway inflammation of rat and mouse. Life Sci, 80, 2263–2269. [DOI] [PubMed] [Google Scholar]
- 181.Lips KS, Volk C, Schmitt BM, Pfeil U, Arndt P, Miska D, Ermert L, Kummer W, & Koepsell H (2005). Polyspecific cation transporters mediate luminal release of acetylcholine from bronchial epithelium. Am J Respir Cell Mol Biol, 33, 79–88. [DOI] [PubMed] [Google Scholar]
- 182.Lips KS, Wunsch J, Zarghooni S, Bschleipfer T, Schukowski K, Weidner W, Wessler I, Schwantes U, Koepsell H, & Kummer W (2007). Acetylcholine and molecular components of its synthesis and release machinery in the urothelium. Eur Urol, 51, 1042–1053. [DOI] [PubMed] [Google Scholar]
- 183.Liu CY, Lin HH, Tang MJ, & Wang YK (2015). Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget, 6, 15966–15983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Liu P, Vikis HG, Wang D, Lu Y, Wang Y, Schwartz AG, Pinney SM, Yang P, de Andrade M, Petersen GM, Wiest JS, Fain PR, Gazdar A, Gaba C, Rothschild H, Mandal D, Coons T, Lee J, Kupert E, Seminara D, Minna J, Bailey-Wilson JE, Wu X, Spitz MR, Eisen T, Houlston RS, Amos CI, Anderson MW, & You M (2008). Familial aggregation of common sequence variants on 15q24–25.1 in lung cancer. J Natl Cancer Inst, 100, 1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Liu Y, & Edwards RH (1997). Differential localization of vesicular acetylcholine and monoamine transporters in PC12 cells but not CHO cells. J Cell Biol, 139, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lockridge O, Norgren RB Jr., Johnson RC, & Blake TA (2016). Naturally Occurring Genetic Variants of Human Acetylcholinesterase and Butyrylcholinesterase and Their Potential Impact on the Risk of Toxicity from Cholinesterase Inhibitors. Chem Res Toxicol, 29, 1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lotfipour S, Mandelkern M, & Brody AL (2011). Quantitative Molecular Imaging of Neuronal Nicotinic Acetylcholine Receptors in the Human Brain with A-85380 Radiotracers. Curr Med Imaging Rev, 7, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lu H, & Jiang Z (2017). Advances in antiangiogenic treatment of small-cell lung cancer. Onco Targets Ther, 10, 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lu L, Zhang X, Zhang B, Wu J, & Zhang X (2013). Synaptic acetylcholinesterase targeted by microRNA-212 functions as a tumor suppressor in non-small cell lung cancer. Int J Biochem Cell Biol, 45, 2530–2540. [DOI] [PubMed] [Google Scholar]
- 190.Luo Z, Alvarado GF, Hatsukami DK, Johnson EO, Bierut LJ, & Breslau N (2008). Race differences in nicotine dependence in the Collaborative Genetic study of Nicotine Dependence (COGEND). Nicotine Tob Res, 10, 1223–1230. [DOI] [PubMed] [Google Scholar]
- 191.Lykhmus O, Gergalova G, Koval L, Zhmak M, Komisarenko S, & Skok M (2014). Mitochondria express several nicotinic acetylcholine receptor subtypes to control various pathways of apoptosis induction. Int J Biochem Cell Biol, 53, 246–252. [DOI] [PubMed] [Google Scholar]
- 192.Lyukmanova EN, Bychkov ML, Sharonov GV, Efremenko AV, Shulepko MA, Kulbatskii DS, Shenkarev ZO, Feofanov AV, Dolgikh DA, & Kirpichnikov MP (2018). Human secreted proteins SLURP-1 and SLURP-2 control the growth of epithelial cancer cells via interactions with nicotinic acetylcholine receptors. Br J Pharmacol, 175, 1973–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lyukmanova EN, Shulepko MA, Buldakova SL, Kasheverov IE, Shenkarev ZO, Reshetnikov RV, Filkin SY, Kudryavtsev DS, Ojomoko LO, Kryukova EV, Dolgikh DA, Kirpichnikov MP, Bregestovski PD, & Tsetlin VI (2013). Water-soluble LYNX1 residues important for interaction with muscle-type and/or neuronal nicotinic receptors. J Biol Chem, 288, 15888–15899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lyukmanova EN, Shulepko MA, Kudryavtsev D, Bychkov ML, Kulbatskii DS, Kasheverov IE, Astapova MV, Feofanov AV, Thomsen MS, Mikkelsen JD, Shenkarev ZO, Tsetlin VI, Dolgikh DA, & Kirpichnikov MP (2016). Human Secreted Ly-6/uPAR Related Protein-1 (SLURP-1) Is a Selective Allosteric Antagonist of alpha7 Nicotinic Acetylcholine Receptor. PLoS One, 11, e0149733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lyukmanova EN, Shulepko MA, Shenkarev ZO, Bychkov ML, Paramonov AS, Chugunov AO, Kulbatskii DS, Arvaniti M, Dolejsi E, Schaer T, Arseniev AS, Efremov RG, Thomsen MS, Dolezal V, Bertrand D, Dolgikh DA, & Kirpichnikov MP (2016). Secreted Isoform of Human Lynx1 (SLURP-2): Spatial Structure and Pharmacology of Interactions with Different Types of Acetylcholine Receptors. Sci Rep, 6, 30698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mai H, May WS, Gao F, Jin Z, & Deng X (2003). A functional role for nicotine in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem, 278, 1886–1891. [DOI] [PubMed] [Google Scholar]
- 197.Maneckjee R, & Minna JD (1994). Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ, 5, 1033–1040. [PubMed] [Google Scholar]
- 198.Martinez-Lopez de Castro A, Nieto-Ceron S, Aurelio PC, Galbis-Martinez L, Latour-Perez J, Torres-Lanzas J, Tovar-Zapata I, Martinez-Hernandez P, Rodriguez-Lopez JN, & Cabezas-Herrera J (2008). Cancer-associated differences in acetylcholinesterase activity in bronchial aspirates from patients with lung cancer. Clin Sci (Lond), 115, 245–253. [DOI] [PubMed] [Google Scholar]
- 199.Martinez-Moreno P, Nieto-Ceron S, Ruiz-Espejo F, Torres-Lanzas J, Tovar-Zapata I, Martinez-Hernandez P, Vidal CJ, & Cabezas-Herrera J (2005). Acetylcholinesterase biogenesis is impaired in lung cancer tissues. Chem Biol Interact, 157–158, 359–361. [DOI] [PubMed] [Google Scholar]
- 200.Martinez-Moreno P, Nieto-Ceron S, Torres-Lanzas J, Ruiz-Espejo F, Tovar-Zapata I, Martinez-Hernandez P, Rodriguez-Lopez JN, Vidal CJ, & Cabezas-Herrera J (2006). Cholinesterase activity of human lung tumours varies according to their histological classification. Carcinogenesis, 27, 429–436. [DOI] [PubMed] [Google Scholar]
- 201.Mastrodicasa MA, Droege CA, Mulhall AM, Ernst NE, Panos RJ, & Zafar MA (2017). Long acting muscarinic antagonists for the treatment of chronic obstructive pulmonary disease: a review of current and developing drugs. Expert Opin Investig Drugs, 26, 161–174. [DOI] [PubMed] [Google Scholar]
- 202.Matsuo A, Bellier JP, Nishimura M, Yasuhara O, Saito N, & Kimura H (2011). Nuclear choline acetyltransferase activates transcription of a high-affinity choline transporter. J Biol Chem, 286, 5836–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, Caporaso NE, Johansson M, Xiao X, Li Y, Byun J, Dunning A, Pooley KA, Qian DC, Ji X, Liu G, Timofeeva MN, Bojesen SE, Wu X, Le Marchand L, Albanes D, Bickeboller H, Aldrich MC, Bush WS, Tardon A, Rennert G, Teare MD, Field JK, Kiemeney LA, Lazarus P, Haugen A, Lam S, Schabath MB, Andrew AS, Shen H, Hong YC, Yuan JM, Bertazzi PA, Pesatori AC, Ye Y, Diao N, Su L, Zhang R, Brhane Y, Leighl N, Johansen JS, Mellemgaard A, Saliba W, Haiman CA, Wilkens LR, Fernandez-Somoano A, Fernandez-Tardon G, van der Heijden HFM, Kim JH, Dai J, Hu Z, Davies MPA, Marcus MW, Brunnstrom H, Manjer J, Melander O, Muller DC, Overvad K, Trichopoulou A, Tumino R, Doherty JA, Barnett MP, Chen C, Goodman GE, Cox A, Taylor F, Woll P, Bruske I, Wichmann HE, Manz J, Muley TR, Risch A, Rosenberger A, Grankvist K, Johansson M, Shepherd FA, Tsao MS, Arnold SM, Haura EB, Bolca C, Holcatova I, Janout V, Kontic M, Lissowska J, Mukeria A, Ognjanovic S, Orlowski TM, Scelo G, Swiatkowska B, Zaridze D, Bakke P, Skaug V, Zienolddiny S, Duell EJ, Butler LM, Koh WP, Gao YT, Houlston RS, McLaughlin J, Stevens VL, Joubert P, Lamontagne M, Nickle DC, Obeidat M, Timens W, Zhu B, Song L, Kachuri L, Artigas MS, Tobin MD, Wain LV, SpiroMeta C, Rafnar T, Thorgeirsson TE, Reginsson GW, Stefansson K, Hancock DB, Bierut LJ, Spitz MR, Gaddis NC, Lutz SM, Gu F, Johnson EO, Kamal A, Pikielny C, Zhu D, Lindstroem S, Jiang X, Tyndale RF, Chenevix-Trench G, Beesley J, Bosse Y, Chanock S, Brennan P, Landi MT, & Amos CI (2017). Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet, 49, 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Meck WH (2006). Temporal memory in mature and aged rats is sensitive to choline acetyltransferase inhibition. Brain Res, 1108, 168–175. [DOI] [PubMed] [Google Scholar]
- 205.Medjber K, Freidja ML, Grelet S, Lorenzato M, Maouche K, Nawrocki-Raby B, Birembaut P, Polette M, & Tournier JM (2015). Role of nicotinic acetylcholine receptors in cell proliferation and tumour invasion in broncho-pulmonary carcinomas. Lung Cancer, 87, 258–264. [DOI] [PubMed] [Google Scholar]
- 206.Mehta N, Musso D, & White H (1985). Water soluble choline acetyltransferase inhibitors: SAR studies. European journal of medicinal chemistry, 20, 443–446. [Google Scholar]
- 207.Mei D, Zhao L, Chen B, Zhang X, Wang X, Yu Z, Ni X, & Zhang Q (2018). alpha-Conotoxin ImI-modified polymeric micelles as potential nanocarriers for targeted docetaxel delivery to alpha7-nAChR overexpressed non-small cell lung cancer. Drug Deliv, 25, 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Merchant N, Nagaraju GP, Rajitha B, Lammata S, Jella KK, Buchwald ZS, Lakka SS, & Ali AN (2017). Matrix metalloproteinases: their functional role in lung cancer. Carcinogenesis, 38, 766–780. [DOI] [PubMed] [Google Scholar]
- 209.Meshorer E, & Soreq H (2006). Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci, 29, 216–224. [DOI] [PubMed] [Google Scholar]
- 210.Meshorer E, Toiber D, Zurel D, Sahly I, Dori A, Cagnano E, Schreiber L, Grisaru D, Tronche F, & Soreq H (2004). Combinatorial complexity of 5’ alternative acetylcholinesterase transcripts and protein products. J Biol Chem, 279, 29740–29751. [DOI] [PubMed] [Google Scholar]
- 211.Meza R, Meernik C, Jeon J, & Cote ML (2015). Lung cancer incidence trends by gender, race and histology in the United States, 1973–2010. PLoS One, 10, e0121323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Miwa JM, Ibanez-Tallon I, Crabtree GW, Sanchez R, Sali A, Role LW, & Heintz N (1999). lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron, 23, 105–114. [DOI] [PubMed] [Google Scholar]
- 213.Montalbano AM, Albano GD, Anzalone G, Bonanno A, Riccobono L, Di Sano C, Gagliardo R, Siena L, Pieper MP, Gjomarkaj M, & Profita M (2014). Cigarette smoke alters non-neuronal cholinergic system components inducing MUC5AC production in the H292 cell line. Eur J Pharmacol, 736, 35–43. [DOI] [PubMed] [Google Scholar]
- 214.More SS, Li S, Yee SW, Chen L, Xu Z, Jablons DM, & Giacomini KM (2010). Organic cation transporters modulate the uptake and cytotoxicity of picoplatin, a third-generation platinum analogue. Mol Cancer Ther, 9, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mori N, Wildes F, Takagi T, Glunde K, & Bhujwalla ZM (2016). The Tumor Microenvironment Modulates Choline and Lipid Metabolism. Front Oncol, 6, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mucchietto V, Crespi A, Fasoli F, Clementi F, & Gotti C (2016). Neuronal Acetylcholine Nicotinic Receptors as New Targets for Lung Cancer Treatment. Curr Pharm Des, 22, 2160–2169. [DOI] [PubMed] [Google Scholar]
- 217.Mucchietto V, Fasoli F, Pucci S, Moretti M, Benfante R, Maroli A, Di Lascio S, Bolchi C, Pallavicini M, Dowell C, McIntosh M, Clementi F, & Gotti C (2018). alpha9- and alpha7-containing receptors mediate the pro-proliferative effects of nicotine in the A549 adenocarcinoma cell line. Br J Pharmacol, 175, 1957–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nakajima K, Tooyama I, Yasuhara O, Aimi Y, & Kimura H (2000). Immunohistochemical demonstration of choline acetyltransferase of a peripheral type (pChAT) in the enteric nervous system of rats. J Chem Neuroanat, 18, 31–40. [DOI] [PubMed] [Google Scholar]
- 219.Nakamura T, Fujiwara R, Ishiguro N, Oyabu M, Nakanishi T, Shirasaka Y, Maeda T, & Tamai I (2010). Involvement of choline transporter-like proteins, CTL1 and CTL2, in glucocorticoid-induced acceleration of phosphatidylcholine synthesis via increased choline uptake. Biol Pharm Bull, 33, 691–696. [DOI] [PubMed] [Google Scholar]
- 220.Narumoto O, Horiguchi K, Horiguchi S, Moriwaki Y, Takano-Ohmuro H, Shoji S, Misawa H, Yamashita N, Nagase T, Kawashima K, & Yamashita N (2010). Down-regulation of secreted lymphocyte antigen-6/urokinase-type plasminogen activator receptor-related peptide-1 (SLURP-1), an endogenous allosteric alpha7 nicotinic acetylcholine receptor modulator, in murine and human asthmatic conditions. Biochem Biophys Res Commun, 398, 713–718. [DOI] [PubMed] [Google Scholar]
- 221.Narumoto O, Niikura Y, Ishii S, Morihara H, Okashiro S, Nakahari T, Nakano T, Matsumura H, Shimamoto C, Moriwaki Y, Misawa H, Yamashita N, Nagase T, Kawashima K, & Yamashita N (2013). Effect of secreted lymphocyte antigen-6/urokinase-type plasminogen activator receptor-related peptide-1 (SLURP-1) on airway epithelial cells. Biochem Biophys Res Commun, 438, 175–179. [DOI] [PubMed] [Google Scholar]
- 222.Nazim M, Masuda A, Rahman MA, Nasrin F, Takeda JI, Ohe K, Ohkawara B, Ito M, & Ohno K (2017). Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms. Nucleic Acids Res, 45, 1455–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nedeljkovic I, Carnero-Montoro E, Lahousse L, van der Plaat DA, de Jong K, Vonk JM, van Diemen CC, Faiz A, van den Berge M, Obeidat M, Bosse Y, Nickle DC, Consortium B, Uitterlinden AG, van Meurs JJB, Stricker BCH, Brusselle GG, Postma DS, Boezen HM, van Duijn CM, & Amin N (2018). Understanding the role of the chromosome 15q25.1 in COPD through epigenetics and transcriptomics. Eur J Hum Genet, 26, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Neufeld G, & Kessler O (2006). Pro-angiogenic cytokines and their role in tumor angiogenesis. Cancer Metastasis Rev, 25, 373–385. [DOI] [PubMed] [Google Scholar]
- 225.Nguyen TH, Pham HV, Pham NK, Quach ND, Pudhom K, Hansen PE, & Nguyen KP (2015). Chemical constituents from Sonneratia ovata Backer and their in vitro cytotoxicity and acetylcholinesterase inhibitory activities. Bioorg Med Chem Lett, 25, 2366–2371. [DOI] [PubMed] [Google Scholar]
- 226.Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, & Nachon F (2003). Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem, 278, 41141–41147. [DOI] [PubMed] [Google Scholar]
- 227.Nieto MA, Huang RY, Jackson RA, & Thiery JP (2016). Emt: 2016. Cell, 166, 21–45. [DOI] [PubMed] [Google Scholar]
- 228.Nishida N, Yano H, Nishida T, Kamura T, & Kojiro M (2006). Angiogenesis in cancer. Vasc Health Risk Manag, 2, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Niu XM, & Lu S (2014). Acetylcholine receptor pathway in lung cancer: New twists to an old story. World J Clin Oncol, 5, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Okuda T and Haga T. (2003) High affinity choline transporter. Neurochemical Res, 3–4 483–488. [DOI] [PubMed] [Google Scholar]
- 231.O’Regan S, Traiffort E, Ruat M, Cha N, Compaore D, & Meunier FM (2000). An electric lobe suppressor for a yeast choline transport mutation belongs to a new family of transporter-like proteins. Proc Natl Acad Sci U S A, 97, 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Oda Y (1999). Choline acetyltransferase: the structure, distribution and pathologic changes in the central nervous system. Pathol Int, 49, 921–937. [DOI] [PubMed] [Google Scholar]
- 233.Pahud G, Bontron S, & Eder-Colli L (2001). Modulation of choline acetyltransferase synthesis by okadaic acid, a phosphatase inhibitor, and KN-62, a CaM kinase inhibitor, in NS-20Y neuroblastoma. Neurochem Int, 38, 75–82. [DOI] [PubMed] [Google Scholar]
- 234.Pandey N, Pal S, Sharma LK, Guleria R, Mohan A, & Srivastava T (2017). SNP rs16969968 as a Strong Predictor of Nicotine Dependence and Lung Cancer Risk in a North Indian Population. Asian Pac J Cancer Prev, 18, 3073–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Paraoanu LE, & Layer PG (2008). Acetylcholinesterase in cell adhesion, neurite growth and network formation. FEBS J, 275, 618–624. [DOI] [PubMed] [Google Scholar]
- 236.Patel SS, Raghuwanshi R, Masood M, Acharya A, & Jain SK (2018). Medicinal plants with acetylcholinesterase inhibitory activity. Rev Neurosci, 29, 491–529. [DOI] [PubMed] [Google Scholar]
- 237.Patocka J, Kuca K, & Jun D (2004). Acetylcholinesterase and butyrylcholinesterase--important enzymes of human body. Acta Medica (Hradec Kralove), 47, 215–228. [PubMed] [Google Scholar]
- 238.Perry C, Sklan EH, & Soreq H (2004). CREB regulates AChE-R-induced proliferation of human glioblastoma cells. Neoplasia, 6, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Pesch B, Kendzia B, Gustavsson P, Jockel KH, Johnen G, Pohlabeln H, Olsson A, Ahrens W, Gross IM, Bruske I, Wichmann HE, Merletti F, Richiardi L, Simonato L, Fortes C, Siemiatycki J, Parent ME, Consonni D, Landi MT, Caporaso N, Zaridze D, Cassidy A, Szeszenia-Dabrowska N, Rudnai P, Lissowska J, Stucker I, Fabianova E, Dumitru RS, Bencko V, Foretova L, Janout V, Rudin CM, Brennan P, Boffetta P, Straif K, & Bruning T (2012). Cigarette smoking and lung cancer--relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int J Cancer, 131, 1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Phillips PA, Yang L, Shulkes A, Vonlaufen A, Poljak A, Bustamante S, Warren A, Xu Z, Guilhaus M, Pirola R, Apte MV, & Wilson JS (2010). Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc Natl Acad Sci U S A, 107, 17397–17402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Picciotto MR, Higley MJ, & Mineur YS (2012). Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron, 76, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Pickett MA, Dush MK, & Nascone-Yoder NM (2017). Acetylcholinesterase plays a non-neuronal, non-esterase role in organogenesis. Development, 144, 2764–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Pieper MP (2012). The non-neuronal cholinergic system as novel drug target in the airways. Life Sci, 91, 1113–1118. [DOI] [PubMed] [Google Scholar]
- 244.Pieper MP, Chaudhary NI, & Park JE (2007). Acetylcholine-induced proliferation of fibroblasts and myofibroblasts in vitro is inhibited by tiotropium bromide. Life Sci, 80, 2270–2273. [DOI] [PubMed] [Google Scholar]
- 245.Pillai S, & Chellappan S (2012). alpha7 Nicotinic Acetylcholine Receptor Subunit in Angiogenesis and Epithelial to Mesenchymal Transition. Curr Drug Targets, 13, 671–679. [DOI] [PubMed] [Google Scholar]
- 246.Pinheiro NM, Miranda CJ, Perini A, Camara NO, Costa SK, Alonso-Vale MI, Caperuto LC, Tiberio IF, Prado MA, Martins MA, Prado VF, & Prado CM (2015). Pulmonary inflammation is regulated by the levels of the vesicular acetylcholine transporter. PLoS One, 10, e0120441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Piperdi B, Merla A, & Perez-Soler R (2014). Targeting angiogenesis in squamous non-small cell lung cancer. Drugs, 74, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Pochini L, Scalise M, Galluccio M, & Indiveri C (2012). Regulation by physiological cations of acetylcholine transport mediated by human OCTN1 (SLC22A4). Implications in the non-neuronal cholinergic system. Life Sci, 91, 1013–1016. [DOI] [PubMed] [Google Scholar]
- 249.Pochini L, Scalise M, Galluccio M, & Indiveri C (2013). OCTN cation transporters in health and disease: role as drug targets and assay development. J Biomol Screen, 18, 851–867. [DOI] [PubMed] [Google Scholar]
- 250.Pochini L, Scalise M, Galluccio M, Pani G, Siminovitch KA, & Indiveri C (2012). The human OCTN1 (SLC22A4) reconstituted in liposomes catalyzes acetylcholine transport which is defective in the mutant L503F associated to the Crohn’s disease. Biochim Biophys Acta, 1818, 559–565. [DOI] [PubMed] [Google Scholar]
- 251.Pohanka M (2011). Cholinesterases, a target of pharmacology and toxicology. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub, 155, 219–229. [DOI] [PubMed] [Google Scholar]
- 252.Prado VF, Roy A, Kolisnyk B, Gros R, & Prado MA (2013). Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem J, 450, 265–274. [DOI] [PubMed] [Google Scholar]
- 253.Proctor RN (2012). The history of the discovery of the cigarette-lung cancer link: evidentiary traditions, corporate denial, global toll. Tob Control, 21, 87–91. [DOI] [PubMed] [Google Scholar]
- 254.Profita M, Bonanno A, Siena L, Bruno A, Ferraro M, Montalbano AM, Albano GD, Riccobono L, Casarosa P, Pieper MP, & Gjomarkaj M (2009). Smoke, choline acetyltransferase, muscarinic receptors, and fibroblast proliferation in chronic obstructive pulmonary disease. J Pharmacol Exp Ther, 329, 753–763. [DOI] [PubMed] [Google Scholar]
- 255.Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J, & Spindel ER (2004). Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology, 145, 2498–2506. [DOI] [PubMed] [Google Scholar]
- 256.Qu X, Wang K, Dong W, Shen H, Wang Y, Liu Q, & Du J (2016). Association between two CHRNA3 variants and susceptibility of lung cancer: a meta-analysis. Sci Rep, 6, 20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Quigley RL, Shafer SH, & Williams CL (1998). Regulation of integrin-mediated adhesion by muscarinic acetylcholine receptors and protein kinase C in small cell lung carcinoma. Chest, 114, 839–846. [DOI] [PubMed] [Google Scholar]
- 258.Ralston JS, Rush RS, Doctor BP, & Wolfe AD (1985). Acetylcholinesterase from fetal bovine serum. Purification and characterization of soluble G4 enzyme. J Biol Chem, 260, 4312–4318. [PubMed] [Google Scholar]
- 259.Ramirez de Molina A, Sarmentero-Estrada J, Belda-Iniesta C, Taron M, Ramirez de Molina V, Cejas P, Skrzypski M, Gallego-Ortega D, de Castro J, Casado E, Garcia-Cabezas MA, Sanchez JJ, Nistal M, Rosell R, Gonzalez-Baron M, & Lacal JC (2007). Expression of choline kinase alpha to predict outcome in patients with early-stage non-small-cell lung cancer: a retrospective study. Lancet Oncol, 8, 889–897. [DOI] [PubMed] [Google Scholar]
- 260.Ray C, Soderblom EJ, Bai Y, Carroll FI, Caron MG, & Barak LS (2017). Probing the Allosteric Role of the alpha5 Subunit of alpha3beta4alpha5 Nicotinic Acetylcholine Receptors by Functionally Selective Modulators and Ligands. ACS Chem Biol, 12, 702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ray R, Mitra N, Baldwin D, Guo M, Patterson F, Heitjan DF, Jepson C, Wileyto EP, Wei J, Payne T, Ma JZ, Li MD, & Lerman C (2010). Convergent evidence that choline acetyltransferase gene variation is associated with prospective smoking cessation and nicotine dependence. Neuropsychopharmacology, 35, 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ready NE, Pang HH, Gu L, Otterson GA, Thomas SP, Miller AA, Baggstrom M, Masters GA, Graziano SL, Crawford J, Bogart J, & Vokes EE (2015). Chemotherapy With or Without Maintenance Sunitinib for Untreated Extensive-Stage Small-Cell Lung Cancer: A Randomized, Double-Blind, Placebo-Controlled Phase II Study-CALGB 30504 (Alliance). J Clin Oncol, 33, 1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Ridgway ND (2013). The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit Rev Biochem Mol Biol, 48, 20–38. [DOI] [PubMed] [Google Scholar]
- 264.Rolfo C, Passiglia F, Ostrowski M, Farracho L, Ondoichova T, Dolcan A, Castiglia M, Remmen R, Papadimitriou K, & Pauwels P (2015). Improvement in lung cancer outcomes with targeted therapies: an update for family physicians. J Am Board Fam Med, 28, 124–133. [DOI] [PubMed] [Google Scholar]
- 265.Romero HK, Christensen SB, Di Cesare Mannelli L, Gajewiak J, Ramachandra R, Elmslie KS, Vetter DE, Ghelardini C, Iadonato SP, Mercado JL, Olivera BM, & McIntosh JM (2017). Inhibition of alpha9alpha10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc Natl Acad Sci U S A, 114, E1825–E1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Rotering S, Deuther-Conrad W, Cumming P, Donat CK, Scheunemann M, Fischer S, Xiong G, Steinbach J, Peters D, Sabri O, Bucerius J, & Brust P (2014). Imaging of alpha7 nicotinic acetylcholine receptors in brain and cerebral vasculature of juvenile pigs with [(18)F]NS14490. EJNMMI Res, 4, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Roth M (2015). Airway and lung remodelling in chronic pulmonary obstructive disease: a role for muscarinic receptor antagonists? Drugs, 75, 1–8. [DOI] [PubMed] [Google Scholar]
- 268.Russell RV (1988). Behavorial Correlates of the pressynaptic events in cholinergic neurotransmission system. In Zucker J (Ed.), Progress in Drug Research (Vol. 32, pp. 43–131). Berlin: Birkhauser Verlag. [DOI] [PubMed] [Google Scholar]
- 269.Russo P, Cesario A, Rutella S, Veronesi G, Spaggiari L, Galetta D, Margaritora S, Granone P, & Greenberg DS (2011). Impact of genetic variability in nicotinic acetylcholine receptors on nicotine addiction and smoking cessation treatment. Curr Med Chem, 18, 91–112. [DOI] [PubMed] [Google Scholar]
- 270.Saccone NL, Culverhouse RC, Schwantes-An TH, Cannon DS, Chen X, Cichon S, Giegling I, Han S, Han Y, Keskitalo-Vuokko K, Kong X, Landi MT, Ma JZ, Short SE, Stephens SH, Stevens VL, Sun L, Wang Y, Wenzlaff AS, Aggen SH, Breslau N, Broderick P, Chatterjee N, Chen J, Heath AC, Heliovaara M, Hoft NR, Hunter DJ, Jensen MK, Martin NG, Montgomery GW, Niu T, Payne TJ, Peltonen L, Pergadia ML, Rice JP, Sherva R, Spitz MR, Sun J, Wang JC, Weiss RB, Wheeler W, Witt SH, Yang BZ, Caporaso NE, Ehringer MA, Eisen T, Gapstur SM, Gelernter J, Houlston R, Kaprio J, Kendler KS, Kraft P, Leppert MF, Li MD, Madden PA, Nothen MM, Pillai S, Rietschel M, Rujescu D, Schwartz A, Amos CI, & Bierut LJ (2010). Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Saeed A, Mahesar PA, Zaib S, Khan MS, Matin A, Shahid M, & Iqbal J (2014). Synthesis, cytotoxicity and molecular modelling studies of new phenylcinnamide derivatives as potent inhibitors of cholinesterases. Eur J Med Chem, 78, 43–53. [DOI] [PubMed] [Google Scholar]
- 272.Sagara Y, Sagara T, Mase T, Kimura T, Numazawa T, Fujikawa T, Noguchi K, & Ohtake N (2002). Cyclohexylmethylpiperidinyltriphenylpropioamide: a selective muscarinic M(3) antagonist discriminating against the other receptor subtypes. J Med Chem, 45, 984–987. [DOI] [PubMed] [Google Scholar]
- 273.Salomon JJ, Muchitsch VE, Gausterer JC, Schwagerus E, Huwer H, Daum N, Lehr CM, & Ehrhardt C (2014). The cell line NCl-H441 is a useful in vitro model for transport studies of human distal lung epithelial barrier. Mol Pharm, 11, 995–1006. [DOI] [PubMed] [Google Scholar]
- 274.Saracino L, Zorzetto M, Inghilleri S, Pozzi E, & Stella GM (2013). Non-neuronal cholinergic system in airways and lung cancer susceptibility. Transl Lung Cancer Res, 2, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Sastry BV, Jaiswal N, Janson V, Day PS, & Naukam RJ (1988). Relationships between chemical structure and inhibition of choline acetyltransferase by 2-(alpha-naphthoyl)ethyltrimethylammonium and related compounds. Pharmacol Res Commun, 20, 751–771. [DOI] [PubMed] [Google Scholar]
- 276.Sastry BV, Jaiswal N, Owens LK, Janson VE, & Moore RD (1988). 2-(alpha-Naphthoyl)ethyltrimethylammonium iodide and its beta-isomer: new selective, stable and fluorescent inhibitors of choline acetyltransferase. J Pharmacol Exp Ther, 245, 72–80. [PubMed] [Google Scholar]
- 277.Schaal C, & Chellappan S (2016). Nicotine-Mediated Regulation of Nicotinic Acetylcholine Receptors in Non-Small Cell Lung Adenocarcinoma by E2F1 and STAT1 Transcription Factors. PLoS One, 11, e0156451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Schuller HM (1992). Nitrosamine-induced lung carcinogenesis and Ca2+/calmodulin antagonists. Cancer Res, 52, 2723s–2726s. [PubMed] [Google Scholar]
- 279.Schuller HM (2002). Mechanisms of smoking-related lung and pancreatic adenocarcinoma development. Nat Rev Cancer, 2, 455–463. [DOI] [PubMed] [Google Scholar]
- 280.Schuller HM (2007). Nitrosamines as nicotinic receptor ligands. Life Sci, 80, 2274–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Schuller HM (2012). Regulatory role of the alpha7nAChR in cancer. Curr Drug Targets, 13, 680–687. [DOI] [PubMed] [Google Scholar]
- 282.Schuller HM, Jull BA, Sheppard BJ, & Plummer HK (2000). Interaction of tobacco-specific toxicants with the neuronal alpha(7) nicotinic acetylcholine receptor and its associated mitogenic signal transduction pathway: potential role in lung carcinogenesis and pediatric lung disorders. Eur J Pharmacol, 393, 265–277. [DOI] [PubMed] [Google Scholar]
- 283.Schuller HM, McGavin MD, Orloff M, Riechert A, & Porter B (1995). Simultaneous exposure to nicotine and hyperoxia causes tumors in hamsters. Lab Invest, 73, 448–456. [PubMed] [Google Scholar]
- 284.Schuller HM, & Orloff M (1998). Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem Pharmacol, 55, 1377–1384. [DOI] [PubMed] [Google Scholar]
- 285.Schuller HM, Plummer HK 3rd, & Jull BA (2003). Receptor-mediated effects of nicotine and its nitrosated derivative NNK on pulmonary neuroendocrine cells. Anat Rec, 270A, 51–58. [DOI] [PubMed] [Google Scholar]
- 286.Schuller HM, Tithof PK, Williams M, & Plummer H 3rd. (1999). The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res, 59, 4510–4515. [PubMed] [Google Scholar]
- 287.Sekhon HS, Song P, Jia Y, Lindstrom J, & Spindel ER (2005). Expression of lynx1 in developing lung and its modulation by prenatal nicotine exposure. Cell Tissue Res, 320, 287–297. [DOI] [PubMed] [Google Scholar]
- 288.Shafferman A, Kronman C, Flashner Y, Leitner M, Grosfeld H, Ordentlich A, Gozes Y, Cohen S, Ariel N, Barak D, & et al. (1992). Mutagenesis of human acetylcholinesterase. Identification of residues involved in catalytic activity and in polypeptide folding. J Biol Chem, 267, 17640–17648. [PubMed] [Google Scholar]
- 289.Shao JX, Wang B, Yao YN, Pan ZJ, Shen Q, & Zhou JY (2016). Autonomic nervous infiltration positively correlates with pathological risk grading and poor prognosis in patients with lung adenocarcinoma. Thorac Cancer, 7, 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Silman I, & Sussman JL (2005). Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr Opin Pharmacol, 5, 293–302. [DOI] [PubMed] [Google Scholar]
- 291.Singh S, Pillai S, & Chellappan S (2011). Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J Oncol, 2011, 456743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Skok M, Gergalova G, Lykhmus O, Kalashnyk O, Koval L, & Uspenska K (2016). Nicotinic acetylcholine receptors in mitochondria: subunit composition, function and signaling. Neurotransmitter, 3, 1–12. [Google Scholar]
- 293.Soldera SV, & Leighl NB (2017). Update on the Treatment of Metastatic Squamous Non-Small Cell Lung Cancer in New Era of Personalized Medicine. Front Oncol, 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Song P, Mark GP, & Spindel ER (2010). Knockdown Of Choline Transporter-like Protein 1 (CTL1) Increases ACh Secretion But Decreases Choline Uptake In Small Cell Lung Carcinoma. In B62. LUNG CANCER BIOMARKERS AND THERAPEUTIC RESPONSE (pp. A3499–A3499).
- 295.Song P, Olivas AS, & Spindel ER (2009). Inhibition of lung cancer cell growth by tiotropium: mechanism of action
- 296.Song P, Olivas AS, & Spindel ER (2010). Tiotropium inhibits growth of squamous cell lung carcinoma (SCC) cell lines in vitro and also inhibits SCC growth in vivo in nude mice by inhalation
- 297.Song P, Rekow SS, Singleton CA, Sekhon HS, Dissen GA, Zhou M, Campling B, Lindstrom J, & Spindel ER (2013). Choline transporter-like protein 4 (CTL4) links to non-neuronal acetylcholine synthesis. J Neurochem, 126, 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Song P, Sekhon HS, Duan J, Mark GP, & Spindel ER (2007). Inhibitory regulation by M2 muscarinic acetylcholine receptors is decreased in lung cancers
- 299.Song P, Sekhon HS, Fu XW, Maier M, Jia Y, Duan J, Proskosil BJ, Gravett C, Lindstrom J, Mark GP, Saha S, & Spindel ER (2008). Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res, 68, 4693–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Song P, Sekhon HS, Jia Y, Keller JA, Blusztajn JK, Mark GP, & Spindel ER (2003). Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res, 63, 214–221. [PubMed] [Google Scholar]
- 301.Song P, Sekhon HS, Lu A, Arredondo J, Sauer D, Gravett C, Mark GP, Grando SA, & Spindel ER (2007). M3 muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer Res, 67, 3936–3944. [DOI] [PubMed] [Google Scholar]
- 302.Song P, Sekhon HS, Proskocil B, Blusztajn JK, Mark GP, & Spindel ER (2003). Synthesis of acetylcholine by lung cancer. Life Sci, 72, 2159–2168. [DOI] [PubMed] [Google Scholar]
- 303.Song P, & Spindel ER (2008). Basic and clinical aspects of non-neuronal acetylcholine: expression of non-neuronal acetylcholine in lung cancer provides a new target for cancer therapy. J Pharmacol Sci, 106, 180–185. [DOI] [PubMed] [Google Scholar]
- 304.Soreq H, & Seidman S (2001). Acetylcholinesterase--new roles for an old actor. Nat Rev Neurosci, 2, 294–302. [DOI] [PubMed] [Google Scholar]
- 305.Spindel ER (2012). Muscarinic receptor agonists and antagonists: effects on cancer. Handb Exp Pharmacol, 451–468. [DOI] [PMC free article] [PubMed]
- 306.Spindel ER (2016). Cholinergic Targets in Lung Cancer. Curr Pharm Des, 22, 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Spitz MR, Amos CI, Dong Q, Lin J, & Wu X (2008). The CHRNA5-A3 region on chromosome 15q24–25.1 is a risk factor both for nicotine dependence and for lung cancer. J Natl Cancer Inst, 100, 1552–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Springer R (2014). Electronic cigarettes--patient safety concerns. Plast Surg Nurs, 34, 165–166. [DOI] [PubMed] [Google Scholar]
- 309.Steinritz D, Emmler J, Hintz M, Worek F, Kreppel H, Szinicz L, & Kehe K (2007). Apoptosis in sulfur mustard treated A549 cell cultures. Life Sci, 80, 2199–2201. [DOI] [PubMed] [Google Scholar]
- 310.Sun H, & Ma X (2015). alpha5-nAChR modulates nicotine-induced cell migration and invasion in A549 lung cancer cells. Exp Toxicol Pathol, 67, 477–482. [DOI] [PubMed] [Google Scholar]
- 311.Sun HJ, Jia YF, & Ma XL (2017). Alpha5 Nicotinic Acetylcholine Receptor Contributes to Nicotine-Induced Lung Cancer Development and Progression. Front Pharmacol, 8, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Swamynathan S, Delp EE, Harvey SA, Loughner CL, Raju L, & Swamynathan SK (2015). Corneal Expression of SLURP-1 by Age, Sex, Genetic Strain, and Ocular Surface Health. Invest Ophthalmol Vis Sci, 56, 7888–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Takiguchi Y, Sekine I, Iwasawa S, Kurimoto R, & Tatsumi K (2014). Chronic obstructive pulmonary disease as a risk factor for lung cancer. World J Clin Oncol, 5, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Tamai I (2013). Pharmacological and pathophysiological roles of carnitine/organic cation transporters (OCTNs: SLC22A4, SLC22A5 and Slc22a21). Biopharm Drug Dispos, 34, 29–44. [DOI] [PubMed] [Google Scholar]
- 315.Tamai I, Ohashi R, Nezu JI, Sai Y, Kobayashi D, Oku A, Shimane M, & Tsuji A (2000). Molecular and functional characterization of organic cation/carnitine transporter family in mice. J Biol Chem, 275, 40064–40072. [DOI] [PubMed] [Google Scholar]
- 316.Taylor P, & Radic Z (1994). The cholinesterases: from genes to proteins. Annu Rev Pharmacol Toxicol, 34, 281–320. [DOI] [PubMed] [Google Scholar]
- 317.Tekinay AB, Nong Y, Miwa JM, Lieberam I, Ibanez-Tallon I, Greengard P, & Heintz N (2009). A role for LYNX2 in anxiety-related behavior. Proc Natl Acad Sci U S A, 106, 4477–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, Thorlacius S, Gudmundsson J, Jonsson T, Jakobsdottir M, Saemundsdottir J, Olafsdottir O, Gudmundsson LJ, Bjornsdottir G, Kristjansson K, Skuladottir H, Isaksson HJ, Gudbjartsson T, Jones GT, Mueller T, Gottsater A, Flex A, Aben KK, de Vegt F, Mulders PF, Isla D, Vidal MJ, Asin L, Saez B, Murillo L, Blondal T, Kolbeinsson H, Stefansson JG, Hansdottir I, Runarsdottir V, Pola R, Lindblad B, van Rij AM, Dieplinger B, Haltmayer M, Mayordomo JI, Kiemeney LA, Matthiasson SE, Oskarsson H, Tyrfingsson T, Gudbjartsson DF, Gulcher JR, Jonsson S, Thorsteinsdottir U, Kong A, & Stefansson K (2008a). A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature, 452, 638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, Thorlacius S, Gudmundsson J, Jonsson T, Jakobsdottir M, Saemundsdottir J, Olafsdottir O, Gudmundsson LJ, Bjornsdottir G, Kristjansson K, Skuladottir H, Isaksson HJ, Gudbjartsson T, Jones GT, Mueller T, Gottsater A, Flex A, Aben KKH, de Vegt F, Mulders PFA, Isla D, Vidal MJ, Asin L, Saez B, Murillo L, Blondal T, Kolbeinsson H, Stefansson JG, Hansdottir I, Runarsdottir V, Pola R, Lindblad B, van Rij AM, Dieplinger B, Haltmayer M, Mayordomo JI, Kiemeney LA, Matthiasson SE, Oskarsson H, Tyrfingsson T, Gudbjartsson DF, Gulcher JR, Jonsson S, Thorsteinsdottir U, Kong A, & Stefansson K (2008b). A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature, 452, 638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Togashi Y, Hayashi H, Okamoto K, Fumita S, Terashima M, de Velasco MA, Sakai K, Fujita Y, Tomida S, Nakagawa K, & Nishio K (2015). Chronic nicotine exposure mediates resistance to EGFR-TKI in EGFR-mutated lung cancer via an EGFR signal. Lung Cancer, 88, 16–23. [DOI] [PubMed] [Google Scholar]
- 321.Toiber D, Berson A, Greenberg D, Melamed-Book N, Diamant S, & Soreq H (2008). N-acetylcholinesterase-induced apoptosis in Alzheimer’s disease. PLoS One, 3, e3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Toiber D, Greenberg DS, & Soreq H (2009). Pro-apoptotic protein-protein interactions of the extended N-AChE terminus. J Neural Transm (Vienna), 116, 1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Tooyama I, & Kimura H (2000). A protein encoded by an alternative splice variant of choline acetyltransferase mRNA is localized preferentially in peripheral nerve cells and fibers. J Chem Neuroanat, 17, 217–226. [DOI] [PubMed] [Google Scholar]
- 324.Tournier JM, & Birembaut P (2011). Nicotinic acetylcholine receptors and predisposition to lung cancer. Curr Opin Oncol, 23, 83–87. [DOI] [PubMed] [Google Scholar]
- 325.Traiffort E, O’Regan S, & Ruat M (2013). The choline transporter-like family SLC44: properties and roles in human diseases. Mol Aspects Med, 34, 646–654. [DOI] [PubMed] [Google Scholar]
- 326.Tsetlin V (1999). Snake venom alpha-neurotoxins and other ‘three-finger’ proteins. Eur J Biochem, 264, 281–286. [DOI] [PubMed] [Google Scholar]
- 327.Tsetlin V, Utkin Y, & Kasheverov I (2009). Polypeptide and peptide toxins, magnifying lenses for binding sites in nicotinic acetylcholine receptors. Biochem Pharmacol, 78, 720–731. [DOI] [PubMed] [Google Scholar]
- 328.Tsetlin VI (2015). Three-finger snake neurotoxins and Ly6 proteins targeting nicotinic acetylcholine receptors: pharmacological tools and endogenous modulators. Trends Pharmacol Sci, 36, 109–123. [DOI] [PubMed] [Google Scholar]
- 329.Tsoukalas N, Aravantinou-Fatorou E, Tolia M, Giaginis C, Galanopoulos M, Kiakou M, Kostakis ID, Dana E, Vamvakaris I, Korogiannos A, Tsiambas E, Salemis N, Kyrgias G, Karameris A, & Theocharis S (2017). Epithelial-Mesenchymal Transition in Non Small-cell Lung Cancer. Anticancer Res, 37, 1773–1778. [DOI] [PubMed] [Google Scholar]
- 330.Uchida Y, Inazu M, Takeda H, Yamada T, Tajima H, & Matsumiya T (2009). Expression and functional characterization of choline transporter in human keratinocytes. J Pharmacol Sci, 109, 102–109. [DOI] [PubMed] [Google Scholar]
- 331.Usdin TB, Eiden LE, Bonner TI, & Erickson JD (1995). Molecular biology of the vesicular ACh transporter. Trends Neurosci, 18, 218–224. [DOI] [PubMed] [Google Scholar]
- 332.Volk C (2014). OCTs, OATs and OCTNs: structure and function of the polyspecific organic ion transporters of SLC22 family. WIREs Membr. Transp. Signal, 3, 1–13. [Google Scholar]
- 333.Waller LL, Miller AA, & Petty WJ (2010). Using erlotinib to treat patients with non-small cell lung cancer who continue to smoke. Lung Cancer, 67, 12–16. [DOI] [PubMed] [Google Scholar]
- 334.Wang S, & Hu Y (2018). alpha7 nicotinic acetylcholine receptors in lung cancer. Oncol Lett, 16, 1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Wang T, Li J, Chen F, Zhao Y, He X, Wan D, & Gu J (2007). Choline transporters in human lung adenocarcinoma: expression and functional implications. Acta Biochim Biophys Sin (Shanghai), 39, 668–674. [DOI] [PubMed] [Google Scholar]
- 336.Wang Z, Dabrosin C, Yin X, Fuster MM, Arreola A, Rathmell WK, Generali D, Nagaraju GP, El-Rayes B, Ribatti D, Chen YC, Honoki K, Fujii H, Georgakilas AG, Nowsheen S, Amedei A, Niccolai E, Amin A, Ashraf SS, Helferich B, Yang X, Guha G, Bhakta D, Ciriolo MR, Aquilano K, Chen S, Halicka D, Mohammed SI, Azmi AS, Bilsland A, Keith WN, & Jensen LD (2015). Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol, 35 Suppl, S224–S243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Ware JJ, van den Bree M, & Munafo MR (2012). From men to mice: CHRNA5/CHRNA3, smoking behavior and disease. Nicotine Tob Res, 14, 1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Weihe E, Tao-Cheng JH, Schafer MK, Erickson JD, & Eiden LE (1996). Visualization of the vesicular acetylcholine transporter in cholinergic nerve terminals and its targeting to a specific population of small synaptic vesicles. Proc Natl Acad Sci U S A, 93, 3547–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Wen L, Jiang K, Yuan W, Cui W, & Li MD (2016). Contribution of Variants in CHRNA5/A3/B4 Gene Cluster on Chromosome 15 to Tobacco Smoking: From Genetic Association to Mechanism. Mol Neurobiol, 53, 472–484. [DOI] [PubMed] [Google Scholar]
- 340.Wenk G, Sweeney J, Hughey D, Carson J, & Olton D (1986). Cholinergic function and memory: extensive inhibition of choline acetyltransferase fails to impair radial maze performance in rats. Pharmacol Biochem Behav, 25, 521–526. [DOI] [PubMed] [Google Scholar]
- 341.Wess J (1996). Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol, 10, 69–99. [DOI] [PubMed] [Google Scholar]
- 342.Wessler I, Kirkpatrick CJ, & Racke K (1998). Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol Ther, 77, 59–79. [DOI] [PubMed] [Google Scholar]
- 343.West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM, Harris C, Belinsky S, & Dennis PA (2003). Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest, 111, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.West KA, Linnoila IR, Belinsky SA, Harris CC, & Dennis PA (2004). Tobacco carcinogen-induced cellular transformation increases activation of the phosphatidylinositol 3’-kinase/Akt pathway in vitro and in vivo. Cancer Res, 64, 446–451. [DOI] [PubMed] [Google Scholar]
- 345.Whiteaker P, Christensen S, Yoshikami D, Dowell C, Watkins M, Gulyas J, Rivier J, Olivera BM, & McIntosh JM (2007). Discovery, synthesis, and structure activity of a highly selective alpha7 nicotinic acetylcholine receptor antagonist. Biochemistry, 46, 6628–6638. [DOI] [PubMed] [Google Scholar]
- 346.Williams CL (2003). Muscarinic signaling in carcinoma cells. Life Sci, 72, 2173–2182. [DOI] [PubMed] [Google Scholar]
- 347.Williams CL, & Lennon VA (1990). Activation of M3 muscarinic acetylcholine receptors inhibits voltage-dependent calcium influx in small cell lung carcinoma. J Biol Chem, 265, 1443–1447. [PubMed] [Google Scholar]
- 348.Williams CL, & Lennon VA (1991). Activation of muscarinic acetylcholine receptors inhibits cell cycle progression of small cell lung carcinoma. Cell Regul, 2, 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Wilson C, Lee MD, & McCarron JG (2016). Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J Physiol, 594, 7267–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Wong SHM, Fang CM, Chuah LH, Leong CO, & Ngai SC (2018). E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit Rev Oncol Hematol, 121, 11–22. [DOI] [PubMed] [Google Scholar]
- 351.Wright SC, Zhong J, Zheng H, & Larrick JW (1993). Nicotine inhibition of apoptosis suggests a role in tumor promotion. Faseb J, 7, 1045–1051. [PubMed] [Google Scholar]
- 352.Wu J, Zhou J, Yao L, Lang Y, Liang Y, Chen L, Zhang J, Wang F, Wang Y, Chen H, & Ma J (2013). High expression of M3 muscarinic acetylcholine receptor is a novel biomarker of poor prognostic in patients with non-small cell lung cancer. Tumour Biol, 34, 3939–3944. [DOI] [PubMed] [Google Scholar]
- 353.Wu JC, Chruscinski A, De Jesus Perez VA, Singh H, Pitsiouni M, Rabinovitch M, Utz PJ, & Cooke JP (2009). Cholinergic modulation of angiogenesis: role of the 7 nicotinic acetylcholine receptor. J Cell Biochem, 108, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Xi HJ, Wu RP, Liu JJ, Zhang LJ, & Li ZS (2015). Role of acetylcholinesterase in lung cancer. Thorac Cancer, 6, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Xiao D, & He J (2010). Epithelial mesenchymal transition and lung cancer. J Thorac Dis, 2, 154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Xu J, Huang H, Pan C, Zhang B, Liu X, & Zhang L (2007). Nicotine inhibits apoptosis induced by cisplatin in human oral cancer cells. Int J Oral Maxillofac Surg, 36, 739–744. [DOI] [PubMed] [Google Scholar]
- 357.Xu R, Shang C, Zhao J, Han Y, Liu J, Chen K, & Shi W (2015). Activation of M3 muscarinic receptor by acetylcholine promotes non-small cell lung cancer cell proliferation and invasion via EGFR/PI3K/AKT pathway. Tumour Biol, 36, 4091–4100. [DOI] [PubMed] [Google Scholar]
- 358.Yang IA, Holloway JW, & Fong KM (2013). Genetic susceptibility to lung cancer and co-morbidities. J Thorac Dis, 5 Suppl 5, S454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Yang K, Song Y, Tang YB, Xu ZP, Zhou W, Hou LN, Zhu L, Yu ZH, Chen HZ, & Cui YY (2014). mAChRs activation induces epithelial-mesenchymal transition on lung epithelial cells. BMC Pulm Med, 14, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Ye X, Zhang C, Chen Y, & Zhou T (2015). Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J Cancer, 6, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Yoo BB, & Mazmanian SK (2017). The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity, 46, 910–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Zakut H, Even L, Birkenfeld S, Malinger G, Zisling R, & Soreq H (1988). Modified properties of serum cholinesterases in primary carcinomas. Cancer, 61, 727–737. [DOI] [PubMed] [Google Scholar]
- 363.Zanini D, Schmatz R, Pelinson LP, Pimentel VC, da Costa P, Cardoso AM, Martins CC, Schetinger CC, Baldissareli J, do Carmo Araujo M, Oliveira L, Chiesa J, Morsch VM, Leal DB, & Schetinger MR (2013). Ectoenzymes and cholinesterase activity and biomarkers of oxidative stress in patients with lung cancer. Mol Cell Biochem, 374, 137–148. [DOI] [PubMed] [Google Scholar]
- 364.Zeidler R, Albermann K, & Lang S (2007). Nicotine and apoptosis. Apoptosis, 12, 1927–1943. [DOI] [PubMed] [Google Scholar]
- 365.Zenko D, & Hislop JN (2018). Regulation and trafficking of muscarinic acetylcholine receptors. Neuropharmacology, 136, 374–382. [DOI] [PubMed] [Google Scholar]
- 366.Zhang B, Lu L, Zhang X, Ye W, Wu J, Xi Q, & Zhang X (2014). Hsa-miR-132 regulates apoptosis in non-small cell lung cancer independent of acetylcholinesterase. J Mol Neurosci, 53, 335–344. [DOI] [PubMed] [Google Scholar]
- 367.Zhang C, Ding XP, Zhao QN, Yang XJ, An SM, Wang H, Xu L, Zhu L, & Chen HZ (2016). Role of alpha7-nicotinic acetylcholine receptor in nicotine-induced invasion and epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Oncotarget, 7, 59199–59208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Zhang C, Yu P, Zhu L, Zhao Q, Lu X, & Bo S (2017). Blockade of alpha7 nicotinic acetylcholine receptors inhibit nicotine-induced tumor growth and vimentin expression in non-small cell lung cancer through MEK/ERK signaling way. Oncol Rep, 38, 3309–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Zhang S, Togo S, Minakata K, Gu T, Ohashi R, Tajima K, Murakami A, Iwakami S, Zhang J, Xie C, & Takahashi K (2010). Distinct roles of cholinergic receptors in small cell lung cancer cells. Anticancer Res, 30, 97–106. [PubMed] [Google Scholar]
- 370.Zhang T, Lu H, Shang X, Tian Y, Zheng C, Wang S, Cheng H, & Zhou R (2006). Nicotine prevents the apoptosis induced by menadione in human lung cancer cells. Biochem Biophys Res Commun, 342, 928–934. [DOI] [PubMed] [Google Scholar]
- 371.Zhang XJ, & Greenberg DS (2012). Acetylcholinesterase involvement in apoptosis. Front Mol Neurosci, 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Zhang Y, Jia Y, Li P, Li H, Xiao D, Wang Y, & Ma X (2017). Reciprocal activation of alpha5-nAChR and STAT3 in nicotine-induced human lung cancer cell proliferation. J Genet Genomics, 44, 355–362. [DOI] [PubMed] [Google Scholar]
- 373.Zhang Y, Jiang M, Li Q, Liang W, He Q, Chen W, & He J (2016). Chromosome 15q25 (CHRNA3-CHRNB4) Variation Indirectly Impacts Lung Cancer Risk in Chinese Males. PLoS One, 11, e0149946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Zhang Y, Kang S, Fang W, Hong S, Liang W, Yan Y, Qin T, Tang Y, Sheng J, & Zhang L (2015). Impact of smoking status on EGFR-TKI efficacy for advanced non-small-cell lung cancer in EGFR mutants: a meta-analysis. Clin Lung Cancer, 16, 144–151 e141. [DOI] [PubMed] [Google Scholar]
- 375.Zhao M, He X, Yang YH, Yu XJ, Bi XY, Yang Y, Xu M, Lu XZ, Sun Q, & Zang WJ (2015). Acetylcholine protects mesenteric arteries against hypoxia/reoxygenation injury via inhibiting calcium-sensing receptor. J Pharmacol Sci, 127, 481–488. [DOI] [PubMed] [Google Scholar]
- 376.Zhao Q, Gu X, Zhang C, Lu Q, Chen H, & Xu L (2015). Blocking M2 muscarinic receptor signaling inhibits tumor growth and reverses epithelial-mesenchymal transition (EMT) in non-small cell lung cancer (NSCLC). Cancer Biol Ther, 16, 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Zhao Q, Yue J, Zhang C, Gu X, Chen H, & Xu L (2015). Inactivation of M2 AChR/NF-kappaB signaling axis reverses epithelial-mesenchymal transition (EMT) and suppresses migration and invasion in non-small cell lung cancer (NSCLC). Oncotarget, 6, 29335–29346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Zhao Y (2016). The Oncogenic Functions of Nicotinic Acetylcholine Receptors. J Oncol, 2016, 9650481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Zheng Y, Ritzenthaler JD, Roman J, & Han S (2007). Nicotine Stimulates Human Lung Cancer Cell Growth by Inducing Fibronectin Expression. Am J Respir Cell Mol Biol [DOI] [PubMed]
- 380.Zhou W, Geng T, Wang H, Xun X, Feng T, Zou H, Kang L, Jin T, & Chen C (2015). CHRNA3 genetic polymorphism and the risk of lung cancer in the Chinese Han smoking population. Tumour Biol, 36, 4987–4992. [DOI] [PubMed] [Google Scholar]
- 381.Zimmermann M (2013). Neuronal AChE splice variants and their non-hydrolytic functions: redefining a target of AChE inhibitors? Br J Pharmacol, 170, 953–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Zoli M, Pucci S, Vilella A, & Gotti C (2018). Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr Neuropharmacol, 16, 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Zovko A, Specici K, & Turk T (2009). New Aspects of the Relationship between Acetylcholinesterase Activity and Cancer:Poly-APS Experiments. WSEAS TRANSACTIONS on BIOLOGY and BIOMEDICINE, 6, 58–69. [Google Scholar]