

Summary
The protist parasite Trypanosoma brucei is an obligate extracellular pathogen that retains its highly-polarized morphology during cell division and has evolved a novel cytokinetic process independent of non-muscle myosin II. The polo-like kinase homolog TbPLK is essential for transmission of cell polarity during division and for cytokinesis. We previously identified a putative TbPLK substrate named Tip of the Extending FAZ 1 (TOEFAZ1) as an essential kinetoplastid-specific component of the T. brucei cytokinetic machinery. We performed a proximity-dependent biotinylation (BioID) screen using TOEFAZ1 as a means to identify additional proteins that are involved in cytokinesis. Using quantitative proteomic methods, we identified nearly 500 TOEFAZ1-proximal proteins and characterized 59 in further detail. Among the candidates, we identified an essential putative phosphatase that regulates the expression level and localization of both TOEFAZ1 and TbPLK, a previously uncharacterized protein that is necessary for the assembly of a new cell posterior, and a microtubule plus-end directed orphan kinesin that is required for completing cleavage furrow ingression. The identification of these proteins provides new insight into T. brucei cytokinesis and establishes TOEFAZ1 as a key component of this essential and uniquely-configured process in kinetoplastids.
Keywords: BioID, cytokinesis, T. brucei
Graphical Abstract
Abbreviated summary
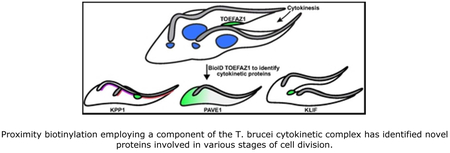
Trypanosoma brucei employs a novel cytokinetic mechanism that proceeds along the long axis of the cell and requires the kinetoplastid-specific protein TOEFAZ1. Proximity biotinylation using TOEFAZ1 as the bait has identified novel and essential proteins involved in T. brucei cytokinesis, including a kinesin, a phosphatase, and a protein associated with the subpellicular microtubules. These components illustrate the unique configuration of T. brucei cytokinesis and will allow a better mechanistic understanding of this process.
Introduction
The protist parasite Trypanosoma brucei is the causative agent of human African trypanosomiasis (HAT) and nagana in livestock, which are significant medical and economic burdens in Sub-Saharan Africa (Fèvre et al., 2008; Hotez and Kamath, 2009; Simarro et al., 2011; Welburn and Maudlin, 2012). The parasite is an obligate extracellular pathogen that has developed several strategies for avoiding detection and clearance by its hosts (Engstler et al., 2007; Hovel-Miner et al., 2015; Mugnier et al., 2015; Bartossek et al., 2017). Among these adaptations is a highly-polarized cell morphology, which is established by a set of microtubules that underlie the cell surface and give the cell body its distinctive tapered shape (Gull, 1999; Vickerman et al., 1969). This elaborate morphology is essential for the trypanosome’s auger-like motility, which is optimized for movement in high-viscosity solutions such as blood and can be configured by Trypanosoma subspecies to suit their different dissemination patterns in their mammalian hosts (Heddergott et al., 2012; Bargul et al., 2016; Wheeler, 2017). While the unique shape of the parasite has been tuned to maximize viability within its hosts, this high degree of specialization has placed unique constraints on other cellular processes such as cell division, which the parasite undergoes while maintaining its polarized layout (Wheeler, Gluenz, et al., 2013). Many eukaryotic cells decrease or entirely lose their polarity during cell division as a means to facilitate this process (Ragkousi and Gibson, 2014). The cleavage furrow, which most organisms form at a midpoint between segregated daughter organelles on the short axis of the cell, originates at the anterior tip of dividing trypanosomes and ingresses towards the posterior end along the long cellular axis, following the helical pattern of the subpellicular microtubules (Hammarton et al., 2007; Farr and Gull, 2012; Wheeler, Scheumann, et al., 2013). This process likely does not employ a contracting actomyosin ring to drive furrow ingression as the non-muscle myosin II motor that provides the force for contractile ring constriction in most organisms is absent in trypanosomes (Richards and Cavalier-Smith, 2005; Foth et al., 2006). The role of actin in cytokinesis is unclear, although significant depletion of the protein in the insect-resident procyclic form of the parasite yielded no growth defect (García-Salcedo et al., 2004). Understanding the T. brucei cytokinetic mechanism would uncover the unique adaptations this parasite has made to survive within its hosts and describe a unique and essential pathway that may be amenable to drug design.
Morphologic analysis has shown that cytokinesis in procyclic trypanosomes is linked to the duplication and positioning of the parasite’s flagellum (Kohl et al., 2003; Davidge et al., 2006). The flagellum is nucleated by a basal body that docks to the base of the flagellar pocket (FP), which is an invagination of the plasma membrane that functions as the sole site of endo- and exo-cytosis (Lacomble et al., 2009; Field and Carrington, 2009). The top of the flagellar pocket is tightly apposed to the flagellar membrane by a series of cytoskeletal structures, including the flagella pocket collar, the hook complex, and the centrin arm (Esson et al., 2012; Morriswood, 2015; Perdomo et al., 2016). Once the flagellum emerges onto the cell surface, it is attached to the cell body by the flagellum attachment zone (FAZ), which forms a series of desmosome-like structures that span the plasma and flagellar membranes to form a tight seal (Vickerman, 1969; Sunter and Gull, 2016). A set of four microtubules known as the microtubule quartet (MtQ) that nucleate at the basal body and are thought to have opposing polarity to the subpellicular microtubules are found next to the FAZ filament (Lacomble et al., 2009; Lacomble et al., 2012). During cell division, the new flagellum extends along the cell body next to the old flagellum; new FAZ formation follows approximately 2 microns behind the new flagellum tip within the cell body (Kohl et al., 1999). Towards the end of cell division, the cytokinetic furrow initiates from the anterior tip of the new FAZ and ingresses towards the posterior of the cell along an indentation known as the cleavage furrow fold (Robinson et al., 1995; Farr and Gull, 2012; Wheeler, Scheumann, et al., 2013). As the furrow ingresses, a new posterior end is formed that contains an organizing center for the subpellicular microtubules (Robinson et al., 1995; Wheeler, Scheumann, et al., 2013). While cytokinesis follows a similar pattern in the mammalian-resident bloodstream form of the parasite, furrow positioning is more symmetric and the basal bodies do not separate as far as they do in procyclics prior to ingression (Wheeler, Scheumann, et al., 2013).
Previous work has shown that perturbing FAZ formation either blocks furrow ingression or leads to mispositioning of the furrow, resulting in shortened daughter cells (LaCount et al., 2002; Vaughan et al., 2008; Zhou et al., 2011). Recent studies have identified a series of proteins that localize to the anterior tip of the new FAZ and play a role in controlling ingression of the cleavage furrow (Sunter et al., 2015; McAllaster et al., 2015). The polo-like kinase homolog in trypanosomes, known as TbPLK, is present in the posterior of the cell early in the cell cycle and is then recruited to the new FAZ tip, where it remains until just prior to furrow ingression (Kumar and Wang, 2006; de Graffenried et al., 2008; Ikeda and de Graffenried, 2012). The Aurora kinase homolog and components of the chromosomal passenger complex migrate from the nucleus to the new FAZ tip once chromosome congression is complete (Li et al., 2008; Li et al., 2009). Proximity biotinylation (BioID) and phosphoproteomics using TbPLK identified several FAZ proteins, including two proteins that associate with the tip of the new FAZ (McAllaster et al., 2015). One of these proteins, which we named Tip of the Extending FAZ 1 (TOEFAZ1), is necessary for cytokinesis and for recruitment of TbPLK to the new FAZ tip (McAllaster et al., 2015; Sinclair-Davis et al., 2017). Subsequent work by others named the protein cytokinetic initiation factor 1 (CIF1) (Zhou, Gu, et al., 2016). TOEFAZ1 is conserved in T. cruzi and Leishmania, which makes it possible that this protein is a unique and essential component of a kinetoplastid-specific cytokinetic complex. Proximity-dependent biotinylation was recently used to identify a single protein with a similar localization pattern, which was named CIF2 (Zhou, Hu, et al., 2016). This protein is essential for TOEFAZ1 recruitment to the tip of the new FAZ and is also required for formation of the cleavage furrow. Several other cytoskeletal components were also identified as potential interactors, but their specific localization pattern was not reported.
In this work, we have performed a BioID screen using TOEFAZ1 as the bait protein with the intent of mapping additional interacting proteins that could be important cytokinetic regulators. We identified almost 500 novel TOEFAZ1 interactors and localized 59 of the candidate proteins by tagging their endogenous loci. Among our candidates, we have identified a putative phosphatase that localizes to the new FAZ tip and regulates the expression levels of TOEFAZ1 and TbPLK, a protein that plays an essential role in the assembly of the new cell posterior, and an orphan kinesin that functions in the completion of cleavage furrow ingression. These proteins provide significant mechanistic insight into the different stages of cytokinesis in trypanosomes and will allow a more thorough analysis of how this divergent and essential process occurs.
Results
Optimized TOEFAZ1 BioID identifies nearly 500 nearby neighbors
To perform BioID, we generated a cell line expressing a triple Ty1-tagged BirA*-TOEFAZ1 fusion protein under doxycline-inducible control. BioID relies on the proximity of interacting proteins to the BirA* fusion, so assuring that the fusion protein is correctly localized is essential (Kim et al., 2014). We titrated the expression level of the Ty1-BirA*-TOEFAZ1 fusion compared to a cell line where both endogenous TOEFAZ1 alleles had been tagged with 3x-Ty1 (McAllaster et al., 2016). We found that expression of the BirA*-TOEFAZ1 fusion at 40 ng/mL of doxycycline was very similar to the double-tagged endogenous levels of protein (Fig. S1A). In doxycycline-treated cells incubated with 50 μM biotin, the BirA*-TOEFAZ1 fusion produced additional bands compared to uninduced controls when probed with streptavidin-HRP (Fig. S1B). The BirA*-TOEFAZ1 fusion localized to a bar-like structure along the dorsal side of dividing cells, marking the tip of the extending FAZ, which also labeled with avidin (Fig. S1C) (McAllaster et al., 2015; Sinclair-Davis et al., 2017). Previous BioID on TOEFAZ1 was performed at induction levels of 500 ng/mL doxycycline, which produces approximately a 3-fold overexpression compared to the double-tagged line and a broader cellular distribution that included a more distal part of the FAZ, along with additional fluorescent avidin labeling at the anterior end of the cell body (Fig. S1A,C) (Zhou, Hu, et al., 2016). While 500 ng/mL doxycycline is 5-fold over the reported saturation level for maximal pLEW100 expression in 29-13 cells, the precise degree of overexpression of the previous C-terminal BirA*-fusion is not known (Wirtz et al., 1999).
With the conditions for TOEFAZ1 BioID established, we performed large-scale experiments with capture on streptavidin magnetic beads so that we could identify proximal proteins by LC-MS/MS. Bound proteins from a small fraction of the beads were eluted in boiling SDS-PAGE loading buffer and blotted with streptavidin-HRP to identify biotinylated products. In lysates where the cells were not treated with doxycycline to induce expression of the BirA* fusion, approximately three bands were present, while multiple strong bands were visible in the lysates from cells where the BirA* fusion had been induced (Fig. S1D). The remaining beads from the control and doxycycline-induced samples were subjected to proteomic analysis by LC-MS/MS. Label-free quantitation was used to identify proteins enriched in the induced fraction, along with SAINTq to assign significant interactions (Choi et al., 2002; Choi et al., 2010). Three independent experimental replicates were used to identify proteins that were significantly enriched in the induced lysates.
In total, we identified 497 statistically significant interactors (P < 0.05), including 78 which were previously identified when the BioID was performed using the C-terminus of TOEFAZ1 at elevated levels of protein expression (Supplemental Table 1) (Zhou, Hu, et al., 2016). Our increased detection levels are most likely due to ion exchange fractionation prior to LC-MS/MS, which significantly increases sensitivity (Delahunty and Yates, 2005). Tagging the N-terminus instead of the C-terminus could also bring more proteins within the diffusion range of the AMP-biotin, as has been seen with nuclear pore components (Kim et al., 2014). Our list included many previously-identified FAZ components, such as FAZ10, FAZ16, SAS4, which localize to the full length of the FAZ or to the anterior tip (Morriswood et al., 2013; Sunter et al., 2015; Huiqing Huxs et al., 2015. We also identified CIF2 and TbPLK, which have been shown to interact with TOEFAZ1 by BioID and co-immunoprecipitation (McAllaster et al., 2015; Zhou, Hu, et al., 2016; Sinclair-Davis et al., 2017). TbSmee1, which localizes to the hook complex and the new FAZ tip during division, was also identified (Perry et al., 2017). A series of proteins associated with the subpellicular microtubule array, such as WCB1, TbAIR9, and XMAP215 were also identified; the latter is present at the posterior end of the cell (Woods et al., 1992; May et al., 2012; Wheeler, Scheumann, et al., 2013). The presence of many FAZ proteins and other FAZ-proximal cytoskeletal compartments suggested that the less characterized candidates contained novel components that merited further study.
We chose 59 proteins to characterize in further detail (Fig. 1). We selected these proteins using the following criteria: 1) Strength of the association based on the SAINTq score; 2) Proteins that were uncharacterized or had no reported localization; 3) Proteins that had domains that could be involved in microtubule-based or cytoskeletal interactions. We included proteins that had been previously identified as TOEFAZ1 interactors but whose localizations were not reported (Zhou, Hu, et al., 2016). Each gene selected for analysis was initially N-terminally tagged at its endogenous locus with three copies of the Ty1 epitope using a Gibson Assembly™ strategy (McAllaster et al., 2016). Clonal cell lines for each protein were tested for correct insertion of the endogenous tag using PCR on isolated genomic DNA with primers specific for each locus and anti-Ty1 western blotting to test the size of the tagged protein. When proteins had putative signal sequences or little to no signal by immunofluorescence or western blotting, we moved the Ty1 tag to the C-terminus to determine if location of the epitope affects expression. Overall, we were able to tag all 59 of our candidate proteins and establish their localization (Fig. S7).
Figure 1.

Table summarizing the localization pattern of the 59 proteins selected for further characterization after being identified as nearby neighbors of Ty1-BirA* TOEFAZ1.
Our candidate proteins localized primarily to compartments that are proximal to TOEFAZ1 or could be involved in cytokinesis. These compartments include the hook complex/centrin arm, the full length of the FAZ, and the tip of the extending FAZ, which are the structures that TOEFAZ1 localizes to during cell division (McAllaster et al., 2015; Zhou, Gu, et al., 2016; Sinclair-Davis et al., 2017). We also identified a series of endomembrane components which are potentially interesting because membrane remodeling likely occurs at the position of the ingressing furrow (Farr and Gull, 2012). Another set of proteins appeared to have polarized distributions along the subpellicular microtubules, either biased towards the anterior or posterior ends. We subsequently focused our attention on three proteins that played distinct roles at different points in the cytokinetic process: a putative phosphatase necessary for TbPLK and TOEFAZ1 expression and recruitment to the new FAZ tip, a protein involved in formation of a new posterior end, and an orphan kinesin that is essential for completion of cleavage furrow ingression.
Putative phosphatase KPP1 regulates expression of cytokinetic components
We first focused on Tb927.5.4380, which has been annotated as a kinetoplastid-specific protein (Ser/Thr) phosphatase. The putative phosphatase has a C-terminal domain with homology to PP1 and PP2 phosphatases. The residues involved in catalytic activity and divalent metal binding appear to be intact, so it is likely that the protein is active (Parsons et al., 2005; Brenchley et al., 2007). In a concurrent paper, another group has named this protein Kinetoplastid-specific Protein Phosphatase 1 (KPP1), so we will maintain this nomenclature (Zhou, JBC, in press). While several kinases such as TbPLK and TbAUK have been implicated in regulating cytokinesis, very little is known about the phosphatases involved in this process. TOEFAZ1 has been proposed to function as a scaffold protein and contains many phosphosites that are likely to influence its ability to recruit different proteins to the new FAZ tip at different points of the cell cycle (McAllaster et al., 2015; Zhou, Gu, et al., 2016).
To establish the localization of KPP1, we tagged one allele of the putative phosphatase with Ty1 and determined the localization of the protein compared to TOEFAZ1 during the cell cycle. KPP1 had a significant cytoplasmic pool throughout the cell cycle, so we confined our analysis to cytoskeletons that had been extracted with low levels of detergent. Once TOEFAZ1 was visible in late 1N1K cells, the putative phosphatase accumulated at the tip of the new FAZ structure while also maintaining its cytoplasmic distribution (Fig. 2A). This partial colocalization persisted until the 2N2K stage, when KPP1 signal diminished from the new FAZ tip but remained in the cytoplasm.
Figure 2.

The putative phosphatase KPP1 localizes to the tip of the new FAZ and is necessary for cytokinesis. A) KPP1 was tagged at its endogenous locus with a Ty1 epitope tag in a cell line that contained a TOEFAZ1 allele tagged with an HA epitope tag. Cells were extracted, fixed, and stained with anti-HA and anti-Ty1 antibody. Empty arrowheads: HA-TOEFAZ1. Filled-in arrowheads: Ty1-KPP1. B) Quantification of DNA state in PFA-fixed KPP1 RNAi cells that were treated with either vehicle control or 1 μg/mL doxycycline to induce RNAi for four and five days.
We depleted KPP1 using a tetracycline-inducible long hairpin RNAi (lhRNAi) in cells where one KPP1 allele was tagged with Ty1 so we could follow depletion of the protein by western blotting (McAllaster et al., 2016). Cells were treated with doxycycline to induce lhRNAi expression or a vehicle control, and cell growth was monitored over the course of eight days (Fig. S2A). Cell growth was indistinguishable between the control and KPP1-depleted samples until the third day of the experiment, at which point the depleted cells rapidly ceased to divide. Western blotting showed that small amounts of protein were visible until the third day of depletion, which suggests that the growth defect appeared rapidly after protein levels dropped below detectable levels (Fig. S2B). We employed DIC imaging on fixed cells stained with DAPI to identify cell cycle and morphological defects on days four and five after initiating RNAi (Fig. S2C). Among the normal cell cycle states, we observed a decrease in 1N1K and 1N2K cells along with an increase in 2N2K cells, which suggests a cytokinetic delay or block (Fig. 2B). Many of the 2N2K cells had detached new flagella tips (Fig. S2D), an event that should not occur until late in the cell cycle after significant ingression of the cleavage furrow, which was also not evident in these cells (Wheeler, Scheumann, et al., 2013; Varga et al., 2017). We noted the appearance of cells with aberrant DNA states, including 2N1K, 0N1K, and multinucleate cells.
Depletion of KPP1 may change the phosphorylation state of TOEFAZ1, which could alter the recruitment of different proteins necessary for cytokinesis. To test this, a Ty1 tag was appended to the N-terminus of TOEFAZ1 in cells carrying the KPP1-lhRNAi plasmid, followed by RNAi induction using doxycycline. We first evaluated TOEFAZ1 expression by western blot to look for changes in electrophoretic mobility as a sign of altered phosphorylation (Fig. 3A). TOEFAZ1 migrates at a significantly higher molecular weight than expected, most likely due to a high degree of phosphorylation that has been detected (McAllaster et al., 2015; Zhou, Gu, et al., 2016). We observed very modest differences in mobility but were surprised to note that the expression level of TOEFAZ1 was significantly decreased. This was especially noticeable beyond 3 days of RNAi induction, which correlates with the onset of full KPP1 depletion and the cell growth phenotype. We next evaluated the localization of TOEFAZ1 in KPP1 RNAi cells (Fig. 3 B,C). In the control sample TOEFAZ1 showed the expected localization pattern, initially appearing near the flagellar pocket as the new FAZ is formed and then moving towards the anterior at the tip of the cell along the new FAZ as it extends (McAllaster et al., 2015). In cells lacking KPP1, TOEFAZ1 signal was significantly decreased at all stages of the cell cycle. In the few remaining cells that did express TOEFAZ1, the protein was no longer effectively localized to the new FAZ tip but was confined to the flagellar pocket region. The assembly of the new FAZ appeared to progress normally, although we did observe detached new flagella tips, as we saw in our DAPI/DIC analysis (Fig. 3B).
Figure 3.

TOEFAZ1 localization is altered in KPP1-depleted cells. A) KPP1 RNAi was induced using 1 μg/mL doxycycline in a cell line in which TOEFAZ1 was tagged at its endogenous locus with a Ty1 epitope tag. Lysates were collected every 24 h and subjected to western blotting using anti-Ty1 antibody. B) Cells from (A) were collected after four days, methanol-fixed, and stained using anti-Ty1 antibody and 1B41 antibody to label the FAZ. Arrowheads:Ty1-TOEFAZ1 localization. Asterisks indicate detached flagellar tips. DAPI was used to stain DNA. C) Quantification of TOEFAZ1 localization in cells from B).
Considering the loss of TOEFAZ1 expression and localization, we looked at the expression of TbPLK to see if it was also affected by the loss of KPP1. We induced RNAi against KPP1 and determined the expression levels of TbPLK by western blotting with antibodies against the native protein (Fig. 4A). Similar to what is observed for TOEFAZ1, depletion of KPP1 caused a concomitant decrease in TbPLK expression levels that manifested at day three. We stained control and KPP1-depleted cells with 1B41 to label the FAZ and antibodies against TbPLK (Fig. 4B). In control cells, the expression of TbPLK mirrored what has been seen previously, with the kinase initially localized on the basal body and the cytoskeletal structures neighboring the flagellar pocket (de Graffenried et al., 2008). Once the new FAZ has formed, TbPLK migrates onto the tip of the new FAZ and remains there until late into the 2N2K stage of the cell cycle (Ikeda and de Graffenried, 2012). In cells lacking KPP1, we noted a significant decrease in the number of cells expressing the kinase at the 1N2K and 2N2K stages (Fig. 4 B,C). This result is also similar to the phenotype observed when TOEFAZ1 itself is depleted or when TbPLK is inhibited with a small molecule, supporting the idea that KPP1 functions in a similar pathway (Lozano-Núñez et al., 2013; McAllaster et al., 2015).
Figure 4.

TbPLK no longer tracks to the tip of the extending FAZ in KPP1-depleted cells. A) KPP1 RNAi was induced and lysates were collected every 24 h, subjected to western blot and stained with anti-TbPLK antibodies. B) Cells from (A) were methanol-fixed after four days of KPP1 RNAi induction and labeled with anti-TbPLK antibody and 1B41 for the FAZ. DAPI was used to label DNA. Arrowheads: TbPLK localization. Asterisks indicate detached flagellar tips. C) Quantification of TbPLK localization from cells in (B).
PAVE1 is necessary to shape the posterior cytoskeleton of T. brucei
Our second candidate gene (Tb927.8.2030) localized to the posterior tip of the cell, with stronger signal along the ventral side of the cell body (Fig. 5A) when one endogenous allele was tagged with Ty1. At later points in the cell cycle, the protein collects at the nascent posterior end and at the lateral fold that forms at the posterior end prior to cleavage furrow ingression (Fig. 5A, asterisk). In cleaving 2N2K cells, the protein is present along the ingressing furrow, where it colocalizes with TOEFAZ1 (Fig. 5B, arrowhead). Based on the localization pattern of this protein, we named it Posterior and Ventral Edge protein 1 (PAVE1). PAVE1 is conserved among other trypanosomatids, though its function has not been studied. Protein folding prediction programs (PHYRE2, COILS) predict that PAVE1 is comprised of a series of coiled coil domains (McDonnell et al., 2006; Mezulis et al., 2015).
Figure 5.

PAVE1 is necessary for proper posterior end formation. A) PAVE1 was tagged at its endogenous locus with a Ty1 epitope tag and cells were methanol-fixed and labeled with anti-Ty1 antibody. Asterisk indicates the nascent posterior of the old-flagellum containing daughter cell. B) Cells in which PAVE1 and TOEFAZ1 were endogenously tagged with a Ty1 and HA epitope tag, respectively, were methanol-fixed and stained with anti-Ty1 and anti-HA antibodies, and DAPI was used for DNA. Arrowhead indicates TOEFAZ1 localization along the cleavage furrow. C) PAVE1 RNAi cells were treated with either vehicle control or 1 μg/mL doxycycline for 24 h. Cells were collected, PFA-fixed, and stained with DAPI for DNA. Arrowheads indicate kinetoplasts located at the posterior edge of the cell. D) Quantification of DNA state of cells from B). E) The kinetoplast-to-nucleus and kinetoplast-to-posterior distance was measured in cells from B). Shaded regions are the S.E.
We depleted PAVE1 using an lhRNAi to observe protein function. As with KPP1, we tagged one allele of PAVE1 with Ty1 in the candidate lhRNAi lines so that protein depletion could be monitored by western blotting. Cell growth began to decline after two days of RNAi induction and subsequent growth was essentially blocked (Fig. S3A). Western blotting with anti-Ty1 antibody showed a significant depletion of PAVE1 protein at days one and two, followed by loss of detectable protein levels at later time points (Fig. S3B). We assessed cell cycle progression in control and doxycycline-induced cells to determine if there were any defects (Fig. 5 C,D). After one day of RNAi induction, a small increase in 1N2K cells was evident. After two days of depletion, a population of anucleate “zoid” (0N1K) cells appeared, along with 2N1K cells and multinucleated cells. Interestingly, the number of 2N1K and multinucleated cells was close to the total number of zoids, which suggested that the zoids were being produced by aberrant cell divisions, perhaps due to misplacement of the cleavage furrow. We also noted a specific phenotype in 1N1K cells where the kinetoplast was located in the extreme posterior end of the cell, similar to its placement in bloodstream form trypanosomes (Fig. 5C).
To further characterize the kinetoplast placement defect, we calculated the distance between the posterior end of the cell and the kinetoplast in control cells and cells lacking PAVE1 at different stages of the cell cycle (Fig. 5E). We employed DIC imaging to mark the edge of the cell and DAPI to label the kinetoplasts. In control 1N1K cells, the median distance between the posterior of the cell and the kinetoplast was 6.7 microns, with 90% of cells falling within 4 and 9 microns. In 1N1K cells lacking PAVE1, the median distance was 5.3 microns, with more than 30% of cells containing kinetoplasts within 3 microns of the posterior end of the cell. It is possible that the posterior localization of the kinetoplast is due to an increased distance between the nucleus and kinetoplast. To see if this was the case in our PAVE1-depleted cells, we measured the distance between the nucleus and kinetoplast in 1N1K cells (Fig. 5E). There was no apparent change in this distance, which strongly suggested that the observed defect was due to a shortening of the posterior of the cell. In 1N2K and 2N2K cells the more posterior kinetoplast was also closer to the cell posterior, suggesting that the shortened posterior end of the cells persisted throughout the cell cycle.
During the characterization of PAVE1, we noted that detergent-extracted cytoskeletons prepared from cells lacking the protein tended to be frayed and disorganized when visualized by DIC. To further characterize this phenotype, we isolated extracted cytoskeletons from PAVE1 RNAi and control cells, stained them with aurothioglucose as a negative stain, and subjected them to whole-mount electron microscopy. Control cells had tapered posteriors produced by a subset of the subpellicular microtubules coming together at a distinct point (Fig. 6A). The basal body and flagellum localized to the anterior side of the tapered portion of the cell. In cells lacking PAVE1, the subpellicular array appeared to terminate abruptly at the same point with minimal tapering, although the spacing between the subpellicular microtubules was maintained. The basal body was in close proximity to the termination point, which is similar to the aberrant kinetoplast positioning we saw by DAPI/DIC imaging (Fig. 5C).
Figure 6.

Cell posteriors are truncated in PAVE1-depleted cells. A) Negative stain TEM of 1N1K control cells and 1N1K cells depleted of PAVE1 for 1 day. B) Negative stain TEM of replicating control cells and cells depleted of PAVE1 for 1 or 2 days. After 2 days of PAVE1 RNAi, 2N1K and 0N1K cells start to appear in the population, most likely due to improper cleavage furrow resolution.
We next focused on the phenotypes of cells lacking PAVE1 that were actively dividing. In control samples, we could identify 2N2K cells where an indentation at the posterior end of the cell marked the position of the nascent posterior end (Fig. 6B). The old posterior end of the cell was tapered and the new flagellum was correctly positioned. In cells lacking PAVE1, we noted 2N2K cells with frayed posterior ends and basal bodies localized at the extreme posterior, similar to the 1N1K cells we saw previously, suggesting that cells with these aberrant structures could still duplicate and arrange their organelles effectively. However, in the absence of PAVE1 cells had detached new flagella tips with no evidence of furrow ingression, which argued that there was a delay in progression through cytokinesis. The indentation that designates the nascent posterior end was shifted towards the posterior compared to wild type. We also observed 2N1K cells with a single flagellum present between the two nuclei, along with a frayed posterior end, and zoids where the basal body was located in the extreme posterior of the cytoplast.
The posterior end of the trypanosome cell body has previously been shown to be enriched for extending microtubules (Sherwin et al., 1987; Sasse and Gull, 1988). Cells lacking PAVE1 may not be able to extend or modify microtubules during assembly of a new cell posterior during cell division, which would explain their inability to duplicate correctly. Newly-formed tubulin contains a C-terminal tyrosine residue that can be selectively removed and re-added by the actions of tubulin tyrosine carboxypeptidase and tubulin tyrosine ligase (Janke and Chloë Bulinski, 2011; Janke, 2014). The anti-tyrosinated tubulin antibody YL1/2 preferentially labels the posterior end of the cell, along with a pool of unpolymerized tubulin monomers that associate with the trypanosome basal body (Kilmartin et al., 1982; Sasse and Gull, 1988; Sherwin and Gull, 1989; Stephan et al., 2007). PAVE1 partially co-localizes with YL1/2 staining at the cell posterior (Fig. S4A). We tested to see if depletion of PAVE1 caused a change in the distribution of tyrosinated tubulin. After 1 or 2 days of PAVE1 depletion, the YL1/2 labeling at the cell posterior had an altered morphology that reflected the rounding and shortening of the posterior end, but antibody labeling was still evident (Fig. S4 B,C). We performed YL1/2 western blots to determine if there was a significant change in the total amount of tyrosinated tubulin in cells lacking PAVE1 (Fig. S4C). After depleting PAVE1 over the course of six days, we were unable to observe any difference in the total amount of tyrosinated tubulin in cells, which shows that the primary defect we are observing is not due to a defect in tubulin synthesis or tyrosination of tubulin in general.
The kinesin KLIF is responsible for cleave furrow resolution at the cell posterior
We also identified an orphan kinesin among our BioID hits that is a member of the TbKinX1 family (Wickstead and Gull, 2006). To determine its localization, we tagged the motor protein with Ty1 at its N-terminus and appended a 3X HA-tag to the N-terminus of TOEFAZ1 in the same cell line. We then performed 4-color immunofluorescence imaging with the FAZ marker 1B41 and DAPI to mark DNA state (Fig. 7A) (Gallo and Précigout, 1988; Gallo et al., 1988). Early in the cell cycle, the kinesin had two discrete localization patterns. Most 1N1K cells had limited signal for either the kinesin or TOEFAZ1, although low levels could be present in the cytosol. TOEFAZ1 became visible as the cells initiated the formation of a new FAZ. No evidence of the kinesin was visible during the 1N2K stage, but it was present in a subset of 2N2K cells with a fully extended new FAZ and colocalized with TOEFAZ1. In 2N2K cells with actively ingressing cleavage furrows, the motor was localized to a focus along the side of the furrow where the old FAZ is present, while TOEFAZ1 was present in a more diffuse pattern along most of the furrow (more examples of the kinesin moving along ingressing furrows are presented in Fig. S5A). This focus persisted in early 1N1K cells near the cell posterior, which likely marks the end point of kinesin migration along the FAZ towards the posterior end of the cell during furrow ingression. The focus disappears soon after the completion of cytokinesis, as the structure was not observed in TOEFAZ1-positive 1N1K cells that are entering cell division. The immunofluorescence experiments show that the kinesin first localizes to the tip of the extending FAZ in late 2N2K cells that are about to initiate furrow ingression, then migrates along the old FAZ towards the cell posterior as the cleavage furrow ingresses, finally concentrating in a single focus near the kinetoplast in 1N1K cells that have just completed cell division. Because of its localization pattern, we name this protein Kinesin Localized to the Ingressing Furrow (KLIF).
Figure 7.

KLIF is required for nascent posterior end formation and resolution of the cleavage furrow. A) Cells containing KLIF and TOEFAZ1 endogenously tagged with Ty1- and HA-epitope tags, respectively, were methanol-fixed and stained with anti-Ty1 and anti-HA antibody, as well as 1B41 to label the FAZ. Empty arrowheads: Ty1-KLIF. Filled-in arrowheads: HA-TOEFAZ1. B) Quantification of DNA state in control and KLIF RNAi cells that were PFA-fixed. C) The number of 2N2K and multinucleated cells containing cleavage furrows were counted in cells from B). To the right are representative images of stalled furrow formation (arrowheads). D) TOEFAZ1 RNAi was induced for 2 days in a cell line containing KLIF tagged at its endogenous locus with a Ty1 epitope tag. Cells were extracted using PEME- 0.25% NP-40, PFA-fixed, and stained with anti-Ty1 antibody. 1B41 was used to label the FAZ. Arrowhead points to Ty1-KLIF localization. In all immunofluorescence panels, cells were stained with DAPI for DNA.
We depleted KLIF to determine if the protein was essential for cleavage furrow ingression. Cells carrying a lhRNAi against KLIF and a Ty1 tag on the N-terminus of one KLIF allele were induced with doxycycline or a vehicle control. Induction of KLIF RNAi led to a rapid decline in the rate of cell division, which first appeared after two days and led to growth arrest after four days (Fig. S5B). The decreased rate of cell division correlated with the loss of protein, which was assessed by Ty1 western blotting (Fig. S5C). KLIF levels were decreased significantly after one day of RNAi and undetectable at later time points. We next determined the DNA state of cells lacking KLIF using DIC and DAPI imaging to identify any morphological or DNA defects (Fig. 7B,S5D). Cultures lacking KLIF rapidly accumulated elevated levels of 2N2K cells along with cells containing aberrant DNA stages, including anucleate zoids and multinucleate cells, suggesting a defect in cytokinesis. We also noticed a preponderance of cells with ingressing cleavage furrows that terminated near the duplicated nuclei (Fig. 7C). Under control conditions, fewer than 5% of 2N2K cells had ingressing furrows, while this number increased to 30% one day after KLIF RNAi. More than 40% of the multinucleated cells in the KLIF RNAi conditions had one or more ingressing furrows. From these results, it appears that KLIF is not essential for the initiation of the cleavage furrow, but instead is necessary for the furrow to completely ingress and for the cells to complete cytokinesis. If the process does not complete, the cells are able to undergo subsequent rounds of organelle replication and partitioning, which then fails at the same point of furrow ingression.
The timing of KLIF recruitment to the tip of the new FAZ could be controlled by TOEFAZ1, which we have proposed to function as a scaffolding protein that recruits different components as the cell cycle progresses (Sinclair-Davis et al., 2017). We depleted TOEFAZ1 in cells where KLIF was tagged with a Ty1 epitope so that we could monitor the localization and expression of the motor. In cells lacking TOEFAZ1, we saw a near-quantitative loss of KLIF recruitment to the tip of the new FAZ and to the ingressing furrow (from 52% in control cells to 3% in cells depleted of KLIF), which strongly suggests that TOEFAZ1 is required for KLIF to localize correctly (Fig. 7D). We assessed the level of KLIF protein expression to determine if the loss of KLIF localization was due to a decrease in protein expression in cells lacking TOEFAZ1 (Fig. S5E). Anti-Ty1 western blotting showed no decline in KLIF expression levels in the presence or absence of TOEFAZ1, which argues that the protein was still expressed but was likely confined to the cytoplasm. The slightly elevated levels of KLIF in cells at day 1.5 and day 2 are due to an increase in cell size at these later time points due to defects in cytokinesis caused by the absence of TOEFAZ1 (McAllaster et al., 2015; Sinclair-Davis et al., 2017).
To gain further mechanistic information into how KLIF functions at the cleavage furrow, we expressed and purified a truncated version of the motor in bacteria and determined its speed and directionality using an ensemble in vitro motility assay. Like conventional kinesin, KLIF can be divided into four distinct domains (Woehlke and Schliwa, 2000). It contains a C-terminal tail that may function in binding adapter proteins or cargo, a long coiled-coil stalk, a neck and a conserved microtubule and MgATP-binding motor domain with a unique ~128 amino acid N-terminal extension. We also expressed the well-studied truncation construct of conventional rat brain kinesin (KIF5c) (Sack et al., 1997). We appended an mClover3 variant of GFP with a His8 tag to the C-terminus of the both motor domains to allow us to purify and immobilize them in a flow chamber (Bajar et al., 2016). The proteins were expressed in BL21 E. coli and purified on nickel-agarose beads, followed by size exclusion in the case of KLIF, to produce homogeneous proteins of the predicted molecular weights (Fig. 8 A,D). KLIF and KIF5c were specifically attached to a GFP antibody-coated flow chamber through its C-terminal mClover3 tag to ensure the heads are oriented to interact with microtubules (Fig. S6). We then added taxol-stabilized, fluorescently-labeled microtubules that had been polymerized under conditions that selectively enhance the fluorescent signal at their minus end (Hyman, 1991). The speed and directionality of microtubule movement was monitored using fluorescence microscopy (Vale et al., 1985; Massow et al., 1989). We found that KLIF moves microtubules with their bright minus-ends leading, indicating that KLIF is a plus-end directed motor, which is typical for most kinesins (Fig. 8B and Supporting Information Movie S1). As a control, we show that the plus-end directed Kif5c also moves polarity-marked microtubules with their bright minus ends leading (Fig. 8E) (Howard and Hyman, 1993). We further quantified the movement of microtubules and found that KLIF generates an average speed of only 10 nm/sec (Fig. 8C). Under the same buffer conditions, KIF5c moves microtubules at much faster speed, with an average rate of 666 nm/sec (Fig. 8F).
Figure 8.

In vitro characterization of expressed KLIF motor domain. A,D) Lane 1, molecular weight protein markers on a 4-12% SDS PAGE gradient gel stained with Coomassie. Lane 2, bacterial expressed motor domain of KLIF (a.a. 1-672) or conventional kinesin Kif5 (a.a. 1-407) fused at the C-terminus with the GFP variant, mClover3 and His8 tag, purified by nickel affinity and size exclusion chromatography. B,E) Epifluorescence images showing movement of polarity marked microtubules on KLIF-mClv3 or Kif5-mClv3 using an in vitro motility assay. The minus-end of the microtubule (arrowhead) is marked with a heavily rhodamine-labeled microtubule seed. Movement of microtubules with the minus-end leading indicates that KLIF and Kif5 are plus-end directed motors. Kif5 has a well-established role in plus-end directed cargo transport. C) Representative speed histogram showing that KLIF moves microtubules with an average velocity of 9.7 ± 0.66 nm/sec (N = 100) at 1 mM MgATP concentrations. Data is representative of 3 independent experiments and two protein preparations. F) Representative speed histogram showing that conventional kinesin moves microtubules with an average velocity of 666 ± 54.2 nm/sec (N = 100) at 1 mM MgATP concentrations. Data is representative of 2 independent experiments and one protein preparation.
Discussion
In this work, we have employed BioID to identify potential TOEFAZ1 interactors as a means to better understand the divergent mechanism of cytokinesis in kinetoplastids. We have found proteins that play a role in key aspects of cytokinesis, including regulation of expression of cytokinetic proteins, the formation of a new cell posterior, and the resolution of the posterior end during cleavage furrow ingression. These proteins will provide a foundation for expanding our understanding of the unique aspects of T. brucei cytokinesis.
Our TOEFAZ1 BioID hits include many components of the FAZ and the subpellicular microtubule array (Figs. 1, S7, & Supplemental Table 1). TOEFAZ1 has been proposed to function as a scaffold for the recruitment of different morphogenic and cytokinetic proteins throughout the extension of the new FAZ and ingression of the cleavage furrow (Sinclair-Davis et al., 2017). For example, TbPLK interacts with TOEFAZ1 at early points of new FAZ extension and then slowly disassociates from the structure, while KLIF1 is recruited just prior to cleavage furrow ingression (Fig. 7A) (McAllaster et al., 2015). This makes it likely that different complexes form at the point depicted by TOEFAZ1 depending on what cellular events are occurring. The preponderance of FAZ components identified suggests that the TOEFAZ1 complex may be contiguous with the FAZ. The distribution of FAZ components along the filament and MtQ is still being established, but our previous work suggests that the TOEFAZ1 complex is likely associated with the tip of the new MtQ (Ikeda and de Graffenried, 2012). It is interesting to note that well-established filament proteins such as FAZ1 are absent from our screen, while proteins associated with the MtQ and FAZ ER such as CC2D are present (Vaughan et al., 2008; Zhou et al., 2011).
The FAZ filament and MtQ are closely associated with the subpellicular microtubules and the proteins that decorate them. Proteins identified in our screen such as WCB and AIR9 are found throughout the corset and play important roles in the maintenance and inheritance of the cytoskeleton during cell division (Baines and Gull, 2008; May et al., 2012). It is possible that a complex containing TOEFAZ1 plays a role in remodeling the subpellicular microtubules during cleavage furrow ingression. Moreover, since several secretory components were discovered in our TOEFAZ1 BioID, ingression likely requires the local remodeling and deposition of plasma membrane components to generate two distinct cell bodies. While membrane budding and fusion is thought to be confined to the flagellar pocket, it is possible that some membrane components are trafficked directly to the furrow to facilitate membrane remodeling, similar to the process that occurs at the midbody in mammalian cells (Baluška et al., 2006; Chi-Kuo Hu et al., 2012).
KPP1 is a member of the kinetoplastid-specific Ser/Thr phosphatases that have not been well studied to date (Brenchley et al., 2007; Szöőr, 2010). The phosphatase Cdc25, which plays a pivotal role in cell division in most eukaryotes, is absent in T. brucei (Szöőr, 2010; Li, 2012). However, the general PP1-PP2A phosphatase inhibitor okadaic acid was shown to block cytokinesis and kinetoplast segregation, arguing that other phosphatases are likely to play similar roles to Cdc25 (Das et al., 1994; Li et al., 2006). Along with KPP1, another PP2A homolog has recently been identified that blocks cell division, although the localization of the phosphatase was not reported and there appeared to be a flagellum duplication defect that we did not observe with KPP1 (Rothberg et al., 2014).
There are several possibilities for how KPP1 is involved in controlling the expression levels of TbPLK and TOEFAZ1 (Figs. 3,4). The putative phosphatase appears to be excluded from the nucleus, so direct control of gene expression seems unlikely, unless a cytosolic substrate must be dephosphorylated prior to undergoing nuclear translocation to promote TbPLK/TOEFAZ1 expression. Both proteins are heavily phosphorylated, so it is possible that they are direct substrates of KPP1 (Urbaniak et al., 2013; McAllaster et al., 2015). In the case of TbPLK, a PEST domain that functions as a degron was recently identified in the linker domain between the N-terminal catalytic domain and the C-terminal polo-box (Huiqing Hu et al., 2017). Deletion of the PEST domain in T. brucei led to the stabilization of TbPLK compared to wild type. It is possible that KPP1 functions within this pathway by removing phosphorylation within the PEST domain that causes TbPLK degradation. However, in overexpressed versions of TbPLK mutagenizing all the potential phosphosites to alanine did not increase TbPLK stability compared to wild type, but this may not represent the physiologically relevant expression level. While TOEFAZ1 does not have a PEST domain, its central IDP domain is necessary for protein turnover at the end of the cell cycle and is also essential for retaining TbPLK at the new FAZ tip (Sinclair-Davis et al., 2017). A subset of the TOEFAZ1 phosphosites may be involved in degrading the protein at the end of the cell cycle and therefore need to be actively removed during early parts of the cell cycle by KPP1.
PAVE1 localizes to the posterior end of the parasite, where it is likely associated with a subset of the subpellicular microtubules (Fig. 5A). Several proteins have been identified that associate with the subpellicular microtubules, including CAP15/17, WCB, and CAP5/5.5 (Hertz-Fowler et al., 2001; Vedrenne et al., 2002; Baines and Gull, 2008). These proteins localize throughout the corset microtubule array and are thought to provide crosslinking and stabilization to the microtubules. Depletion or overexpression tends to cause a defect in the integrity of the array and in cell division, usually resulting in the production of anucleate cells. The AIR9 homolog in trypanosomes is associated with the corset and appears to control nuclear positioning and cleavage furrow ingression (May et al., 2012). PAVE1 shares a similar localization pattern with GB4, a small repetitive protein that was found to localize primarily in the cell posterior, although its function has not been identified (Rindisbacher et al., 1993).
PAVE1 is necessary for formation of the tapered posterior end of the trypanosome. At early time points, cells lacking PAVE1 are able to divide to produce cells with correct DNA content, but these cells have disordered posterior ends that include basal bodies and kinetoplasts abutting the extreme posterior terminus of the cell body (Fig. 5E,6A). The aberrant placement of these structures is due to the absence of the tapered posterior end and not due to a defect in the normal position of the basal body and kinetoplast with regard to the rest of the cellular organelles. Subsequent cell division events lead to the production of zoids and multinucleate cells, most likely due to a defect in positioning of the cytokinetic furrow caused by either the disorganization of the microtubule corset or the proximity of the basal body to the cell posterior (Figs. 5C,6B). The subpellicular array is highly crosslinked to provide stability, with a constant spacing between the microtubules (Robinson et al., 1995). The tapering of the posterior end must therefore include a reduction in the number of microtubules to allow for the narrowing of the cell body (Wheeler, Scheumann, et al., 2013). One possible function of PAVE1 is to cap or bind to microtubule termini so that they are organized in a manner that allows the cell body to taper its posterior.
We have identified a novel orphan kinesin that is essential for the completion of furrow ingression and is downstream from the function of TOEFAZ1. Other orphan kinesins have been identified that appear to have an indirect effect on cytokinesis, but the localization of KLIF to the tip of the new FAZ and its subsequent track along the ingressing furrow point to a more direct function (Fig. 7A) (Li et al., 2008; Huiqing Hu et al., 2012). KLIF cytoplasmic signal is evident throughout the cell cycle, so it is likely that there is a signal that recruits the kinesin to the new FAZ tip when furrow ingression initiates. A possible candidate for this signal is a change in phospho-state of TOEFAZ1, either by the addition or removal of specific phosphorylation that generates a binding site for the kinesin or another component of a motor complex. Once loaded onto the point of furrow ingression, KLIF is present specifically on the side of the furrow that is proximal to the old FAZ and not the ventral side of the daughter cell (Fig. 7A). This suggests that the extension of the new FAZ may function to deliver KLIF to the site where cleavage furrow ingression initiates, while the old FAZ may provide positional information to restrict the motor to the cleavage furrow during ingression.
Cells depleted of KLIF were able to initiate furrow ingression but failed to complete cytokinesis multiple times (Fig 7C). The furrow frequently stalled at a position in the cell body between the two nuclei, near where the new tapered posterior end would begin to form. This location is similar to the very posterior end of the old FAZ, where the MtQ passes between the centrin arm and hook complex, then wraps around the flagellar pocket (Esson et al., 2012). It is possible that either the track the cleavage furrow follows or the specific mode of furrow ingression changes at this point to require the force producing properties of KLIF. Thus, our data indicates that there are two distinct processes required to complete cytokinesis: an initial KLIF-independent pathway that initiates furrow ingression, followed by a KLIF-dependent process required for completion of cytokinesis. Recent work has suggested that T. brucei can employ an alternate cytokinetic pathway in the absence of TOEFAZ1 or when CIF2 is overexpressed, during which a cleavage furrow initiates from the posterior end of the cell (Zhou, Gu, et al., 2016; Zhou, Hu, et al., 2016). If this pathway were a viable alternative to conventional cytokinesis, it should be able to compensate for the absence of KLIF because all that remains to be done to complete division in cells lacking the kinesin is the resolution of the cell posterior. It is possible that KLIF is essential for the unconventional cytokinetic pathway and that its depletion blocks the process. However, it should be noted that TOEFAZ1 RNAi does not affect KLIF levels but blocks kinesin recruitment to the new FAZ tip and subsequently to the ingressing furrow, which would make it unlikely to participate in the unconventional mechanism. Since the primary instance of unconventional cytokinesis is in the absence of TOEFAZ1, it would be difficult to assign KLIF a role in this process.
Why does KLIF localize to the cleavage furrow throughout cytokinesis when it is only needed late in this process? One possibility is that KLIF is carried along the furrow to position the motor at the posterior to complete the final stages of cytokinesis. Alternatively, the motor may play a regulatory role, influencing the positioning or rate of cleavage furrow ingression (Figs. S5D,8C). KLIF is a positive-end directed kinesin (Fig. 8B) and therefore may engage with the subpellicular microtubules that have their growing ends facing the cell posterior in order to move with the direction of furrow ingression. KLIF moves microtubules at a significantly slower rate than other kinesins, such as conventional kinesin KIF5c (Fig 8 C,F) (Sack et al., 1997). This diminished rate may reflect KLIF function in cytokinesis, as furrow ingression occurs at a much slower rate than other motor-mediated process such as vesicular transport. Thus, the rate of the motor could be tuned for its function in cytokinesis and may be important for regulating the timing and speed of ingression.
Among the three candidate proteins we characterized in detail, two of them appear to play roles in the final stages of cytokinesis that involve the formation of a new cell posterior and completion of furrow ingression. Considering that KLIF depletion causes cleavage furrow ingression to stall, while cells lacking TOEFAZ1 can still remodel their posterior ends to some extent, it is likely that the late stages of cytokinesis represent a cellular process that is distinct from the initiation of the cleavage furrow (Zhou, Gu, et al., 2016; Sinclair-Davis et al., 2017). Thus, we propose that trypanosome furrow ingression takes place in two stages: first, the initial ingression that begins at the very anterior tip of the new FAZ and progresses towards the cell posterior to separate the cell body attached to the flagellum; and second, the resolution of the subpellicular microtubules near the nascent posterior end to create the two distinct and tapered posteriors of the daughter cells. Determining how proteins such as KLIF and PAVE1 function in the formation of a new cell posterior will provide novel insights into the regulation of microtubule dynamics that provides not only the characteristic shape of the trypanosome cell, but also produces the force to complete the unique cytokinetic process in T. brucei.
Experimental Procedures
Antibodies
Antibodies were sourced as follows. Anti-biotin was purchased from Abcam (clone SB58c, cat# ab79111), YL1/2 was purchased from ThermoFisher (cat# MA1-80017), streptavidin-HRP was purchased from BioLegend, Dylight Avidin-488 was purchased from ThermoFisher Scientific, and anti-HA was purchased from Sigma-Aldrich (clone 3F10). The anti-Ty1 antibody is from Cynthia He (National University of Singapore, Singapore); and 1B41 from Linda Kohl (Centre National de la Recherche Scientifique, Paris, France). Anti-GFP, anti-GRASP, anti-TbPLK, anti-TbCentrin4, and anti-TbCentrin2 have been described previously (Ho et al 2006; de Graffenried et al 2008; Ikeda and de Graffenried, 2012).
Cell Culture
Cells used in all experiments were either procyclic T. brucei brucei strain 427 or the doxycycline-inducible strain 29-13. 427-based cell lines were cultured at 27 °C in Beck’s Medium (Hyclone, GE Healthcare, Logan, Utah) supplemented with 500 μg/mL penicillin-streptomycin-glutamine (Hyclone), 10% fetal calf serum (Gemini Bioproducts, West Sacramento, CA), and 10 μg/mL gentamycin (ThermoFisher Scientific, Waltham, MA). 29-13-based lines were cultured at 27°C in Beck’s Medium supplemented with 500 μg/mL penicillin-streptomycin-glutamine, 15% doxycycline-free fetal calf serum (Clontech), 10 μg/mL gentamycin, 50 μg/mL hygromycin (ThermoFisher Scientific), and 15 μg/mL neomycin (Sigma-Aldrich, St. Louis, MO). Cell concentration was determined using a Z2 Coulter Counter particle counter (Beckman Coulter, Brea, CA).
Cloning and Cell Line Assembly
All DNA constructs were assembled using PCR amplification of inserts followed by either restriction-ligation or Gibson Assembly as previously described (McAllaster et al., 2016). Constructs were transfected into cells using an electroporator (GenePulser xCell, Bio-Rad, Hercules, CA). Clonal cell lines were isolated by limiting dilution and selection with the appropriate resistance marker. All cell lines were verified using loci PCR and western blotting.
Overexpression and RNAi Constructs
Briefly, the Ty1-BirA*-TOEFAZ1 construct was created by Gibson Assembly in the doxycycline-inducible vector pLEW100 using the complete TOEFAZ1 coding sequence amplified from 427 genomic DNA, along with Ty1 epitope tag and BirA* sequences amplified from previously-described plasmids (Morriswood et al., 2013).
Target sequences for RNAi were obtained using the RNAit software (trypanofan.bioc.cam.ac.uk/software/RNAit.html) and assembled into a modified pLEW100 vector as previously described (Redmond et al., 2003; McAllaster et al., 2016). 400-600 bp of the coding region were targeted for each of the proteins selected for RNAi: bp 1311-1772 for KLIF, bp 97-543 for PAVE1, and bp 994-1576 for KPP1. Doxycycline-inducible 29-13 cells were transfected with the Ty1-BirA*-TOEFAZ1 or RNAi pLEW100 vectors after linearization with NotI (NEB). Clones were selected with 40 μg/mL Zeocin (Life Technologies).
Endogenous Tagging Constructs
Gibson Assembly was also used to rapidly generate endogenous tagging constructs into a sequencing vector (PCR4Blunt). N-terminal Ty1 epitope tags were targeted to endogenous loci using approximately 500 bp of the 5’ UTR and the first 500 bp of the coding sequence. For C-terminal Ty1 epitope tags, targeting of endogenous loci was obtained using the last 500 bp of the 3’ coding sequence and the first 500 bp of the 3’ UTR(Aslett et al., 2009). Endogenous tagging constructs were excised using PacI and NsiI (NEB), transfected into the 427 cell line, cloned by limiting dilution, and selected with 20 μg/mL blasticidin.
Proximity-dependent biotinylation and immunoprecipitation
Cells carrying inducible Ty1 BirA*-TOEFAZ1 were seeded at a concentration of 3.0 × 106 cells/mL in 240 mL of media were either induced with 40 ng/mL doxycycline or treated with 70% ethanol as a vehicle control. After 24 h, cells were reseeded at 4.0 × 106 cells/mL in 300 mL media, and a stock solution of dissolved biotin was added to the cell media to a final concentration of 50 μM. 18 h after the addition of biotin, a total of 3.0 × 109 cells per condition were harvested by centrifugation, washed with PBS, and snap frozen in liquid nitrogen for short-term storage. The pulldown of biotinylated proteins onto streptavidin magnetic beads was performed as previously reported (Morriswood et al., 2013; McAllaster et al., 2015)). The beads were then washed five times with 50 mM ammonium bicarbonate for 5 minutes and stored at −80 °C. The LC-MS/MS was performed at the Proteomics Core Facility at Sanford-Burnham Medical Research Institute in La Jolla, CA.
2DLC-MS/MS
Sample Preparation
Following immunoprecipitation, proteins were digested directly on-beads. Briefly, proteins bound to the beads were resuspended with 8M urea and 50 mM ammonium bicarbonate; cysteine disulfide bonds were reduced with 10 mM tris(2-carboxyethyl) phosphine (TCEP) at 30 °C for 60 min followed by cysteine alkylation with 30 mM iodoacetamide (IAA) in the dark at RT for 30 min. Following alkylation, urea was diluted to 1 M using 50 mM ammonium bicarbonate, and proteins were finally subjected to overnight digestion with mass spec grade Trypsin/Lys-C mix (Promega, Madison, WI). Finally, beads were pulled down and the solution with peptides collected into a new tube. The beads were then washed once with 50mM ammonium bicarbonate to increase peptide recovery. The digested samples were desalted using a C18 TopTip (PolyLC, Columbia, MD), and the organic solvent was removed in a SpeedVac concentrator prior to LC-MS/MS analysis.
2DLC-MS/MS Analysis
Dried samples were reconstituted in 100 mM ammonium formate pH ~10 and analyzed by 2DLC-MS/MS using a 2D nanoACQUITY Ultra Performance Liquid Chromatography (UPLC) system (Waters Corp., Milford, MA) coupled to a Q-Exactive Plus mass spectrometer (ThermoFisher Scientific). Peptides were loaded onto the first-dimension column, XBridge BEH130 C18 NanoEase (300 μm × 50 mm, 5 μm) equilibrated with solvent A (20mM ammonium formate pH 10, first dimension pump) at 2 μL/min. The first fraction was eluted from the first dimension column at 17% of solvent B (100% acetonitrile) for 4 min and transferred to the second dimension Symmetry C18 trap column 0.180 × 20 mm (Waters Corp., Milford, MA) using a 1:10 dilution with 99.9% second dimensional pump solvent A (0.1% formic acid in water) at 20 μL/min. Peptides were then eluted from the trap column and resolved on the analytical C18 BEH130 PicoChip column 0.075 × 100 mm, 1.7μm particles (NewObjective, MA) at low pH by increasing the composition of solvent B (100% acetonitrile) from 2 to 26% over 94 min at 400 nL/min. Subsequent fractions were carried with increasing concentrations of solvent B. The following 4 first dimension fractions were eluted at 19.5, 22, 26, and 65% solvent B. The mass spectrometer was operated in positive data-dependent acquisition mode. MS1 spectra were measured with a resolution of 70,000, an AGC target of 1e6 and a mass range from 350 to 1700 m/z. Up to 12 MS2 spectra per duty cycle were triggered, fragmented by HCD, and acquired with a resolution of 17,500 and an AGC target of 5e4, an isolation window of 2.0 m/z and a normalized collision energy of 25. Dynamic exclusion was enabled with duration of 20 sec.
Data analysis
All mass spectra from were analyzed with MaxQuant software (2008) version 1.5.5.1. MS/MS spectra were searched against the Trypanosoma brucei strain 427 Uniprot protein sequence database (version June 2016) and GPM cRAP sequences (commonly known protein contaminants). Precursor mass tolerance was set to 20ppm and 4.5ppm for the first search where initial mass recalibration was completed and for the main search, respectively. Product ions were searched with a mass tolerance 0.5 Da. The maximum precursor ion charge state used for searching was 7. Carbamidomethylation of cysteines was searched as a fixed modification, while oxidation of methionines and acetylation of protein N-terminal were searched as variable modifications. Enzyme was set to trypsin in a specific mode and a maximum of two missed cleavages was allowed for searching. The target-decoy-based false discovery rate (FDR) filter for spectrum and protein identification was set to 1%.
Statistical confidence of prey-bait interactions was scored by the SAINTq algorithm (Teo et al., 2016) using MaxQuant-calculated peptide intensities (Cox et al., 2014) as the input information. A confidence threshold was set to SAINTq scores >0.9 corresponding to a Bayesian false discovery rate <1%.
Peptide hits with a p-value of at most 0.01 were prioritized, taking into account previous published works, annotation in TriTrypDB, and protein domains as determined by PFAM (Sonnhammer et al., 1997; Sonnhammer et al., 1998). The top 59 highest priority proteins were characterized by endogenous tagging.
Immunofluorescence
Cells were harvested by centrifugation at 2400 × g for 5 min and washed in PBS. Cells were then spun onto coverslips and fixed in −20 °C methanol for 20 min followed by air-drying or immediate rehydration in PBS. Coverslips were washed 3 times in PBS and blocked in blocking buffer (3% bovine serum albumin or 5% goat serum in PBS). Coverslips were incubated in primary antibody diluted in blocking buffer for 1 hr RT, after which cells were washed three times in PBS. Coverslips were then incubated with secondary antibody conjugated to Alexa Fluor −488, −568, or −647 (Life Technologies, Carlsbad, CA). After a final set of 3 washes in PBS, coverslips were mounted in Fluoromount G with DAPI (Southern Biotech, Birmingham, AL).
For cytoskeletal isolation, cells were collected as above and coverslips were incubated in PEME extraction buffer (0.1 M PIPES pH 6.9, 2 mM EGTA, 1 mM MgSO4, 0.1 mM EDTA, 0.25% NP-40) for 2 minutes, washed three times in PBS, and fixed for 10 min in −20 °C methanol or 20 mins in PFA at RT. Immunofluorescence was then performed as above.
For observation of DNA states and cellular morphology quantification, cells were harvested as above and fixed on coverslips with 4% paraformaldehyde in PBS for 20 min RT. Cells were then permeabilized with 0.5% NP-40 in PBS for 5 min RT, washed three times in PBS, and either mounted directly in Fluoromount G with DAPI or stained for immunofluorescence as above.
Images were taken using a Zeiss Axio Observer.Z1 microscope (Carl Zeiss Microscopy, Oberkochen, Germany) equipped with an ORCA-Flash 4.0 CMOS camera (Hamamatsu, Shizuoka, Japan) using a Plan-Apochromat 100x/1.4 NA oil lens. Images were acquired with ZEN 2 PRO (Zeiss), analyzed with ImageJ (National Institutes of Health, Bethesda, MD), and assembled for publication in Adobe Photoshop and Illustrator (CC 2017).
Western and Streptavidin Blotting
Cells were harvested by centrifugation at 2400 × g for 5 min, washed in PBS, lysed in SDS-PAGE loading buffer, and incubated at 99 °C for 10 minutes. The lysate equivalent of 3.0 × 106 cells/lane was separated using SDS-PAGE and transferred to a nitrocellulose membrane. Blots undergoing streptavidin-HRP staining were blocked with 2.5% BSA and 0.4% Triton X-100 in PBS. Streptavidin conjugated to horseradish peroxidase (BioLegend, San Diego, CA) was diluted 1:4000 in blocking buffer and incubated with blots at 4 °C overnight. Streptavidin blots were then washed 4 times in blocking buffer, and twice in 0.4% Triton X-100. Blots to be stained with anti-biotin were blocked with 3% BSA in TBS with 0.1% Tween-20. Anti-biotin (Abcam, Cambridge, UK) was diluted 1:1000 into blocking buffer and incubated with blots overnight at 4 °C.
All other blots were blocked for 1 h RT with 5% (w/v) non-fat dried milk dissolved in TBS containing 0.1% Tween-20. Blots were then incubated at 4 °C overnight in primary antibody diluted in blocking buffer. The blots were then washed three times in TBS containing 0.1% Tween-20 and incubated for 1 h RT with secondary antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA). Blots were washed three final times and imaged using Clarity Western ECL Substrate (BioRad) and a BioRad Gel Doc XR+ system. When needed, blots were stripped with Restore Western Blot Stripping Buffer (ThermoFisher Scientific), washed, re-blocked and stained as above.
RNAi
To induce the RNAi, cells carrying pTrypson-based lhRNAi plasmids were seeded at 106 cells/mL and induced with 1 μg/mL doxycycline (ThermoFisher Scientific) or treated with 70% ethanol as a vehicle control. Cell growth was monitored every 24 h unless otherwise noted, and samples were collected for western blotting and immunofluoresence microscopy as above. Every 48 h, cultures were reseeded in fresh medium and doxycycline or ethanol as above. The generation plots created for the RNAi timecourses are representative of three independent experiments, and the error bars are the S.D.
Transmission Electron Microscopy
Cells were harvested by centrifugation at 800 × g for 10 min RT. Cells were then washed twice and resuspended in PBS and spotted onto parafilm in a humidified chamber. Glow-discharged carbon-coated formvar grids (EMS, Hatfield, PA) were floated on the drops for 5 min RT. Grids were moved through two subsequent drops of PEME extraction buffer (0.1 M PIPES pH 6.9, 2 mM EGTA, 1 mM MgSO4, 0.1 mM EDTA, 1.0% NP-40) for 3 min RT each. Grids were washed in PEME buffer without detergent, fixed for 5 min RT with 2.5% glutaraldehyde, rinsed in ultra-pure water, and negatively stained with 1.5% aurothioglucose (Sigma Aldrich). Images were taken on a Phillips 410 transmission electron microscope at 100kV equipped with an 1k × 1k Advantage HR CCD camera from Advanced Microscopy Techniques (AMT) using AMT imaging software.
Recombinant Expression and Protein Purification
For recombinant expression in bacteria, KLIF (Tb927.8.4950) was amplified from T. brucei genomic DNA using PCR and truncated at amino acid 672, followed by the green fluorescent protein variant mClover3 and 8x HIS tag for purification. This fusion construct was inserted into the pET30A vector for bacterial expression and purification. For recombinant expression of conventional kinesin, Kif5C (kinesin heavy chain isoform 5C) from rat (Sequence ID NP_001101200.1) was amplified and truncated at amino acid 407 to produce a constitutively activated kinesin, followed by mClover3 and 8x HIS tag.
KLIF and Kif5 bacterial overexpression constructs were transformed into BL21 (DE3) bacteria. 0.5 L of cells were grown to mid-log (OD ~0.5) at 37 °C and induced for overexpression from the T7 promoter using 0.4 mM IPTG at 16 °C in a shaking incubator overnight. Cells were harvested by centrifugation and cell pellets were immediately frozen at −20 °C for short term storage. Cells were thawed and lysed in 40 mL of Buffer A (10 mM NaPO4, pH 7.4, 300 mM NaCl, 0.5% glycerol) supplemented with 7% sucrose, 0.5% NP40, 0.5 mM DTT, 0.5 mM PMSF, 1x cOmplete ULTRA protease inhibitors (Roche) and 1mM grade II MgATP (Sigma Aldrich) and clarified for 45 min at 17,000 g. The clarified supernatant was batch incubated with 3 mL of Ni-NTA agarose resin (ThermoFisher Scientific) that was prewashed with 3 column volumes of 0.5 M imidazole, pH 7.4 followed by 20 column volumes of buffer A, for 45 min at 4°C with gentle rocking. The protein-bound resin was then passed over a column and washed with 20 column volumes of Buffer B (10 mM NaPO4, pH 7.4, 300 mM NaCl, 0.5% glycerol, 10 mM Imidazole, 0.5 mM DTT, 0.2 mM grade II MgATP), followed by 20 column volumes of Buffer C (10 mM NaPO4, pH 7.4, 300 mM NaCl, 0.5% glycerol, 30 mM Imidazole, 0.5 mM DTT, 0.2 mM grade II MgATP) and the protein was eluted in 1 mL elution fractions with Buffer D (10 mM NaPO4, pH 7.4, 300 mM NaCl, 0.5% glycerol, 200 mM Imidazole, 0.5 mM DTT, 0.2 mM grade II MgATP). Fractions containing purified protein was then dialyzed against storage buffer (25 mM Imidazole, 300 mM NaCl, 50% glycerol, 1 mM DTT, 0.2 mM Grade I ATP (Sigma)) overnight and stored at −20 °C. For KLIF, the protein was further purified by gel filtration using a Sephacryl S-300 26/60 gel filtration column to remove a ~35 kDa degradation product. Protein preparations typically yielded ~5-10 mg of pure protein that was stable over several months at −20 °C.
In vitro Motility Assay
Rhodamine-labeled microtubules were polymerized from tubulin (Cytoskeleton, T333P) and rhodamine-labeled tubulin (Cytoskeleton, TL590M). For assays that used polarity marked microtubules, microtubules were made containing a bright microtubule seed at the minus-end as described previously (Howard and Hyman, 1993). In vitro motility assays were performed using flow chambers made from glass coverslips that are bound to a custom anti-GFP serum to enrich KLIF mClv3 and Kif5C mClv3 to the surface of the flow chamber such that the heads are oriented to interact optimally with microtubules. All reagents were diluted into a modified BRB80 buffer (80 mM PIPES pH 7, 2 mM MgCl2, 0.5 mM EGTA, 10 mM DTT) and incubated in flow chambers at room temperature. Flow chambers were prepared by introducing the following: (1) one passage of anti-GFP serum diluted 1/5 (5 min incubation), (2) three passages of 2 mg/mL BSA (5 min incubation), (3) two passages of 0.125 mg/mL kinesin (5 min incubation), (4) three passages of BRB80, (5) two passages of 0.9 μM taxol stabilized rhodamine-labeled microtubules (5 min incubation), (6) three passages of BRB80 + 10 μM taxol. Finally, microtubule gliding was initiated with the addition of Go buffer (BRB80, 10 mM DTT, 10 μM taxol, 1 mM grade I MgATP and an oxygen-scavenging system (3 mg/ml glucose, 0.1 mg/ml glucose oxidase, and 0.18 mg/ml catalase). In vitro motility was observed at room temperature (22 °C) using epifluorescence. For Kif5C mClv3, frames were captured every 1 second for 2 minutes. For KLIF mClv3, images were captured every 10 seconds for 20 minutes. The speed of microtubules moving along the surface of the flow chamber was measured using the ImageJ plugin mTrackJ (Meijering et al., 2012).
Quantification and Statistics
For all cellular quantification, 200-300 cells were quantified from three independent experiments. All quantification and measurements were performed in ImageJ. Error bars represent the standard deviation of three independent experiments unless otherwise noted. Statistics and graphs were assembled in Excel (Microsoft), and figures were compiled in Adobe Photoshop and Illustrator.
Supplementary Material
Figure S1 Matching Ty1-BirA* TOEFAZ1 expression to endogenous TOEFAZ1 levels. A) Western blot comparing the expression levels between a cell line in which both alleles of TOEFAZ1 are tagged with a Ty1 epitope tag (dTag TF1) and the Ty1-BirA* cell line induced overnight with either 40 ng/mL or 500 ng/mL doxycycline. B) The Ty1-BirA* cell line was induced as above, followed by incubation with 50 μM biotin. Lysates from the induced Ty1-BirA* TOEFAZ1 cells were collected and separated by SDS-PAGE followed by transfer to nitrocellulose for western blotting with streptavidin conjugated to HRP. C) The Ty1-BirA* cell line was induced and incubated with biotin as above. Cells were methanol-fixed and stained with anti-Ty1 antibody and streptavidin. Arrowheads: Ty1-BirA* TOEFAZ1 localization. Asterisks indicate spurious localization. DAPI was used to stain DNA. D) The Ty1-BirA* TOEFAZ1 cell line was induced as in B) with 40 ng/mL doxycycline, followed by lysis and capture of biotinylated proteins, which were eluted and blotted using anti-biotin antibody.
Figure S2 KPP1 is required for cell division. A) KPP1 RNAi was induced for 8 d using 1 μg/mL doxycycline, and cell concentration was counted every 24 h. B) Cell lysates were collected every 24 h from the RNAi timecourse in A), western blotted, and labeled with anti-Ty1 antibody. C) Cells carrying a KPP1 RNAi construct were treated with either vehicle control or 1 μg/mL doxycycline to induce RNAi for four days. Cells were PFA-fixed and stained with DAPI for DNA. Asterisk indicates a detached flagellar tip. D) Quantification of detached new flagella tips in control and KPP1 RNAi cells in which KPP1 has been depleted for 4 d.
Figure S3 PAVE1 is necessary for cell division. A) PAVE1 RNAi cells were treated with either vehicle control or 1 μg/mL doxycycline for 6 days and cell number was monitored every 24 h. B) Cell lysates were collected every 24 h from the time course in A). Lysates were western blotted and stained with anti-Ty1 antibody.
Figure S4 YL1/2 staining is not altered in PAVE1 RNAi cells. A) Immunofluorescence of methanol-fixed wherein PAVE1 was tagged at its endogenous locus with a Ty1 tag. Cells were stained for YL1/2 to detect tyrosinated tubulin, an indicator of growing microtubules, and Ty1 to label PAVE1. B) PAVE1 RNAi was induced with 1 μg/mL doxycycline for 1 and 2 d. Cells were methanol fixed and stained with anti-tubulin antibody and YL1/2. DAPI was used to stain DNA in all immunofluorescence panels. C) Dividing 2N2K cells from the experiment described in B). D) Lysates from the RNAi time course in Fig. S3A were western blotted and labeled with YL1/2 antibody.
Figure S5 KLIF is required for cell division. A) Cells in which KLIF was tagged at its endogenous locus with a Ty1 epitope tag were methanol-fixed and stained with anti-Ty1 antibody and 1B41 to label the FAZ. DAPI was used to label DNA. Arrowheads indicate Ty1-KLIF localization at the cleavage furrow. B) Cells containing the KLIF RNAi construct were treated with either vehicle control or 1 μg/mL doxycycline. Cell concentration was counted every 24 h. C) Lysates were collected every 24 h from the RNAi timecourse in B), western blotted, and stained with anti-Ty1 antibody. D) Cells containing a KLIF RNAi construct were treated with either vehicle control or 1 μg/mL doxycycline for 3 d. Cells were collected and PFA-fixed for morphological analysis. DAPI was used to stain DNA. E) TOEFAZ1 RNAi was induced in a cell line containing KLIF tagged at its endogenous locus with a Ty1 epitope tag. Lysates were collected every 12 h, western blotted, and stained with anti-Ty1 antibody.
Figure S6 Schematic of the in vitro motility assay. KLIF (purple) containing a mClover3 tag (mClv3, green) is bound to an α-GFP coated flow chamber (light blue) and blocked. Then, rhodamine-labeled polarity marked microtubules (red) containing enhanced fluorescent signal at their minus-ends are introduced to the flow chamber to interact with KLIF in the presence of MgATP.
Figure S7 Characterization of curated TOEFAZ1 BioID hits. A Ty1 epitope tag was integrated at either the N- or C-terminus of the endogenous locus of each hit from the TOEFAZ1 BioID screen. The size of each protein was determined by anti-Ty1 western blotting, and the correct integration of the Ty1 tag at the endogenous locus was established by loci PCR. Immunofluorescence analysis was conducted on each protein using anti-Ty1 antibody and, where applicable, using antibodies against known organelles for co-localization.
Supporting Movie 1 In vitro motility assay showing that KLIF moves labeled polarity marked microtubules with their brighter minus-ends leading. The movie is 200-fold real time.
Supplemental Table 1 Table of the unique peptides identified in this screen.
Acknowledgements
We would like to thank Bill Wickstead and Lorenzo Brusini for sharing data prior to publication, Alexandre Rosa Campos for performing the BioID, and all the laboratories that provided antibodies. This work was supported and the National Institutes of Health (NIGMS P20 GM104317, NIAID R01AI112953, NIAID R21AI115089-01).
Abbreviations:
- BioID
proximity-dependent biotin identification
- FAZ
flagellum attachment zone
- TOEFAZ1
Tip of the Extending FAZ protein 1
- FP
flagellar pocket
- FPC
flagellar pocket collar
- HC
hook complex
- MtQ
microtubule quartet
- PCF
procyclic form
- TbPLK
Trypanosoma brucei polo-like kinase
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as an ‘Accepted Article’, doi: 10.1111/mmi.13986
References
- Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, et al. (2009) TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res 38: D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines A, and Gull K (2008) WCB is a C2 Domain Protein Defining the Plasma Membrane – Sub-Pellicular Microtubule Corset of Kinetoplastid Parasites. Protist 159: 115–125. [DOI] [PubMed] [Google Scholar]
- Bajar BT, Wang ES, Lam AJ, Kim BB, Jacobs CL, Howe ES, et al. (2016) Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci Rep 6: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluška F, Menzel D, and Barlow PW (2006) Cytokinesis in plant and animal cells: Endosomes “shut the door.” Developmental Biology 294: 1–10. [DOI] [PubMed] [Google Scholar]
- Bargul JL, Jung J, McOdimba FA, Omogo CO, Adung’a VO, Krüger T, et al. (2016) Species-Specific Adaptations of Trypanosome Morphology and Motility to the Mammalian Host. PLoS Pathog 12: e1005448–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartossek T, Jones NG, Schäfer C, Cvitković M, Glogger M, Mott HR, et al. (2017) Structural basis for the shielding function of the dynamic trypanosome variant surface glycoprotein coat. Nature Microbiology 267: 1–12. [DOI] [PubMed] [Google Scholar]
- Brenchley R, Tariq H, McElhinney H, Szöőr B, Huxley-Jones J, Stevens R, et al. (2007) The TriTryp Phosphatome: analysis of the protein phosphatase catalytic domains. BMC Genomics 8: 434–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin Z-Y, Breitkreutz A, Mellacheruvu D, Fermin D, et al. (2010) SAINT: probabilistic scoring of affinity purification–mass spectrometry data. Nat Meth 8: 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Liu G, Mellacheruvu D, Tyers M, Gingras A-C, and Nesvizhskii AI (2002) Analyzing Protein-Protein Interactions from Affinity Purification-Mass Spectrometry Data with SAINT. John Wiley & Sons, Inc, Hoboken, NJ, USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Gale M, Carter V, and Parsons M (1994) The protein phosphatase inhibitor okadaic acid induces defects in cytokinesis and organellar genome segregation in Trypanosoma brucei. J Cell Sci 107 (Pt 12): 3477–3483. [DOI] [PubMed] [Google Scholar]
- Davidge JA, Chambers E, Dickinson HA, Towers K, Ginger ML, McKean PG, and Gull K (2006) Trypanosome IFT mutants provide insight into the motor location for mobility of the flagella connector and flagellar membrane formation. J Cell Sci 119: 3935–3943. [DOI] [PubMed] [Google Scholar]
- de Graffenried CL, Ho HH, and Warren G (2008) Polo-like kinase is required for Golgi and bilobe biogenesis in Trypanosoma brucei. J Cell Biol 181: 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahunty C, and Yates JR III (2005) Protein identification using 2D-LC-MS/MS. Methods 35: 248–255. [DOI] [PubMed] [Google Scholar]
- Engstler M, Pfohl T, Herminghaus S, Boshart M, Wiegertjes G, Heddergott N, and Overath P (2007) Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell 131: 505–515. [DOI] [PubMed] [Google Scholar]
- Esson HJ, Morriswood B, Yavuz S, Vidilaseris K, Dong G, and Warren G (2012) Morphology of the Trypanosome Bilobe, a Novel Cytoskeletal Structure. Eukaryotic Cell 11: 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr H, and Gull K (2012) Cytokinesis in trypanosomes. Cytoskeleton (Hoboken). [DOI] [PubMed] [Google Scholar]
- Fèvre EM, Wissmann BV, Welburn SC, and Lutumba P (2008) The burden of human African trypanosomiasis. PLoS Negl Trop Dis 2: e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MC, and Carrington M (2009) The trypanosome flagellar pocket. Nat Rev Microbiol 7: 775–786. [DOI] [PubMed] [Google Scholar]
- Foth BJ, Goedecke MC, and Soldati D (2006) New insights into myosin evolution and classification. Proc Natl Acad Sci USA 103: 3681–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo JM, and Précigout E (1988) Tubulin expression in trypanosomes. Biol Cell 64: 137–143. [DOI] [PubMed] [Google Scholar]
- Gallo JM, Précigout E, and Schrével J (1988) Subcellular sequestration of an antigenically unique beta-tubulin. Cell Motil Cytoskeleton 9: 175–183. [DOI] [PubMed] [Google Scholar]
- García-Salcedo JA, Pérez-Morga D, Gijón P, Dilbeck V, Pays E, and Nolan DP (2004) A differential role for actin during the life cycle of Trypanosoma brucei. EMBO J 23: 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gull K (1999) The cytoskeleton of trypanosomatid parasites. Annu Rev Microbiol 53: 629–655. [DOI] [PubMed] [Google Scholar]
- Hammarton TC, Monnerat S, and Mottram JC (2007) Cytokinesis in trypanosomatids. Curr Opin Microbiol 10: 520–527. [DOI] [PubMed] [Google Scholar]
- Heddergott N, Krüger T, Babu SB, Wei A, Stellamanns E, Uppaluri S, et al. (2012) Trypanosome motion represents an adaptation to the crowded environment of the vertebrate bloodstream. PLoS Pathog 8: e1003023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Fowler C, Ersfeld K, and Gull K (2001) CAP5.5, a life-cycle-regulated, cytoskeleton-associated protein is a member of a novel family of calpain-related proteins in Trypanosoma brucei. Mol Biochem Parasitol 116: 25–34. [DOI] [PubMed] [Google Scholar]
- Hotez PJ, and Kamath A (2009) Neglected Tropical Diseases in Sub-Saharan Africa: Review of Their Prevalence, Distribution, and Disease Burden. PLoS Negl Trop Dis 3: e412–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovel-Miner G, Mugnier M, Papavasiliou FN, Pinger J, and Schulz D (2015) A Host-Pathogen Interaction Reduced to First Principles: Antigenic Variation in T. brucei. Results Probl Cell Differ 57: 23–46. [DOI] [PubMed] [Google Scholar]
- Howard J, and Hyman AA (1993) Preparation of marked microtubules for the assay of the polarity of microtubule-based motors by fluorescence microscopy. Methods Cell Biol 39: 105–113. [DOI] [PubMed] [Google Scholar]
- Hu C-K, Coughlin M, and Mitchison TJ (2012) Midbody Assembly and its Regulation during Cytokinesis. Mol Biol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Hu L, Yu Z, Chasse AE, Chu F, and Li Z (2012) An orphan kinesin in trypanosomes cooperates with a kinetoplastid-specific kinesin to maintain cell morphology by regulating subpellicular microtubules. J Cell Sci 125: 4126–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Zhou Q, and Li Z (2015) SAS-4 Protein in Trypanosoma bruceiControls Life Cycle Transitions by Modulating the Length of the Flagellum Attachment Zone Filament. J Biol Chem 290: 30453–30463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Zhou Q, Han X, and Li Z (2017) CRL4WDR1 Controls Polo-like Kinase Protein Abundance to Promote Bilobe Duplication, Basal Body Segregation and Flagellum Attachment in Trypanosoma brucei. PLoS Pathog 13: e1006146–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA (1991) Preparation of marked microtubules for the assay of the polarity of microtubule-based motors by fluorescence. J Cell Sci Suppl 14: 125–127. [DOI] [PubMed] [Google Scholar]
- Ikeda KN, and de Graffenried CL (2012) Polo-like kinase is necessary for flagellum inheritance in Trypanosoma brucei. J Cell Sci 125: 3173–3184. [DOI] [PubMed] [Google Scholar]
- Janke C (2014) The tubulin code: Molecular components, readout mechanisms, and functions. J Cell Biol 206: 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, and Chloë Bulinski J (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12: 773–786. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Wright B, and Milstein C (1982) Rat monoclonal antitubulin antibodies derived by using a new nonsecreting rat cell line. J Cell Biol 93: 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Birendra KC, Zhu W, Motamedchaboki K, Doye V, and Roux KJ (2014) Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci USA 111: E2453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl L, Robinson D, and Bastin P (2003) Novel roles for the flagellum in cell morphogenesis and cytokinesis of trypanosomes. EMBO J 22: 5336–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl L, Sherwin T, and Gull K (1999) Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J Eukaryot Microbiol 46: 105–109. [DOI] [PubMed] [Google Scholar]
- Kumar P, and Wang CC (2006) Dissociation of cytokinesis initiation from mitotic control in a eukaryote. Eukaryotic Cell 5: 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacomble S, Vaughan S, Deghelt M, Moreira-Leite FF, and Gull K (2012) A Trypanosoma brucei protein required for maintenance of the flagellum attachment zone and flagellar pocket ER domains. Protist 163: 602–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacomble S, Vaughan S, Gadelha C, Morphew MK, Shaw MK, McIntosh JR, and Gull K (2009) Three-dimensional cellular architecture of the flagellar pocket and associated cytoskeleton in trypanosomes revealed by electron microscope tomography. J Cell Sci 122: 1081–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCount DJ, Barrett B, and Donelson JE (2002) Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J Biol Chem 277: 17580–17588. [DOI] [PubMed] [Google Scholar]
- Li Z (2012) Regulation of the cell division cycle in Trypanosoma brucei. Eukaryotic Cell 11: 1180–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Tu X, and Wang CC (2006) Okadaic acid overcomes the blocked cell cycle caused by depleting Cdc2-related kinases in Trypanosoma brucei. Exp Cell Res 312: 3504–3516. [DOI] [PubMed] [Google Scholar]
- Li Z, Umeyama T, and Wang CC (2008) The chromosomal passenger complex and a mitotic kinesin interact with the Tousled-like kinase in trypanosomes to regulate mitosis and cytokinesis. PLoS ONE 3: e3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Umeyama T, and Wang CC (2009) The Aurora Kinase in Trypanosoma brucei plays distinctive roles in metaphase-anaphase transition and cytokinetic initiation. PLoS Pathog 5: e1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Núñez A, Ikeda KN, Sauer T, and de Graffenried CL (2013) An analogue-sensitive approach identifies basal body rotation and flagellum attachment zone elongation as key functions of PLK in Trypanosoma brucei. Mol Biol Cell 24: 1321–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massow, von A, Mandelkow EM, and Mandelkow E (1989) Interaction between kinesin, microtubules, and microtubule-associated protein 2. Cell Motil Cytoskeleton 14: 562–571. [DOI] [PubMed] [Google Scholar]
- May SF, Peacock L, Almeida Costa CIC, Gibson WC, Tetley L, Robinson DR, and Hammarton TC (2012) The Trypanosoma brucei AIR9-like protein is cytoskeleton-associated and is required for nucleus positioning and accurate cleavage furrow placement. Mol Microbiol 84: 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllaster MR, Ikeda KN, Lozano-Núñez A, Anrather D, Unterwurzacher V, Gossenreiter T, et al. (2015) Proteomic identification of novel cytoskeletal proteins associated with TbPLK, an essential regulator of cell morphogenesis in Trypanosoma brucei. Mol Biol Cell 26: 3013–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllaster MR, Sinclair-Davis AN, Hilton NA, and de Graffenried CL (2016) A unified approach towards Trypanosoma brucei functional genomics using Gibson assembly. Mol Biochem Parasitol 210: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell AV, Jiang T, Keating AE, and Berger B (2006) Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22: 356–358. [DOI] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, and Smal I (2012) Methods for Cell and Particle Tracking. 1st ed., Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- Mezulis S, Yates CM, Wass MN, Sternberg MJE, and Kelley LA (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B (2015) Form, Fabric, and Function of a Flagellum-Associated Cytoskeletal Structure. Cells 4: 726–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B, Havlicek K, Demmel L, Yavuz S, Sealey-Cardona M, Vidilaseris K, et al. (2013) Novel Bilobe Components in Trypanosoma brucei Identified Using Proximity-Dependent Biotinylation. Eukaryotic Cell 12: 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnier MR, Cross GAM, and Papavasiliou FN (2015) The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science 347: 1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Worthey EA, Ward PN, and Mottram JC (2005) Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 6: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomo D, Bonhivers M, and Robinson DR (2016) The Trypanosome Flagellar Pocket Collar and Its Ring Forming Protein-TbBILBO1. Cells 5: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JA, Sinclair-Davis AN, McAllaster MR, and de Graffenried CL (2017) TbSmee1 regulates hook complex morphology and the rate of flagellar pocket uptake in Trypanosoma brucei. Mol Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragkousi K, and Gibson MC (2014) Cell division and the maintenance of epithelial order. J Cell Biol 207: 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond S, Vadivelu J, and Field MC (2003) RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol Biochem Parasitol 128: 115–118. [DOI] [PubMed] [Google Scholar]
- Richards TA, and Cavalier-Smith T (2005) Myosin domain evolution and the primary divergence of eukaryotes. Nature 436: 1113–1118. [DOI] [PubMed] [Google Scholar]
- Rindisbacher L, Hemphill A, and Seebeck T (1993) A repetitive protein from Trypanosoma brucei which caps the microtubules at the posterior end of the cytoskeleton. Mol Biochem Parasitol 58: 83–96. [DOI] [PubMed] [Google Scholar]
- Robinson DR, Sherwin T, Ploubidou A, Byard EH, and Gull K (1995) Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. J Cell Biol 128: 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg KG, Jetton N, Hubbard JG, Powell DA, Pandarinath V, and Ruben L (2014) Identification of a protein phosphatase 2A family member that regulates cell cycle progression in Trypanosoma brucei. Mol Biochem Parasitol 194: 48–52. [DOI] [PubMed] [Google Scholar]
- Sack S, Müller J, Marx A, Thormählen M, Mandelkow E-M, Brady ST, and Mandelkow E (1997) X-ray Structure of Motor and Neck Domains from Rat Brain Kinesin †,‡. Biochemistry 36: 16155–16165. [DOI] [PubMed] [Google Scholar]
- Sasse R, and Gull K (1988) Tubulin post-translational modifications and the construction of microtubular organelles in Trypanosoma brucei. J Cell Sci 90 (Pt 4): 577–589. [DOI] [PubMed] [Google Scholar]
- Sherwin T, and Gull K (1989) Visualization of detyrosination along single microtubules reveals novel mechanisms of assembly during cytoskeletal duplication in trypanosomes. Cell 57: 211–221. [DOI] [PubMed] [Google Scholar]
- Sherwin T, Schneider A, Sasse R, Seebeck T, and Gull K (1987) Distinct localization and cell cycle dependence of COOH terminally tyrosinolated alpha-tubulin in the microtubules of Trypanosoma brucei brucei. J Cell Biol 104: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simarro PP, Diarra A, Ruiz Postigo JA, Franco JR, and Jannin JG (2011) The human African trypanosomiasis control and surveillance programme of the World Health Organization 2000-2009: the way forward. PLoS Negl Trop Dis 5: e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair-Davis AN, McAllaster MR, and de Graffenried CL (2017) A functional analysis of TOEFAZ1 uncovers protein domains essential for cytokinesis in Trypanosoma brucei. J Cell Sci 130: 3918–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Eddy SR, and Durbin R (1997) Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins 28: 405–420. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, Eddy SR, Birney E, Bateman A, and Durbin R (1998) Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res 26: 320–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan A, Vaughan S, Shaw MK, Gull K, and McKean PG (2007) An essential quality control mechanism at the eukaryotic basal body prior to intraflagellar transport. Traffic 8: 1323–1330. [DOI] [PubMed] [Google Scholar]
- Sunter JD, and Gull K (2016) The Flagellum Attachment Zone: “The Cellular Ruler” of Trypanosome Morphology. Trends in Parasitology 32: 309–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunter JD, Varga V, Dean S, and Gull K (2015) A dynamic coordination of flagellum and cytoplasmic cytoskeleton assembly specifies cell morphogenesis in trypanosomes. J Cell Sci 128: 1580–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöőr B (2010) Trypanosomatid protein phosphatases. Mol Biochem Parasitol 173: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo G, Koh H, Fermin D, Lambert J-P, Knight JDR, Gingras A-C, and Choi H (2016) SAINTq: Scoring protein-protein interactions in affinity purification - mass spectrometry experiments with fragment or peptide intensity data. Proteomics 16: 2238–2245. [DOI] [PubMed] [Google Scholar]
- Urbaniak MD, Martin DMA, and Ferguson MAJ (2013) Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J Proteome Res 12: 2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Reese TS, and Sheetz MP (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga V, Moreira-Leite F, Portman N, and Gull K (2017) Protein diversity in discrete structures at the distal tip of the trypanosome flagellum. Proc Natl Acad Sci USA 114: E6546–E6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan S, Kohl L, Ngai I, Wheeler RJ, and Gull K (2008) A repetitive protein essential for the flagellum attachment zone filament structure and function in Trypanosoma brucei. Protist 159: 127–136. [DOI] [PubMed] [Google Scholar]
- Vedrenne C, Giroud C, and Robinson DR (2002) Two Related Subpellicular Cytoskeleton-associated Proteins in Trypanosoma brucei Stabilize Microtubules. Mol Biol Cell 13: 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickerman K (1969) On the surface coat and flagellar adhesion in trypanosomes. J Cell Sci 5: 163–193. [DOI] [PubMed] [Google Scholar]
- Vickerman K, Press TPA, London, 1979. Comparative Cell Biology of the Kinetoplastid Flagellates Biology of the Kinetoplastida. Vol. 1 Lumsden WHR, and Evans D, editors. [Google Scholar]
- Welburn SC, and Maudlin I (2012) Priorities for the elimination of sleeping sickness. Adv Parasitol 79: 299–337. [DOI] [PubMed] [Google Scholar]
- Wheeler RJ (2017) Use of chiral cell shape to ensure highly directional swimming in trypanosomes. PLoS Comput Biol 13: e1005353–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler RJ, Gluenz E, and Gull K (2013) The Limits on Trypanosomatid Morphological Diversity. PLoS ONE 8: e79581–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler RJ, Scheumann N, Wickstead B, Gull K, and Vaughan S (2013) Cytokinesis in Trypanosoma brucei differs between bloodstream and tsetse trypomastigote forms: implications for microtubule-based morphogenesis and mutant analysis. Mol Microbiol 90: 1339–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, and Gull K (2006) A “holistic” kinesin phylogeny reveals new kinesin families and predicts protein functions. Mol Biol Cell 17: 1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, and Cross GA (1999) A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99: 89–101. [DOI] [PubMed] [Google Scholar]
- Woehlke G, and Schliwa M (2000) Walking on two heads: the many talents of kinesin. Nat Rev Mol Cell Biol 1: 50–58. [DOI] [PubMed] [Google Scholar]
- Woods A, Baines AJ, and Gull K (1992) A high molecular mass phosphoprotein defined by a novel monoclonal antibody is closely associated with the intermicrotubule cross bridges in the Trypanosoma brucei cytoskeleton. J Cell Sci 103 (Pt 3): 665–675. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Gu J, Lun Z-R, Ayala FJ, and Li Z (2016) Two distinct cytokinesis pathways drive trypanosome cell division initiation from opposite cell ends. Proc Natl Acad Sci USA 113: 3287–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Hu H, and Li Z (2016) An EF-hand-containing Protein in Trypanosoma brucei Regulates Cytokinesis Initiation by Maintaining the Stability of the Cytokinesis Initiation Factor CIF1. Journal of Biological Chemistry 291: 14395–14409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Liu B, Sun Y, and He CY (2011) A coiled-coil- and C2-domain-containing protein is required for FAZ assembly and cell morphology in Trypanosoma brucei. J Cell Sci 124: 3848–3858. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Matching Ty1-BirA* TOEFAZ1 expression to endogenous TOEFAZ1 levels. A) Western blot comparing the expression levels between a cell line in which both alleles of TOEFAZ1 are tagged with a Ty1 epitope tag (dTag TF1) and the Ty1-BirA* cell line induced overnight with either 40 ng/mL or 500 ng/mL doxycycline. B) The Ty1-BirA* cell line was induced as above, followed by incubation with 50 μM biotin. Lysates from the induced Ty1-BirA* TOEFAZ1 cells were collected and separated by SDS-PAGE followed by transfer to nitrocellulose for western blotting with streptavidin conjugated to HRP. C) The Ty1-BirA* cell line was induced and incubated with biotin as above. Cells were methanol-fixed and stained with anti-Ty1 antibody and streptavidin. Arrowheads: Ty1-BirA* TOEFAZ1 localization. Asterisks indicate spurious localization. DAPI was used to stain DNA. D) The Ty1-BirA* TOEFAZ1 cell line was induced as in B) with 40 ng/mL doxycycline, followed by lysis and capture of biotinylated proteins, which were eluted and blotted using anti-biotin antibody.
Figure S2 KPP1 is required for cell division. A) KPP1 RNAi was induced for 8 d using 1 μg/mL doxycycline, and cell concentration was counted every 24 h. B) Cell lysates were collected every 24 h from the RNAi timecourse in A), western blotted, and labeled with anti-Ty1 antibody. C) Cells carrying a KPP1 RNAi construct were treated with either vehicle control or 1 μg/mL doxycycline to induce RNAi for four days. Cells were PFA-fixed and stained with DAPI for DNA. Asterisk indicates a detached flagellar tip. D) Quantification of detached new flagella tips in control and KPP1 RNAi cells in which KPP1 has been depleted for 4 d.
Figure S3 PAVE1 is necessary for cell division. A) PAVE1 RNAi cells were treated with either vehicle control or 1 μg/mL doxycycline for 6 days and cell number was monitored every 24 h. B) Cell lysates were collected every 24 h from the time course in A). Lysates were western blotted and stained with anti-Ty1 antibody.
Figure S4 YL1/2 staining is not altered in PAVE1 RNAi cells. A) Immunofluorescence of methanol-fixed wherein PAVE1 was tagged at its endogenous locus with a Ty1 tag. Cells were stained for YL1/2 to detect tyrosinated tubulin, an indicator of growing microtubules, and Ty1 to label PAVE1. B) PAVE1 RNAi was induced with 1 μg/mL doxycycline for 1 and 2 d. Cells were methanol fixed and stained with anti-tubulin antibody and YL1/2. DAPI was used to stain DNA in all immunofluorescence panels. C) Dividing 2N2K cells from the experiment described in B). D) Lysates from the RNAi time course in Fig. S3A were western blotted and labeled with YL1/2 antibody.
Figure S5 KLIF is required for cell division. A) Cells in which KLIF was tagged at its endogenous locus with a Ty1 epitope tag were methanol-fixed and stained with anti-Ty1 antibody and 1B41 to label the FAZ. DAPI was used to label DNA. Arrowheads indicate Ty1-KLIF localization at the cleavage furrow. B) Cells containing the KLIF RNAi construct were treated with either vehicle control or 1 μg/mL doxycycline. Cell concentration was counted every 24 h. C) Lysates were collected every 24 h from the RNAi timecourse in B), western blotted, and stained with anti-Ty1 antibody. D) Cells containing a KLIF RNAi construct were treated with either vehicle control or 1 μg/mL doxycycline for 3 d. Cells were collected and PFA-fixed for morphological analysis. DAPI was used to stain DNA. E) TOEFAZ1 RNAi was induced in a cell line containing KLIF tagged at its endogenous locus with a Ty1 epitope tag. Lysates were collected every 12 h, western blotted, and stained with anti-Ty1 antibody.
Figure S6 Schematic of the in vitro motility assay. KLIF (purple) containing a mClover3 tag (mClv3, green) is bound to an α-GFP coated flow chamber (light blue) and blocked. Then, rhodamine-labeled polarity marked microtubules (red) containing enhanced fluorescent signal at their minus-ends are introduced to the flow chamber to interact with KLIF in the presence of MgATP.
Figure S7 Characterization of curated TOEFAZ1 BioID hits. A Ty1 epitope tag was integrated at either the N- or C-terminus of the endogenous locus of each hit from the TOEFAZ1 BioID screen. The size of each protein was determined by anti-Ty1 western blotting, and the correct integration of the Ty1 tag at the endogenous locus was established by loci PCR. Immunofluorescence analysis was conducted on each protein using anti-Ty1 antibody and, where applicable, using antibodies against known organelles for co-localization.
Supporting Movie 1 In vitro motility assay showing that KLIF moves labeled polarity marked microtubules with their brighter minus-ends leading. The movie is 200-fold real time.
Supplemental Table 1 Table of the unique peptides identified in this screen.