

Abstract
Motivation
The correct identification of ligands in crystal structures of protein complexes is the cornerstone of structure-guided drug design. However, cognitive bias can sometimes mislead investigators into modeling fictitious compounds without solid support from the electron density maps. Ligand identification can be aided by automatic methods, but existing approaches are based on time-consuming iterative fitting.
Results
Here we report a new machine learning algorithm called CheckMyBlob that identifies ligands from experimental electron density maps. In benchmark tests on portfolios of up to 219 931 ligand binding sites containing the 200 most popular ligands found in the Protein Data Bank, CheckMyBlob markedly outperforms the existing automatic methods for ligand identification, in some cases doubling the recognition rates, while requiring significantly less time. Our work shows that machine learning can improve the automation of structure modeling and significantly accelerate the drug screening process of macromolecule-ligand complexes.
Availability and implementation
Code and data are available on GitHub at https://github.com/dabrze/CheckMyBlob.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The interpretation of macromolecular electron density maps generated by X-ray crystallography is a complicated process. Given a 3D map of the experimental electron density, a chemist or biologist has to model the structure of the crystallized molecules, usually to extract important information about the protein’s function. This process is applied not only to X-ray data, but also to similar electrostatic potential maps generated by high-resolution cryo-electron microscopy. With existing model building software (Cowtan, 2006; Perrakis et al., 1999; Terwilliger, 2003) integrated with multi-tasking systems (Adams et al., 2002; Minor et al., 2006; Winn et al., 2011), the regions of macromolecular structure corresponding to polypeptide or polynucleotide chains can be built with high accuracy and speed. On the other hand, small-molecule ligands are usually modeled manually, and their correct identification often requires good judgment and expertise. This process is time-consuming and prone to human error, as small ligands are often difficult to distinguish from one another on the basis of electron density alone. The recognition process is particularly challenging when the resolution of the diffraction data is not very high (2.0 Å or worse), there is local disorder, or the ligand is bound to only a fraction of the molecules. The often-questionable, subjective assignment of ligands to electron density ‘blobs’ and the notorious problem of fictitious ligands modeled without support of experimental evidence (Pozharski et al., 2013) show that improved automatic methods for ligand recognition free from cognitive bias are greatly needed (Adams et al., 2016).
Several approaches are used for automated fitting of known ligands to electron density maps. They are typically based on ligand core recognition followed by iterative element addition (Oldfield, 2001; Terwilliger et al., 2006), principal axes alignment and Metropolis-type optimization (Debreczeni and Emsley, 2012), or combinations of similar techniques (Evrard et al., 2007; Langer et al., 2013; Zwart et al., 2004). Those methods can be adapted to identify unknown ligands by iteratively fitting a moiety from a predefined list of candidates to a given unmodeled electron density blob. Indeed, Terwilliger et al. (2007) combined iterative fitting with fingerprint correlations and achieved 48% accuracy in recognizing instances of the 200 most frequently observed ligands in structures stored in the Protein Data Bank (PDB) (Berman et al., 2000). However, such an approach can be prohibitively slow for large-scale experiments, as it necessitates fitting trials of all candidate ligands.
Alternatives to time-consuming iterative fitting approaches focus mainly on the use of mathematical descriptions of 3D electron density map fragments. Methods from this group range from simple comparisons of ligand bounding boxes (Langer et al., 2013) to the use of more advanced shape descriptors, such as moment invariants (Sommer et al., 2007), three-dimensional Zernike moments (Gunasekaran et al., 2009), chirality indices (Hattne and Lamzin, 2011), pseudo-atomic graph representations (Aishima et al., 2005), or their combinations (Carolan and Lamzin, 2014). However, the best approaches from this group are capable of achieving only 30–32% accuracy in identifying the correct ligand from a list of ∼100 popular ligand structures (Carolan and Lamzin, 2014; Gunasekaran et al., 2009). Nevertheless, these methods are much faster than iterative ligand fitting and perform fairly well when predicting the ten most probable ligand candidates.
We designed an approach called CheckMyBlob that uses machine learning algorithms to identify ligands in electron density maps. In contrast to existing methods, CheckMyBlob learns to generalize ligand descriptions from sets of moieties deposited in the PDB, rather than comparing density maps to theoretical models, graphs, or selected template structures. The ligand descriptors used in our method can be rapidly calculated as they are based on features that take into account the moiety’s shape, volume, chemical environment and resolution of the data. Moreover, in contrast to existing methods, we assume no human intervention during the ligand recognition process. Therefore, we present a method for a completely automatic initial interpretation of residual electron density blobs after structure determination and preliminary biopolymer (polypeptide or/and nucleic acid chains) refinement. We cross-validated the proposed machine learning approach by applying it to 219 931 instances of the 200 ligands most frequently observed in PDB structures and achieved significantly higher recognition rates than current iterative fitting or description comparison methods.
2 Materials and methods
2.1 System overview
CheckMyBlob is a system that learns to generalize ligand descriptions from electron density maps and uses that knowledge to detect and identify ligands in previously unseen density (Fig. 1). In the learning phase, uninterpreted blobs are first cut out from electron density maps generated using the polymer-only portions of PDB structures. Next, each blob is described by a set of numerical features, which are fed to a machine learning algorithm (classifier). The classifier automatically creates a function (classification model) that predicts the best ligand based on the blob’s numerical features. In the identification phase, this classification model is used to recognize ligands in previously unseen electron density maps.
Fig. 1.

CheckMyBlob ligand recognition pipeline. In the training phase (upper arrows), CheckMyBlob uses ligands from structures deposited in the PDB to create a classification model. After training, in the productive runs (lower arrows), CheckMyBlob is capable of automatically detecting and recognizing ligands from unmodeled electron density blobs
2.2 Structure factors and electron density maps
We downloaded all PDB entries as of May 1, 2017 from rsync.ebi.ac.uk and converted entries with structure factors from mmCIF to mtz format, using the cif2mtz program from the CCP4 suite version 7.0.039 (Winn et al., 2011). Out of the 105 726 converted files, 101 538 were successfully processed and standardized with zero cycles of REFMAC, version 5.8.0158 (Murshudov et al., 2011). The remaining files could not be processed with REFMAC due to various data-related errors.
As input to the ligand identification pipeline, we calculated Fo-Fc electron density maps, with 0.2 Å grid spacing, based on data in the mtz files and on atomic coordinates of the main-chain and side-chain atoms with explicit exclusion of all small-molecule moieties and solvent molecules. To reduce the ‘memory’ of these small molecules in the calculated structure factors, the partial models were refined with five cycles of REFMAC using restrained maximum likelihood targets and the following settings: hydrogen atoms included, individual isotropic B-factors, simple anisotropic scaling, bulk solvent correction, no TLS, local automatic NCS included and automatic restraint weights. We did not use TLS or twinning refinement in order to process as many files as possible automatically, i.e. we sacrificed model quality in favor of robustness of the overall processing. Moreover, we wanted to prepare a model that would be close to the initial stage of a typical structure refinement protocol. As a result, out of the 101 538 input files, 99 983 passed the refinement and Fo-Fc electron density map generation. The remaining files could not be processed due to errors in third party libraries. In 82 337 of the successfully processed cases, at least one ligand was present.
2.3 Ligand detection and label assignment
As CheckMyBlob is meant to work automatically on partially modeled structures, it does not use model information to identify ligand fragments in the electron density maps. Instead, CheckMyBlob finds all positive electron density peaks within the Fo-Fc map and for each peak calculates its volume limited by the 2.8σ isosurface computed with a 0.2 Å grid. The contour level is lower than the usual 3σ since water, ions and organic molecules are intentionally deleted from the model in the training phase, thereby increasing the standard deviation of the map with respect to that of a fully refined complete model. Since any peak or fragment of the map that could be assigned to an atom (even a disordered one) is vital in the ligand detection phase, CheckMyBlob considers all residual electron density peaks with a volume greater than Vmin = 0.25 Å3. We denote unmodeled patches of density fulfilling this criterion as blob candidates (Fig. 2A).
Fig. 2.
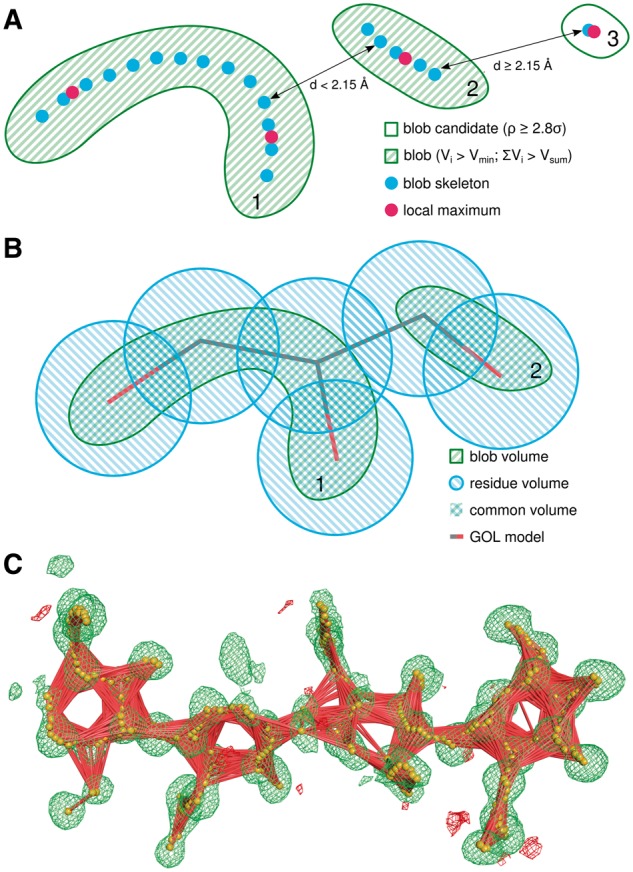
Blob Recognition. (A) Illustration of the blob detection procedure. Blob candidates 1 and 2 are combined into a single blob, whereas candidate 3 is rejected due to insufficient volume. (B) Blob labeling procedure. The volume of the intersection of a detected blob and a modeled moiety determines the ligand label used during the training phase. (C) Skeletonization (orange spheres) of Fo-Fc map (green mesh, isosurface at 2.8σ) and skeletonization graph (red lines) of a NAG-NAG-NAG-NAG blob assigned to PDB structure 3WH1, chain A, residues 301–304 (Color version of this figure is available at Bioinformatics online.)
High resolution maps require special consideration. At ≤1.5 Å resolutions, and particularly at the full atomic resolutions of <1.2 Å (Sheldrick, 1990), electron density contoured at 2.8σ is often split into discrete atomic peaks, even for covalent moieties. To mitigate this problem, we developed a method that detects local maxima and skeletonizes the electron density within the isosurface of each blob candidate in a way similar to that described by Zwart et al. (2004). After skeletonization, adjacent blob candidates are combined into one blob if the distance between the local maxima or skeleton nodes is less than 2.15 Å (Fig. 2A). This distance was chosen halfway between the length of a single C–C bond (1.54 Å) and the distance of two hydrogen-bonded water molecules (2.76 Å). Blobs with volumes smaller than Vsum = 2.14 Å3 are discarded from further analysis, as density blobs this small are usually not modeled, even as water molecules, in atomic resolution structures. The proposed Vsum was determined experimentally as a compromise between detection of all small molecules and reduction of computational cost. Finally, any fragments of electron density in the blob isosurface that overlap with the isosurface of the modeled biopolymer atoms are cut out from the blob. In practice, CheckMyBlob is capable of detecting ligands consisting of tens of blob candidates (Fig. 2C).
Training a machine learning algorithm to recognize ligands in electron density maps requires a set of labeled training examples. In the case of CheckMyBlob, the examples are presented as electron density blobs, and the labels are given in the form of ligand names assigned to each blob. In theory, one could directly use the information from the model structure to identify ligand names; however, since CheckMyBlob does not use model information to extract the blobs, they might not overlap exactly with the model ligands. Moreover, due to the naming conventions used by the PDB, polymers are stored as linked monomers. For example, a NAG-NAG disaccharide is encoded in the PDB as two separate NAG (N-acetyl-d-glucosamine) molecules.
Thus, to label the detected blobs we quantified the volume shared by the blob and the residues modeled in the initial PDB entry. For this purpose, we define (model) residue volume as the volume within 1.05 Å of the residue atom centers. The more natural van der Waals radii were not used to prevent single large atoms (usually metal ions) from dominating the residue volume. The volume of the intersection of the blob and residue volumes, divided by the blob volume, defines blob coverage, while the intersection volume divided by the residue volume defines residue coverage. These values were used for ligand labeling and selection (Fig. 2B). Each blob was labeled with the residue code (e.g. GOL, TRS, etc.) with significant residue coverage. If the blob volume intersects with more than one residue, the label assigned to the blob is an alphabetically sorted list of all codes with residue coverage above 30%. As a result, unique labels are given to common polymers, e.g. NAG-NAG, and moieties whose electron densities are merged at low resolution, e.g. ADP-MG. Although combinations such as ADP-MG do not represent a covalently bonded unit, they do correspond to practical electron density recognition cases. Using 82 337 structures containing in total 608 467 ligands (∼7.39 ligands per structure), the process of blob detection resulted in 591 042 examples of moieties found in the PDB.
2.4 The numerical feature descriptors
The detected and labeled ligands were initially described by 382 numerical features (Supplementary Table S1). Although 382 is a feasible number of descriptors for a machine learning algorithm to process, a relatively high number of features compared to the number of training examples can impede generalization, a phenomenon known in machine learning as the curse of dimensionality (Keogh and Mueen, 2017). Therefore, an automatic feature selection algorithm called recursive feature elimination (Guyon et al., 2002) was run on a random subset of 10 000 ligand binding sites to reduce the number of descriptors. After analyzing the results, 60 features were finally selected as blob descriptors in the machine learning process (Fig. 3).
Fig. 3.
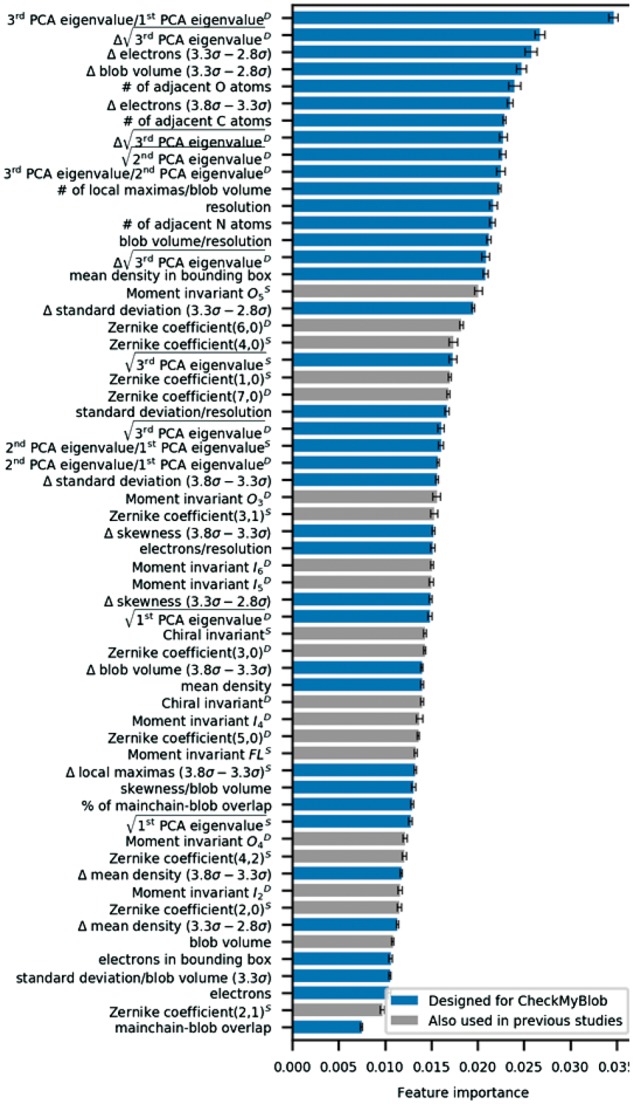
Feature importance ranking. Importance of numerical features used by CheckMyBlob as computed by the GBM algorithm on the CMB dataset. D, density mask; S, shape mask
Map resolution is a feature that requires additional explanation. It must be stressed that resolution is the only ‘global’ map attribute, i.e. it has the same value for all blobs in a given PDB entry. When using such global features, one runs into the risk of overfitting or information leak, which in this case means making the algorithm learn or ‘remember’ irrelevant features of the pdb file that the ligand came from, rather than learning the characteristics of the ligand itself. To eliminate this risk, we applied discretization, i.e. we made the resolution discrete by rounding it to the nearest 0.1 Å. After discretization, the number of examples with the same value of the parameter increases and the classifier is not able to recognize a pdb file from the numerical value of its resolution.
2.5 Ligand datasets
The 591 042 examples were further filtered using various quality criteria to produce three datasets: CMB (CheckMyBlob), TAMC (Terwilliger, Adams, Moriarty, Cohn) and CL (Carolan, Lamzin). The CMB dataset was designed for the present study to specifically test CheckMyBlob, whereas the remaining two datasets attempt to reproduce the setups used in previous automatic ligand recognition studies (albeit with vastly expanded subsets of the PDB).
The CMB dataset consists of ligands with at least 2 non-H atoms from X-ray diffraction experiments of at least 4.0 Å resolution. We also eliminated all suspicious structures according to various quality criteria, such as: RSCC < 0.6, real space Zobs (RSZO) < 1.0, real space Zdiff (RSZD) ≥ 6.0, R factor > 0.3, or occupancy < 0.3. The details of the filtering process are described in the Supplementary Material. Connected PDB ligands were labeled as single, alphabetically-ordered strings of residue codes, excluding unknown species, water molecules, standard amino acids and nucleotides. Finally, the dataset was limited to the 200 most frequently observed ligands (Supplementary Table S2). The resulting dataset consisted of 219 931 examples of ligand binding sites, with individual ligand counts ranging from 48 490 examples for SO4 (sulfate) to 105 for BRU (5-bromo-2′-deoxyuridine-5′-monophosphate).
The TAMC dataset replicates the experimental set of Terwilliger et al. (2007). It consists of ligands from X-ray diffraction experiments with 6–150 non-H atoms. Connected PDB ligands were labeled as single, alphabetically-ordered strings of residue codes, excluding unknown species, water molecules, standard amino acids and nucleotides. Finally, the dataset was limited to 200 most frequently observed ligands (Supplementary Table S2). The resulting dataset consisted of 161 758 examples with individual ligand counts ranging from 36 535 examples for GOL (glycerol) to 114 for LMG (1,2-distearoyl-monogalactosyl-diglyceride).
The CL dataset replicates the experimental set used by Carolan and Lamzin (2014). It consists of ligands from X-ray diffraction experiments with 1.0–2.5 Å resolution. Adjacent PDB ligands were not connected. Ligands were labeled according to the PDB naming conventions. Finally, the dataset was limited to the 82 ligand types (Supplementary Table S2) listed by Carolan and Lamzin (2014). The resulting dataset consisted of 121 360 examples with ligand counts ranging from 42 622 examples for SO4 to 16 for SPO (spheroidene). Links to the CMB, TAMC and CL datasets are available at https://github.com/dabrze/CheckMyBlob.
2.6 Machine learning methods
Blobs described by numerical features were used as training examples for classification algorithms, with the assigned ligand residue codes serving as class labels (Supplementary Fig. S1). It is worth noting that, contrary to previous approaches, CheckMyBlob does not use any predefined scores or distance measures that estimate which ligand candidate is most similar to the analyzed blob. It is the task of the learning algorithm to automatically find a complex function of blob descriptors (classification model) that is best suited for ligand recognition.
To evaluate the recognition rate of the trained classification models, the collected ligand datasets were divided into training and testing sets. In an attempt to make full use of the processed PDB data and to provide reliable estimates of the models’ recognition rates and their standard deviations, we used stratified 10-fold cross-validation (Supplementary Fig. S2). Stratification is of particular importance in this study, as the collected datasets have a strongly skewed ligand type distribution (Supplementary Fig. S3), and purely random, non-stratified folds would produce unreliable error estimates.
Considering the numerous reports warning of problematic interpretations of ligand electron density in many PDB entries (Kleywegt, 2007; Pozharski et al., 2013), we decided to perform additional automatic outlier and noise removal on the training data using the isolation forest algorithm (Liu et al., 2012) parameterized to remove 0.5% training examples, a value based on the proportion of the smallest to the largest class in the CMB dataset. Moreover, ligands with multiple conformations in the wwPDB validation reports were removed from the training data to reduce the noise in ligand descriptions. The discussed restrictions were only applied to the training data and did not affect the testing folds. Additionally, to prevent bias toward features with larger ranges of numerical values, all the features were normalized using min-max [0-1] scaling (Tan et al., 2005) calibrated on the training data.
We evaluated the performance of three popular classifiers (Breiman, 2001; Fix and Hodges, 1951; Friedman, 2001): k-nearest neighbors (k-NN), random forest (RF), and gradient boosting machine (GBM) (Table 1). These classifiers were selected because of their ability to learn non-linear relationships among features and their computational efficiency. Additionally, we evaluated the combination of these three algorithms using stacked generalization (Stacking) with five cross-validation folds (Wolpert, 1992). Stacking involves training a learning algorithm (called a combiner) to aggregate the predictions of several component classifiers. In our stacking implementation, we used k-NN, RF and GBM as components, and GBM as a combiner trained to make a final prediction based on ligand probabilities from the components.
Table 1.
Cross-validation results
| Algorithm | Testing examples | Accuracy (recognition rate) | Top-5 accuracy | Top-10 accuracy | Top-20 accuracy | Macro-averaged recall | Cohen’s Kappa | |
|---|---|---|---|---|---|---|---|---|
| CMB | CheckMyBlob: k-NN | 0.523 (13) | 0.816 (8) | 0.874 (6) | 0.901 (4) | 0.261 (12) | 0.462 (14) | |
| CheckMyBlob: RF | 219 931 | 0.563 (12) | 0.836 (8) | 0.896 (5) | 0.933 (3) | 0.327 (11) | 0.511 (14) | |
| CheckMyBlob: GBM | 0.572 (12) | 0.849 (7) | 0.910 (5) | 0.949 (3) | 0.366 (14) | 0.523 (13) | ||
| CheckMyBlob: Stacking | 0.575 (11) | 0.852 (8) | 0.913 (5) | 0.950 (3) | 0.391 (13) | 0.526 (12) | ||
| TAMC | Terwilliger et al. (15) | 200 | 0.485 | 0.780 | 0.870 | 0.925 | – | – |
| CheckMyBlob: k-NN | 0.500 (10) | 0.751 (8) | 0.814 (6) | 0.856 (5) | 0.226 (13) | 0.430 (11) | ||
| CheckMyBlob: RF | 161 758 | 0.551 (9) | 0.778 (7) | 0.842 (5) | 0.892 (5) | 0.287 (17) | 0.496 (10) | |
| CheckMyBlob: GBM | 0.560 (7) | 0.790 (7) | 0.857 (6) | 0.910 (4) | 0.320 (17) | 0.508 (9) | ||
| CheckMyBlob: Stacking | 0.563 (8) | 0.796 (7) | 0.861 (6) | 0.912 (5) | 0.346 (19) | 0.513 (9) | ||
| CL | Carolan et al. (21) | 1100 | 0.320 | – | 0.840 | 0.940 | – | – |
| CheckMyBlob: k-NN | 0.686 (12) | 0.901 (4) | 0.932 (2) | 0.947 (3) | 0.339 (19) | 0.589 (15) | ||
| CheckMyBlob: RF | 121 360 | 0.715 (13) | 0.913 (4) | 0.945 (2) | 0.966 (1) | 0.415 (20) | 0.628 (17) | |
| CheckMyBlob: GBM | 0.722 (11) | 0.920 (4) | 0.953 (2) | 0.975 (1) | 0.440 (18) | 0.639 (15) | ||
| CheckMyBlob: Stacking | 0.725 (12) | 0.921 (4) | 0.954 (3) | 0.976 (2) | 0.483 (20) | 0.645 (16) |
The best values in each test group are highlighted in bold. Note: Average performance metrics and standard deviations (in parentheses, in the unit of the last significant digit of the mean value) for different algorithms on three ligand datasets: CMB, TAMC and CL.
To preprocess the data, remove outliers and generate k-NN, random forest and Stacking classifiers, we used scikit-learn v.0.18.1 (Pedregosa, 2011). For GBM we used Microsoft’s LightGBM package. The classifiers’ parameters were tuned only on the training folds using two shuffled repetitions of stratified 5-fold cross validation (2x5 CV). Repeated cross-validation was used, as it was shown to provide more reliable results for parameter selection than standard cross-validation (Dietterich, 1998). Employing cross-validation for both parameter tuning and model evaluation is a rigorous machine learning procedure called nested cross-validation (Japkowicz and Shah, 2011). The parameter values considered during classifier tuning are listed at https://github.com/dabrze/CheckMyBlob.
The classifiers were evaluated using the following metrics: classification accuracy, top-5/10/20 accuracy, Cohen’s kappa, and macro-averaged recall. Classification accuracy is the proportion of correctly recognized ligands to all testing examples. Top-n accuracy is the proportion of cases where the correct ligand was among the n highest-ranked hits in the classifier’s prediction. Cohen’s kappa is a measure that corrects accuracy for chance predictions. Macro-averaged recall is the (unweighted) arithmetic mean of the recognition rates for each class. Accuracy and top-n accuracy were chosen because they were also reported by Terwilliger et al. (2007) as well as by Carolan and Lamzin (2014). The remaining metrics are measures commonly used in machine learning to evaluate classifiers on datasets with skewed class distributions (Japkowicz and Shah, 2011). The evaluation was conducted on an Amazon EC2 r4.8xlarge virtual machine equipped with 32 vCPUs and 244 GB of RAM.
3 Results
3.1 Importance of novel ligand description attributes
Apart from popular 3D shape descriptors used in previous studies (Carolan and Lamzin, 2014; Novotni and Klein, 2003; Sommer et al., 2007), such as geometric, chiral and Zernike moment invariants, CheckMyBlob uses novel attributes to describe ligands. The new attributes include, in particular, blob volume and number of electrons, map statistics, principal component analysis (PCA) eigenvalues based on positive peaks from Fo-Fc maps, and differences between these features at several contour levels (2.8σ, 3.3σ and 3.8σ) (Supplementary Table S1). Additionally, CheckMyBlob utilizes chemical information, such as the number of adjacent biopolymer atoms and the degree of biopolymer-blob overlap. Several of the descriptors are calculated for both what we call the blob shape mask and the density mask. The blob shape mask is the blob fragment of the electron density where values above the cutoff threshold are set to 1 or set to 0 otherwise. The density mask is similar, but with actual electron density values instead of 1, and 0 otherwise.
The results of automatic importance analysis performed on features used by CheckMyBlob (Fig. 3) emphasize the influence of the new ligand descriptors introduced in this study. In particular, features based on PCA eigenvalues provide a means of encoding the relationships between the principal blob dimensions and give a rotation-invariant description of a ligand. Moreover, features that report the differences (deltas) between the estimated number of electrons for consecutive contour levels are also highly relevant, as they capture the dynamics of changing contour levels and, indirectly, encode information about a blob surface. Finally, the number of modeled biopolymer atoms (O, N, C) in the proximity of a blob adds chemical information to the blob description.
3.2 Experimental comparison with existing approaches
CheckMyBlob showed high classification accuracy on a dataset selected for this study (CMB), and outperformed existing approaches on two datasets (TAMC, CL) that replicate experimental setups from previous studies (Table 1). While analyzing the results, it must be noted that the methods that CheckMyBlob was compared against do not detect ligands automatically and were tested on electron density fragments labeled by human experts as ligands in the PDB deposits. Moreover, the iterative fitting approach of Terwilliger et al. (2007) was tested on electron densities of fully modeled structures, as opposed to the method of Carolan and Lamzin (2014) and our own approach, which use electron density generated by re-refinement of macromolecular models with all small molecule components stripped out. Our automatic ligand detection and erasure of model ‘memory’ through re-refinement correspond to a much more challenging, yet more realistic, evaluation scenario.
All four classifiers used with CheckMyBlob [k-nearest neighbors (k-NN), random forest (RF), gradient boosting machine (GBM) and stacked generalization (Stacking)] achieved much higher recognition rates than those recorded by previous studies (Table 1). The improvement is particularly substantial on the CL dataset, where CheckMyBlob reaches 72% accuracy compared to 32% reported by Carolan and Lamzin (2014). Moreover, experiments conducted in this study involved over a hundred times more testing examples than previous analyses (Table 1), making the performance estimates more reliable. Apart from achieving better recognition rates on larger testing sets, CheckMyBlob was also found to be significantly faster than the competing algorithms (Table 2, Supplementary Material).
Table 2.
Single-core execution time for identifying one ligand, averaged over 30 PDB deposits in parentheses, standard deviations in the unit of the last significant digit of the mean value; >3600 denotes consistent timeouts after 1 hour; differences between CheckMyBlob (k-NN, RF, GBM, Stacking) and the competing algorithms are statistically significant at α = 0.01 according to the Friedman and Nemenyi tests (Supplementary Material)
| Algorithm | Running time [s] |
|---|---|
| CheckMyBlob: k-NN | 103 (64) |
| CheckMyBlob: RF | 118 (68) |
| CheckMyBlob: GBM | 106 (65) |
| CheckMyBlob: Stacking | 121 (66) |
| Terwilliger et al. | >3600 |
| Carolan et al. | 252 (132) |
The best performance is highlighted in bold.
The analysis of recognition rates for different density map resolutions, real-space correlations (RSCC) and ligand sizes (Fig. 4A–C) underlines the value of well-defined, high resolution structures. Moreover, CheckMyBlob’s classification is well calibrated, i.e. the prediction probability corresponds linearly with the recognition rate (Fig. 4D). In contrast to scores outputted by template-based methods (Carolan and Lamzin, 2014; Terwilliger et al., 2007), the prediction probability can be interpreted on its own without looking at the second-best prediction. If a ligand is predicted to be glycerol with 80%, it means that in 80% of such cases it will be indeed GOL, regardless of whether the second best prediction has 15 or 5% probability. This shows that the CheckMyBlob methodology can be applied in user-oriented tools, where the probability can be interpreted as the prediction’s reliability or used to set an acceptable rate of false identifications.
Fig. 4.

Recognition rates. CheckMyBlob ligand recognition rates (accuracy, shown as points) and ligand distributions (number of examples, shown as histograms) versus (A) real-space correlation coefficient (RSCC); (B) ligand size; (C) resolution; (D) prediction probability. The tests were run on three datasets, CMB, TAMC and CL, as explained in the text
3.3 Ligand validation
To additionally verify its performance (Table 1), we analyzed the ability of CheckMyBlob to recognize ligands in a set of example structures including the PDB entries: 1OGV, which was examined by Terwilliger et al. (2007); 3MB5 and 4IUN, which were used by Carolan and Lamzin (2014); 5N0H, which illustrates the recognition of buffer components; 4Y1U, which highlights the problem of missing density; and 2PDT, 1FPX, 4RK3 and 1KWN, which showcase correct prediction of ligands that were misidentified in the original PDB deposits.
CheckMyBlob was able to recognize large, distinctive molecules such as bacteriochlorophyll A (1OGV; BCL M 1303; resolution 2.35 Å) (Fig. 5A), as well as medium-size moieties such as SAM (S-adenosyl-l-methionine) (3MB5; SAM A 301; resolution 1.6 Å) and thymidine-3′,5′-diphosphate (4IUN; THP A 202; resolution 1.6 Å) (Fig. 5B and C). Moreover, the system recognizes common buffer or cryo-protectant components, such as glycerol (5N0H; GOL B 303, resolution 1.9 Å) (Fig. 5D).
Fig. 5.
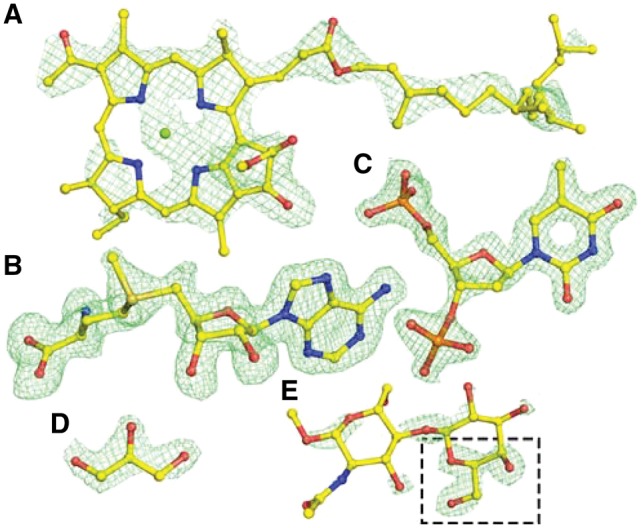
Examples of ligand identification in PDB deposits using CheckMyBlob. (A) 1OGV, bacteriochlorophyll A (BCL M 1303). (B) 3MB5, S-adenosyl-l-methionine (SAM A 301). (C) 4IUN, thymidine-3′,5′-diphosphate (THP A 202). (D) 5N0H, glycerol (GOL B 303). (E) 4Y1U, β-d-galactose (GAL B 201), misclassified by Check MyBlob as GOL (black frame). For each example, shown in green mesh are the isosurfaces of Fo-Fc maps contoured at 2.8σ, calculated after removal of solvent and other small molecules (including the ligand) from the model and five cycles of REFMAC5 (Murshudov et al., 2011) refinement. Atomic coordinates were taken from the PDB deposits (Color version of this figure is available at Bioinformatics online.)
As expected, CheckMyBlob works best when the resolution is better than 2 Å and the electron density is well defined, i.e. there is no missing density and the noise level is low. CheckMyBlob may not recognize a ligand if the electron density is poorly defined, as in 4Y1U (GAL B 201; resolution 1.76 Å), where the blob that was labeled as β-d-galactose (GAL) was misidentified by CheckMyBlob as glycerol (GOL; Fig. 5E). The system recognized glycerol because the highest Fo-Fc peak is similar to a glycerol isosurface and missing electron density prevented the connection of adjacent blob candidates.
However, there are also cases where CheckMyBlob most probably identified the ligand correctly, but the original authors of the PDB deposit either mislabeled a molecule or simply modeled it incorrectly. An example of the former case can be seen in the PDB entry 2PDT (FAD D 204; resolution 2.20 Å) (Fig. 6A), where the authors labeled a ligand as flavin-adenine dinucleotide (FAD) without modeling the adenine moiety. In reality, the electron density corresponds to flavin mononucleotide (FMN), which is the label identified by CheckMyBlob. The primary citation of 2PDT (Zoltowski et al., 2007) mentions that the sample was treated with phosphodiesterase, which would be expected to hydrolyze FAD to FMN. CheckMyBlob also detected incorrect chirality for 1KWN (TAR A 501; resolution 1.20 Å). The modeled atoms have the configuration of l(+)-tartaric acid (TLA) as identified by CheckMyBlob, whereas the deposition authors labeled this moiety as d(−)-tartaric acid (TAR) (Fig. 6B). An example of incorrect modeling was found in 1FPX (SAM A 1699; resolution 1.65 Å), where the authors modeled S-adenosyl-l-methionine instead of S-adenosyl-l-homocysteine (SAH) (Fig. 6C). Another example of incorrect modeling was found in 4RK3 (GOL A 401; resolution 1.80 Å), where the authors modeled disordered glycerol molecules in electron density, most likely corresponding to tris(hydroxymethyl)aminomethane (TRS) (Fig. 6D) which was also present in the crystallization buffer. The above cases can be viewed as interactive visualizations in Molstack (Porebski et al., 2018) at: http://molstack.bioreproducibility.org/collection/view/YskIjr2eiLoQelKrwnIG/.
Fig. 6.

Examples of misidentified ligands detected in the PDB (left panels) with CheckMyBlob-assigned labels (right). (A) 2PDT (FAD D 204), flavin-adenine dinucleotide, reinterpreted by CheckMyBlob as flavin mononucleotide; the missing adenine fragment is shown in dark gray. (B) 1KWN (TAR A 501), the modeled atoms have the configuration of l(+)-tartaric acid (TLA) as identified by CheckMyBlob, whereas the deposition authors labeled this moiety (inconsistently with the real configuration of the ligand coordinates) as d(−)-tartaric acid (TAR), whose correct chirality is visualized in dark gray. (C) 1FPX (SAM A 1699), modeled as S-adenosyl-l-methionine (SAM), reinterpreted as S-adenosyl-l-homocysteine (SAH); the crystalized protein is an isoflavone O-methyltransferase, therefore, both the substrate and product may be present in the ligand binding site; however, there is a negative peak of Fo-Fc electron density (shown in red at -2.8σ) near the CE methyl carbon atom, making SAH much more probable. (D) 4RK3 (GOL A 401), electron density modeled as disordered glycerol and the same electron density interpreted as TRIS buffer. For each example, shown in this figure, the green mesh represents the isosurfaces of Fo-Fc maps contoured at 2.8σ, calculated after the removal of solvent molecules and other small-molecule moieties (including the ligand) from the model, followed by five cycles of REFMAC5 (Murshudov et al., 2011) refinement. Ligands identified by CheckMyBlob have been manually fitted using COOT (Debreczeni and Emsley, 2012) (Color version of this figure is available at Bioinformatics online.)
We have re-refined the above four sample structures and discussed the changes with the original authors, who welcomed the improvements in all four cases. Jointly with the original authors, we deposited all four corrected structures in the PDB to replace the original entries. As the re-deposition summary in Table 3 shows, sometimes improvements of model quality were quite dramatic (e.g. 5% improvement of Rfree or drop of clashscore by 20 points). This demonstrates that CheckMyBlob can correctly identify ligands, even in sub-optimally modeled structures. Moreover, it is worth noting that structure-remediation servers, such as PDB_REDO (Joosten et al., 2009), do not handle ligand misinterpretation; therefore, for all these four cases they produced ‘corrected’ structures with incorrect ligands.
Table 3.
Summary of refinement and structure quality statistics for original and re-refined structures
| Pdb codeO | Wrong ligand | ResolutionO [Å] | R/RfreeO | ClashscoreO | RMSD bonds O [Å] | Pdb codeR | Correct ligand | ResolutionR [Å] | R/RfreeR | ClashscoreR | RMSD bondsR [Å] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2PDT | FAD | 2.20 | 0.234/0.266 | 21.4 | 0.008 | 6CNY | FMN | 2.10 | 0.163/0.204 | 1.5 | 0.014 |
| 1KWN | TAR | 1.20 | 0.127/0.145 | 4.7 | 0.016 | 6COA | TLA | 1.20 | 0.103/0.117 | 0.6 | 0.011 |
| 1FPX | SAM | 1.65 | 0.218/0.235 | 8.3 | 0.021 | 6CIG | SAH | 1.65 | 0.146/0.174 | 4.2 | 0.013 |
| 4RK3 | GOL | 1.80 | 0.157/0.200 | 1.4 | 0.019 | 6CHK | TRS | 1.80 | 0.140/0.190 | 0.7 | 0.014 |
Note: All four re-refined structures were re-deposited in the PDB to supersede the original entries.
OOriginal deposit.
RAfter re-refinement.
4 Discussion
CheckMyBlob is a machine learning approach to ligand identification that requires minimal human intervention and autonomously detects ligands in partially modeled structures based on experimental electron density maps. Our study shows that the abundance of structures deposited in the PDB makes it possible to automatically learn ligand representations and that this knowledge can be used to recognize small molecules during determination of novel crystallographic structures and to validate the existing PDB deposits. Current approaches to ligand identification rely on fitting, template matching, or graph comparisons, which are all based on measuring similarity between pairs of ligands. Our approach shows that machine learning algorithms offer better recognition rates than template-based methods, in a fraction of the computational time. From the recognition rates of our method (Fig. 4A–C), it is also apparent that high-resolution structures with well-defined density are of significant value not only to human experts, but also to automatic recognition systems.
However, the performance of machine learning systems also relies on the number of available training examples. Indeed, CheckMyBlob requires training data in the form of previous observations of any particular target ligand. Therefore, in order to detect ligands with limited or no examples in the training data, using the proposed pipeline one can only predict moieties that are structurally similar to the target ligand. Such an approach was suggested also for template-based methods where the authors proposed to cluster ligands and predict ligand groups rather than individual compounds (Terwilliger et al., 2007). In practice, this approach requires human supervision or explicit information about compounds in the crystallization cocktail. An alternative to predicting ligand groups could be offered by methods from the field of one-shot learning (Fei-Fei et al., 2006).
Evaluating CheckMyBlob on different ligand complexes raises the question of quality criteria which should be used to select the ligands for such studies. In the dataset chosen for this study (CMB), we concentrated on selecting examples with decent resolution (dmin ≤ 4.0 Å), RSCC ≥ 0.6, real space Zobs (RSZO) ≥ 1.0, real space Zdiff (RSZD) < 6.0, R factor ≤ 0.3 and occupancy ≥ 0.3. These cutoffs were chosen to ensure sufficient quality of the training examples and to minimize the risk of using incorrectly labeled examples for evaluation, while still maintaining a very large pool of examples. It must be noted that the main challenge in predicting ligands in electron density maps lies in the variance of the training data. Since X-ray electron density maps are noisy by nature, eliminating all noisy examples would remove most of the training data and, in consequence, would impede the machine learning process. In the future, it will be possible to tighten these relatively generous selection criteria as the number of examples in the PDB grows, and retrain CheckMyBlob only on the highest quality examples. Indeed, an important aspect of learning systems is that the performance will be dramatically improved with more training examples. The rapidly growing number of PDB deposits (Berman et al., 2013) and the simultaneous elimination of ‘bad apples’, by efforts such as the re-refinements in this study (Table 3), ensure that machine learning approaches to ligand identification will significantly improve with time.
The case of flexible ligands—those capable of assuming several conformations—needs special comment. In the training set, all rotamers of a flexible ligand are tagged with the same label. In the predictive mode, this labeling ‘ambiguity’ is no weakness at all, because precise rotamer fit is not our objective; a given ligand, once correctly recognized, would be optimized (refined) in its electron density by appropriate tools, such as COOT (Debreczeni and Emsley, 2012). Also, the existence of rotamers indistinguishable in their training electron density is not a concern. It would produce somewhat noisier, but also more populous, clusters of examples. However, if the flexibility significantly affects the overall shape of the ligand, it would be beneficial for the predictive performance of classifiers to subdivide ligands with large conformational variability to stereochemically more consistent subclasses.
Finally, in contrast to manual identification of possible ligands, automatic machine learning procedures are not biased by ‘wishful thinking’ on the part of an experimenter often convinced that soaking a crystal in a particular solution must result in the presence of that particular, ordered ligand in the structure. Thus, the use of CheckMyBlob should produce fewer fictitious ligands modeled according to wish rather than electron density.
CheckMyBlob can be further advanced by designing new ligand descriptors, including graph features (Aishima et al., 2005), pseudo-atomic representations (Carolan and Lamzin, 2014) and deep learning (LeCun et al., 2015) on 3D electron density data. As our ongoing work, we are designing an online tool that will employ the CheckMyBlob methodology to help users detect and validate ligands. To achieve this goal, challenges concerning the predicted ligands’ taxonomy, result visualization and user interaction must be addressed.
The methodology presented here can be directly applied to drug screening protocols by training CheckMyBlob on ligand sets corresponding to drug screening cocktails. In this context, ligand identification probabilities returned by the classification model will facilitate and prioritize further work by telling human experts which ligands are most likely to fit into a set of structures of interest. As this study shows, machine learning methods can provide such suggestions significantly faster than the existing approaches. Moreover, by providing a method for autonomous detection of blobs corresponding to small moieties and buffer molecules, CheckMyBlob is also a step toward fully automated model building and refinement, with applications not only in protein crystallography but also in high-resolution cryo-electron microscopy.
Funding
This work was supported in part by NIH grants HG008424, GM117325 and GM117080, funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health and the Department of Health and Human Services under contract nos. HHSN272201200026C and HHSN272201700060C, as well as PUT Institute of Computing Science Statutory Funds.
Conflict of Interest: none declared.
Supplementary Material
References
- Adams P.D., et al. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr., 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Adams P.D., et al. (2016) Outcome of the first wwPDB/CCDC/D3R ligand validation workshop. Structure, 24, 502–508. (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aishima J., et al. (2005) Automated crystallographic ligand building using the medial axis transform of an electron-density isosurface. Acta Crystallogr. D Biol. Crystallogr., 61, 1354–1363. [DOI] [PubMed] [Google Scholar]
- Berman H.M., et al. (2000) The protein data bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., et al. (2013) Trendspotting in the Protein Data Bank. FEBS Lett., 587, 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Carolan C.G., Lamzin V.S. (2014) Automated identification of crystallographic ligands using sparse-density representations. Acta Crystallogr. D Biol. Crystallogr., 70, 1844–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr., 62, 1002–1011. [DOI] [PubMed] [Google Scholar]
- Debreczeni J.É., Emsley P. (2012) Handling ligands with Coot. Acta Crystallogr. D Biol. Crystallogr., 68, 425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietterich T.G. (1998) Approximate statistical tests for comparing supervised classification learning algorithms. Neural Comput., 10, 1895–1923. [DOI] [PubMed] [Google Scholar]
- Evrard G.X., et al. (2007) Assessment of automatic ligand building in ARP/wARP. Acta Crystallogr. D Biol. Crystallogr., 63, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei-Fei L., et al. (2006) One-shot learning of object categories. IEEE Trans. Pattern Anal. Mach. Intell., 28, 594–611. [DOI] [PubMed] [Google Scholar]
- Fix E., Hodges J.L. (1951) Discriminatory analysis, nonparametric discrimination: consistency properties. In: US Air Force School of Aviation Medicine Technical Report 4, p. 477. [Google Scholar]
- Friedman J. (2001) Greedy function approximation: a gradient boosting machine. Ann. Stat., 29, 1189–1232. [Google Scholar]
- Gunasekaran P., et al. (2009) Ligand electron density shape recognition using 3D zernike descriptors. In: Pattern Recognition in Bioinformatics, pp. 125–136. [Google Scholar]
- Guyon I., et al. (2002) Gene selection for cancer classification using support vector machines. Mach. Learn., 46, 389–422. [Google Scholar]
- Hattne J., Lamzin V.S. (2011) A moment invariant for evaluating the chirality of three-dimensional objects. J. R. Soc. Interface, 8, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R.P., et al. (2009) PDB_REDO: automated re-refinement of X-ray structure models in the PDB. J. Appl. Crystallogr., 42, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Japkowicz N., Shah M.. (2011). Evaluating Learning Algorithms: A Classification Perspective. Cambridge University Press, New York, NY, USA. [Google Scholar]
- Keogh E., Mueen A. (2017)Curse of dimensionality. In: Sammut C., Webb G.I. (eds.) Encyclopedia of Machine Learning and Data Mining. Springer, Boston, MA, USA, pp. 314–315. [Google Scholar]
- Kleywegt G.J. (2007) Crystallographic refinement of ligand complexes. Acta Crystallogr. D Biol. Crystallogr., 63, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G.G., et al. (2013) Visual automated macromolecular model building. Acta Crystallogr. D Biol. Crystallogr., 69, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y., et al. (2015) Deep learning. Nature, 521, 436–444. [DOI] [PubMed] [Google Scholar]
- Liu F.T., et al. (2012) Isolation-based anomaly detection. ACM Trans. Knowl. Discov. Data, 6, 3:1–3:39. [Google Scholar]
- Minor W., et al. (2006) HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., et al. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr., 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotni M., Klein R. (2003)3D zernike descriptors for content based shape retrieval. In: Proc. Eighth ACM Symp. Solid Model. Appl., pp. 216–225. [Google Scholar]
- Oldfield T.J. (2001) X-LIGAND: an application for the automated addition of flexible ligands into electron density. Acta Crystallogr. D Biol. Crystallogr., 57, 696–705. [DOI] [PubMed] [Google Scholar]
- Pedregosa F., et al. (2011) Scikit-learn: machine learning in Python. J. Mach. Learn. Res., 12, 2825–2830. [Google Scholar]
- Perrakis A., et al. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Porebski P.J., et al. (2018) Molstack-interactive visualization tool for presentation, interpretation, and validation of macromolecules and electron density maps. Protein Sci., 27, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozharski E., et al. (2013) Techniques, tools and best practices for ligand electron-density analysis and results from their application to deposited crystal structures. Acta Crystallogr. D Biol. Crystallogr., 69, 150–167. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. (1990) Phase annealing in SHELX-90: direct methods for larger structures. Acta Crystallogr. A Found. Crystallogr., 46, 467–473. [Google Scholar]
- Sommer I., et al. (2007) Moment invariants as shape recognition technique for comparing protein binding sites. Bioinformatics, 23, 3139–3146. [DOI] [PubMed] [Google Scholar]
- Tan P.N., et al. (2005)Introduction to Data Mining .Addison Wesley, Boston, MA, USA. [Google Scholar]
- Terwilliger T.C. (2003) Solve and resolve: automated structure solution and density modification. Methods Enzymol., 374, 22–37. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C., et al. (2006) Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D Biol. Crystallogr., 62, 915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C., et al. (2007) Ligand identification using electron-density map correlations. Acta Crystallogr. D Biol. Crystallogr., 63, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.D., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr., 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpert D.H. (1992) Stacked generalization. Neural Netw., 5, 241–260. [Google Scholar]
- Zoltowski B.D., et al. (2007) Conformational switching in the fungal light sensor vivid. Science, 316, 1054–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart P.H., et al. (2004) Modelling bound ligands in protein crystal structures. Acta Crystallogr. D Biol. Crystallogr., 60, 2230–2239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.