

Abstract
The broadly neutralizing antibody against HIV-1, bl2, binds to the CD4 binding site (CD4bs) on the outer domain (OD) of the gp120 subunit of HIV-1 Env. We have previously reported the design of an E.coli expressed fragment of HIV-1 gp120, b122a, containing about 70% of the b12 epitope with the idea of focusing the immune response to this structure. Since the b122a structure was found to be only partially folded, as assessed by circular dichroism and protease resistance, we attempted to stabilize it by the introduction of additional disulfide bonds. One such mutant, b122al-b showed increased stability and bound bl2 with 30-fold greater affinity as compared to b122a. Various b122a and OD fragment proteins were displayed on the surface of Qβ virus-like particles. Sera raised against these particles in six-month long rabbit immunization studies could neutralize Tierl viruses across different subtypes with the best results observed with b122a1-b displayed particles. Significantly higher amounts of antibodies directed towards the CD4bs were also elicited by particles displaying b122a1-b. This study highlights the ability of fragment immunogens to focus the antibody response to the conserved CD4bs of HIV-1.
Keywords: protein stability, immune focusing, neutralizing antibodies, vaccine, nanoparticles
INTRODUCTION
HIV-1 is the causative agent for AIDS. About 37 million people are living with HIV worldwide. There are many circulating HIV-1 strains, which are further divided into subtypes [1] [2]. When more than one strain infects an individual, circulating recombinant forms are produced. Partly because of this high diversity, a successful vaccine remains elusive. The virus infects cells of the immune system when the envelope glycoprotein of HIV-1 binds to CD4 receptors on T helper cells [3]. The envelope glycoprotein (Env) is the primary target of the humoral immune response elicited against HIV-1. Env is synthesized as a single polypeptide precursor (gp160) which is processed in the Golgi to produce two chains, gp120 and gp41. These associate in a non-covalent manner to form a trimer of heterodimers of surface-exposed gp120 and membrane-anchored gp41 chains. gp120 contains the conserved CD4 receptor binding site and gp41 drives the fusion of viral and host cell membranes [4, 5].
In natural infection, most of the antibodies produced at any given time are unable to efficiently neutralize the virus present [6–8]. Viral sequence diversity and the presence of misfolded/unfolded forms of Env shift the focus of the antibody response away from conserved sites [9]. A large amount of glycosylation on the surface of gp120 also occludes the exposure of conserved sites [10]. In an effort to focus the immune response on conserved epitopes of the CD4 binding site, we previously reported the design of a non-glycosylated, bacterially expressed outer domain fragment of HIV-1 Env which was folded and could bind its cognate ligands b12, CD4 and VRC01 [11, 12]. In rabbit immunization studies, it elicited neutralization of primarily Tier 1 viruses. We also designed a small fragment of gp120, called b122a, comprised of a compact beta barrel located on the lower part of the outer domain (Figure S1A) [13]. When bacterially expressed, b122a was found to be only partially folded but able to bind the broadly neutralizing antibody b12 with micromolar affinity. Upon immunization in rabbits and subsequent boosting with gp120, the anti-sera showed broad, albeit moderate neutralization of a 16 virus panel. Competition experiments and serum depletion studies showed that the neutralization was mediated by CD4 binding site antibodies.
Since b122a was only partially structured as assessed by CD and protease resistance [13], we attempted to enhance its ability to elicit neutralizing antibodies by stabilizing the structure of the gp120 fragment immunogen with additional disulfides, and by displaying it on the surface of simple virus-like particles (VLPs) to present the epitope in polyvalent fashion [14–17]. The design, production, and immunogenic properties of these constructs are described here.
Foreign polypeptide antigens can be incorporated into VLPs to create “chimeric” structures, either as fusions to either end of the VLP capsid (coat) protein (CP) or as additions to well-presented loops [18]. This kind of direct fusion to the coat is usually good for small peptides, however, larger proteins may not be properly folded. In such cases, chemical conjugation to the VLP surface, usually through reactive amino or thiol groups, can be a better option [19, 20] as the folding of the coupled antigen is not dependent on how the VLP monomer folds and the site of attachment can be suitably engineered so that it does not cover important epitopes. The platform used here, derived from bacteriophage QP is a 133 amino acid CP (14.3 kDa), 180 copies of which self-assemble into icosahedral VLPs when expressed in E. coli. There are four externally-exposed amino groups on each subunit, which are available for conjugation to foreign antigens. Engineered versions of the Qβ CP have been used as vaccine candidates, with variable efficacy but consistently high safety [21, 22].
In this work, the gp120 fragment proteins were displayed either by conjugation of the purified protein on the surface of the VLPs using click chemistry, or by direct display as a fusion to the coat protein of the phage. When used for rabbit immunization studies (with and without adjuvant), these protein-particle structures elicited high priming immunogen specific titers. Significant anti-gpl20 titers were observed only after boosting animals with gpl20. However, the resulting sera primarily induced a Tier 1 neutralization response. Weak, sporadic Tier 2 neutralization was seen in antisera raised by VLP’s displaying b122a1-b.
MATERIALS AND METHODS
Brief descriptions are below. More information is provided in supplementary materials.
Protein Purification and Characterization
The K383F mutation was introduced in the background of b122a, referred to here as b122a1. Cysteine mutations were introduced in the background of b122a1. b122a1 with 293–448 disulfide bond is referred to as b122a1-b. For conjugation to Qβ virus like particles, the free cysteine in b122a was introduced as an insertion in the pET15b plasmid encoded sequence after residue 19 (b122a-19iC). Proteins were expressed and purified from E. coli BL21(DE3) cells essentially as described previously [13]. The ODEcprotein was expressed and purified as described previously [11]. Biophysical characterization was done by Far-UV CD, Fluorescence Spectroscopy, Gel Filtration, Iodoacetamide labeling and Reverse phase HPLC.
Genetic Screen to assess in vivo stability of b122a and b122a1-b
b122a and b122a1-b were PCR amplified from the pET 15b vector and cloned in pBR322bla-link* vector in between residues 196 and 197 of β-lactamase gene (bla) using BamHl and Xhol restriction sites and henceforth referred to as pBR.322bla-link*bl 22a and pBR322bla-Iink*b122al-b respectively. This plasmid is a derivative of pBR322 plasmid where a 33 residue Gly-Ser-Ala linker was introduced between residues 196 and 197 of the bla gene. The fusion proteins were monitored for antibiotic resistance as described previously [23]. Briefly, 1 ml of mid log phase cells were taken and the A6oo was adjusted to 1.0. The cells were serially diluted 10 fold in 170 mM NaCl under sterile conditions. 2 μl of each dilution was spotted onto LB Agar supplemented with different concentrations of PenG. Plates were incubated at 37°C for 18–20 hours.
Binding affinity studies using surface plasmon resonance
All binding studies were performed with a Biacore 2000 (Biacore, Uppsala, Sweden) optical biosensor at 25°C as described previously [11]. Four different concentrations of gpl20, b122a, b122a1, ODEc or the disulfide and cysteine insertion mutants were passed across each sensor surface in a running buffer of PBS, pH 7.4, containing 0.005% P20 surfactant. The kinetic parameters were obtained by fitting the data to the simple 1:1 Langmuir interaction model using BIA EVALUATION 3.1 software.
Qβ particles displaying fragment immunogens as coat protein extensions.
b122a, ODEc, and b122a1-b were individually cloned immediately downstream of the Qβ coat protein gene with an 8 amino acid hydrophilic spacer separating the two domains. Particles were purified and characterized as previously described [16], the latter was carried out by FPLC size exclusion chromatography on Superose 6, dynamic light scattering (DLS) (Wyatt Dynapro), and microfluidic gel electrophoresis (Agilent Bioanalyzer 2100, Series II Protein 80 chips). The number of extended coat proteins per particle was determined by integration of the electrophoretic bands in the Bioanalyzer software (Table S2).
Synthetic Manipulations of Virus-Like Particles-Synthesis of Qβ-b122a
The alkyne-bearing Qβ particle was prepared as described previously (Figure 2A) [24]. Azide-functionalized b122a-19iC was generated by reacting the engineered free Cys with an azido-modified succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) linker. A solution of b122a-19iC-azide was reacted with Qβ-alkyne particles under ligand-accelerated copper catalysed azide-alkyne cycloaddition conditions [25]. The reaction was allowed to proceed for 2 hours at room temperature and the product was purified using a 10–40% sucrose density gradient. The purified protein was resuspended in phosphate-buffered saline (PBS) and characterized using DLS, FPLC, and microfluidic protein quantification.
Figure 2:
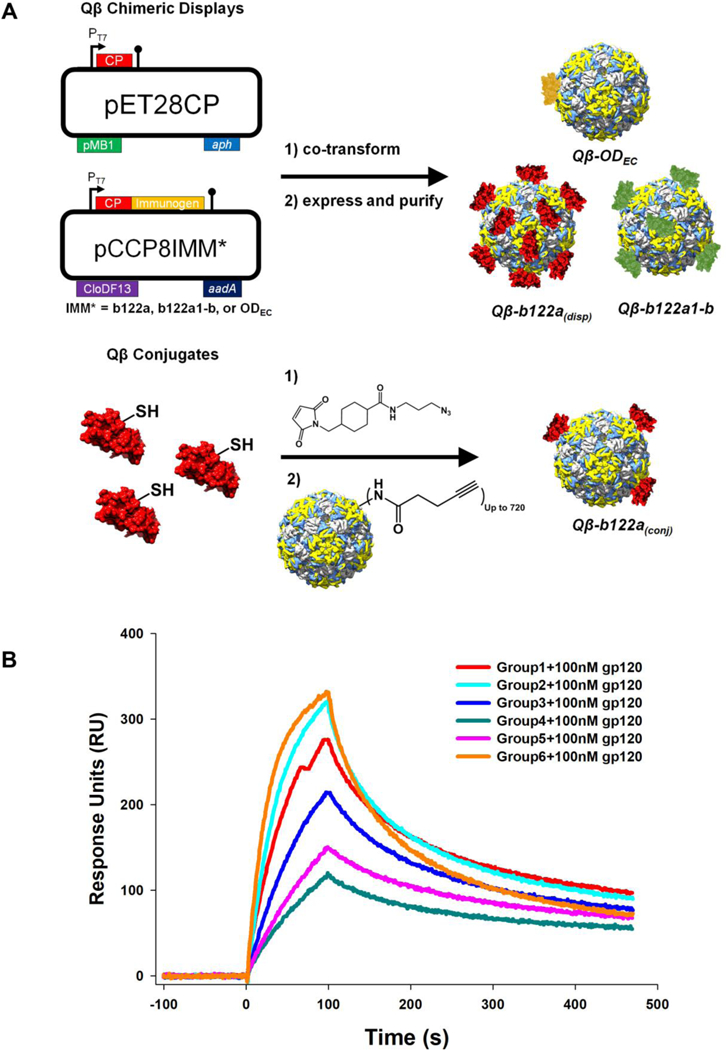
Generation of VLP-based immunogens and competition binding studies of corresponding sera. (A) Schematic representation of particle design and production. Top, plasmids used for chimeric VLP display; bottom, chemical conjugation to display b122a. (B) Competition assay between sera and monoclonal antibody b12 for binding to CD4bs on gp120. Binding was monitored by surface plasmon resonance. Pre-binding of gp120 to sera from groups 4 and 5 results in reduced binding to b12, compared to other groups indicating presence of substantial amounts of CD4bs directed antibodies in the former two groups. Sera from all groups were normalized for gp120 titers.
Immunization studies in rabbits
This study C0913–11 was performed at Covance, Inc., Denver, PA. The facility is iAAALAC accredited and all work was approved by the Covance Institutional Animal Care and Use Committee.
Six groups, each having four rabbits (New Zealand White, female, 2 months old) were used for immunization studies. All rabbits were injected intramuscularly with 50μg of the VLP/protein immunogen. Priming was done at weeks 0 and 4 and boosts were given at weeks 12 and 20. Four weeks following the last boost, the rabbits were terminated. Serum samples were collected at weeks 0 and two weeks post each immunization, heat inactivated and stored in aliquots for further analysis.
Determination of Serum Antibody Titers
Antibody titers against test immunogens were determined by ELISA. Antibody titer was defined as the reciprocal of the highest dilution that gave an OD value above the mean plus two standard deviations of control wells.
Neutralization Assay
Neutralizing antibody activity in sera was measured in a standardized in vitro assay in TZM-bl cells using Tat-regulated luciferase reporter gene expression to quantify reductions in virus infection in TZM-bl cells. Assays were carried out with Env-pseudotyped viruses as described previously [26]. Neutralization titers (ID50) was calculated as the sample dilution at which relative luminescence units (RLU) were reduced by 50% compared to RLU in virus control wells after subtraction of background RLU in cell control wells.
Competition experiments
Competition ELISA between the week 22 sera and antibodies bl2, VRC01, VRC-PG04, FI05 and PGT128 was carried out as described elsewhere [27]. Briefly, 96-well plates (Nunc MaxiSorp) were coated with 300 ng of gpl20 in 50 μL PBS and kept overnight at 4 °C. Plates were then washed with PBST and blocked for 1 h with PBSB. 50 μL of week 22 sera was added to each well starting at a 1:50 dilution followed by a threefold serial dilution in PBSB. As a negative control, pre-immune sera from each group was used starting at a 1:50 dilution followed by a threefold serial dilution in PBSB. After 2 h of incubation, the plates were washed with PBST. The competing antibody was then added to each well at a fixed concentration (10 times EC50, as determined from the titration curve of the antibodies with gpl20) of lμg/ml for bl2 and F105, 2μg/ml for both VRC01 and PGT 128, 4μg/ml for VRC-PG04. After 2 h of incubation with competing antibody, the plates were washed with PBST. The wells were then probed with 50 μL of ALP-conjugated goat anti-human antibody (Sigma) at a predetermined dilution (1:10,000) to detect the bound competing antibody. The plates were washed and developed using the chromogenic substrate p-nitrophenyl phosphate (Sigma). The optical density was measured at 405 nm (SPECTRAmax Plus 384L; Molecular Devices). The percent competition was calculated as follows: % competition = [(PI-W22)/PI] x 100, where PI is the signal of competing antibody binding to gpl20 in the presence of pre-immune sera and W22 is the binding signal of competing antibody to gpl20 in the presence of week 22 sera for that group. All SPR competition experiments were carried out on a BioRad ProteOn instrument. Approximately 6000 RUs of monoclonal antibody bl2 were immobilized on the GLC chip surface. The week 22 serum from all groups was normalized for gpl20 titers and was incubated with lOOnM of JRFL gpl20 for 30 min at room temperature. JRFL gpl20 alone or with the pre-incubated serum was passed over the surface.
RESULTS
b122al
The b122a construct had a lysine residue that was buried in the corresponding gpl20 structure. As buried charges are destabilizing, a mutant was made where this residue was converted to phenylalanine. This mutant is referred to as b122al (Table S3). The protein was soluble and the CD spectrum showed slightly improved secondary structure compared to the parent construct b122a (Figure 1C). The intrinsic fluorescence spectrum showed a red shift upon denaturation (Figure S2A). From gel filtration studies, it appeared to be a monomer (Figure 1A, 1B). In reverse-phase HPLC studies, it eluted primarily as a single major species under both oxidized and reduced conditions. (Figure S2B). The KD for binding of b122a1 to b12, was 2.4 μΜ (Figure S3B and Table 1) exhibiting approximately 6 times greater affinity than b122a.
Figure 1:
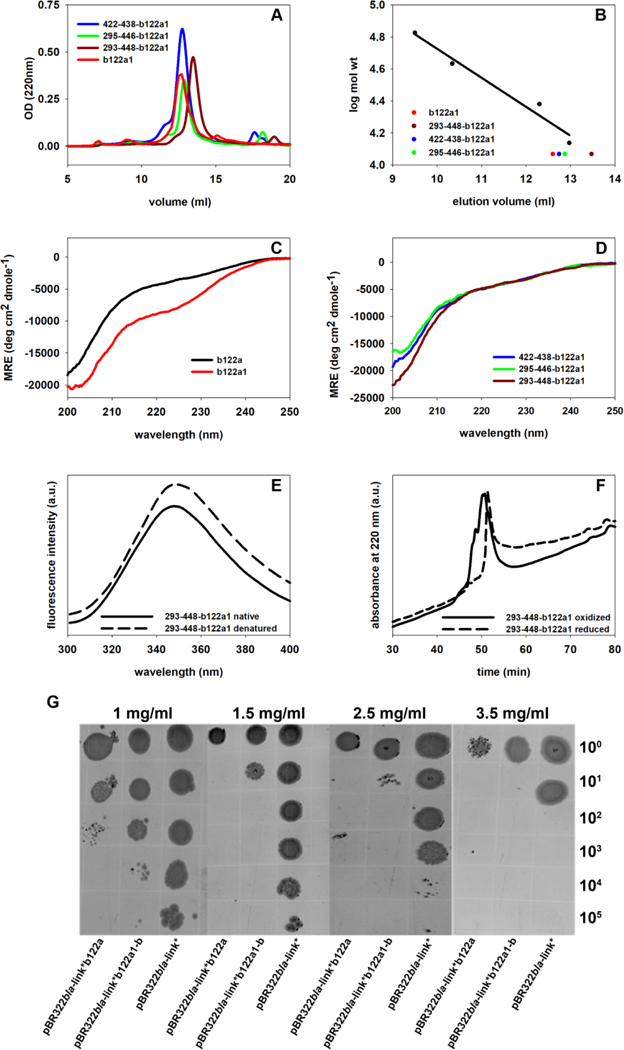
In Vitro and in vivo characterization of designed immunogens. (A) Comparison of the analytical gel filtration elution profiles of b122a1, 422–438-b122a1, 295–446-b122a1 and 293– 448-b122a1 (b122a1-b). The absorbance at 220nm is shown as a function of the elution volume. (B) Calibration curve for Superdex-75 column with standard proteins. The positions of b122a1 and the three disulfide mutants are shown in different colored symbols. The 293 −448-b122a1 (b122a1-b) mutant has the lowest elution volume and is therefore the most compact. (C) Overlay of the CD spectra of b122a and b122a1. The latter shows a more pronounced dip in the range of 220–230nm, indicating increased secondary structure. (D) Overlay of the CD spectra of 422–438, 295–446 and 293–448 disulfide mutants of b122a1. (E) The intrinsic fluorescence emission spectra of native and guanidine hydrochloride denatured 293–448-b122a1 (b122a1-b). (F) Reverse phase HPLC profiles for 293–448-b122a1 (b122a1-b) in both oxidized and reduced condition. The oxidized protein eluted at a lower acetonitrile concentration than the reduced one, indicating that is more compact. (G) Increased in vivo stability of b122a1-b compared to b122a. Cells transformed with the indicated constructs were grown to mid log phase, and the A600 was adjusted to 1.0 in 170 mM NaCl. Five 10-fold dilutions were made for each construct. 2μl of each dilution was spotted on LB agar supplemented with 1.0 mg/ml, 1.5 mg/ml, 2.5 mg/ml and 3.5 mg/ml of PenG. At all concentrations of PenG, pBR322bla-link* b122a1-b show growth at a higher dilution than pBR322bla-link* b122a, indicating increased antibiotic resistance and thus implying increased stability of b122a1-b relative to b122a.
Table 1:
SPR derived kinetic parameters for binding of gp120 and disulfide mutants of b12 binding site constructs to IgG b12
| Protein | b12 |
||
|---|---|---|---|
| kon (M−1 s−1) | koff (s−1) | KD (μΜ) | |
| gp120 | 1.4*105 | 5.2*10−3 | 0.036 ± 0.002 |
| b122a | 5.3*102 | 7.5*10−3 | 14.2 ± 1.4 |
| b122a1a | 4.0*103 | 9.6*10−3 | 2.4 ± 1.1 |
| b122a1–422C-438C | 8.8*102 | 2*10−2 | 22.7 ± 3.3 |
| b122a1–295C-446C | 6.4*102 | 1.3*10−2 | 20.3 ± 1.3 |
| b122a1-bb (293C-448C) | 1.2*104 | 6.3*10−3 | 0.5 ± 0.04 |
| b122a-19iC | 1.9 * 103 | 2.2 * 10−2 | 11.5 ± 2.2 |
b122a with K383F mutation
b122a with K383F, 293C-448C mutations
b122a1 disulfide mutants
In an attempt to impart structural rigidity and further increase the stability of b122a1, disulfides were introduced in various regions of the molecule [28, 29]. The possible disulfide positions were chosen based on the high-resolution structure of b12 bound to gp120 (2NY7), which was used as input for the program MODIP (Modeling Disulfide bridges in Proteins) [30, 31]. The list of these positions is given in Supplementary Table S1; some were not suitable because they were located at hydrogen bonding positions of anti-parallel beta strands or were in close proximity to the b12 binding site (Table S1). In addition, several other exposed sites on anti-parallel beta strands were chosen by visual inspection, inspired by a study showing most naturally occurring cross-strand disulfides in anti-parallel beta-strands to be located at non-hydrogen bonded positions [32]. The positions finally chosen were 422–438, 295–446, and 293–448, indicating the amino acids substituted with cysteine to form disulfide bonds between them (Figure S1B) in the background of b122a1. These mutant proteins were produced and purified from the soluble fraction by the above procedure, giving a typical yield of approximately 20 mg per liter. No increase in protein mass was observed upon iodoacetamide treatment in the absence of reducing agent, confirming the spontaneous formation of disulfide linkages for these polypeptides. The same treatment after disulfide reduction showed the incorporation of eight iodoacetamide molecules per protein. However, far UV CD (Figure 1D) measurements showed that all the mutants had structure similar to b122al indicating that all the proteins were only partially folded. All the three proteins showed a small red shift upon denaturation, conforming the presence of a partially folded conformation prior to denaturation (Figure 1E, S2C, S2E). By this measure, the introduction of disulfide bonds did not lead to a substantial gain of protein secondary structure.
The proteins were also analyzed for their oligomeric state using gel filtration on a Superdex-75 column (Figure 1A). Comparison to a calibration curve (Figure 1B) showed all the disulfide mutants to have higher elution volumes than the parent construct, indicating that they are more compact. Furthermore, mixtures of peaks for the oxidized proteins were observed on reversed phase chromatography, indicating that they consist of more than one combination of disulfide bonded isomers (Figure 1F, S2D, S2F). In contrast, the reduced mutant proteins eluted as a single peak and at a higher acetonitrile concentration than the oxidized protein. Disulfide mutants 422–438 and 295–446 in the background of b122al were found to exhibit no improvement in binding to immobilized bl2 IgG by SPR (Figure S3C, S3D and Table 1) compared to b122al. However, the 293–448 disulfide mutant in the background of b122al bound to bl2 about five times better than b122al (Figure S3E and Table 1); this construct is referred to as b122al-b (Table S3).
The b122al-b protein was found to be significantly more stable in E. coli cells than the parent b122a protein, as assessed by the split β-lactamase assay [23]. Thus, cells containing pBR322bla-link*b122a, pBR322bla-link*b122al-b, and wild type β-lactamase (see Experimental section for descriptions) were spotted on LB agar containing different concentrations of penicillin G ranging from 1 mg/ml to 3.5 mg/ml. Figure 1G shows that pBR322bla-link*b122al-b confers higher antibiotic resistance than pBR322bla-link*b122a, suggesting greater stability of the folded structure owing to introduction of an additional disulfide.
Cysteine Insertion Mutant of b122a
For conjugation to Qβ virus like particles, the free cysteine in b122a was introduced as an insertion in the pET15b plasmid encoded sequence after residue 19. This construct is referred to as b122a-19iC (Table S3). Protein b122a-19iC was purified from the soluble fraction of the E. coli BL21(DE3) cell lysate. The protein was about 90% pure, and the identity was confirmed by mass spectrometry. The far-UV CD spectrum of the protein was similar to the b122a construct (Figure S4A) indicating that introduction of the single cysteine close to the N-terminus did not perturb the protein structure. Gel filtration studies confirmed that the protein eluted at the same volume as the wild-type protein (Figure S4B), indicating that it is not aggregated. Iodoacetamide labeling studies under native conditions, confirmed the presence of one free cysteine. This mutant bound to bl2 IgG [33] with KD in the range of 8–12 μΜ (Figure S4C and Table 1), similar to b 122a.
Immunization and neutralization studies
Rabbit immunization studies were carried out with fragment immunogens b122a and b122al-b displayed on Qβ virus like particles (Table S4). In addition, the previously described construct ODEc, consisting of residues 255–474 of the gpl20 outer domain [11] was displayed on the surface of Qβ virus like particles as a fusion to the coat protein (Figure 2A). A prime-boost rabbit immunization study was employed as described previously [13] which involved priming with the b122a/ODEc/b122al-b protein fragments displayed on the particles at weeks 0,4 and boosting with full-length gp120 at weeks 12, 20. Sera were collected at two weeks after each immunization. Briefly, we hypothesized that the designed fragments may not adopt exactly the same conformation as the corresponding regions in the whole molecule, in the absence of antibody b12/VRC01. Hence boosting with gp120 might elicit gp120 cross-reactive antibodies some of which could be targeted to the broadly neutralizing antibody epitopes that are present in the priming immunogen.
Group 1 was primed with b122a conjugated to the particles. To test the difference between the display strategies, group 2 rabbits were primed with b122a fragment expressed as a fusion to the coat protein on the surface of the particles and compared to group1. Our earlier immunization studies with soluble ODEC showed modest neutralization [11]. In an attempt to improve the immune response, group 3 was primed with outer domain based construct ODEC displayed on particles as a fusion to the coat protein. Group 4 was primed with the b122a1-b displayed on particles as a fusion to coat protein. We expect to see a better immune response generated by this protein as it showed better biophysical properties in terms of stability as well as significantly improved binding to antibody b12 when compared to b122a. VLPs can generally elicit a high immune response without adjuvant. However, to see if adjuvant can further enhance titers, group 5 rabbits were primed with b122a conjugated to the particles, administered with the adjuvant and compared to group 1. Group 6 animals were primed with empty particles. All groups were boosted with full length gp120 in the presence of adjuvant.
Week 2 and week 4 ELISA titers obtained after particle immunizations, did not have any significant cross-reactivity to gp120. After one boost with full-length gp120, at least one rabbit from all groups showed weak gp120 titers (~400), except group 6 which received empty particles for priming. After two boosts, the week 22 sera showed good gp120 ELISA titers in the range of 105 (Table 2). Comparable gp120 and b122a
Table 2:
ELISA titers of sera from all groups for study C0913–11 against either gpl20 or the priming immunogen. ‘W’ refers to the week number. Following priming at weeks 0 and 4, all animals were boosted with JRFL gp120 at weeks 12 and 20.
| Group | Group Details | Animal ID | W2 | W6 | W14 | W22 | W24 Terminal | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gp120 | ab122a/ODEc | gpl20 | ab122a/ODEc | gpl20 | ab122a/ODEc | gpl20 | ab122a/ODEc | gpl20 | ab122a/ODEc | |||
| 1 | particles conjugated to b122a-19iCfor prime, full length JRFL gpl20 for boost | #874 | <100 | <100 | <100 | 6400 | 100 | 400 | 102400 | 400 | 51200 | 1600 |
| #875 | <100 | <100 | <100 | 400 | 400 | <100 | 102400 | <100 | 51200 | <100 | ||
| #876 | <100 | <100 | <100 | 1600 | 400 | 400 | 51200 | <100 | 12800 | <100 | ||
| #877 | <100 | <100 | 100 | 400 | 100 | <100 | 204800 | <100 | 51200 | <100 | ||
| 2 | particles displaying b122a for prime, full length JRFL gpl20 for boost | #878 | <100 | 1600 | 100 | 6400 | 100 | 1600 | 25600 | 1600 | 12800 | 1600 |
| #879 | <100 | 6400 | <100 | 6400 | 100 | 1600 | 51200 | 400 | 51200 | 400 | ||
| #880 | <100 | 1600 | <100 | 1600 | 400 | <100 | 51200 | <100 | 25600 | <100 | ||
| #881 | <100 | 1600 | <100 | 1600 | 100 | 400 | 25600 | 400 | 12800 | 400 | ||
| 3 | particles displaying ODEc for prime, full length JRFL gpl20 for boost | #882 | <100 | <100 | <100 | <100 | 400 | <100 | 51200 | <100 | 25600 | <100 |
| #883 | <100 | <100 | <100 | 400 | 100 | <100 | 102400 | <100 | 51200 | <100 | ||
| #884 | <100 | <100 | <100 | <100 | 100 | <100 | 51200 | <100 | 25600 | <100 | ||
| #885 | <100 | <100 | <100 | <100 | 100 | <100 | 102400 | <100 | 25600 | <100 | ||
| 4 | particles displaying b122al-b for prime, full length JRFL gpl20 for boost | #886 | <100 | 400 | <100 | 1600 | 1600 | <100 | 409600 | <100 | 102400 | <100 |
| #887 | <100 | 6400 | <100 | 25600 | 100 | 1600 | 102400 | 1600 | 25600 | 400 | ||
| #888 | <100 | 400 | <100 | 400 | <100 | 400 | 102400 | 400 | 25600 | 400 | ||
| #889 | <100 | 1600 | <100 | 1600 | 400 | <100 | 51200 | <100 | 25600 | <100 | ||
| 5 | particles conjugated to b122a-19iC(ADJUPLEX) for prime. full length JRFL gpl20 for boost | #890 | 100 | 1600 | 100 | 409600 | 400 | 25600 | 204800 | 25600 | 51200 | 25600 |
| #891 | <100 | 1600 | <100 | 409600 | <100 | 25600 | 102400 | 25600 | 51200 | 6400 | ||
| #892 | <100 | 1600 | <100 | 102400 | 100 | 1600 | 102400 | 1600 | 25600 | 1600 | ||
| #893 | <100 | <100 | <100 | 409600 | 400 | 25600 | 102400 | 25600 | 25600 | 6400 | ||
| 6 | empty particles for prime, full length JRFL gp120 for boost | #894 | <100 | <100 | <100 | <100 | 100 | <100 | 25600 | <100 | 6400 | <100 |
| #895 | <100 | <100 | 100 | <100 | 100 | <100 | 51200 | <100 | 25600 | <100 | ||
| #896 | <100 | <100 | <100 | <100 | <100 | <100 | 51200 | <100 | 25600 | <100 | ||
| #897 | <100 | <100 | <100 | <100 | 100 | <100 | 51200 | <100 | 25600 | <100 | ||
Priming Immunogen titers were determined using b122a for Groups 1,2,4,5,6; ODEc for Group3, 6. For Group 6, titers against both b122a and ODEc were less than 100
ELISA titers were seen for group 1 and group 2 showing that the display strategy (genetic fusion vs. covalent coupling) did not contribute to immunogenicity of the particles. The mean gp120 titers for group 1 and group 5 showed a very small difference but b122a titers were significantly increased for group 5 which persisted even after 2 boosts with gp120 showing the presence of antibodies with higher cross reactivity to the b12 epitope. This was also reflected in the neutralization ID50 values (Table 3), where group 5 sera showed significantly higher titers when compared to group 1 sera both against subtype B and subtype C tier 1 pseudoviruses.
Table 3:
Neutralization ID50 values obtained with sera from wk0 and and wk22 in a standard TZMbl assay. Pre immune sera failed to neutralize any of the tested viruses.
| Group | Group details | Animal ID | Bleed Week | Neutralization ID50 | ||||
|---|---|---|---|---|---|---|---|---|
| SVA-MLV Neg. Ctrl. | MN.3 Clade B Tier 1 | JR-FL Clade B Tier 2 | SF162.LS Clade B Tier 1 | MW965.26 Clade C Tier 1 | ||||
| 1 | particles conjugated to b122a-19iC for prime, full length JRFL gp120 for boost | #874 | 22 | 12 | 118 | <10 | 62 | 23 |
| #875 | 22 | <10 | 13 | <10 | 39 | <10 | ||
| #876 | 22 | <10 | 16 | <10 | 11 | 11 | ||
| #877 | 22 | <10 | 53 | <10 | 10 | 13 | ||
| 2 | particles displaying b122a for prime, full length JRFL gp120 for boost | #878 | 22 | 14 | 15 | 11 | <10 | 15 |
| #879 | 22 | <10 | 29 | <10 | 14 | 20 | ||
| #880 | 22 | <10 | 28 | <10 | 18 | <10 | ||
| #881 | 22 | 12 | 11 | <10 | <10 | 10 | ||
| 3 | particles displaying ODec for prime, full length JRFL gp120 for boost | #882 | 22 | <10 | 4502 | <10 | 179 | 23 |
| #883 | 22 | <10 | 21 | <10 | 11 | 11 | ||
| #884 | 22 | <10 | 56 | <10 | <10 | 12 | ||
| #885 | 22 | 11 | 12 | <10 | 830 | 64 | ||
| 4 | particles displaying b122a1-b for prime, full length JRFL gp120 for boost | #886 | 22 | 12 | 351 | <10 | 1149 | 108 |
| #887 | 22 | <10 | 427 | <10 | 74 | 24 | ||
| #888 | 22 | 12 | 121 | 40 | 615 | 576 | ||
| #889 | 22 | 14 | 19 | 20 | 17 | <10 | ||
| 5 | particles conjugated to b122a-19iC (ADJUPLEX) for prime, full length JRFL gp120 for boost | #890 | 22 | <10 | 376 | <10 | 238 | 187 |
| #891 | 22 | <10 | 737 | <10 | 35 | 40 | ||
| #892 | 22 | <10 | 49 | <10 | 18 | 152 | ||
| #893 | 22 | 11 | 14 | <10 | 764 | 1111 | ||
| 6 | empty particles for prime, full length JRFL gp120 for boost | #894 | 22 | <10 | 10 | <10 | <10 | <10 |
| #895 | 22 | <10 | 17 | <10 | 45 | <10 | ||
| #896 | 22 | <10 | 17 | <10 | <10 | 23 | ||
| #897 | 22 | <10 | 20 | <10 | 19 | <10 | ||
The mean gp120 titer for the week 22 sera was highest for group 4, which received particles displaying a stabilized disulfide mutant of b122a1 (293–448) as the priming immunogen. Priming immunogen ELISA titers were high for group 4 when compared to other groups except group 5 (Table 2). This could be possibly due to use of b122a instead of b122a1-b to measure priming immunogen titers. Neutralization titers against a panel of four HIV-1 pseudoviruses were also highest for group 4. Two of four animals from Group 4 also showed weak neutralization of Tier 2 subtype B JRFL strain (Table 3). ELISA titers and neutralization titers were lowest for group 6 (primed with empty particles) showing the specificity of the immune response generated against our molecules.
To test for the presence of CD4bs antibodies, competition experiments were carried out using SPR to probe for competition between b12 antibody and the week 22 sera from all groups, for the CD4bs on gp120. gp120 binding to b12 was measured in the presence and absence of sera. Addition of sera containing CD4bs/b12-like antibodies should result in reduction of gp120 binding to b12. However an increase in binding was observed (Figure 2B) presumably because antibodies to other binding sites on gp120 would also have been elicited following the boosts with gp120. Such antibodies can be expected to bind to gp120 and the entire complex will then bind to b12 on the chip surface, thus showing an increase in RU in SPR. Sera eliciting the highest titer of CD4bs antibodies are expected to show the lowest increase. Consistent with ELISA and neutralization data described above, gp120 incubated with group 4 sera show the least increase upon binding to b12 followed by group 5 sera indicating the presence of a higher fraction of CD4bs directed antibodies than other groups. Group 6 sera showed the highest increase in binding, indicating the lack of a CD4bs focused response, consistent with the absence of a CD4bs focused primary immunogen in this group (Figure 2B). To further characterize the antisera, competition ELISAs were carried out to probe for competition between various CD4bs antibodies and the week 22 sera from all groups, for the CD4bs on gp120. Table 4 lists percent competition at 1:50 dilution of week 22 sera. As a negative control, competition was carried out with the V3 epitope targeting broadly neutralizing antibody PGT128. None of the sera showed any significant competition (<15%). Group 4 sera show the highest competition with antibody b12 compared to other groups consistent with ELISA (Table 2) and neutralization data (Table 3). This was observed for competition experiments performed with SPR also (Figure 2B). We also performed competition with other CD4bs antibodies like VRC01, VRC-PG04 and F105. For both VRC01 and VRC-PG04 all groups show very little competition. It is known that residues in loop D and variable loop five are part of the VRC01 but not the b12 epitope and hence are not present in b122a based immunogens [34]. From Figure S1A it is clear that b122a contains a small part of the VRC01 epitope and so its lack of VRC01 binding is expected. Consistent with this, sera raised against b122a immunogens do not compete with VRC01 and VRC-PG04. ODEc does contain the VRC01 epitope but the sera did not compete well with VRC01 possibly due to low valency display of this antigen on the VLP. Competition studies were also performed with the CD4bs non-neutralizing antibody F105 whose epitope significantly overlaps with bl2 [35, 36]. The data show that Group 4 sera which show the highest competition with bl2, also compete with FI05.
Table 4:
Sera competition binding studies using ELISA.
| Percent competition at 1:50 dilution of week 22 seraa | |||||
|---|---|---|---|---|---|
| b12 | F105 | VRC-PG04 | VRC01 | PGT128 | |
| Group1 | 10±2 | 38±1 | 9±2 | 14±2 | 2±1 |
| Group2 | 12±0.5 | 26±2 | 14±1 | 12±1 | 4±2 |
| Group3 | 17±2 | 28±2 | 22±1 | 11±1 | 14±1 |
| Group4 | 42±2b | 57±1b | 26±2 | 21±1 | 15±1 |
| Group5 | 21±2 | 29±1 | 26±1 | 18±2 | 14±1 |
| Group6 | 19±0.5 | 18 | 14±1 | 18±1 | 4±1 |
Data shown represent percent competition from two independent experiments and are represented as mean±SE.
Percent competition for Group 4 sera with antibodies b12 and F105 is significantly higher compared to other groups, P<0.01 (individual means compared by Welch’s t-test).
DISCUSSION
In many cases, protective immunity against various viral and bacterial diseases has been achieved using live attenuated vaccines, because they closely mimic natural infection. However, for certain infections like HIV, live attenuated viruses are not considered safe, both because of viral integration into the chromosome and because of mutations that can reactivate the virus. In such cases, virus like particles or VLPs are an attractive alternative, as they resemble intact virions in terms of size and presentation of antigens, but are non-pathogenic, non-replicative and hence are safe for administration. Chimeric VLPs are an efficient way to display foreign epitopes in multivalent form and are able to elicit a robust immune response without adjuvant [37]. In this study, we displayed various designed gpl20 fragment proteins on the surface of Qβ virus like particles. We improved the stability and bl2 affinity of our previously described fragment immunogen b122a [13] by incorporating the K383F mutation with additional disulfides. Construct b122al-b resulted in the largest improvement in affinity to bl2. No significant improvement was observed in the secondary structure of the protein as assessed by CD spectroscopy, but increased in vivo stability was seen in a β-lactamase activity assay [23]. For b122a-19iC conjugated particles, the final gpl20 ELISA titers were the same both in absence and presence of adjuvant but increased titers against priming immunogen and neutralization was seen with the latter. This indicated that the adjuvant increased the immunogenicity of the particles. Sera from group 4 primed with the stabilized protein b122al-b showed higher neutralization activity against Tier 1 viruses and also showed weak neutralization of the Tier 2, subtype B JRFL strain. Sera from the group primed with empty particles, showed low gpl20 and priming immunogen titers, indicating that priming with the fragment immunogens is beneficial. Competition assays using both SPR and ELISA showed an increased CD4 binding site directed response in week 22 sera from group primed with Qβ VLPs displaying b122al-b compared to other groups (Table 4, Figure 2B). Collectively the data demonstrate that priming with a CD4bs directed fragment immunogen results in some degree of immune focusing to the CD4bs and improving immunogen stability and bl2 binding affinity resulted in enhanced elicitation of neutralizing antibodies. However, a more native-like boosting immunogen than gpl20 is likely required to elicit a higher proportion of neutralizing antibodies.
Various studies attempting to focus the immune response to a particular epitope on HIV-1 Env have been carried out. Most of the immunogens described in these studies are based on the outer domain of gpl20. An OD construct (OD1) containing residues, 252–482 from YU2 strain of gpl20 was expressed in Drosophila S2 cells. This construct was glycosylated and also retained VIV2 and V3 variable loops, but the sera obtained after rabbit immunizations did not neutralize homologous YU2 virus [38]. In another study, mice were immunized with clade C OD as a fusion to human IgGl Fc domain, but failed to elicit any neutralizing response. [39, 40]. Another study described two OD immunogens (monomeric and trimeric) based on group M consensus sequence. The immunogens were able to induce heterologous Tier 1 neutralizing responses although sera immunized with monomeric gpl20 showed higher neutralization titers for all viruses [41]. The OD3 construct based on subtype C strain 1084i having stabilizing disulfides and a cavity filling mutation was able to elicit a Tier 1 and a weak Tier 2 neutralizing response in guinea pig immunization studies [42], In the present work we were able to show elicitation of a strong cross-clade Tier 1 response with Qβ VLPs displaying either ODEc or the stabilized b122al-b even in the absence of adjuvant. b122a conjugated to Qβ VLP’s elicited a similar response when immunized in the presence of adjuvant. Sera elicited by Qβ VLP’s displaying the stabilized construct, b122al-b, also elicited a weak Tier 2 response. One possible reason we did not see a better elicitation of antibodies that could neutralize multiple Tier 2 viruses in case of b122a VLP’s is that the displayed antigens might not have been conformationally intact. Another reason could be the fact that the valency of antigens displayed on the virus like particles (Table S2) was lower than expected, resulting in reduced immunogenicity. Boosting with more native like trimeric gpl40 [43] or trimeric gpl20 [44, 45] instead of monomeric gpl20 could prove beneficial, and can be used in future immunization studies to improve elicitation of neutralizing antibodies. VLP’s are known to enhance cell mediated responses. In this study we focused only on humoral responses and hence cannot comment on elicitation of T cell responses, which have been shown to substantially contribute to control of HIV-1 infection.
Supplementary Material
Highlights.
gpl20 fragments with enhanced stability, affinity to CD4bs antibody bl2 were designed.
Stabilized fragments were displayed on Qβ VLP’s and used in rabbit immunizations.
The most stable fragment elicited cross clade Tier 1 and weak Tier 2 neutralization.
Priming with the fragments resulted in an epitope focused response to the CD4bs.
Acknowledgements
Monoclonal antibodies b12 and VRC01 were obtained from the Neutralizing Antibody Consortium of IAVI. pBR322bla-link* vector to determine in vivo stability of immunogens was kindly provided by Dr. Linda Foit and Dr. James C. A. Bardwell. The assistance of Nonavinakere Seetharam Srilatha is duly acknowledged for the SPR experiments. MP and SB acknowledge the support of Council of Scientific and Industrial Research, Government of India. This work was funded by the International Aids Vaccine Initiative grant (grant number COATOD00107) and a grant from National Institutes of Health (grant number R01AI118366– 01, DT.15/7/2015) to RV. We also acknowledge funding for infrastructural support from the following programs of the Government of India: DST-FIST, UGC Center for Advanced Study, and the DBT-IISc Partnership Program and of a JC Bose Fellowship from DST to RV.
Abbreviations
- CD4bs
CD4 binding site
- OD
outer domain
- Env
envelope glycoprotein
- CP
capsid protein
- VLPs
virus-like particles
- PBS
phosphate buffer saline
- Cys
cysteine
- SMCC
succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate
- RLU
relative luminescence units
- RU
response units
- MODIP
Modeling Disulfide bridges in Proteins
- SPR
surface plasmon resonance
- PBST
phosphate buffer saline with 0.05% Tween-20
- PBSB
phosphate buffer saline with 0.05% Tween-20 and 3% BSA
- ELISA
Enzyme-linked immune sorbent assay
Footnotes
Conflict of Interest Statement
The authors declare that they have no conflicts of interest with the contents of this article
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Robertson DL, et al. , HIV-1 nomenclature proposal. Science, 2000. 288(5463): p. 55–6. [DOI] [PubMed] [Google Scholar]
- 2.Buonaguro L, Tornesello ML, and Buonaguro FM, Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J Virol, 2007. 81(19): p. 10209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalgleish AG, et al. , The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature, 1984. 312(5996): p. 763–7. [DOI] [PubMed] [Google Scholar]
- 4.Chan DC and Kim PS, HIV entry and its inhibition. Cell, 1998. 93(5): p. 681–4. [DOI] [PubMed] [Google Scholar]
- 5.Wyatt R and Sodroski J, The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science, 1998. 280(5371): p. 1884–8. [DOI] [PubMed] [Google Scholar]
- 6.Richman DD, et al. , Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci U S A, 2003. 100(7): p. 4144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mikell I, et al. , Characteristics of the earliest cross-neutralizing antibody response to HIV-1. PLoS Pathog, 2011. 7(1): p. e1001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alter G and Moody MA, The humoral response to HIV-1: new insights, renewed focus. J Infect Dis, 2010. 202 Suppl 2: p. S315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connor RI, et al. , Immunological and virological analyses of persons infected by human immunodeficiency virus type 1 while participating in trials of recombinant gp120 subunit vaccines. J Virol, 1998. 72(2): p. 1552–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leonard CK, et al. , Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem, 1990. 265(18): p. 10373–82. [PubMed] [Google Scholar]
- 11.Bhattacharyya S, et al. , Design of a non-glycosylated outer domain-derived HIV-1 gp120 immunogen that binds to CD4 and induces neutralizing antibodies. J Biol Chem, 2010. 285(35): p. 27100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rathore U, et al. , Glycosylation of the core of the HIV-1 envelope subunit protein gp120 is not required for native trimer formation or viral infectivity. J Biol Chem, 2017. 292(24): p. 10197–10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharyya S, et al. , Design of an Escherichia coli expressed HIV-1 gp120 fragment immunogen that binds to b12 and induces broad and potent neutralizing antibodies. J Biol Chem, 2013. 288(14): p. 9815–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Astronomo RD, et al. , Defining criteria for oligomannose immunogens for HIV using icosahedral virus capsid scaffolds. Chemistry & biology, 2010. 17(4): p. 357–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banerjee D, et al. , Multivalent display and receptor-mediated endocytosis of transferrin on virus-like particles. Chembiochem : a European journal of chemical biology, 2010. 11(9): p. 1273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown SD, Fiedler JD, and Finn MG, Assembly of hybrid bacteriophage Qbeta virus-like particles. Biochemistry, 2009. 48(47): p. 11155–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen Gupta S, et al. , Accelerated bioorthogonal conjugation: a practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconjugate chemistry, 2005. 16(6): p. 1572–9. [DOI] [PubMed] [Google Scholar]
- 18.Pumpens P and Grens E, HBV core particles as a carrier for B cell/T cell epitopes. Intervirology, 2001. 44(2–3): p. 98–114. [DOI] [PubMed] [Google Scholar]
- 19.Chackerian B, Lowy DR, and Schiller JT, Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. The Journal of clinical investigation, 2001. 108(3): p. 415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jegerlehner A, et al. , A molecular assembly system that renders antigens of choice highly repetitive for induction of protective B cell responses. Vaccine, 2002. 20(25–26): p. 3104–12. [DOI] [PubMed] [Google Scholar]
- 21.Maurer P, et al. , A therapeutic vaccine for nicotine dependence: preclinical efficacy, and Phase I safety and immunogenicity. Eur J Immunol, 2005. 35(7): p. 2031–40. [DOI] [PubMed] [Google Scholar]
- 22.Low JG, et al. , Safety and immunogenicity of a virus-like particle pandemic influenza A (H1N1) 2009 vaccine: results from a double-blinded, randomized Phase I clinical trial in healthy Asian volunteers. Vaccine, 2014. 32(39): p. 5041–8. [DOI] [PubMed] [Google Scholar]
- 23.Foit L, et al. , Optimizing protein stability in vivo. Mol Cell, 2009. 36(5): p. 861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pokorski JK, et al. , Functional virus-based polymer-protein nanoparticles by atom transfer radical polymerization. J Am Chem Soc, 2011. 133(24): p. 9242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pokorski JK, Hovlid ML, and Finn MG, Cell targeting with hybrid Qbeta virus-like particles displaying epidermal growth factor. Chembiochem, 2011. 12(16): p. 2441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M, et al. , Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. Journal of virology, 2005. 79(16): p. 10108–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallajosyula VV, et al. , Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc Natl Acad Sci U S A, 2014. 111(25): p. E2514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Creighton TE, Disulphide bonds and protein stability. BioEssays : news and reviews in molecular, cellular and developmental biology, 1988. 8(2): p. 57–63. [DOI] [PubMed] [Google Scholar]
- 29.Harrison PM and Sternberg MJ, Analysis and classification of disulphide connectivity in proteins. The entropic effect of cross-linkage. Journal of molecular biology, 1994. 244(4): p. 448–63. [DOI] [PubMed] [Google Scholar]
- 30.Dani VS, Ramakrishnan C, and Varadarajan R, MODIP revisited: re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein engineering, 2003. 16(3): p. 187–93. [DOI] [PubMed] [Google Scholar]
- 31.Sowdhamini R, et al. , Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis. Protein engineering, 1989. 3(2): p. 95–103. [DOI] [PubMed] [Google Scholar]
- 32.Indu S, et al. , Conformational analysis and design of cross-strand disulfides in antiparallel beta-sheets. Proteins, 2011. 79(1): p. 244–60. [DOI] [PubMed] [Google Scholar]
- 33.Zhou T, et al. , Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature, 2007. 445(7129): p. 732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, et al. , Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J Virol, 2011. 85(17): p. 8954–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, et al. , Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science, 2009. 326(5956): p. 1123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pantophlet R, et al. , Fine mapping of the interaction of neutralizing and nonneutralizing monoclonal antibodies with the CD4 binding site of human immunodeficiency virus type 1 gp120. J Virol, 2003. 77(1): p. 642–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bachmann MF and Jennings GT, Virus-Like Particles: Combining Innate and Adaptive Immunity for Effective Vaccination, in Novel Vaccination Strategies. 2004, Wiley-VCH Verlag GmbH & Co. KGaA. p.415–432. [Google Scholar]
- 38.Yang X, et al. , Characterization of the outer domain of the gp120 glycoprotein from human immunodeficiency virus type 1. J Virol, 2004. 78(23): p. 12975–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H, Xu X, and Jones IM, Immunogenicity of the outer domain of a HIV-1 clade C gp120. Retrovirology, 2007. 4: p. 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H, et al. , Mapping the immune response to the outer domain of a human immunodeficiency virus-1 clade C gp120. J Gen Virol, 2008. 89(Pt 10): p. 2597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin Y, et al. , Eliciting neutralizing antibodies with gp120 outer domain constructs based on M-group consensus sequence. Virology, 2014. 462–463: p. 363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu D, et al. , Tryptophan 375 stabilizes the outer-domain core of gp120 for HIV vaccine immunogen design. Vaccine, 2017. 35(23): p. 3067–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torrents de la Pena, A., et al. , Improving the Immunogenicity of Native-like HIV-1 Envelope Trimers by Hyperstabilization. Cell Rep, 2017. 20(8): p. 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones AT, et al. , A Trimeric HIV-1 Envelope gp120 Immunogen Induces Potent and Broad Anti-V1V2 Loop Antibodies against HIV-1 in Rabbits and Rhesus Macaques. J Virol, 2018. 92(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kesavardhana S, et al. , Structure-based Design of Cyclically Permuted HIV-1 gp120 Trimers That Elicit Neutralizing Antibodies. J Biol Chem, 2017. 292(1): p. 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.