

SUMMARY
Thiazolidinedione drugs (TZDs) target the transcriptional activity of PPARγ to reverse insulin resistance in type 2 diabetes, but side effects limit their clinical use. Here, using human adipose stem cell-derived adipocytes, we demonstrate that single-nucleotide polymorphisms (SNPs) were enriched at sites of patient-specific PPARγ binding, which correlated with the individual-specific effects of TZD rosiglitazone (rosi) on gene expression. Rosi induction of ABCA1, which regulates cholesterol metabolism, was dependent upon SNP rs4743771, which modulated PPARγ binding by influencing the genomic occupancy of its cooperating factor NFIA. Conversion of rs4743771 from the inactive SNP allele to the active one by CRISPR/Cas9-mediated editing rescued PPARγ binding as well as rosi-induction of ABCA1 expression. Moreover, rs4743771 is a major determinant of undesired serum cholesterol increases in rosi-treated diabetics. These data highlight human genetic variation that impacts PPARγ genomic occupancy and patient responses to antidiabetic drugs, with implications for developing personalized therapies for metabolic disorders.
Graphical Abstract
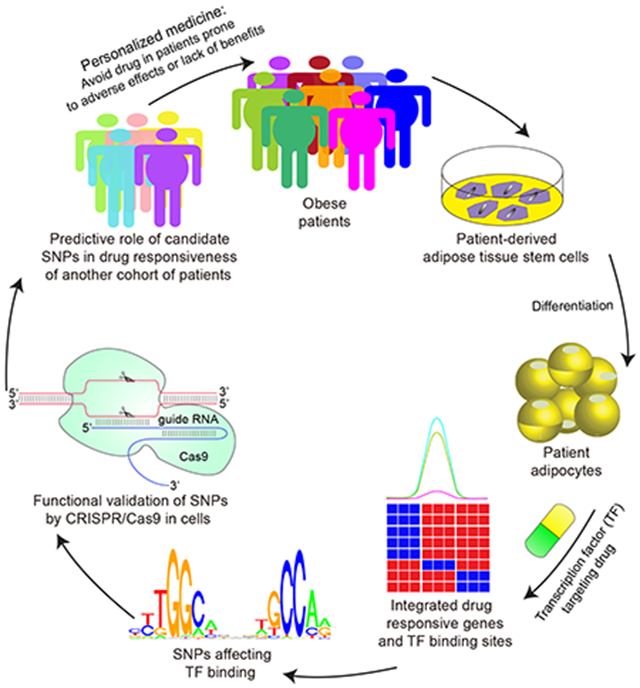
In Brief:
Lazar and colleagues utilize patient adipose stem cell-derived adipocytes to reveal single-nucleotide polymorphisms that modulate the effects of anti-diabetic drugs by controlling genomic binding of PPARγ.
INTRODUCTION
Obesity has reached global epidemic proportions and is a major risk factor for type 2 diabetes (Caballero, 2007). The nuclear receptor Peroxisome Proliferator Activated Receptor γ (PPARγ) is required for adipogenesis and is the target of antidiabetic thiazolidinedione (TZD) drugs (Chawla and Lazar, 1994). TZDs are the only drugs that reverse the insulin resistance central to the pathophysiology of type 2 diabetes and can prevent the development of diabetes and ameliorate cardiovascular complications (Soccio et al., 2014). However, a barrier to progress is that TZDs have notable side effects that limit their routine use, including weight gain, edema, and bone loss (Soccio et al., 2014). Moreover, individuals respond to TZDs differently, such that 20%-30% of diabetic patients fail to respond to TZDs (Sears et al., 2009). Thus, understanding the underlying mechanism(s) driving a differential response to TZDs could inform personalized and precision approaches to the treatment of type 2 diabetes and associated metabolic diseases.
Here, we used patient adipose stem cell (hASC)-derived adipocytes to demonstrate that genetic variation modulates human TZD responses by affecting the genomic occupancy of PPARγ.
RESULTS
hASC-derived adipocytes differentially respond to rosiglitazone treatment
We initially isolated ASCs from subcutaneous adipose tissue of five obese patients (Figure 1A and S1A). hASCs were differentiated to adipocytes in well-defined adipogenic differentiation medium for 2 weeks and then cultured in maintenance medium for 1 week (Figure 1B). After 21 days, we evaluated their adipogenic differentiation efficiency based on morphology, lipid content and gene expression. Neutral lipid content, measured by Oil Red O staining, was comparable in all five hASC-derived adipocytes (Figure S1B). The adipocyte marker genes fatty acid binding protein 4 (FABP4) and PPARγ were also expressed at similar levels (Figure S1B and S1C). These five hASC-derived adipocytes with similar properties were used as models to test antidiabetic drug response.
Figure 1. Differential rosi responsiveness and PPARγ genomic occupancy in patient-specific hASC-derived adipocytes.
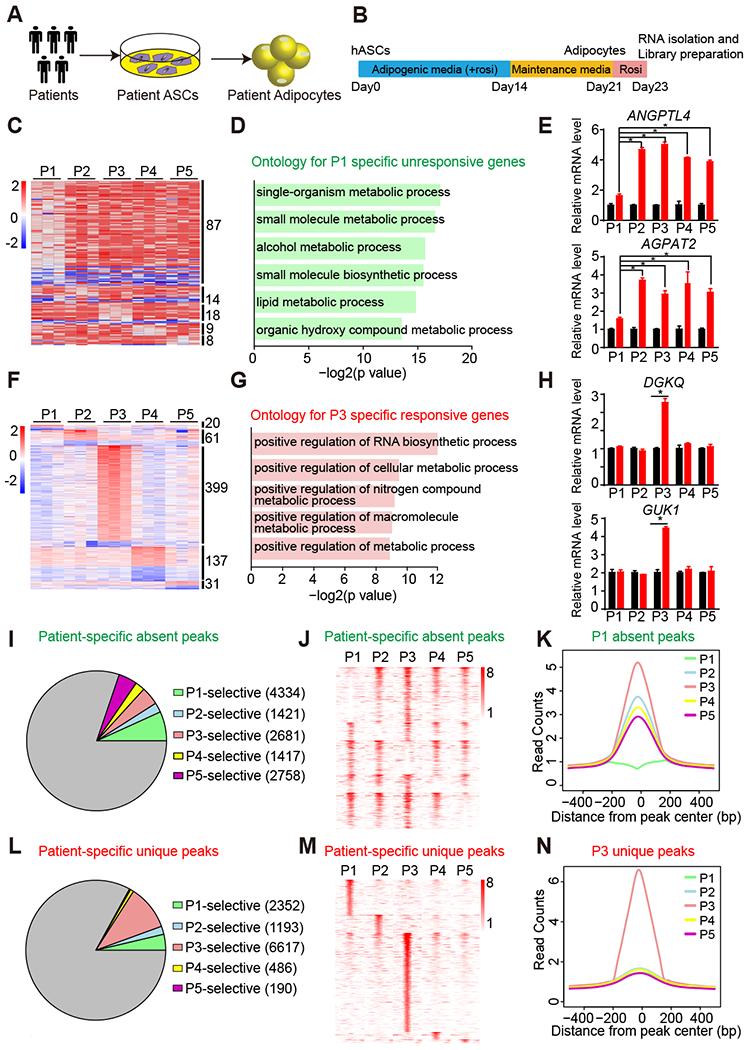
(A) Experimental design of derivation of patient adipocytes.
(B) Scheme of adipogenic differentiation procedure and rosi treatment.
(C) Heat map of patient-specific unresponsive genes that are not regulated by rosi in only one patient.
(D) Gene ontology for patient P1-specific unresponsive genes.
(E) mRNA expression of patient P1-specific unresponsive genes ANGPTL4 and AGPAT2 in adipocytes from five patients, normalized to HPRT, DMSO was set to 1, as measured by RT-qPCR.
(F) Heat map of patient-specific responsive genes that are significantly regulated by rosi in only one patient.
(G) Gene ontology for patient P3-specific responsive genes.
(H) mRNA expression of patient P3-specific responsive genes DGKQ and GUK1 in adipocytes from five patients, normalized to HPRT, DMSO was set to 1, as measured by RT-qPCR.
(I and J) Proportion (I) and Heat map (J) of patient-specific absent peaks that are specifically absent in only one patient, with at least 2-fold less reads in one patient compared to other four patients.
(K) For P1-specific absent peaks, the average binding profiles are shown in 1 kb windows across patients.
(L and M) Proportion (G) and Heat map (H) of patient-specific unique peaks that are specifically unique in only one patient, with at least 2-fold more reads in one patient compared to other four patients.
(N) For P3-specific unique peaks, the average binding profiles are shown in 1 kb windows across patients.
RT-qPCR data are expressed as mean ± SEM. (*) p < 0.05 in Student’s t-test. n = 3 per group.
We next treated the hASC-derived adipocytes with the potent TZD rosiglitazone (rosi) for 48h beginning at day 21 (Figure 1B). Biological replicates of adipocytes differentiated from different aliquots of hASCs from each patient on different days had reproducible transcriptomes (Figure S2A), with 304 genes commonly regulated by rosi (Figure S2B and S2E). GO and KEGG analysis showed that, as expected, the up-regulated genes were enriched for lipid metabolic process and PPAR signaling pathway (Figure S2C and S2H). Among the notable genes commonly induced by rosi, we verified classic PPARγ-responsive genes FABP4 and ADIPOQ using RT-qPCR (Figure S2D).
Remarkably, besides the commonly regulated genes, we also found 136 genes that were unresponsive to rosi treatment in adipocytes derived from only one of the five patients (Figure 1C and S2F). Patient P1 had the greatest number of patient-specific unresponsive genes, and these 87 genes were enriched for multiple metabolic processes and pathways (Figure 1D and S2I). Of note, genes regulating glucose homeostasis and lipid metabolism, Angiopoietin Like 4 (ANGPTL4) and 1-Acylglycerol-3-Phosphate O-Acyltransferase 2 (AGPAT2), were less-responsive in patient P1 (Figure 1E). Conversely, we found 648 genes were uniquely regulated by rosi, 399 of which specifically responded to rosi in patient P3 (Figure 1F and S2G). GO and KEGG analysis showed that P3-specific responsive genes were enriched in metabolic processes and pathways (Figure 1G and S2J). We further confirmed that two metabolic genes, Diacylglycerol Kinase Theta (DGKQ) and Guanylate Kinase 1 (GUK1), were specifically induced by rosi in patient P3 adipocytes using RT-qPCR (Figure 1H). Collectively, these data demonstrate that patient-derived adipocytes differentially respond to rosi treatment, recapturing individual differences of the effects of rosi in patients.
Patient-specific PPARγ cistromes in hASC-derived adipocytes
Since rosi is a potent activating ligand of PPARγ, we next examined whether PPARγ genomic binding is different in these patient hASC-derived adipocytes. PPARγ ChIP-seq in the hASC-derived adipocytes from five patients revealed 12600 PPARγ binding sites shared among patients (Figure S3A), with comparable binding intensity (Figure S3B and S3C). The genes near these common sites were enriched for PPAR signaling pathway and fatty acid metabolism (Fig S3D). Strikingly, genomic binding of PPARγ was also patient-specific. In each patient’s adipocytes, we can found some PPARγ binding sites were absent in specific patient (Figure 1I-1K). Notably, patient P1 had the greatest number of sites at which PPARγ binding was uniquely absent (Figure 1I), consistent with the unresponsive gene activation in patient P1 adipocytes based on the earlier transcriptome analysis (Figure 1C).
In contrast, we also identified unique PPARγ binding sites in each patient’s adipocytes. Specifically, patient P3 had most unique PPARγ binding sites (Figure 1L-1N), which is also consistent with our previous finding that patient P3 adipocytes activate more genes in response to rosi treatment (Figure 1F). KEGG pathway analysis showed that the genes near these patient P1 and P3-specific sites were enriched for insulin resistance and adipocytokine signaling pathway respectively (Figure S3E and S3F). We also found many sites are unique to two or three patients (Figure S3G-S3I). Overall, these studies revealed that a fraction of genomic PPARγ binding is different among patient adipocytes, which correlate with their responsiveness to rosi treatment.
To test whether patient-specific hASC-derived adipocytes faithfully reflect adipose tissue biology, we performed transcriptome profiling and PPARγ ChIP-seq on subcutaneous adipose tissue from the same patients. Patient-specific genes and PPARγ cistromes were closely correlated between isogenic adipose tissues and corresponding adipocytes (Figure S3J and S3K), confirming the utility of studying hASC-derived adipocytes as a platform for patient-specific transcription factor binding and drug response.
Differential PPARγ binding drives patient-specific rosi response
To determine whether the differential genomic PPARγ binding affects the individual rosi response, we associated patient-specific peaks with patient-specific genes. Interestingly, patient-specific rosi-unresponsive genes, especially patient P1-specific rosi-unresponsive genes, were much more likely be near PPARγ absent binding sites (Figure 2A). As an example, the SLC7A10 gene, which has a metabolic function in adipocytes (Ussar et al., 2014), did not respond to rosi in patient P1 adipocytes (Figure 2B), consistent with a weaker upstream PPARγ binding site (Figure 2C). Conversely, patient-specific rosi-responsive genes tended to have patient-specific PPARγ binding sites nearby (Figure 2D). For example, the gene FAM160B2 was specifically induced by rosi in patient P3 (Figure 2E). Consistent with this, we also found that upstream PPARγ binding was also stronger in P3 compared to that in other patient adipocytes (Figure 2F). Genome-wide, integration of the single nucleotide polymorphisms (SNPs) from dbSNP150 with patient-specific PPARγ binding sites revealed that SNPs occurred more frequently than would be expected by chance in individual patient-specific sites of absent binding and unique binding, but not at common PPARγ binding sites where SNPs were, if anything, less frequent than by chance (Figure S4A). These data indicate that patient-specific responses to rosi are determined by individual differences in PPARγ genomic binding.
Figure 2. Differential PPARγ binding drives patient-specific rosi response.
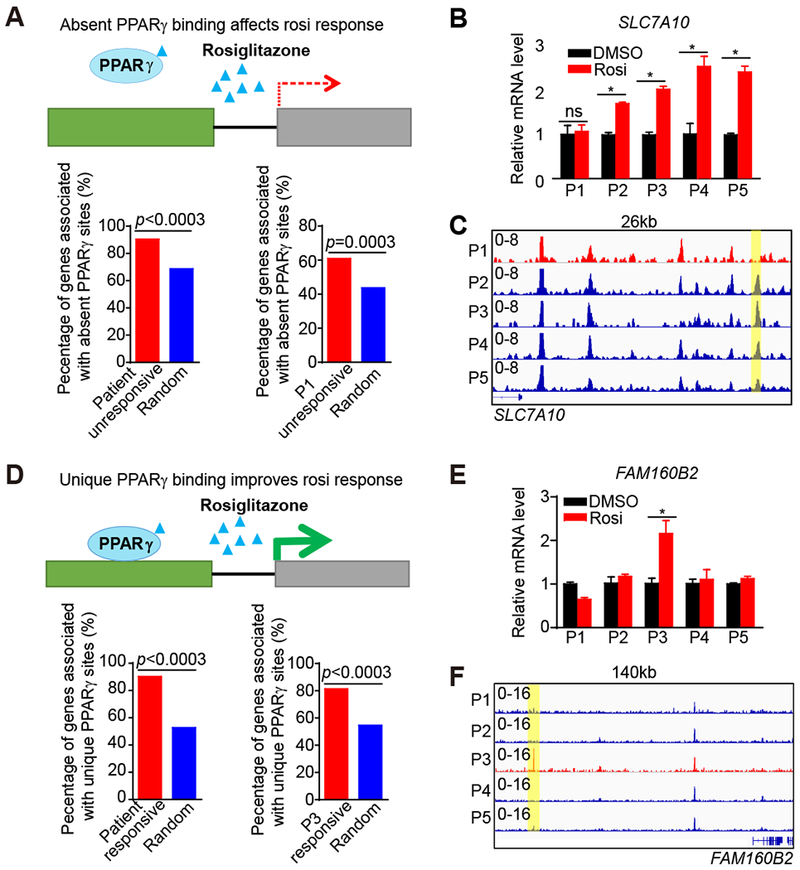
(A) Diagram depicting the association between patient-specific absent PPARγ peaks and patient-specific unresponsive genes (upper panel). Percent of patient-specific unresponsive genes surrounded by patient-specific absent peaks within 200 kb (bottom panel). P-values are determined by random test.
(B) mRNA expression of SLC7A10 in adipocytes from five patients, normalized to HPRT, DMSO was set to 1, as measured by RT-qPCR.
(C) Visualization of a Pi-specific absent PPARγ peak region (yellow box) at SLC7A10 loci across patients using Integrative Genomics Viewer (IGV).
(D) Diagram depicting the association between patient-specific unique PPARγ peaks and patient-specific responsive genes (upper panel). Percent of patient-specific responsive genes surrounded by patient-specific unique peaks within 200 kb (bottom panel). P-value is determined by random test as control.
(E) mRNA expression of FAM160B2 in adipocytes from five patients, normalized to HPRT, DMSO was set to 1, as measured by RT-qPCR.
(F) Visualization of a P3-specific PPARγ peak region (yellow box) at FAM160B2 loci across patients.
RT-qPCR data are expressed as mean ± SEM. (*) p < 0.05, (ns) p > 0.05 in Student’s t-test. n = 3 per group.
Rosi-response of cholesterol metabolism gene ABCA1 is determined by SNP rs4743771
We identified a total of 129 SNPs that were located in PPARγ binding sites that were absent in P1 or unique to P3, and were also predicted to have a strong effect on the binding motif for PPARγ or transcription factors previously shown to cooperatively support PPARγ binding (C/EBPα, NFI, or GR) (Soccio et al., 2015). The SNP rs4743771 was of particular interest because it had a major effect on PPARγ binding and was in the vicinity of the PPARγ-regulated ABCA1 gene (Chawla et al., 2001), whose protein product regulates reverse cholesterol transport (Oram and Lawn, 2001). We therefore focused on this SNP as proof-of-concept for functional testing of genetic variants in individual response to drug therapies.
Patient P1 was uniquely homozygous for the minor A allele (A/A) (minor allele frequency [MAF], A=0.4012, 1000 Genomes), while all of the other four patients had at least one C allele, with 3 being C/C (Figure 3A). Patient P1 had a weaker PPARγ peak in the genomic region spanning rs4743771, and this result was further confirmed by PPARγ ChIP-qPCR (Figure 3B). Rosi had very little effect on PPARγ binding in all patients, as has been previously found in mouse adipocytes (Step et al., 2014). Remarkably, however, rosi induced ABCA1 gene expression in every patient except patient P1, who carried the A/A genotype (Figure 3C). Chromosome conformation capture (3C) in patient-derived adipocytes demonstrated the existence of a chromatin loop connecting the region near rs4743771 and the ABCA1 gene promoter (Figure 3D). The chromatin looping was stronger in C/C patient adipocytes (Figure 3E), and while this did not alter basal gene expression, it may contribute to the ability of rosi to induce transcription from this PPARγ-bound enhancer.
Figure 3. Genetic variants determine differential PPARγ occupancy by modulating binding of NFIA.
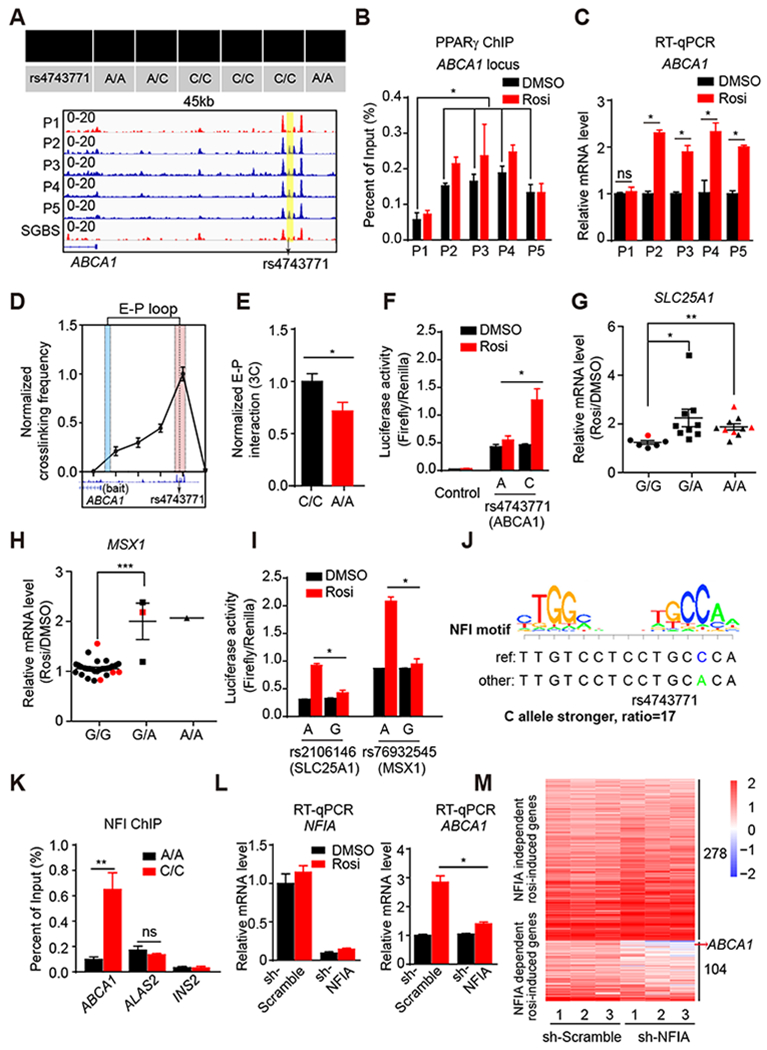
(A) The genotype of rs4743771 in all five patients and SGBS cell line (upper panel). Visualization of a Pi-specific absent peak region (yellow box) at ABCA1 loci across patients and SGBS cell line (bottom panel). Black arrow indicates the position of rs4743771.
(B) PPARγ ChIP-qPCR for ABCA1 in all five patient-adipocytes treated with DMSO or Rosi (n=3).
(C) mRNA expression of ABCA1 in five patient-adipocytes treated with DMSO and Rosi (n=3), normalized to HPRT, DMSO was set to 1, as measured by RT-qPCR.
(D) Enhancer-promoter loop (E-P loop) identified between the region near rs4743771(red) and ABCA1 gene promoter (blue) by 3C in C/C patient adipocytes (n=4).
(E) E-P loop in A/A and C/C patient adipocytes (n=5). E-P interactions were normalized to the intragenic interaction at the TBP locus.
(F) The activities of luciferase reporters with the different alleles for rs4743771 and control reporter PGL4.24 in 3T3-L1 adipocytes treated with DMSO or 1μM rosi.
(G and H) mRNA expression of SLC25A1 (G) and MSX1 (H) in hASC-derived adipocytes from 25 patients. Red dots represent patients P1-P5. Data are expressed as mean ± SEM of Rosi/DMSO fold change.
(I) The activities of luciferase reporters with the different alleles for rs2106146 and rs76932545 in 3T3-L1 adipocytes treated with DMSO or 1μM rosi.
(J) The putative effect of rs4743771 on NFI binding.
(K) NFI ChIP-qPCR for ABCA1, ALAS2 and INS2 in 3 patient-adipocytes carrying A/A and 3 patient-adipocytes carrying C/C.
(L) mRNA expression of NFIA and ABCA1 in NFIA knocked-down adipocytes treated with rosi (n=3), normalized to HPRT, as measured by RT-qPCR.
(M) Heat map of rosi-induced genes in control adipocytes and NFIA-depleted adipocytes treated with either DMSO or 1 μM Rosi.
RT-qPCR, ChIP-qPCR, luciferase reporter and 3C data are expressed as mean ± SEM. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (ns) p > 0.05 in Student’s t-test.
See also Figure S4.
The transcriptional importance of rs4743771 was assessed using luciferase reporter assays in mouse 3T3-L1 adipocytes. Consistent with the activity of the C allele in the native gene, this sequence conferred rosi-responsiveness to a reporter gene carrying the C allele, whereas the A did not (Figure 3F). Since the PPARγ binding-dependent genotype/rosi-response phenotype relationship was not unique to the ABCA1 locus, we tested additional examples, including the A allele at SNP rs2106146 (MAF, G=0.4083, 1000 Genomes) which conferred rosi responsiveness to the SLC25A1 gene (Figure 3G) and the A allele at SNP rs76932545 (MAF, A=0.0266, 1000 Genomes) which was critical for rosi responsiveness of the MSX1 gene (Figure 3H). In both cases, the allele correlating with rosi-induced gene expression was found to be uniquely active in the ability of rosi to drive expression of a reporter gene (Figure 3I). These results clearly demonstrate predictable modulation of PPARγ occupancy and rosi response by genetic variation.
Variation in rs4743771 controls PPARγ function by modulating binding of NFIA
To understand how rs4743771 regulates PPARγ genomic binding, we examined its potential effects on the PPARγ motif as well as the motifs for nearby PPARγ cooperating transcription factors. Although rs4743771 does not affect a PPARγ motif, it has a major effect on a motif predicted to be a binding site for NFI, a transcription factor that has been shown to cooperate with PPARγ at the genome (Hiraike et al., 2017; Soccio et al., 2015). Intriguingly, the score of agreement with the NFI consensus motif was ~17 greater for the C allele than for the A (Figure 3J). To test this, we performed NFI ChIP-qPCR in adipocytes from 3 A/A patients and 3 C/C patients. Indeed, adipocytes with the C/C genotype displayed much greater NFI binding at this site in the ABCA1 locus than adipocytes from A/A patients (Figure 3K). In contrast, NFI binding was genotype-independent on an unrelated NFI binding region (ALAS2). Moreover, knockdown of NFIA impaired the rosi-induced ABCA1 gene expression (Figure 3L). The effect of NFIA was genome-wide, such that NFIA deletion abrogated the induction of 104 genes in rosi-treated adipocytes (Figure 3M). Consistent with this, ~8% of patient-specific PPARγ binding sites contained SNPs which altered NFI binding motifs, which was similar to the percentage of patient-specific PPARγ binding at sites where the SNP affected the PPARγ binding motifs itself (Figure S3L).
Genome editing confers PPARγ binding and rosi response at the ABCA1 locus.
To determine whether the C allele is sufficient to convey rosi-responsiveness to the ABCA1 gene, we next used clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 to edit rs4743771. For this purpose we used the SGBS human preadipocyte cell line (Fischer-Posovszky et al., 2008), which is more highly proliferative and thus more amenable to gene editing than patient-derived ASCs. We found SGBS to be homozygous for the rosi-unresponsive A/A genotype, and PPARγ binding was weak in the genomic region spanning rs4743771, similar to what was observed in adipocytes from patient P1 (Figure 3A). Importantly, we also noted that ABCA1 gene expression was not induced by rosi in SGBS-differentiated adipocytes (Figure S4B). The replacement of A allele of SGBS cells into the C allele (A>C) was confirmed by Sanger sequencing (Figure 4A). The A>C edited SGBS cells retained the ability of SGBS to differentiate into adipocytes (Figure S4C). Remarkably, SGBS cells that underwent A>C editing acquired PPARγ binding at the ABCA1 locus with no change at positive and negative control sites (Figure 4B). Moreover, they also acquired NFI binding selectively at this locus (Figure 4C), again with no change at an unrelated binding site near the ALAS2 gene as well as a negative control site, consistent with the conclusion that this genetic variant directly alters binding of this PPARγ cooperating factor which in turn affects responsiveness to rosi.
Figure 4. Genetic variants determine and predict individual differences in rosi responsiveness.
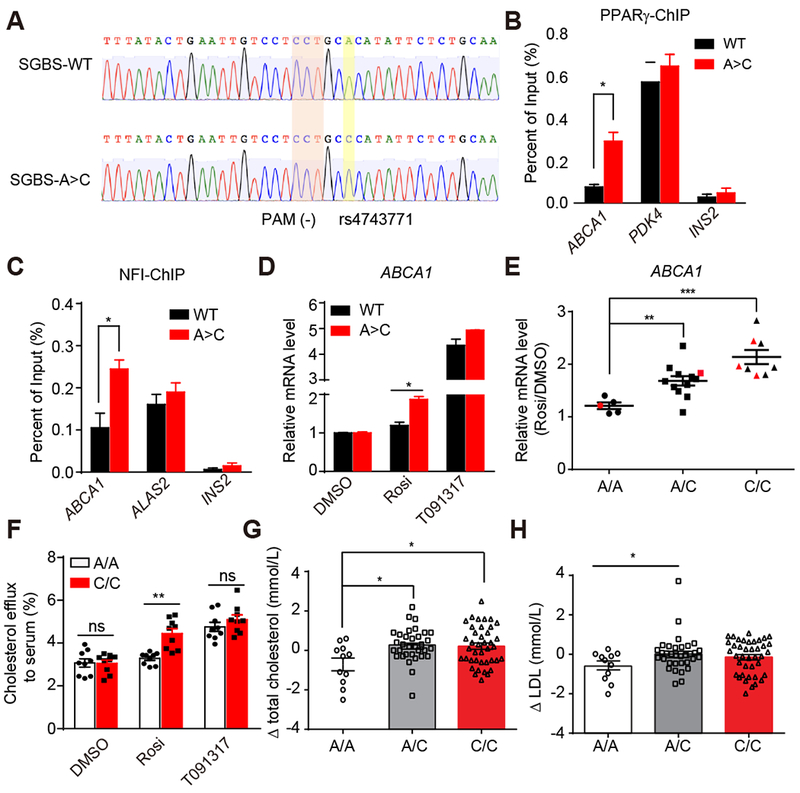
(A) Sanger sequencing validation of the correction of A/A allele to C/C allele.
(B and C) PPARγ (B) and NFI (C) ChIP-qPCR for ABCA1, PDK4, ALAS2, and INS2 in SGBS WT and SGBS A>C adipocytes.
(D) mRNA expression of ABCA1 in SGBS WT and SGBS A>C adipocytes treated with DMSO, 1μM rosi or 10μM T091317 (n=3).
(E) mRNA expression of ABCA1 in hASC-derived adipocytes from 25 patients. Red dots represent patients P1-P5. Data are expressed as mean ± SEM of Rosi/DMSO fold change.
(F) Cholesterol efflux to serum in 3 patient-adipocytes carrying A/A and 3 patient-adipocytes carrying C/C treated with DMSO, 1μM rosi or 10μM T091317 (n=3).
(G and H) The change of total cholesterol (G) and LDL (H) levels after rosi treatment in patients carrying different rs4743771 genotypes.
RT-qPCR, ChIP-qPCR data are expressed as mean ± SEM. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (ns) p > 0.05 in Student’s t-test.
See also Figure S4.
Functionally, A>C edited SGBS cells acquired the ability for rosi to induce ABCA1 expression (Figure 4D). By contrast, basal expression of ABCA1 was unchanged, and LXR agonist T091317, a known inducer of ABCA1 by a different mechanism (Chawla et al., 2001), induced ABCA1 similarly in both wild type and edited adipocytes (Figure 4D). Moreover, the genome editing did not affect the ability of rosi to induce FABP4, another PPARγ target gene (Figure S4D). Thus, the C allele of rs4743771 is sufficient to support NFIA and PPARγ binding, and to convey rosi-responsiveness to the ABCA1 gene.
Rosi transcriptional responses can be predicted from genome information
Based on the above results, we prospectively tested whether SNP rs4743771 is predictive for rosi-responsiveness of ABCA1 in 20 additional patients (4 A/A, 11 A/C, and 5 C/C patients). As predicted from the analysis of the initial 5 patients, C/C patient adipocytes consistently responded to rosi with ~2-fold induction of ABCA1, while A/A patients did not (Figure 4E). Moreover, A/C heterozygotes had intermediate responses to rosi, indicative of gene dosage effects of this allele. By contrast rs4743771 genotypes were unrelated to basal ABCA1 gene expression (Figure S4E), indicating that other factors other than PPARγ likely drive basal ABCA1 transcription.
Genetic variation at rs4743771 controls rosi effects on cholesterol metabolism in patient adipocytes
Since ABCA1 is regulator of cholesterol efflux, we next explored whether SNP rs4743771 also affects cholesterol metabolism. Notably, we found that rosi induced cholesterol efflux from adipocyte to serum in C/C patient adipocytes, but was ineffective in A/A patient adipocytes (Figure 4F). As a control, T091317 similarly induced cholesterol efflux in patient adipocytes of either genotype. Thus, the SNP that is permissive for rosi induction of ABCA1 by altering PPARγ binding predictably controlled whether a PPARγ ligand altered lipid metabolism, whereas the effects of an LXR ligand were independent of the specific rs4743771 allele.
Similarly, as SLC25A1 encodes a citrate transporter which is important for mitochondrial metabolism (Hlouschek et al., 2018), we also examined whether the effect of rosi on the mitochondrial function of patient-derived adipocytes was differentially affected by the rs2106146 genotype. Indeed, the G/G patient-derived adipocytes with reduced PPARγ binding and rosi induction of SLC25A1 exhibited an impaired effect of rosi on both basal (Figure S4F) and maximal mitochondrial respiration (Figure S4G).
Genetic variation at rs4743771 controls effects of rosi treatment on cholesterol metabolism in diabetic patients
In genome-wide association studies, A-allele of rs4743771 was associated with higher body fat percentage (P=0.006, effect = 0.017 SD/allele), as previously observed in a large-scale meta-analysis on body fat percentage (N = 74,388) (Lu et al., 2016). While no association was observed with BMI in the overall population (P=0.18) (Locke et al., 2015), we found that the rs4743771 A-allele was associated with higher BMI in physically inactive individuals (P = 0.003, effect = 0.030 SD/allele, N = 42,066), and this association was even more pronounced among women (P = 0.0002, effect = 0.048 SD/allele, N = 26,836) (Graff et al., 2017).
These findings are intriguing, and suggest a role of the rs4743771 in regulating fat cell mass, but do not address the more specific question of whether genetic variation in rs4743771 has relevance to the effects of rosi on serum cholesterol levels. It is well established that rosi treatment leads to increases in total as well as LDL cholesterol (Rosenblit, 2016). The mechanism is not well understood, but this hypercholesterolemic response is thought to be an adverse effect of rosi that may contribute to myocardial infarctions and strokes among patients taking the drug (Graham et al., 2010).
We interrogated the clinical relevance of SNP rs4743771 in a cohort of 84 diabetic patients whose serum chemistries were studied before and after treatment with rosi for 48 weeks (Wang et al., 2008). The patients were blindly genotyped, and then genotype-phenotype analysis was performed. Remarkably, diabetic patients whose genomes contained one or two copies of the rs4743771 C allele increased their total cholesterol (Figure 4G) and LDL cholesterol (Figure 4H) in response to rosi to a greater extent than patients with A allele. Increased cholesterol levels are a well-described adverse effect of rosi, which has been mechanistically linked to increased ABCA1 gene expression in preclinical studies (Vaisman et al., 2001). No rs4743771 genotype altered the beneficial effects of rosi on hemoglobin A1c (Figure S4H) and fasting glucose levels (Figure S4I). Thus patients with the rs4743771 A/A genotype get the glycemic benefits of rosi with less likelihood of experiencing the adverse consequence of elevated cholesterol, suggesting a way forward to identifying individuals with lower risks associated with rosi treatment, which is a cornerstone of personalized pharmacotherapy.
DISCUSSION
By applying unbiased “omics” approaches to patient-specific adipose stem cell models we have determined that genetic variation is enriched at PPARγ sites whose absence or presence is specific to individuals. Importantly, a single SNP that affected rosi-regulation of cholesterol metabolism identified patients at risk for adverse effects of rosi in a clinical study. Thus, our data and methodology provide a proof of principle for mechanistic understanding of how natural genetic variants control individual responses to anti-diabetic drugs. These principles could be extended to different cell types such as macrophages, where PPARγ is relatively abundant and TZDs generally favor an anti-inflammatory alternative activation phenotype (Nelson, 2018).
Genetic variants modulate the response to environmental stimuli and drugs in the context of complex diseases (Lee et al., 2014; Soccio et al., 2015). Indeed, several variant annotations in the Pharmacogenomics Knowledgebase have been identified as modulating rosi responsiveness (e.g., in CYP2C8, LPIN1, PAX4 and SLCO1B1). Mechanistically, genetic variants in SLO1B1 and CYP2C8 affect patient plasma rosi concentration (Dawed et al., 2016), as rosi is thought to be transported into the liver by OATP1B1 (encoded by SLCO1B1) and metabolized by CYP450 2C8 enzyme (encoded by CYP2C8). However, the precise biological mechanisms for other genetic variants are not known. Non-coding SNPs in regulatory regions may affect transcription factor binding and gene expression, thus contributing to the response to drugs. Here, we employed a genome-wide experimental pipeline with integrative analysis of patient hASC-derived adipocytes, which revealed noncoding SNPs that affect PPARγ genomic binding and the response to TZDs.
Our detailed analysis of one PPARγ binding-disrupting SNP, rs4743771, revealed its association with rosi-induction of the ABCA1 gene. ABCA1 is responsible for the efflux of cholesterol to APOA1 and small high density lipoprotein (HDL) particles (McNeish et al., 2000), and deletion of ABCA1 in mouse adipose tissue alters systemic lipid and glucose metabolism (Cuffe et al., 2018). Interestingly, a previous report has also described a coding variant at the ABCA1 locus that affects the efficacy of rosiglitazone monotherapy in type 2 diabetes patients (Wang et al., 2008). This highlights the power of our approach to identify new non-coding genetic variants controlling drug responses. Rosi has been associated with increased LDL and total cholesterol in diabetic patients (Rosenblit, 2016), and these adverse lipid changes have been suggested to promote myocardial infarction and stroke in patients treated with rosi. We find that rosi did not increase LDL and total cholesterol in A/A patients, while maintaining its beneficial effects on glucose metabolism.
With a minor allele frequency of 0.4, the A/A genotype of rs4743771 represents ~16% of patients. Considering the millions of patients with diabetes in the US alone, this represents a large number of patients for whom rosi therapy might be considered because of the predicted lack of adverse effects on cholesterol with retention of amelioration of insulin resistance. We have also tested the association between the identified genetic variants (rs2106146 and rs76932545) and individual responsiveness to the rosiglitazone treatment. However, there are only four G/G patients for rs2106146 and no A/A patients for rs76932545 in the cohort of 84 patients that we examined, so there is insufficient power to examine whether these two SNPs are associated to responsiveness to rosi and the synergic effect of these three SNPs on rosi response.
In sum, our study presents an advanced strategy using stem cell-derived adipocytes to identify human genetic variation that determines patient response to antidiabetic drugs. While the overall effects of drugs are clearly polygenic, the present work demonstrates that individual SNPs can have predictable effects on gene expression and metabolic phenotype.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PPARγ | Santa Cruz Biotechnology | Cat# sc-7196, RRID: AB_654710 |
| FABP4 | R & D Systems | Cat# AF3150, RRID: AB_2278261 |
| NFI | Santa Cruz Biotechnology | Cat# SC-74445X, RRID: AB_2153046 |
| Tubulin-HRP | Abcam | Cat# ab21058, RRID: AB_ 727045 |
| Chemicals and Reagents | ||
| DMEM | Thermo Fisher Scientific | Cat# 11995065 |
| DMEM/F12 | Thermo Fisher Scientific | Cat# 11330032 |
| Fetal bovine serum | Atlanta Biologicals | Cat# S11150 |
| Insulin solution human | Sigma | Cat# I9278 |
| 3-Isobutyl-1-methylxanthine | Sigma | Cat# I5879 |
| Dexamethasone | Sigma | Cat# D4902 |
| Indomethacin | Sigma | Cat# I7378 |
| Rosiglitazone | Sigma | Cat# R2408 |
| Biotin | Sigma | Cat# B4639 |
| Pantothenate | Sigma | Cat# P5155 |
| Insulin-Transferrin-Selenium | Thermo Fisher Scientific | Cat# 41400045 |
| Hydrocortisone | Sigma | Cat# H0888 |
| Triiodothyronine | Sigma | Cat# T6397 |
| T091317 | Sigma | Cat# T2320 |
| Paraformaldehyde Solution, 4% | Affymetrix | Cat# 19943 1 LT |
| SuperScript II Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18064014 |
| cOmplete, EDT-free Protease Inhibitor Cocktail | Roche | Cat# 11873580001 |
| Chloroform | Sigma | Cat# C2432-500ML |
| RNase A (DNase and Protease Free) | Fermentas Life Sciences | Cat# EN0531 |
| Phenol/Chloroform/Isoamyl Alcohol | Fisher Scientific | Cat# BP1753I-400 |
| ChIP DNA Clean & Concentrator-Capped column | Zymo Research | Cat# D5205 |
| DNA LoBind Microcentrifuge Tube | Eppendorf | Cat# 30108.051 |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat# N0447S |
| Phusion Hot Start II DNA Polymerase | Thermo Fisher Scientific | Cat# F-549L |
| UltraPure Glycogen | Thermo Fisher Scientific | Cat# 10814010 |
| T4 DNA Polymerase | New England Biolabs | Cat# M0203S |
| Klenow Fragment | New England Biolabs | Cat# M0212S |
| Klenow DNA Polymerase | New England Biolabs | Cat# M0210S |
| T4 Polynucleotide Kinase | New England Biolabs | Cat# M0201S |
| ExoSAP-IT | Affymetrix | Cat# 78201.1.ML |
| Calf Intestinal Phosphatase | New England Biolabs | Cat# M0290S |
| Human serum | Millipore Sigma | Cat# S1-100ML |
| [1,2-3H(N)]-Cholesterol | Perkin Elmer | Cat# NET139250UC |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000008 |
| EcoRI-HF | New England Biolabs | Cat# R3101L |
| T4 DNA Ligase | New England Biolabs | Cat# M0202 |
| Sodium Pyruvate | Thermo Fisher Scientific | Cat# 11360070 |
| D-(+)-Glucose solution | Sigma | Cat# G8769 |
| L-Glutamine | Thermo Fisher Scientific | Cat# 25030081 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368813 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| Agilent high sensitivity DNA assay | Agilent | Cat# 5067-4626 |
| Agencourt AMPure XP beads | Beckman Coulter | Cat# A63881 |
| Illumina TruSeq stranded Total RNA Library Prep Kit | Illumina | Cat# RS-122-2303 |
| SNaPshot Multiplex Kit | Thermo Fisher Scientific | Cat# 4323159 |
| Seahorse XF cell mito stress test kit | Agilent Technologies | Cat# 103015-100 |
| TaqMan™ Gene Expression Master Mix | Thermo Fisher Scientific | Cat# 4369016 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat# E1910 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Deposited Data | ||
| RNA-seq | This study | GEO: GSE 115421 |
| PPARγ ChIP-seq | This study | GEO: GSE 115421 |
| PPARγ ChIP-seq in SGBS | (Soccio et al., 2011) | GEO: GSE25836 |
| Recombinant DNA | ||
| pGuide | Addgene | Cat# 44719 |
| pCas9_GFP | Addgene | Cat# 64711 |
| pGL4.24 Vector | Promega | Cat# E842A |
| TRC Lentiviral Non-targeting shRNA Control | Dharmacon | Cat# RHS6848 |
| TRC Lentiviral Human NFIA shRNA | Dharmacon | Cat# RHS4533-EG4774 |
| psPAX2 | Addgene | Cat# 12260 |
| pCMV-VSV-G | Addgene | Cat# 8454 |
| BAC ABCA1 | CHORI | Cat# RP11-1N10 |
| BAC TBP | CHORI | Cat# RP11-794H3 |
| Oligonucleotides | ||
| See Table S1 | N/A | N/A |
| Software and Algorithms | ||
| Bowtie 2.3.0 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml |
| SAMtools 1.8 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| Bedtools 2.26.0 | (Quinlan and Hall, 2010) | http://bedtools.readthedocs.org/en/latest/ |
| Homer v4.9 | (Heinz et al., 2010) | http://homer.salk.edu/homer/ |
| R 3.3.2 | www.r-project.org/ | |
| StringTie 1.3.3b | (Pertea et al., 2015) | https://ccb.jhu.edu/software/stringtie/ |
| Hisat2 2.0.5 | (Kim et al., 2015) | https://ccb.jhu.edu/software/hisat2/index.shtml |
| IGV 2.4 | (Robinson et al., 2011) | http://software.broadinstitute.org/software/igv |
| MEME SUITE 4.12.0 | (Grant et al., 2011) | http://meme-suite.org/ |
| FeatureCounts 1.5.1 | (Liao et al., 2014) | http://bioinf.wehi.edu.au/featureCounts/ |
| DAVID Bioinformatics Resources 6.8 | (Huang da et al., 2009) | https://david.ncifcrf.gov/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mitchell A. Lazar, M.D., Ph.D. (lazar@pennmedicine.upenn.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary hASCs
For hASCs isolation, abdominal subcutaneous adipose tissues around the umbilical area were obtained from obese individuals with participant informed consent obtained after the nature and possible consequences of the studies were explained under protocols approved by the Institutional Review Boards of the Perelman School of Medicine at the University of Pennsylvania. The fat biopsies were digested using 0.1% collagenase type IA at 37 °C for 30-60 min. Afterwards, the aliquots of the infranatant containing the stromal vascular fraction (SVF) were pelleted at 1,200 × g for 10 min. The pellet was resuspended in DMEM (ThermoFisher, 11995-065) supplemented with 10% fetal bovine serum (Atlanta Biologicals, S11150) and penicillin/streptomycin, and flited through a 100-μm filter (Falcon). The cells were maintained in DMEM medium at 37 °C with 5% CO2 and passaged 3-4 times before adipogenic differentiation.
SGBS cell line
SGBS preadipocytes were cultured in DMEM/F12 (ThermoFisher, 11330-032) supplemented with 10% fetal bovine serum (Atlanta Biologicals, S11150), 1% penicillin/streptomycin, 33 μM biotin and 17 μM pantothenate.
Patients and study design
Patients and study design was as previously described (Wang et al., 2008). The study was approved by the institutional review board of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital (Shanghai, China). Each patient provided written informed consent before participating in the study.
A total of 105 newly diagnosed patients with type 2 diabetes, defined according to the World Health Organization criteria, were derived from the outpatient clinics at 10 hospitals in Shanghai. All patients were naive to prior antidiabetic therapy and treated with rosiglitazone for 48 weeks. Enrolled patients were 30–70 years of age, glycated hemoglobin ≥6.5%, and a body mass index (BMI) ≥18.5 kg/m2. For the female patients, postmenopause, surgical sterilization, or effective contraception was required. Exclusion criteria were: (i) type 1 diabetes, gestational diabetes, or other specific types; (ii) acute or chronic complications in need of insulin therapy; (iii) significant cardiocerebral, hepatic or nephric disease; (iv) malignant tumor, hematological disease, autoimmune disease, psychiatric disease, or significant digestion and absorption disturbances; (v) current exposure to medication affecting glucose metabolism, such as glucocorticoid; (vi) long-term alcohol or drug abuse; (vii) fasting plasma glucose >13 mmol/L (234 mg/dL) and/or 2 h post-load plasma glucose >18 mmol/L (364 mg/dL); and (viii) blood pressure >180/110 mmHg.
The initial dose was 4 mg/d and escalated to 8 mg/d in patients who failed to attain glycemic targets of fasting plasma glucose >7 mmol/L (126 mg/dL) and/or 2 h plasma glucose >11 mmol/L (200 mg/dL). Patients with glycated hemoglobin was ≥8% or fasting plasma glucose >13 mmol/L (234 mg/dL) or 2 h plasma glucose >18 mmol/L (364 mg/dL) twice (a maximal interval of 6 d) were withdrawn from the study, so the results of 93 patients were included in the original study (Wang et al., 2008). The present study analyzed 84 patients, because two patients’ genomic DNA was not available after 10 years’ use and seven patients did not have lipids profile data.
METHOD DETAILS
Adipogenic differentiation of hASCs and SGBS
hASCs at passage P3-P4 were cultured in DMEM medium. Confluent hASCs were then transferred into adipogenic medium for 14 days. Adipogenic medium formulation was as follows: DMEM with 10% FBS, 1% penicillin/streptomycin, dexamethasone (1 μM), IBMX (0.5 mM), indomethacin (0.2mM), Insulin (10 μg/ml) and rosiglitazone (1 μM). Then, cells were further cultured in maintenance medium for another 7 days. Maintenance medium formulation was as follows: DMEM with 10% FBS, 1% penicillin/streptomycin, dexamethasone (1 μM), IBMX (0.5 mM), indomethacin (0.2mM) and Insulin (10 μg/ml).
For adipogenic differentiation of SGBS cells, confluent SGBS cells were cultured in DMEM/F12 (ThermoFisher, 11330-032) supplemented with 1% penicillin/streptomycin, 33 μM biotin, 17 μM pantothenate, 1% Insulin-Transferrin-Selenium (ThermoFisher, 41400-045), 100 nM hydrocortisone, 0.2 nM triiodothyronine, 25 nM dexamethasone, 0.5 mM IBMX and 1 μM rosiglitazone. After 4 days, the medium was changed to differentiation medium without dexamethasone, IBMX and rosiglitazone for 10 days. Medium was changed every 2-3 days.
Oil Red O staining and Immunostaining
For Oil Red O staining, adipocytes were fixed by 4% PFA solution for 60 min at room temperature (RT). Fixed cells were stained with 0.24% Oil Red O in 40% 2-propanol for 15 min.
For Immunostaining, cells plated on glass coverslips were fixed by 4% PFA solution for 10 min at room temperature. Then cells were incubated in blocking buffer (1% bovine serum albumin in PBS) with 0.5% Triton X-100 for 30 min at RT. Afterwards, samples were incubated with primary antibodies at 4 °C overnight and then with appropriate fluorescen t probe-conjugated secondary antibodies for 1 h at RT. Images were captured with fluorescence microscope.
Western blot and gene expression analysis
For western blot, adipocytes were washed with cold PBS and lysed with Laemmli’s sample buffer, and then cell lysates were separated on SDS-PAGE, transferred onto nitrocellulose membrane and blotted with indicated primary antibodies. The membrane was detected by secondary antibody conjugated to HRP.
For gene expression analysis, total RNA samples were collected with RNeasy kit (QIAGEN) according to manufacturer’s instructions. The RNA for each reaction was reverse-transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit. Quantitative real-time PCR was subsequently conducted with specific primers and Power SYBR Green PCR Master Mix (Applied Biosystems). The relative expression levels were normalized against the internal control (HPRT). Primers used were listed in Supplement information, Table 1.
RNA-seq and data processing
The RNA samples from independent experiments were processed with the Ribo-Zero rRNA Removal Kit (Epicentre Biotechnologies) to reduce ribosomal RNA abundance, prepared with the TruSeq RNA Sample Prep Kit (Illumina) and sequenced single-end at 100bp read length on Illumina HiSeq2000 by the Functional Genomics Core of the Penn Diabetes Research Center or sequenced paired-end at 150bp read length by Novogene.
RNA-seq reads were aligned to human reference genome (hg19) using Hisat2 with default parameters. Only unique mapped reads were considered for further analysis. Normalized expression value, fragments per kilobase of exon per million reads mapped (FPKM), was calculated for each gene using StringTie. A gene was considered expressed if its expression value is larger than 1 in at least one subject, and there were 16,961 expressed genes (12001 expressed protein-coding genes). For differential expression analysis, raw read counts were measured within Ensembl genes (GRCH37.75) using featureCounts, and then edgeR pipeline was used with adjusted p value (Benjamini Hochberg) ≤ 0.01 and fold change > 1.5 from samples treated with rosi against samples treated with DMSO for each patient. Differentially expressed genes with fold change > 1.5 in only one patient were defined as patient-specific responsive genes. In contrast, differentially expressed genes with fold change < 1.5 in only one patient were defined as patient-specific unresponsive genes. Genes with fold change > 1.5 in all five patients were defined as common responsive genes. GO and KEGG enrichment analysis were performed using DAVID Bioinformatics Resources v6.8. The same pipeline was used for RNA-seq data from NFIA-depleted samples. Differentially expressed genes were identified between samples treated with rosi against samples treated with DMSO with adjusted p value ≤ 0.01 and fold change > 1.5.
To compare gene expression pattern in adipocytes and adipose tissues, FPKM value of each gene in each sample was transformed by log2 before analysis. Differentially expressed genes between groups were identified by one-way analysis of variance (ANOVA) with p value < 0.02, followed by principal component analysis (PCA) analysis.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed as described previously (Dispirito et al., 2013). Briefly, mature adipocytes or human adipose tissues were crosslinked in 1% formaldehyde for 15 min followed by quenching with 1/20 volume of 2.5 M glycine solution for 5 min. Soluble chromatin was prepared following sonication and then was incubated with anti-PPARγ antibody (Santa-Cruz) or anti-NFI antibody (Santa-Cruz). Crosslinking was reversed overnight at 65°C in SDS buffer (50 mM Tris-HCL, 10 mM EDTA, 1% SDS at pH 8), and DNA was purified using phenol/chloroform/isoamyl alcohol. Precipitated DNA was analyzed by qPCR or next-generation sequencing.
ChIP-seq and data processing
ChIP experiments were performed independently on mature human adipocytes or human adipose tissues. DNA was amplified according to the ChIP-seq sample preparation guide provided by Illumina using adaptor oligo and primers from Illumina, enzymes from New England Biolabs, and PCR purification kit and MinElute kit from Qiagen. ChIP-seq libraries were sequenced single-end at 50bp read length on Illumina HiSeq2000 by the Functional Genomics Core of the Penn Diabetes Research Center.
ChIP-seq reads were aligned to human reference genome (hg19) using Bowtie2. Only unique mapped reads were considered for further analysis. Aligned reads from biological replicates were pooled together and peak calling was performed by HOMER with normalized tag count ≥6, p value < 0.00001 and fold change ≥ 4. Afterwards, peaks from each patient were merged together using BEDTools, and then resized to 200 bp. Normalized read counts was calculated using HOMER for each peak in each patient. Peaks were defined as patient-specific peaks only if the read counts in one patient is at least 2 times stronger than all other four patients. In contrast, peaks were defined as patient-specific absent peaks only if the read counts in one patient is at least 0.5 times weaker than all other four patients. Peaks with normalized reads counts larger than 8 in all five patients were defined as common PPARγ peaks. GO and KEGG enrichment analysis were performed using DAVID Bioinformatics Resources v6.8 based on the nearby genes within 10kb. Browser tracks were processed by Homer v4.9 and visualized on Integrative Genomics Viewer (IGV). Each differential peak was associated with the closest TSS (within 200 kb) of patient-specific genes identified in RNA-seq as described above. A random test was performed by shuffling gene/peak label 3000 times to compare the differential peak-gene association. To assess if SNPs are enriched in patient-specific PPARγ peaks, a random test by shuffling peak label 3000 times was performed. To compare PPARγ binding sites in adipocytes and adipose tissues, normalized tag counts of PPARγ binding site in each sample were transformed by log2 before analysis. Differentially PPARγ binding sties between groups were identified by one-way ANOVA with p value < 0.05, followed by PCA analysis.
Motif analysis
To find SNPs within patient-specific unique/absent PPARγ binding sites affecting PPARγ motif or its cooperating factors NFI, CEBPα and GR motifs, the position weighted matrixes (PWMs) of these motifs coming from Homer and MEME were used. For each SNP in patient-specific unique/absent PPARγ binding sites, the hg19 reference sequence for 20bp on either side was retrieved, and this was modified to generate the new sequences by replacing the base at reference sequence with its counterpart alleles. A FASTA file with these sequences was interrogated for each PWMs using FIMO algorithm in MEME suite with p-value < 0.001. FIMO assigns p-value to each sequence for PWMs and points out the coordinate of motifs at each sequence. The SNPs with position weight at a PWM larger than 0.3 and with the ratio of the position weights between two genotypes of the SNP larger than 3 were considered.
SNaPshot genotyping assay
Genotyping were carried out with SNaPshot Multiplex Kit (Applied Biosystem) according to manufacturer’s instructions. Briefly, each region of interest flanking the SNP was PCR amplified, and 5μl of PCR product was purified by ExoSAP-IT reagent (Affmetrix). Primer extension was performed by adding 1μl of purified PCR product to a mix of 2.5μl SNaPshot reagent, 1μl water and 0.2pmol extension primer for 25 cycles on Thermocycler. To remove unincorporated fluorescent dNTPs post-extension, each reaction was incubated 1U of Calf Intestinal Phosphatase (New England BioLabs). Samples were sequenced by the Penn DNA Sequencing Facility on an ABI 3730, and genotypes were identified manually using Peak Scanner Software (Life Technologies).
CRISPR/Cas9 editing of SGBS
The CRISPR/Cas9 expression vector pCas9_GFP and the sgRNA-expression vector pGuide were gifts from Dr. Kiran Musunuru (Addgene plasmid #44719 and #64711). Guide RNAs were designed by manual inspection of the genomic sequences flanking rs4743771, and then constructed in the pGuide plasmid. Next, the pGuide-rs4743771 vector, the pCas9_GFP vector, and the ssODN containing C allele (Integrated DNA Technologies) were co-transfected into the SGBS preadipocyte cell line using the Amaxa-Nucleofector device (program U-033) and the basis nucleofector kit for primary mammalian fibroblasts (Lonza). Cells were dissociated with trypsin 48 h post-transfection, and GFP-positive cells were isolated by FACS (FACSAriaII, BD Biosciences) and replated onto 6-cm dishes. Subsequently, selected clones were genotyped by PCR and Sanger sequencing.
Chromatin conformation capture (3C)
In situ chromosome conformation capture (3C) samples were prepared as described previously with modifications (Rao et al., 2014). Briefly, 5 million cells were crosslinked with 1% formaldehyde and quenched by 2.5 M glycine. Cells were collected and resuspended in Hi-C lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% NP-40) with proteinase inhibitor (Sigma). Isolated nuclei were digested with EcoR1 and then ligated. After ligation, the supernatant was removed, the pellet containing nuclei resuspended in Hi-C lysis buffer and residual EcoRI enzymes were denatured by incubating at 65°C for 30 min. The nuclei were spun down for 5 min at 600g, after which the supernatant was discarded, and the pellet containing nuclei was resuspended again in Hi-C lysis buffer. The nuclei were reversed crosslinked and treated with proteinase overnight. DNA was isolated using phenol/chloroform/isoamyl alcohol and second chloroform wash. Precipitated DNA was dissolved in water, and 100ng of DNA was used for each technical replicate for quantitative PCR with specific TaqMan probes. Standards were prepared using EcoRI digested and randomly ligated DNA fragments from BAC (the CHORI BACPAC Resource Center) spanning an entire locus to be probed. All interactions are normalized to the intragenic interaction at the TBP locus to control for DNA amounts and crosslinking efficiency. The BAC, primers and probes used are listed in Supplement information, Table 1.
Luciferase reporter assay
~200bp human DNA fragments encompassing SNP rs4743771 in the ABCA1 locus, rs2106146 in SLC25A1 locus and rs76932545 in MSX1 locus were synthesized from Integrated DNA Technologies with different SNP genotypes. They were cloned into the XhoI and BglII restriction sites of the pGL4 luciferase reporter (Promega) and sequence-verified. Transient transfections of 3T3-L1 cells 2 days post differentiation were performed in 24-well plates, n=3 wells per condition, using Lipofectamine 3000 (Invitrogen) to add 400ng of pGL4 luciferase reporter and 2ng of renilla luciferase for normalization. One day later, the cells were treated with 1μM rosi for 48 hours. The Dual-Luciferase Kit (Promega) was used to measure luciferase activities on a Synergy HT plate reader (Biotek).
Seahorse assay
Cells were uniformly plated in XF96 plates and differentiated for 21 days, then treated with DMSO or 1 μM Rosi for 48 hours. OCR was measured with the Seahorse XF96 extracellular flux analyzer (Agilent). Experiments were conducted in XF medium (non-buffered Seahorse XF base medium supplemented with 10 mM glucose (Sigma), 2 mM L-glutamine (Invitrogen), 2 mM sodium pyruvate (Invitrogen), with OCR measured basally and in response to sequential addition of 2 μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone + antimycin A (Agilent). All Seahorse XF data were normalized to total well protein quantified with Pierce BCA protein kit (Thermo Fisher).
Cholesterol efflux assay
Cholesterol efflux assay was performed as described previously (Pospisilik et al., 2010). Briefly, mature adipocytes derived from different patients were labeled with 3H-cholesterol (2μCi/mL) (Perkin-Elmer Analytical Sciences) overnight. The labeling medium was removed and human adipocytes were then equilibrated for an additional 24 h period in the presence or in the absence of either 1 μM rosiglitazone or 10 μM T0901317. 3H-cellular cholesterol efflux to 5% human serum was assessed in serum-free medium for a 4-hour in the presence or absence of either 1 μM rosiglitazone or 10 μM T0901317. Cell lipid was extracted with isopropanol and total cellular 3H-cholesterol was measured by liquid scintillation counting. The efficiency of cholesterol efflux was calculated as 100 × (medium cpm) / (medium cpm + cell cpm).
Clinical laboratory tests
Blood samples were collected after an overnight fast and 2 h after a 75 g oral glucose tolerance test (OGTT). Plasma glucose concentrations were measured using the glucose oxidase-peroxidase method with commercial kits (Shanghai Biological Products Institution, Shanghai, China). Glycated hemoglobin values were determined by high-performance liquid chromatography performed on a Bio-Rad Variant II hemoglobin testing system (Bio-Rad Laboratories, Hercules, CA, USA). Serum lipid profiles, including total cholesterol, triglyceride, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) were measured with a type 7600-020 Automated analyzer (Hitachi, Tokyo, Japan). The serum levels of insulin and proinsulin were measured in duplicate at 0, 2, 4, and 6 min after an intravenous injection of 50 mL arginine solution at the concentration of 10%, using radioimmunoassay (Linco Research, St Charles, MO, USA).
Genome-wide association studies (GWAS) analysis
To examine whether SNPs are associated with other traits, we searched publicly available data from previously published large-scale genome-wide association studies (GWAS), using PhenoScanner. PhenoScanner is a comprehensive, curated database that catalogues 65 billion genetic associations, for 150 million unique genetic variants.
Genotyping
Genomic DNA was extracted from peripheral blood leucocytes in the whole-blood samples, amplified and genotyped using Sanger sequencing. The genotyping was performed blindly without knowledge of patient phenotypes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Error bars represent the standard error to mean (SEM), and statistical significance was determined by unpaired two tailed Student t-test or one-way ANOVA; a p-value of ≤ 0.05 was considered significant. Statistical tests were performed using Prism 6.
DATA AND SOFTWARE AVAILABILITY
The GEO accession number for the RNA-seq and ChIP-seq data reported in this paper is GSE 115421.
Supplementary Material
HIGHLIGHTS.
Patient stem-cell-derived adipocytes differentially respond to antidiabetic drugs
Drug responses are governed by genomic binding of PPARγ
Patient-specific PPARγ genomic binding is controlled by genetic variation
Genetic variation determines and predicts individual drug responsiveness
ACKNOWLEDGEMENTS
We thank Daniel Cohen, Victoria Nelson, Yong Hoon Kim and members of the Lazar lab for technical support and valuable discussions. We thank Amrith Rodrigues and Daniel Rader for technical help with the cholesterol efflux assay. We thank Benjamin Voight for early help with GWAS analysis. We thank the Functional Genomics Core of the Penn Diabetes Research Center (P30 DK19525) for next-generation sequencing. For access to adipose tissue from de-identified, clinically-phenotyped subjects, we thank Dr. Gary B. Korus of the Penn Metabolic and Bariatric Surgery Program, and William Patterson and Julian Hernandez of the Human Metabolic Tissue Resource of the Penn Institute for Diabetes, Obesity, and Metabolism. This work was supported by the Cox Medical Institute, the JPB Foundation (to M.A.L.) as well as by National Institutes of Health grants (R01-DK049780 to M.A.L.; R01-DK098542 to D.J.S.; K08-DK094968 to R.E.S.; F32-DK116519 to D.G.). W.H. was supported by American Diabetes Association Training grant #1-18-PDF-132.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.A.L. is an advisory board member for Eli Lilly and Pfizer Inc., and consultant to Madrigal Pharmaceuticals and Novartis. G.N.N. is cofounder, board member, and consultant to Renalytix AI, and consultant to PulseData and BioVie Inc. The remaining authors declare no competing financial interests.
REFERENCES
- Caballero B (2007). The global epidemic of obesity: an overview. Epidemiologic reviews 29, 1–5. [DOI] [PubMed] [Google Scholar]
- Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, et al. (2001). A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molecular cell 7, 161–171. [DOI] [PubMed] [Google Scholar]
- Chawla A, and Lazar MA (1994). Peroxisome proliferator and retinoid signaling pathways co-regulate preadipocyte phenotype and survival. Proceedings of the National Academy of Sciences of the United States of America 91, 1786–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuffe H, Liu M, Key CC, Boudyguina E, Sawyer JK, Weckerle A, Bashore A, Fried SK, Chung S, and Parks JS (2018). Targeted Deletion of Adipocyte Abca1 (ATP-Binding Cassette Transporter A1) Impairs Diet-Induced Obesity. Arteriosclerosis, thrombosis, and vascular biology 38, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawed AY, Donnelly L, Tavendale R, Carr F, Leese G, Palmer CN, Pearson ER, and Zhou K (2016). CYP2C8 and SLCO1B1 Variants and Therapeutic Response to Thiazolidinediones in Patients With Type 2 Diabetes. Diabetes care 39, 1902–1908. [DOI] [PubMed] [Google Scholar]
- Dispirito JR, Fang B, Wang F, and Lazar MA (2013). Pruning of the adipocyte peroxisome proliferator-activated receptor gamma cistrome by hematopoietic master regulator PU.1. Mol Cell Biol 33, 3354–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Posovszky P, Newell FS, Wabitsch M, and Tornqvist HE (2008). Human SGBS cells - a unique tool for studies of human fat cell biology. Obesity facts 1, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff M, Scott RA, Justice AE, Young KL, Feitosa MF, Barata L, Winkler TW, Chu AY, Mahajan A, Hadley D, et al. (2017). Genome wide physical activity interactions in adiposity - A meta-analysis of 200,452 adults. PLoS genetics 13, e1006528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, Ali F, Sholley C, Worrall C, and Kelman JA (2010). Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone. Jama 304, 411–418. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraike Y, Waki H, Yu J, Nakamura M, Miyake K, Nagano G, Nakaki R, Suzuki K, Kobayashi H, Yamamoto S, et al. (2017). NFIA co-localizes with PPARgamma and transcriptionally controls the brown fat gene program. Nat Cell Biol 19, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlouschek J, Hansel C, Jendrossek V, and Matschke J (2018). The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia. Frontiers in oncology 8, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, et al. (2014). Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Day FR, Gustafsson S, Buchkovich ML, Na J, Bataille V, Cousminer DL, Dastani Z, Drong AW, Esko T, et al. (2016). New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nature communications 7, 10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish J, Aiello RJ, Guyot D, Turi T, Gabel C, Aldinger C, Hoppe KL, Roach ML, Royer LJ, de Wet J, et al. (2000). High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP-binding cassette transporter-1. Proceedings of the National Academy of Sciences of the United States of America 97, 4245–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson VL, Nguyen HB, Garcia-Canaveras JC, Briggs ER, Ho WY, DiSpirito JR, Marinis JM,Hill DA, Lazar MA (2018). PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes & Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF, and Lawn RM (2001). ABCA1. The gatekeeper for eliminating excess tissue cholesterol. Journal of lipid research 42, 1173–1179. [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilik JA, Schramek D, Schnidar H, Cronin SJ, Nehme NT, Zhang X, Knauf C, Cani PD, Aumayr K, Todoric J, et al. (2010). Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 140, 148–160. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nature biotechnology 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblit PD (2016). Common medications used by patients with type 2 diabetes mellitus: what are their effects on the lipid profile? Cardiovascular diabetology 15, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears DD, Hsiao G, Hsiao A, Yu JG, Courtney CH, Ofrecio JM, Chapman J, and Subramaniam S (2009). Mechanisms of human insulin resistance and thiazolidinedione-mediated insulin sensitization. Proceedings of the National Academy of Sciences of the United States of America 106, 18745–18750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, and Lazar MA (2014). Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell metabolism 20, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Fang B, Everett LJ, et al. (2015). Genetic Variation Determines PPARgamma Function and Antidiabetic Drug Response In Vivo. Cell 162, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Tuteja G, Everett LJ, Li Z, Lazar MA, and Kaestner KH (2011). Species-specific strategies underlying conserved functions of metabolic transcription factors. Mol Endocrinol 25, 694–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Step SE, Lim HW, Marinis JM, Prokesch A, Steger DJ, You SH, Won KJ, and Lazar MA (2014). Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARgamma-driven enhancers. Genes Dev 28, 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Lee KY, Dankel SN, Boucher J, Haering MF, Kleinridders A, Thomou T, Xue R, Macotela Y, Cypess AM, et al. (2014). ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Science translational medicine 6, 247ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman BL, Lambert G, Amar M, Joyce C, Ito T, Shamburek RD, Cain WJ, Fruchart-Najib J, Neufeld ED, Remaley AT, et al. (2001). ABCA1 overexpression leads to hyperalphalipoproteinemia and increased biliary cholesterol excretion in transgenic mice. J Clin Invest 108, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Bao YQ, Hu C, Zhang R, Wang CR, Lu JX, Jia WP, and Xiang KS (2008). Effects of ABCA1 variants on rosiglitazone monotherapy in newly diagnosed type 2 diabetes patients. Acta pharmacologica Sinica 29, 252–258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GEO accession number for the RNA-seq and ChIP-seq data reported in this paper is GSE 115421.