

A novel painkiller nanomedicine triggers the specific delivery of enkephalin neuropeptide into inflamed tissues.
Abstract
The clinical use of endogenous neuropeptides has historically been limited due to pharmacokinetic issues, including plasma stability and blood-brain barrier permeability. In this study, we show that the rapidly metabolized Leu-enkephalin (LENK) neuropeptide may become pharmacologically efficient owing to a simple conjugation with the lipid squalene (SQ). The corresponding LENK-SQ bioconjugates were synthesized using different chemical linkers in order to modulate the LENK release after their formulation into nanoparticles. This new SQ-based nanoformulation prevented rapid plasma degradation of LENK and conferred on the released neuropeptide a notable antihyperalgesic effect that lasted longer than after treatment with morphine in a rat model of inflammation (Hargreaves test). The biodistribution study as well as the use of brain-permeant and -impermeant opioid receptor antagonists indicated that LENK-SQ NPs act through peripherally located opioid receptors. This study represents a novel nanomedicine approach, allowing the specific delivery of LENK neuropeptide into inflamed tissues for pain control.
INTRODUCTION
Pain represents an important global health challenge for many reasons, including high prevalence, serious associated sequelae, and the relative lack of efficient treatment, especially for neuropathic pain alleviation. Pain-relevant disorders such as arthritis, cancer, and pathological changes in the nervous system are highly prevalent and bring great inconvenience and distress to the patients (1). Chronic pain has a significant impact not only on the patients themselves but also on the broader community and economy. With the activation of μ-opioid receptors, morphine and the related synthetic opioids become the most powerful and most widely used painkillers in current clinical practice. However, morphinic treatments are associated with severe side effects, such as respiratory depression and addiction linked to the development of opioid tolerance and dependence (2). According to the National Vital Statistics System of the U.S. Centers for Disease Control and Prevention/National Center for Health Statistics (3), every day, more than 115 people in the United States die after overdosing on opioids. The misuse of and addiction to opioids, especially morphine, is a serious national crisis in the United States (and probably also in other countries) that affects public health as well as social and economic welfare. This highlights the need to urgently find new painkillers. In this context, endogenous neuropeptides, such as enkephalin, remain an attractive option. Enkephalins activate both μ- and δ-opioid receptors, but with a 10-fold higher affinity toward δ-opioid receptors (4). Compared with μ-opioid receptor agonists, δ-opioid receptor ligands are believed to have a much lower abuse potential (5), as well as reduced respiratory (6), gastrointestinal (7), and cognitive (8) impairments. However, enkephalins have historically been limited because of pharmacokinetic issues and rapid plasma metabolization.
To date, the two main approaches to enhancing the analgesic activity of opioid peptides relied on (i) the increase of the stability of endogenous peptides using enkephalinase inhibitors or (ii) the chemical synthesis of exogenous peptides with enhanced lipophilicity and degradation resistance. However, because of insufficient enzymatic specificity, the enkephalinase inhibitors are often endowed with poorly tolerated side effects (9). In addition, the derivatization of peptides often ends up with biologically inactive compounds, and the same applies to neuropeptides covalently linked to transport vectors for crossing the blood-brain barrier (BBB) (10). This explains why none of the research efforts performed decades ago has resulted in marketed medicines.
On the other hand, although nanoparticulate drug delivery systems are an efficient approach for protecting drug molecules from rapid metabolization, only a few were applied to enkephalins and enkephalin derivatives. In a primary study, Kreuter et al. (11) showed that intravenously injected dalargin-loaded polybutylcyanoacrylate nanoparticles (NPs) coated with a non-ionic surfactant induced time- and dose-dependent antinociceptive effects. In subsequent studies, positively charged nanocarriers were also used for the delivery of opiate-related drugs and peptides into the brain (12–15). Nevertheless, these approaches have met with limited success because, in general, the amount of NPs able to cross the BBB remains very low (less than 1% of the injected dose) (16). In addition, the toxicity and elimination of NPs from the brain parenchyma remain a major issue; this is especially true for the abovementioned cationic nanodrugs.
Thus, the design of safe analgesic nanoformulations capable of restricting their activity peripherally and optimizing drug concentration at the site of injury may overcome these issues. Another advantage of targeting peripheral opioid receptors is that it prevents and reverses the effects of multiple excitatory agents expressed in damaged tissue (17).
Here, we report a very simple and easy way to use the currently unusable Leu-enkephalin (LENK) as an analgesic drug following intravenous injection. To this goal, a new nanoformulation was achieved, which proved capable of precise and efficient delivery of LENK for pain control. Practically, LENK was conjugated to squalene (SQ), a natural and biocompatible lipid, through various chemical linkers, resulting in a library of LENK lipidic prodrugs, which allowed the controlled release of the peptide. As shown previously with anticancer compounds (18), the linkage of LENK with SQ triggered the spontaneous self-assembly of the bioconjugates into LENK-squalene nanoparticles (LENK-SQ NPs) in water, which was attributed to the dynamically folded conformation of the natural lipid. The analgesic effect of these LENK-SQ NPs was evaluated in a carrageenan-induced pain model using a thermal nociception test (Hargreaves) to assess hyperalgesia. Pain sensitivity was rated in response to a hot stimulus on the inflamed hind paw of rats. In addition, the in vivo biodistribution of NPs was investigated in mice using in vivo fluorescence imaging for assessing the ability of the LENK-SQ NPs to target the inflamed tissue. Last, a toxicological study was also performed to ensure the safety of these NPs.
RESULTS
Synthesis of LENK-SQ conjugates
In this study, various LENK-SQ conjugates were designed with different linkers using bioconjugation (Fig. 1). Conjugation of squalene to LENK was performed by exploiting two sites: C-terminal acid and N-terminal amine. To modulate the release kinetics of LENK from NPs, we used three linkers with different sensitivity to hydrolysis in the following order: dioxycarbonyl (LENK-SQ-Diox also called “sensitive bound”) > diglycolic (LENK-SQ-Dig) > amide (LENK-SQ-Am).
Fig. 1. Chemical structures of the bioconjugates.
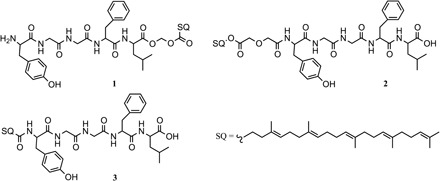
(1) Leu-enkephalin-squalene with dioxycarbonyl linker (LENK-SQ-Diox), (2) Leu-enkephalin-squalene with diglycolic linker (LENK-SQ-Dig), and (3) Leu-enkephalin-squalene with amide linker (LENK-SQ-Am).
Practically, the squalenic acid was coupled to C-terminal LENK using a dioxycarbonyl linker (LENK-SQ-Diox, conjugate 1) or squalenol was conjugated to N-terminal LENK through a diglycolic spacer (LENK-SQ-Dig, conjugate 2). Starting from squalenic acid, it was also possible to perform the linkage to N-terminal LENK using a simple amide bond (LENK-SQ-Am, conjugate 3).
The LENK-SQ-Diox conjugate was synthesized by alkylation of the carboxylate function of the peptide with the chloromethyl ester of squalenic acid, which was prepared upon treatment of squalenic acid with chloromethyl chlorosulfate. To avoid N-terminal conjugation, the Fmoc (9-fluorenylmethoxycarbonyl) strategy was first adopted for the protection of the primary amino group of LENK, but due to the early release of the peptide from Fmoc-LENK-SQ during the deprotection step, this approach was abandoned in favor of the Alloc (allyloxycarbonyl) strategy. Thus, LENK was protected with an Alloc group on its N-terminal amine before reacting with the chloromethyl ester of squalenic acid. Subsequent deprotection of Alloc-LENK-SQ under neutral conditions was then achieved via the catalytic transfer hydrogenation method using triethylsilane (TES) and 10% Pd-C (19), affording pure LENK-SQ-Diox in 9.5% yield.
The LENK-SQ-Dig prodrug with a 2′-diglycolate spacer was synthesized by the reaction of squalenol with diglycolic anhydride before reaction with the condensing agent and LENK, resulting in 69% yield. The oxa moiety of the linker was intended to enhance the susceptibility to hydrolysis by increasing the distance between LENK and SQ and, thus, the accessibility to the linkage. Direct conjugation between LENK and squalenic acid via single amide bond was achieved by acid activation using ethyl chloroformate, affording LENK-SQ-Am in 73% yield.
Preparation and characterization of LENK-SQ NPs
All bioconjugates showed the capability to self-assemble as NPs in aqueous solution after nanoprecipitation from LENK-SQ ethanolic solutions. When measured by dynamic light scattering (DLS), the size of the NPs varied from 60 to 120 nm, depending on the linkage between squalene and enkephalin (Fig. 2). The difference in NP zeta potential was related to the nature of the exposed amino acids onto the NP surface. In the case of the LENK-SQ-Diox bioconjugate, the SQ conjugation on the C-terminal LENK peptide let its N-terminal site free (primary amino group), leading to a net positive charge. In contrast, the zeta potential became negative when the conjugation with SQ was performed on the N-terminal LENK peptide (LENK-SQ-Dig and LENK-SQ-Am). Drug loadings (Fig. 2) ranged between 53 and 60%, which was much higher than in conventional nanoparticles or liposomes, which amounted to a maximum of 5% (20, 21). Figure 2 shows representative cryogenic transmission electron microscopy (cryo-TEM) images of the LENK-SQ NPs at a concentration of 4 mg/ml in Milli-Q water. They displayed spherical and monodisperse structures with sizes ranging from 50 to 100 nm. The slight discrepancy between DLS and cryo-TEM size measurements could be attributed to the known hydrodynamic radius-related differences (22). The sizes and the surface charges of the LENK-SQ NPs were found to be quite stable at +4°C (fig. S6).
Fig. 2. NP characterization.
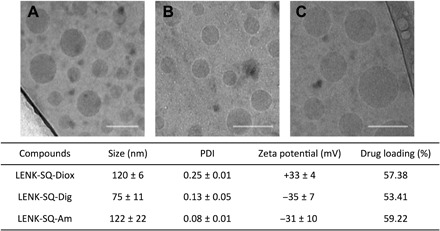
Representative cryo-TEM images showing the formation of NPs from different bioconjugates: (A) LENK-SQ-Diox NPs, (B) LENK-SQ-Dig NPs, and (C) LENK-SQ-Am NPs. Scale bars, 100 nm. Physicochemical characteristics of NPs [i.e., size, polydispersity index (PDI), zeta potential, and % drug loading] are shown in the table.
In vitro release of LENK from NPs in serum
The incubation of LENK-SQ-Diox in serum resulted in a decrease of the bioconjugate, which correlated well with the release of the peptide (Fig. 3A). The concentration of the bioconjugate decreased gradually till 7 hours, while LENK-SQ-Diox NPs progressively released the free LENK peptide. The peptide was then slowly degraded by the peptidases of the serum but still lasted beyond 10 hours after incubation (Fig. 3A). The incubation of LENK-SQ-Dig in serum resulted in a decrease of the bioconjugate until complete disappearance at 2 hours, but no presence of free peptide was detected. The reverse-phase high-performance liquid chromatography (RP-HPLC) analyses, however, highlighted a slow release of the peptide still attached to its linker. This release reached a maximum at 45 min, followed by progressive degradation of the peptide-linker fragment that could still be detected over 10 hours (Fig. 3B). In contrast, LENK-SQ-Am remained stable in serum, without a significant decrease for a period of 48 hours, and no peptide was released in the course of the experiment (Fig. 3C). It was observed that the degradation of the free LENK peptide was very fast (half-life, 2 min), whereas the LENK-SQ bioconjugate was unaffected during the course of the experiment (60 min) (fig. S7).
Fig. 3. In vitro bioconversion of LENK-SQ bioconjugates into LENK in the presence of serum.
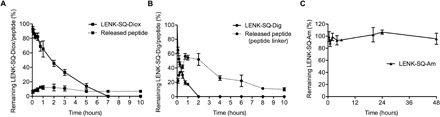
(A) LENK-SQ with dioxycarbonyl linker, (B) LENK-SQ with diglycolic linker, and (C) LENK-SQ with amide bond. Solid lines and dashed lines represent the bioconjugates and the released peptides, respectively.
Analgesic efficacy of LENK-SQ NPs
The antihyperalgesic effect of LENK-SQ NPs was determined in a carrageenan-induced paw edema model in rats. Baseline measurements of paw withdrawal latencies (PWLs) were made before carrageenan injection using the Hargreaves test (23) and presented a mean value ± SEM (n = 8) of 6.65 ± 0.37 s. Then, thermal sensitivities were evaluated 3 hours after carrageenan injection into the right hind paw, which corresponded to the peak inflammatory response. We assessed antihyperalgesic effects using the same test at various times after acute administration of the different drug treatments at this 3-hour inflammation peak (Fig. 4).
Fig. 4. Experimental design for algesimetry.
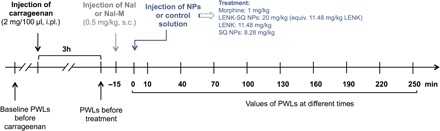
The antinociceptive effect of NPs was tested in a pathophysiological context induced by an intraplantar carrageenan injection (2% saline, 100 μl). Involvement of central or peripheral opioid receptors was performed using a brain-permeant opioid antagonist, naloxone (Nal), and a brain-impermeant opioid receptor antagonist, naloxone methiodide (Nal-M). NP suspensions or control solutions were injected intravenously with a dose volume of 10 ml/kg during 30 s. The Hargreaves test was performed 10 min after the NP administration and then every 30 min up to a period of 250 min. The dose of LENK-SQ NPs (20 mg/kg) was equivalent to LENK (11.48 mg/kg) and to SQ NPs (8.28 mg/kg) and corresponded to 20.66 mmol/kg for both LENK-SQ and LENK. s.c., subcutaneous; i.pl., intraplantar.
Effect of intraplantar λ-carrageenan injection on thermal sensitivity
Intraplantar injection of λ-carrageenan into the right hind paw induced a local inflammatory response characterized by marked edema, hyperthermia, and hyperalgesia restricted to the injected right hind paw. Thermal hypersensitivity was developed in all the rats, with a mean decrease of 52.48% PWL compared to the basal PWLs in naïve rats. (P < 0.001; see Fig. 5).
Fig. 5. Antihyperalgesic effect of LENK-SQ NPs and morphine.
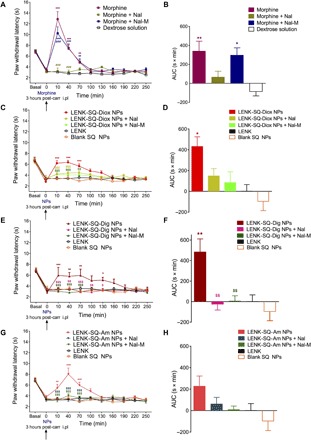
Antihyperalgesic effects of acute treatment with morphine (A and B), LENK-SQ-Diox NPs (C and D), LENK-SQ-Dig NPs (E and F), and LENK-SQ-Am NPs (G and H) in λ-carrageenan–induced inflammatory pain injected rats. Administration of morphine, LENK-SQ NPs, Nal, Nal-M, LENK, blank SQ NPs, or dextrose solution (vehicle) was performed (arrows, 0 on abscissa) 3 hours after λ-carrageenan injection into the right hind paw. Morphine (A), LENK-SQ-Diox NPs (C), LENK-SQ-Dig NPs (E), and LENK-SQ-Am NPs (G) induced an increase in PWL (in seconds, means ± SEM of independent determinations in five to nine animals per group) in the Hargreaves test. *P < 0.05, **P < 0.01, ***P < 0.001, compared to dextrose solution or LENK solution; ###P < 0.001, compared to morphine; $P < 0.05, $$P < 0.01, $$$P < 0.001, compared to LENK-SQ NPs. Two-way analysis of variance (ANOVA) with repeated measures, Bonferroni post test. Nal or Nal-M was administered 15 min before morphine or LENK-SQ NP injection. Basal on abscissa: Control (naïve) rats (before λ-carrageenan injection). (B, D, F, and H) Bars are the means ± SEM of AUCs (seconds × minutes) of the cumulative durations derived from the time course changes (A, C, E, and G) in PWL after the various treatments. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA, Tukey post test, compared to dextrose (vehicle) or LENK solution; $P < 0.05, $$P < 0.01, $$$P < 0.001, compared to LENK-SQ NPs.
Effect of morphine on thermal hyperalgesia
The acute treatment with morphine (1 mg/kg; Fig. 5A) reduced the thermal hyperalgesia, as shown by the resulting significant increase in PWLs. Ten minutes after morphine injection, the PWL reached 12.87 ± 1.38 s, while it remained at 3.05 ± 0.20 s after treatment with a control dextrose solution (Fig. 5A). However, morphine antihyperalgesic pharmacological activity disappeared rapidly, and no significant effect was observed as soon as 100 min after morphine administration (Fig. 5A).
Effect of LENK-SQ NPs on thermal hyperalgesia
We evaluated the antihyperalgesic effect of LENK-SQ NPs with the three different linkers during 4 hours after their administration (Fig. 5, C to H). All injected rats with LENK-SQ NPs displayed significant reduction of thermal hyperalgesia, as expressed by a marked increase of the respective area under the curve (AUC) values in comparison with λ-carrageenan–treated rats injected with either the free LENK peptide or the blank SQ NPs (Fig. 5, D, F, and H). In particular, the antihyperalgesic activity was significant at all time points from 10 to 130 min in rats injected with LENK-SQ Diox NPs or LENK-SQ Am NPs (Fig. 5, C and G). As shown in Fig. 5E, LENK-SQ-dig NPs also displayed a significant antihyperalgesic effect, with a maximum increase in PWL maintained from 10 to 130 min after injection, and a progressive decline down to baseline at 220 min. Maximal PWL values reached after the administration of LENK-SQ NPs in λ-carrageenan–treated rats corresponded to basal PWL values measured in control naïve rats, before λ-carrageenan treatment (Fig. 5, C, E, and G), indicating a pure antihyperalgesic action of these NPs. In contrast, morphine injection in λ-carrageenan–treated rats resulted in PWL values twice as high as those found in control naïve rats (Fig. 5A), as expected from not only an antihyperalgesic effect but also the well-established analgesic effect of the opiate agonist. In addition, blank SQ NPs (without the LENK) did not demonstrate any antihyperalgesic activity (Fig. 5), which indicated that the analgesic response to LENK-SQ NP administration resulted from the release of the LENK peptide.
Effects of opioid receptor blockade using naloxone and naloxone methiodide
To ascertain the involvement of central or peripheral opioid receptors during the antihyperalgesic effect of LENK-SQ NPs, we subcutaneously injected naloxone (Nal, a brain-permeant opioid receptor antagonist) or naloxone methiodide (Nal-M, a brain-impermeant opioid receptor antagonist) (24) 15 min before the injection of morphine or NPs (Fig. 4). Preadministration of the nonselective opioid receptor antagonist Nal (0.5 mg/kg subcutaneously) abolished the amplitude and the duration of the antihyperalgesic effect of morphine (PWL, 3.20 ± 0.59 s versus 12.87 ± 1.38 s at 10 min) and decreased the corresponding AUC value by 81% in comparison with the morphine group (Fig. 5B). The peripheral opioid receptor antagonist, Nal-M, was markedly less effective since it reduced the morphine’s effect by only 13% (Fig. 5, A and B). Preadministration of either Nal or its quaternary derivative Nal-M abrogated the antihyperalgesic effect of the three LENK-SQ NPs (Fig. 5, C to H). Nal pretreatment caused a reduction of 66, 105, or 73% AUC values compared to these found in rats injected with LENK-SQ-Diox, LENK-SQ-Dig, and LENK-SQ-Am NPs alone, respectively. The corresponding reductions in AUC values with Nal-M reached 81, 99, and 96%, respectively, indicating that the selective blockade of only the peripheral opioid receptors was enough to abrogate the antihyperalgesic effects of LENK-SQ NPs.
Biodistribution of LENK-SQ NPs
We assessed the in vivo biodistribution of LENK-SQ-Am NPs after intravenous injection of fluorescent DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt)–labeled LENK-SQ-Am NPs in a murine λ-carrageenan–induced paw edema model (right hind paw). The fluorescence in tissues was monitored up to 24 hours, noninvasively, from the abdomen side using an IVIS Lumina (Fig. 6). Mice injected with saline into the paw were used as non-inflamed control. The real-time in vivo imaging showed, in comparison with the healthy paw, an increase of two to three times that of the average radiant efficiency within the inflamed paw after intravenous injection of fluorescent LENK-SQ-Am NPs (Fig. 6, A, D, and F). In a control experiment, when the λ-carrageenan–administered mice were intravenously injected with a single DiD solution, no significant accumulation of fluorescence was observed in the inflamed paw (Fig. 6, C and F). In another control experiment, mice were injected locally with saline in the hind paw and intravenously treated with fluorescent LENK-SQ NPs. No significant accumulation of fluorescence at the hind paw level was observed under this condition (Fig. 6, B and E), showing that the accumulation of fluorescence in the λ-carrageenan–inflamed paw was not due to the local hind paw injection per se. In an additional experiment, it was shown that the incubation in serum of LENK-SQ NPs containing the fluorescent dye (DiD) and a fluorescence quencher [DiR (1,1′-dioctadecyltetramethyl indotricarbocyanine iodide)] resulted in the progressive appearance of fluorescence, the dynamics of which indicate a relatively slow dissociation of LENK-SQ NPs in serum: 20% after 5 min and 50% after 30 min. This suggested that, under our in vivo conditions, a significant proportion of intact NPs could reach the inflamed tissue (fig. S8).
Fig. 6. IVIS Lumina scan of mice and of their organs after intravenous administration of fluorescent LENK-SQ-Am NPs or control fluorescent dye solution (ventral view).
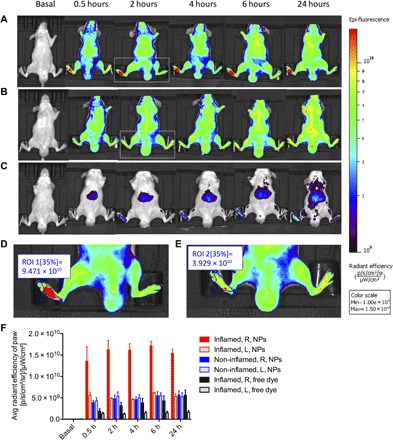
(A) Biodistribution of fluorescent LENK-SQ-Am NPs in mice with inflamed right hind paw. (B) Biodistribution of fluorescent LENK-SQ-Am NPs in mice with non-inflamed hind paw (saline injected only into the right hind paw). (C) Biodistribution of free dye in mice with inflamed right paw. (D) Zoom of group A at 2 hours. (E) Zoom of group B at 2 hours. (F) Quantitative analysis of the paws with the same region of interest (ROI). R, right hind paw; L, left hind paw.
Last, in a separate experiment, 4 hours after the intravenous injection of fluorescent NPs or DiD solution, animals were euthanized and transcardially perfused with 40 ml of saline to remove the fluorescence from the blood. After collection of tissues, a strong ex vivo fluorescence signal was again observed not only in the inflamed paw but also in the liver, the spleen, and the lungs, whereas no detectable accumulation of fluorescence occurred in the brain of the animals (fig. S9).
Toxicity study
The overall toxicity of LENK-SQ NPs was investigated 24 and 48 hours after their intravenous administration (20 mg/kg) in rats and compared to control animals injected with 5% dextrose solution. The levels of aspartate transaminase (AST; fig. S10A) and alanine transaminase (ALT; fig. S10B) were not increased in the LENK-SQ NP group, indicating no toxicity toward the liver, which was confirmed by histological analysis of this tissue at 24 and 48 hours (fig. S10, C to F). The observations of the spleen (fig. S10, G to J), the kidneys (fig. S10, K to N), the lungs (fig. S10, O to R), and the heart (fig. S10, S to V) did not show any morphological damage after LENK-SQ NP administration either. Together, these results show that the LENK-SQ NPs may be considered as safe upon systemic intravenous administration at the therapeutic dose of 20 mg/kg.
DISCUSSION
Peptides and proteins have great potential as therapeutic macromolecules. However, their use in clinical practice is generally hampered by poor serum stability and rapid metabolization (25). In this context, the “squalenoylation” technology should be of great interest, as we showed here that it allows the delivery of therapeutic amounts of LENK neuropeptide for efficient pain control. The so-called squalenoylation approach, which refers to the linkage of a drug to the squalene, has already been proposed for small molecules, mainly for anticancer compounds, such as gemcitabine, doxorubicin, or cisplatin (26), or for other molecules, such as adenosine, ddI, or ddC (27). Nevertheless, the conjugation of a peptide to SQ has been an innovative but tricky achievement for the following reasons: (i) Peptides are unstable biomolecules, and their chemical engineering is not easy, and (ii) peptides are hydrophilic molecules, whereas SQ is a lipid, insoluble in water. This makes the chemical reaction rather uncertain, and (iii) the chemical modification of a peptide often results in a loss of its pharmacological activity. The chemical approach used in the present study has overcome all these complications, allowing the design of a small library of innovative enkephalin-squalene bioconjugates with a preserved pharmacological activity. For the synthesis of these bioconjugates, we took advantage of the remarkable, dynamically folded conformation of SQ to chemically conjugate this natural lipid with the neuropeptide using different linkers. Although it is well known that the N terminus of LENK is required for binding to opioid receptors (28), the conjugation on N terminus was also achieved, based on the fact that the amide bond is susceptible to be cleaved by overexpressed peptidases within an inflammation site (29). Thus, we synthesized the resulting LENK-SQ prodrugs with either direct amide bond or diglycolic or dioxycarbonyl linker to investigate the possible influence of the linkage stability on the peptide release. It was expected that LENK-SQ-Diox released faster than LENK-SQ-Dig, and LENK-SQ-Am was supposed to trigger the slower release as reported in the literature (30). Both LENK-SQ-Am and LENK-SQ-Dig were obtained in good yields (around 70%). The third one with dioxycarbonyl linker led to a lower yield due to an additional step for removal of the Alloc group, followed by two successive purifications.
The LENK-SQ bioconjugates were then formulated as nanoparticles in dextrose solution (2 mg/ml) with impressively high drug payload (i.e., 53 to 59%) using a simple nanoprecipitation technique without the aid of any surfactant. It is noteworthy that this drug payload was markedly higher than that in liposomes (20) or PLGA [poly(lactic-co-glycolic acid)] (21) enkephalin-loaded nanoparticles (0.4 and 4.75%, respectively, drug loading), which might explain why the latter two nanoformulations had never been used for in vivo pharmacological studies. The size of the NPs varied from 61 to 112 nm, depending on the peptide and the conjugation site (Fig. 2). The NPs displayed spherical and monodisperse structures with net positive or negative surface charge (Fig. 2), which could be attributed to the free terminal function of the peptide depending on the bioconjugation mode. Free N-terminal amine function led to a net positive surface charge, while free C-terminal acid function resulted in a net negative surface charge.
To ascertain that the free LENK peptide could be released from the LENK-SQ NPs, we tested the chemical stability of the different linkers (i.e., direct amide or dioxycarbonyl or diglycolate spacers) after incubation of the LENK-SQ NPs with mouse serum (Fig. 3). Corresponding experiments showed that the LENK peptide was released from LENK-SQ-Diox and LENK-SQ-Dig but not from LENK-SQ-Am (Fig. 3). In the case of LENK-SQ-Diox and LENK-SQ-Dig, the release of the respective LENK and LENK linker fragment was followed by a progressive degradation of the peptide, due to serum enkephalinases (Fig. 3, A and B). In the case of the LENK-SQ-Diox bioconjugate, both the LENK and the SQ moieties were each linked to the dioxycarbonyl linker through an ester bond. In the case of the LENK-SQ-Dig bioconjugate, the diglycolate linker was attached on one side to squalene by an ester bond and on the other side to the LENK through an amide bond; in the LENK-SQ-Am bioconjugate, a direct amide bond connected the LENK to the SQ moiety. The absence of release of LENK from LENK-SQ-Am was expected given that, in general, an amide bond is chemically and enzymatically more stable than an ester bond (31, 32). Furthermore, it is well known that N-terminal modification of linear peptides increases the peptidase resistance, which is the case for LENK-SQ-Am and LENK-SQ-Dig (33). Last, as mouse serum is particularly rich in esterases, it was expected that the release of LENK from LENK-SQ NPs mainly resulted from enzymatic hydrolysis of the ester bond (31). All three conjugates were then recruited for antihyperalgesia experiments. It was expected that the more aggressive in vivo enzymatic content, particularly rich in proteolytic enzymes at the inflammation site, will contribute to the release of LENK from all the bioconjugates (29). The presence of high concentrations of proteolytic enzymes such as chymotrypsin, cathepsin D, and other proteases in inflammatory exudates has been reported, which also indicates their important role in the inflammatory process (29).
Then, the antihyperalgesic properties of LENK-SQ NPs were evaluated using an animal model of inflammatory hyperalgesia that mimics human clinical pain conditions (34). We assessed the antihyperalgesic activity after a single intravenous administration of the different LENK-SQ NPs in the λ-carrageenan–induced inflammatory paw model using the Hargreaves test. All LENK-SQ NPs displayed a significant antihyperalgesic effect on inflamed hind paw. The antihyperalgesic effect was less intense than after morphine treatment, but NPs evidenced a much longer-lasting effect. Unexpectedly, LENK-SQ-Am NPs, which was expected to release the peptide slower in comparison with the other two NPs, exhibited a stronger effect with a shorter duration, probably because the enzymatic serum capability is not predictive of the enzymatic ecosystem in the inflamed paw. LENK-SQ-Dig and LENK-SQ-Diox NPs had nearly the same antihyperalgesic profile with prolonged effect, resulting in a significantly higher AUC than morphine and LENK-SQ-Am NPs. In particular, the LENK-SQ-Dig NPs showed an antihyperalgesic effect that lasted twice as long as morphine. In addition, as expected for an analgesic compound, PWL values after morphine treatment in λ-carrageenan–treated rats exceeded basal values in naïve healthy rats, whereas, in contrast, under the conditions used here, PWL values after LENK-SQ NPs just reached these basal values (Fig. 5). This would suggest that LENK NPs are devoid of analgesic properties but are especially potent to counteract hyperalgesia in subjects suffering from chronic pain. However, further studies are required to assess this hypothesis.
Pretreatment with Nal (a prototypical opioid antagonist) prevented the increase in PWL evoked by LENK-SQ NPs and morphine, indicating that the antihyperalgesic effect of all these compounds was mediated by opioid receptors. On the other hand, pretreatment with Nal-M [which does not cross the BBB (24)] only marginally decreased the antihyperalgesic effect of morphine, suggesting that the drug acted mainly through central and, to a lesser extent, peripheral opioid receptors. In contrast, Nal-M pretreatment abolished the antihyperalgesic effect of LENK-SQ NPs, which demonstrated that all three LENK-SQ NPs acted exclusively through peripherally located opioid receptors.
To investigate the ability of LENK-SQ NPs to address the peptide toward the inflamed tissue, biodistribution studies were performed using in vivo fluorescence imaging in a mouse carrageenan-induced paw edema model. Our data highlighted the ability of the NPs to gain access to the peripheral inflamed tissue. Intravenous injection of fluorescent DiD–labeled LENK-SQ NPs in inflammation-bearing mice resulted in a marked increase of fluorescence within the inflamed hind paw, up to a level that is threefold higher than that in the contralateral non-inflamed paw or in the paw of mice treated with saline only (instead of λ-carrageenan). The very low accumulation of fluorescence in the non-inflamed paw and in the brain confirmed that the antihyperalgesic effect resulted from the targeting of LENK-SQ NPs toward peripheral opioid receptors in inflamed tissue rather than toward central opioid receptors. It is likely that the LENK-SQ distributed within the inflamed area as NPs rather than in a single LENK-SQ molecular form. Whole-body imaging in Fig. 6 shows strong fluorescence in the inflamed paw, which was not the case when the fluorescent dye was injected as a free compound. In addition, after incubation of LENK-SQ NPs in serum, a significant proportion of NPs remained intact (fig. S8). Last, safety of LENK-SQ NPs after intravenous injection was confirmed by normal levels of transaminases and normal histology of vital organs.
Thus, the novelty of the approach resulted from the unexpected ability of the peptidic NPs to target the small area of the body where inflammation and nociception occur. As this resulted in an effective pain alleviation, which lasted even longer than with morphine and avoided any diffusion into the CNS, such properties of LENK-SQ NPs might open novel perspectives for pain management.
CONCLUSION
On the basis of the bioconjugation of LENK to squalene, we describe here a new nanoformulation capable of precise and efficient delivery of LENK for pain control associated with inflammatory events. Our data demonstrated that the antihyperalgesic activity of LENK-SQ NPs took place at the level of peripheral opioid receptors. The experimental approach to making these nanoparticles is simple and easy (i.e., it does not require any complicated nanoparticle surface functionalization), which should facilitate further pharmaceutical development and clinical translation. Although further studies are needed to more precisely determine how dosage, administration frequency, and timing of treatment with LENK-SQ may affect the clinical outcome, this study opens a new exciting perspective for an efficient treatment of intense pain, which evades the severe side effects associated with morphine or related synthetic opioids. Last, because of the versatility of the approach, the application of this delivery system to other therapeutic peptide molecules may be reasonably envisioned.
MATERIALS AND METHODS
Materials
All the chemicals used were of analytical grade. Squalene (SQ), diglycolic anhydride, ethyl chloroformate, chloromethyl chlorosulfate, ammonium acetate, n-Bu4NHSO4, TES, and triethylamine (TEA) were purchased from Sigma-Aldrich (France). Pd-C was obtained from Alfa Aesar (France). DiD and DiR were purchased, respectively, from PromoKine (Germany) and Interchim (France). Roti-Histofix 4% (formaldehyde) was provided by Roth (Germany).
All the drugs used were of analytical grade. Morphine sulfate salt pentahydrate, λ-carrageenan, naloxone hydrochloride (Nal, μ, δ, and κ opioid receptors antagonist), and naloxone methiodide (Nal-M, a nonspecific opioid receptor antagonist that does not cross the BBB) were purchased from Sigma-Aldrich (Saint-Quentin-Fallavier, France). Ketamine and xylazine were purchased from Centravet (Maisons-Alfort, France). LENK and Alloc-LENK were purchased from Ontores Biotechnologies (Zhejiang, China).
General information on chemicals
Analytical thin-layer chromatography was performed on Merck silica gel 60F254 glass precoated plates (0.25 mm layer). Column chromatography was performed on Merck silica gel 60 (230-400 mesh). HPLC water was purified using a Milli-Q system (Millipore, France). Tetrahydrofuran (THF) was distilled from sodium/benzophenone ketyl. Dimethylformamide (DMF), dichloromethane (DCM), and pyridine were dried on calcium hydride (CaH2) before distillation under an argon atmosphere. Methanol (MeOH) was dried over magnesium and distilled. All reactions involving air- or water-sensitive compounds were routinely conducted in glassware, which was flame-dried under a positive pressure of nitrogen. HPLC-grade acetonitrile (ACN), MeOH, ethanol (EtOH), and ethyl acetate (AcOEt) were provided by Carlo Erba (Rodano, Italy).
Synthesis of LENK-SQ-Diox
1,1′,2-Tris-norsqualenic acid chloromethyl ester
1,1′,2-Tris-norsqualenic acid was synthesized by oxidation of 1,1′,2-tris-norsqualenic aldehyde by Jones reagent as previously reported (35, 36). A solution of KHCO3 (300 mg, 3.0 mmol) in water (2 ml) was added to a solution of 1,1′,2-tris-norsqualenic acid (400 mg, 1 mmol) and n-Bu4NHSO4 (34 mg, 0.1 mmol) in DCM (2 ml). The reaction mixture was vigorously stirred, and chloromethyl chlorosulfate (185 mg, 1.15 mmol) was added dropwise. After stirring for 1 hour, DCM (10 ml) was added to extract the product. The organic phase was separated, washed with brine, dried over magnesium sulfate, and concentrated under reduced pressure to afford a pale yellow oil that was used in the following step without further purification.
Alloc-Leu-enkephalin-squalene (Alloc-LENK-SQ-Diox)
The 1,1′,2-tris-norsqualenic acid chloromethyl ester (200 mg, 0.445 mmol) was added into a mixture of Alloc-LENK (285 mg, 0.445 mmol) and NaHCO3 (37 mg, 0.4 mmol) in 3 ml of DMF. The reaction mixture was stirred at 40°C under argon for 4 days. The final reaction mixture was concentrated in vacuo, and the residue was purified by flash column chromatography on silica gel DCM/EtOH (100:0 to 97:3) to afford the title compound as a pale yellow oil (168 mg, 40% yield).
Leu-enkephalin-squalene with dioxycarbonyl linker (LENK-SQ-Diox)
TES (1215 mg, 10 mmol) was added dropwise neat to a stirred solution of Alloc-LENK-SQ (110 mg, 0.1 mmol) and 10% Pd-C (20% by weight of Alloc-LENK-SQ-Diox) in MeOH (11 ml) under argon. When the reaction was completed, the mixture was filtered through celite to remove the Pd-C, and the residual TES and solvent were removed by evaporation. The residue was first purified by flash column chromatography on silica gel with DCM/EtOH (90:10). The resulting product was dissolved in 200 μl of EtOH before undergoing a second purification via the semi-preparative RP-HPLC system (Waters, MA, USA) on an Uptisphere C18 column (100 mm by 21.2 mm; pore size, 5 μm; Interchim, CA, USA) to obtain the pure product (23 mg; 23% yield). HPLC was then performed using a gradient elution with the mobile phase composed of an ammonium acetate buffer (20 mM) and ACN. Elution was carried out at a flow rate of 21 ml/min for 10 min with the linear gradient from 10 to 100% ACN, and then, the system was held at 100% ACN with isocratic flow for 10 min. Temperature was set at 30°C, and ultraviolet (UV) detection was monitored at 280 and 257 nm. The retention time was 15 min, and the total yield of the pure product, after coupling and deprotection steps, corresponded to 9.5%.
Synthesis of LENK-SQ-Dig
1,1′,2-Tris-norsqualenol was synthesized from squalene via 1,1′,2-tris-norsqualenic aldehyde according to previously reported methods (35, 36). Diglycolic anhydride (150 mg, 1.29 mmol) was added to a solution of 1,1′,2-tris-norsqualenol (200 mg, 0.52 mmol) in 3 ml of dry pyridine. The reaction was stirred overnight at room temperature. The solvent was removed and the residue was extracted with DCM from dilute hydrochloric acid and brine. Conversion to the squalene–diglycolic acid, monitored by TLC, was approximately 100%. The resultant product was dried under vacuum and used in the following step without further purification. Ethyl chloroformate (10.8 mg, 0.1 mmol) was added to a solution of squalene–diglycolic acid (50 mg, 0.1 mmol) and TEA (12 mg, 0.12 mmol) in 1 ml of anhydrous THF under argon at 0°C. The reaction was stirred for 1 hour at room temperature, and a solution of LENK (55 mg, 0.1 mmol) in 1 ml anhydrous DMF was added. The mixture was maintained at 40°C for 2 days with stirring under argon. The solvents were removed in vacuo, and the crude product was purified using silica gel chromatography (purified with gradient eluent DCM/EtOH: 100:0 to 90:10). Then, ammonium salt was eliminated by simple filtration on silica using EtOH/AcOEt (40:60) as solvents. The pure bioconjugate was obtained with 69% yield.
Synthesis of LENK-SQ-Am
1,1′,2-Tris-norsqualenic acid (100 mg, 0.25 mmol) and TEA (34.79 mg, 0.3 mmol) were dissolved in 1.5 ml of anhydrous THF under argon, and ethyl chloroformate (27 mg, 0.25 mmol) was added to the mixture at 0°C. The reaction was allowed to warm at room temperature and kept under stirring for 1 hour. A solution of LENK (138 mg, 0.25 mmol) in 1.5 ml of anhydrous DMF was then added to the reaction, and the mixture was kept under stirring for 2 days. The solvents were removed in vacuo, and the crude product was purified twice using silica gel chromatography [purification with gradient eluent DCM/EtOH (100:0 to 90:10) and then simple filtration with EtOH/AcOEt (40:60) to remove the ammonium salt]. The pure bioconjugate was obtained with 73% yield.
Preparation and characterization of LENK-SQ NPs
Preparation of nanoparticles
LENK-SQ NPs were prepared using the nanoprecipitation methodology. Briefly, the LENK-SQ bioconjugate (i.e., LENK-SQ-Diox, LENK-SQ-Dig, or LENK-SQ-Am) was dissolved in EtOH (8 mg/ml) and added dropwise under stirring (500 rpm) into a 5% aqueous dextrose solution (EtOH/dextrose solution volume ratio, 1:4). The solution became spontaneously turbid with a Tyndall effect, indicating the formation of the nanoparticles. EtOH was then completely evaporated using a Rotavapor (80 rpm, 30°C, 30 mbar) to obtain an aqueous suspension of pure LENK-SQ NPs (final concentration, 2 mg/ml). Blank SQ NPs (LENK-free NPs) were prepared by the same method as described above by adding dropwise an ethanolic solution of squalenic acid into 5% aqueous dextrose solution. Fluorescently labeled LENK-SQ NPs were also obtained by the same procedure, except that the fluorescent probe DiD was solubilized in the ethanolic phase together with the LENK-SQ-Am bioconjugate (DiD/LENK-SQ-Am ratio was 4 wt %) before addition to the dextrose solution. Fluorescence quencher LENK-SQ NPs were also prepared by the same way using DiR as a fluorescent probe (DiD/DiR/LENK-SQ-Am ratio was 2:2:100 wt). The peptide drug loadings into the NPs were expressed as percentage (%), calculated from the ratio between LENK peptide Mw and LENK-SQ bioconjugate Mw. The LENK-SQ NPs were regularly observed by cryo-TEM. All the NPs were freshly prepared and used within 2 hours (conservation at 4°C) before in vivo experiments.
DLS measurements
The mean particle size, polydispersity index (PDI), and zeta potential were primarily evaluated by DLS (Nano ZS, Malvern; 173° scattering angle at 25°C). The measurements were performed in triplicate following appropriate dilution of the NPs in water (DLS) or in 0.1 mM KCl (zeta potential). The results represent the mean and SD of three repeated sample preparations or more.
Cryo-TEM
The morphology of the LENK-SQ NPs was investigated by cryo-TEM. NPs were vitrified using a chamber designed and set up in the laboratory where both humidity and temperature could be controlled. Four microliters of solution of LENK-SQ NPs (4 mg/ml in Milli-Q water) was deposited onto a perforated carbon film mounted on a 200-mesh electron microscopy grid. The homemade carbon film hole dimensions were about 2 mm in diameter. Most of the drop was removed with a blotting filter paper, and the residual thin films remaining within the holes were quick-frozen by plunging them into liquid ethane cooled with liquid N2. The specimen was then transferred, using liquid N2, to a cryo-specimen holder and observed using a JEOL FEG-2010 electron microscope. Micrographs were recorded at 200 kV under low-dose conditions at a magnification of ×40,000 on SO-163 Kodak films. Micrographs were digitized using a film scanner (Super Coolscan 8000 ED, Nikon), and analyses were made using the ImageJ software.
In vitro LENK release from NPs in serum
Frozen serum of male SWISS mice (900 μl) was quickly thawed and then preincubated at 37°C for 30 min before the addition of 300 μl of LENK-SQ-Dig NPs or LENK-SQ-Am NPs (2 mg/ml). In the case of LENK-SQ-Diox NPs, diluted serum (30% in 5% dextrose solution) was used for the release study. At various time intervals, aliquots (80 μl) were collected and added into 320 μl of ACN to denature and precipitate the enzymes and proteins of the serum, to remove them after centrifugation (3000g for 15 min). To quantify the residual LENK-SQ bioconjugate and the released LENK, the resulting supernatants (150 μl) were evaporated to dryness at 40°C under nitrogen flow and then solubilized in 150 μl of Milli-Q water. Free peptide quantification was performed using RP-HPLC on an Uptisphere Strategy C18HQ column (4.6 mm by 100 mm, 5 μm; Interchim), a 1525 Binary LC Pump (Waters), a 2707 Auto-sampler (Waters), and a 2998 PDA detector (Waters). The HPLC was carried out using a gradient elution with the mobile phase composed of 5 mM ammonium acetate in Milli-Q water (phase A) and 5 mM ammonium acetate in ACN (phase B). Elution was carried out at a flow rate of 1 ml/min for 13 min with the linear gradient from 10 to 100% of B; then, the system was held at 100% of B with isocratic flow for 10 min. Temperature was set at 35°C, and UV detection was monitored at 257 nm. The detection limit of the HPLC technique was 0.39 μg/ml for the peptide. This method exhibited linearity (R2 = 0.99998) over the assayed concentration range (0.39 to 200 μg/ml) and demonstrated good precision with relative SD being all less than 2.01%. The accuracy corresponded to 96 ± 5%.
General information on in vivo study
Animals
Adult male Sprague-Dawley rats (200 to 220 g on arrival, 280 to 300 g at the time of experiments) and adult male Swiss mice (18 to 20 g on arrival, 22 to 25 g at the time of experiments) were purchased from JANVIER LABS (France) for algesimetry tests and biodistribution, respectively. They were housed in a standard controlled environment (22° ± 1°C, 60% relative humidity, 12-hour light/12-hour dark cycle, lights on at 8:00 a.m.) with food and water available ad libitum, without any handling for at least 1 week before being used for experiments. In all cases, experiments were performed in conformity with the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain (37) and approved by the Animal Care Committee of the University Paris-Sud in accordance with the principles of laboratory animal care and the European legislation 2010/63/EU. All efforts were made to reduce animal numbers and minimize their suffering, as defined in the specific agreement (registered under no. 7493-2016102520355414).
Carrageenan-induced paw edema model
Since the demonstration that indomethacin reduces inflammation caused by intraplantar injection of λ-carrageenan, this acute model is widely accepted for screening compounds with anti-inflammatory potentialities (38). λ-Carrageenan was dissolved in physiological saline (NaCl, 0.9%) just before injection. Rats or mice received a single intraplantar injection of λ-carrageenan solution in the plantar region of the right hind paw (23, 39) to induce inflammation. The injected λ-carrageenan dose corresponded to 100 μl (2% solution, w/v) for rat and 20 μl (3% solution, w/v) for mice. Inflammation reached its maximum 3 hours after λ-carrageenan injection. Thermal nociceptive test was then performed on the ipsilateral inflamed hind paw.
Nociceptive behavioral study in rats
Thermal nociceptive test
Hypersensitivity to thermal nociceptive stimuli was assessed using the Hargreaves test (23). Rats were placed individually in an open Plexiglas cylindrical chamber (20 cm in diameter, 35 cm high) on a 3-mm-thick transparent glass floor and allowed to habituate for at least 20 min before testing. A movable radiant heat source (Model 7370; Ugo Basile Plantar Test, Italy) was positioned under the glass floor directly beneath the plantar surface of the right hind paw, and the time (in seconds) that elapsed from switching on the radiant heat until paw withdrawal was measured automatically. A cutoff time of 20 s was established to prevent tissue damage. Each trial was repeated three times with 5-min intervals for basal threshold and two times spaced (2-min intervals) after NP treatments 3 hours after λ-carrageenan injection. The average of PWLs was calculated and expressed as means ± SEM.
Experimental design for algesimetry test
Basal responses to thermal stimuli were obtained on the day before the λ-carrageenan injection. On the basis of previous studies, acute pharmacological treatments were performed 3 hours after carrageenan injection, which corresponded to the peak inflammatory response. The efficacy of these treatments on thermal hyperalgesia was evaluated by measurement of PWLs using the Hargreaves test at regular time intervals after drug or vehicle administration, first at 10 min and then each at 30 min for a period of 4 hours (Fig. 4).
Acute pharmacological treatments
Morphine and LENK were dissolved in 5% dextrose solution, whereas Nal and Nal-M were dissolved in physiological saline (NaCl, 0.9%). All these drugs and NP suspensions were prepared just before administration. All acute treatments were performed 3 hours after λ-carrageenan intraplantar injection according to Fig. 4. Nal and Nal-M were injected subcutaneously, whereas the intravenous route in the tail vein was used for LENK-SQ NPs, LENK, and their controls. The antagonist (Nal or Nal-M) was administered 15 min before the agonist (morphine or tested NPs). A single dose of morphine (1 mg/kg), Nal (0.5 mg/kg), and Nal-M (0.5 mg/kg) was administered on the basis of literature data (40). A single intravenous dose of LENK-SQ NPs (20 mg/kg, equivalent to 11.48 mg/kg of LENK) or control unconjugated SQ NPs (8.28 mg/kg) was used based on the maximal volume of LENK-SQ NPs that could be injected.
Biodistribution study in mice
In vivo imaging biodistribution studies were performed after intravenous injection of fluorescent LENK-SQ-Am NPs (250 μl, 2 mg/ml containing 4% DiD) or control fluorescent DiD solution (250 μl, 80 μg/ml in 5% dextrose solution) in shaved mice bearing λ-carrageenan–induced inflammation. In parallel, control non-inflamed shaved mice (injected with 20 μl of saline into the right hind paw instead of λ-carrageenan) also received injection of fluorescent LENK-SQ NPs. The biodistribution of the NPs was recorded at 0.5, 2, 4, 6, and 24 hours with the IVIS Lumina LT Series III system (Caliper Life Sciences) using 640 nm excitation and 695 to 775 nm emission filters, respectively. During imaging, the mice were kept on the imaging stage under anesthesia with 2% isoflurane gas in oxygen flow (1 liter/min) and were imaged in ventral position. Images and measures of fluorescence signals were acquired and analyzed with Living Imaging. To measure photon radiance, regions of interest (ROIs; threshold of 35%) were selected on the paw of the mice, and average radiant efficiency values were used for quantification. Threshold of ROI for the inflamed paw was then pasted on the non-inflamed paw to compare the radiance with the same region.
In a separate experiment, fluorescent LENK-SQ NP–injected mice were deeply anesthetized with a mixture of ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) before euthanasia by transcardiac perfusion of 40 ml of saline (8 ml/min), until the fluid exiting the right atrium was entirely clear. Then, liver, spleen, kidneys, heart, lungs, brain, and inflamed right hind paw were excised and immediately imaged with the imager. The fluorescence emitted was quantified with Living Image software over the ROI (threshold of 20%).
Toxicity study in rats
Adult male Sprague-Dawley rats were injected with either LENK-SQ-Am NPs (20 mg/kg) or 5% dextrose (n = 3 animals per group). At 24 or 48 hours after injection, the animals were anesthetized with 2% isoflurane gas in oxygen flow (1 liter/min), and blood was collected from tail blood vessels by anticoagulant (heparin, 500 UI/ml)–treated syringes. The blood samples were centrifuged at 1000g for 15 min, and the plasma was collected and stored at −20°C before analysis. The AST and ALT levels in plasma were analyzed by Cerba Vet, France.
In a separate experiment, 24 or 48 hours after injection, rats were deeply anesthetized by Dolethal. Liver, kidneys, spleen, heart, and lungs were then excised, fixed by 4% formaldehyde, paraffin-embedded, and cut into 5-μm-thick sections. Hematoxylin and eosin staining was performed on all the organs for analysis of the morphology (Zeiss).
Statistical analyses
All values are expressed as means ± SEM, and statistical analyses were made with GraphPad Prism 6 software (San Diego, CA, USA). A two-way analysis of variance (ANOVA) was used with or without repeated measures as appropriate (see Materials and Methods and legends to figures). The comparison between groups was performed using the Bonferroni post hoc test. A one-way ANOVA followed by a Tukey post test was used to compare three or more groups in AUC bars. AUCs were calculated using the trapezoidal rule. For all analyses, statistical significance was set at P ≤ 0.05.
Supplementary Material
Acknowledgments
We thank V. Domergue for animal housing and care at the Animex facility, IPSIT, Châtenay-Malabry, France, and J. Mougin (Institut Galien Paris-Sud, Châtenay-Malabry, France) and G. Frébourg (Electron Microscopy Facility/FR 3631-CNRS-UPMC) are acknowledged for their contribution to cryo-TEM. We thank M. Varna-Pannerec (Institut Galien Paris-Sud, Châtenay-Malabry, France) and D. Courilleau (CIBLOT Plateforme, Châtenay-Malabry, France) for help concerning the histological study. C. Dejean (BioCIS, Châtenay-Malabry, France) is acknowledged for help with the nuclear magnetic resonance interpretations. Funding: J.F. is a fellow of the Chinese Scholarship Council (CSC). Part of this work was supported by the RBUCE-UP grant agreement (no. 00001002483/78) between the ERC and Université Paris-Sud, by the ERC under the Framework Program FP7/2007-2013 (grant agreement no. 249835), and by the Centre National de la Recherche Scientifique. UMR 8612 (P.C. team) is a member of the laboratory of excellence NANOSACLAY. Author contributions: P.C. and S.L.-M. were involved in planning and supervised the work. J.F. and S.L.-M. conceived and designed the bioconjugates. P.C. and S.L.-M. conceived and planned the experiments. J.F., S.L.-M., and P.C. developed the methodology. M.H. and A.G. developed the experimental design for algesimetry and F.C. attended meetings. J.F. carried out the experiments. A.G. participated in algesimetry experiments, and S.M. and C.C. participated in biodistribution studies. P.C., J.F., and S.L.-M. contributed to the analysis and interpretation of the results. M.H. contributed to the interpretation of the results of the behavioral study. J.F. wrote the manuscript (in consultation with S.L.-M. and P.C.). S.L.-M. and P.C. revised the manuscript. M.H. and A.G. revised the nociceptive behavioral study part. S.M. and C.C. participated in the manuscript revision. Competing interests: P.C., J.F., and S.L.-M. are inventors on a European Application patent related to this work filed by CNRS and the University of Paris-Sud (no. 18306002.9, filed 23 July 2018). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaau5148/DC1
Supplementary Text
Fig. S1. Synthesis of LENK-SQ-Diox.
Fig. S2. Synthesis of LENK-SQ-Dig.
Fig. S3. Synthesis of LENK-SQ-Am.
Fig. S4. 1H spectrum of LENK-SQ bioconjugates.
Fig. S5. 13C spectrum of LENK-SQ bioconjugates.
Fig. S6. Size and zeta potential of LENK-SQ NPs kept at +4°C.
Fig. S7. Hydrolysis of LENK or LENK-SQ-Am NPs in the presence of serum.
Fig. S8. In vitro colloidal stability of LENK-SQ-Am NPs in mouse serum.
Fig. S9. Biodistribution of fluorescent LENK-SQ-Am NPs or control fluorescent dye solution in mice with or without inflamed paw.
Fig. S10. Toxicity study of LENK-SQ-Am NPs upon systemic administration.
REFERENCES AND NOTES
- 1.Goldberg D. S., McGee S. J., Pain as a global public health priority. BMC Public Health 11, 770 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kieffer B. L., Gavériaux-Ruff C., Exploring the opioid system by gene knockout. Prog. Neurobiol. 66, 285–306 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Adams J. M., Increasing naloxone awareness and use: The role of health care practitioners. JAMA 319, 2073–2074 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Anna J., Jakub F., Tomasz J., Opioid receptors and their ligands. Curr. Top. Med. Chem. 4, 1–17 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Contet C., Kieffer B. L., Befort K., Mu opioid receptor: A gateway to drug addiction. Curr. Opin. Neurobiol. 14, 370–378 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Kiritsy-Roy J. A., Marson L., Van Loon G. R., Sympathoadrenal, cardiovascular and blood gas responses to highly selective mu and delta opioid peptides. J. Pharmacol. Exp. Ther. 251, 1096–1103 (1989). [PubMed] [Google Scholar]
- 7.Tavani A., Petrillo P., La Regina A., Sbacchi M., Role of peripheral mu, delta and kappa opioid receptors in opioid-induced inhibition of gastrointestinal transit in rats. J. Pharmacol. Exp. Ther. 254, 91–97 (1990). [PubMed] [Google Scholar]
- 8.Dykstra L., Granger A. L., Allen R. M., Zhang X., Rice K. C., Antinociceptive effects of the selective delta opioid agonist SNC80 alone and in combination with mu opioids in the squirrel monkey titration procedure. Psychopharmacology (Berl) 163, 420–429 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Campbell D. J., Long-term neprilysin inhibition—Implications for ARNIs. Nat. Rev. Cardiol. 14, 171–186 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Goodwin D., Simerska P., Toth I., Peptides as therapeutics with enhanced bioactivity. Curr. Med. Chem. 19, 4451–4461 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Kreuter J., Petrov V. E., Kharkevich D. A., Alyautdin R. N., Influence of the type of surfactant on the analgesic effects induced by the peptide dalargin after its delivery across the blood–brain barrier using surfactant-coated nanoparticles. J. Control. Release 49, 81–87 (1997). [Google Scholar]
- 12.Chen Y.-C., Hsieh W.-Y., Lee W.-F., Zeng D.-T., Effects of surface modification of PLGA-PEG-PLGA nanoparticles on loperamide delivery efficiency across the blood–brain barrier. J. Biomater. Appl. 27, 909–922 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Lalatsa A., Schätzlein A. G., Garrett N. L., Moger J., Briggs M., Godfrey L., Iannitelli A., Freeman J., Uchegbu I. F., Chitosan amphiphile coating of peptide nanofibres reduces liver uptake and delivers the peptide to the brain on intravenous administration. J. Control. Release 197, 87–96 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Popov M., Abu Hammad I., Bachar T., Grinberg S., Linder C., Stepensky D., Heldman E., Delivery of analgesic peptides to the brain by nano-sized bolaamphiphilic vesicles made of monolayer membranes. Eur. J. Pharm. Biopharm. 85, 381–389 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Lalatsa A., Lee V., Malkinson J. P., Zloh M., Schätzlein A. G., Uchegbu I. F., A prodrug nanoparticle approach for the oral delivery of a hydrophilic peptide, leucine5-enkephalin, to the brain. Mol. Pharm. 9, 1665–1680 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Wohlfart S., Gelperina S., Kreuter J., Transport of drugs across the blood–brain barrier by nanoparticles. J. Control. Release 161, 264–273 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Stein C., Schäfer M., Machelska H., Attacking pain at its source: New perspectives on opioids. Nat. Med. 9, 1003–1008 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Maksimenko A., Dosio F., Mougin J., Ferrero A., Wack S., Reddy L. H., Weyn A.-A., Lepeltier E., Bourgaux C., Stella B., Cattel L., Couvreur P., A unique squalenoylated and nonpegylated doxorubicin nanomedicine with systemic long-circulating properties and anticancer activity. Proc. Natl. Acad. Sci. U.S.A. 111, E217–E226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandal P. K., McMurray J. S., Pd−C-induced catalytic transfer hydrogenation with triethylsilane. J. Org. Chem. 72, 6599–6601 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Betageri G. V., Vutla N. B., Banga A. K., Liposomal formulation and characterization of the opioid peptide leucine enkephalin. Pharm. Pharmacol. Commun. 3, 587–591 (2011). [Google Scholar]
- 21.Chen Y., Wang F., Benson H. A. E., Effect of formulation factors on incorporation of the hydrophilic peptide dalargin into PLGA and mPEG-PLGA nanoparticles. Biopolymers 90, 644–650 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Chernyshev V. S., Rachamadugu R., Tseng Y. H., Belnap D. M., Jia Y., Branch K. J., Butterfield A. E., Pease L. F. III, Bernard P. S., Skliar M., Size and shape characterization of hydrated and desiccated exosomes. Anal. Bioanal. Chem. 407, 3285–3301 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Hargreaves K., Dubner R., Brown F., Flores C., Joris J., A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32, 77–88 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Buller K. M., Hamlin A. S., Osborne P. B., Dissection of peripheral and central endogenous opioid modulation of systemic interleukin-1β responses using c-fos expression in the rat brain. Neuropharmacology 49, 230–242 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Sato A. K., Viswanathan M., Kent R. B., Wood C. R., Therapeutic peptides: Technological advances driving peptides into development. Curr. Opin. Biotechnol. 17, 638–642 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Couvreur P., Stella B., Reddy L. H., Hillaireau H., Dubernet C., Desmaële D., Lepêtre-Mouelhi S., Rocco F., Dereuddre-Bosquet N., Clayette P., Rosilio V., Marsaud V., Renoir J.-M., Cattel L., Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 6, 2544–2548 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Gaudin A., Yemisci M., Eroglu H., Lepetre-Mouelhi S., Turkoglu O. F., Dönmez-Demir B., Caban S., Sargon M. F., Garcia-Argote S., Pieters G., Loreau O., Rousseau B., Tagit O., Hildebrandt N., Le Dantec Y., Mougin J., Valetti S., Chacun H., Nicolas V., Desmaële D., Andrieux K., Capan Y., Dalkara T., Couvreur P., Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat. Nanotechnol. 9, 1054–1062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Büscher H. H., Hill R. C., RÖMer D., Cardinaux F., Closse A., Hauser D., Pless J., Evidence for analgesic activity of enkephalin in the mouse. Nature 261, 423–425 (1976). [DOI] [PubMed] [Google Scholar]
- 29.Viswanatha Swamy A. H. M., Patil P. A., Effect of some clinically used proteolytic enzymes on Inflammation in rats. Indian J. Pharm. Sci. 70, 114–117 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sémiramoth N., Di Meo C., Zouhiri F., Saïd-Hassane F., Valetti S., Gorges R., Nicolas V., Poupaert J. H., Chollet-Martin S., Desmaële D., Gref R., Couvreur P., Self-assembled squalenoylated penicillin bioconjugates: An original approach for the treatment of intracellular infections. ACS Nano 6, 3820–3831 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Simões M. F., Valente E., Gómez M. J. R., Anes E., Constantino L., Lipophilic pyrazinoic acid amide and ester prodrugs stability, activation and activity against M. tuberculosis. Eur. J. Pharm. Sci. 37, 257–263 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Wong P. T., Choi S. K., Mechanisms of drug release in nanotherapeutic delivery systems. Chem. Rev. 115, 3388–3432 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Oliyai R., Prodrugs of peptides and peptidomimetics for improved formulation and delivery. Adv. Drug Deliv. Rev. 19, 275–286 (1996). [Google Scholar]
- 34.Burma N. E., Leduc-Pessah H., Fan C. Y., Trang T., Animal models of chronic pain: Advances and challenges for clinical translation. J. Neurosci. Res. 95, 1242–1256 (2016). [DOI] [PubMed] [Google Scholar]
- 35.van Tamelen E. E., Curphey T. J., The selective oxidation of the terminal double bonds in squalene. Tetrahedron Lett. 3, 121–124 (1962). [Google Scholar]
- 36.Skarbek C., Lesueur L. L., Chapuis H., Deroussent A., Pioche-Durieu C., Daville A., Caron J., Rivard M., Martens T., Bertrand J.-R., Le Cam E., Vassal G., Couvreur P., Desmaele D., Paci A., Preactivated oxazaphosphorines designed for isophosphoramide mustard delivery as bulk form or nanoassemblies: Synthesis and proof of concept. J. Med. Chem. 58, 705–717 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Zimmermann M., Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16, 109–110 (1983). [DOI] [PubMed] [Google Scholar]
- 38.Winter C. A., Risley E. A., Nuss G. W., Anti-inflammatory and antipyretic activities of indomethacin, 1-(p-chlorobenzoyl)-5-methoxy-2-methyl-indole-3-acetic acid. J. Pharmacol. Exp. Ther. 141, 369–376 (1963). [PubMed] [Google Scholar]
- 39.Meller S. T., Cummings C. P., Traub R. J., Gebhart G. F., The role of nitric oxide in the development and maintenance of the hyperalgesia produced by intraplantar injection of carrageenan in the rat. Neuroscience 60, 367–374 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Brasch H., Zetler G., Caerulein and morphine in a model of visceral pain. Naunyn Schmiedebergs Arch. Pharmacol. 319, 161–167 (1982). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaau5148/DC1
Supplementary Text
Fig. S1. Synthesis of LENK-SQ-Diox.
Fig. S2. Synthesis of LENK-SQ-Dig.
Fig. S3. Synthesis of LENK-SQ-Am.
Fig. S4. 1H spectrum of LENK-SQ bioconjugates.
Fig. S5. 13C spectrum of LENK-SQ bioconjugates.
Fig. S6. Size and zeta potential of LENK-SQ NPs kept at +4°C.
Fig. S7. Hydrolysis of LENK or LENK-SQ-Am NPs in the presence of serum.
Fig. S8. In vitro colloidal stability of LENK-SQ-Am NPs in mouse serum.
Fig. S9. Biodistribution of fluorescent LENK-SQ-Am NPs or control fluorescent dye solution in mice with or without inflamed paw.
Fig. S10. Toxicity study of LENK-SQ-Am NPs upon systemic administration.